ZusammensetzungWirkstoffe
Lenalidomidum.
Hilfsstoffe
Dosisstärke Hartkapsel
2.5 mg 5 mg 7.5 mg 10 mg 15 mg 20 mg 25 mg
Lactose (als wasserf 73,5 mg 147,0 mg 144,5 mg 294,0 mg 289,0 mg 244,5 mg 200,0 mg
reie Lactose)
Mikrokristalline Cellulose, Magnesiumstearat, Croscarmellose-Natrium.
Kapselhülle: Gelatine, Titandioxid, Eisenoxid (nur bei Hartkapseln 2,5 mg, 7,5 mg, 10 mg und 20 mg), Indigocarmin E132 (nur bei Hartkapseln 2,5 mg, 10 mg, 15 mg und 20 mg).
Drucktinte: Shellack, Propylenglycol, Eisenoxid, Kaliumhydroxid.
Eine Hartkapsel enthält max. 0,2 mg (Hartkapseln 2,5 mg) bzw. max. 0,4 mg (Hartkapseln 5 mg und 7,5 mg) bzw. max. 0,8 mg (Hartkapseln 10 mg, 15 mg, 20 mg und 25 mg) Natrium.
Darreichungsform und Wirkstoffmenge pro EinheitHartkapseln zu 2,5 mg, 5 mg, 7,5 mg, 10 mg, 15 mg, 20 mg und 25 mg.
Indikationen/AnwendungsmöglichkeitenRevlimid in Kombination mit Bortezomib und Dexamethason ist indiziert zur Behandlung von erwachsenen Patienten mit unbehandeltem multiplem Myelom.
Revlimid ist indiziert zur Behandlung von erwachsenen Patienten mit multiplem Myelom als Erhaltungstherapie nach autologer Stammzelltransplantation.
Revlimid in Kombination mit Dexamethason oder Revlimid in Kombination mit Melphalan und Prednison, jeweils gefolgt von einer Revlimid Erhaltungstherapie, ist indiziert zur Behandlung erwachsener Patienten mit unbehandeltem multiplem Myelom, die nicht transplantierbar sind.
Revlimid in Kombination mit Dexamethason ist indiziert zur Behandlung von Patienten mit multiplem Myelom, die wenigstens eine vorangegangene medikamentöse Therapie erhalten haben.
Revlimid ist indiziert zur Behandlung von Patienten mit transfusionsabhängiger Anämie infolge von myelodysplastischem Syndrom mit niedrigem oder intermediärem Risiko 1 in Verbindung mit einer zytogenetischen Deletion 5q-Anomalie mit oder ohne weitere zytogenetische Anomalien.
Revlimid ist indiziert zur Behandlung von Patienten mit rezidiviertem oder refraktärem Mantelzell-Lymphom (MCL) nach vorangegangener Therapie, welche Bortezomib und Chemotherapie/Rituximab umfasste.
Revlimid in Kombination mit Rituximab (Anti-CD20-Antikörper) ist indiziert zur Behandlung von erwachsenen Patienten mit einem rezidivierten oder refraktären follikulären Lymphom (Grad 1-3A) (siehe "Eigenschaften/Wirkungen" ).
Dosierung/AnwendungDie Behandlung muss von einem erfahrenen Hämatologen oder Onkologen begonnen und überwacht werden.
Multiples Myelom
Revlimid in Kombination mit Bortezomib und Dexamethason bei Patienten mit unbehandeltem multiplem Myelom
-Initiale Therapie: Revlimid in Kombination mit Bortezomib und Dexamethason
Die Behandlung mit Revlimid in Kombination mit Bortezomib und Dexamethason darf nicht begonnen werden, wenn die absolute Neutrophilenzahl (ANC) < 1,0 x 109/l und/oder die Thrombozytenzahl < 50 x 109/l ist.
Die empfohlene Initialdosis Revlimid beträgt 25 mg oral einmal täglich entweder
a.an den Tagen 1-14 jedes 21-tägigen Behandlungszyklus oder
b.an den Tagen 1-21 jedes 28-tägigen Behandlungszyklus.
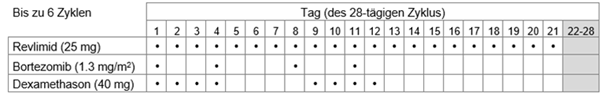
Bortezomib soll als subkutane Injektion (1,3 mg/m2 Körperoberfläche) zweimal wöchentlich an den Tagen 1, 4, 8 und 11 jedes 21-Tage- oder 28-Tage-Zyklus gegeben werden.
Die empfohlene Dosis Dexamethason beträgt
a.20 mg oral einmal täglich an den Tagen 1, 2, 4, 5, 8, 9, 11 und 12 oder
b.40 mg oral einmal täglich an den Tagen 1 bis 4 und 9 bis 12 jedes Zyklus.
Es werden bis zu acht 21-Tages- oder sechs 28-Tages-Zyklen (24-wöchige initiale Therapie) empfohlen.
Tabelle 1: Empfohlenes Dosierungsschema für Revlimid in Kombination mit Bortezomib und Dexamethason

Oder
-Fortsetzung der Behandlung bei Patienten, die keine Stammzelltransplantation erhalten: Revlimid in Kombination mit Dexamethason bis zur Progression der Erkrankung
Weiterbehandlung mit 25 mg Revlimid oral einmal täglich an den Tagen 1-21 der sich wiederholenden 28-Tages-Zyklen in Kombination mit Dexamethason. Die empfohlene Dosis Dexamethason beträgt 40 mg oral einmal täglich an den Tagen 1, 8, 15 und 22 der sich wiederholenden 28-Tages-Zyklen. Die Behandlung kann bis zur Progression der Erkrankung oder Unverträglichkeit fortgesetzt werden.
-Fortsetzung der Behandlung: Autologe Stammzelltransplantation
Bei Patienten, deren Behandlung mit einer autologen Stammzelltransplantation fortgesetzt wird, sollte innerhalb der ersten 4 Zyklen der initialen Therapie eine Mobilisierung der hämatopoietischen Stammzellen erfolgen.
Revlimid bei Patienten nach autologer Stammzelltransplantation
Nach autologer Stammzelltransplantation ist die Erhaltungstherapie mit Revlimid nach angemessener hämatologischer Erholung einzuleiten. Die Behandlung mit Revlimid darf nicht begonnen werden, wenn die ANC < 1,0 x 109/l und/oder die Zahl der Thrombozyten < 75 x 109/l ist.
Empfohlene Dosierung
Die empfohlene Initialdosis beträgt 10 mg Revlimid oral einmal täglich kontinuierlich (an den Tagen 1-28 der sich wiederholenden 28-Tage-Zyklen) und wird bis zur Progression der Erkrankung oder Unverträglichkeit fortgesetzt. Nach drei 28-Tage-Zyklen kontinuierlicher Erhaltungstherapie mit Revlimid kann die Dosis bei Verträglichkeit auf 15 mg oral einmal täglich erhöht werden.
Revlimid in Kombination mit Dexamethason bis zur Progression der Erkrankung bei unbehandelten Patienten, die nicht transplantierbar sind
Die Behandlung mit Revlimid darf nicht begonnen werden, wenn die ANC < 1,0 x 109/l und/oder die Zahl der Thrombozyten < 50 x 109/l ist.
Empfohlene Dosierung
Die empfohlene Initialdosis beträgt 25 mg Revlimid oral einmal täglich an den Tagen 1 - 21 der sich wiederholenden 28-Tage-Zyklen. Die empfohlene Dosis von Dexamethason beträgt 40 mg oral einmal täglich an den Tagen 1, 8, 15 und 22 der sich wiederholenden 28-Tage-Zyklen. Die Behandlung mit Revlimid und Dexamethason kann bis zur Progression der Erkrankung oder Unverträglichkeit fortgesetzt werden.
Revlimid in Kombination mit Melphalan und Prednison gefolgt von einer Erhaltungsmonotherapie bei unbehandelten, nicht transplantierbaren Patienten
Die Behandlung mit Revlimid darf nicht begonnen werden, wenn die ANC < 1,5 x 109/l und/oder die Zahl der Thrombozyten < 75 x 109/l ist.
Empfohlene Dosierung
Die empfohlene Initialdosis beträgt 10 mg/Tag Revlimid oral an den Tagen 1 - 21 der sich wiederholenden 28-Tage-Zyklen über bis zu 9 Zyklen, Melphalan 0,18 mg/kg oral an den Tagen 1 - 4 der sich wiederholenden 28-Tage-Zyklen, Prednison 2 mg/kg oral an den Tagen 1 - 4 der sich wiederholenden 28-Tage-Zyklen.
Patienten, die 9 Zyklen abgeschlossen haben oder die die Kombinationstherapie wegen Unverträglichkeit nicht zu Ende führen können, erhalten Revlimid allein, 10 mg/Tag oral an den Tagen 1 - 21 der sich wiederholenden 28-Tage-Zyklen, bis zur Progression der Erkrankung.
Revlimid in Kombination mit Dexamethason bei Patienten mit multiplem Myelom, die wenigstens eine vorangegangene Therapie erhalten haben
Die empfohlene Anfangsdosis beträgt 25 mg Revlimid oral einmal täglich an den Tagen 1–21 der sich wiederholenden 28-tägigen Behandlungszyklen. Während der ersten 4 Behandlungszyklen beträgt die empfohlene Dosis Dexamethason 40 mg oral einmal täglich an den Tagen 1–4, 9–12 und 17–20 eines jedes 28-Tage-Zyklus und anschliessend 40 mg einmal täglich an den Tagen 1–4. Die Behandlung sollte bis zur Progredienz oder bis zum Auftreten inakzeptabler Toxizitäten weitergeführt werden.
Myelodysplastisches Syndrom
Die empfohlene Anfangsdosis beträgt 10 mg Revlimid oral einmal täglich an den Tagen 1–21 der sich wiederholenden 28-tägigen Behandlungszyklen. Lässt sich 16 Wochen nach Beginn der Revlimid-Therapie nicht zumindest ein geringfügiges Ansprechen d.h. mindestens eine 50%ige Verbesserung nachweisen, wird ein Absetzen der Behandlung wegen mangelnder Wirksamkeit empfohlen.
Rezidiviertes oder refraktäres Mantelzell-Lymphom
Die empfohlene Anfangsdosis beträgt täglich 25 mg Revlimid oral an den Tagen 1–21 der sich wiederholenden 28-tägigen Behandlungszyklen. Die Behandlung sollte bis zum Progress oder bis zum Auftreten inakzeptabler Toxizitäten weitergeführt werden.
Follikuläres Lymphom (FL) in Kombination mit Rituximab (R2 Regime)
Die Behandlung mit Revlimid darf nicht begonnen werden, wenn die absolute Neutrophilenzahl (ANC) < 1 x 109/l beträgt und/oder die Thrombozytenzahl < 50 x 109/l, sofern dies nicht die Folge einer Infiltration des Knochenmarks durch das Lymphom ist.
Die empfohlene Anfangsdosis von Revlimid beträgt 20 mg oral einmal täglich an den Tagen 1–21 der sich wiederholenden 28-tägigen Behandlungszyklen für bis zu 12 Behandlungszyklen. Die empfohlene Anfangsdosis von Rituximab beträgt 375 mg/m² intravenös (i.v.) einmal wöchentlich in Zyklus 1 (Tag 1, 8, 15 und 22) und an Tag 1 jedes 28-tägigen Zyklus für Zyklen 2 bis einschliesslich 5.
Dosisanpassung
Die Dosierung von Revlimid oder anderen Arzneimitteln, die im Rahmen einer Kombinationsbehandlung angewendet werden (Dexamethason, Melphalan, Prednison, Bortezomib, Rituximab) ist auf der Grundlage von klinischen Befunden und Laborwerten anzupassen.
Bezüglich toxizitätsbedingter Dosisanpassungen bei anderen Arzneimitteln als Revlimid, die im Rahmen einer Kombinationsbehandlung angewendet werden, ist die Fachinformation des jeweiligen Arzneimittels zu konsultieren.
Hämatotoxizität
Empfohlene Dosisanpassungen während der Behandlung und bei Wiederaufnahme der Behandlung
Für den Umgang mit einer Grad-3- oder Grad-4-Thrombozytopenie oder -Neutropenie, sowie jeder anderen Grad-3- oder Grad-4-Toxizität, die als Lenalidomid-bedingt bewertet wird, werden Dosisanpassungen, wie unten je nach Indikation beschrieben, empfohlen.
Revlimid in Kombination mit Bortezomib und Dexamethason bei unbehandelten Patienten mit multiplem Myelom
Schritte zur Dosisreduktion
Lenalidomid
Initialdosis 25 mg
Dosisstufe -1 20 mg
Dosisstufe -2 15 mg
Dosisstufe -3 10 mg
Dosisstufe -4 5 mg
Dosisstufe -5 2,5 mg täglich oder 5 mg alle 48 h
Thrombozytopenie
Veränderung Thrombozytenzahl Empfohlene Vorgehensweise
Abfall auf < 30 x 109/l Unterbrechung der Lenalidomid-Behandlung und wöchentliche
Überwachung des vollständigen Blutbildes
Wiederanstieg auf ≥50 x 109/l Fortsetzung von Lenalidomid auf der Dosisstufe -1
Bei jedem weiteren Abfall unter Unterbrechung der Lenalidomid-Behandlung Fortsetzung von
< 30 x 109/l Wiederanstieg auf Lenalidomid auf der nächst niedrigeren Dosisstufe. Nicht unter
≥50 x 109/l 2,5 mg einmal täglich dosieren.
Neutropenie
Veränderung Neutrophilenzahl Empfohlene Vorgehensweise a
Erster Abfall auf < 0,5 x 109/l oder Unterbrechung der Lenalidomid-Behandlung und
febrile Neutropenie (Fieber ≥38 °C; < 1 x wöchentliche Überwachung des vollständigen Blutbildes
109/l) Wiederanstieg auf ≥1 x 109/l Fortsetzung von Lenalidomid auf der Dosisstufe -1
Bei jedem weiteren Abfall unter < 0,5 x Unterbrechung der Lenalidomid-Behandlung Fortsetzung
109/l oder febrile Neutropenie von Lenalidomid auf der nächst niedrigeren Dosisstufe.
Wiederanstieg auf ≥1 x 109/l Nicht unter 2,5 mg einmal täglich dosieren.
a Wenn bei einer beliebigen Dosisstufe Neutropenie als einzige Toxizität auftritt, so wird nach Ermessen des Arztes, unter Beibehaltung der Dosisstufe von Lenalidomid, der Granulozytenkolonie stimulierende Faktor (G-CSF) gegeben.
Revlimid bei Patienten nach autologer Stammzelltransplantation
Schritte zur Dosisre
duktion
Initialdosis (10 mg) Falls Dosis erhöht (15 mg)a
Dosisstufe -1 5 mg einmal täglich, kontinuierlich 10 mg einmal täglich,
kontinuierlich
Dosisstufe -2 5 mg einmal täglich an Tagen 1-21 der 5 mg einmal täglich,
28-Tage-Zyklen kontinuierlich
Dosisstufe -3 Nicht zutreffend 5 mg einmal täglich an Tagen
1-21 der 28-Tage-Zyklen
An Tagen 1-21 der 28-Tage-Zyklen nicht
unter 5 mg einmal täglich dosieren
a Nach drei 28-Tage-Zyklen kontinuierlicher Erhaltungstherapie mit Revlimid kann die Dosis bei Verträglichkeit auf 15 mg oral einmal täglich erhöht werden.
Thrombozytopenie
Veränderung Thrombozytenzahl Empfohlene Vorgehensweise
Abfall auf < 30 x 109/l Unterbrechung der Lenalidomid-Behandlung und wöchentliche
Überwachung des vollständigen Blutbildes
Wiederanstieg auf ≥30 x 109/l Fortsetzung von Lenalidomid auf der Dosisstufe -1
Bei jedem weiteren Abfall Unterbrechung der Lenalidomid-Behandlung
unter < 30 x 109/l
Wiederanstieg auf ≥30 x 109l/l Fortsetzung von Lenalidomid auf der nächst niedrigeren Dosisstufe
Neutropenie
Veränderung Neutrophilenzahl Empfohlene Vorgehensweisea
Abfall auf < 0,5 x 109/l Unterbrechung der Lenalidomid-Behandlung und wöchentliche
Überwachung des vollständigen Blutbildes
Wiederanstieg auf ≥0,5 x 109/l Fortsetzung von Lenalidomid auf der Dosisstufe -1
Bei jedem weiteren Abfall Unterbrechung der Lenalidomid-Behandlung
unter < 0,5 x 109/l
Wiederanstieg auf ≥0,5 x 109/l Fortsetzung von Lenalidomid auf der nächst niedrigeren Dosisstufe
a Wenn bei einer beliebigen Dosisstufe Neutropenie als einzige Toxizität auftritt, so wird nach Ermessen des Arztes, unter Beibehaltung der Dosisstufe von Lenalidomid, der Granulozytenkolonie stimulierende Faktor (G-CSF) gegeben.
Revlimid in Kombination mit Dexamethason bei unbehandelten Patienten, die nicht transplantierbar sind
Schritte zur Dosisreduktion
Lenalidomid Dexamethason
Initialdosis 25 mg 40 mg
Dosisstufe -1 20 mg 20 mg
Dosisstufe -2 15 mg 12 mg
Dosisstufe -3 10 mg 8 mg
Dosisstufe -4 5 mg 4 mg
Dosisstufe -5 2,5 mg täglich oder 5 mg alle 48 h Nicht zutreffend
Thrombozytopenie
Veränderung Thromboz Empfohlene Vorgehensweise
ytenzahl
Abfall auf < 25 x Unterbrechung der Lenalidomid-Behandlung für den Rest des Zyklusa
109/l
Wiederanstieg auf Fortsetzung von Lenalidomid mit einer 5 mg niedrigeren Dosis als die vorige
≥50 x 109/l Dosis. Nach einer Dosis von 5 mg Fortsetzung von Lenalidomid mit einer Dosis
von 2,5 mg täglich oder 5 mg alle 48 Stunden. Keine Dosis darf unter 2,5 mg
täglich oder 5 mg alle 48 Stunden liegen.
ª Bei Auftreten einer dosislimitierenden Toxizität (DLT) an Tag 15 eines Zyklus wird die Lenalidomid-Behandlung mindestens für den Rest des jeweiligen 28-Tage-Zyklus unterbrochen.
Neutropenie
Veränderung Neutrophilenzahl Empfohlene Vorgehensweisea
Erster Abfall auf < 0,5 x 109/l oder febrile Unterbrechung der Lenalidomid-Behandlung
Neutropenie (Fieber ≥38 °C; < 1 x 109/l) Wiederanstieg Fortsetzung von Lenalidomid mit der
auf ≥1 x 109/l bei Neutropenie als einzige beobachtete Initialdosis einmal täglich
Toxizität
Wiederanstieg auf ≥0,5 x 109/l bei Beobachtung anderer Fortsetzung von Lenalidomid auf der
dosisabhängiger hämatologischer Toxizitäten ausser Dosisstufe -1 einmal täglich
Neutropenie
Bei jedem weiteren Abfall unter < 0,5 x 109/l Unterbrechung der Lenalidomid-Behandlung
Wiederanstieg auf ≥0,5 x 109/l Fortsetzung von Lenalidomid auf der
nächst niedrigeren Dosisstufe einmal
täglich.
a Wenn bei einer beliebigen Dosisstufe Neutropenie als einzige Toxizität auftritt, so wird nach Ermessen des Arztes, unter Beibehaltung der Dosisstufe von Lenalidomid, der Granulozytenkolonie stimulierende Faktor (G-CSF) gegeben.
Wenn die Lenalidomid-Dosis wegen einer hämatologischen DLT reduziert wurde, kann die Lenalidomid-Dosis im Ermessen des behandelnden Arztes auf die nächst höhere Dosisstufe (bis zur Initialdosis) wieder gesteigert werden, sofern die fortgesetzte Lenalidomid- / Dexamethason-Therapie zu einer verbesserten Knochenmarksfunktion geführt hat (keine DLT über mindestens 2 aufeinanderfolgende Zyklen und eine ANC ≥1'500/µl bei einer Thrombozytenzahl ≥100'000/µl zu Beginn eines neuen Zyklus auf der aktuellen Dosisstufe).
Revlimid in Kombination mit Melphalan und Prednison gefolgt von einer Erhaltungsmonotherapie bei nicht transplantierbaren Patienten
Schritte zur Dosisreduktion
Lenalidomid Melphalan Prednison
Initialdosis 10 mg 0,18 mg/kg 2 mg/kg
Dosisstufe -1 7,5 mg täglich oder 15 mg alle 48 h 0,14 mg/kg 1 mg/kg
Dosisstufe -2 5 mg 0,10 mg/kg 0,5 mg/kg
Dosisstufe -3 2,5 mg täglich oder 5 mg alle 48 h Nicht zutreffend 0,25 mg/kg
Thrombozytopenie
Veränderung Thrombozytenzahl Empfohlene Vorgehensweise
Erster Abfall auf < 25 x 109/l Unterbrechung der Lenalidomid-Behandlung Fortsetzung von
Wiederanstieg auf ≥25 x 109/l Lenalidomid und Melphalan auf der Dosisstufe -1
Bei jedem weiteren Abfall unter Unterbrechung der Lenalidomid-Behandlung Fortsetzung von
30 x 109/l Wiederanstieg auf Lenalidomid auf der nächst niedrigeren Dosisstufe (Dosisstufe -2
≥30 x 109/l oder -3) einmal täglich.
Neutropenie
Veränderung Neutrophilenzahl Empfohlene Vorgehensweisea
Erster Abfall auf < 0,5 x 109/l Wiederanstieg Unterbrechung der Lenalidomid-Behandlung
auf ≥0,5 x 109/l bei Neutropenie als einzige Fortsetzung von Lenalidomid mit der Initialdosis
beobachtete Toxizität einmal täglich
Wiederanstieg auf ≥0,5 x 109/l bei Beobachtung Fortsetzung von Lenalidomid auf der Dosisstufe -1
anderer dosisabhängiger hämatologischer einmal täglich
Toxizitäten ausser Neutropenie
Bei jedem weiteren Abfall unter < 0,5 x 109/l Unterbrechung der Lenalidomid-Behandlung
Wiederanstieg auf ≥0,5 x 109/l Fortsetzung von Lenalidomid auf der nächst
niedrigeren Dosisstufe einmal täglich.
a Wenn bei einer beliebigen Dosisstufe Neutropenie als einzige Toxizität auftritt, so wird nach Ermessen des Arztes, unter Beibehaltung der Dosisstufe von Lenalidomid, der Granulozytenkolonie stimulierende Faktor (G-CSF) gegeben.
Multiples Myelom mit mindestens einer Vortherapie, myelodysplastisches Syndrom und Mantelzell-Lymphom
Für die Indikationen MM nach mindestens einer Vortherapie oder MDS, bei einer Thrombozytopenie mit Abfall der Werte auf < 25 x 109/l oder bei einer Neutropenie mit Abfall der Werte auf < 0,5 x 109/l sollte die Behandlung mit Lenalidomid unterbrochen werden.
Für die Indikation MCL, bei einer Thrombozytopenie mit Abfall der Werte auf < 50 x 109/l oder bei einer Neutropenie mit Abfall der Werte auf < 0,5 x 109/l oder mit Abfall auf < 1 x 109/l während mindestens 7 Tagen oder mit Abfall auf < 1 x 109/l mit einer damit verbundenen Temperatur von ≥38,5 °C, sollte die Behandlung mit Lenalidomid unterbrochen werden.
Nach Normalisierung der Thrombozyten/Neutrophilenzahlen sollte die Behandlung mit der nächst niedrigeren Dosisstärke fortgeführt werden. Bei Wiederauftreten ist die Dosis weiter zu reduzieren. Bei Toxizität unter der niedrigsten Dosisstärke ist die Behandlung mit Lenalidomid abzubrechen.
Bei MM nach mindestens einer Vortherapie ist die erste Dosisreduktion 15 mg täglich, bei erneuter Toxizität 10 mg und dann 5 mg täglich.
Bei MDS ist die erste Dosisreduktion 5 mg täglich, bei erneutem Auftreten wird eine zweite Dosisreduktion von 2,5 mg täglich oder 5 mg alle 2 Tage empfohlen. Eine dritte Dosisreduktion auf 5 mg zweimal wöchentlich wird empfohlen beim Wiederauftreten der Toxizität.
Bei MCL-Patienten, welche über mehr als 3 Monate unter einer tieferen Dosis von Revlimid kein Ansprechen gezeigt haben, ist der Wechsel auf eine andere Therapie in Erwägung zu ziehen.
Follikuläres Lymphom (FL)
Schritte zur Dosisredukti
on
Lenalidomid
Initialdosisa 20 mg einmal täglich an den Tagen 1-21 alle 28 Tage
Dosistufe -1 15 mg einmal täglich an den Tagen 1-21 alle 28 Tage
Dosisstufe -2 10 mg einmal täglich an den Tagen 1-21 alle 28 Tage
Dosisstufe -3 5 mg einmal täglich an den Tagen 1-21 alle 28 Tage
Dosisstufe -4 b 2,5 mg einmal täglich an den Tagen 1-21 alle 28 Tage oder 5 mg alle 48
Stunden
a Für Initialdosis bei Patienten mit Nierenfunktionsstörungen siehe Abschnitt unten.
b Nur für angepasste Initialdosis bei Patienten mit mässiggradiger Niereninsuffizienz.
Thrombozytopenie
Veränderung Thromboz Empfohlene Vorgehensweise
ytenzahl
Erster Abfall auf < Lenalidomid-Behandlung unterbrechen und vollständiges Blutbild wöchentlich
50 x 109/l Wiederans bestimmen Wiederaufnahme von Lenalidomid auf Dosisstufe -1 Tag 1-21 des
tieg auf ≥50 x 109/l 28-tägigen Zyklus.)
Bei jedem weiteren Lenalidomid-Behandlung unterbrechen und vollständiges Blutbild wöchentlich
Abfall < 50 x 109/l kontrollieren. Wiederaufnahme von Lenalidomid auf der nächst niedrigeren
Wiederanstieg auf Dosisstufe (Dosistufe -2 oder -3 einmal täglich). Nicht niedriger als
≥50 x 109/l Dosisstufe -3 dosieren. Wenn die Initialdosis 10 mga war, nicht niedriger als
Dosisstufe -4 dosieren.
a siehe Abschnitt "Patienten mit Nierenfunktionsstörungen"
Neutropenie
Veränderung Neutrophilenzahl Empfohlene Vorgehensweisea
Erster Abfall auf < 1,0 x 109/l für Unterbrechung der Lenalidomid-Behandlung und Kontrolle
mindestens 7 Tage ODER Abfall auf < des vollständigen Blutbildes mindestens alle 7 Tage
1,0 x 109/l in Verbindung mit Fieber
≥38,5°C) ODER Abfall auf < 0,5 x 109/l
Wiederanstieg auf ≥1,0 x 109/l Wiederaufnahme von Lenalidomid auf der nächst niedrigeren
Dosisstufe (Dosisstufe -1)
Bei jedem weiteren Abfall auf unter Unterbrechung der Lenalidomid-Behandlung und Erstellen
1,0 x 109/l für mindestens 7 Tage oder eines vollständigen Blutbildes mindestens alle 7 Tage
einem Abfall auf < 1,0 x 109/l Wiederaufnahme von Lenalidomid auf der nächst niedrigeren
verbunden mit Fieber (Körpertemperatur Dosisstufe (Dosistufe -2 oder -3). Nicht niedriger als
≥38,5°C) oder Abfall auf < 0,5 x 109/l Dosisstufe -3 dosieren. Wenn die Initialdosis 10 mgb war,
Wiederanstieg auf ≥0,5 x 109/l nicht niedriger als Dosisstufe -4 dosieren.
ª Wenn bei einer beliebigen Dosisstufe Neutropenie als einzige Toxizität auftritt, so wird nach Ermessen des Arztes, unter Beibehaltung der Dosisstufe von Lenalidomid, der Granulozytenkolonie stimulierende Faktor (G-CSF) gegeben.
b siehe Abschnitt "Patienten mit Nierenfunktionsstörungen"
Andere Gründe
Beim Auftreten von nicht-schuppendem Hautausschlag (mit Blasenbildung) Grad 3, von Neuropathie Grad 3, oder von einer allergischen Reaktion Grad 2, muss die Therapie unterbrochen werden. Eine Wiederaufnahme kann nach entsprechender Rückbildung bis ≤ Grad 1 mit der nächst niedrigeren Dosisstufe erfolgen.
Beim Auftreten von schuppendem Hautausschlag (mit Blasenbildung), von nicht-schuppendem Hautausschlag (mit Blasenbildung) Grad 4, von Neuropathie Grad 4, oder von einer allergischen Reaktion ≥ Grad 3 muss Revlimid abgesetzt werden.
Beim Auftreten einer Obstipation (≥ Grad 3) muss die Therapie unterbrochen und die Behandlung der Obstipation eingeleitet werden. Eine Wiederaufnahme kann nach Rückbildung der Obstipation bis ≤ Grad 2 mit der nächst niedrigeren Dosisstufe erfolgen.
Beim Auftreten einer Venenthrombose/Embolie (≥ Grad 3) muss die Therapie unterbrochen und die Antikoagulation eingeleitet werden. Eine Wiederaufnahme der Therapie liegt im ärztlichen Ermessen (Beibehaltung der Dosisstufe).
Bei Angioödem, Anaphylaxie, Grad-4-Hautausschlag, exfoliativem oder bullösem Hautausschlag, bei Verdacht auf Stevens-Johnson-Syndrom (SJS), toxischer epidermaler Nekrolyse (TEN) oder Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS) muss Revlimid abgesetzt werden; nach Abbruch aufgrund dieser Reaktionen sollte die Behandlung nicht wiederaufgenommen werden.
Weitere Toxizitäten 3./4. Grades
Bei Auftreten weiterer Toxizitäten 3./4. Grades, die Revlimid zugeschrieben werden, ist die Behandlung abzubrechen und nach Abklingen der Toxizität auf ≤ Grad 2 nach Ermessen des Arztes mit der nächst niedrigeren Dosierung fortzuführen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit Leberfunktionsstörungen wurde Revlimid nicht untersucht, und es gibt keine speziellen Dosierungsempfehlungen.
Patienten mit Nierenfunktionsstörungen
Bei leichter Niereninsuffizienz (ClCr 80-60 ml/min) ist keine Dosisanpassung erforderlich.
Für die Behandlung von MM-Patienten mit Anfangsdosis 25 mg, für Patienten mit follikulärem Lymphom mit Anfangsdosis 20 mg und für MM-Patienten bzw. MDS-Patienten mit Anfangsdosis 10 mg werden zu Beginn und im Verlauf der Therapie bei mässiggradiger (30 ≤ ClCr < 60 ml/min) oder schwerer Niereninsuffizienz (ClCr < 30 ml/min) oder Niereninsuffizienz im Endstadium folgende Dosisanpassungen empfohlen
Nierenfunktion Dosisanpassung
(ClCr)
Initiale Dosis 25 mg Initiale Dosis 20 mg Initiale Dosis 10 mg
Normale Nierenfunkti 25 mg täglich 20 mg täglich 10 mg täglich
on/Milde Niereninsuf
fizienz (ClCr ≥60
ml/min)
Mässige Niereninsuff 10 mga täglich 10 mgc täglich 5 mg täglich
izienz (30 ≤ ClCr<
60 ml/min)
Schwere Niereninsuff 7,5 mg täglich oder 5 mg täglich 2,5 mg täglich oder 5 mg alle
izienz (ClCr < 30 15 mgb alle 48 48 Stunden
ml/min, keine Stunden
Dialyse nötig)
Niereninsuffizienz 5 mg täglich; An 5 mg täglich An 2,5 mg täglich oder 5 mg
im Endstadium (ClCr Dialysetagen ist die Dialysetagen ist die dreimal wöchentlich; An
< 30 ml/min, Dialyse Dosis nach der Dosis nach der Dialysetagen ist die Dosis
nötig) Dialyse zu verabreich Dialyse zu verabreich nach der Dialyse zu
en en verabreichen
a Die Dosis kann nach 2 Zyklen auf 15 mg täglich gesteigert werden, falls der Patient nicht auf die Behandlung anspricht und das Medikament verträgt.
b Die Dosis kann auf 10 mg täglich gesteigert werden, falls der Patient das Medikament verträgt.
c Die Dosis kann nach 2 Zyklen auf 15 mg täglich gesteigert werden, falls der Patient das Medikament verträgt.
Bei der Behandlung von MCL–Patienten wird ein Einfluss der Nierenfunktion auf den Plasmaspiegel der aktiven Substanz Revlimid analog zum beobachteten Effekt in MM-, MDS- und FL-Patienten erwartet. Eine entsprechende Dosisreduktion sollte in MCL-Patienten mit einer Störung der Nierenfunktion in Betracht gezogen werden. Man beachte, dass bei MCL-Patienten mit Creatinin-Clearance zwischen 30 und 60 ml/min die Anfangsdosis 10 mg nicht überschritten werden darf.
Ältere Patienten
Dosisanpassungen sind nicht erforderlich. Da bei älteren Patienten mit einer verminderten Nierenfunktion gerechnet werden muss, sollte bei diesen die Nierenfunktion regelmässig überwacht werden. Revlimid wurde in klinischen Studien bei Patienten bis zu einem Alter von 95 Jahren eingesetzt.
Patienten mit unbehandeltem multiplen Myelom, die nicht transplantierbar sind
Bei Patienten über 75 Jahren, die mit Lenalidomid in Kombination mit Dexamethason behandelt werden, beträgt die Initialdosis von Dexamethason 20 mg/Tag an den Tagen 1, 8, 15 und 22 eines jeden 28-tägigen Behandlungszyklus.
Für Patienten über 75 Jahre, die mit Lenalidomid in Kombination mit Melphalan und Prednison behandelt werden, wird keine Dosisanpassung empfohlen.
Kinder und Jugendliche
Revlimid wurde bei pädiatrischen Patienten nicht untersucht. Daher sollte Revlimid in dieser Altersgruppe nicht eingesetzt werden.
Art der Anwendung
Revlimid Kapseln sollten jeweils etwa zur gleichen Tageszeit unabhängig von einer Mahlzeit mit etwas Wasser eingenommen werden. Die Kapseln sollen nicht geöffnet oder zerkaut werden. Unmittelbar nach dem Kontakt mit den Kapseln sollen die Hände gewaschen werden. Es ist darauf zu achten, dass das in den Kapseln enthaltene Pulver (z.B. bei einer Beschädigung einer Kapsel) nicht eingeatmet wird und nicht mit der Haut oder Schleimhaut in Kontakt kommt. Falls es zu einem Hautkontakt kommt, ist die Stelle mit Wasser und Seife zu waschen, bei Augenkontakt ist mit Wasser zu spülen.
Wurde die Einnahme einer Dosis von Revlimid vergessen und sind seit der versäumten Einnahme weniger als 12 Stunden vergangen, kann die Dosis noch eingenommen werden. Liegt der übliche Einnahmezeitpunkt mehr als 12 Stunden zurück, soll die Dosis nicht mehr eingenommen werden. Der Patient soll bis zum nächsten Tag warten und dann die nächste Dosis zur gewohnten Zeit einnehmen. Es dürfen nicht 2 Dosen auf einmal eingenommen werden.
KontraindikationenSchwangerschaft.
Gebärfähige Frauen, ausser wenn alle Bedingungen des Schwangerschaftsverhütungsprogramms erfüllt sind (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Überempfindlichkeit gegenüber Lenalidomid oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenSchwangerschaftsverhütungsprogramm
Programm bei Patientinnen
Die Bedingungen des Schwangerschaftsverhütungsprogramms müssen bei allen Patientinnen erfüllt sein, ausser wenn die Patientin erwiesenermassen nicht schwanger werden kann.
Kriterien zur Abklärung des Schwangerschaftspotentials
Eine Patientin oder Partnerin eines männlichen Patienten wird als gebärfähig klassifiziert, ausser sie erfüllt mindestens eine der folgenden Bedingungen:
-Alter ≥50 Jahre und spontan amenorrhoisch während ≥1 Jahr*
-Bestätigtes vorzeitiges Ovarialversagen
-Vorhergehende beidseitige Salpingo-Oophorektomie, Tubensterilisation oder Hysterektomie
-XY-Genotyp, Turner Syndrom, Uterus-Aplasie
* Eine Amenorrhoe nach Krebstherapie schliesst Gebärfähigkeit nicht aus.
Beratung
Bei gebärfähigen Frauen ist Lenalidomid kontraindiziert, wenn nicht alle der folgenden Bedingungen erfüllt sind:
-Die Patientin versteht das zu erwartende teratogene Risiko für das ungeborene Kind.
-Sie versteht die Notwendigkeit einer wirksamen Schwangerschaftsverhütung ohne Unterbrechung 4 Wochen vor Behandlungsbeginn, während der ganzen Behandlungsdauer inkl. Behandlungsunterbrüchen und 4 Wochen nach dem Ende der Behandlung.
-Sogar wenn eine gebärfähige Patientin amenorrhoisch ist, muss sie alle Empfehlungen zu einer wirksamen Kontrazeption befolgen.
-Sie soll fähig sein, sich an wirksame kontrazeptive Massnahmen zu halten.
-Sie ist informiert und versteht die Konsequenzen einer Schwangerschaft und die Notwendigkeit, rasch medizinischen Rat zu suchen, falls eine Schwangerschaft vermutet wird.
-Sie versteht die Notwendigkeit, und ist bereit, Schwangerschaftstests alle 4 Wochen durchführen zu lassen.
-Sie hat bestätigt, dass sie die Gefahren und notwendigen Sicherheitsmassnahmen im Zusammenhang mit der Einnahme von Lenalidomid verstanden hat.
Der verschreibende Arzt muss bei gebärfähigen Frauen sicherstellen, dass
die Patientin die obenstehenden Bedingungen erfüllt.
die Patientin die Bedingungen zur Schwangerschaftsverhütung einhält, einschliesslich der Bestätigung eines genügenden Verständnisses.
die Patientin ausreichende kontrazeptive Massnahmen während mindestens 4 Wochen vor Beginn der Behandlung angewendet hat und wirksame kontrazeptive Massnahmen während der ganzen Behandlungszeit inkl. Behandlungsunterbrüchen und während mindestens 4 Wochen nach Abschluss der Behandlung weiterführen wird. Bei Patientinnen, bei welchen eine sofortige Behandlung mit Lenalidomid notwendig ist, muss eine adäquate Kontrazeption inkl. Verwendung von Kondomen während 7 Tagen vor Beginn der Behandlung durchgeführt werden.
ein negatives Resultat eines Schwangerschaftstests vor Beginn der Behandlung vorliegt.
Kontrazeption
Gebärfähige Frauen müssen während 4 Wochen vor Beginn der Behandlung, während der ganzen Behandlungszeit inkl. Behandlungsunterbrüchen und während 4 Wochen nach Abschluss der Behandlung wirksame kontrazeptive Methoden anwenden. Bei Patientinnen, bei denen eine sofortige Behandlung mit Lenalidomid notwendig ist, muss während 7 Tagen vor Beginn der Behandlung eine wirksame Kontrazeption inkl. Verwendung von Kondomen durchgeführt werden. Falls nicht schon vorher wirksame kontrazeptive Methoden angewendet wurden, muss die Patientin an eine medizinische Beratungsstelle überwiesen werden, wo sie eine umfassende Beratung betreffend wirksamer kontrazeptiver Methoden erhält.
Die folgenden Verfahren können als wirksame kontrazeptive Methoden angesehen werden:
- von der Patientin unabhängige Methoden:
-Implantat
-Medroxyprogesteron-Acetat-Depot
-Sterilisation
- von der Patientin abhängige Methoden:
-Abstinenz von heterosexuellem Geschlechtsverkehr
-Heterosexueller Geschlechtsverkehr nur mit einem vasektomierten männlichen Partner; die Vasektomie muss durch zweimalige negative Spermauntersuchung bestätigt werden
-Orale Kontrazeptiva nur Progesteron enthaltend.
Wegen des erhöhten Risikos venöser Thromboembolien unter Lenalidomid werden kombinierte orale Kontrazeptiva nicht empfohlen. Falls eine Patientin bereits kombinierte orale Kontrazeptiva verwendet, sollte ein Wechsel zu einer anderen kontrazeptiven Methode in Betracht gezogen werden. Das Risiko venöser Thromboembolien bleibt während 4-6 Wochen nach Abschluss der Behandlung mit kombinierten oralen Kontrazeptiva bestehen. Falls andere Methoden nicht angewendet werden können, sollte eine Thromboseprophylaxe während der weiteren Verwendung der kombinierten oralen Kontrazeptiva in Betracht gezogen werden. Die Patientin sollte angemessen über das Risiko einer venösen Thromboembolie informiert werden.
Intrauterine Systeme haben ein erhöhtes Risiko von Infektionen beim Einsetzen und können zu unregelmässigen vaginalen Blutungen führen. Diese Methoden werden daher nicht empfohlen.
Schwangerschaftstests
Es müssen Schwangerschaftstests mit einer Empfindlichkeit von mindestens 25 IU/ml hCG bei gebärfähigen Frauen durchgeführt werden.
Jeder Fall einer Patientin mit einem positiven Schwangerschaftstest muss unverzüglich dem Swiss Teratogen Information Service (STIS), Lausanne mit dem Swissmedic-Formular "Meldung einer vermuteten unerwünschten Arzneimittelwirkung (UAW)" gemeldet werden.
- Vor Beginn einer Behandlung
Ein Schwangerschaftstest muss während der Konsultation, bei welcher Lenalidomid verschrieben wird, oder innerhalb von drei Tagen vor dem Besuch des verschreibenden Arztes durchgeführt werden, nachdem die Patientin während mindestens 4 Wochen eine wirksame Kontrazeption durchgeführt hat. Der Test soll sicherstellen, dass die Patientin bei Beginn der Behandlung mit Lenalidomid nicht schwanger ist.
- Vor Beginn der Behandlung, wenn sofortige Behandlung notwendig ist
Ein quantitativer hCG-Test im Serum sollte sofort durchgeführt werden. Nach wirksamer Kontrazeption inkl. Verwendung eines Kondoms während 7 Tagen muss dieser Test wiederholt werden. Falls beide Tests bestätigen, dass die Patientin nicht schwanger ist, kann mit der Behandlung begonnen werden.
- Während und bei Abschluss der Behandlung
Ein Schwangerschaftstest muss alle 4 Wochen, einschliesslich 4 Wochen nach Abschluss der Behandlung, wiederholt werden. Diese Schwangerschaftstests sollten während der Arztbesuche zur Verschreibung von Lenalidomid oder in den drei Tagen vor dem Arztbesuch durchgeführt werden.
Am besten sollten Schwangerschaftstests, Verschreibung und Abgabe von Lenalidomid am gleichen Tag erfolgen. Die Abgabe von Lenalidomid muss innerhalb von maximal 7 Tagen nach der Verschreibung erfolgen.
Programm bei Patienten
Klinische Daten belegen, dass es bei männlichen Patienten während der Einnahme von Revlimid zum Übertritt dieses Wirkstoffs in das Sperma kommt. Patienten mit gebärfähigen Partnerinnen sollten deshalb während der Behandlung mit Revlimid und mindestens für 7 Tage nach Beendigung der Behandlung beim Geschlechtsverkehr Kondome benutzen. Männer, welche Revlimid einnehmen, müssen folgende Bedingungen erfüllen:
-Sie müssen das zu erwartende teratogene Risiko verstehen, falls sie mit einer gebärfähigen Frau Geschlechtsverkehr haben.
-Sie müssen verstehen und damit einverstanden sein, während der ganzen Behandlungsdauer, inklusive Behandlungsunterbrüchen und während 7 Tagen nach Abschluss der Behandlung ein Kondom zu benützen, wenn sie mit einer gebärfähigen Frau Geschlechtsverkehr haben.
Der verschreibende Arzt muss sicherstellen, dass männliche Patienten die Notwendigkeit der Verwendung eines Kondoms während der ganzen Behandlungsdauer, inkl. Behandlungsunterbrüchen und während 7 Tagen nach Abschluss der Behandlung verstehen und damit einverstanden sind, wenn sie mit einer gebärfähigen Frau Geschlechtsverkehr haben.
Die Patienten dürfen während der Behandlung mit Revlimid und 7 Tage danach kein Sperma spenden.
Zusätzliche Vorsichtsmassnahmen
Die Patienten und Patientinnen müssen angewiesen werden, dieses Arzneimittel niemals anderen Personen zu geben und nicht verwendete Kapseln ihrem Arzt oder Apotheker nach Beendigung der Therapie zurück zu bringen.
Die Patienten sollten während der Therapie und für mindestens 1 Woche nach Absetzen von Lenalidomid kein Blut spenden.
Andere Warnhinweise und Vorsichtsmassnahmen
Neutropenie und Thrombozytopenie
Neutropenie und Thrombozytopenie gehören zu den wichtigsten dosislimitierenden Toxizitäten von Lenalidomid. Deshalb sollte ein vollständiges Blutbild mit Differenzialblutbild, Thrombozytenzahl, Hämoglobinkonzentration und Hämatokrit angefertigt werden.
Eine Unterbrechung der Behandlung und/oder Dosisreduktion kann erforderlich sein (siehe "Dosierung/Anwendung" ). Patienten mit Neutropenie sollten auf Zeichen einer Infektion überwacht werden. Patienten und Ärzte werden aufgefordert, auf Zeichen und Symptome von Blutungen, einschliesslich Petechien und Nasenbluten, zu achten, besonders bei gleichzeitiger Anwendung von Medikamenten, die das Blutungsrisiko erhöhen können. Wird eine solche Toxizität beobachtet, sollten entsprechende Massnahmen erfolgen.
Bei Patienten mit unbehandeltem multiplem Myelom, die transplantierbar sind und die Revlimid in Kombination mit Bortezomib und Dexamethason einnehmen, sollte die Beurteilung eines vollständigen Blutbildes während des ersten Behandlungszyklus alle 7 Tage (einmal wöchentlich) erfolgen und danach vor Beginn jedes anschliessenden Zyklus. Bei Fortsetzung der Behandlung mit Revlimid in Kombination mit Dexamethason sind monatliche Kontrollen (alle 4 Wochen) erforderlich.
Bei Patienten mit multiplem Myelom nach autologer Stammzelltransplantation, die Revlimid einnehmen, sollte die Beurteilung eines vollständigen Blutbildes alle 7 Tage (einmal wöchentlich) in den ersten beiden 28-Tage-Zyklen, alle 2 Wochen (Tag 1 und Tag 15) im dritten 28-Tage-Zyklus und danach alle 28 Tage (4 Wochen) erfolgen.
Bei Patienten mit unbehandeltem multiplem Myelom die nicht transplantierbar sind und die Revlimid in Kombination mit Melphalan und Prednison einnehmen, sollte die Beurteilung des vollständigen Blutbildes alle 7 Tage (1 Woche) im ersten Zyklus (28 Tage), alle 14 Tage (2 Wochen) bis zum Abschluss von 9 Zyklen, und alle 28 Tage (4 Wochen) danach erfolgen.
Das vollständige Blutbild sollte bei Patienten mit unbehandeltem multiplen Myelom, die nicht transplantierbar sind und die Revlimid in Kombination mit Dexamethason einnehmen, alle 7 Tage (wöchentlich) während der ersten 2 Zyklen, an Tag 1 und Tag 15 von Zyklus 3, und danach alle 28 Tage (4 Wochen) kontrolliert werden.
Bei Patienten mit multiplem Myelom, die mindestens eine Vorbehandlung erhalten haben und die Revlimid in Kombination mit Dexamethason einnehmen, sollte in den ersten 12 Wochen der Therapie alle 14 Tage (2 Wochen) eine Kontrolle des vollständigen Blutbildes erfolgen und danach einmal monatlich.
Bei Patienten, die Revlimid wegen MDS mit Deletion 5q-Anomalie einnehmen, sollte in den ersten 8 Wochen der Therapie einmal wöchentlich eine Kontrolle des vollständigen Blutbildes erfolgen und danach einmal monatlich.
Bei Patienten, die Revlimid wegen MCL einnehmen, sollte die Beurteilung des vollständigen Blutbildes im ersten Zyklus (28 Tage) einmal wöchentlich, während der Zyklen 2 - 4 alle 2 Wochen und anschliessend einmal monatlich erfolgen.
Bei Patienten mit vorbehandeltem follikulärem Lymphom, die mit Revlimid und Rituximab behandelt wurden, soll die Überwachung in den ersten 3 Wochen von Zyklus 1 (28 Tage) wöchentlich erfolgen, dann 14-tägig während Zyklus 2 bis einschliesslich 4 und danach zu Beginn jedes anschliessenden Zyklus.
Infektionen mit oder ohne Neutropenie
Patienten mit multiplem Myelom sind anfällig für die Entwicklung von Infektionen, einschliesslich Pneumonie. Unter Lenalidomid in Kombination mit Dexamethason wurde eine höhere Rate von Infektionen beobachtet als unter MPT. Infektionen Grad ≥3 traten im Rahmen von Neutropenien bei weniger als einem Drittel der Patienten auf. Patienten mit bekannten Risikofaktoren für das Auftreten von Infektionen müssen engmaschig überwacht werden. Alle Patienten sind anzuweisen, beim ersten Anzeichen einer Infektion (z.B. Husten, Fieber etc.) sofort einen Arzt aufzusuchen, um so durch eine frühzeitige Behandlung eine Verminderung des Schweregrades zu ermöglichen.
In seltenen Fällen wurde bei Patienten, die Lenalidomid erhielten und zuvor mit dem Hepatitis-B-Virus (HBV) infiziert worden waren, über eine Reaktivierung von Hepatitis B berichtet. In einigen Fällen führte dies zu einem akuten Leberversagen, was ein Absetzen von Lenalidomid und eine adäquate antivirale Behandlung erforderte. Der Hepatitis-B-Virus-Status ist vor Beginn der Behandlung mit Lenalidomid abzuklären. Bei Patienten, die positiv auf eine HBV-Infektion getestet wurden, sollte ein Arzt mit Erfahrung in der Behandlung von Hepatitis B herangezogen werden. Entsprechende Vorsicht ist geboten, wenn Lenalidomid bei zuvor mit HBV infizierten Patienten angewendet wird. Diese Patienten müssen während der gesamten Behandlung engmaschig auf Anzeichen und Symptome einer aktiven HBV-Infektion überwacht werden.
Wenn Lenalidomid bei Patienten über 75 Jahren, mit ISS-Stadium III, ECOG PS ≥2 oder CLcr < 60 ml/min im Rahmen einer Kombinationstherapie angewendet wurde, war die Rate von Unverträglichkeiten (unerwünschte Ereignisse Grad 3 oder 4, schwerwiegende unerwünschte Ereignisse, Behandlungsabbrüche) erhöht. Die Patienten sind unter Berücksichtigung von Alter und weiteren Komorbiditäten auf ihre Eignung, eine Lenalidomid-Kombinationstherapie zu tolerieren, sorgfältig zu beurteilen.
Venöse und arterielle thromboembolische Ereignisse (VTE/ATE)
Bei Patienten mit multiplem Myelom ist die Kombination von Lenalidomid mit Dexamethason oder anderen Chemotherapien (z.B. Melphalan und Prednison) mit einem erhöhten Risiko für venöse thromboembolische Ereignisse (vorwiegend tiefe Venenthrombosen und pulmonale Embolie) verbunden. Das Risiko von VTE ist bei der Erhaltungstherapie im multiplen Myelom nach autologer Stammzelltransplantation (ASZT) sowie bei MDS-, MCL-Patienten unter Lenalidomid-Monotherapie und FL-Patienten unter R2-Therapie geringer.
Es besteht ein erhöhtes Risiko für arterielle thromboembolische Ereignisse (vorwiegend Myokardinfarkt und zerebrovaskuläre Ereignisse) bei Patienten mit multiplem Myelom, die eine Kombinationstherapie mit Lenalidomid und Dexamethason erhalten und im geringeren Umfang bei Kombination mit Lenalidomid, Melphalan und Prednison.
Das Risiko für ATE ist bei Patienten mit multiplem Myelom, die Lenalidomid als Erhaltungstherapie nach autologer Stammzelltransplantation erhalten, geringer als bei Patienten mit multiplem Myelom, die mit einer Kombinationstherapie von Lenalidomid (entweder mit Dexamethason oder Melphalan und Prednison) behandelt werden.
Patienten mit bekannten Risikofaktoren für das Auftreten einer Thromboembolie – einschliesslich einer früher aufgetretenen Thrombose – müssen daher engmaschig überwacht werden. Die Patienten müssen deshalb angewiesen werden, bei Symptomen wie beispielsweise Kurzatmigkeit, Husten, Brustschmerzen oder Schmerzen und/oder Schwellungen an Armen und Beinen ärztliche Hilfe in Anspruch zu nehmen. Es sollten Massnahmen ergriffen werden, um alle beeinflussbaren Risikofaktoren (z.B. Rauchstopp, Kontrolle von Hypertonie und Hyperlipidämie) zu minimieren.
Die gleichzeitige Gabe von erythropoesestimulierenden Substanzen oder thromboembolische Ereignisse in der Vorgeschichte erhöhen möglicherweise bei diesen Patienten auch das Thromboserisiko. Daher sollten erythropoesestimulierende Substanzen oder andere Substanzen, die das Thromboserisiko erhöhen können, wie zum Beispiel eine Hormonersatztherapie, bei Patienten mit multiplem Myelom, die Lenalidomid mit Dexamethason erhalten, mit Vorsicht angewendet werden. Eine Hämoglobin-Konzentration von mehr als 11 g/dl sollte zum Absetzen der erythropoesestimulierenden Substanzen führen.
Die Anwendung von Arzneimitteln zur Thrombose-Prophylaxe sollte insbesondere für Patienten mit zusätzlichen thromboembolischen Risikofaktoren empfohlen werden.
Die Entscheidung für Massnahmen zur Thrombose Prophylaxe sollte nach sorgfältiger Beurteilung für jeden Patienten individuell getroffen werden.
Bei Auftreten eines thromboembolischen Ereignisses ist die Behandlung mit Lenalidomid abzubrechen und eine Standard-Antikoagulationstherapie zu beginnen. Sobald sich der Zustand des Patienten stabilisiert hat, kann die Lenalidomid-Behandlung falls erforderlich unter Beibehaltung der Antikoagulation fortgesetzt werden.
Pulmonal-arterielle Hvpertonie
Bei Patienten, die mit Lenalidomid behandelt wurden, wurde über Fälle von pulmonal-arterieller Hypertonie mit z. T. tödlichem Ausgang berichtet. Die Patienten sollten daher vor Beginn und auch während einer Lenalidomid-Therapie auf Anzeichen und Symptome einer kardiopulmonalen Grunderkrankung untersucht werden.
Myokardinfarkt
Es liegen Berichte über Myokardinfarkte bei Patienten vor, die mit Lenalidomid behandelt wurden, insbesondere von Patienten mit bekannten Risikofaktoren. Patienten mit bekannten Risikofaktoren – einschliesslich einer früher aufgetretenen Thrombose – sind engmaschig zu überwachen und es sollten Massnahmen ergriffen werden, um alle beeinflussbaren Risikofaktoren (wie z.B. Rauchen, Hypertonie und Hyperlipidämie) zu minimieren.
Sekundäre Primärmalignome (SPM)
Auf der Grundlage einer geringen Anzahl von Fällen wurde in klinischen Studien an vorbehandelten Patienten mit multiplem Myelom unter Lenalidomid/Dexamethason im Vergleich zu den Kontrollen ein numerisches Ungleichgewicht beobachtet, wobei es sich in erster Linie um Basalzell- oder Plattenepithelkarzinome der Haut handelte.
In klinischen Studien an Patienten mit neu diagnostiziertem multiplem Myelom wurde ein Anstieg invasiver sekundärer Primärmalignome einschliesslich AML und MDS beobachtet, wobei die diagnostizierten Fälle in Patienten, die Lenalidomid in Kombination mit Melphalan (Häufigkeit von 5,3 %) oder unmittelbar nach hochdosierter Melphalantherapie und ASZT (Häufigkeit von 7,5 %) erhielten, auftraten. Die beobachtete Häufigkeit der AML und MDS Fälle im Lenalidomid/Dexamethason Arm betrug 0,4 %.
Fälle von B-Zell-Malignomen (einschliesslich Morbus Hodgkin) wurden in klinischen Studien beobachtet, in denen die Patienten Revlimid nach ASZT erhielten.
Es wurde ein Anstieg solider SPMs beobachtet bei Patienten, die Lenalidomid unmittelbar nach hochdosiertem intravenösem Melphalan (HDM) und ASZT erhielten (Häufigkeit von 7,7 %).
Bei Patienten mit neu diagnostiziertem multiplem Myelom, die Lenalidomid in Kombination mit Bortezomib und Dexamethason erhielten, betrug die Häufigkeit von hämatologischen SPMs 0,0 % bis 0,8 % und die Häufigkeit von soliden SPMs 0,4 % bis 4,5 %.
Bei FL-Patienten, die mit einer Kombination aus Lenalidomid und Rituximab behandelt wurden, betrug die Häufigkeit von hämatologischen SPMs 0,7 % und die Häufigkeit von soliden SPMs 1,4 %.
Vor Beginn der Behandlung mit Lenalidomid ist sowohl der mit Lenalidomid erzielte Nutzen als auch das Risiko sekundärer Primärmalignome zu berücksichtigen. Der Arzt/die Ärztin sollten die Patienten vor und während der Behandlung mithilfe der üblichen Massnahmen zur Krebsfrüherkennung hinsichtlich des Auftretens sekundärer Primärmalignome sorgfältig untersuchen und gegebenenfalls eine Therapie einleiten.
Lebererkrankungen
Bei Patienten, die eine Lenalidomid-Behandlung in Kombination mit Dexamethason erhielten, wurde über das Auftreten von Leberinsuffizienz, darunter Fälle mit tödlichem Verlauf, berichtet: akute Leberinsuffizienz, toxische Hepatitis, zytolytische Hepatitis, cholestatische Hepatitis und gemischte zytolytische/cholestatische Hepatitis wurden gemeldet. Die Mechanismen der schweren arzneimittelbedingten Hepatotoxizität sind nach wie vor unbekannt, obwohl in manchen Fällen eine vorbestehende virale Lebererkrankung, erhöhte Ausgangswerte der Leberenzyme und möglicherweise eine Antibiotikabehandlung Risikofaktoren sein können.
Es wurde häufig über abnormale Leberfunktionswerte berichtet, die generell asymptomatisch und nach Therapieunterbrechung reversibel waren. Sobald die Leberfunktionsparameter zum Ausgangsniveau zurückgekehrt sind kann eine Behandlung mit einer niedrigeren Dosis in Betracht gezogen werden.
Lenalidomid wird über die Nieren ausgeschieden. Bei Patienten mit eingeschränkter Nierenfunktion, ist es wichtig, eine Dosisanpassung vorzunehmen, um Plasmaspiegel zu verhindern, die das Risiko für häufigere hämatologische Nebenwirkungen oder eine Hepatotoxizität erhöhen könnten. Eine Überwachung der Leberfunktion wird daher empfohlen, insbesondere bei gleichzeitig bestehenden oder in der Vorgeschichte vorkommenden viralen Leberinfektionen oder wenn Lenalidomid in Kombination mit Medikamenten verabreicht wird, von denen bekannt ist, dass sie mit Leberfunktionsstörungen assoziiert sind.
Allergische Reaktionen und schwere Hautreaktionen
Es wurde über das Auftreten von Angioödem, Anaphylaxie und schweren dermatologischen Reaktionen, einschliesslich Stevens-Johnson-Syndrom (SJS), toxischer epidermaler Nekrolyse (TEN) und Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS) berichtet. Das DRESS-Syndrom kann sich in Form einer Hautreaktion (wie Hautausschlag oder exfoliative Dermatitis) in Verbindung mit Eosinophilie, Fieber und/oder Lymphadenopathie mit systemischen Komplikationen wie Hepatitis, Nephritis, Pneumonitis, Myokarditis und/oder Perikarditis zeigen. Diese Ereignisse können tödlich sein. Bei einem Grad-2 oder Grad-3-Hautausschlag sollte eine Unterbrechung oder das Absetzen von Revlimid erwogen werden. Bei Angioödem, Anaphylaxie, Grad-4-Hautausschlag, exfoliativem oder bullösem Hautausschlag, bei Verdacht auf SJS, TEN oder DRESS muss Revlimid abgesetzt werden; nach Abbruch aufgrund dieser Reaktionen sollte die Behandlung nicht wiederaufgenommen werden. Patienten mit schwerem Hautausschlag Grad 4 in Zusammenhang mit einer Thalidomid-Behandlung sollten nicht mit Revlimid behandelt werden.
Tumorlyse-Syndrom
Es kann ein Tumorlyse-Syndrom (TLS) auftreten, einschliesslich bei Lymphompatienten. Während der Behandlung mit Revlimid wurde über tödliche Fälle von TLS berichtet.
Gefährdet sind Patienten mit einer hohen Tumorlast vor Behandlungsbeginn. Diese Patienten sind engmaschig zu überwachen, insbesondere während des ersten Zyklus oder bei einer Dosiseskalation, und es müssen geeignete Vorsichtsmassnahmen getroffen werden.
Tumor-Flare-Reaktion
Eine sorgfältige Überwachung und Untersuchung auf eine Tumor-Flare-Reaktion (TFR) wird empfohlen. Ein Tumor-Flare kann ein Fortschreiten der Erkrankung (Progress, PD) vortäuschen und kann tödlich sein. In der Zulassungsstudie MCL-001 kam es bei etwa 10 % der Patienten zu einer TFR; alle Fälle wurden als Schweregrad 1 oder 2 eingestuft und als behandlungsbedingt beurteilt.
Die TFR-Rate in der Studie NHL-007 lag bei 13,0 %, wovon ein Ereignis einem Grad 3 Ereignis entsprach. In der Studie NHL-008 lag die Rate bei 4,0 % mit einem schwerwiegenden Ereignis unter den übrigen Grad 1-2 Ereignissen. Die meisten der Ereignisse traten in Zyklus 1 auf. Bei Patienten mit TFR Grad 1 und 2 kann die Behandlung mit Lenalidomid im Ermessen des Arztes ohne Unterbruch oder Modifikation fortgesetzt werden. Zur Behandlung der TFR-Symptome wurden in den klinischen Studien MCL-001, NHL-007 und NHL-008 Patienten mit TFR Grad 1 und 2 mit Kortikosteroiden, nichtsteroidalen Antiphlogistika (NSAIDs) und/oder narkotischen Analgetika therapiert werden. Die Entscheidung betreffend therapeutischer Massnahmen sollte nach sorgfältiger klinischer Beurteilung des einzelnen Patienten getroffen werden. Bei Patienten mit TFR Grad 3 oder 4 ist die Behandlung mit Lenalidomid auszusetzen, bis sich die TFR auf ≤ Grad 1 zurückgebildet hat. Die Patienten können zur Behandlung der Symptome entsprechend den für TFR Grad 1 und 2 gegebenen Hinweisen therapiert werden.
Vorzeitiger Tod bei MCL-Patienten
In der Studie MCL-002 wurde insgesamt ein sichtbarer Anstieg der vorzeitigen Todesfälle (innerhalb von 20 Wochen) beobachtet. Patienten mit hoher Tumorlast bei Behandlungsbeginn haben ein erhöhtes Risiko eines vorzeitigen Todes, im Lenalidomid-Arm waren es 20 % (16/81) und im Kontrollarm 7 % (2/28). Im 52-Wochen-Zeitraum lagen die entsprechenden Zahlen bei 40 % (32/81) und 21 % (6/28).
Abstossungsreaktionen nach Organtransplantation
Im Rahmen von Erfahrungen nach der Marktzulassung wurde über Fälle von Organtransplantatabstossung bei Verwendung von Revlimid berichtet, wovon manche tödlich ausgingen. In der Mehrzahl der Fälle trat die Abstossungsreaktion innerhalb der ersten 2 Monate nach Therapiebeginn mit Revlimid auf. Mögliche Faktoren, die in den berichteten Fällen zur Abstossung des Organtransplantats beigetragen haben, sind die Grunderkrankung (z.B. Amyloidose), gleichzeitig auftretende Infektionen und kürzliches Absetzen oder Reduzieren der immunsuppressiven Therapie. Die Inzidenzrate von Abstossungsreaktionen bei Organtransplantaten kann aufgrund der Einschränkung der nach Marktzulassung erhobenen Sicherheitsdaten nicht zuverlässig abgeschätzt werden. In der Regel wurde Revlimid nach Auftreten der Abstossungsreaktion dauerhaft abgesetzt. Vor Therapiebeginn mit Revlimid sollte der Nutzen einer Behandlung mit Revlimid gegenüber dem Risiko einer möglichen Organtransplantatabstossung bei Empfängern von Organtransplantaten abgewogen werden.
Störungen der Schilddrüsenfunktion
Unter der Behandlung mit Lenalidomid wurden sowohl Hypothyreose als auch Hyperthyreose beobachtet (siehe "Unerwünschte Wirkungen" ). Daher wird empfohlen, vor Beginn der Revlimid-Behandlung für eine optimale Einstellung von Begleiterkrankungen zu sorgen, die einen Einfluss auf die Schilddrüsenfunktion haben können. Zu Behandlungsbeginn und während der Behandlung wird eine Überwachung der Schilddrüsenfunktion empfohlen.
Kardiale Elektrophysiologie
Unter Behandlung mit Lenalidomid sind im EKG Verlängerungen der QTc-Zeit beobachtet worden. Eine gleichzeitige Behandlung mit die QT-Zeit verlängernden Arzneimitteln und eine Behandlung bei Patienten mit Long-QT-Syndrom sollte nur unter grosser Vorsicht und regelmässiger EKG-Kontrolle erfolgen (siehe "Eigenschaften/Wirkungen" ).
Immunsuppressive Wirkung
Lenalidomid hat eine stark immunsuppressive Wirkung. Daher sollte die gemeinsame Einnahme mit anderen immunmodulierenden Wirkstoffen nur mit Vorsicht erfolgen. Die Wirkung von Impfungen kann beeinträchtigt sein. Impfungen mit Lebendorganismen sollten wegen der Gefahr einer Infektion nicht während der Behandlung mit Lenalidomid durchgeführt werden.
Kombinationstherapie
Hinweise zu anderen Arzneimitteln, die in Kombination mit Lenalidomid angewendet werden, sind der Fachinformation des jeweiligen Arzneimittels zu entnehmen.
Laktose-Intoleranz
Revlimid Kapseln enthalten Laktose. Patienten mit einer seltenen erblichen Galaktose-Intoleranz, Lapp-Laktase-Mangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Kapsel, d.h. es ist nahezu "natriumfrei" .
InteraktionenDa Lenalidomid nicht über Phase-I-Enzyme metabolisiert und nur gering an Plasmaproteine gebunden wird, sind Interaktionen über Cytochrom-P450 und über die Proteinbindung unwahrscheinlich.
Da Lenalidomid aktiv tubulär sezerniert wird, sind Wechselwirkungen mit anderen Arzneimitteln, die aktiv tubulär sezerniert werden, möglich. Erfahrungen mit erhöhten Harnsäurewerten sind gering.
Lenalidomid (10 mg) hatte keine Auswirkungen auf die Pharmakokinetik einer gleichzeitig verabreichten Einzeldosis von R- und S-Warfarin. Eine Einzeldosis von 25 mg Warfarin hatte keine Auswirkungen auf die Pharmakokinetik von gleichzeitig verabreichtem Lenalidomid. Es ist jedoch nicht bekannt, ob in der klinischen Anwendung Wechselwirkungen auftreten. Daher wird zu einer engmaschigen Überwachung der Warfarinkonzentration während der Behandlung geraten.
Eine Behandlung mit Coumarinen ist bei dem hohen Risiko einer Thrombozytopenie nicht empfohlen.
Dexamethason (40 mg/Tag) hatte keinen Einfluss auf die Pharmakokinetik von Revlimid.
Die begleitende Gabe von 10 mg Lenalidomid/Tag erhöhte die Plasmaverfügbarkeit von Digoxin (0,5 mg, Einzeldosis) um 14 % mit einem 90 %-KI (Konfidenzintervall) [0,52% - 28,2%]. Es ist nicht bekannt, ob der Effekt in der Therapiesituation (höhere Lenalidomid-Dosen und begleitende Therapie mit Dexamethason) abweicht. Daher ist während der Behandlung mit Lenalidomid eine Überwachung der Digoxin-Konzentration angezeigt.
Die gleichzeitige Verabreichung mehrerer Dosen des P-gp-Inhibitors Chinidin (600 mg zweimal täglich) hat keine klinisch relevante Auswirkung auf die Pharmakokinetik von Lenalidomid (25 mg). Die gleichzeitige Verabreichung von Lenalidomid (25 mg) und des P-gp-Inhibitors/Substrate Temsirolimus (25 mg) verändert die Pharmakokinetik beider Medikamente nicht.
Erythropoese stimulierende Wirkstoffe oder andere Wirkstoffe, welche das Thromboserisiko erhöhen können, wie z.B. eine Hormonersatztherapie, sollen bei Patientinnen und Patienten mit multiplem Myelom, die Lenalidomid mit Dexamethason erhalten, nur mit Vorsicht angewendet werden.
Schwangerschaft, StillzeitSchwangerschaft
Für Lenalidomid liegen keine klinischen Daten zur Anwendung bei Schwangeren vor. Lenalidomid ist strukturverwandt zu Thalidomid. Thalidomid ist eine bekanntermassen beim Menschen teratogen wirkende Substanz, die schwere lebensbedrohende Geburtsschäden verursacht. In einer embryofetalen Entwicklungsstudie bei trächtigen weiblichen Affen führte Lenalidomid zu Missbildungen bei den Nachkommen (siehe auch "Präklinische Daten" ). Ein teratogener Effekt von Lenalidomid ist beim Menschen zu erwarten. Details zum Schwangerschaftsverhütungsprogramm: siehe "Warnhinweise und Vorsichtsmassnahmen" .
Betreffend die Behandlung von männlichen Patienten siehe "Warnhinweise und Vorsichtsmassnahmen" .
Stillzeit
Es ist nicht bekannt, ob Lenalidomid in die Muttermilch übergeht. Daher sollte Revlimid nicht bei stillenden Frauen eingesetzt oder abgestillt werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen durchgeführt. Unter Revlimid kann es zu unerwünschten Wirkungen wie Müdigkeit, Benommenheit, Somnolenz und Verschwommensehen kommen. Daher wird zur Vorsicht geraten, wenn Patienten am Verkehr teilnehmen oder Maschinen bedienen.
Unerwünschte WirkungenMultiples Myelom
Patienten mit unbehandeltem multiplem Myelom, die transplantierbar sind und Lenalidomid in Kombination mit Bortezomib und Dexamethason erhielten
In den Studien PETHEMA GEM2012 (gepoolte Arm A und B (RVd), n=458) und IFM 2009 (Arm A (RVd), n=356) wurde folgende schwerwiegende unerwünschte Wirkung unter Lenalidomid in Kombination mit Bortezomib und Dexamethason am häufigsten (≥5 %) beobachtet:
-Pneumonie (5,9 %) in PETHEMA GEM2012.
In der PETHEMA GEM2012 Studie waren die unter Lenalidomid in Kombination mit subkutanem Bortezomib und Dexamethason am häufigsten beobachteten unerwünschten Wirkungen periphere Neuropathie (35,2 %), Neutropenie (31,9 %) und Thrombozytopenie (25,3 %).
In der IFM 2009 Studie waren die unter Lenalidomid in Kombination mit intravenösem Bortezomib und Dexamethason am häufigsten beobachteten unerwünschten Wirkungen periphere Neuropathie (54,8 %) und Lymphopenie (52,2 %).
Patienten mit multiplem Myelom, die nach autologer Stammzelltransplantation mit Lenalidomid behandelt wurden
In zwei doppelblinden, placebokontrollierten, zweiarmigen Phase-III-Studien (IFM 2005-02 und CALGB 100104) erhielten 517 Patienten Lenalidomid und 501 Patienten Placebo. Die Nebenwirkungen aus der Studie CALGB 100104 beinhalteten nicht nur Ereignisse aus der Erhaltungstherapiephase, sondern auch Ereignisse, die nach HDM/ASZT berichtet wurden. In der Studie IFM 2005-02 beziehen sich die Angaben zu den Nebenwirkungen nur auf die Erhaltungstherapiephase.
Die schwerwiegenden unerwünschten Wirkungen, die unter der Lenalidomid-Erhaltungstherapie häufiger (≥5 %) als unter Placebo beobachtet wurden, waren:
-Pneumonien (10,6 %, Sammelbegriff)
-Lungeninfektionen (9,4 %)
Die unerwünschten Wirkungen, die in beiden Studien unter der Lenalidomid-Erhaltungstherapie häufiger als unter Placebo beobachtet wurden, waren: Neutropenie (79,0 %), Thrombozytopenie (72,3 %), Diarrhoe (54,5 %), Bronchitis (47,4 %), Nasopharyngitis (34,8 %), Muskelspasmen (33,4 %), Hautausschlag (31,7 %), Leukopenie (31,7 %), Asthenie (29,7 %), Husten (27,3 %), Infektionen der oberen Atemwege (26,8 %), Fatigue (22,8 %), Gastroenteritis (22,5 %), Anämie (21,0 %) und Fieber (20,5 %).
Patienten mit unbehandeltem multiplem Myelom, die nicht transplantierbar sind und mit Lenalidomid in Kombination mit Bortezomib und Dexamethason behandelt wurden
In der Studie SWOG S0777 (Arm B (RVd), n=262) waren die schwerwiegenden unerwünschten Wirkungen, die unter Lenalidomid in Kombination mit intravenösem Bortezomib und Dexamethason häufiger (≥5 %) als unter Lenalidomid in Kombination mit Dexamethason beobachtet wurden:
-Hypotonie (6,5 %), Lungeninfektion (5,7 %), Dehydratation (5,0 %).
Unerwünschte Wirkungen, die unter Lenalidomid in Kombination mit Bortezomib und Dexamethason häufiger beobachtet wurden als unter Lenalidomid in Kombination mit Dexamethason waren Fatigue (73,7 %), periphere Neuropathie (71,8 %), Thrombozytopenie (57,6 %), Obstipation (56,1 %) und Hypokalzämie (50,0 %).
Patienten mit unbehandeltem multiplem Myelom, die mit Lenalidomid in Kombination mit niedrig dosiertem Dexamethason behandelt wurden
In einer offenen 3-armigen Phase III Studie erhielten 535 Patienten eine Kombination aus Lenalidomid und niedrig dosiertem Dexamethason bis zur Progression der Erkrankung (Rd), 541 Patienten die Kombination aus Lenalidomid und niedrig dosiertem Dexamethason bis zum Abschluss von achtzehn 28 Tages-Zyklen (Rd18) und 547 Patienten die Kombination aus Melphalan, Prednison und Thalidomid (MPT).
Schwerwiegenden Nebenwirkungen, die unter Lenalidomid in Kombination mit niedrig dosiertem Dexamethason (Rd und Rd18) häufiger (≥5 %) als unter Melphalan, Prednison und Thalidomid (MPT) beobachtet wurden, waren:
-Pneumonie (9,8 %)
-Nierenversagen (auch akutes; 6,3 %)
Nebenwirkungen, die unter Rd bzw. Rd18 häufiger als unter MPT beobachtet wurden, waren: Diarrhoe (45,5 %), Fatigue (32,8 %), Rückenschmerzen (32,0 %), Asthenie (28,2 %), Schlaflosigkeit (27,6 %), Hautausschlag (24,3 %), Appetitlosigkeit (23,1 %), Husten (22,7 %), Fieber (21,4 %) und Muskelkrämpfe (20,5 %).
Patienten mit unbehandeltem multiplem Myelom, die mit Lenalidomid in Kombination mit Melphalan und Prednison behandelt wurden
In einer doppelblinden, placebokontrollierten 3-armigen Phase III Studie wurden die Sicherheit und Wirksamkeit der Kombinationstherapie aus Melphalan, Prednison und Lenalidomid (MPR) gefolgt von einer Monotherapie mit Lenalidomid als Erhaltungstherapie beurteilt. 152 Patienten erhielten eine Induktionstherapie mit der oral verabreichten MPR-Kombination, gefolgt von Lenalidomid als Erhaltungstherapie (MPR+R), 153 Patienten erhielten eine Induktionstherapie mit der oral verabreichten MPR-Kombination, gefolgt von Erhaltungstherapie mit einem Placebo (MPR+p), und 154 Patienten erhielten eine Induktionstherapie mit der oral verabreichten MPp (MP + Placebo)-Kombination, gefolgt von Erhaltungstherapie mit einem Placebo (MPp+p).
Schwerwiegenden Nebenwirkungen, die unter Melphalan, Prednison und Lenalidomid gefolgt von einer Lenalidomid-Erhaltungstherapie (MPR+R) bzw. unter Melphalan, Prednison und Lenalidomid gefolgt von Placebo (MPR+p) häufiger (≥5 %) als unter Melphalan, Prednison und Placebo gefolgt von Placebo (MPp+p) beobachtet wurden, waren:
-Febrile Neutropenie (6,0 %)
-Anämie (5,3 %)
Nebenwirkungen, die unter MPR+R bzw. MPR+ p häufiger als unter MPp+p beobachtet wurden, waren: Neutropenie (83,3 %), Anämie (70,7 %), Thrombozytopenie (70,0 %), Leukopenie (38,8 %), Obstipation (34,0 %), Diarrhoe (33,3 %), Hautausschlag (28,9 %), Fieber (27,0 %), periphere Ödeme (25,0 %), Husten (24,0 %), Appetitlosigkeit (23,7 %) und Asthenie (22,0 %).
Patienten mit multiplem Myelom, die wenigstens eine vorangegangene Therapie erhalten haben
In placebokontrollierten Phase-III-Studien erhielten 353 Patienten die Kombination Lenalidomid/Dexamethason und 350 Patienten die Kombination Placebo/Dexamethason. Bei 325 Patienten (92 %) in der Lenalidomid/Dexamethason-Gruppe wurde mindestens eine Nebenwirkung beobachtet, verglichen mit 288 Patienten (82 %) in der Placebo/Dexamethason-Gruppe.
Die schwerwiegendsten beobachteten unerwünschten Wirkungen waren venöse Thromboembolien (tiefe Venenthrombose, Lungenembolie) und Neutropenie Grad 4.
Die am häufigsten beobachteten unerwünschten Wirkungen in der Lenalidomid/Dexamethason-Gruppe waren Neutropenie (39,4 %; Grad 4: 5,1 %), Thrombozytopenie (18,4 %, Grad 3/4: 9,9 %), Fatigue (27,2 %), Obstipation (23,5 %), Muskelkrämpfe (20,1 %), Asthenie (17,6 %), Anämie (17,0 %), Diarrhoe (14,2 %) und Hautausschlag (10,2 %), Schlaflosigkeit (26,7 %) und Muskelschwäche (10,1 %). Neutropenie und Thrombozytopenie traten hauptsächlich dosisabhängig auf und liessen sich durch eine Dosisreduzierung erfolgreich behandeln.
Myelodysplastisches Syndrom
In einer placebokontrollierten Phase-III-Studie erhielten 69 Patienten einmal täglich 10 mg Lenalidomid und 67 Patienten Placebo.
Die schwerwiegendsten beobachteten unerwünschten Wirkungen waren venöse Thromboembolien (tiefe Venenthrombose, Lungenembolie), Grad 3-4 Neutropenie, febrile Neutropenie und Grad 3-4 Thrombozytopenie.
Die am häufigsten beobachteten unerwünschten Wirkungen in der Lenalidomid-Gruppe waren Neutropenie (76,8 %; Grad 3-4: 75,4 %), Thrombozytopenie (49,3 %; Grad 3-4: 40,6 %), Diarrhoe (37,7 %), Pruritus (27,5 %), Übelkeit (20,3 %), Fatigue (18,8 %), Obstipation (17,4 %), Muskelspasmen (17,4 %), Fieber (15,9 %), Nasopharyngitis (14,5 %), Bronchitis (14,5 %) und Kopfschmerzen (14,5 %). Neutropenie und Thrombozytopenie traten hauptsächlich dosisabhängig auf und liessen sich durch eine Dosisreduzierung erfolgreich behandeln.
Mantelzell-Lymphom
In der MCL-Zulassungsstudie erhielten insgesamt 134 Patienten mindestens eine Dosis Revlimid. Infektionen waren die häufigste Art schwerwiegender unerwünschter Ereignisse.
Bei den schwerwiegenden Infektionen wurde am häufigsten über Pneumonie berichtet.
Die am häufigsten beobachteten unerwünschten Wirkungen waren Pneumonie (14,2 %; Grad 3-4: 9 %), Infektionen der oberen Atemwege (12,7 %), Neutropenie (48,5 %; Grad 3-4: 43,3 %), Thrombozytopenie (35,8 %, Grad 3-4: 27,6 %), Anämie (30,6 %; Grad 3-4: 11,2 %), Leukopenie (14,9 %; Grad 3-4: 6,7 %), Verminderter Appetit (14,2 %), Hypokaliämie (12,7 %; Grad 3-4: 2,2 %), Gewichtsabnahme (12,7 %), Husten (28,4 %), Dyspnoe (17,9 %; Grad 3-4: 6 %), Diarrhoe (31,3 %; Grad 3-4: 6 %), Übelkeit (29,9 %), Obstipation (15,7 %), Erbrechen (11,9 %), Hautausschlag (22,4 %; Grad 3-4: 1,5 %), Pruritus (17,2 %), Rückenschmerzen (13,4 %; Grad 3-4: 1,5 %), Muskelspasmen (12,7 %), Fatigue (33,6 %; Grad 3-4: 6,7 %), Fieber (23,1 %; Grad 3-4: 2,2 %), periphere Ödeme (15,7 %) und Asthenie (14,2 %; Grad 3-4: 3 %).
Follikuläres Lymphom (FL)
Das allgemeine Sicherheitsprofil von Lenalidomid in Kombination mit Rituximab bei Patienten mit vorbehandeltem follikulärem Lymphom basiert auf Daten von 146 Patienten aus der Studie NHL-007 und 177 Patienten aus der Studie NHL-008.
Die schwerwiegendsten beobachteten unerwünschten Wirkungen waren: febrile Neutropenie (2,7 %), Lungenembolie (2,7 %) und Pneumonie (2,7 %).
Die am häufigsten beobachteten unerwünschten Wirkungen in der Lenalidomid-Rituximab Gruppe waren Neutropenie (58,2 %), Diarrhoe (30,8 %), Leukopenie (28,8 %), Obstipation (21,9 %), Husten (21,9 %) und Müdigkeit (21,9 %).
Die Nebenwirkungen, die bei Patienten mit multiplem Myelom, myelodysplastischem Syndrom, Mantelzell-Lymphom und follikulärem Lymphom beobachtet wurden, sind nachstehend nach Organsystem und Häufigkeit aufgelistet. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Häufigkeitsangaben: Sehr häufig (≥1/10); häufig (≥1/100, < 1/10); gelegentlich (≥1/1'000, < 1/100); selten (≥1/10'000, < 1/1'000); sehr selten (< 1/10'000).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Bronchitis (47,4 %), Nasopharyngitis (34,8 %), Infektionen der oberen Atemwege (26,8 %), Gastroenteritis (22,5 %), neutropenische Infektionen (17,9 %), Pneumonien (17,1 %), Rhinitis (15,0 %), Sinusitis (14,0 %), Influenza (13,3 %), Harnwegsinfektionen (11,6 %).
Häufig: Bakteriämie, Sepsis, lokale und systemische Infektionen (bakterielle, virale oder Mykosen), Cellulitis, orale Candidiasis, Infektion der Atemwege, Infektion der Lunge, Infektion der unteren Atemwege, infektiöse Enterokolitis.
Gelegentlich: Atypische Pneumonie, Pneumocystis-carinii-Pneumonie, subakute Endokarditis, ophthalmischer Herpes, Herpes Zoster, Ohreninfektionen, ösophageale Candidiasis, Virus-Reaktivierung* (Hepatitis-B-Virus oder Herpes Zoster).
Sehr selten: Progressive multifokale Leukoenzephalopathie*.
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
Sehr häufig: Tumor-Flare-Reaktion (13,0 %).
Häufig: Akute myeloische Leukämie, myelodysplastisches Syndrom, Plattenepithelkarzinom der Haut, Basalzellkarzinom, Tumorlyse-Syndrom.
Gelegentlich: Akute T-Zell-Leukämie.
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig: Neutropenie (79,0 %), Thrombozytopenie (72,3 %), Lymphopenie (52,2 %), Anämie (43,8 %), Leukopenie (36,0 %), febrile Neutropenie (17,4 %).
Häufig: Panzytopenie.
Gelegentlich: Granulozytopenie, hämolytische Anämie, verlängerte Koagulation, Monozytopenie, Leukozytose, Lymphadenopathie.
Erkrankungen des Immunsystems
Gelegentlich: Erworbene Hypogammaglobulinämie, Angioödem*, akute Graft-versus-Host-Reaktion*.
Selten: Anaphylaxie*.
Unbekannt: Organtransplantatabstossung*.
Endokrine Erkrankungen
Häufig: Cushing Syndrom.
Gelegentlich: Adrenale Insuffizienz, Hypothyreose, Hyperthyreose, erhöhtes oder reduziertes TSH, Hirsutismus.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hypokalzämie (50,0 %), verminderter Appetit (34,4 %), Hyponatriämie (30,5 %), Hypokaliämie (29,0 %), Dehydration (16,4 %), Gewichtsabnahme (13,5 %), Hyperglykämie (11,7 %), Hypoglykämie (10,7 %).
Häufig: Anorexie, Hypomagnesiämie, Flüssigkeitsretention, Gewichtszunahme, Eisenüberladung, Hypophosphatämie, Hyperkalzämie, Hyperurikämie.
Gelegentlich: Metabolische Azidose, Diabetes mellitus, Hypoalbuminämie, Kachexie, Gicht, Hyperphosphatämie, gesteigerter Appetit.
Psychiatrische Erkrankungen
Sehr häufig: Schlaflosigkeit (32,8 %), Depressionen (10,9 %).
Häufig: Verwirrtheitszustände, Halluzinationen, Stimmungsschwankungen, Angst, Irritierbarkeit, Schläfrigkeit.
Gelegentlich: Psychotische Störungen, Hypomanie, Wahnvorstellungen, verminderte Libido, Persönlichkeitsveränderungen, Nervosität, Aggression, Albträume.
Erkrankungen des Nervensystems
Sehr häufig: Periphere Neuropathie (71,8 %), gestörte Geschmacksempfindung (30,2 %), Schwindel (29,4 %), Parästhesie (22,5 %), Kopfschmerzen (15,4 %).
Häufig: Hirndurchblutungsstörung, Synkope, Benommenheit, Zittern, Gedächtnisstörungen, Neuralgie, Dysästhesie, periphere sensorische Neuropathie.
Gelegentlich: Cerebraler Insult, Leukoencephalopathie, Sprechstörungen, Aufmerksamkeitsstörung, Gleichgewichtsstörung, Bewegungsstörung, orale Parästhesie, psychomotorische Hyperaktivität, Anosmie, Ataxie, Dyskinesie, motorische Dysfunktion, myasthenisches Syndrom.
Augenerkrankungen
Sehr häufig: Verschwommenes Sehen (16,0 %), Katarakt (13,7 %).
Häufig: Sehstörungen, vermehrter Tränenfluss, Konjunktivitis.
Gelegentlich: Erblindung, retinale Arteriosklerose, retinale Venenthrombose, Keratitis, Augenirritation, trockene Augen.
Erkrankungen des Ohrs und des Labyrinths
Häufig: Vertigo.
Gelegentlich: Taubheit, Gehörverminderung, Tinnitus, Ohrenschmerzen.
Herzerkrankungen
Häufig: Vorhofflimmern, Myokardinfarkt*, Herzversagen.
Gelegentlich: Stauungsherzinsuffizienz, Herzklappeninsuffizienz, Vorhofflattern, ventrikuläre Trigeminie, Bradykardie, Tachykardie, QT-Verlängerung, Lungenödeme, Arrhythmie.
Gefässerkrankungen
Sehr häufig: Hypotonie (16,4 %), tiefe Venenthrombosen (10,2 %).
Häufig: Hypertonie, Flushing, Hämatom.
Gelegentlich: Kreislaufkollaps, Ischämie, Phlebitis.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Dyspnoe (30,5 %), Husten (29,4 %).
Häufig: Lungenembolie, Atemnot, pleuritische Schmerzen, Hypoxie, oropharyngeale Schmerzen, Epistaxis, Rhinorrhoe, Dysphonie, Heiserkeit, Schluckauf.
Gelegentlich: Asthma, Brustschmerzen, pulmonal-arterielle Hypertonie.
Selten: Interstitielle Pneumonitis.
Leber- und Gallenerkrankungen
Sehr häufig: Abnormale Leberfunktionstests wie erhöhte Alaninaminotransferase (ALT; 25,6 %), erhöhte Aspartataminotransferase (AST; 21,4 %) oder Hyperbilirubinämie (15,2 %); alkalische Phosphatase im Blut erhöht (25,2 %).
Häufig: Leberzellschädigung, Hepatotoxizität, erhöhte Bilirubinwerte im Blut.
Gelegentlich: Leberinsuffizienz.
Unbekannt: Akute Leberinsuffizienz, toxische Hepatitis, zytolytische Hepatitis, cholestatische Hepatitis, gemischte zytolytische/cholestatische Hepatitis.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Obstipation (56,1 %), Diarrhoe (54,5 %), Übelkeit (37,4 %), Dyspepsie (19,1 %), Erbrechen (17,6 %), Bauchschmerzen (14,7 %), Stomatitis (12,2 %), Mundtrockenheit (11,5 %).
Häufig: Dünndarmobstruktion, Gastritis, Abdominal-Distension, Oberbauchschmerzen, Blähungen.
Gelegentlich: Gastrointestinale Blutungen, Kolitis, Proktitis, Dysphagie, Hämorrhoiden, Schmerzen im Mundbereich, Zahnfleischbluten.
Selten: Pankreatitis*.
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr häufig: Hautausschlag (31,7 %), Pruritus (27,5 %), trockene Haut (10,6 %).
Häufig: Gesichtsödeme, Erytheme, Follikulitis, Hyperpigmentierung, Exantheme, vermehrtes Schwitzen, Haarausfall, nächtliche Schweissausbrüche.
Gelegentlich: Erythema nodosum, Urtikaria, Ekzeme, Hyperkeratose, Hautfissuren, Akne, Lichen sklerosus, Lichtempfindlichkeitsreaktion, Brennen der Haut, Hautabschuppungen.
Selten: Stevens-Johnson-Syndrom*, toxische epidermale Nekrolyse*.
Sehr selten: Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen*.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Muskelspasmen (33,4 %), Rückenschmerzen (33,2 %), Muskelschwäche (24,4 %), Muskelkrämpfe (20,1 %), Arthralgie (19,0 %), Gliederschmerzen (17,9 %), Myalgie (14,9 %), Schmerzen der Skelettmuskulatur (14,8 %), Knochenschmerzen (11,8 %), muskuloskelettale Brustschmerzen (11,3 %).
Häufig: Myopathie, periphere Schwellungen, Nackenschmerzen.
Gelegentlich: Osteonekrose, Muskelatrophie, Spondylitis, Gelenkschwellungen, Steifheit der Skelettmuskulatur, lokale Schwellungen.
Erkrankungen der Nieren und Harnwege
Häufig: Niereninsuffizienz, Nierenversagen (auch akut), akuter Nierenschaden.
Gelegentlich: Häufiges Wasserlassen, Nierentubulusnekrose, Harnverhalt, erworbenes Fanconi-Syndrom, Harninkontinenz.
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Häufig: Erektile Dysfunktion, Gynäkomastie, Metrorrhagie, Brustwarzenschmerz.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Fatigue (73,7 %), periphere Ödeme (46,6 %), Asthenie (29,7 %), Fieber (23,1 %).
Häufig: Sturz, Schüttelfrost, nicht-kardiale Brustschmerzen, Prellung, Malaise.
Gelegentlich: Durst, Kältegefühl.
* = Erfahrung nach Marktzulassung
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn den Studien war die dosislimitierende Toxizität hauptsächlich hämatologischer Art. Im Fall einer Überdosierung sind Kontrollen (Klinik, Labor) sowie unterstützende Massnahmen angezeigt.
Lenalidomid ist nur wenig dialysierbar.
Eigenschaften/WirkungenATC-Code
L04AX04
Wirkungsmechanismus
Lenalidomid ist ein Derivat von Thalidomid und liegt als Racemat vor. Es besitzt sowohl immunmodulierende als auch anti-angiogenetische Eigenschaften.
Lenalidomid bindet an das intrazelluläre Protein Cereblon (CRBN). Dieses ist Teil des Ubiquitin-Ligase-Komplexes E3, welcher das Desoxyribonukleinsäure (DNA) Damage-Binding Protein 1 (DDB1), Cullin 4 (CUL4) und Roc1 umfasst. E3-Ubiquitinligasen sind für die Polyubiquitination einer Reihe verschiedener Substratproteine verantwortlich und können möglicherweise die unter der Behandlung mit Lenalidomid zu beobachtenden pleiotropen zellulären Wirkungen erklären.
Lenalidomid hemmt die Freisetzung von proinflammatorischen Zytokinen einschliesslich Tumornekrosefaktor α (TNF-α), Interleukin-1β (IL-1β), IL-6 und IL-12 aus den Lipopolysaccharid-(LPS-)stimulierten mononukleären Zellen des peripheren Bluts und steigert die Bildung des antiinflammatorischen Zytokins IL-10 in LPS-stimulierten Zellen.
Es induziert die Produktion von IL-2 und Interferon-1γ (IFN-1γ) und steigert die Proliferation der T-Zellen sowie die zytotoxische Aktivität der natürlichen Killer-Zellen.
Lenalidomid hemmt die Proliferation verschiedener hämatopoetischer Tumorzelllinien.
Die Kombination von Lenalidomid mit Rituximab erhöht beim follikulären Lymphom die NK-vermittelte, die antikörperabhängige und zellvermittelte Zytotoxizität (ADCC), die Bildung von Immunsynapsen und die direkte Apoptose, was zu einer erhöhten Antitumoraktivität der Kombination im Vergleich zu Monotherapien führt.
In in-vitro-Angiogenese-Modellen hemmt Lenalidomid die Angiogenese, indem es die Ausbildung von Mikrogefässen und endothelialen Zellkanälen wie auch die Migration von endothelialen Zellen verhindert. Lenalidomid hemmt ausserdem die Bildung des proangiogenetischen Faktors VEGF in PC-3-Prostatatumor-Zellen.
Pharmakodynamik
Kardiale Elektrophysiologie QT Studie
Bei Einmalgabe von Lenalidomid in einer Dosis von 10 mg oder 50 mg wurde bei gesunden männlichen Probanden keine Verlängerung des QTc-Intervalls festgestellt.
Klinische Wirksamkeit
Klinische Erfahrung mit Lenalidomid in Kombination mit Bortezomib und Dexamethason bei unbehandelten Patienten mit multiplem Myelom, die transplantierbar sind
Die Wirksamkeit (gemäss Ansprechkriterien der International Myeloma Working Group, IMWG) und Sicherheit von Lenalidomid in Kombination mit Bortezomib und Dexamethason (RVd) wurde in zwei multizentrischen klinischen Studien der Phase 3 bewertet: PETHEMA GEM2012 und IFM 2009.
PETHEMA GEM2012
Bei der PETHEMA GEM2012 Studie handelte es sich um eine randomisierte, kontrollierte, offene Multizenterstudie der Phase 3, in der 2 vor Transplantation verabreichte Konditionierungsregime (Busulfan-Melphalan und MEL200) bei Patienten, die RVd als initiale Therapie erhalten hatten, miteinander verglichen wurden. RVd wurde in sechs 4-wöchigen Zyklen (24 Wochen) gegeben. Die Patienten erhielten Lenalidomid 25 mg/Tag oral an den Tagen 1-21, subkutanes Bortezomib 1,3 mg/m2 an den Tagen 1, 4, 8 und 11 sowie Dexamethason 40 mg/Tag oral an den Tagen 1-4, 9-12 der sich wiederholenden 28-Tage-Zyklen. Im Anschluss an die initiale Therapie erhielten die Patienten entweder ein Konditionierungsregime mit Busulfan-Melphalan oder mit MEL200 (1:1 Randomisierung) und eine ASZT. Ausserdem erhielten die Patienten zwei zusätzliche Behandlungszyklen (8 Wochen) mit RVd im Anschluss an die ASZT. In die Studie wurden insgesamt 458 Patienten aufgenommen.
In der PETHEMA GEM2012 Studie betrug am Ende der initialen Therapie mit RVd die ≥ VGPR-Rate 67 %, die CR-Rate 33 % und 47 % (217/458) der Studienteilnehmer waren MRD-negativ. Von den Studienteilnehmern mit ≥ VGPR waren 64 % (196/305) MRD-negativ (10-4 Sensitivität). Die ≥ VGPR-Rate nach der Transplantation betrug 75 %, die CR-Rate 44 % und 59 % (287/458) der Studienteilnehmer waren MRD-negativ. Von den Studienteilnehmern mit ≥ VGPR waren 79 % (271/344) MRD-negativ (10-4 Sensitivität).
IFM 2009
Die IFM 2009 Studie war eine randomisierte, kontrollierte, offene Multizenterstudie der Phase 3 zum Vergleich von RVd mit und ohne ASZT als initiale Therapie bei transplantierbaren Patienten mit nicht vorbehandeltem multiplem Myelom. Die Patienten erhielten Lenalidomid 25 mg/Tag oral an den Tagen 1-14, intravenöses Bortezomib 1,3 mg/m2 an den Tagen 1, 4, 8 und 11 sowie Dexamethason 20 mg/Tag oral an den Tagen 1, 2, 4, 5, 8, 9, 11 und 12 der sich wiederholenden 21-Tages-Zyklen. RVd wurde als acht 3-wöchige Zyklen (24 Wochen) ohne unmittelbare ASZT (Arm A) oder als drei 3-wöchige Zyklen (9 Wochen) vor der ASZT (Arm B) gegeben. Die Patienten in Arm B erhielten ausserdem zwei zusätzliche 3-wöchige Zyklen von RVd nach der ASZT. In die Studie wurden insgesamt 700 Patienten aufgenommen.
In der IFM 2009 Studie lag die ≥ VGPR-Rate am Ende der initialen Therapie bei 68 % und die CR-Rate bei 31 %. Von den Studienteilnehmern mit ≥ VGPR waren 57 % (136/237) MRD-negativ (10-4 Sensitivität).
Klinische Erfahrung von Lenalidomid bei Patienten mit multiplem Myelom nach autologer Stammzelltransplantation
Die Wirksamkeit und Sicherheit von Lenalidomid wurde in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten, zweiarmigen Parallelgruppenstudien der Phase III untersucht: CALGB 100104 und IFM 2005-02. Der primäre Endpunkt beider Studien war das progressionsfreie Überleben (PFS).
CALGB 100104
Patienten zwischen 18 und 70 Jahren mit aktivem behandlungsbedürftigem multiplem Myelom und ohne bisherige Progression nach der initialen Therapie wurden in die Studie eingeschlossen.
Die Patienten wurden im Verhältnis 1:1 innerhalb von 90-100 Tagen nach ASZT auf die Lenalidomid-Erhaltungstherapie oder den Placebo-Arm randomisiert. Die Lenalidomid-Erhaltungsdosis betrug 10 mg einmal täglich an den Tagen 1-28 der sich wiederholenden 28-Tage-Zyklen (und wurde bei Verträglichkeit nach 3 Monaten auf 15 mg einmal täglich erhöht), und die Behandlung wurde bis zur Progression der Erkrankung fortgesetzt.
Insgesamt wurden 460 Patienten randomisiert: 231 Patienten auf Lenalidomid und 229 Patienten auf Placebo. Beide Arme waren hinsichtlich demographischer und krankheitsbezogener Charakteristika vergleichbar.
Die Studie wurde nach Erreichen einer vordefinierte PFS-Schwelle in einer geplanten Zwischenauswertung auf Empfehlung des Data Monitoring Committee entblindet. Nach der Entblindung war es den Patienten im Placebo-Arm, vor der Progression der Erkrankung, gestattet in den Lenalidomid-Behandlungsarm zu wechseln.
Die mediane Nachbeobachtungszeit betrug beim Daten Cut-off 01. Februar 2016 81,9 Monate. Das Risiko einer Progression oder Tod war zugunsten von Lenalidomid um 39 % vermindert (HR = 0,61, 95%-KI = 0,48 - 0,76; p < 0.001). Das mediane progressionsfreie Überleben betrug 56,9 Monate im Lenalidomid-Arm im Vergleich zu 29,4 Monaten im Placebo-Arm.
In der OS-Analyse betrug die beobachtete HR 0,61 (95%-KI = 0,46 - 0,81) für Lenalidomid gegenüber Placebo und wies auf ein um 39 % vermindertes Sterberisiko hin. Das mediane Gesamtüberleben betrug 111,0 Monate im Lenalidomid-Arm, verglichen von 84,2 Monaten im Placebo-Arm.
IFM 2005-02
Patienten, die bei Diagnosestellung unter 65 Jahre alt waren und sich einer Hochdosis-Chemotherapie mit nachfolgender ASZT unterzogen und zum Zeitpunkt der hämatologischen Erholung zumindest eine Stabilisierung der Erkrankung erreicht hatten, wurden in die Studie eingeschlossen.
Innerhalb von 6 Monaten nach der ASZT wurden die Patienten auf die Lenalidomid-Erhaltungstherapie oder den Placebo-Arm randomisiert. Nach zwei Lenalidomid-Konsolidierungszyklen (25 mg/Tag an den Tagen 1-21 eines 28-Tage-Zyklus) betrug die Lenalidomid-Erhaltungsdosis 10 mg einmal täglich (1-28 eines 28-Tage-Zyklus; und wurde bei Verträglichkeit nach 3 Monaten auf 15 mg einmal täglich erhöht). Die Behandlung wurde bis zur Progression der Erkrankung fortgesetzt.
Insgesamt wurden 614 Patienten randomisiert: 307 Patienten auf Lenalidomid und 307 Patienten auf Placebo.
Die Behandlung wurde bei den verbliebenen 119 Studienteilnehmenden, welche die Lenalidomid-Erhaltungstherapie (Mindestbehandlungsdauer 27 Monate) erhielten, wegen einer beobachteten ungleichen Verteilung aufgetretener SPM beendet.
Die mediane Nachbeobachtungszeit betrug beim Daten Cut-off 01. Februar 2016 96,7 Monate. Das Risiko einer Progression der Erkrankung oder Tod war um 43 % zugunsten von Lenalidomid vermindert (HR = 0,57, 95%-KI = 0,42-0,76; p < 0.001). Das mediane PFS betrug 44,4 Monate im Lenalidomid-Arm im Vergleich zu 23,8 Monaten im Placebo-Arm.
In der OS-Analyse betrug die beobachtete HR 0,90 (95%-KI = 0,72 – 1,13) für Lenalidomid gegenüber Placebo. Das mediane Gesamtüberleben betrug 105.9 Monate im Lenalidomid-Arm, verglichen mit 88,1 Monaten im Placebo-Arm.
Klinische Erfahrung mit Lenalidomid in Kombination mit Bortezomib und Dexamethason bei unbehandelten Patienten mit multiplem Myelom, die nicht transplantierbar sind
Die SWOG S0777 Studie bewertete die zusätzliche Anwendung von Bortezomib zu einer Basisbehandlung mit Lenalidomid und Dexamethason als initiale Therapie, gefolgt von einer Weiterbehandlung mit Rd bis zur Progression der Erkrankung bei Patienten mit nicht vorbehandeltem multiplem Myelom, denen keine Stammzelltransplantation unmittelbar bevorstand.
Die Patienten im Behandlungsarm mit Lenalidomid, Bortezomib und Dexamethason (RVd) erhielten Lenalidomid 25 mg/Tag oral an den Tagen 1-14, intravenöses Bortezomib 1,3 mg/m2 an den Tagen 1, 4, 8 und 11 sowie Dexamethason 20 mg/Tag oral an den Tagen 1, 2, 4, 5, 8, 9, 11 und 12 der sich wiederholenden 21-Tage-Zyklen für bis zu acht 21 Tage-Zyklen (24 Wochen). Die Patienten im Behandlungsarm mit Lenalidomid und Dexamethason (Rd) erhielten Lenalidomid 25 mg/Tag oral an den Tagen 1-21 und Dexamethason 40 mg/Tag oral an den Tagen 1, 8, 15 und 22 der sich wiederholenden 28-Tage-Zyklen für bis zu sechs 21 Tage-Zyklen (24 Wochen). Die Patienten in beiden Behandlungsarmen nahmen kontinuierlich das Rd-Schema ein: Lenalidomid 25 mg/Tag oral an den Tagen 1-21 und Dexamethason 40 mg/Tag oral an den Tagen 1, 8, 15 und 22 der sich wiederholenden 28-Tage-Zyklen. Es war vorgesehen, die Behandlung bis zur Progression der Erkrankung fortzusetzen.
Der primäre Wirksamkeitsendpunkt der Studie war das progressionsfreie Überleben (PFS). Es wurden insgesamt 523 Patienten in die Studie aufgenommen, davon wurden 263 auf RVd randomisiert und 260 auf Rd. Die demographischen Daten und die krankheitsbezogenen Ausgangsmerkmale der Patienten waren zwischen den Behandlungsarmen gut ausgewogen.
Die Ergebnisse für das PFS (IRAC-Prüfung, EMA-Zensurregeln), mit Datenstichtag 01. Dezember 2016 und einem medianen Verlaufsbeobachtungszeitraum der überlebenden Studienteilnehmer von 60,6 Monaten, zeigten eine 24 %ige Reduktion des Risikos für eine Progression der Erkrankung oder Tod zugunsten von RVd (HR = 0,76; 95 % KI 0,62; 0,94). Das mediane Gesamt-PFS betrug 41,7 Monate (95 % KI 33,1; 51,5) im RVd-Arm versus 29,7 Monate (95 % KI 24,2; 37,8) im Rd-Arm.
Bei den Teilnehmern im RVd-Arm wurde eine 28 %ige Reduktion des Risikos für Tod im Vergleich zum Rd-Arm beobachtet (HR = 0,72; 95 % KI = 0,56 bis 0,94). Das mediane OS lag bei insgesamt 89,1 Monaten (95 % KI 76,1; nicht beurteilbar) im RVd-Arm, verglichen mit 67,2 Monaten (95 % KI 58,4; 90,8) im Rd-Arm. Ebenso war die ≥ VGPR-Rate im RVd-Arm (58 %) höher als im Rd-Arm (32 %).
Klinische Erfahrung von Lenalidomid in Kombination mit Dexamethason bei unbehandelten Patienten, die nicht transplantierbar sind
Die Sicherheit und Wirksamkeit von Lenalidomid wurde in einer multizentrischen, randomisierten, unverblindeten, 3-armigen Studie der Phase III (MM-020) an Patienten untersucht, die entweder mindestens 65 Jahre alt waren oder bei denen, wenn sie jünger als 65 Jahre waren, eine Stammzelltransplantation nicht durchgeführt werden konnte, weil sie diese ablehnten oder weil eine Stammzelltransplantation dem Patienten aus Kostengründen oder anderen Gründen nicht zur Verfügung stand. In der Studie (MM-020) wurde Lenalidomid plus Dexamethason (Rd) über 2 unterschiedlich lange Anwendungsdauern (d.h. bis zur Progression der Erkrankung [Arm Rd] oder über bis zu achtzehn 28-Tage-Zyklen [72 Wochen, Arm Rd18]) mit Melphalan, Prednison plus Thalidomid (MPT) über maximal zwölf 42-Tage-Zyklen (72 Wochen) verglichen. Die Patienten wurden (1:1:1) auf einen der drei Behandlungsarme randomisiert. Die Patienten wurden bei der Randomisierung nach Alter (≤75 Jahre versus > 75 Jahre), Stadium (ISS-Stadien I und II versus Stadium III) und Land stratifiziert. Die Patienten in den Armen Rd und Rd18 nahmen an den Tagen 1 bis 21 der 28-Tage-Zyklen einmal täglich 25 mg Lenalidomid ein. Dexamethason 40 mg wurde einmal täglich an den Tagen 1, 8, 15 und 22 eines jeden 28-Tage-Zyklus eingenommen. Initialdosis und Therapieschema für Rd und Rd18 wurden nach Alter und Nierenfunktion angepasst. Patienten über 75 Jahren erhielten eine Dexamethason-Dosis von 20 mg einmal täglich an den Tagen 1, 8, 15 und 22 eines jeden 28-Tage-Zyklus. Alle Patienten erhielten während der Studie eine prophylaktische Antikoagulation (niedermolekulares Heparin, Warfarin, Heparin, niedrig dosierte Acetylsalicylsäure).
Der primäre Wirksamkeitsendpunkt der Studie war das progressionsfreie Überleben (progression free survival, PFS). Insgesamt wurden 1623 Patienten in die Studie aufgenommen, wobei 535 Patienten auf Rd, 541 Patienten auf Rd18 und 547 Patienten auf MPT randomisiert wurden. Die demographischen und krankheitsbedingten Charakteristika der Patienten waren zu Studienbeginn zwischen den 3 Armen ausgewogen. Generell war die Erkrankung bei den Studienteilnehmern in einem fortgeschrittenen Stadium: 41 % des gesamten Studienkollektivs wiesen ein ISS-Stadium III auf und 9 % hatten eine schwere Niereninsuffizienz (Kreatinin-Clearance [CLcr] < 30 ml/min). Das mediane Alter lag in den 3 Armen bei 73 Jahren.
Das PFS war unter Rd (26,0 Monate) signifikant länger als unter MPT (21,9 Monate): HR von 0,69 (95%iges KI: 0,59-0,80; p = 0.001) was auf eine 31%ige Reduktion des Risikos einer Progression oder Tod hinweist. In den beiden Behandlungsarmen trugen Todesfälle während der Laufzeit der Studie denselben Anteil (10 %) zum PFS bei. Im Rd-Arm ergab sich im Vergleich zum MPT-Arm eine Verbesserung der medianen PFS-Zeit um 4,3 Monate. Die Ansprechrate des Myeloms war unter Rd signifikant höher als unter MPT (75,1 % versus 62,3 %; p < 0,00001), wobei bei 15,1 % der Patienten im Rd-Arm ein vollständiges Ansprechen vorlag, im Vergleich zu 9,3 % der Patienten im MPT-Arm. Die mediane Zeit bis zum ersten Ansprechen betrug im Rd-Arm 1,8 Monate und im MPT-Arm 2,8 Monate.
Für die Analyse des Gesamtüberlebens (OS) betrug die mediane Beobachtungszeit für alle überlebenden Patienten 37,0 Monate, mit 574 Todesereignissen bei einem 64%igen Auftreten (574/896) der letzten OS-Ereignisse. Die beobachtete HR betrug 0,78 zugunsten Rd gegenüber MPT (95%iges KI = 0,64, 0,96; nominaler p-Wert= 0,01685) bei einer 22%igen Reduzierung des Sterberisikos.
Klinische Erfahrung von Lenalidomid in Kombination mit Melphalan und Prednison bei unbehandelten Patienten, die nicht transplantierbar sind
Die Sicherheit und Wirksamkeit von Lenalidomid wurde in einer multizentrischen, randomisierten, doppelblinden, 3-armigen Studie der Phase-III (MM-015) an Patienten beurteilt, die mindestens 65 Jahre alt waren und ein Serumkreatinin < 2,5 mg/dl aufwiesen. In der Studie wurde Lenalidomid in Kombination mit Melphalan und Prednison (MPR) mit oder ohne Lenalidomid-Erhaltungsmonotherapie bis zur Progression der Erkrankung verglichen mit der Kombination Melphalan plus Prednison über maximal 9 Zyklen. Die Patienten wurden im Verhältnis 1:1:1 in einen von drei Behandlungsarmen randomisiert: Arm MPR+R- Induktionstherapie mit der oral verabreichten MPR-Kombination, gefolgt von Lenalidomid als Erhaltungstherapie; Arm MPR+ p- Induktionstherapie mit der oral verabreichten MPR-Kombination, gefolgt von Erhaltungstherapie mit einem Placebo; Arm MPp+ p – oral MPp (MP + Placebo)- Induktionstherapie mit der oral verabreichten MPp (MP + Placebo)-Kombination, gefolgt von Erhaltungstherapie mit einem Placebo (MPp+p).
Das progressionsfreie Überleben war gemäss dem verblindeten unabhängigen Review für MPR+R signifikant länger als für MPp+p, bei einer HR von 0,388 (95%iges KI = 0,274, 0,550), die für MPR+R auf eine 61%ige Reduktion des Risikos einer Progression der Erkrankung im Vergleich zu MPp+p hinweist.
Klinische Erfahrungen bei Patienten mit multiplem Myelom, die wenigstens eine vorangegangene Therapie erhalten haben
In zwei multizentrischen, randomisierten, placebokontrollierten, parallelgruppenkontrollierten Doppelblindstudien mit gleichem Design (MM-009 in den USA und Kanada bzw. MM-010 in Europa, Israel und Australien) wurden 353 bzw. 351 mit einem oder mehreren Chemotherapieschemata vorbehandelte Patienten mit multiplem Myelom entweder mit Lenalidomid plus Dexamethason oder mit Dexamethason behandelt.
In einer gepoolten Auswertung beider Studien betrug die mediane Zeit bis zur Progression bei Patienten unter Lenalidomid/Dexamethason 48 Wochen (95% KI: 41,1; 60,1) und bei Patienten unter Placebo/Dexamethason 20,1 Wochen (95% Kl: 19,9; 20,7). Die mediane Dauer des progressionsfreien Überlebens betrug 47,3 Wochen (95% KI: 36,9; 58,4) versus 20,1 Wochen (95% KI: 18,1; 20,3). Die Gesamtüberlebenszeit war bei Lenalidomid/Dexamethason mit 90,3 vs. 80,2 Wochen, p = 0,015, signifikant höher (die Patienten im Placeboarm konnten nach Progression bzw. nach Entblindung zum Verum wechseln; 50 % wurden mit Lenalidomid/Dexamethason behandelt). Die mediane Behandlungsdauer betrug 28,1 Wochen (min: 0,1; max: 110,7).
Klinische Erfahrungen beim myelodysplastischen Syndrom
In einer multizentrischen, einarmigen, offenen Phase-II-Studie (MDS-003 in Deutschland und den USA) wurden 120 Patienten mit gesicherter Erythrozytentransfusionsabhängigkeit auf dem Boden eines MDS mit niedrigem Risiko oder intermediärem Risiko 1 mit einer zytogenetischen Deletion 5q-Anomalie mit oder ohne weitere zytogenetische Anomalien mit Lenalidomid 10 mg behandelt. Die Therapiedauer lag im Median bei 52,5 Wochen. Die Rate der Transfusionsunabhängigkeit (> 56 Tage) betrug 62,8 %. Der Anstieg des Hämoglobinwerts lag im Median bei 5,9 g/dl. Die mediane Ansprechdauer betrug 97 Wochen. Ein deutliches zytogenetisches Ansprechen wurde bei 34,6 % der Patienten und ein weniger deutlich ausgeprägtes zytogenetisches Ansprechen bei 38,5 % der Patienten beobachtet.
In einer multizentrischen, doppelblinden, placebokontrollierten, dreiarmigen Phase-III-Studie (MDS-004 in Europa und Israel) wurden 138 Patienten mit gesicherter Erythrozytentransfusionsabhängigkeit auf dem Boden eines MDS mit niedrigem Risiko oder intermediärem Risiko 1 mit einer zytogenetischen Deletion 5q-Anomalie mit oder ohne weitere zytogenetische Anomalien randomisiert mit Lenalidomid 10 mg, Lenalidomid 5 mg oder Placebo behandelt. Die Dauer der Doppelblindphase betrug 16 - 52 Wochen. Die Rate der Transfusionsunabhängigkeit (> 182 Tage) lag in der 10 mg-Gruppe bei 56,1 %. Die entsprechenden Transfusionsunabhängigkeitsraten in der 5 mg- und Placebogruppe betrugen 41,3 % bzw. 5,9 %. Die mediane Ansprechdauer lag in der 10 mg-Gruppe bei 106 Wochen; in der 5 mg- und Placebogruppe liess sie sich hingegen nicht ermitteln. Ein deutliches bzw. ein weniger deutlich ausgeprägtes zytogenetisches Ansprechen wurde bei 24,0 % bzw. 17,1 % (10 mg); 10,9 % bzw. 6,5 % (5 mg); und 0 % bzw. 0 % (Placebo) der Patienten beobachtet.
Die Rate der Transfusionsunabhängigkeit (> 56 Tage) lag in der 10 mg-Gruppe bei 61,0 %, mit einem medianen Anstieg des Hämoglobinwerts um 6,3 g/dl. Die entsprechenden Transfusionsunabhängigkeitsraten und Hämoglobinanstiege in der 5 mg- und Placebogruppe betrugen 50,0 % bzw. 7,8 % und 5,1 g/dl bzw. 2,3 g/dl.
Klinische Erfahrungen beim Mantelzell-Lymphom
Bei MCL-001 handelte es sich um eine multizentrische unkontrollierte Phase-2-Studie zur Lenalidomid-Monotherapie mit dem Ziel, die Sicherheit und Wirksamkeit von Lenalidomid bei Patienten mit Mantelzell-Lymphom zu beurteilen, bei denen es nach der Behandlung mit Bortezomib oder einem Bortezomib-haltigen Schema zu einem Rezidiv gekommen war resp. die gegen diese Behandlung refraktär waren. Nur Patienten mit nachgewiesener Translokation oder Zyklin-Überexpression sowie Patienten welche für eine Stammzelltransplantation nicht in Frage kommen wurden in die Studie eingeschlossen. Lenalidomid wurde an den Tagen 1–21 der sich wiederholenden 28-tägigen Behandlungszyklen bis zum Progress oder Auftreten einer inakzeptablen Toxizität oder Widerruf der Einwilligung verabreicht.
Voraussetzung für die Studienteilnahme war, dass die Patienten bereits mit einem Anthracyclin oder Mitoxantron, Cyclophosphamid, Rituximab und Bortezomib allein oder in Kombination vorbehandelt waren.
Eingeschlossen wurden Patienten mit einer absoluten Neutrophilenzahl (ANZ) ≥1'500 Zellen/mm3, einer Thrombozytenzahl ≥60'000 Zellen/mm3, SGOT/AST bzw. SGPT/ALT im Serum < 3,0 x ULN (obere Normgrenze) ausser bei dokumentierten Anhaltspunkten für einen Leberbefall durch das Lymphom, Gesamtbilirubin im Serum < 1,5 x ULN ausser bei Gilbert-Syndrom oder dokumentiertem Leberbefall durch das Lymphom sowie eine (nach der Cockcroft-Gault-Formel) rechnerisch ermittelte Kreatinin-Clearance > 30 ml/min.
Die primären Wirksamkeitsendpunkte der Studie MCL-001 waren die Gesamtansprechrate (ORR) und die Ansprechdauer (DOR). Ein Überblick über die Ergebnisse zur Wirksamkeit für das ITT (Intent to Treat)-Kollektiv entsprechend der Befundung durch das IRC (Independent Review Committee) findet sich in nachstehender Tabelle. Die mediane Zeit bis zum Ansprechen war 2,2 Monate (1,7 bis 13,1 Monate). Das mediane Gesamtüberleben war 19,0 Monate (95% Cl 12,5; 22,9 Monate). Das progressionsfreie Überleben bei der gesamten Studienpopulation lag bei 3,95 Monaten.
Auswertung der Ansprechrate (N = 134) N (%) 95% Cl
Gesamtansprechrate (IWRC) (CR+CRu+PR) 37 (28) (20,2; 36,0)
Komplette Remission (CR+CRu) 10 (7) (3,6; 13,3)
CR 2 (1)
CRu 8 (6)
Partielle Remission (PR) 27 (20)
Stabile Erkrankung (SD) 39 (29)
Dauer der Remissionen (Monate) Median 95 % CI
Dauer des Gesamtansprechens (CR + CRu + PR) N = 37 16,6 (7,7; 26,7)
In der Studie MCL-002 wurde in der ITT-Population insgesamt ein sichtbarer Anstieg der Todesfälle innerhalb von 20 Wochen im Lenalidomid-Arm 13 % (22/170) gegenüber dem Kontrollarm 7 % (6/84) beobachtet. Bei Patienten mit hoher Tumorlast lagen die entsprechenden Zahlen bei 20 % (16/81) und 7 % (2/28).
Klinische Erfahrungen beim follikulären Lymphom
NHL-007
Bei der Studie CC-5013-NHL-007 (AUGMENT) handelt es sich um eine Phase 3, multizentrische, doppelblinde, randomisierte Studie. Untersucht wurde die Wirksamkeit und Sicherheit von Lenalidomid in Kombination mit Rituximab (R2) versus Rituximab plus Placebo bei Patienten mit rezidivierendem/refraktärem indolentem Lymphom.
Insgesamt 358 Patienten im Alter von mindestens 18 Jahren mit FL von Grad 1, 2 oder 3A (N = 295) oder histologisch gesichertem Marginalzellen-Lymphom (MZL) wurden im Verhältnis 1:1 randomisiert. Die Patienten waren zuvor mit mindestens einer systemischen Chemotherapie, Immuntherapie oder Immun-Chemotherapie behandelt worden. Die Patienten mussten mindestens 2 vorherige Dosen Rituximab erhalten haben und durften nicht Rituximab-refraktär sein.
Lenalidomid wurde in einer oralen Dosis von 20 mg einmal täglich an den ersten 21 Tagen der sich wiederholenden 28-tägigen Behandlungszyklen gegeben und zwar über 12 Zyklen oder bis zum Auftreten einer nicht akzeptablen Toxizität. Die Dosis Rituximab betrug 375 mg/m2 einmal wöchentlich in Zyklus 1 (Tag 1, 8, 15 und 22) und an Tag 1 jedes 28-tätigen Zyklus ab Zyklus 2 bis einschliesslich Zyklus 5.
Der primäre Wirksamkeitsendpunkt der Studie war das progressionsfreie Überleben (PFS). Das mediane progessionsfreie Überleben (PFS) war im R2-Arm bei Patienten mit FL signifikant länger (39,4 Monate; 95% KI 25,1; NE) als im Kontrollarm (13,8 Monate; 95% KI: 11,2; 16,0); das Rezidivrisiko wurde um 60 % reduziert (HR 0,40; 95% KI: 0,29; 0,55). Das Ergebnis des primären Endpunkts war klinisch und statistisch signifikant.
Darüber hinaus zeigen FL-Patienten im R2-Arm eine höhere Gesamtansprechrate (ORR) (ORR 80,3 %; 95% CI: 72,9; 86,4) im Vergleich zur Rituximab-Monotherapie (ORR 55,4; 95% CI: 47,0; 63,6). Die mediane Ansprechzeit betrug 36,6 Monate im R2-Arm und 15,5 Monate im Kontrollarm. Die Mortalität, gemessen als Gesamtüberlebensrate (OS) nach 2 Jahren, wurde im R2-Arm um 55 % (HR 0,45; 95% CI:0,22; 0,92) reduziert, das heisst nach 2 Jahren lebten 94,8 % der Patienten im R2 Arm verglichen mit 85,8 % bei Patienten unter Rituximab-Monotherapie.
NHL-008
Die Studie NHL-008 ist eine offene und randomisierte Phase 3 Studie in Patienten (N = 232) mit rezidiviertem oder refraktärem FL (Grad 1-3B), MZL oder MCL. Im Unterschied zu Studie NHL-007 schloss Studie NHL-008 Patienten ein, die gegenüber Rituximab refraktär waren das heisst entweder nicht auf die Behandlung ansprachen, oder innerhalb von 6 Monaten nach der Rituximab-Behandlung ein Rezidiv zeigten, oder die sowohl gegenüber Rituximab als auch gegenüber der Chemotherapie refraktär waren.
Nach einer initialen gemeinsamen Behandlungsphase mit Rituximab + Lenalidomid (R2) über 12 Zyklen wurden die Patienten für die anschliessende Erhaltungstherapie randomisiert, um entweder die Kombinationstherapie R2 (oder nach Zyklus 18 optional Lenalidomid-Monotherapie) oder Rituximab-Monotherapie zu erhalten.
Während der Induktionsbehandlung wurde Lenalidomid in einer Dosis von 20 mg an den Tagen 1-21 der sich wiederholenden 28-tägigen-Zyklen gegeben und zwar für bis zu 12 Zyklen oder bis eine nicht akzeptable Toxizität auftrat oder die Einwilligung zur Teilnahme an der Studie zurückgezogen wurde. Die Dosis Rituximab betrug 375 mg/m2 pro Woche in Zyklus 1 (Tag 1, 8, 15 und 22) und an Tag 1 jedes zweiten 28-tägigen Zyklus (Zyklen 3, 5, 7, 9 und 11) für bis zu 12 Behandlungszyklen
Der primäre Wirksamkeitsendpunkt der Induktionsphase der Studie war die Gesamtansprechrate (ORR) unter Verwendung der modifizierten Response-Kriterien der International Working Group (IWGRC) von 1999. Die vorliegenden Ergebnisse basieren auf der Interimsanalyse aus der initialen R2-Behandlungsphase.
Nach der Induktionsphase von 12 Zyklen betrug der ORR aller Studienteilnehmer mit FL (n=148) 70,3 %; Rituximab-refraktäre-Patienten (N = 60) hatten einen ORR von 58,3 %, während Rituximabnicht-refraktäre Patienten (N = 88) einen ORR von 79,3% hatten.
PharmakokinetikAbsorption
Lenalidomid wird schnell absorbiert mit einer Tmax von 1 Stunde. Die orale Bioverfügbarkeit beträgt etwa 70 %. Die Pharmakokinetik von Lenalidomid verläuft dosisproportional.
Bei gleichzeitiger Verabreichung mit einer fettreichen Mahlzeit wird bei gesunden Freiwilligen das Ausmass der Resorption vermindert, so dass es zu einer etwa 20%igen Abnahme der Fläche unter der Konzentrations-Zeit-Kurve (AUC) und einer 50%igen Abnahme der Cmax im Plasma kommt.
Populationspharmakokinetische Analysen zeigen, dass die orale Absorptionsrate von Lenalidomid bei MCL-Patienten mit der bei Patienten mit MM oder MDS beobachteten vergleichbar ist.
Distribution
Die Bindung von Lenalidomid an Plasmaproteine ist gering (< 30 %). Ob Lenalidomid die Blut-Hirn-Schranke passiert, ist nicht untersucht.
Nach Verabreichung einer Tagesdosis von 25 mg tritt Lenalidomid in das Sperma über (< 0,01 % der Dosis). Drei Tage nach dem Absetzen des Arzneimittels ist Lenalidomid in der Samenflüssigkeit gesunder Probanden nicht mehr nachweisbar.
Metabolismus
Der Metabolismus von Lenalidomid ist gering und erfolgt nicht über Phase-I-Enzyme. Die beim Menschen in vivo im Blut auftretende Hauptkomponente ist unverändertes Lenalidomid. Als Metaboliten wurden 5-Hydroxyl-Lenalidomid und N-Acetyl-Lenalidomid identifiziert, von denen jeder weniger als 5 % der Blutspiegel der Muttersubstanz erreicht.
Elimination
Ungefähr zwei Drittel einer Lenalidomid-Dosis werden unverändert über die Nieren ausgeschieden.
Die renale Clearance von Lenalidomid übersteigt die glomeruläre Filtrationsrate; daher wird Lenalidomid zumindest in gewissem Umfang aktiv sezerniert.
In den therapeutischen Dosierungen (bis 25 mg/Tag) beträgt die Halbwertszeit im Plasma bei gesunden Freiwilligen etwa 3 Stunden und liegt bei Patienten zwischen 3 und 5 Stunden.
Steady-State-Konzentrationen werden an Tag 4 erreicht. Es kommt nicht zu einer Akkumulation bei Mehrfachdosierung.
Kinetik spezieller Patientengruppen
Daten zur Pharmakokinetik bei pädiatrischen Patienten liegen nicht vor.
Lenalidomid wird hauptsächlich als unveränderter Wirkstoff über glomeruläre Filtration und aktive tubuläre Sekretion eliminiert. Nach einer Einzeldosis von 25 mg ist bei leichtgradiger Niereninsuffizienz (ClCr 80-50 ml/min) die AUC um 25 % erhöht, bei mässiggradiger Niereninsuffizienz (ClCr 50-30 ml/min) ist die AUC um das 3-fache und bei schwerer Niereninsuffizienz (ClCr < 30 ml/min) und/oder dialysepflichtiger Niereninsuffizienz (Interdialyse Periode) um das 4 bis 5-fache erhöht. Die Eliminationshalbwertszeit verlängert sich bei mässiggradiger Niereninsuffizienz um das 3-fache auf 9-10 Stunden.
Leberfunktionsstörungen
Populationspharmakokinetische Analysen schlossen Patienten mit leicht eingeschränkter Leberfunktion (N = 16, Gesamtbilirubin > 1,0 bis ≤1,5 x ULN oder AST > ULN) mit ein und zeigen, dass eine leicht eingeschränkte Leberfunktion die Disposition von Lenalidomid nicht beeinflusst. Zu Patienten mit mässig bis stark eingeschränkter Leberfunktion liegen keine Daten vor.
Präklinische DatenKurzzeit- / Langzeittoxizität
Lenalidomid weist ein geringes Potenzial für akute Toxizität auf; bei Nagetieren betrugen die niedrigsten letalen Dosen nach oraler Verabreichung mehr als 2'000 mg/kg. Die Langzeitverabreichung von Lenalidomid führte bei Ratten, am auffälligsten bei weiblichen Tieren, zu einer Mineralisation des Nierenbeckens. Die Dosis, bei der keine Nebenwirkungen auftreten (no observed adverse effect level, NOAEL), wird für Ratten auf weniger als 75 mg/kg geschätzt und ist damit, basierend auf der AUC, um etwa das 25-fache höher als die menschliche Tagesexposition bei einer Dosis von 25 mg/Tag. Bei Affen führten wiederholte orale Gaben zu einer dosisabhängigen Abnahme der Neutrophilen-Zahl; dieser Effekt ist durch die pharmakodynamische Wirkung des Wirkstoffes bedingt. Wiederholte orale Gaben von 4 und 6 mg/kg an Affen über einen Zeitraum von bis zu 20 Wochen führten zu Mortalität und erheblicher Toxizität (deutliche Gewichtsabnahme, Abnahme der Zahl roter und weisser Blutkörperchen sowie der Thrombozytenzahl, multiple Organblutungen, Entzündungen des Gastrointestinaltraktes, Atrophie des Lymphgewebes und des Knochenmarks). Die Verabreichung von 1 und 2 mg/kg/Tag über 52 Wochen führte bei Affen zu Veränderungen im Zellanteil des Knochenmarks, einer leichten Abnahme des Verhältnisses myeloider zu erythroiden Zellen und zu Thymusatrophie. Bei 1 mg/kg/Tag wurde eine geringe Suppression der Leukozytenzahl beobachtet. Der NOAEL lag bei 1 mg/kg/Tag. Die AUC-Exposition entspricht bei dieser Dosis der human-therapeutischen Exposition bei 25 mg/Tag.
Mutagenität / Karzinogenität
In-vitro- (bakterielle Mutationen, menschliche Lymphozyten, Mauslymphom, embryonale Zelltransformation beim Syrischen Hamster) und in-vivo- (Mikronukleustest an Ratten) Mutagenitätsstudien zeigten keine wirkstoffbedingten Effekte, weder auf genetischer noch auf chromosomaler Ebene. Kanzerogenitätsstudien mit Lenalidomid wurden nicht durchgeführt.
Reproduktionstoxizität
An Ratten, Kaninchen und Affen wurden Studien zur Entwicklungstoxizität (embryo-fötale Toxizität/Teratogenität) durchgeführt. In einer Studie an Affen wurde Lenalidomid in Dosen von bis zu 4 mg/kg/Tag verabreicht. Die Studienergebnisse zeigen, dass die Gabe von Lenalidomid an trächtige weibliche Affen zu Missbildungen bei den Nachkommen führte, welche vergleichbar zu Thalidomidmissbildungen waren.
Bei Kaninchen, welche eine orale Gabe von 3, 10 und 20 mg/kg/Tag erhielten, war die Entwicklungstoxizität bei Dosierungen von 10 und 20 mg/kg/Tag charakterisiert durch leicht reduziertes Körpergewicht der Feten, häufigere Postimplantationsverluste (frühe und späte Resorptionen und intrauterine Todesfälle), sowie makroskopische äussere Befunde bei den Feten, verbunden mit Morbidität und pharmakotoxischen Effekten durch Lenalidomid (violette Verfärbung der Haut am ganzen Körper). Bei 10 mg und 20 mg/kg/Tag wurden bei den Feten Veränderungen von Weichteilen und Skelett beobachtet, die jedoch für den verwendeten Kaninchenstamm typisch sind. Die maternale und entwicklungsbezogenen NOAELs für Lenalidomid betrugen bei Kaninchen 3 mg/kg/Tag.
Wie aus früheren Thalidomiduntersuchungen an Ratten bekannt, zeigte auch eine embryo-fötale Entwicklungsstudie an Ratten bei Lenalidomid-Gaben bis zu 500 mg/kg/Tag keine teratogene Wirkung. Bei Gaben von 100, 300 oder 500 mg/kg/Tag zeigte sich eine minimale maternale Toxizität, unter anderem geringe, vorübergehende Reduzierung der durchschnittlichen Körpergewichtszunahme und Nahrungsaufnahme.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
In der Originalverpackung, nicht über 25 °C und für Kinder unzugänglich aufbewahren.
Hinweise für die Handhabung
Wie bei den Zytostatika ist auch bei der Handhabung und Entsorgung von Revlimid besondere Vorsicht geboten (siehe auch "Dosierung/Anwendung" ).
Zulassungsnummer57712 (Swissmedic)
PackungenRevlimid 2,5 mg: 21 Hartkapseln (A)
Revlimid 5 mg: 21 Hartkapseln (A)
Revlimid 7,5 mg: 21 Hartkapseln (A)
Revlimid 10 mg: 21 Hartkapseln (A)
Revlimid 15 mg: 21 Hartkapseln (A)
Revlimid 20 mg: 21 Hartkapseln (A)
Revlimid 25 mg: 21 Hartkapseln (A)
ZulassungsinhaberinBristol-Myers Squibb SA, Steinhausen
Stand der InformationAugust 2022
|