ZusammensetzungWirkstoffe
Alteplasum ADNr.
Hilfsstoffe
·In der Injektionsflasche mit Lyophilisat: Polysorbatum 80, Argininum, Acidum phosphoricum.
·In der Lösungsmittelflasche: Aqua ad iniectabilia.
Indikationen/Anwendungsmöglichkeiten1.Thrombolytische Therapie bei akutem Myokardinfarkt
2.Thrombolytische Behandlung bei Patienten mit akuter massiver Lungenembolie und hämodynamischer Instabilität
3.Thrombolytische Behandlung bei akutem ischämischem Hirnschlag
Dosierung/AnwendungDosierung
Die Gabe von Actilyse sollte durch einen mit dieser Therapie erfahrenen Arzt erfolgen und entsprechend überwacht werden.
Actilyse sollte so früh wie möglich nach Eintritt der Symptome appliziert werden.
Die rekonstituierte Lösung wird intravenös angewendet und soll unmittelbar nach der Zubereitung verabreicht werden (siehe auch «Art der Anwendung» und «Sonstige Hinweise»).
Akuter Myokardinfarkt
a) 90-Minuten-Dosierungsschema für Patienten mit akutem Myokardinfarkt, bei denen die Therapie innerhalb von 6 Stunden nach Symptomeintritt begonnen werden kann.
Bei Patienten mit einem Körpergewicht ≥65 kg:
·15 mg als intravenöser Bolus, unmittelbar gefolgt von
·50 mg als intravenöse Infusion über die ersten 30 Minuten, unmittelbar gefolgt von einer intravenösen Infusion von
·35 mg über 60 Minuten, bis die Maximaldosis von insgesamt 100 mg erreicht ist.
Bei Patienten mit einem Körpergewicht < 65 kg sollte die Gesamtdosis nach folgendem Schema gewichtsabhängig angepasst werden:
·15 mg als intravenöser Bolus, unmittelbar gefolgt von
·0,75 mg/kg Körpergewicht als intravenöse Infusion über die ersten 30 Minuten (maximal 50 mg), unmittelbar gefolgt von einer intravenösen Infusion von
·0,5 mg/kg über 60 Minuten (bis maximal 35 mg).
b) 3-Stunden-Dosierungsschema für Patienten mit akutem Myokardinfarkt, bei denen die Therapie innerhalb von 6-12 Stunden nach Symptomeintritt begonnen werden kann. Das folgende Schema entspricht dem bisher empfohlenen langsamen Ablauf der Verabreichung.
Bei Patienten mit einem Körpergewicht ≥65 kg:
·10 mg als intravenöser Bolus, unmittelbar gefolgt von
·50 mg als intravenöse Infusion über die erste Stunde, unmittelbar gefolgt von einer intravenösen Infusion von
·40 mg über 2 Stunden, bis die Maximaldosis von insgesamt 100 mg erreicht ist.
Bei Patienten mit einem Körpergewicht < 65 kg:
·10 mg als intravenöser Bolus, unmittelbar gefolgt von
·einer intravenösen Infusion über 3 Stunden bis zu einer maximalen Gesamtdosis von 1,5 mg/kg Körpergewicht.
Begleittherapie:
Es wird empfohlen die antithrombotische Begleittherapie entsprechend den derzeitigen internationalen Richtlinien zur Behandlung von Myokardinfarkt mit ST-Hebung durchzuführen.
Akute massive Lungenembolie
Bei Patienten mit einem Körpergewicht ≥65 kg:
Eine Gesamtdosis von 100 mg sollte über 2 Stunden verabreicht werden. Am meisten Erfahrungen wurden mit folgendem Dosierungsschema gesammelt:
·10 mg als intravenöser Bolus über 1-2 Minuten, unmittelbar gefolgt von
·90 mg als intravenöse Infusion über 2 Stunden bis die maximale Gesamtdosis von 100 mg erreicht ist.
Bei Patienten mit einem Körpergewicht < 65 kg:
·10 mg als intravenöser Bolus über 1-2 Minuten, unmittelbar gefolgt von
·einer intravenösen Infusion über 2 Stunden bis zu einer maximalen Gesamtdosis von 1,5 mg/kg Körpergewicht.
Begleittherapie:
Nach der Actilyse-Behandlung sollte die Heparin-Therapie einsetzen (oder fortgesetzt werden), wenn die aPTT-Werte unter dem doppelten oberen Grenzwert der Normalwerte liegen. Die Infusion sollte auf einen aPTT-Erhaltungswert von 50 – 70 Sekunden (1,5- bis 2,5-facher Referenzwert) eingestellt werden.
Akuter ischämischer Hirnschlag
Die empfohlene Gesamtdosis beträgt 0,9 mg Wirkstoff/kg Körpergewicht (insgesamt höchstens 90 mg), beginnend mit 10% der Gesamtdosis als initialer intravenöser Bolus, unmittelbar gefolgt vom Rest der Gesamtdosis als intravenöse Infusion über 60 Minuten.
Die Behandlung muss so früh wie möglich innerhalb von 4,5 Stunden nach Symptomeintritt eingeleitet werden (siehe «Warnhinweise und Vorsichtsmassnahmen»). Vor der Behandlung muss durch geeignete bildgebende Verfahren (z.B. Computertomographie des Schädels oder ein anderes bildgebendes Verfahren, das in der Lage ist, eine Blutung nachzuweisen) eine intrakranielle Blutung ausgeschlossen werden. Die Wirkung der Behandlung ist zeitabhängig. Das bedeutet, dass eine frühzeitigere Behandlung die Wahrscheinlichkeit für einen günstigen Verlauf erhöht (siehe auch «Kontraindikationen» und «Warnhinweise und Vorsichtsmassnahmen»).
|
DOSIERUNGSTABELLE BEI AKUTEM ISCHÄMISCHEM HIRNSCHLAG
Bei Anwendung der empfohlenen Standardkonzentration von 1 mg/ml entspricht das
anzuwendende Volumen (ml) der empfohlenen Dosierung (mg)
| |
Gewicht
(kg)
|
Gesamtdosis
(mg)
|
Bolusdosis
(mg)
|
Infusionsdosis*
(mg)
| |
40
|
36.0
|
3.6
|
32.4
| |
42
|
37.8
|
3.8
|
34.0
| |
44
|
39.6
|
4.0
|
35.6
| |
46
|
41.4
|
4.1
|
37.3
| |
48
|
43.2
|
4.3
|
38.9
| |
50
|
45.0
|
4.5
|
40.5
| |
52
|
46.8
|
4.7
|
42.1
| |
54
|
48.6
|
4.9
|
43.7
| |
56
|
50.4
|
5.0
|
45.4
| |
58
|
52.2
|
5.2
|
47.0
| |
60
|
54.0
|
5.4
|
48.6
| |
62
|
55.8
|
5.6
|
50.2
| |
64
|
57.6
|
5.8
|
51.8
| |
66
|
59.4
|
5.9
|
53.5
| |
68
|
61.2
|
6.1
|
55.1
| |
70
|
63.0
|
6.3
|
56.7
| |
72
|
64.8
|
6.5
|
58.3
| |
74
|
66.6
|
6.7
|
59.9
| |
76
|
68.4
|
6.8
|
61.6
| |
78
|
70.2
|
7.0
|
63.2
| |
80
|
72.0
|
7.2
|
64.8
| |
82
|
73.8
|
7.4
|
66.4
| |
84
|
75.6
|
7.6
|
68.0
| |
86
|
77.4
|
7.7
|
69.7
| |
88
|
79.2
|
7.9
|
71.3
| |
90
|
81.0
|
8.1
|
72.9
| |
92
|
82.8
|
8.3
|
74.5
| |
94
|
84.6
|
8.5
|
76.1
| |
96
|
86.4
|
8.6
|
77.8
| |
98
|
88.2
|
8.8
|
79.4
| |
100+
|
90.0
|
9.0
|
81.0
| |
*angewendet in einer Konzentration von 1 mg/ml über 60 min
|
Begleittherapie:
Sicherheit und Wirksamkeit dieses Dosierungsregimes in Kombination mit Heparin oder Thrombozytenaggregationshemmern wie Acetylsalicylsäure innerhalb der ersten 24 Stunden nach Symptombeginn wurde nicht genügend untersucht. Daher sollte innerhalb der ersten 24 Stunden nach der Behandlung mit Actilyse wegen eines erhöhten Blutungsrisikos die Gabe von intravenösem Heparin oder Thrombozytenaggregationshemmern wie Acetylsalicylsäure vermieden werden. Sofern Heparin anderweitig indiziert erscheint (z.B. zur Prophylaxe von tiefen Beinvenenthrombosen), darf die Dosis 10'000 I.E. täglich subkutan nicht überschreiten.
Spezielle Dosierungsanweisungen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Actilyse bei Kindern und Jugendlichen ist bisher noch nicht erwiesen.
Actilyse ist zur Therapie des akuten Schlaganfalls bei Kindern und Jugendlichen unter 16 Jahren kontraindiziert (siehe «Kontraindikationen»). Für Jugendliche über 16 Jahren siehe «Warnhinweise und Vorsichtsmassnahmen».
Ältere Patienten
Zur Therapie des akuten Schlaganfalls bei Erwachsenen, die älter als 80 Jahre sind siehe «Warnhinweise und Vorsichtsmassnahmen».
Patienten mit eingeschränkter Nieren- oder Leberfunktion
Es liegen keine klinischen Daten vor.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Art der Anwendung
Die rekonstituierte Lösung sollte intravenös angewendet werden und ist für die unmittelbare Anwendung bestimmt.
Für die Anleitung zur Rekonstitution der Lösung und Anwendung bitte auch die Angaben in der Rubrik «Sonstige Hinweise» unter «Hinweise für die Handhabung» beachten.
KontraindikationenActilyse ist kontraindiziert bei:
·Patienten mit bekannter Überempfindlichkeit gegenüber dem Wirkstoff Alteplase oder einem der Hilfsstoffe.
·Fällen, in denen ein erhöhtes Blutungsrisiko besteht; dies gilt insbesondere bei:
·manifester oder während der letzten 6 Monate eingetretener schwerer oder gefährlicher Blutung, bekannter hämorrhagischer Diathese
·Patienten, die eine Therapie mit oralen Antikoagulantien, die mit klinisch relevanten gerinnungshemmenden Wirkungen assoziiert sind, erhalten wie z.B. Acenocoumarol oder Phenprocoumon mit einem INR-Wert >1,3
·jedem Hinweis auf Schädel-Hirn-Trauma in den vergangenen 3 Monaten, intrakranieller oder spinaler Chirurgie oder anderen Affektionen des ZNS wie intrakranieller Neoplasie, arteriovenöser Missbildung oder Aneurysma
·früher erlittener Hirnblutung oder aktuellem Verdacht auf Hirnblutung
·subarachnoidaler oder Verdacht auf subarachnoidale Blutung
·unkontrollierter schwerer arterieller Hypertonie
·grösserem chirurgischem Eingriff oder Trauma während der letzten 10 Tage (einschliesslich möglichem Trauma im Zusammenhang mit dem aktuellen Infarkt)
·kurz zurückliegender (weniger als 10 Tage) traumatischer äusserlicher Herzmassage, Entbindung, oder Punktion eines nicht abdrückbaren Blutgefässes (z.B. Vena subclavia oder jugularis)
·schwerer Lebererkrankung einschliesslich Leberversagen, Zirrhose, Pfortaderhochdruck (Ösophagusvarizen) und aktiver Hepatitis
·bakterieller Endokarditis, Perikarditis.
·akuter Pankreatitis.
·Dokumentierter ulzerativer Magendarmerkrankung in den letzten 3 Monaten, arteriellen Aneurysmen, arteriellen/venösen Missbildungen.
·Neoplasma mit erhöhtem Blutungsrisiko.
Bei akutem Herzinfarkt oder akuter massiver Lungenembolie ist zudem folgende Kontraindikation zu beachten:
·Hämorrhagischer Hirnschlag oder Hirnschlag unbekannter Genese zu irgendeiner Zeit
·Ischämischer Hirnschlag oder transitorische ischämische Attacke (TIA) in den letzten 6 Monaten, ausgenommen der aktuelle akute Hirnschlag (aufgetreten innerhalb der letzten 4.5 Stunden)
Beim akuten ischämischen Hirnschlag sind zusätzlich und insbesondere folgende Kontraindikationen zu beachten:
·Patienten, bei denen der Zeitpunkt des Einsetzens der Symptome länger als 4.5 Stunden vor dem Infusionsstart zurückliegt oder unbekannt ist.
·Früher erlittener Hirnschlag
·Symptomatik die vor Beginn der Infusion mild ist oder sich rasch verbessert hat.
·Klinisch nachgewiesener schwerer Hirnschlag (z.B. NIHSS >25) und/oder bildtechnisch nachgewiesen.
·Bei Kindern und Jugendlichen unter 16 Jahren (für Jugendliche über 16 Jahren siehe «Warnhinweise und Vorsichtsmassnahmen»).
·Bei epileptischem Anfall zu Beginn des Hirnschlags ist vor einer allfälligen Thrombolyse der Verschluss des der Klinik entsprechenden Hirngefässes neuroradiologisch nachzuweisen, um nicht Patienten mit isolierter postiktaler Parese, jedoch ohne Hirnschlag, unnötig der Gefahr einer Thrombolyse auszusetzen.
·Dies gilt auch bei Verdacht auf Hypo- oder Hyperglykämie oder andere metabolische Störung, die ein fokal betontes neurologisches Defizit erklären kann.
·Gabe von Heparin innerhalb von 48 Stunden vor Symptombeginn und verlängerte aktivierte partielle Thromboplastinzeit (aPTT) bei Spitaleintritt.
·Thrombozyten <100'000/mm3.
·Zu Beginn der Thrombolyse soll eine Blutprobe zwecks Gerinnungsbestimmung entnommen und die Actilyseinfusion gestartet werden. Ergeben die Resultate eine Prothrombinzeit (PT) über 15 Sekunden oder eine verlängerte aPTT, sollte die Infusion abgebrochen werden.
·Systolischer Blutdruck >185 mmHg oder diastolischer Blutdruck >110 mmHg, bzw. Notwendigkeit einer aggressiven Behandlung, um diese Werte zu unterschreiten.
·Blutzuckerwerte <50 mg/dl oder >400 mg/dl (<2.8 mmol/l oder >22.2 mmol/l).
·Die angegebenen Maximaldosen dürfen nicht überschritten werden, da sonst das Risiko intrakranieller Blutungen ansteigt.
Warnhinweise und VorsichtsmassnahmenActilyse steht zur Behandlung von akutem Myokardinfarkt, akuter massiver Lungenembolie und akutem ischämischem Hirnschlag in unterschiedlichen Dosierstärken zur Verfügung.
Die geeignete Dosisstärke von Actilyse muss sorgfältig entsprechend der vorgesehenen Anwendung gewählt werden. Die Dosisstärke 2 mg von Actilyse Cathflo 2 mg ist nicht für die Anwendung bei akutem Myokardinfarkt, akuter massiver Lungenembolie oder akutem ischämischem Hirnschlag indiziert (wegen des Risikos einer massiven Unterdosierung). Bei diesen Anwendungsgebieten sind nur die Dosisstärken 10 mg, 20 mg und 50 mg von Actilyse indiziert.
Es wird empfohlen, dass die Behandlung mit Actilyse unter Reanimationsbedingungen erfolgt.
Hypersensitivität
Immunvermittelte Überempfindlichkeitsreaktionen in Verbindung mit der Gabe von Actilyse können durch den Wirkstoff Alteplase oder einen der Hilfsstoffe (siehe Kontraindikationen) hervorgerufen werden.
Es wurde keine anhaltende Bildung von Antikörpern gegen das rekombinante humane gewebespezifische Plasminogenaktivator-Molekül nach der Behandlung beobachtet. Es liegen keine systematischen Erfahrungen zur erneuten Gabe von Actilyse vor.
Ausserdem besteht ein Risiko für nicht immunologisch vermittelte Überempfindlichkeitsreaktionen.
Angioödeme sind die am häufigsten beobachtete Überempfindlichkeitsreaktion im Zusammenhang mit Actilyse. Dieses Risiko kann bei der Indikation «akuter ischämischer Hirnschlag» und/oder durch die begleitende Behandlung mit ACE-Hemmern erhöht sein (siehe Abschnitt «Interaktionen»).
Für alle Indikationen sind die Patienten während und bis zu 24 Stunden nach der Infusion im Hinblick auf Angioödeme zu überwachen.
Im Fall einer schweren Überempfindlichkeitsreaktion (z.B. Angioödem) sollte unverzüglich die Infusion beendet und eine geeignete Behandlung (ggf. inklusive Intubation) eingeleitet werden.
Blutungen
Das Risiko intrazerebraler Blutungen ist bei älteren Patienten erhöht. Da der therapeutische Nutzen allerdings ebenfalls gerade bei älteren Patienten höher sein kann, muss eine sehr sorgfältige Risiko-Nutzen-Abwägung erfolgen.
Wie bei allen Thrombolytika ist bei der Anwendung von Actilyse der erwartete therapeutische Nutzen sehr sorgfältig gegen allfällige Risiken abzuwägen; dies gilt insbesondere bei
·Hämorrhagischer Retinopathie, z.B. bei Diabetes (Sehstörungen können auf eine hämorrhagische Retinopathie hinweisen) oder anderen hämorrhagischen Augenleiden.
·kleineren frischen Traumen wie Biopsien, Punktion grösserer Gefässe, intramuskulären Injektionen, Herzmassage zur Reanimation,
·Situationen mit erhöhter Blutungsgefahr, die im Abschnitt «Kontraindikationen» nicht erwähnt sind.
Bei Patienten, die mit oralen Antikoagulantien vortherapiert sind, muss der Gerinnungsstatus vor Beginn der Actilyse Therapie bestimmt werden. Eine Therapie mit Actilyse kann in Betracht gezogen werden, wenn keine klinisch relevante gerinnungshemmende Wirkung nachgewiesen werden kann.
Der Gebrauch von starren Kathetern und unnötige Manipulation des Patienten sind zu vermeiden, um Gefässe nicht zu verletzen.
Sollte eine bedrohliche Blutung (insbesondere Hirnblutung) auftreten, so ist die fibrinolytische Therapie abzubrechen und eine begleitende Heparingabe ist sofort zu beenden. Eine Substitution der Gerinnungsfaktoren ist allerdings wegen der kurzen Halbwertszeit von Actilyse und wegen des geringen Einflusses auf die systemischen Gerinnungsfaktoren im Allgemeinen nicht notwendig. Bei den meisten Patienten, die eine Blutung erleiden, genügt es, die Thrombolyse- und Antikoagulantien-Therapie abzusetzen, den Volumenverlust auszugleichen und die Blutgefässe zu komprimieren. Falls Heparin innerhalb von 4 Stunden vor Blutungsbeginn oder noch anschliessend gegeben wurde, ist die Gabe von Protamin zu erwägen. Bei den wenigen Patienten, die auf diese konservativen Massnahmen nicht ansprechen, kann eine sorgfältige Anwendung von Transfusionspräparaten angezeigt sein. Zu erwägen sind Gaben von Kryopräzipitat, frisch gefrorenem Plasma und Thrombozyten, wobei nach jeder Verabreichung die klinischen Parameter und die Laborwerte zu kontrollieren sind. Bei Verabreichung von Kryopräzipitat soll ein Fibrinogenspiegel von 1 g/l angestrebt werden. Als weitere Alternative sind Antifibrinolytika verfügbar.
Die angegebenen Maximaldosen dürfen nicht überschritten werden, da diese mit einem Anstieg des Risikos intrakranieller Blutungen einhergeht (siehe auch «Unerwünschte Wirkungen»).
Zusätzlich sind für die Therapie des akuten Myokardinfarkts und der akuten massiven Lungenembolie die folgenden Vorsichtsmassnahmen und Warnhinweise zu beachten:
·Systolischer Blutdruck >160 mmHg (siehe «Kontraindikationen»).
Zusätzlich sind für die Therapie des akuten Myokardinfarkts die folgenden Vorsichtsmassnahmen und Warnhinweise zu beachten:
·Koronare Thrombolyse kann Arrhythmien assoziiert mit Reperfusion auslösen. Reperfusionsarrhythmien können zum Herzstillstand führen, lebensbedrohlich sein und eine konventionelle antiarrhythmische Therapie erfordern.
·Der gleichzeitige Gebrauch von GPIIb/IIIa Rezeptorantagonisten erhöht das Blutungsrisiko.
·Bei Patienten mit einem Linksherzthrombus kann der Gebrauch von Thrombolitika das Risiko thromboembolistischer Ereignisse erhöhen (z.B. Mitralstenose, Vorhofflimmern).
Zusätzlich sind für die Therapie des akuten ischämischen Hirnschlags die folgenden Vorsichtsmassnahmen und Warnhinweise zu beachten:
Blutungen
Die intrazerebrale Blutung stellt die häufigste Nebenwirkung dar (bis zu 15% der Patienten), jedoch ohne einen relevanten Anstieg von Gesamtmorbidität oder Gesamtmortalität in einer Studie mit 624 Patienten (NINDS Studie).
Verglichen mit anderen Anwendungsgebieten tragen Patienten mit einem akuten ischämischen Hirnschlag, der mit Actilyse behandelt wird, ein signifikant höheres Risiko intrakranieller Blutungen, zumal die Blutungen vorwiegend in das vom Infarkt betroffene Gebiet hinein erfolgen. Dies gilt insbesondere unter folgenden Bedingungen:
·alle Situationen, die unter «Kontraindikationen» aufgeführt sind, sowie ganz allgemein alle Situationen, die ein hohes Blutungsrisiko einschliessen,
·Später Behandlungsbeginn
·Patienten, die mit Acetylsalicylsäure (ASS) vorbehandelt sind, können ein höheres intrakranielles Blutungsrisiko haben, besonders wenn die Behandlung mit Actilyse sich verzögert.
·verglichen mit jüngeren Patienten ist der Behandlungserfolg einer Thrombolyse bei Patienten in einem fortgeschrittenen Alter (über 80 Jahre) unabhängig von der Behandlung möglicherweise etwas schlechter und es kann im Zuge der Thrombolyse ein höheres Risiko für intrazerebrale Blutungen bestehen. Im Allgemeinen bleibt das Nutzen-Risiko-Verhältnis einer Thrombolyse bei Patienten in fortgeschrittenem Alter positiv. Der Nutzen einer Thrombolyse bei Patienten mit akutem ischämischem Hirnschlag sollte individuell gegen die erwarteten Risiken abgewogen werden.
Die Behandlung der Patienten soll nicht später als 4.5 Stunden nach Einsetzen der Symptome begonnen werden (siehe «Kontraindikationen»), da sich sonst ein ungünstiges Nutzen-Risiko-Verhältnis ergibt, das sich wie folgt erklärt:
·die erwünschten Effekte der Behandlung nehmen mit der Zeit ab,
·besonders bei Patienten, die zuvor mit ASS behandelt wurden, steigt die Sterblichkeitsrate,
·das Risiko symptomatischer Blutungen erhöht sich.
Überwachung des Blutdrucks
Die Überwachung des Blutdrucks während, sowie bis zu 24 Stunden nach der Verabreichung des Arzneimittels ist sinnvoll. Eine intravenöse Hochdruckbehandlung wird bei einem systolischen Blutdruck über 180 mmHg bzw. einem diastolischen Blutdruck über 105 mmHg empfohlen.
Spezielle Patientengruppen mit einem verminderten Nutzen gegenüber den Risiken
Der therapeutische Nutzen ist bei Patienten mit einem Hirnschlag in der Vorgeschichte (siehe «Kontraindikationen») oder einem schlecht eingestellten bzw. unbehandelten Diabetes von vornherein vermindert, dennoch ist auch bei diesen Patienten das Nutzen-Risiko-Verhältnis als positiv anzusehen.
Bei Patienten mit ausgedehnten Infarkten (z.B. mehr als 1/3 des Versorgungsgebiets der mittleren Gehirnarterie, Masseneffekt, Verschiebung der Mittellinie) besteht eine grössere Gefahr eines ungünstigen Ausgangs einschliesslich schwerwiegender Blutungen und des Todes. Für diese Patienten sollte das Nutzen-Risiko-Verhältnis besonders eingehend erwogen werden.
Bei Schlaganfall-Patienten sinkt die Wahrscheinlichkeit für einen günstigen Ausgang mit der Zeitspanne, die zwischen Behandlungsbeginn und Auftreten der Symptome vergangen ist, zunehmendem Lebensalter, zunehmendem Schweregrad des Schlaganfalls sowie erhöhtem Blutglucosespiegel zum Zeitpunkt der stationären Aufnahme; die Wahrscheinlichkeit für bleibende schwerwiegende Behinderungen und Tod oder symptomatische intrakranielle Blutungen steigen dabei unabhängig von der Art der Behandlung.
Gehirnödem
Die Reperfusion des ischämischen Gebietes kann in der Infarktzone ein Gehirnödem auslösen. Patienten mit langjährig ungenügend behandelter arterieller Hypertonie, solche mit Vorhofflimmern oder mit Leukenzephalopathie, weisen möglicherweise ein erhöhtes Risiko für einen Hirnschlag mit Behinderung oder Todesfolge und/oder mittlerer bis schwerer Gehirnblutung auf. Bei diesen Patienten soll die Indikation zur Thrombolyse besonders sorgfältig gegen die Risiken abgewogen werden.
Patienten mit Hirnschlag und deren Angehörige sollen über Risiken und potentiellen Nutzen einer Thrombolyse sehr sorgfältig informiert werden.
Kinder und Jugendliche
Zur Anwendung von Actilyse bei Kindern und Jugendlichen liegen bisher nur begrenzte Erfahrungen vor.
Bei Jugendlichen ≥16 Jahre sollte der Nutzen sorgfältig individuell gegen die Risiken abgewogen werden.
Jugendliche ≥16 Jahre sollten gemäss den Behandlungsleitlinien für Erwachsene behandelt werden, nachdem ein thromboembolischer arterieller ischämischer Schlaganfall bestätigt wurde (Auschluss von «Stroke Mimics»).
InteraktionenWirkstoffe mit Auswirkungen auf die Blutgerinnung/Thrombozytenfunktion
Kumarinderivate, Thrombozytenaggregationshemmer, Heparin und andere Arzneimittel, welche die Blutgerinnung beeinflussen, können das Blutungsrisiko vor, während und nach der Actilyse Therapie, erhöhen und sollten in den ersten 24 Stunden nach der Behandlung eines akuten ischämischen Schlaganfalls vermieden werden (siehe «Kontraindikationen»).
Actilyse ist ein Enzym, das unter In-vitro-Bedingungen in den Blutproben in pharmakologischen Konzentrationen aktiv bleibt. Dies kann zum Abbau von Fibrinogen in den Blutproben führen, der teilweise durch Zugabe von Aprotinin gehemmt werden kann (150-200 Einheiten/ml). Daher können Gerinnungsteste oder Messungen der fibrinolytischen Aktivität während oder kurz nach der Actilyse-Therapie nicht sicher verwendbare Werte ergeben.
ACE-Hemmer
Die gleichzeitige Behandlung mit ACE-Hemmern kann das Risiko für eine Überempfindlichkeitsreaktion erhöhen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Schwangerschaft, StillzeitSchwangerschaft
Die Erfahrungen mit der Anwendung von Actilyse bei schwangeren Frauen ist begrenzt. Tierexperimentelle Untersuchungen mit höheren als beim Menschen verabreichten Dosen haben neben den bekannten pharmakologischen Wirkungen, fötale Unreife und/oder Embryotoxizität gezeigt. Im Falle einer akuten lebensbedrohenden Erkrankung ist der Nutzen gegen das potentielle Risiko abzuwägen.
Stillzeit
Es ist nicht bekannt, ob Alteplase in die Muttermilch übergeht.
Vorsicht ist geboten, wenn Actilyse einer stillenden Frau verabreicht wird, und es muss entschieden werden, ob das Stillen in den ersten 24 Stunden nach der Verabreichung von Actilyse unterbrochen werden sollte.
Fertilität
Klinische Daten zur Fertilität mit Alteplase liegen nicht vor. Tierstudien mit Alteplase haben keine negativen Auswirkungen auf die Fertilität gezeigt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenNicht zutreffend
Unerwünschte WirkungenDie unerwünschten Wirkungen bei den Indikationen Myokardinfarkt, akuter ischämischer Hirnschlag und Lungenembolie wurden aufgrund den Daten der Assent-2-Studie (Studie mit 8'299 Patienten welche mit Actilyse behandelt wurden) ausgewertet. Bei der Indikation akuter ischämischer Hirnschlag basieren die Häufigkeiten der Nebenwirkungen intrakranielle Blutungen auf den Daten der NINDS- und ECASS III-Studien (Studien mit 312 bzw 418 Patienten welche in einem Intervall von 0-4.5 Stunden mit Actilyse behandelt wurden).
Die häufigsten unerwünschten Wirkungen bei der Anwendung von Actilyse sind schwere Blutungen die zu lebensbedrohlichen Situationen, bleibender Behinderung oder zum Tod führen können.
Neurologische Symptome wie Somnolenz, Aphasie, Hemiparese, Konvulsion können mit intrakraniellen Blutungen assoziiert sein.
Die verwendeten Frequenzen sind wie folgt definiert: «Sehr häufig» (≥1/10), «häufig» (≥1/100 bis <1/10), «gelegentlich» (≥1/1'000 bis <1/100), «selten» (≥1/10'000 bis <1/1'000), «sehr selten» (<1/10'000).
Erkrankungen des Immunsystems
Selten: anaphylaktoide Reaktionen (wie Hautausschlag, Urtikaria, Bronchospasmen, Hypotonie, Angio-Ödem, Schock).
Erkrankungen des Nervensystems
Die folgenden Frequenzen sind spezifisch für die Indikationen:
·akuter Myokardinfarkt und akute massive Lungenembolie:Häufig: intrakranielle Blutungen (wie Gehirnblutung, zerebrales Hämatom, hämorrhagischer Hirnschlag, hämorrhagische Transformation des Hirninsultes, intrakranielles Hämatom, subarachnoidale Blutung).
·akuter ischämischer Hirnschlag:Sehr häufig: intrakranielle Blutungen (NINDS: 15.4%; ECASS III: 14.1%) wie Gehirnblutung, zerebrales Hämatom, hämorrhagischer Hirnschlag, hämorrhagische Transformation des Hirninsultes, intrakranielles Hämatom, subarachnoidale Blutung.
Die Frequenzen dieser Nebenwirkungen basieren auf den Daten der NINDS- und ECASS III-Studien.
Augenerkrankungen
Selten: Blutungen im Auge, insbesondere bei Patienten mit einer diabetischen Retinopathie.
Herzerkrankungen
Selten: perikardiale Blutung.
Die folgenden unerwünschten Wirkungen und Frequenzen sind spezifisch für die Indikation akuter Myokardinfarkt:
Gelegentlich: Arrhythmien während der Reperfusion (wie Arrhythmie, Extrasystolen, Vorhofflimmern/-flattern, AV-Block I bis zum kompletten AV-Block, Bradykardie, Tachykardie, ventrikuläre Arrhythmie, Kammerflimmern, ventrikuläre Tachykardie, elektromechanische Entkopplung EMD). Reperfusionsarrhythmien können zu einem Herzstillstand führen, lebensbedrohlich sein und eine konventionelle antiarrhythmische Therapie benötigen.
Gefässerkrankungen
Sehr häufig: Blutungen wie Hämatom (17.1%).
Selten: thrombotische Embolie.
Unbekannte Frequenz*: Blutungen parenchymatöser Organe (wie Leberblutung).
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Blutungen in den Atemwegen (wie Rachenblutungen).
Gelegentlich: Hämoptyse, Nasenbluten.
Selten: Lungenblutung.
Erkrankungen des Gastrointestinaltrakts
Häufig: Blutungen im gastrointestinalen Bereich (wie Magenblutung, Blutung eines Magengeschwürs, rektale Blutung, Hämatemesis, Melaena, Blutung im Mund, Zahnfleischbluten).
Selten: Blutungen im retroperitonealen Bereich (wie retroperitoneales Hämatom), Übelkeit.
Unbekannte Frequenz*: Erbrechen.
Ausserdem können Übelkeit und Erbrechen als Symptome eines Myokardinfarkts auftreten.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Ekchymosen.
Erkrankungen der Nieren und Harnwege
Häufig: Blutungen im urogenitalen Bereich (wie Hämaturie, Harnwegsblutung).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Blutungen an der Injektionsstelle bzw. Punktionsstelle wie Katheter-Hämatom, Blutung an der Katheterstelle.
Untersuchungen
Gelegentlich: Blutdrucksenkung.
Unbekannte Frequenz*: Erhöhung der Körpertemperatur.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Unbekannte Frequenz*: Fettembolien (Embolien durch Cholesterinkristalle).
Chirurgische und medizinische Eingriffe
Unbekannte Frequenz*: Notwendigkeit der Gabe einer Bluttransfusion.
* Diese unerwünschten Reaktionen stammen aus Spontanmeldungen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungSymptome
Bei Überschreiten der empfohlenen Höchstdosis steigt das Risiko für intrakranielle Blutungen an.
Bei Überdosierung kann es zu einer klinisch relevanten Verminderung von Fibrinogen und anderen Blutgerinnungskomponenten kommen.
Behandlung
In der Regel kann in solch einem Falle die physiologische Neusynthese dieser Faktoren nach Absetzen der Actilyse-Therapie abgewartet werden. Tritt eine schwere Blutung auf, wird die Gabe von frisch gefrorenem Plasma oder Frischblut empfohlen und sofern notwendig, können synthetische Antifibrinolytika verabreicht werden.
Eigenschaften/WirkungenATC-Code
B01AD02
Pharmakotherapeutische Gruppe: Antithrombotisches Mittel.
Wirkungsmechanismus
Der Wirkstoff von Actilyse ist ein gentechnologisch hergestelltes humanes Glykoprotein, das Plasminogen direkt zu Plasmin aktiviert. Bei intravenöser Verabreichung bleibt Actilyse im Kreislauf relativ inaktiv, bis es an Fibrin bindet. Durch die Bindung an Fibrin wird Actilyse aktiviert, induziert die Umwandlung von Plasminogen zu Plasmin und führt damit zur Auflösung des Fibringerinnsels.
Pharmakodynamik
Aufgrund seiner relativen Fibrinspezifität bewirkt Actilyse bei Verabreichung einer Gesamtdosis von 100 mg eine mässige Senkung der zirkulierenden Fibrinogen-Werte auf ca. 60% nach 4 Stunden, wobei diese nach 24 Stunden wieder auf 80% ansteigen. Plasminogen und Alpha-2-Antiplasmin fallen nach 4 Stunden auf 20% bzw. 35% ab, und steigen nach 24 Stunden wieder auf über 80% an. Eine ausgeprägte und langdauernde Senkung des Fibrinogenspiegels im Blut ist nur bei wenigen Patienten zu beobachten.
Klinische Wirksamkeit
Patienten mit akutem Myokardinfarkt
Bei Patienten mit akutem Myokardinfarkt wurden zwei Dosisschemata von Actilyse untersucht. Es erfolgte keine vergleichende Beurteilung der Wirksamkeit dieser beiden Schemata.
Beschleunigte Infusion bei Patienten mit akutem Myokardinfarkt
Die beschleunigte Infusion von Actilyse wurde im Rahmen einer internationalen, multizentrischen Studie (GUSTO) untersucht, in der 41021 Patienten mit akutem Myokardinfarkt randomisiert vier Thrombolyse-Schemata zugeteilt wurden. Die Verabreichung von 100 mg Actilyse über 90 Minuten, in Kombination mit einer gleichzeitigen intravenösen Heparin-Infusion, hatte eine geringere Mortalität nach 30 Tagen (6,3 %) zur Folge als die Verabreichung von Streptokinase, 1,5 Millionen IE über 60 Minuten mit subkutanem oder intravenösem Heparin (7,3 %). Die absolute Reduktion der 30-Tages-Mortalität unter Actilyse gegenüber Streptokinase von 1 % war statistisch signifikant (p = 0,001).
Mit Actilyse behandelte Patienten wiesen 60 und 90 Minuten nach der Thrombolyse eine höhere Durchgängigkeitsrate von Infarkt-bezogenen Gefässen auf als mit Streptokinase behandelte Patienten. Nach 180 Minuten oder später wurden keine Unterschiede bei der Durchgängigkeitsrate beobachtet.
Eine grosse Mortalitätsstudie (ASSENT 2) mit etwa 17000 Patienten zeigte, dass Alteplase und Tenecteplase im Hinblick auf die Reduktion der Mortalität therapeutisch äquivalent sind (6,2 % für beide Behandlungen, nach 30 Tagen). Die Anwendung von Tenecteplase war mit einer signifikant niedrigeren Inzidenz von nicht-intrakraniellen Blutungen verbunden als die Anwendung von Alteplase (26,4 % versus 28,9 %, p = 0,0003). Die Verringerung des Blutungsrisikos steht wahrscheinlich im Zusammenhang mit der höheren Fibrin-Spezifität von Tenecteplase und dem gewichtsabhängigen Dosisschema.
3-stündige Infusion bei Patienten mit akutem Myokardinfarkt
In einer doppelblinden, randomisierten Studie (5013 Patienten), die Actilyse mit Placebo verglich (ASSET-Studie), zeigten Patienten, denen innerhalb von 5 Stunden nach Beginn der Symptome eines akuten Myokardinfarkts Actilyse infundiert wurde, eine Verbesserung des 30-Tages-Überlebens gegenüber mit Placebo behandelten Patienten. Nach 1 Monat betrug die allgemeinen Mortalitätsrate in der Actilyse-Behandlungsgruppe 7,2 % und in der Placebogruppe 9,8 % (p = 0,001). Dieser Vorteil in der Actilyse-Gruppe gegenüber der Placebogruppe blieb über 6 Monate erhalten (10,4 % vs. 13,1 %, p = 0,008).
In einer doppelblinden, randomisierten Studie (721 Patienten), die Actilyse mit Placebo verglich, hatten Patienten, denen innerhalb von 5 Stunden nach Symptombeginn Actilyse infundiert wurde, 10 – 22 Tage nach der Behandlung eine bessere linksventrikuläre Funktion als die Patienten der Placebogruppe, wenn die globale Ejektionsfraktion mittels Kontrastventrikulographie bestimmt wurde (50,7 % versus 48,5 %, p = 0,01). Bei den mit Actilyse behandelten Patienten fiel der Infarkt signifikant um 20 % kleiner aus [bestimmt über die kumulative freigesetzte HBD-(α-Hydroxybutyratdehydrogenase)-Aktivität] als bei den mit Placebo behandelten Patienten (p = 0,001). Mit Actilyse behandelte Patienten hatten weniger Episoden von kardiogenem Schock, Kammerflimmern und Perikarditis. Zudem war die 14-Tages-Mortalität bei den mit Actilyse behandelten Patienten auf 2,8 % verringert, gegenüber 5,7 % bei den mit Placebo behandelten Patienten (nicht signifikant). In der Actilyse Gruppe waren 6 Patienten mit dokumentierten hämorrhagischen Schlaganfall und 1 Patient mit einem ischämischen Schlaganfall, während in der Placebo Gruppe keine Schlaganfälle auftraten. Diese Daten belegen zwar nicht eindeutig eine signifikante Reduktion der Mortalität in dieser Studie, weisen aber auf eine Tendenz hin, die durch die Ergebnisse der ASSET-Studie unterstützt wird.
In einer placebokontrollierten Studie (LATE) mit 5711 Patienten mit akutem Myokardinfarkt und 6 bis 24 Stunden zurückliegendem Symptombeginn wurde die Infusion von 100 mg Actilyse über 3 Stunden mit Placebo verglichen. Unter Actilyse wurde eine nicht-signifikante Reduktion der 30-Tages-Mortalität um 14,1 % (95%-KI: 0 – 28,1 %; p > 0,05) beobachtet. In einer vom Steering Committee verlangten Überlebensanalyse zu Patienten, die innerhalb von 12 Stunden nach Symptombeginn behandelt wurden, wurde eine signifikante Reduktion der Mortalität um 25,6 % zugunsten von Actilyse (95%-KI: 6,3 – 45 %; p = 0,023) beobachtet.
Patienten mit akuter massiver Lungenembolie
In einer randomisierten Vergleichsstudie zwischen Alteplase und Urokinase bei 63 Patienten mit angiographisch dokumentierter akuter massiver Lungenembolie war in beiden Behandlungsgruppen eine signifikante Reduktion der einer durch die Lungenembolie induzierten pulmonalen Hypertonie zu beobachten. Der pulmonäre arterielle Mitteldruck (PAPm) sank in beiden Gruppen innert 12 Std. von 28±7 mmHg auf 17±6 mmHg. Die pulmonale Hämodynamik verbesserte sich unter Actilyse signifikant schneller als unter Urokinase (Behandlung x Zeit-Interaktion: p=0.0006).
Patienten mit akutem ischämischem Hirnschlag
Zum akuten ischämischen Hirnschlag wurden mehrere Studien durchgeführt. Die NINDS-Studie ist die einzige Studie ohne obere Altersbegrenzung, d.h. sie schliesst auch Patienten über 80 Jahren ein. Alle anderen Studien schlossen Patienten über 80 Jahren aus. Der Nutzen einer Thrombolyse bei Patienten mit akutem ischämischem Hirnschlag sollte individuell gegen die erwarteten Risiken abgewogen werden.
Zwei placebokontrollierte, doppelblinde Studien (NINDS t-PA Stroke Trial, Teil 1 und Teil 2) schlossen Patienten mit messbarem neurologischem Defizit ein, die innerhalb von 3 Stunden nach Symptombeginn ein Screening absolvieren und die Behandlung beginnen konnten. Vor der Behandlung wurde eine Computertomographie (CT) des Schädels angefertigt, um eine symptomatische intrakranielle Blutung auszuschliessen. Ausserdem wurden Patienten ausgeschlossen, die mit einem erhöhten Blutungsrisiko einhergehende Störungen, ein geringes neurologisches Defizit, eine sich schnell bessernde Symptomatik vor Beginn der Studienbehandlung oder einen Blutzuckerspiegel < 50 mg/dl oder > 400 mg/dl aufwiesen. Die Patienten wurden randomisiert einer Behandlung mit 0,9 mg/kg Actilyse (maximal 90 mg) oder Placebo zugeteilt. Actilyse wurde als initialer Bolus von 10 % der Dosis über 1 Minute mit anschliessender intravenöser Dauerinfusion der restlichen Dosis über 60 Minuten verabreicht.
Die initiale Studie (NINDS-Teil 1, n = 291) beurteilte die neurologische Besserung 24 Stunden nach Eintritt des Hirnschlags. Der primäre Endpunkt, der Anteil der Patienten mit einer Besserung des Punktwertes auf der Hirnschlagskala der National Institutes of Health (NIHSS-Skala) um 4 Punkte oder mehr oder vollständiger Wiederherstellung (NIHSS-Score = 0), fiel zwischen den Behandlungsgruppen nicht signifikant unterschiedlich aus. Eine sekundäre Analyse wies anhand der folgenden Skalen für die Beurteilung des Hirnschlags auf ein besseres Behandlungsergebnis nach 3 Monaten im Zusammenhang mit der Actilyse-Behandlung hin: Barthel-Index, Modifizierte Rankin-Skala (mRS), Glasgow Outcome Scale und NIHSS-Skala. In einer zweiten Studie (NINDS-Teil 2, n = 333) war das Behandlungsergebnis nach 3 Monaten der primäre Endpunkt. Ein positives Behandlungsergebnis war definiert als minimale oder keine Behinderung auf den vier zur Beurteilung des Hirnschlags eingesetzten Skalen: Barthel-Index (Punktwert ≥95), Modifizierte Rankin-Skala (Punktwert ≤1), Glasgow Outcome Scale (Punktwert = 1) und NIHSS-Skala (Punktwert ≤1). Die Odds-Ratio für ein positives Behandlungsergebnis betrug in der Actilyse-Gruppe 1,7 (95%-KI: 1,2 – 2,6). Im Vergleich zu Placebo betrug die absolute Zunahme der Anzahl der Patienten mit minimaler oder keiner Behinderung 13 % (mRS 0 – 1) (OR: 1,7; 95%-KI: 1,1 – 2,6). Darüber hinaus war auch auf anderen neurologischen Skalen und Invaliditäts-Skalen ein einheitlicher Nutzen von Actilyse zu beobachten. Sekundäre Analysen zeigten auf allen vier zur Beurteilung des Hirnschlags eingesetzten Skalen auf der Grundlage der Mediane der Punktwerte einheitlich eine funktionelle und neurologische Besserung. Diese Ergebnisse wiesen eine hohe Übereinstimmung mit den 3-Monats-Daten zur Wirkung der Behandlung in Teil 1 der Studie auf. Nach Behandlung mit Actilyse wurden signifikant mehr symptomatische intrakranielle Blutungen (gemäss NINDS-Definition) innerhalb von 36 Stunden beobachtet als unter Placebo (Actilyse 6,4 %; Placebo 0,65 %). Hingegen kam es bei den mit Actilyse-behandelten Patienten im Vergleich zu Placebo nicht zu einer erhöhten 90-Tages-Mortalität oder erhöhten Inzidenz von schwerwiegenden Behinderungen (Actilyse 20,5 %; Placebo 17,3 %).
Eine gepoolte Analyse mit 2775 Patienten aus 6 grösseren randomisierten klinischen Studien (NINDS Teil 1 und 2, zwei ECASS-Studien und ATLANTIS Teil A und B) untersuchte den Behinderungsstatus von mit Actilyse oder Placebo behandelten Patienten. In dieser Analyse nahm die Chance auf ein positives Behandlungsergebnis nach 3 Monaten zu, je kürzer die Zeit bis zur Actilyse-Behandlung gewesen war. Symptomatische intrakranielle Blutungen waren bei 5,9 % der mit Actilyse behandelten Patienten gegenüber 1,1 % der Kontrollen zu beobachten (p < 0,0001) und zeigten einen Zusammenhang mit dem Alter, nicht jedoch mit der Zeit bis zur Behandlung. Diese Analyse bietet starke Unterstützung für die Feststellung, dass eine schnelle Behandlung mit Actilyse mit einem besseren Behandlungsergebnis nach 3 Monaten verbunden ist. Darüber hinaus liefert sie Evidenz, dass das therapeutische Fenster bis auf 4,5 Stunden ausgedehnt werden kann.
In einer grossen Beobachtungsstudie (SITS-MOST: The Safe Implementation of Thrombolysis in Stroke-Monitoring Study) wurde die Sicherheit und Wirksamkeit von Actilyse bei der Behandlung von akuten Hirnschlägen innerhalb von 3 Stunden im routinemässigen klinischen Alltag bestimmt und mit den Ergebnissen randomisierter klinischer Studien (RCT) verglichen. Bei allen Patienten mussten die Bedingungen der europäischen Fachinformation (EU-SPC) für Actilyse eingehalten werden. Es wurden Behandlungs- und Verlaufsdaten von 6483 Patienten aus 285 Zentren in 14 europäischen Ländern erhoben. Primärer Endpunkt waren symptomatische intrakranielle Blutungen innerhalb von 24 Stunden und die Mortalität nach 3 Monaten. Die in der SITS-MOST-Studie beobachtete Rate von symptomatischen intrakraniellen Blutungen zum Tage 7 war der aus randomisierten Studien beschriebenen Rate ähnlich: 7,3 % (95%KI: 6,7 – 8,0) in der SITS-MOST-Studie versus 8,6 % (95%-KI: 6,1 – 11,1) in RCT. Die Mortalität betrug in der SITS-MOST-Studie 11,3 % (95%-KI: 10,5 – 12,1) gegenüber 17 % (95%-KI: 13,9 – 20,7) in RCT. Die Ergebnisse der SITS-MOST-Studie weisen darauf hin, dass die innerhalb von 3 Stunden nach Eintreten des Hirnschlags erfolgende Anwendung von Actilyse im routinemässigen klinischen Alltag ebenso sicher ist wie in klinischen Studien beschrieben.
Die ECASS-III-Studie war eine placebokontrollierte, doppelblinde Studie bei Patienten mit akutem Hirnschlag und einem Behandlungs-Zeitfenster von 3 bis 4,5 Stunden. Die Studie schloss Patienten mit messbarem neurologischem Defizit ein, bei denen die Bedingungen der europäischen Fachinformation (EU-SPC) mit Ausnahme des Zeitfensters eingehalten wurden. Nach Ausschluss einer Hirnblutung oder eines grösseren Infarkts mittels Computertomographie wurden Patienten mit akutem ischämischem Hirnschlag randomisiert und doppelblind im Verhältnis 1:1 entweder intravenöser Alteplase (0,9 mg/kg Körpergewicht) oder Placebo zugeteilt. Primärer Endpunkt war eine Behinderung nach 90 Tagen, aufgeteilt nach günstigem (modifizierte Rankin-Skala [mRS] 0 bis 1) oder ungünstigem (mRS 2 bis 6) Verlauf. Wichtigster sekundärer Endpunkt war eine globale Verlaufsanalyse anhand der Kombination aus vier neurologischen Skalen und Invaliditäts-Skalen. Sicherheitsendpunkte umfassten Mortalität, symptomatische intrakranielle Blutungen und schwerwiegende unerwünschte Ereignisse. Insgesamt wurden 821 Patienten (418 Alteplase/403 Placebo) randomisiert. Mehr der mit Alteplase (52,4 %) als der mit Placebo (45,2 %; Odds-Ratio [OR]: 1,34; 95%-KI 1,02 – 1,76; p = 0,038) behandelten Patienten erzielten ein günstiges Ergebnis. Auch bei der globalen Analyse ergab sich ein besseres Behandlungsergebnis unter Alteplase (OR: 1,28; 95%-KI: 1,00 – 1,65; p = 0,048). Die mit Alteplase behandelten Patienten wiesen eine höhere Inzidenz von (symptomatischen / nicht-Symptomatischen) intrakraniellen Blutungen auf als die mit Placebo behandelten Patienten (alle intrakraniellen Blutungen: 27,0 % versus 17,6 %, p = 0,0012; symptomatische intrakranielle Blutungen gemäss NINDS-Definition: 7,9 % versus 3,5 %, p = 0,006; symptomatische intrakranielle Blutungen gemäss ECASS-III-Definition 2.4% versus 0.2 %, p = 0.008). Die Mortalität war gering und fiel unter Alteplase (7,7 %) und Placebo nicht signifikant unterschiedlich aus (8,4 %; p = 0,681). Die Ergebnisse der ECASS-III-Studie zeigen, dass Actilyse bei Verabreichung zwischen 3 und 4,5 Stunden nach Symptombeginn den klinischen Verlauf bei Patienten mit akutem ischämischem Hirnschlag signifikant verbessert.
Die Sicherheit und Wirksamkeit von Actilyse bei der Behandlung von akuten ischämischen Hirnschlägen im Zeitfenster von bis zu 4,5 Stunden zwischen Erstsymptomen und Behandlungsbeginn (onset to treatment time, OTT) wurde anhand eines laufenden Registers zu akuten ischämischen Hirnschlägen untersucht (SITS-ISTR: The Safe Implementation of Thrombolysis in Stroke registry). Es wurden die Daten für das primäre Behandlungsergebnis und Mortalitätsdaten von 21566 Patienten, die im Zeitfenster 0 bis 3 Stunden behandelt wurden, mit den Daten von 2376 Patienten, die 3 bis 4,5 Stunden nach Eintreten des akuten ischämischen Hirnschlags (Daten von 2010) behandelt wurden verglichen. Die Inzidenz von symptomatischen intrazerebralen Blutungen (gemäss SITS-MOST-Definition) war im Zeitfenster 3 bis 4,5 Stunden etwas höher (2.2 %) als im Zeitfenster bis 3 Stunden (1.7 %). Die Mortalitätsrate nach 3 Monaten fiel für das Zeitfenster 3 bis 4,5 Stunden (12,0 %) vergleichbar hoch aus wie für das Zeitfenster 0 bis 3 Stunden (12,3 %).
Ältere Patienten (> 80 Jahren)
Metaanalysen von adjustierten Daten von 6756 Patienten, darunter auch Personen über 80 Jahren, aus neun randomisierten Studien, in denen Alteplase mit Placebo oder unverblindeten Kontrollen verglichen wurde, untersuchten das Nutzen-Risiko-Verhältnis von Alteplase bei Patienten > 80 Jahren.
Die Wirkung der Alteplase-Behandlung war bei Patienten bis 80 Jahren [mittlere Behandlungsverzögerung 4,1 Stunden: 990/2512 (39 %) mit Alteplase behandelte Patienten vs. 853/2515 (34 %) Patienten in der Kontrollgruppe erzielten ein gutes Behandlungsergebnis an Tag 90/180; OR 1,25, 95 %-KI 1,10 – 1,42] ähnlich wie bei jenen über 80 Jahren [mittlere Behandlungsverzögerung 3,7 Stunden: 155/879 (18 %) mit Alteplase behandelte Patienten vs. 112/850 (13 %) Patienten in der Kontrollgruppe erzielten ein gutes Behandlungsergebnis; OR 1,56, 95 %-KI 1,17 – 2,08].
Bei Patienten über 80 Jahren, die bis zu 3 Stunden nach dem Ereignis mit Alteplase behandelt wurden, wurde ein gutes Behandlungsergebnis bei 55/302 (18,2 %) im Vergleich zu 30/264 (11,4 %) in der Kontrollgruppe erzielt (OR 1,86, 95 %-KI 1,11 – 3,13); bei jenen Patienten, die zwischen 3 und 4,5 Stunden nach dem Ereignis mit Alteplase behandelt wurden, war das Behandlungsergebnis bei 58/342 (17,0 %) gut, im Vergleich zu 50/364 (13,7 %) in der Kontrollgruppe (OR 1,36, 95 %-KI 0,87 – 2,14).
Eine weitere Metaanalyse, die auf Basis derselben Datenbank mit einer Kohorte von Patienten, welche die Auswahlkriterien in der Fachinformation erfüllen, durchgeführt wurde, zeigte, dass die Wahrscheinlichkeit eines guten Ergebnisses nach Schlaganfall (mRS 0–1 an Tag 90/180) bis zu 4,5 Stunden nach Einsetzen der Schlaganfall-Symptome erhöht war, wobei der Nutzen einer früheren Behandlung grösser war (p-Wert für Interaktion 0,0203), und nicht vom Alter abhing (p-Wert = 0,7383).
Eine parenchymatöse Blutung Typ 2 trat innerhalb von 7 Tagen bei 231 (6,8 %) der 3391 mit Alteplase behandelten Patienten auf; in der Kontrollgruppe lag die Zahl bei 44 (1,3 %) von 3365 Patienten (OR 5,55, 95 %-KI 4,01 – 7,70).
Eine parenchymatöse Blutung Typ 2 mit tödlichem Ausgang trat innerhalb von 7 Tagen bei 91 (2,7 %) der mit Alteplase behandelten Patienten auf; in der Kontrollgruppe lag die Zahl bei 13 (0,4 %) Patienten (OR 7,14, 95 %-KI 3,98 – 12,79).
Bei Patienten über 80 Jahren, die mit Alteplase behandelt wurden, trat eine intrakranielle Blutung mit tödlichem Ausgang innerhalb von 7 Tagen bei 32/879 (3,6 %) auf, im Vergleich zu 4/850 (0,5 %) in der Kontrollgruppe (OR 7,95, 95 %-KI 2,79 – 22,60).
Unter den mit Alteplase behandelten Patienten im Alter bis einschliesslich 80 Jahren trat eine intrakranielle Blutung mit tödlichem Ausgang innerhalb von 7 Tagen bei 59/2512 Patienten (2,3 %) gegenüber 9/2515 Patienten (0,4 %) in der Kontrollgruppe auf (OR 6,93, 95%-KI 3,42 – 14,02).
Bei Patienten über 80 Jahren mit einem akuten ischämischen Schlaganfall besteht aufgrund ihres Alters und eines höheren NIHSS-Ausgangswerts ein höheres Risiko für ein schlechtes Ergebnis.
Die Behandlung mit Alteplase erhöht bei Patienten über 80 Jahren mit einem akuten ischämischen Schlaganfall die Wahrscheinlichkeit eines guten Behandlungsergebnisses (mRS-Score 0 – 1) an Tag 90/180 ohne Anstieg der Mortalität nach 90 Tagen im Vergleich zur Kontrollgruppe (267/879 [30,4 %] gegenüber 259/850 [30,5 %], OR 0,95, 95%-KI 0,76 – 1,18).
Die Mortalität nach 90 Tagen ist in der jüngeren Patientengruppe (≤80 Jahre) unabhängig von der Behandlung niedriger (341/2512 [13,6 %] für die mit Alteplase behandelten Patienten ≤80 Jahren gegenüber 297/2515 [11,8 %] für die Kontrollgruppe ≤80 Jahren, OR 1,25, 95%-KI 1,05 – 1,49).
In der älteren Population (> 80 Jahre) mit akutem ischämischem Schlaganfall, die mit Alteplase behandelt wurde, trat eine symptomatische intrakranielle Blutung bei 78/879 (8,9 %) auf, im Vergleich zu 17/850 (2 %) in der Kontrollgruppe (OR 4,80, 95%-KI 2,81 – 8,20).
In der mit Alteplase behandelten jüngeren Population (≤80 Jahre) mit akutem ischämischem Schlaganfall trat eine symptomatische intrakranielle Blutung bei 153/2512 Patienten (6,1 %) gegenüber 27/2515 Patienten (1,1 %) in der Kontrollgruppe auf (OR 6,20, 95%-KI 4,09 – 9,38).
Das SITS-ISTR-Register umfasst 8658 Patienten > 80 Jahren, die < 4,5 Stunden nach Auftreten eines Schlaganfalls behandelt wurden. Hieraus wurden die Daten von 2157 Patienten, die > 3 bis 4,5 Stunden nach Auftreten eines Schlaganfalls behandelt wurden mit denen von 6501 Patienten, die < 3 Stunden behandelt wurden, verglichen.
Die funktionelle Unabhängigkeit (mRS-Score 0 – 2) nach 3 Monaten betrug 36 bzw. 37 % (adjustierte OR 0,79, 95 %-KI 0,68 – 0,92), die Mortalität lag bei 29,0 % bzw. 29,6 % (adjustierte OR 1,10, 95 %-KI 0,95 – 1,28) und sICH (gemäss SITS-MOST-Definition) traten bei 2,7 % bzw. 1,6 % (adjustierte OR 1,62, 95 %-KI 1,12 – 2,34) auf.
Kinder und Jugendliche
Daten aus einer nicht-randomisierten Registerstudie ohne Vergleichsgruppe zu Schlaganfallpatienten im Alter von 16 – 17 Jahren mit bestätigter Alteplase-Therapie stammen aus dem SITS-ISTR (Safe Implementation of Treatments in Stroke – International Stroke Thrombolysis Register, ein unabhängiges internationales Register).
Bei einer Zwischenanalyse der laufenden Registerstudie wurden Daten von insgesamt 32 pädiatrischen Patienten mit einer bestätigten Anwendung von Alteplase in der Altersgruppe von 16–17 Jahren in das SITS-Register aufgenommen. Die mediane Alteplase-Dosis in dieser Altersgruppe betrug 0,9 mg/kg (0,83 – 0,99 mg/kg). Bei 30 von 32 Patienten wurde die Behandlung innerhalb des Zeitfensters von 4,5 Stunden nach Auftreten der ersten Schlaganfallsymptome eingeleitet (22 innerhalb von 3 h; 8 innerhalb von 3 – 4,5 h; 1 innerhalb von 5 – 5,5 h; 1 Fall: keine Angaben). Das Körpergewicht betrug 58 – 90 kg. Bei den meisten Patienten lag ein mittelschwerer oder ein mittelschwerer bis schwerer Schlaganfall mit einem medianen NIHSS-Ausgangswert von 9,0 vor (1 – 30).
Bei 24 von 32 Patienten waren mRS-Scores am Tag 90 vorhanden. Am Tag 90 wiesen 16 von 24 Patienten einen mRS-Score von 0 – 1 auf (keine Symptome oder keine wesentliche Behinderung), und 5 weitere Patienten hatten mRS=2 (leichte Behinderung). Das heisst, dass 21 von 24 Patienten (annähernd 90%) am Tag 90 gemäss mRS ein günstiges Endergebnis erreicht hatten. Bei den übrigen 3 Patienten war das gemeldete Endergebnis eine mittelschwere (mRS=3; n=1), eine mittelschwere bis schwere Behinderung (mRS=4; n=1) bzw. war innerhalb von 7 Tagen der Tod eingetreten (mRS=6; n=1).
Bei sieben Patienten war kein mRS-Score am Tag 90 angegeben. Laut den letzten verfügbaren Informationen hatten 3 dieser 7 Patienten am Tag 7 mRS=2, 3 der 7 Patienten berichteten am Tag 7 über eine eindeutige allgemeine Verbesserung und 1 der 7 Patienten mit einem mittelschweren bis schweren Schlaganfall bei Studienbeginn hatte am Tag 7 mRS=4.
Das Register verfügte auch über Sicherheitsdaten zu den Nebenwirkungen Blutungen und zerebralem Ödem bei 31 von 32 Patienten. Bei keinem der 31 Patienten in der Alterskategorie 16 – 17 Jahre waren symptomatische intrazerebrale Blutungen (sICH, ICH-Typ PH2) aufgetreten. In 5 Fällen entwickelte sich nach der Alteplase-Behandlung ein zerebrales Ödem. Bei 4 von 5 Patienten mit zerebralem Ödem wurde entweder ein mRS zwischen 0 und 2 am Tag 90 angegeben oder sie zeigten am Tag 7 nach der Behandlung eine allgemeine Verbesserung. Bei einem Patienten wurde am Tag 90 mRS=4 (mittelschwere bis schwere Behinderung) angegeben. Keiner dieser Fälle verlief tödlich.
Zusammengefasst enthielt das SITS-Register 31 Berichte über Patienten zwischen 16 und 17 Jahren mit akutem ischämischem Schlaganfall, die gemäss den Empfehlungen für Erwachsene mit Alteplase behandelt worden waren. Obwohl die geringe Fallzahl keine statistische Analyse zulässt, zeigen die Gesamtergebnisse eine positive Tendenz, wobei diese Patienten die entsprechende Dosis für Erwachsene erhalten hatten. Im Vergleich zu Erwachsenen lassen die Daten kein erhöhtes Risiko für symptomatische intrazerebrale Blutungen oder Ödeme erkennen.
PharmakokinetikAbsorption
Die relevante Halbwertszeit T½ alpha beträgt 3,5-5 Minuten. Das bedeutet, dass nach 20 Minuten weniger als 10% des Ausgangswertes im Plasma vorhanden ist.
Distribution
Das Distributionsvolumen beträgt 2,8-4,4 Liter (V1) bzw. 8-9 Liter (Vss).
Metabolismus / Elimination
Alteplase wird rasch aus dem Blutkreislauf ausgeschieden und hauptsächlich über die Leber metabolisiert (Plasma-Clearance 550-680 ml/Min.). Unter physiologischen Bedingungen weist der Grossteil von Alteplase im Kreislauf eine Inhibitorbindung auf. Die hepatische Clearance von Alteplase wird durch das Vorliegen anderer Proteine, einschliesslich Alteplase-Inhibitoren, nicht gestört. Komplexe aus Alteplase und ihrem Inhibitor werden als freie Alteplase eliminiert. Für die in einem tiefen Kompartiment verbleibende Restmenge wurde eine beta-Halbwertszeit von rund 40 Minuten gemessen.
Präklinische DatenIn Mutagenitäts-Tests zeigten sich keine Hinweise auf ein mutagenes Potential.
Bei trächtigen Tieren wurden nach intravenöser Infusion bzw. pharmakologisch wirksamen Dosen keine teratogenen Wirkungen beobachtet. Bei Kaninchen führten Tagesdosen über 3 mg/kg/Tag zu Embryotoxizität (Embryoletalität, Wachstumsverzögerung). Bei Ratten wurde bei Dosen bis zu 10 mg/kg/Tag kein Einfluss auf die peri- und postnatale Entwicklung oder auf die Fruchtbarkeitsparameter beobachtet.
Sonstige HinweiseInkompatibilitäten
Actilyse darf nicht mit anderen Arzneimitteln gemischt werden, weder in derselben Infusionsflasche noch über denselben Venenzugang (auch nicht mit Heparin).
Haltbarkeit / Besondere Lagerungshinweise
Den gefriergetrockneten Stoff bis zur Zubereitung vor Licht geschützt in der Originalverpackung aufbewahren.
Nicht über 25°C lagern.
Es wurde eine chemische und physikalische Stabilität der rekonstituierten Lösung von 24 Stunden bei 2-8°C und von 8 Stunden bei 30°C nachgewiesen.
Aus mikrobiologischer Sicht sollte die gebrauchsfertige Lösung sofort nach ihrer Herstellung verwendet werden. Falls aseptisch hergestellt sollten üblicherweise 24 Stunden bei 2-8°C nicht überschritten werden.
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Hinweise für die Handhabung
Korrekte Art der Anwendung:
Der Inhalt einer Injektionsflasche Actilyse (10 mg, 20 mg oder 50 mg) Trockensubstanz wird unter aseptischen Bedingungen mit sterilisiertem Wasser für Injektionszwecke (10 ml, 20 ml oder 50 ml, je nach Grösse der Actilyse-Flasche) zu einer Konzentration von 1 mg Actilyse pro ml gelöst.
Für diesen Zweck enthalten die Packungen à 20 mg und 50 mg eine Überleitungskanüle. Bei der Packung à 10 mg sollte eine sterile Spritze benutzt werden.
Anweisungen für die Rekonstitution von Actilyse
|
1
|
Unmittelbar vor der Verabreichung rekonstituieren.
|
|
| |
2
|
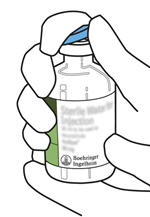
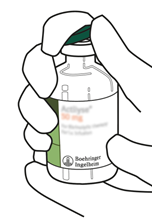
Entfernen Sie die Schutzkappen von den zwei Durchstechflaschen, die sterilisiertes Wasser und Actilyse-Trockensubstanz enthalten, indem Sie sie mit dem Daumen hochdrücken.
|
|
|
| |
3
|
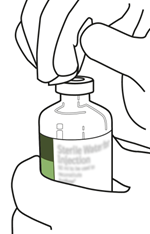
Desinfizieren Sie den Gummistopfen jeder Durchstechflasche mit einem Alkoholtupfer.
|
|
|
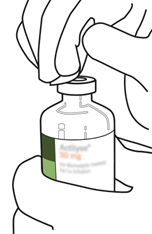
| |
4
|
Entnehmen Sie die Überleitungskanüle* aus der Umhüllung. Desinfizieren oder sterilisieren Sie die Überleitungskanüle nicht; sie ist bereits steril. Nehmen Sie eine Kappe ab.
(*falls eine Überleitungskanüle in der Packung enthalten ist. Die Rekonstitution kann auch mit Hilfe einer Spritze und einer Nadel erfolgen.)
|
|
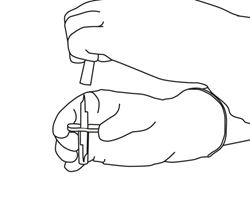
| |
5
|
Stellen Sie die Flasche mit sterilisiertem Wasser aufrecht auf eine stabile Oberfläche. Stechen Sie den Dorn der Überleitungskanüle senkrecht von oben direkt in die Mitte des Gummistopfens, indem Sie vorsichtig, aber fest drücken ohne zu drehen.
|
|
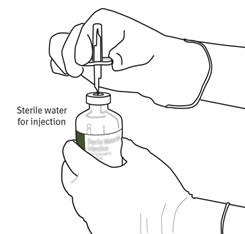
| |
6
|
Halten Sie die Flasche mit sterilisiertem Wasser und der Überleitungskanüle mit einer Hand an den beiden seitlichen Flügeln fest.
Entfernen Sie die obere Kappe der Überleitungskanüle.
|
|
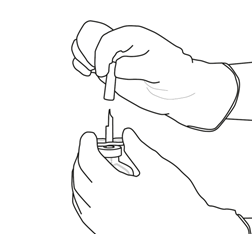
| |
7
|
Halten Sie die Flasche mit sterilisiertem Wasser und der Überleitungskanüle mit einer Hand an den beiden seitlichen Flügeln fest.
Halten Sie die Durchstechflasche mit Actilyse-Trockensubstanz umgekehrt vertikal über die Überleitungskanüle und positionieren Sie die Spitze der Überleitungskanüle genau in die Mitte des Stopfens.
|
|
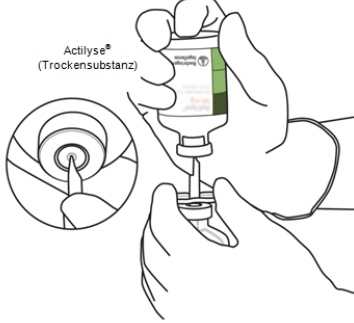
| |
|
Drücken Sie die Durchstechflasche mit der Trockensubstanz senkrecht von oben direkt auf die Überleitungskanüle hinunter und stechen Sie vorsichtig, aber fest senkrecht in den Gummistopfen ein, ohne zu drehen.
|
|
| |
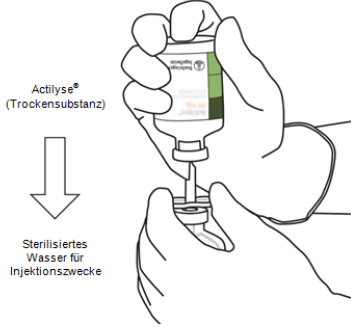
8
|
Drehen Sie die beiden Durchstechflaschen zusammen um und lassen Sie das Wasser für Injektionszwecke vollständig in die Trockensubstanz einlaufen.
|
|
| |
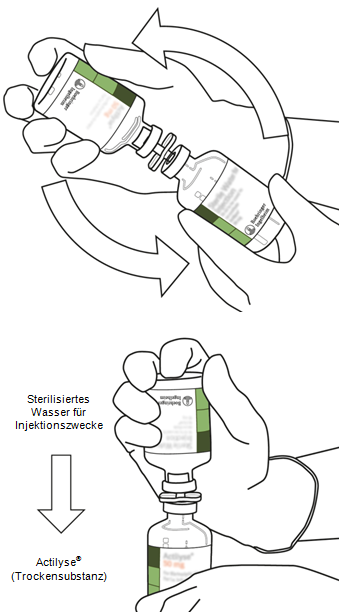
9
|
Entfernen Sie die entleerte Wasserflasche zusammen mit der Überleitungskanüle und verwerfen Sie sie.
|
|
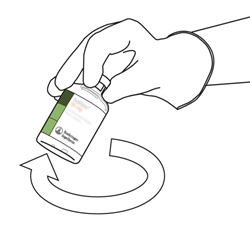
| |
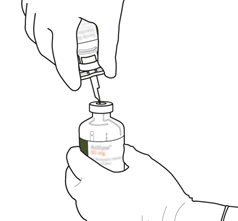
10
|
Nehmen Sie die Durchstechflasche mit rekonstituiertem Actilyse und schwenken Sie sie sanft hin und her, um eventuell noch vorhandenes Pulver zu lösen. Nicht schütteln, da dies zu Schaumbildung führen kann.
Bei Blasenbildung lassen Sie die Lösung einige Minuten ruhig stehen, bis sich die Bläschen aufgelöst haben.
|
|
|

| |
11
|
Die rekonstituierte Lösung enthält 1 mg/ml Actilyse. Sie muss klar und farblos bis leicht gelblich sein und darf keine Partikel enthalten.
| |
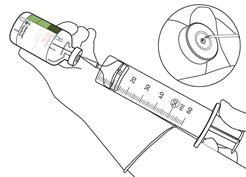
12
|
Entnehmen Sie die erforderliche Menge nur mit Hilfe einer sterilen Nadel und einer sterilen Spritze.
Nutzen Sie dabei nicht die Einstichöffnung der Überleitungskanüle, um ein Austreten zu vermeiden.
|
|
| |
13
|
Sofort verwenden.
Nicht verwendete Lösung ist zu verwerfen.
|
Die rekonstituierte 1 mg/ml Lösung kann mit steriler Natriumchlorid-Injektionslösung mit 9 mg/ml (0,9 %) bis auf eine Mindestkonzentration von 0,2 mg/ml weiter verdünnt werden, wobei das Auftreten einer Trübung der rekonstituierten Lösung nicht ausgeschlossen werden kann.
Eine weitere Verdünnung der rekonstituierten 1 mg/ml Lösung mit sterilem Wasser für Injektionszwecke oder die Verwendung von kohlenhydrathaltigen Infusionslösungen (z.B. Dextrose) wird aufgrund einer erhöhten Trübungsbildung nicht empfohlen.
Actilyse darf nicht mit anderen Arzneimitteln gemischt werden, weder in derselben Infusionsflasche noch über denselben Venenzugang (auch nicht mit Heparin).
Um eine genaue Dosierung zu erreichen, sollte Actilyse mit Perfusoren/Infusions-Pumpen verabreicht werden. Sind diese nicht verfügbar, kann ein anderes Infusionssystem verwendet werden, das ebenfalls eine genaue Einstellung der Infusionsgeschwindigkeit gewährleistet.
Um sicherzustellen, dass der Patient die volle Dosis Actilyse bekommt, sollte das Residualvolumen in dem Infusionssystem auf ein Minimum beschränkt werden.
Zulassungsnummer48313 (Swissmedic)
PackungenPackung mit 1 Injektionsflasche mit 10 mg Wirkstoff und 1 Flasche mit 10 ml Wasser für Injektionszwecke.
Packung mit 1 Injektionsflasche mit 20 mg Wirkstoff und 1 Flasche mit 20 ml Wasser für Injektionszwecke.
Packung mit 1 Injektionsflasche mit 50 mg Wirkstoff und 1 Flasche mit 50 ml Wasser für Injektionszwecke.
Abgabekategorie: B
ZulassungsinhaberinBoehringer Ingelheim (Schweiz) GmbH, Basel
Stand der InformationFebruar 2023
|