ZusammensetzungWirkstoffe
Eculizumab, hergestellt durch rekombinante DNA-Technologie in NS0-Zelllinien.
Hilfsstoffe
Natriumphosphat monobasisch monohydrat
Natriumphosphat dibasisch heptahydrat
Natriumchlorid
Polysorbat 80 (hergestellt aus gentechnisch verändertem Mais)
Wasser für Injektionszwecke
Eine Dosis (Durchstechflasche) enthält 115 mg Natrium.
Darreichungsform und Wirkstoffmenge pro EinheitKonzentrat zur Herstellung einer Infusionslösung.
Jede Durchstechflasche mit 30 ml enthält 300 mg Eculizumab (10 mg/ml).
Nach Verdünnung beträgt die endgültige Konzentration der zu infundierenden Lösung 5 mg/ml.
Indikationen/AnwendungsmöglichkeitenSoliris (Eculizumab) ist indiziert zur Behandlung
von Erwachsenen, Kindern und Jugendlichen mit
paroxysmaler nächtlicher Hämoglobinurie (PNH)
Der klinische Nutzen ist bei Patienten mit Hämolyse und klinischen Symptomen einer hohen Krankheitsaktivität nachgewiesen, unabhängig von der Transfusionshistorie (siehe "Eigenschaften/Wirkungen" ).
atypischem Hämolytisch-Urämischem Syndrom (aHUS) (siehe "Eigenschaften / Wirkungen" )
von Erwachsenen, Kindern ab 6 Jahren und Jugendlichen mit
refraktärer generalisierter Myasthenia gravis (gMG), die Azetylcholinrezeptor (AChR)-Antikörper-positiv sind (siehe "Eigenschaften/Wirkungen" ).
von Erwachsenen mit
-Neuromyelitis-optica-Spektrumerkrankungen (NMOSD), die positiv für Anti-Aquaporin-4(AQP4)-Antikörper sind und einen schubförmigen Krankheitsverlauf zeigen (siehe "Eigenschaften/Wirkungen" ).
Dosierung/AnwendungSoliris muss von einer medizinischen Fachperson und unter der Aufsicht eines mit der Behandlung von Patienten mit hämatologischen Erkrankungen oder neuromuskulären oder neuroinflammatorischen Erkrankungen erfahrenen Arztes verabreicht werden. Die behandelnden Ärzte müssen mit den Patienten über Nutzen und Risiken der Soliris-Behandlung sprechen und ihnen die Patienten-Informationsbroschüre und die Patientenkarte aushändigen.
Die Therapie mit Soliris darf nicht ohne vorherige Impfung gegen Neisseria meningitidis eingeleitet werden, die mindestens 2 Wochen vor Beginn der Behandlung verabreicht werden muss. Patienten unter 2 Jahren und solche, die Soliris eher als 2 Wochen nach einer Meningokokkenimpfung erhalten, müssen bis 2 Wochen nach der Impfung eine geeignete Antibiotikaprophylaxe erhalten (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Für Patienten, welche die Infusionen im Krankenhaus gut vertragen haben, kann eine Heiminfusion in Betracht gezogen werden. Die Entscheidung darüber, ob ein Patient Heiminfusionen erhalten kann, sollte nach entsprechender Prüfung auf Empfehlung des behandelnden Arztes erfolgen. Heiminfusionen müssen von einer qualifizierten medizinischen Fachkraft verabreicht werden.
Paroxysmale nächtliche Hämoglobinurie (PNH) bei Erwachsenen
Das Dosierungsschema zur Behandlung der PNH bei Erwachsenen (≥18 Jahre) besteht aus einer 4-wöchigen Initialphase, an die sich eine Erhaltungsphase anschliesst:
-Initialphase: 600 mg Soliris als intravenöse Infusion, die über 25–45 Minuten (35 ± 10 Minuten) einmal wöchentlich in den ersten 4 Wochen verabreicht wird.
- Erhaltungsphase: 900 mg Soliris als intravenöse Infusion, die über 25–45 Minuten (35 ± 10 Minuten) in Woche 5 verabreicht wird, gefolgt von 900 mg Soliris als intravenöse Infusion, die über 25-45 Minuten (35 ± 10 Minuten) alle 14 ± 2 Tage verabreicht wird (siehe "Eigenschaften / Wirkungen" ).
Atypisches Hämolytisch-Urämisches Syndrom (aHUS), refraktäre generalisierte Myasthenia gravis (gMG) und Neuromyelitis-optica-Spektrumerkrankungen (NMOSD) bei Erwachsenen
Das Dosierungsschema zur Behandlung von aHUS, refraktärer gMG und NMOSD bei Erwachsenen (≥18 Jahre) besteht aus einer 4-wöchigen Induktionsphase, an die sich eine Erhaltungsphase anschliesst:
-Induktionsphase: 900 mg Soliris als intravenöse Infusion, die über 25-45 Minuten (35 ± 10 Minuten) einmal wöchentlich in den ersten 4 Wochen verabreicht wird.
-Erhaltungsphase: 1200 mg Soliris als intravenöse Infusion, die über 25-45 Minuten (35 ± 10 Minuten) in Woche 5 verabreicht wird, gefolgt von 1200 mg Soliris als intravenöse Infusion, die über 25-45 Minuten (35 ± 10 Minuten) alle 14 ± 2 Tage verabreicht wird (siehe "Eigenschaften / Wirkungen" ).
Refraktäre gMG
Die verfügbaren Daten lassen darauf schliessen, dass in der Regel nach 12 Wochen Soliris-Behandlung ein klinisches Ansprechen erzielt wird. Wenn sich bei einem Patienten nach 12 Wochen kein therapeutischer Nutzen zeigt, sollte erwogen werden, die Behandlung abzubrechen.
Kinder und Jugendliche mit PNH, aHUS oder refraktärer gMG:
Kinder und Jugendliche mit PNH, aHUS oder refraktärer gMG (siehe Klinische Wirksamkeit) mit einem Körpergewicht von 40 kg und darüber werden mit der Dosierung für Erwachsene behandelt.
Bei Kindern und Jugendlichen mit PNH, aHUS oder refraktärer gMG (siehe Klinische Wirksamkeit) mit einem Körpergewicht von unter 40 kg wird Soliris folgendermassen dosiert:
Körpergewicht des Induktionsphase Erhaltungsphase
Patienten
30 bis < 40 kg 600 mg wöchentlich in den ersten 2 900 mg in Woche 3; dann 900 mg alle
Wochen 2 Wochen
20 bis < 30 kg 600 mg wöchentlich in den ersten 2 600 mg in Woche 3; dann 600 mg alle
Wochen 2 Wochen
10 bis < 20 kg 600 mg als Einmaldosis in Woche 1 300 mg in Woche 2; dann 300 mg alle
2 Wochen
5 bis < 10 kg 300 mg als Einmaldosis in Woche 1 300 mg in Woche 2; dann 300 mg alle
3 Wochen
Soliris wurde bei Patienten mit PNH oder refraktärer gMG, die weniger als 40 kg wiegen, nicht untersucht. Die Dosierung für Soliris bei Kindern und Jugendlichen mit PNH oder refraktärer gMG und einem Körpergewicht unter 40 kg entspricht der gewichtsbasierten Dosierungsempfehlung für Kinder und Jugendliche mit aHUS. Auf der Grundlage der pharmakokinetischen (PK)/pharmakodynamischen (PD) Daten, die bei mit Soliris behandelten Patienten mit aHUS oder PNH vorliegen, wird erwartet, dass dieses auf dem Körpergewicht basierende Dosierungsschema bei Kindern und Jugendlichen zu einem ähnlichen Wirksamkeits- und Sicherheitsprofil wie bei Erwachsenen führt.
Für Patienten mit refraktärer gMG und einem Körpergewicht unter 40 kg wird erwartet, dass das auf dem Körpergewicht basierende Dosierungsschema zu einem ähnlichen Wirksamkeits- und Sicherheitsprofil wie bei Erwachsenen führt.
Soliris wurde nicht bei Kindern und Jugendlichen mit NMOSD untersucht.
Bei gleichzeitiger Plasmapherese (PP), gleichzeitigem Plasmaaustausch (PE) oder Infusion mit Fresh Frozen Plasma (PI) ist eine zusätzliche Gabe von Soliris erforderlich, wie unten beschrieben:
Art der Plasmainterv Letzte Soliris- Dosis Zusätzliche Soliris-Dosi Zeitpunkt der zusätzlichen
ention s nach jeder Interventio Soliris- Dosis
n mit PP/PE/PI
Plasmapherese oder 300 mg 300 mg nach jeder Innerhalb von 60 Minuten
Plasmaaustausch Plasmapherese- oder nach jeder Plasmapherese-
Plasmaaustausch-Sitzung oder Plasmaaustausch-Sitzu
ng
≥ 600 mg 600 mg nach jeder
Plasmapherese- oder
Plasmaaustausch-Sitzung
Infusion mit Fresh ≥ 300 mg 300 mg pro Infusion mit 60 Minuten vor jeder
Frozen Plasma Fresh Frozen Plasma Infusion mit Fresh Frozen
Plasma
Abkürzungen: PP/PE/PI = Plasmapherese/Plasmaaustausch/Plasmainfusion
Bei gleichzeitiger Behandlung mit intravenösem Immunglobulin (IVIg) ist eine zusätzliche Gabe von Soliris erforderlich, wie unten beschrieben (siehe "Interaktionen" ).
Letzte Soliris-Dosis Zusätzliche Soliris-Dosis Zeitpunkt der zusätzlichen Soliris-Dosis
≥ 900 mg 600 mg pro IVIg-Zyklus So bald wie möglich nach dem IVIg-Zyklus
≤ 600 mg 300 mg pro IVIg-Zyklus
Abkürzung: IVIg = intravenöses Immunglobulin
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Soliris wurde bei Patienten mit Leberfunktionsstörung nicht untersucht.
Patienten mit Nierenfunktionsstörungen
Für Patienten mit Nierenfunktionsstörungen ist eine Dosisanpassung nicht erforderlich (siehe "Eigenschaften / Wirkungen" ).
Ältere Patienten
Soliris kann an ältere Patienten (≥65 Jahren) verabreicht werden. Es gibt keine Hinweise, die darauf hindeuten, dass besondere Vorsichtsmassnahmen bei der Behandlung älterer Menschen erforderlich sind – obwohl die Erfahrungen mit Soliris in dieser Patientenpopulation noch begrenzt sind.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Soliris bei Kindern mit refraktärer gMG im Alter von unter 6 Jahren ist nicht erwiesen.
Die Sicherheit und Wirksamkeit von Soliris bei Kindern mit NMOSD im Alter von unter 18 Jahren ist nicht erwiesen.
Art der Anwendung
Anweisungen zur Zubereitung der verdünnten Lösungen sind in "Sonstige Hinweise" aufgeführt.
Nicht als intravenöse Druck- oder Bolusinjektion verabreichen. Soliris darf nur wie nachstehend beschrieben durch intravenöse Infusion verabreicht werden.
Die verdünnte Soliris-Lösung wird intravenös als Tropfinfusion oder mittels einer Spritzenpumpe oder einer volumetrischen Infusionspumpe über 25-45 Minuten (35 ± 10 Minuten) bei erwachsenen und 1-4 Stunden bei pädiatrischen Patienten im Alter unter 18 Jahren mittels Schwerkraftinfusion, mit einer Spritzenpumpe oder einer Infusionspumpe verabreicht werden. Es ist nicht erforderlich, die verdünnte Soliris-Lösung während der Verabreichung am Patienten vor Licht zu schützen.
Die Patienten sollen nach der Infusion eine Stunde lang überwacht werden. Falls während der Verabreichung von Soliris eine Nebenwirkung auftritt, kann die Infusion nach Ermessen des Arztes verlangsamt oder abgesetzt werden. Wenn die Infusion verlangsamt wird, darf die Gesamtinfusionsdauer bei Erwachsenen und Jugendlichen zwei Stunden und bei pädiatrischen Patienten unter 18 Jahren vier Stunden nicht überschreiten.
Es liegen begrenzte unterstützende Sicherheitsdaten zu Infusionen im häuslichen Umfeld vor. Zusätzliche Vorsichtsmassnahmen im häuslichen Umfeld, wie z.B. die Verfügbarkeit einer Notfallversorgung bei Infusionsreaktionen oder Anaphylaxie, werden empfohlen. Infusionsreaktionen werden in den Abschnitten "Warnhinweise und Vorsichtsmassnahmen" und "Unerwünschte Wirkungen" beschrieben.
Monitoring der Behandlung: Patienten mit aHUS sollen hinsichtlich Anzeichen und Symptomen einer thrombotischen Mikroangiopathie (TMA) beobachtet werden (siehe "Labormedizinische Überwachung" ).
Die Soliris-Behandlung ist als lebenslange Behandlung empfohlen, es sei denn ein Absetzen ist aus medizinischen Gründen indiziert (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
KontraindikationenÜberempfindlichkeit gegen Eculizumab, murine Proteine oder einen der in Abschnitt "Zusammensetzung" genannten sonstigen Bestandteile.
Die Therapie mit Soliris darf nicht eingeleitet werden bei Patienten (siehe "Warnhinweise und Vorsichtsmassnahmen" ):
mit nicht ausgeheilter Infektion mit Neisseria meningitidis
ohne aktuellen Impfschutz gegen Neisseria meningitidis (es sei denn, sie erhalten eine geeignete Antibiotikaprophylaxe bis zwei Wochen nach Impfung)
mit hereditären Komplementdefekten
Warnhinweise und VorsichtsmassnahmenEs wird nicht damit gerechnet, dass sich Soliris bei PNH-Patienten auf die aplastische Komponente der Anämie auswirkt.
Meningokokkeninfektion:
Aufgrund seines Wirkungsmechanismus erhöht Soliris die Anfälligkeit des Patienten für eine Meningokokkeninfektion (Neisseria meningitidis). Eine Meningokokkenerkrankung kann durch jede Serogruppe auftreten. Zur Verringerung des Infektionsrisikos müssen alle Patienten mindestens 2 Wochen vor der Verabreichung von Soliris geimpft werden es sei denn, das Risiko, das mit einer Verzögerung der Soliris-Therapie verbunden wäre, wiegt schwerer als die Risiken einer Meningokokkeninfektion. Patienten, die eine Behandlung mit Soliris früher als 2 Wochen nach einer tetravalenten Meningokokkenimpfung beginnen, müssen bis 2 Wochen nach der Impfung eine geeignete Antibiotikaprophylaxe erhalten. Impfstoffe gegen die Serotypen A, C, Y, W 135 werden empfohlen, um Infektionen mit den häufig pathogenen Meningokokken-Serogruppen zu verhindern. Sofern verfügbar, wird auch eine Impfung gegen die Serogruppe B empfohlen. Patienten müssen eine Impfung gemäss den nationalen Impfempfehlungen erhalten.
Eine Impfung kann das Komplement zusätzlich aktivieren. Folglich können sich bei Patienten mit komplementvermittelten Erkrankungen, einschliesslich PNH, aHUS, refraktärer gMG und NMOSD, die Anzeichen und Symptome der Grunderkrankung verstärken, wie z.B. Hämolyse (PNH), TMA (aHUS) oder Exazerbation der MG (refraktäre gMG) oder Schub (NMOSD). Daher sollten die Patienten im Anschluss an die empfohlene Impfung engmaschig auf Krankheitssymptome überwacht werden.
Eine Impfung ist unter Umständen nicht ausreichend, um eine Meningokokkeninfektion zu verhindern. Die offiziellen Empfehlungen zur indikationsgerechten Anwendung von Antibiotika sollten berücksichtigt werden. Es wurde über Fälle von schwerwiegenden oder tödlich verlaufenden Meningokokkeninfektionen bei mit Soliris behandelten Patienten berichtet. Sepsis ist eine häufige Folge von Meningokokkeninfektion bei Patienten, die mit Soliris behandelt werden (siehe "Unerwünschte Wirkungen" ). Alle Patienten sollten auf Frühzeichen einer Meningokokkeninfektion überwacht, bei Infektionsverdacht sofort untersucht und, falls erforderlich, mit geeigneten Antibiotika behandelt werden. Die Patienten sollten über diese Anzeichen und Symptome sowie die für eine sofortige ärztliche Behandlung einzuleitenden Schritte informiert werden. Behandelnde Ärzte müssen mit den Patienten über Nutzen und Risiken der Soliris-Behandlung sprechen und ihnen die Patienten-Informationsbroschüre und die Patientenkarte aushändigen.
Infektionen:
Aufgrund seines Wirkungsmechanismus sollte eine Therapie mit Soliris bei Patienten mit akuten systemischen Infektionen nur mit Vorsicht durchgeführt werden. Patienten könnten eine erhöhte Anfälligkeit gegenüber Infektionen, insbesondere mit Neisseria und bekapselten Bakterien, aufweisen. Es wurden schwerwiegende Infektionen mit Neisseria-Arten (zusätzlich zu Neisseria meningitidis) einschliesslich disseminierter Gonokokkeninfektionen berichtet. Ärzte sollten die Patienten dahingehend beraten, wie man einer Gonorrhoe vorbeugen kann.
Im Fall einer schweren oder schwerwiegenden Infektion unter der Therapie mit Eculizumab sollten der Nutzen und die Risiken einer Fortsetzung der Therapie mit Eculizumab unter Berücksichtigung eines möglichen Wiederauftretens von Anzeichen und Symptomen der Grunderkrankung abgewogen werden.
Infusionsreaktionen:
Wie bei allen therapeutischen Proteinen, kann die Verabreichung von Soliris zu Infusionsreaktionen oder Immunogenität führen, die allergische Reaktionen oder Überempfindlichkeitsreaktionen (einschliesslich Anaphylaxie) verursachen könnten. In klinischen Studien kam es bei 1 (0.9 %) Patienten mit refraktärer gMG zu einer Infusionsreaktion, die das Absetzen von Soliris erforderte. Bei keinem der Kinder und Jugendlichen mit PNH, aHUS, refraktärer gMG oder NMOSD kam es zu einer Infusionsreaktion, die das Absetzen von Soliris erforderte. Bei allen Patienten, bei denen schwere Infusionsreaktionen auftreten, muss die Verabreichung von Soliris unterbrochen und eine geeignete medizinische Behandlung durchgeführt werden.
Patienten müssen über dieses Risiko und die Behandlungsmöglichkeiten schwerer allergischer Reaktionen informiert werden. Dabei sollte auch die Abgabe und Instruktion eines Notfallsets (inklusive Adrenalin-Autoinjektor) erwogen werden. Patienten sollten angewiesen werden, bei schweren systemischen allergischen Reaktionen, Angioödem, Schluckschwierigkeiten, Atemschwierigkeiten, Stimmveränderungen oder Klossgefühl im Hals nebst Anwendung des Notfallsets sofort einen Arzt zu konsultieren.
Immunogenität:
In allen klinischen Studien wurden in seltenen Fällen Antikörperreaktionen bei den mit Soliris behandelten Patienten beobachtet. In placebo-kontrollierten Studien bei PNH wurden Reaktionen mit niedrigen Antikörperwerten mit einer Häufigkeit vergleichbar zu Patienten unter Placebo beobachtet (3,4 % vs. 4,8 %). Bei Patienten mit aHUS, die mit Soliris behandelt wurden, konnten mit einem ECL-Test bei 3 von 100 Patienten (3 %) gegen Soliris gerichtete Antikörper festgestellt werden.
Einer von 100 Patienten mit aHUS (1 %) hatte schwach positive Werte von neutralisierenden Antikörpern.
In einer placebo-kontrollierten refraktäre-gMG-Studie wurde in den 26 Wochen der aktiven Behandlung bei keinem (0/62) der mit Soliris behandelten Patienten eine Antikörperreaktion gegen das Arzneimittel beobachtet, wogegen bei einer refraktären gMG-Verlängerungsstudie insgesamt 3/117 (2.6 %) bei mindestens einem Besuch nach Studienbeginn (Baseline) positiv für ADA waren. Positive ADA-Ergebnisse schienen vorübergehend zu sein, da bei nachfolgenden Besuchen keine positiven Titer beobachtet wurden und bei diesen Patienten keine klinischen Befunde vorlagen, die auf eine Wirkung positiver ADA-Titer hindeuteten.
In einer placebo-kontrollierten Studie bei NMOSD zeigten 2 von 95 (2.1 %) der mit Soliris behandelten Patienten nach Studienbeginn (Baseline) eine Antikörperreaktion gegen das Arzneimittel (ADA). Beide Patienten waren negativ für neutralisierende Antikörper. Positive Proben zeigten ADA mit niedrigem Titer, die transient waren. Es war keine Korrelation von Antikörperentwicklung und klinischem Ansprechen oder unerwünschten Ereignissen zu beobachten.
Immunisierung:
Vor Beginn der Therapie mit Soliris wird empfohlen, dass Patienten mit PNH, aHUS, refraktärer gMG und NMOSD die gemäss den geltenden Impfrichtlinien empfohlenen Impfungen erhalten. Darüber hinaus müssen alle Patienten mindestens 2 Wochen vor Verabreichung von Soliris gegen Meningokokkeninfektionen geimpft werden, es sei denn, das Risiko, das mit einer Verzögerung der Soliris-Therapie verbunden wäre, wiegt schwerer als das Risiko einer Meningokokkeninfektion. Patienten, die eine Behandlung mit Soliris früher als 2 Wochen nach einer tetravalenten Meningokokkenimpfung beginnen, müssen bis 2 Wochen nach der Impfung eine geeignete Antibiotikaprophylaxe erhalten. Impfstoffe gegen die Serogruppen A, C, Y und W 135 werden empfohlen, um Infektionen mit den häufig pathogenen Meningokokken-Serogruppen zu verhindern. Sofern verfügbar, wird auch eine Impfung gegen die Serogruppe B empfohlen (siehe Abschnitt Meningokokkeninfektion).
Patienten unter 18 Jahren müssen gegen Haemophilus influenzae und Pneumokokken geimpft werden. Dabei müssen die nationalen Impfempfehlungen für die jeweiligen Altersgruppen streng eingehalten werden.
Eine Impfung kann das Komplement zusätzlich aktivieren. Folglich können sich bei Patienten mit komplementvermittelten Erkrankungen, einschliesslich PNH, aHUS, refraktärer gMG und NMOSD, die Anzeichen und Symptome der Grunderkrankung verstärken, wie z.B. Hämolyse (PNH), TMA (aHUS), Exazerbation der MG (refraktäre gMG) oder Schub (NMOSD). Daher sollten die Patienten im Anschluss an die empfohlene Impfung engmaschig auf Krankheitssymptome überwacht werden.
Therapie mit Antikoagulantien:
Unter der Behandlung mit Soliris sollte die Therapie mit Antikoagulantien nicht verändert werden.
Therapie mit Immunsuppressiva und Cholinesterasehemmern:
Refraktäre gMG
Patienten, bei denen die Dosis von Immunsuppressiva und Cholinesterasehemmern verringert wird oder diese abgesetzt werden, müssen engmaschig auf Anzeichen einer Exazerbation der Erkrankung überwacht werden.
Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD)
Wenn die Immunsuppressiva-Therapie reduziert oder abgesetzt wird, sind die Patienten engmaschig auf Anzeichen und Symptome eines potenziellen NMOSD-Schubes zu beobachten.
Labormedizinische Überwachung bei PNH:
PNH-Patienten sollten auf Anzeichen und Symptome einer intravaskulären Hämolyse einschliesslich der Laktatdehydrogenase (LDH)-Spiegel im Serum überwacht werden. PNH-Patienten, die mit Soliris behandelt werden, sollten ebenfalls durch Messung der LDH-Spiegel auf eine intravaskuläre Hämolyse überwacht werden und gegebenenfalls ist eine Dosisanpassung des empfohlenen Dosisplanes von 14±2 Tagen während der Erhaltungsphase auf 12 Tage erforderlich.
Labormedizinische Überwachung bei aHUS:
aHUS-Patienten, die mit Soliris behandelt werden, sollten durch Messung der Thrombozytenzahl, der Serum-LDH-Spiegel und der Serum-Kreatinin-Spiegel auf Anzeichen einer thrombotischen Mikroangiopathie überwacht werden. Gegebenenfalls kann eine Dosisanpassung innerhalb des empfohlenen Dosierungsschemas von 14±2 Tagen während der Erhaltungsphase (bis zu alle 12 Tage) erforderlich sein.
Behandlungsabbruch bei PNH:
Ein Absetzen der Therapie muss medizinisch begründet sein. Wenn die Behandlung mit Soliris bei Patienten mit PNH abgesetzt wird, sollten sie auf Anzeichen und Symptome einer schweren intravaskulären Hämolyse überwacht werden. Eine schwere Hämolyse ist an höheren Serum-LDH-Spiegeln als vor der Behandlung in Verbindung mit Folgendem erkennbar: absolute Abnahme der Grösse des PNH-Klons um mehr als 25 % (nicht eingerechnet Verdünnungseffekte aufgrund von Transfusionen) innerhalb einer Woche oder weniger; ein Hämoglobin-Spiegel von < 5 g/dl oder eine Abnahme von > 4 g/dl innerhalb einer Woche oder weniger; Veränderung des Geisteszustandes; Anstieg des Serum-Kreatinin-Spiegels um 50 % oder das Auftreten von Thrombosen. Jeder Patient, der Soliris absetzt, muss mindestens 8 Wochen überwacht werden, um eine schwere Hämolyse oder andere Reaktionen zu detektieren.
Wenn nach Absetzen von Soliris eine schwere Hämolyse auftritt, sind folgende Vorgehen/Therapien in Erwägung zu ziehen: Bluttransfusionen (Erythrozytenkonzentrat) oder Austauschtransfusionen, falls mittels Durchflusszytometrie festgestellt wird, dass die PNH-Erythrozyten > 50 % der Erythrozyten insgesamt ausmachen; Antikoagulantien; Kortikosteroide oder Wiederaufnahme der Solirisbehandlung. In klinischen PNH-Studien haben 16 Patienten die Behandlung mit Soliris unterbrochen, wobei keine einzige schwere Hämolyse beobachtet wurde.
Behandlungsabbruch bei aHUS:
Ein Absetzen der Therapie muss medizinisch begründet sein.
Bei einigen Patienten konnten 4 Wochen nach Abbruch der Behandlung mit Soliris und bis zu 127 Wochen danach Komplikationen einer thrombotischen Mikroangiopathie (TMA) beobachtet werden. Eine Unterbrechung der Behandlung sollte ausschliesslich in solchen Fällen in Betracht gezogen werden, in denen sie medizinisch begründet ist.
In klinischen Studien bei aHUS unterbrachen 61 Patienten (21 pädiatrische Patienten) die Behandlung mit Soliris, mit einer anschliessenden Folgeüberwachung von im Median 24 Wochen. Bei 12 Patienten konnten 15 schwere Komplikationen einer TMA nach Behandlungsabbruch beobachtet werden. Bei weiteren 2 Patienten, die eine geringere Dosis Solaris ausserhalb des zugelassenen Dosierungsschemas erhielten, traten 2 weitere schwere Komplikationen einer thrombotischen Mikroangiopathie auf. Schwere Komplikationen einer thrombotischen Mikroangiopathie (TMA) traten bei Patienten unabhängig von identifizierten genetischen Mutationen, hoch riskanten Polymorphismen oder Autoantikörpern auf. Weitere schwere medizinische Komplikationen, wie z.B. starke Verschlechterung der Nierenfunktion, krankheitsbedingte Spitaleinweisungen und Fortschreiten der Erkrankung bis zum terminalen Nierenversagen mit Dialysepflicht, traten bei diesen Patienten auf. Trotz Wiederaufnahme der Soliris-Therapie kam es bei einem Patienten zum terminalen Nierenversagen.
Wenn die Behandlung mit Soliris bei Patienten mit aHUS abgesetzt wird, sollten sie daher engmaschig auf Anzeichen von schweren Komplikationen einer thrombotischen Mikroangiopathie überwacht werden.
Wenn die Behandlung mit Soliris bei aHUS-Patienten abgesetzt wird, kann die Überwachung zur Prognose oder Verhinderung von schweren Komplikationen einer TMA nicht ausreichend sein.
Schwere Komplikationen einer thrombotischen Mikroangiopathie nach Absetzen sind zu erkennen (i) am Auftreten einer Kombination von zwei oder wiederholter Messung eines der folgenden Parameter:
Verringerung der Thrombozytenzahl um ≥ 25% im Vergleich zum Ausgangswert vor Soliris-Behandlung oder zur maximalen Thrombozytenzahl während der Soliris-Behandlung; Anstieg des Serum-Kreatinins um ≥ 25% im Vergleich zum Ausgangswert vor Soliris-Behandlung oder zum Tiefstwert (Nadir) während der Soliris-Behandlung; Anstieg der Serum-LDH um ≥25% im Vergleich zum Ausgangswert vor Soliris-Behandlung oder zum Tiefstwert (Nadir) während der Soliris-Behandlung oder (ii) am Auftreten eines der folgenden Anzeichen/Symptome: Veränderung des Geisteszustands oder Krampfanfälle; Angina pectoris oder Dyspnoe oder einer Thrombose.
Wenn nach Absetzen von Soliris schwere Komplikationen einer thrombotischen Mikroangiopathie auftreten, sollten eine Fortsetzung der Soliris-Behandlung, unterstützende Massnahmen durch Plasmaaustausch/Plasmainfusion oder geeignete supportive Massnahmen wie eine Dialyse zur Unterstützung der Nierenfunktion, eine mechanische Beatmung zur Unterstützung der Atemfunktion oder eine Behandlung mit Antikoagulanzien in Betracht gezogen werden.
Behandlungsabbruch bei refraktärer gMG:
Die Anwendung von Soliris zur Behandlung von refraktärer gMG wurde nur im Rahmen einer dauerhaften Verabreichung untersucht. Patienten, bei denen die Soliris-Behandlung abgebrochen wird, sollten sorgfältig auf Anzeichen und Symptome einer Exazerbation der Krankheit überwacht werden.
Behandlungsabbruch bei NMOSD:
Die Anwendung von Soliris zur Behandlung von NMOSD wurde nur im Rahmen einer dauerhaften Anwendung untersucht und die Wirkung des Absetzens von Soliris wurde nicht beschrieben. Patienten, bei denen die Soliris-Behandlung abgesetzt wird, sollten sorgfältig auf Anzeichen eines möglichen NMOSD-Schubs überwacht werden.
Informationen zu Soliris:
Alle Ärzte, die beabsichtigen Soliris zu verschreiben, müssen mit der Informationsbroschüre für Fachkreise zur Behandlung von Soliris vertraut sein. Sie müssen Nutzen und Risiken einer Soliris-Behandlung mit den Patienten besprechen und ihnen die Informationsbroschüre für Patienten zur Behandlung mit Soliris und die Patientenkarte aushändigen. Die Patienten müssen darüber aufgeklärt werden, dass sie sich bei Auftreten von Fieber, Kopfschmerzen zusammen mit Fieber und/oder Nackensteifigkeit oder Lichtempfindlichkeit umgehend an einen Arzt wenden müssen, da dieses Anzeichen für eine Meningokokkeninfektion sein können.
Hilfsstoffe:
Dieses Arzneimittel enthält 5,00 mmol Natrium pro Dosis (1 Durchstechflasche), entsprechend 6% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
Bei Umstellung der Therapie auf eine andere Darreichungsform und/oder ein anderes Arzneimittel mit gleichem Wirkstoff ist Vorsicht geboten. Der Patient sollte adäquat kontrolliert werden.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt. Aufgrund der potenziellen Hemmwirkung von Eculizumab auf die komplementabhängige Zytotoxizität von Rituximab kann Eculizumab die erwarteten pharmakodynamischen Wirkungen von Rituximab mindern.
Es wurde gezeigt, dass Plasmaaustausch (PE), Plasmapherese (PP), Infusion von Fresh Frozen Plasma (PI) und intravenösem Immunglobulin (IVIg) die Eculizumab-Serumspiegel senken. In diesen Fällen ist eine zusätzliche Dosis Eculizumab erforderlich. Hinweise zur gleichzeitigen Behandlung mit PE, PP, PI oder IVIg finden Sie in Abschnitt "Dosierung/Anwendung" .
Die gleichzeitige Anwendung von Eculizumab mit neonatalen Fc-Rezeptorblockern (FcRn) kann die systemische Exposition verringern und die Wirksamkeit von Eculizumab reduzieren.
In allen diesen Fällen ist genau auf eine verminderte Wirksamkeit von Eculizumab zu achten.
Eine Langzeitbehandlung mit intravenösem humanem Immunglobulin (IVIg) kann den durch den endosomalen neonatalen Fc-Rezeptor (FcRn) vermittelten Recyclingmechanismus von monoklonalen Antikörpern wie Eculizumab beeinträchtigen und so zu einer Abnahme der Eculizumab-Konzentration im Serum führen.
Schwangerschaft, StillzeitBei gebärfähigen Frauen sollte die Anwendung einer geeigneten Verhütungsmethode zur Verhinderung einer Schwangerschaft während der Behandlung und mindestens 5 Monate nach der letzten Eculizumab-Dosis in Betracht gezogen werden.
Schwangerschaft
Es liegen keine gut kontrollierten Studien an Schwangeren vor, die mit Eculizumab behandelt wurden. Daten über eine begrenzte Zahl von exponierten Schwangeren (Ergebnisse von weniger als 300 Schwangerschaften) deuten nicht auf ein erhöhtes fetales Fehlbildungsrisiko oder eine fetale/neonatale Toxizität von Eculizumab hin. Aufgrund des Fehlens gut kontrollierter Studien bleibt jedoch eine gewisse Unsicherheit bestehen. Daher wird empfohlen, vor Beginn und während einer Behandlung mit Eculizumab bei Schwangeren eine individuelle Nutzen-Risiko-Analyse durchzuführen. Sollte diese Behandlung während einer Schwangerschaft für notwendig erachtet werden, wird zu einer strengen Überwachung von Mutter und Fetus entsprechend den nationalen Leitlinien geraten.
Es wurden keine Reproduktionsstudien an Tieren mit Eculizumab durchgeführt (siehe "Präklinische Daten" ).
Humanes IgG passiert bekanntlich die Plazentaschranke und demzufolge kann Eculizumab potenziell eine terminale Komplementinhibition im fetalen Kreislauf verursachen. Deshalb sollte Soliris einer Schwangeren nur dann gegeben werden, wenn dies eindeutig erforderlich ist.
Stillzeit
Es sind keine Auswirkungen auf gestillte Neugeborene/Kinder zu erwarten, da aus den verfügbaren begrenzten Daten hervorgeht, dass Eculizumab nicht in die Muttermilch übergeht. Aufgrund der Einschränkungen der verfügbaren Daten sollte jedoch der Nutzen des Stillens für Entwicklung und Gesundheit des Kindes zusammen mit dem klinischen Bedarf der Mutter für Eculizumab und potenziellen unerwünschten Wirkungen auf das gestillte Kind durch Eculizumab oder die Grunderkrankung der Mutter in Betracht gezogen werden.
Fertilität
Es wurden keine spezifischen Studien zur Fertilität mit Eculizumab durchgeführt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und das Bedienen von Maschinen durchgeführt.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Unterstützende Sicherheitsdaten wurden in 33 klinischen Studien erhoben, in denen 1555 Patienten mit komplement-vermittelten Erkrankungen, einschliesslich PNH, aHUS, refraktärer gMG und NMOSD, mit Eculizumab behandelt wurden. Die häufigste Nebenwirkung war Kopfschmerz (trat am häufigsten in der Induktionsphase der Behandlung auf). Die schwerwiegendste Nebenwirkung war Meningokokken-Infektion.
Tabellarische Auflistung der Nebenwirkungen
Tabelle 1 enthält Nebenwirkungen aus Spontanberichten und abgeschlossenen klinischen Studien mit Eculizumab, einschliesslich Studien bei PNH, aHUS, refraktärer gMG und NMOSD. Sehr häufige (≥ 1/10), häufige (≥ 1/100, < 1/10), gelegentliche (≥ 1/1000, < 1/100) oder seltene (≥ 1/10.000, < 1/1.000) Nebenwirkungen von Eculizumab sind geordnet nach Organklassen und bevorzugter Bezeichnung aufgeführt. Die Nebenwirkungen sind innerhalb jeder Häufigkeitsgruppe nach abnehmender Schwere gelistet.
Die häufigsten Nebenwirkungen waren:
-Kopfschmerzen, Schwindelgefühl, Übelkeit und Fieber mit einer Häufigkeit von 5 % oder mehr in den klinischen Studien zur Behandlung der PNH. Kopfschmerzen waren in den meisten Fällen auf die erste Phase der Verabreichung von Soliris beschränkt.
-Leukopenie mit einer Häufigkeit von 10 % oder mehr in den klinischen Studien zur Behandlung des aHUS.
Tabelle 1: Nebenwirkungen, die bei Patienten aus den gesamten klinischen Studien mit Eculizumab, einschliesslich PNH, aHUS, refraktärer gMG und NMOSD, und seit der Markteinführung berichtet wurden
MedDRA Systemorgan-k Sehr häufig (≥ 1/10) Häufig (≥ 1/100, < Gelegentlich (≥ Selten (≥ 1/10.000,
lassen 1/10) 1/1000, < 1/100) < 1/1.000)
Infektionen und Pneumonie, Infektion Meningokokken- Aspergillus-Infektio
parasitäre Erkrankun der oberen Atemwege Infektionb, Sepsis, nc, bakterielle
gen , Bronchitis, septischer Schock, Arthritisc, Gonokokk
Nasopharyngitis, Peritonitis, Infekti en-Infektion des
Harnwegsinfektion, on der unteren Urogenitaltrakts,
Lippenherpes Atemwege, Pilzinfekt Haemophilus-influenz
ion, Virusinfektion, ae- Infektion,
Abszessa, Zelluliti Impetigo
s, Grippe, gastroint
estinale Infektion,
Zystitis, Infektion,
Sinusitis, Zahnflei
schentzündung
Gutartige, bösartige Malignes Melanom,
und unspezifische Myelodysplastisches
Neubildungen (inklus Syndrom
ive Zysten und
Polypen)
Erkrankungen des Leukopenie, Anämie Thrombozytopenie, Hämolyse*, abnormer
Blutes und des Lymphopenie Gerinnungsfaktor,
Lymphsystems Erythrozyten-Aggluti
nation, Koagulopathi
e
Erkrankungen des Anaphylaktische
Immunsystems Reaktion, Hypersensi
tivität
Endokrine Erkrankung Morbus Basedow
en
Stoffwechsel- und Appetitverlust
Ernährungsstörungen
Psychiatrische Insomnie Angst, Depression, Abnorme Träume
Erkrankungen Stimmungsschwankunge
n, Schlafstörungen
Erkrankungen des Kopfschmerzen Schwindelgefühl Parästhesie, Tremor,
Nervensystems Veränderung der
Geschmackswahrnehmun
g, Synkope
Augenerkrankungen Verschwommenes Sehen Bindehautreizung
Erkrankungen des Tinnitus, Vertigo
Ohres und des
Labyrinths
Herzerkrankungen Palpitation
Gefässerkrankungen Hypertonie Akzelerierte Hyperto Hämatom
nie, Hypotonie,
Hitzewallungen,
Venenerkrankung
Erkrankungen der Oropharyngeale Dyspnoe, Nasenbluten
Atemwege, des Schmerzen, Husten , Halsreizung,
Brustraums und verstopfte Nase,
Mediastinums Rhinorrhoe
Erkrankungen des Bauchschmerzen, Obstipation, Dyspeps Gastroösophageale
Gastrointestinaltrak Diarrhoe, Übelkeit, ie, abdominales Refluxkrankheit,
ts Erbrechen Spannungsgefühl schmerzendes Zahnfle
isch
Leber- und Gallenerk Ikterus
rankungen
Erkrankungen der Hautausschlag, Urtikaria, Erythem, Depigmentstörung
Haut und des Unterha Pruritus, Alopezie Petechien, Hyperhidr der Haut
utzellgewebes ose, trockene Haut,
Dermatitis
Skelettmuskulatur-, Arthralgie, Myalgie, Muskelkrämpfe, Trismus, Gelenkschwe
Bindegewebs- und Schmerzen in Knochenschmerzen, llung
Knochenerkrankungen Extremitäten Rückenschmerzen,
Nackenschmerzen
Erkrankungen der Nierenschädigung,
Nieren und Harnwege Dysurie, Hämaturie
Erkrankungen der Spontanerektion Menstruations-störun
Geschlechtsorgane gen
und der Brustdrüse
Allgemeine Erkrankun Fieber, Fatigue, Ödeme, Thorax-Beschw Extravasat, Parästhe
gen und Beschwerden grippeähnliche erden, Asthenie, sie an der Infusions
am Verabreichungsort Erkrankung Schmerzen im Brustra stelle, Wärmegefühl
um, Schmerzen an
der Infusionsstelle,
Schüttelfrost,
Untersuchungen Alanin-Aminotransfer Coombs-Test positivc
ase erhöht, Aspartat
-Aminotransferase
erhöht, γ-Glutamyltr
ansferase erhöht,
Hämatokrit erniedrig
t, Hämoglobin
erniedrigt
Verletzungen, Infusionsbedingte
Vergiftungen und Reaktion
durch Eingriffe
bedingte Komplikatio
nen
Einbezogene Studien: Asthma (C07-002), aHUS (C08-002, C08-003, C10-003, C10-004), Dermatomyositis (C99-006), refraktäre gMG (C08-001, ECU-MG-301, ECU-MG-302, ECU-MG-303), Neuromyelitis-optica- Spektrumerkrankungen (ECU-NMO-301, ECU-NMO-302), IMG (C99-004, E99-004), PNH (C02-001, C04-001, C04-002, C06-002, C07-001, E02-001, E05-001, E07-001, M07-005, X03-001, X03-001A), Psoriasis (C99-007), RA (C01-004, C97-001, C99-001, E01-004, E99-001), STEC-HUS (C11-001), SLE (C97-002). MedDRA Version 24.1.
*Siehe Abschnitt "Beschreibung ausgewählter Nebenwirkungen"
a Abszess umfasst die folgende Gruppe von Preferred Terms (PTs): Abszess Gliedmasse, Kolonabszess, Nierenabszess, subkutaner Abszess, Zahnabszess, hepatosplenischer Abszess, perirektaler Abszess, Rektalabszess.
b = Meningokokkeninfektion umfasst die folgende Gruppe von „Preferred Terms“: Meningokokken-Infektion, Meningokokken-Sepsis, Meningokokken-Meningitis, Neisseria-Infektion
c = Seit der Markteinführung identifizierte Nebenwirkungen
Beschreibung ausgewählter Nebenwirkungen
Die schwerwiegendste unerwünschte Arzneimittel-Wirkung aller klinischen Studien war das Auftreten einer Meningokokken-Sepsis, welche eine häufige Form einer Meningokokkeninfektion bei Patienten ist, die mit Soliris behandelt werden (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Andere Fälle von Neisseria-Arten wurden berichtet, einschliesslich Sepsis durch Neisseria gonorrhoeae, Neisseria sicca/subflava, und unspezifizierten Neisseria spp.
Bei 2 % der PNH-Patienten wurden mit einem ELISA-Test, bei 3 % der aHUS-Patienten und 2 % der NMOSD-Patienten mit einem ECL-Test gegen Soliris gerichtete Antikörper nachgewiesen. In placebo-kontrollierten Studien bei refraktärer gMG wurden keine Antikörper gegen das Arzneimittel beobachtet. Wie bei allen Proteinen besteht ein Potenzial für Immunogenität.
Nach ausgesetzten oder verspäteten Soliris-Gaben in klinischen Studien zur Behandlung der PNH wurden Fälle von Hämolyse berichtet (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Nach ausgesetzten oder verspäteten Soliris-Gaben in klinischen Studien zur Behandlung des aHUS wurden Fälle von TMA berichtet (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Kinder und Jugendliche
Das in der pädiatrischen PNH-Studie M07-005 beobachtete Sicherheitsprofil bei 7 Kindern und Jugendlichen (Alter 11 bis unter 18 Jahre) mit PNH erschien vergleichbar mit dem Sicherheitsprofil bei Erwachsenen. Die häufigste Nebenwirkung bei Kindern und Jugendlichen war Kopfschmerz.
Bei Kindern mit aHUS (Patientenalter 2 Monate bis < 18 Jahre), die in den Studien C08-002, C08-003, C09-001r und C10-003 untersucht wurden, erscheint das beobachtete Sicherheitsprofil vergleichbar mit dem Sicherheitsprofil bei Erwachsenen mit aHUS.
Bei Kindern scheint das Sicherheitsprofil in den unterschiedlichen Alters-Untergruppen ähnlich zu sein.
Das in der pädiatrischen Studie ECU-MG-303 beobachtete Sicherheitsprofil bei Kindern und Jugendlichen (Alter 12 bis unter 18 Jahre) mit refraktärer gMG erschien vergleichbar mit dem Sicherheitsprofil bei Erwachsenen mit refraktärer gMG.
Soliris wurde nicht bei Kindern und Jugendlichen mit NMOSD untersucht.
Ältere Patienten
Es wurden insgesamt keine Unterschiede in Bezug auf die Sicherheit zwischen älteren (≥ 65 Jahre) und jüngeren (< 65 Jahre) Patienten mit refraktärer gMG berichtet (siehe "Eigenschaften/Wirkungen" ).
Sicherheitsdaten aus anderen klinischen Studien
Unterstützende Sicherheitsdaten wurden in 12 abgeschlossenen klinischen Studien erhoben, in denen 934 Patienten, mit sechs anderen Erkrankungen als PNH, aHUS, refraktärer gMG oder NMOSD mit Eculizumab behandelt wurden. Bei einem ungeimpften Patienten mit idiopathischer membranöser Glomerulonephropathie trat eine Meningokokkenmeningitis auf. Die bei Patienten mit anderen Erkrankungen als PNH, aHUS, refraktärer gMG oder NMOSD gemeldeten Nebenwirkungen waren ähnlich denen bei Patienten mit PNH, aHUS, refraktärer gMG oder NMOSD (siehe Tabelle 1 oben). Aus diesen klinischen Studien gingen keine spezifischen Nebenwirkungen hervor.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/WirkungenATC-Code
L04AJ01
Soliris ist ein rekombinanter humanisierter monoklonaler IgG2/4k-Antikörper, der an das humane Komplementprotein C5 bindet und die Aktivierung des terminalen Komplements hemmt. Der Soliris-Antikörper enthält humanisierte konstante Regionen und murine Komplementarität bestimmende Regionen, die auf die variablen Regionen der leichten und schweren Ketten des menschlichen Gerüsts aufgesetzt sind. Soliris besteht aus zwei schweren Ketten mit 448 Aminosäuren und zwei leichten Ketten mit 214 Aminosäuren und hat ein Molekulargewicht von ca. 148 kDa.
Soliris wird in einem murinen Myelom-(NS0-Zelllinien-) Expressionssystem hergestellt und wird durch Affinitäts- und Ionenaustauschchromatographie gereinigt. Der Herstellungsprozess des Arzneistoffs umfasst ebenfalls spezifische Virusinaktivierungs- und -suppressionsschritte.
Wirkungsmechanismus
Eculizumab, der Wirkstoff in Soliris, ist ein terminaler Komplementinhibitor, der spezifisch und mit hoher Affinität an das Komplementprotein C5 bindet und dadurch dessen Spaltung in die Fragmente C5a und C5b blockiert und somit die Bildung des terminalen Komplementkomplexes C5b-9 verhindert. Eculizumab erhält die frühen Komponenten der Komplementaktivierung, die von wesentlicher Bedeutung für die Opsonisierung von Mikroorganismen und die Elimination (Clearance) von Immunkomplexen sind.
Bei PNH-Patienten werden die unkontrollierte terminale Komplementaktivierung und die daraus resultierende komplementvermittelte intravaskuläre Hämolyse durch die Behandlung mit Soliris blockiert.
Bei den meisten PNH-Patienten reichen Eculizumab-Serumkonzentrationen von etwa 35 µg/ml für eine praktisch vollständige Hemmung der terminalen komplementvermittelten intravaskulären Hämolyse aus.
Die dauerhafte Verabreichung von Soliris bei PNH führte zu einer raschen und nachhaltigen Verringerung der komplementvermittelten hämolytischen Aktivität. Bei aHUS-Patienten werden die unkontrollierte terminale Komplementaktivierung und die daraus resultierende komplementvermittelte thrombotische Mikroangiopathie durch die Behandlung mit Soliris blockiert.
Alle aHUS-Patienten, die nach dem empfohlenen Dosierungsschema mit Soliris behandelt wurden, erreichten eine rasche und anhaltende Abnahme der terminalen Komplementaktivität. Bei allen Patienten mit aHUS reichen Eculizumab-Serumkonzentrationen von etwa 50-100 µg/ml für eine praktisch vollständige Hemmung der terminalen Komplementaktivität aus.
Die dauerhafte Verabreichung von Soliris bei aHUS führte zu einer raschen und nachhaltigen Verringerung der komplementvermittelten thrombotischen Mikroangiopathie.
Bei Patienten mit refraktärer gMG verursacht die unkontrollierte terminale Komplementaktivierung eine vom Membranangriffskomplex (MAC) abhängige Lyse und eine C5a-abhängige Entzündung an der neuromuskulären Endplatte, was zum Ausfall der neuromuskulären Übertragung führt. Die dauerhafte Verabreichung von Soliris führt zu einer sofortigen, vollständigen und nachhaltigen Hemmung der terminalen Komplementaktivität (Eculizumab-Serumkonzentrationen ≥116 µg/ml).
Bei Patienten mit NMOSD führt eine durch Autoantikörper gegen AQP4 verursachte unkontrollierte Aktivierung des terminalen Komplements zur Entstehung der MAC- und C5a-abhängigen Entzündung, die Astrozytennekrose und eine erhöhte Durchlässigkeit der Blut-Hirn-Schranke sowie das Absterben der umgebenden Oligodendrozyten und Neuronen zur Folge hat. Die dauerhafte Anwendung von Soliris führt zu einer sofortigen, vollständigen und anhaltenden Hemmung der terminalen Komplementaktivität (Eculizumab-Serumkonzentrationen ≥116 µg/ml).
Pharmakodynamik
siehe "Wirkungsmechanismus"
Klinische Wirksamkeit
Paroxysmale nächtliche Hämoglobinurie
Die Sicherheit und Wirksamkeit von Soliris bei PNH-Patienten mit Hämolyse wurden in einer 26-wöchigen randomisierten, doppelblinden, placebokontrollierten Studie (C04-001) untersucht. PNH-Patienten wurden ebenfalls in einer 52-wöchigen einarmigen Studie (C04-002) sowie in einer Langzeit-Fortsetzungsstudie (E05-001) mit Soliris behandelt. Die Patienten erhielten vor Verabreichung von Soliris eine Meningokokkenimpfung. In allen Studien betrug die Eculizumab-Dosis 600 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 900 mg 7 ± 2 Tage später und anschliessend 900 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 25 bis 45 Minuten (35 ± 10 Minuten) verabreicht. Zusätzlich wurde ein nicht-interventionelles Beobachtungsregister (M07-001) bei PNH Patienten initiiert, um den natürlichen Verlauf der PNH bei unbehandelten Patienten, sowie die klinischen Ergebnisse unter Soliris-Therapie zu charakterisieren.
In die Studie C04-001 (TRIUMPH) wurden PNH-Patienten mit mindestens 4 Transfusionen in den vorangegangenen 12 Monaten, einem mittels Durchflusszytometrie bestätigten Anteil von mindestens 10 % PNH-Zellen und einer Thrombozytenzahl von mindestens 100.000/Mikroliter randomisiert entweder der Soliris- (n = 43) oder der Placebo-Behandlung (n = 44) zugeordnet. Vor der Randomisierung durchliefen alle Patienten eine anfängliche Beobachtungsphase, um den Bedarf an Erythrozytentransfusion zu bestätigen und die Hämoglobinkonzentration (den „Sollwert“) zu ermitteln, welche die Hämoglobinstabilisierung und die Transfusionsergebnisse jedes Patienten bestimmen würde. Der Hämoglobin-Sollwert war bei Patienten mit Symptomen ≤ 9 g/dl und bei Patienten ohne Symptome ≤ 7 g/dl. Primäre Endpunkte waren die Hämoglobin-Stabilisierung (Patienten, die eine über dem Hämoglobinsollwert liegende Hämoglobinkonzentration aufrechterhielten und ohne weitere Erythrozytentransfusion während des gesamten 26-wöchigen Zeitraums auskamen) und der Bedarf an Bluttransfusion. Fatigue und Quality of Life (krankheitsbedingte Lebensqualität) waren relevante sekundäre Endpunkte. Die Hämolyse wurde hauptsächlich durch Messung der LDH-Spiegel im Serum überwacht, und der Anteil der PNH-Erythrozyten wurde mittels Durchflusszytometrie kontrolliert. Patienten, die Antikoagulantien und systemische Kortikosteroide zu Beginn erhielten, setzten die Einnahme dieser Medikamente fort. Die wichtigsten Ausgangsmerkmale waren in beiden Behandlungsarmen vergleichbar (siehe Tabelle 2).
In der nicht-kontrollierten Studie C04-002 (SHEPHERD) erhielten PNH-Patienten mit mindestens einer Transfusion in den vorausgegangenen 24 Monaten und mindestens 30.000 Thrombozyten/Mikroliter Soliris über einen 52-wöchigen Zeitraum. Zu den Begleittherapien gehörten Antithrombotika bei 63 % der Patienten und systemische Kortikosteroide bei 40 % der Patienten. Die Ausgangsparameter sind in Tabelle 2 dargestellt.
Tabelle 2: Demografische Patientendaten und parameter in den Studien C04-001 und C04-002
C04-001 C04-002
Parameter PlaceboN = 44 SolirisN = 43 SolirisN = 97
Mittleres Alter (SD) 38,4 (13,4) 42,1 (15,5) 41,1 (14,4)
Geschlecht - weiblich (%) 29 (65,9) 23 (53,5) 49 (50,5)
Aplastische Anämie oder MDS in 12 (27,3) 8 (18,7) 29 (29,9)
der Anamnese (%)
Begleitmedikation Antikoagulantien 20 (45,5) 24 (55,8) 59 (61)
(%)
Begleitmedikation Steroiden/Immuns 16 (36,4) 14 (32,6) 46 (47,4)
uppressiva (%)
Behandlungsabbruch 10 2 1
Erythrozytenkonzentrate in den 17,0 (13,5/25,0) 18,0 (12,0/24,0) 8,0 (4,0/24,0)
vorangegangenen 12 Monaten
(Median (Q1, Q3))
Mittlerer Hb-Spiegel (g/dl) am 7,7 (0,75) 7,8 (0,79) n.a.
Sollwert (SD)
Prätherapeutische LDH-Spiegel 2234,5 2032,0 2051,0
(Median, U/l)
Freies Hämoglobin bei 46,2 40,5 34,9
Studienbeginn (Median, mg/dl)
In der TRIUMPH Studie zeigten die mit Soliris behandelten Patienten eine signifikante Verminderung der Hämolyse (p<0,001), was Verbesserungen der Anämie bewirkte, was sich durch eine erhöhte Hämoglobin-Stabilisierung und einen verminderten Bedarf an Erythrozyten-Transfusionen im Vergleich zu den mit Placebo behandelten Patienten äusserte (siehe Tabelle 3). Die Verbesserung konnte in allen 3 Erythrozyten-Transfusion-Untergruppen (4–14 Einheiten; 15–25 Einheiten; > 25 Einheiten) nachgewiesen werden. Nach 3 Wochen Soliris-Therapie zeigten die Patienten eine Verminderung der Fatigue und eine verbesserte Quality of Life (gesundheitsbezogene Lebensqualität). Aufgrund des Stichprobenumfangs und der Dauer der Studie konnten die Wirkungen von Soliris auf thrombotische Ereignisse nicht ermittelt werden. In der SHEPHERD-Studie beendeten 96 der 97 eingeschlossenen Patienten die Studie (ein Patient starb nach einem thrombotischen Ereignis). Die Reduktion intravaskulärer Hämolyse, die anhand der LDH-Spiegel im Serum gemessen wurde, hielt während des Behandlungszeitraums an und führte zu einer erhöhten Transfusionsvermeidung, einem verringerten Bedarf an Erythrozyten-Transfusion und zu geringerer Fatigue (siehe Tabelle 3).
Tabelle 3: Wirksamkeitsergebnisse in den Studien C04-001 und C04-002
C04-001 C04-002*
PlaceboN = 44 SOLIRISN = 43 p-Wert SOLIRISN = 97 p-Wert
Prozentualer Anteil 0 49 < 0,001 n.a.
der Patienten mit
stabilisierten
Hämoglobinspiegeln
am Ende der Studie
Transfundierte 10 0 < 0,001 0 < 0,001
Einheiten Erythrozyt
enkonzentrat während
der Behandlung
(Median)
Verminderter Bedarf 0 51 < 0,001 51 < 0,001
an Transfusionen
während der Behandlu
ng (%)
LDH-Spiegel am Ende 2167 239 < 0,001 269 < 0,001
der Studie (Median,
U/l)
LDH-AUC am Ende der 411822 58587 < 0,001 -632264 < 0,001
Studie (Median, U/l
x Tag)
Freies Hämoglobin 62 5 < 0,001 5 < 0,001
am Ende der Studie
(Median, mg/dl)
FACIT-Fatigue 1,12 < 0,001 1,14 < 0,001
(Effektgrösse)
* Die Ergebnisse aus der Studie C04-002 beziehen sich auf Vergleiche vor und nach der Behandlung.
195 mit Soliris behandelte Patienten aus den Studien C04-001, C04-002 in anderen Anfangsstudien wurden Langzeit-Fortsetzungs-Studie (E05-001) eingeschlossen. Bei allen Patienten konnte eine Reduktion der intravaskulären Hämolyse während der gesamten Dauer der Soliris-Behandlung von 10 bis 54 Monaten nachgewiesen werden. Im Vergleich zur gleichen Zeitdauer vor der Behandlung traten unter der Behandlung mit Soliris weniger thrombotische Ereignisse auf. Dieses Ergebnis wurde jedoch in einer nicht-kontrollierten klinischen Studie festgestellt.
Das PNH Register (M07-001) wurde genutzt, um die Wirksamkeit von Soliris bei PNH-Patienten ohne Transfusionen mit Erythrozytenkonzentraten in der Historie zu beurteilen. Diese Patienten hatten eine hohe Krankheitsaktivität, die durch eine erhöhte Hämolyse (LDH ≥1,5xONW) und das Vorhandensein eines oder mehrerer der damit verbundenen klinischen Symptome definiert ist: Fatigue, Hämoglobinurie, abdominelle Schmerzen, Kurzatmigkeit (Dyspnoe), Anämie (Hämoglobin <100 g/l), schwere unerwünschte vaskuläre Ereignisse (einschliesslich Thrombosen), Dysphagie oder erektile Dysfunktion.
Im PNH Register wurde, bei mit Soliris behandelten Patienten, eine Reduktion der Hämolyse und der damit verbundene Symptome beobachtet. Nach 6 Monaten hatten die mit Soliris behandelten Patienten ohne Transfusionen mit Erythrozytenkonzentraten in der Historie signifikant (p<0,001) reduzierte LDH-Spiegel (medianer LDH-Spiegel von 305 U/l; Tabelle 4). Weiterhin erfuhren 74% der mit Soliris behandelten Patienten ohne Transfusionshistorie klinisch relevante Verbesserungen im FACIT-Fatigue Score (d.h. Erhöhung um 4 oder mehr Punkte) und 84% im EORTC Fatigue Score (d.h. Abnahme um 10 oder mehr Punkte).
Tabelle 4: Ergebnisse zur Wirksamkeit (LDH-Spiegel und FACIT-Fatigue) bei PNH-Patienten ohne Transfusionshistorie im M07-001
M07-001
Parameter Soliris Keine Transfusion
LDH Spiegel zu Beginn der Studie(Median, U/l) N=43 1447
LDH Spiegel nach 6 Monaten (Median, U/l) N=36 305
FACIT-Fatigue Score zu Beginn der Studie (Median) N=25 32
FACIT-Fatigue Score der letzten verfügbaren Auswertung (Median) N=31 44
FACIT-Fatigue wird auf einer Skala von 0-52 ermittelt, wobei höhere Werte auf weniger Fatigue hinweisen
Atypisches Hämolytisch-Urämisches Syndrom
Die Wirksamkeit von Soliris in der Behandlung des aHUS wurde in vier prospektiven kontrollierten klinischen Studien mit 100 Patienten (drei Studien bei Erwachsenen und Jugendlichen (C08-002A/B, C08-003A/B, C10-004), einer Studie bei Kindern und Jugendlichen (C10-003) und einer retrospektiven Studie (C09-001r) mit 30 Patienten untersucht.
Bei Studie C08-002A/B handelte es sich um eine prospektive, kontrollierte, offene Studie, in die Patienten in der Frühphase eines aHUS mit Anzeichen einer klinisch manifestierten thrombotischen Mikroangiopathie (Thrombozytenzahl von ≤ 150 x 109/l trotz Plasmaaustausch/Plasmainfusion und LDH und Serum-Kreatinin oberhalb der oberen Grenze des Normalbereichs) eingeschlossen wurden. Bei Studie C08-003A/B handelte es sich um eine prospektive, kontrollierte, offene Studie, in die Patienten mit länger bestehendem aHUS ohne offensichtliche Hinweise auf eine klinisch manifestierte thrombotische Mikroangiopathie eingeschlossen wurden. Diese Patienten hatten über längere Zeit Plasmaaustausch/Plasmainfusionen (PA/PI) erhalten (≥ 1 PA/PI-Sitzung alle zwei Wochen und nicht mehr als 3 PA/PI-Sitzungen/Woche über mindestens 8 Wochen vor der ersten Dosis). In beiden prospektiven Studien wurden die Patienten über 26 Wochen mit Soliris behandelt. Die meisten dieser Patienten wurden danach in eine offene Verlängerungsstudie aufgenommen. Alle Patienten, die in die beiden prospektiven Studien aufgenommen wurden, hatten einen ADAMTS-13-Wert über 5 %.
Vor Therapieeinleitung mit Soliris wurden die Patienten gegen Meningokokken geimpft oder erhielten eine geeignete Antibiotikaprophylaxe bis 2 Wochen nach Impfung. In allen Studien betrug die Soliris-Dosis bei Erwachsenen und Jugendlichen mit aHUS 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1200 mg 7 ± 2 Tage später, dann 1200 mg alle 14 ± 2 Tage über die gesamte Studiendauer. Soliris wurde als intravenöse Infusion über 35 Minuten verabreicht. Das Dosierungsschema bei Kindern und Jugendlichen mit einem Körpergewicht unter 40 kg wurde mithilfe einer pharmakokinetischen Simulation festgelegt, mit der die empfohlene Dosis und das Dosierungsschema auf Basis des Körpergewichts ermittelt wurde (siehe "Dosierung / Anwendung" ).
Zu den primären Endpunkten gehörte in Studie C08-002A/B die Änderung der Thrombozytenzahl gegenüber dem Ausgangswert und in Studie C08-003A/B die Abwesenheit von Ereignissen einer thrombotischen Mikroangiopathie (TMA). Weitere Endpunkte waren TMA-Interventionsrate, Normalisierung hämatologischer Parameter, vollständiges Ansprechen der TMA, Änderungen der LDH, Nierenfunktion und gesundheitsbezogenen Lebensqualität. Die Abwesenheit von TMA-Ereignissen war definiert als Abwesenheit folgender Ereignisse über mindestens 12 Wochen: Abnahme der Thrombozytenzahl von > 25% gegenüber dem Ausgangswert, Plasmaaustausch/Plasmainfusion und neu eingeleitete Dialyse. TMA-Interventionen waren definiert als Plasmaaustausch/Plasmainfusion oder neu eingeleitete Dialyse. Normalisierung hämatologischer Parameter war definiert als Normalisierung der Thrombozytenzahl und der LDH-Spiegel während ≥ 2 aufeinanderfolgender Messungen während ≥ 4 Wochen. Vollständiges Ansprechen der TMA war definiert als Normalisierung hämatologischer Parameter und anhaltende Abnahme des Serumkreatinins um ≥ 25% in ≥ 2 aufeinanderfolgenden Messungen über ≥ 4 Wochen.
Die Ausgangsparameter für die jeweiligen Studien sind in Tabelle 5 dargestellt.
Tabelle 5: Demografische Patientendaten und -parameter in den Studien C08-002A/B und C08-003A/B
Parameter C08-002A/B C08-003A/B
Soliris N = 17 Soliris N = 20
Zeit von der Erstdiagnose bis zum Screening, Median in 10 (0,26/236) 48 (0,66/286)
Monaten (min/max)
Zeit von der Manifestierung der bestehenden TMA bis zum < 1 (< 1/4) 9 (1/45)
Screening, Median in Monaten (min/max)
Anzahl der Sitzungen für Plasmaaustausch/Plasmainfusion 17 (2/37) 62 (20/230)
zur Behandlung einer bestehenden TMA, Median (min/max)
Anzahl der Sitzungen für Plasmaaustausch/Plasmainfusion 6 (0/7) 2 (1/3)
innerhalb 7 Tagen vor der ersten Eculizumab-Verabreichun
g, Median (min/max)
Thrombozytenzahl, Ausgangswert (× 109/l), Mittelwert 109 (32) 228 (78)
(SD)
LDH Ausgangswert (U/l), Mittelwert (SD) 323 (138) 223 (70)
Patienten ohne identifizierte Mutation, n (%) 4 (24) 6 (30)
Patienten in der aHUS-Studie C08-002A/B wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung im Rahmen einer Verlängerungsstudie fort. In der aHUS-Studie C08-002A/B betrug die mediane Behandlungsdauer mit Soliris 100 Wochen (Spanne: 2 - 145 Wochen).
Nach Beginn der Soliris-Behandlung waren eine Abnahme der terminalen Komplementaktivität sowie ein Anstieg der Thrombozytenzahlen im Vergleich zu den Ausgangswerten zu beobachten. Die Abnahme der terminalen Komplementaktivität war bei allen Patienten nach Beginn der Soliris-Behandlung zu beobachten. Daten zur Wirksamkeit von Soliris in Studie C08-002A/B sind in Tabelle 6 zusammengefasst. Sämtliche Wirksamkeitsparameter haben sich während der 2 Behandlungsjahre verbessert oder erhalten. Das vollständige Ansprechen der TMA blieb bei allen Respondern aufrechterhalten. Von den Patienten, bei denen die Behandlung länger als 26 Wochen durchgeführt wurde, haben 2 weitere ein vollständiges Ansprechen der TMA erreicht und aufrechterhalten, was mit der Normalisierung des LDH-Werts (1 Patient) und der Senkung des Serum-Kreatinins (2 Patienten) zusammenhängt.
Unter Behandlung mit Soliris verbesserte und hielt sich die Nierenfunktion, gemessen als geschätzte glomeruläre Filtrationsrate (eGFR). Vier der 5 Patienten, die zu Studienbeginn dialysepflichtig waren, konnten die Dialyse für die Dauer der Soliris-Behandlung unterbrechen. Ein Patient benötigte eine neue Dialyse. Die Patienten berichteten eine Verbesserung ihrer gesundheitsbezogenen Lebensqualität (QoL).
In der aHUS-Studie C08-002A/B zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren kodieren, ein vergleichbares Ansprechen auf Soliris.
Patienten in der aHUS-Studie C08-003A/B wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung im Rahmen einer Verlängerungsstudie fort. In der aHUS-Studie C08-003A/B betrug die mediane Behandlungsdauer mit Soliris 114 Wochen (Spanne: 26-129 Wochen). Daten zur Wirksamkeit von Soliris in Studie C08-003A/B sind in Tabelle 6 zusammengefasst.
In der aHUS-Studie C08-003A/B zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren kodieren, ein vergleichbares Ansprechen auf Soliris. Bei allen Patienten wurde nach Beginn der Soliris Behandlung eine Abnahme der terminalen Komplementaktivität beobachtet. Sämtliche Wirksamkeitsparameter verbesserten oder hielten sich während der 2 Behandlungsjahre. Ein vollständiges Ansprechen der TMA blieb bei allen Respondern erhalten. Von den Patienten, bei denen die Behandlung länger als 26 Wochen durchgeführt wurde, erreichten 6 weitere ein vollständiges Ansprechen der TMA und konnten es erhalten, was mit der Senkung des Serum-Kreatinins zusammenhängt. Kein Patient benötigte eine neue Dialyse. Die Nierenfunktion, gemessen als geschätzte glomeruläre Filtrationsrate (eGFR), verbesserte sich unter Soliris-Behandlung.
Tabelle 6: Ergebnisse zur Wirksamkeit in den prospektiven aHUS-Studien C08-002A/B und C08-003A/B
C08-002A/B N = 17 C08-003A/B N = 20
Nach 26 Wochen Nach 2 Jahren1 Nach 26 Wochen Nach 2 Jahren1
Normalisierung der 14 (82) (57-96) 15 (88) (64-99) 18 (90) (68-99) 18 (90) (68-99)
Thrombozytenzahl 13/15 (87) 13/15 (87) 3/20 (15) 1/3 (33)
Alle Patienten, n
(%) (95 % KI)
Patienten mit
abnormem Ausgangswer
t, n/n (%)
Abwesenheit von 15 (88) (64-99) 15 (88) (64-99) 16 (80) (56-94) 19 (95) (75-99)
TMA-Ereignissen, n
(%) (95 % KI)
TMA Interventionsrat 0,88 (0,04/1,59) 0 0,88 (0,04/1,59) 0 0,23 (0,05/1,09) 0 0,23 (0,05/1,09) 0
e, Median pro Tag (0/0,31) p < 0,0001 (0/0,31) p <0,0001 p < 0,0001 P<0,0001
(min/max) -
prä-Eculizumab -
unter Eculizumab
p-Wert
CKD Verbesserung um 10 (59) (33-82) 12 (71) (44-90) 7 (35) (15-59) 12 (60) (36-81)
≥ 1 Stadium n (%)
(95 % KI)
eGFR Veränderung 20 (-1;98) 28 (3;82) 5 (-1;20) 11(-42;30)
ml/min/1,73 m2:
Median (Spanne)
eGFR Veränderung ≥ 8 (47) (23-72) 10 (59) (33-82) 1 (5) (0-25) 8 (40) (19-64)
15 ml/min/1,73m2; n
(%) (95 % KI)
Veränderung des Hb 11 (65) (38-86)2 13 (76) (50-93) 9 (45) (23-68)3 13 (65) (41-85)
> 20g/l, n (%) (95
% KI)
Normalisierung der 13 (76) (50-93) 15 (88) (64-99) 18 (90) (68-99) 18 (90) (68-99)
hämatologischen
Parameter, n (%)
(95 % KI)
Vollständiges 11 (65) (38-86) 13 (76) (50-93) 5 (25) (9-49) 11 (55) (32-77)
Ansprechen der TMA,
n (%) (95 % KI)
1 bei cut-off (20.April 2012)
2 Studie C008-002: 3 Patienten erhielten Erythropoiese stimulierende Substanzen, die nach Beginn der Soliris-Behandlung abgesetzt wurden.
3 Studie C008-003: 8 Patienten erhielten Erythropoiese stimulierende Substanzen, die bei 3 Patienten während der Soliris-Behandlung abgesetzt wurden.
Die Studie C10-004 untersuchte 41 Patienten mit Anzeichen von thrombotischer Mikroangiopathie (TMA). Einschlusskriterien waren: Thrombozytenzahl niedriger als die untere Grenze des Normalbereichs, Anzeichen für eine Hämolyse wie erhöhter Serum-LDH-Spiegel und Serum-Kreatinin-Spiegel oberhalb der oberen Grenze des Normalbereichs, ohne chronischen Dialysebedarf. Das mediane Alter der Patienten betrug 35 Jahre (zwischen 18 und 80 Jahre). Alle in die Studie C10-004 aufgenommenen Patienten hatten einen ADAMTS-13-Wert über 5 %. 51 % der Patienten hatten eine festgestellte Mutation eines Komplement-regulierenden Faktors oder Autoantikörper. Insgesamt erhielten 35 Patienten eine Plasmainfusion oder einen Plasmaaustausch, oder eine Soliris-Gabe vor Beginn der Behandlung mit Eculizumab. Tabelle 7 fasst die klinischen Eigenschaften und die Eigenschaften, die in Zusammenhang mit der Erkrankung der Patienten bei Beginn der Studie C10-004 stehen, zusammen.
Tabelle 7: Eigenschaften der Patienten bei Beginn der klinischen Studie zu aHUS C10-004
Parameter Studie aHUS C10-004n
= 41
Zeit von der Erstdiagnose der aHUS bis zur ersten Studienmedikation (Monat), 0,79 (0,03 – 311)
Median (min, max)
Zeit von der Manifestierung der bestehenden TMA und erster verabreichter 0,52 (0,03 – 19)
Dosis innerhalb der Studie (Monat), Median (min, max)
Thrombozytenzahl bei Screening (× 109/l), Median (min, max) 125 (16 – 332)
LDH Ausgangswert (U/l), Median (min, max) 375 (131 – 3318)
eGFR, Ausgangswert (ml/min/1- 73m3) Median (min; max) 10 (6; 53)
Die Patienten der Studie C10-004 wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung dauerhaft fort. Bei cut-off betrug die mediane Behandlungsdauer mit Soliris ungefähr 50 Wochen (Spanne: 13-86 Wochen).
Nach dem Beginn der Behandlung mit Soliris konnten eine Abnahme der terminalen Komplementaktivität und ein Anstieg der Thrombozytenzahl gegenüber dem Studieneinschluss beobachtet werden. Soliris reduzierte die Anzeichen einer Komplement-vermittelten TMA, was der Anstieg der medianen Thrombozytenzahl zwischen Studieneinschluss und Woche 26 zeigt. In der Studie stieg die mediane Thrombozytenzahl von 119 ± 66 x109/l bei Studieneinschluss auf 200 ± 84 x109/l nach Woche 1; diese Entwicklung konnte 26 Wochen lang aufrechterhalten werden (mediane Thrombozytenzahl in Woche 26: 252 ± 70 x109/l). Die Nierenfunktion, die anhand der medianen eGFR bewertet wurde, konnte unter Soliris verbessert werden. 20 der 24 Patienten, die vor Studienbeginn auf eine Dialyse angewiesen waren, konnten während der Dauer der Solirisbehandlung die Dialysebehandlung unterbrechen. Tabelle 8 fasst die Ergebnisse aus Studie C10-004 hinsichtlich der Wirksamkeit zusammen.
Tabelle 8: Ergebnisse zur Wirksamkeit in der prospektiven aHUS Studie C10-004
Wirksamkeitsparameter Studie aHUS C10-004 (n =
41) Bis 26 Wochen
Veränderung der Thrombozytenzahl zwischen Screening und Woche 26 111 (-122; 362)
(109/l)
Normalisierung der Blutwerte, n (%)Dauer bis zur Normalisierung der 36 (88) 46 (10; 74)
Blutwerte, Median in Wochen (min, max)
Vollständiges Ansprechen der TMA, n (%)Dauer bis zum vollständigen 23 (56) 42 (6; 74)
Ansprechen der TMA, Median in Wochen (min, max)
Fehlende Anzeichen von TMA, n (%) KI 95 % 37 (90) 77; 97
TMA Interventionsrate, Median pro Tag (min, max): -prä-Eculizumab 0,63 (0; 1,38) 0 (0; 0,58)
-unter Eculizumab
1 Bei cut-off (4. September 2012), bei medianer Dauer der Soliris-Therapie von 50 Wochen (Spanne: 13 bis 86 Wochen)
Eine Langzeitbehandlung mit Soliris (Median 52 Wochen, im Bereich von 15 bis 126 Wochen) war mit einem erhöhten Anteil von klinisch bedeutsamen Verbesserungen bei erwachsenen aHUS-Patienten verbunden. Als die Soliris-Therapie länger als 26 Wochen beibehalten wurde, erreichten 3 weitere Patienten (63 % der Patienten insgesamt) ein vollständiges MAT-Ansprechen. Weitere 4 Patienten (98 % der Patienten insgesamt) erreichten eine Normalisierung der hämatologischen Parameter. Bei der letzten Auswertung erreichten 25 der 41 Patienten (61 %) eine Verbesserung der eGFR ≥ 15 ml/min / 1,73 m2 im Vergleich zum Ausgangswert.
Refraktäre generalisierte Myasthenia gravis
Die Wirksamkeit von Soliris bei der Behandlung von Patienten mit refraktärer gMG wurde aus den Daten von 139 Patienten in zwei prospektiven kontrollierten klinischen Studien (C08-001 und ECU-MG-301) und einer offenen Verlängerungsstudie (ECU-MG-302) ermittelt.
Bei der Studie ECU-MG-301 (REGAIN) handelte es sich um eine 26-wöchige doppelblinde, randomisierte, placebo-kontrollierte, multizentrische Phase-3-Studie mit Soliris bei Patienten, die auf vorangegangene Therapien nicht angesprochen hatten und weiterhin symptomatisch waren. Einhundertachtzehn (118) der 125 (94 %) Patienten schlossen die 26-wöchige Behandlungsphase ab und 117 (94 %) Patienten wurden anschliessend in die Studie ECU-MG-302 aufgenommen, eine offene, multizentrische Verlängerungsstudie zur Untersuchung der langfristigen Wirksamkeit und Sicherheit, bei der alle Patienten eine Behandlung mit Soliris erhielten.
In der Studie ECU-MG-301 wurden gMG-Patienten mit einem positiven serologischen Test auf Anti-AChR-Antikörper, mit klinischer MGFA-Klassifizierung (Myasthenia Gravis Foundation of America) der Klassen II bis IV und einem MG-ADL-Gesamtscore von mindestens 6 randomisiert entweder Soliris (n = 62) oder Placebo (n = 63) zugeordnet. Alle in die Studie eingeschlossenen Patienten hatten refraktäre gMG und erfüllten die folgenden im Vorfeld festgelegten Kriterien:
1) Seit mindestens einem Jahr erfolglose Behandlung mit 2 oder mehr immunsuppressiven Therapien (entweder in Kombination oder als Monotherapie), d.h. Patienten, deren Alltagsaktivitäten trotz immunsuppressiver Therapien weiterhin eingeschränkt waren
ODER
2) Erfolglose Behandlung mit mindestens einer immunsuppressiven Therapie und Notwendigkeit eines dauerhaften Plasmaaustauschs oder von intravenösen Immunglobulinen (IVIg) zur Kontrolle der Symptome, d.h. die Patienten benötigten mindestens alle 3 Monate über die letzten 12 Monate regelmässig Plasmaaustausch oder IVIg zur Behandlung von Muskelschwäche.
Vor Beginn der Behandlung mit Soliris wurden die Patienten gegen Meningokokken geimpft oder erhielten eine geeignete Antibiotikaprophylaxe bis 2 Wochen nach der Impfung. In den Studien ECU-MG-301 und ECU-MG-302 betrug die Soliris-Dosis bei erwachsenen Patienten mit refraktärer gMG 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1.200 mg in Woche 5 ± 2 Tage und anschliessend 1.200 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 35 Minuten verabreicht. Die Tabelle 9 zeigt die Ausgangsparameter der in die Studie ECU-MG-301 aufgenommenen Patienten mit refraktärer gMG.
Tabelle 9: Demografische Patientendaten und -parameter in der Studie ECU-MG-301
Soliris (n=62) Placebo (n=63)
Alter zum Zeitpunkt der MG-Diagnose (in Jahren), 38,0 (5,9; 70,8) 38,1 (7,7; 78,0)
Mittelwert (Min, Max)
Weiblich, n (%) 41 (66,1) 41 (65,1)
Dauer der MG (Jahre), Mittelwert (Min, Max) 9,9 (1,3; 29,7) 9,2 (1,0; 33,8)
Ausgangswert MG-ADL-Score
Mittelwert (SD) 10,5 (3,06) 9,9 (2,58)
Median 10,0 9,0
Ausgangswert QMG-Score
Mittelwert (SD) 17,3 (5,10) 16,9 (5,56)
Median 17,0 16,0
≥3 vorangegangene immunsuppressive Therapien* seit der 31 (50,0) 34 (54,0)
Diagnose, n (%)
Anzahl Patienten mit vorangegangenen Exazerbationen 46 (74,2) 52 (82,5)
seit der Diagnose, n (%)
Anzahl Patienten mit vorangegangener MG-Krise seit der 13 (21,0) 10 (15,9)
Diagnose, n (%)
Vorangegangene künstliche Beatmung seit der Diagnose, n 15 (24,2) 14 (22,2)
(%)
Vorangegangene Intubation seit der Diagnose 11 (17,7) 9 (14,3)
(MGFA-Klasse V), n (%)
* Immunsuppressiva waren unter anderem Kortikosteroide, Azathioprin, Mycophenolat, Methotrexat, Ciclosporin, Tacrolimus und Cyclophosphamid.
Der primäre Endpunkt der Studie ECU-MG-301 war die Veränderung des MG-ADL-Gesamtscores (ADL, Activities of Daily Living Profile – eine vom Patienten gemeldete Outcome-Variable, die bei gMG bewertet wird) in Woche 26 gegenüber dem Ausgangswert. Die primäre Analyse des MG-ADL war eine Worst-Rank-ANCOVA mit einem durchschnittlichen Rang von 56,6 für Soliris und 68,3 für Placebo, basierend auf 125 Patienten in der Studie (p = 0,0698).
Der wichtigste sekundäre Endpunkt war die Veränderung des Gesamtscores des Quantitative MG Scoring System (QMG – eine vom Arzt gemeldete Outcome-Variable, die bei gMG bewertet wird) in Woche 26 gegenüber dem Ausgangswert. Die primäre Analyse des QMG war eine Worst-Rank-ANCOVA mit einem durchschnittlichen Rang von 54,7 für Soliris und 70,7 für Placebo, basierend auf 125 Patienten der Studie (p = 0,0129).
Die Ergebnisse zur Wirksamkeit für die zuvor festgelegten Analysen der primären und sekundären Endpunkte mit wiederholten Messungen sind in Tabelle 10 aufgeführt.
Tabelle 10: ECU-MG-301 Ergebnisse zur Wirksamkeit: Veränderung in Woche 26 gegenüber dem Ausgangswert
Wirksamkeitsendpunkt Soliris (n=62) (SEM) Placebo (n=63) (SEM) Veränderung unter p-Wert (mittels
e: Veränderung des Soliris gegenüber Analyse mit wiederho
Gesamtscores in Placebo – Differenz lten Messungen)
Woche 26 gegenüber der Kleinste-Quadrat
dem Ausgangswert e-Mittelwerte (95%
KI)
MG-ADL -4,2 (0,49) -2,3 (0,48) -1,9 (-3,3; -0,6) 0,0058
QMG -4,6 (0,60) -1,6 (0,59) -3,0 (-4,6; -1,3) 0,0006
MGC -8,1 (0,96) -4,8 (0,94) -3,4 (-6,0; -0,7) 0,0134
MG-QoL-15 -12,6 (1,52) -5,4 (1,49) -7,2 (-11,5; -3,0) 0,0010
SEM = Standardfehler des Mittelwertes; KI = Konfidenzintervall; MGC = Myasthenia Gravis Composite; MG-QoL15 = Myasthenia Gravis Quality of Life 15
In der Studie ECU-MG-301 wurde für einen klinischen Responder beim MG-ADL-Gesamtscore eine Verbesserung um mindestens 3 Punkte definiert. Der Anteil der klinischen Responder, die bis einschliesslich Woche 26 keine Notfallbehandlung erhielten, betrug unter Soliris 59,7 %, verglichen mit 39,7 % unter Placebo (p = 0,0229). In Studie ECU-MG-301 wurde für einen klinischen Responder beim QMG-Gesamtscore eine Verbesserung um mindestens 5 Punkte definiert. Der Anteil der klinischen Responder, die in Woche 26 keine Notfallbehandlung erhielten, betrug bei Soliris 45,2 % verglichen mit 19 % unter Placebo (p = 0,0018).
Tabelle 11 zeigt eine Übersicht der Patienten, die in den 26 Wochen über eine klinische Verschlechterung berichteten, und der Patienten, die in diesem Zeitraum eine Notfallbehandlung benötigten.
Tabelle 11: Klinische Verschlechterung und Notfallbehandlung in ECU-MG-301
Variable Statistik Placebo (N=63) Soliris (N=62)
Gesamtzahl Patienten, die über n (%) 15 (23,8) 6 (9,7)
eine klinische Verschlechterung
berichteten
Gesamtzahl Patienten, die eine n (%) 12 (19,0) 6 (9,7)
Notfallbehandlung benötigten
Von den 125 Patienten der Studie ECU-MG-301 wurden 117 Patienten anschliessend in die Langzeit-Verlängerungsstudie (Studie ECU-MG-302) aufgenommen, in der alle Patienten Soliris erhielten. Patienten, die zuvor in der Studie ECU-MG-301 mit Soliris behandelt worden waren, zeigten bei allen Zielgrössen (MG-ADL, QMG, MGC und MG-QoL15) über einen zusätzlichen Eculizumab-Behandlungszeitraum von 130 Wochen weiterhin eine nachhaltige Wirkung von Soliris. Bei Patienten, die in der Studie ECU-MG-301 (Placebo/Eculizumab-Arm der Studie ECU-MG-302) Placebo erhielten, trat nach Beginn der Behandlung mit Eculizumab eine Verbesserung ein, die in der Studie ECU-MG-302 über mehr als 130 Wochen aufrechterhalten wurde. Abbildung 1 zeigt die Veränderung gegenüber dem Ausgangswert bei MG-ADL (A) und QMG (B) nach 26wöchiger Behandlung in Studie ECU-MG-301 und nach 130-wöchiger Behandlung (n = 80 Patienten) in Studie ECU-MG-302.
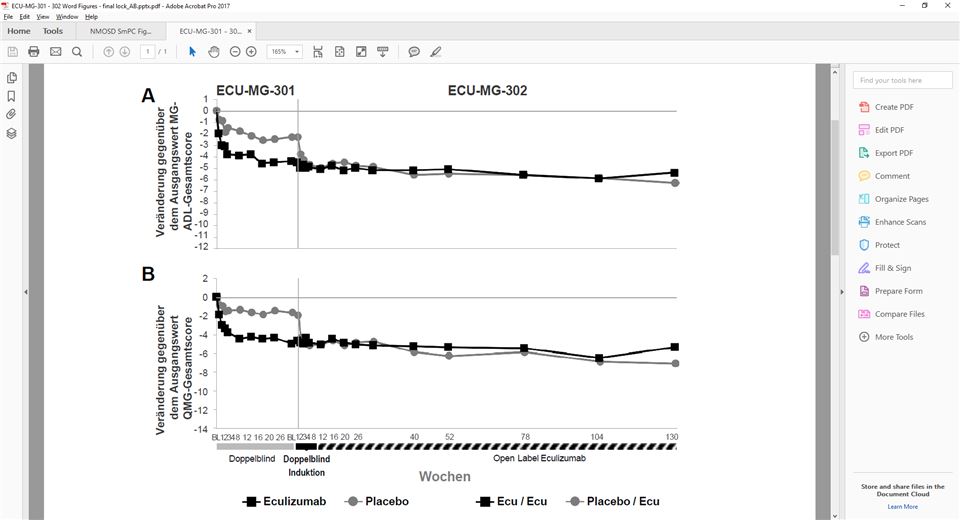
Abbildung 1: Durchschnittliche Veränderungen gegenüber dem Ausgangswert bei MG-ADL (1A) und QMG (1B) in den Studien ECU-MG-301 und ECU-MG-302
In der Studie ECU-MG-302 hatten Ärzte die Möglichkeit, bestehende Hintergrundtherapien mit Immunsuppressiva anzupassen. In diesem Rahmen verringerten 65,0% der Patienten ihre tägliche Dosis von mindestens 1 immunsuppressiven Therapie (IST); 43,6% der Patienten brachen eine bestehende IST ab. Der häufigste Grund für eine Änderung der IST-Therapie war die Verbesserung der MG-Symptome.
Neuromyelitis-optica-Spektrumerkrankungen
Daten von 143 Patienten in einer kontrollierten Studie (ECU-NMO-301) und von 119 Patienten, die in einer offenen Verlängerungsstudie (Studie ECU-NMO-302) weiterbehandelt wurden, wurden verwendet, um die Wirksamkeit und Sicherheit von Soliris bei der Behandlung von Patienten mit NMOSD zu beurteilen.
Studie ECU-NMO-301 war eine doppelblinde, randomisierte, placebokontrollierte, multizentrische Phase-III-Studie mit Soliris bei Patienten mit NMOSD.
In Studie ECU-NMO-301 wurden NMOSD-Patienten mit positivem Serumtest auf Anti-AQP4-Antikörper, einer Anamnese mit mindestens 2 Schüben in den letzten 12 Monaten oder 3 Schüben in den letzten 24 Monaten und mindestens 1 Schub in den 12 Monaten vor dem Screening sowie einem Expanded Disability Status Scale (EDSS) Score von ≤ 7 im Verhältnis 2:1 auf Soliris (n = 96) oder Placebo (n = 47) randomisiert.
Den Patienten war es gestattet, während der Studie eine Hintergrundbehandlung mit Immunsuppressiva in stabiler Dosis, mit Ausnahme von Rituximab und Mitoxantron anzuwenden.
Die Patienten erhielten entweder mindestens 2 Wochen vor Beginn der Behandlung mit Soliris eine Meningokokken-Impfung oder bis 2 Wochen nach der Impfung eine prophylaktische Behandlung mit geeigneten Antibiotika. In dem klinischen Entwicklungsprogramm für Eculizumab bei NMOSD betrug die Soliris-Dosis bei erwachsenen NMOSD-Patienten 900 mg alle 7 ± 2 Tage über 4 Wochen, gefolgt von 1200 mg in Woche 5 ± 2 Tage und anschliessend 1200 mg alle 14 ± 2 Tage für die Dauer der Studie. Soliris wurde als intravenöse Infusion über 35 Minuten gegeben.
Die Mehrheit (90,9 %) der Patienten war weiblich. Ungefähr die Hälfte der Patienten (49,0 %) war weiss. Das Durchschnittsalter bei der ersten Gabe des Studienmedikaments betrug 45 Jahre.
Tabelle 12: Krankheitsanamnese und Ausgangsparameter der Patienten in Studie ECU-NMO-301
Variable Statistik Placebo (N = 47) Eculizumab (N = 96) Insgesamt (N = 143)
NMOSD-Anamnese
Alter bei der Mittelwert (SD) 38,5 (14,98) 35,8 (14,03) 36,6 (14,35)
ersten klinischen
Manifestation der
NMOSD (Jahre)
Median 38,0 35,5 36,0
Min, Max 12; 73 5; 66 5; 73
Zeitraum von der Mittelwert (SD) 6,601 (6,5863) 8,156 (8,5792) 7,645 (7,9894)
ersten klinischen
Manifestation der
NMOSD bis zur
Anwendung der
ersten Dosis des
Studienmedikaments
(Jahre)
Median 3,760 5,030 4,800
Min, Max 0,51; 29,10 0.41, 44.85 0,41; 44,85
Anamnestische Mittelwert (SD) 2,07 (1,037) 1,94 (0,896) 1,99 (0,.943)
annualisierte
Schubrate innerhalb
von 24 Monaten vor
dem Screening
Median 1,92 1,85 1,92
Min, Max 1,0; 6,4 1.0, 5.7 1,0; 6,4
Ausgangsparameter
EDSS-Ausgangsscore Mittelwert (SD) 4,26 (1,510) 4,15 (1,646) 4,18 (1,598)
Median 4,00 4,00 4,00
Min, Max 1,0; 6,5 1,0; 7,0 1,0; 7,0
Keine IST-Anwendung n (%) 13 (27,7) 21 (21,9) 34 (23,8)
zu Studienbeginn
Abkürzungen: EDSS = Expanded Disability Status Scale; IST = Immunsupressiva -Therapie; Max = Maximum; Min = Minimum; NMOSD = Neuromyelitis-optica-Spektrum-Erkrankungen; SD = Standardabweichung (standard deviation).
Der primäre Endpunkt von Studie ECU-NMO-301 war die Dauer bis zum ersten Schub während der Studie gemäss der Bestätigung durch ein unabhängiges und für die Behandlung verblindetes Komitee. Während der Studie wurde ein signifikanter Effekt von Eculizumab im Vergleich zu Placebo bezüglich der Dauer bis zum Auftreten des ersten bestätigten Schubes beobachtet (relative Risikoreduktion 94 %; Hazard Ratio 0,058; p<0,0001) (Abbildung 2). Die mit Soliris behandelten Patienten zeigten eine ähnliche Verbesserung der Dauer bis zum ersten bestätigten Schub während der Studie mit oder ohne begleitende IST-Therapie.
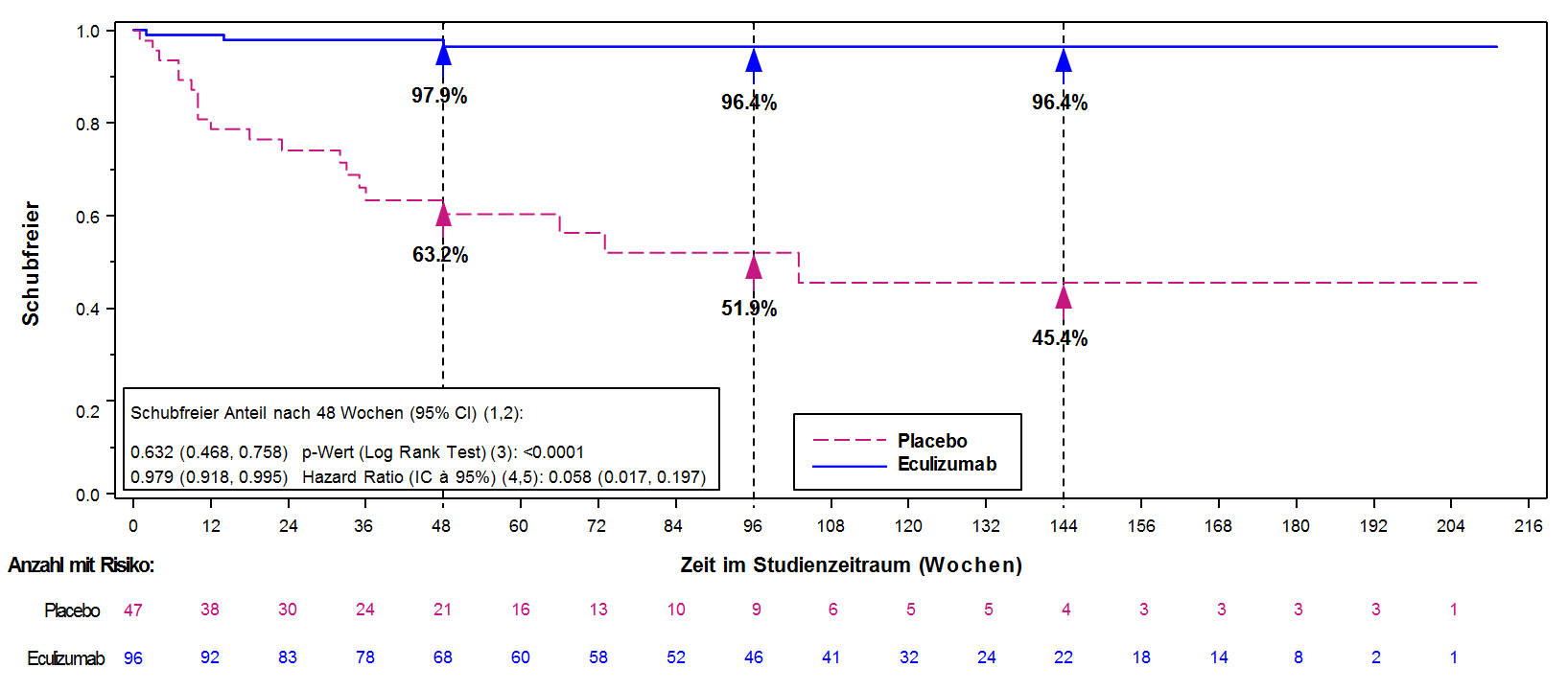
Abbildung 2: Kaplan-Meier-Kurve für die Zeit bis zum ersten bestätigten Schub während der Studie ECU-NMO-301 – vollständiges Analyseset
Hinweis: Patienten, bei denen kein bestätigter Schub während der Studie auftrat, wurden am Ende des Studienzeitraums zensiert.
Stratifizierte Analysen basieren auf vier Randomisierungsstraten:
(i) niedriger EDSS bei Randomisierung (<= 2,0), (ii) hoher EDSS (> = 2,5 bis <= 7) und nicht vorbehandelt bei Randomisierung, (iii) hoher EDSS (> = 2,5 bis <= 7) und Fortsetzung derselben IST(s) seit dem letzten Schub bei der Randomisierung, (iv) hoher EDSS (> = 2,5 bis <= 7) und Änderungen bei den IST(s) seit dem letzten Schub bei der Randomisierung.
1 Basierend auf der Kaplan-Meier-Methode.
2 Basierend auf der komplementären Log-Log-Transformation.
3 Basierend auf einem stratifizierten Log-Rank-Test.
4 Basierend auf einem stratifizierten Cox-Proportional-Hazards-Modell.
5 Wald-Konfidenzintervall.
Abkürzungen: KI = Konfidenzintervall (confidence interval); EDSS = Expanded Disability Status Scale; IST = Immunsuppressiva-Therapie
Das Verhältnis der bestätigten jährlichen Schubrate (annualized relapse rate, ARR) während der Studie (95% KI) für Eculizumab im Vergleich zu Placebo betrug 0,045 (0,013; 0,151). Dies entspricht einer relativen Reduktion der bestätigten ARR während der Studie von 95,5% bei Patienten, die mit Eculizumab behandelt wurden, im Vergleich zu Placebo (p <0,0001) (Tabelle 13).
Tabelle 13: Bestätigte annualisierte Schubrate während der Studie – vollständiges Analyseset
Variable Statistik Placebo (N = 47) Eculizumab (N = 96)
Gesamtzahl der Schübe Summe 21 3
Gesamtzahl der Patientenjahre im n 52,41 171,32
Studienzeitraum
Adjustierte bestätigte ARRa Rate 0,350 0,016
95% KI 0,199; 0,616 0,005; 0,050
Behandlungseffekta Rate Ratio (Eculizum … 0,045
ab/Placebo)
95% KI … 0,013; 0,151
p-Wert … <0,0001
a Basierend auf einer
Poisson-Regression, angepasst um
Randomisierungsstraten und die
historische ARR in 24 Monaten vor
dem Screening. Abkürzungen: ARR =
annualisierte Schubrate; KI =
Konfidenzintervall.
Verglichen mit Patienten, die Placebo erhielten, wiesen die mit Soliris behandelten Patienten niedrigere annualisierte Raten auf für stationäre Behandlungen (0,04 für Soliris versus 0,31 für Placebo), intravenöse Kortikosteroid-Anwendungen zur Behandlung von akuten Schüben (0,07 für Soliris versus 0,42 für Placebo) und Plasmaaustausch-Behandlungen (0,02 für Soliris versus 0,19 für Placebo).
Die Messung der Veränderungen von Studienbeginn bis Studienende bei den sekundären Endpunkten neurologische Behinderungen (EDSS-Score [p-Wert = 0,0597] und mRS [nominaler p-Wert = 0,0154]), Funktionseinschränkung (HAI [nominaler p-Wert = 0,0002]) und Lebensqualität (EQ-5D VAS [nominaler p-Wert = 0,0309] und EQ-5D Index [nominaler p-Wert = 0,0077]) fiel zu Gunsten von Eculizumab gegenüber Placebo aus.
Die finale Analyse der Studie ECU-NMO-302 (mediane Behandlungsdauer 20.00 Wochen [0.1;198.4 Wochen]) zeigte eine statistisch signifikante und klinisch bedeutsame Abnahme der ARR während der Studie (gemäss Entscheidung des behandelnden Arztes) unter der Eculizumab-Behandlung, basierend auf der medianen (Min, Max) Veränderung (-1,825 [-6,38; 1,02], p <0,0001) gegenüber der anamnestischen ARR (24 Monate vor dem Screening in der Studie ECU-NMO-301). Insgesamt liegen für die Behandlung mit Soliris über 20 Wochen nur limitierte Daten zu Wirksamkeit und Sicherheit vor.
In der Studie ECU-NMO-302 hatten Ärzte die Möglichkeit, bestehende Hintergrundtherapien mit Immunsuppressiva anzupassen. In diesem Rahmen war die häufigste Änderung der immunsuppressiven Therapie eine Verringerung der Immunsuppressiva-Dosis, die bei 21,0 % der Patienten erfolgte. Zudem brachen 15,1 % der Patienten eine bestehende IST-Therapie ab.
Soliris (Eculizumab) wurde nicht im Hinblick auf die Behandlung von akuten Schüben bei NMOSD-Patienten untersucht.
Sicherheit und Wirksamkeit bei älteren Patienten
Refraktäre gMG
Zweiundzwanzig (22) (17,6 %) der älteren Patienten mit refraktärer gMG (> 65 Jahre) wurden im Rahmen der klinischen Studien mit Soliris behandelt. Dabei wurden keine wesentlichen Unterschiede in Bezug auf Sicherheit und Wirksamkeit in Zusammenhang mit dem Lebensalter festgestellt.
Neuromyelitis-optica-Spektrumerkrankungen
Soliris wurde nur in insgesamt 9 älteren Patienten mit NMOSD (3 unter Placebo, 6 unter Eculizumab untersucht, so dass keine verlässlichen Aussagen über Sicherheit und Wirksamkeit in dieser Population getroffen werden können.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Paroxysmale Nächtliche Hämoglobinurie
In der Studie M07-005 wurden insgesamt 7 pädiatrische Patienten mit PNH mit einem medianen Körpergewicht von 57,2 kg (Spanne 48,6 bis 69,8 kg) und im Alter von 11 bis 17 Jahren (medianes Alter: 15,6 Jahre) mit Soliris behandelt.
Die Behandlung mit Eculizumab entsprechend dem Dosierungsschema für Kinder und Jugendliche führte zu einer Verminderung der intravaskulären Hämolyse, gemessen anhand des Serum-LDH Spiegels. Die Behandlung führte auch zu einer deutlichen Reduktion oder Elimination des Bedarfes an Bluttransfusionen und tendenziell zu einer generellen Verbesserung des Allgemeinzustandes. Die Wirksamkeit einer Behandlung mit Eculizumab bei pädiatrischen Patienten mit PNH scheint mit der Wirksamkeit bei Erwachsenen mit PNH übereinzustimmen, die in den Zulassungsstudien (C04-001 und C04-002) behandelt wurden (Tabelle 3 und 14).
Tabelle 14: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten mit PNH in Studie M07-005
P – Wert
Mittelwert (SD) Wilcoxon-Vorzeichen-Rang
-Test
Veränderung LDH-Wert nach 12 Wochen gegenüber dem -771 (914) 0,0156
Ausgangswert (U/l)
LDH-AUC(U/l x Tag) -60.634 (72.916) 0,0156
Atypisches Hämolytisch-Urämisches Syndrom
In Studie C09-001r wurden insgesamt 15 pädiatrische Patienten (Alter 2 Monate bis <12 Jahre) mit Soliris behandelt. Siebenundvierzig Prozent dieser Patienten hatten eine nachgewiesene Mutation eines Komplementregulierenden Faktors oder Autoantikörper. Die mediane Zeit von der aHUS-Diagnose bis zur ersten Verabreichung von Soliris betrug 14 Monate (Spanne <1 bis 110 Monate). Die mediane Zeit von der Manifestation der bestehenden thrombotischen Mikroangiopathie bis zur ersten Verabreichung von Soliris betrug 1 Monat (Spanne <1 bis 16 Monate). Die mediane Dauer der Soliris-Behandlung betrug in der Gruppe der Patienten unter 2 Jahren 16 Wochen (Spanne 4 bis 70 Wochen; n = 5), in der Gruppe der 2 bis < 12-jährigen Patienten 31 Wochen (Spanne 19-63 Wochen; n = 10).
Insgesamt erscheinen die Ergebnisse zur Wirksamkeit bei diesen pädiatrischen Patienten konsistent mit den Ergebnissen der Patienten in den aHUS Pivot-Studien C008-002 und C008-003 (Tabelle 6). Keiner der pädiatrischen Patienten benötigte eine neue Dialyse.
Tabelle 15: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten in Studie C09-001r
Wirksamkeits-Parameter < 2 Jahre (n = 5) 2 bis < 12 Jahre (n < 12 Jahre (n = 15)
= 10)
Patienten mit Normalisierung der 4 (80) 10 (100) 14 (93)
Thrombozytenzahl, n (%)
Vollständiges Ansprechen der TMA, 2 (40) 5 (50) 7 (50)
n (%)
TMA Interventionsrate, Median pro 1 (0/2) <1 (0/<1) <1 (0,07/1.46) 0 <1 (0/2) 0 (0/<1)
Tag (Spanne) -prä-Eculizumab (0/<1)
-unter Eculizumab
Patienten mit eGFR Veränderung 2 (40) 6 (60) 8 (53)
≥15 ml/min/1,73 m2, n (%)
Bei pädiatrischen Patienten mit einer kürzeren Dauer einer bestehenden schweren klinischen Manifestation der thrombotischen Mikroangiopathie (TMA) vor Eculizumab-Behandlung wurden eine Kontrolle der TMA und eine Verbesserung der Nierenfunktion unter Eculizumab-Behandlung erreicht (Tabelle 15).
Bei pädiatrischen Patienten mit einer längeren Dauer einer bestehenden schweren klinischen Manifestation der thrombotischen Mikroangiopathie (TMA) vor Eculizumab-Behandlung wurde eine Kontrolle der TMA unter Eculizumab-Behandlung erreicht. Die Nierenfunktion verbesserte sich jedoch auf Grund der bereits bestehenden irreversiblen Nierenschädigung nicht (Tabelle 16).
Tabelle 16: Ergebnisse zur Wirksamkeit bei pädiatrischen Patienten in Studie C09-001r in Bezug auf die Dauer der aktuellen schweren klinischen Manifestation einer thrombotischen Mikroangiopathie (TMA)
Dauer der aktuellen schweren
klinischen Manifestation einer TMA
< 2 Monate(N = 10) % > 2 Monate(N= 5) %
Patienten mit Normalisierung der 9 (90) 5 (100)
Thrombozytenzahl, n (%)
Abwesenheit von TMA-Ereignissen, n (%) 8 (80) 3 (60)
Vollständiges Ansprechen der TMA, n 7 (70) 0
(%)
Patienten mit eGFR Veränderung ≥15 7 (70) 0*
ml/min/1,73 m2, n (%)
*Ein Patient erreichte eine Verbesserung der eGFR nach Nierentransplantation
Insgesamt erhielten 22 Kinder oder Jugendliche (im Alter zwischen 5 Monaten und 17 Jahren) eine Behandlung mit Soliris im Rahmen der Studie C10-003.
Um in die Studie aufgenommen werden zu können, mussten die Patienten eine Thrombozytenzahl aufweisen, die unterhalb der unteren Grenze des Normalbereichs lag, zudem Anzeichen für eine Hämolyse wie einen erhöhten Serum-LDH-Spiegel oberhalb des oberen Normalbereichs und einen Serum-Kreatinin-Spiegel ≥ 97. Perzentile im Verhältnis zum Alter ohne chronischen Dialysebedarf. Das mediane Alter der Patienten lag bei 6,5 Jahren (Spanne: 5 Monate bis 17 Jahre). Die Patienten, die in die Studie zu aHUS C10-003 aufgenommen wurden, hatten einen ADAMTS-13-Wert über 5 %. 50 % der Patienten hatten eine festgestellte Mutation eines Komplement-regulierenden Faktors oder Autoantikörper. Insgesamt erhielten 10 Patienten eine Plasmainfusion, einen Plasmaaustausch oder eine Soliris-Gabe vor Beginn der Behandlung mit Soliris. Tabelle 17 fasst die klinischen Eigenschaften und die Eigenschaften, die in Zusammenhang mit der Erkrankung der Patienten bei Beginn der Studie C10-003 stehen, zusammen.
Tabelle 17: Eigenschaften der pädiatrischen Patienten bei Beginn der Studie zu aHUS C10-003
Parameter 1 Monat bis < 12 Alle Patienten
Jahre(n=18) (n=22)
Zeit von der Erstdiagnose der aHUS bis zur ersten 0,51 (0,03 – 58) 0,56 (0,03-191)
Studienmedikation (Monat) Median (min, max)
Zeit von der Manifestierung der bestehenden TMA und 0,23 (0,03 – 4) 0,2 (0,03-4)
erster verabreichter Dosis innerhalb der Studie,
(Monat) Median (min, max)
Thrombozytenzahl bei Screening (× 109/l), Median (min, 110 (19-146) 91 (19-146)
max)
LDH-Ausgangswert (U/l), Median (min, max) 1510 (282-7164) 1244 (282-7164)
eGFR, Ausgangswert (ml/min/1.73m2) Median (min, max) 22 (10,105) 22 (10, 105)
Patienten der Studie C10-003 wurden mindestens 26 Wochen mit Soliris behandelt. Nach Abschluss der initialen 26-wöchigen Behandlungsphase führten die meisten Patienten die Behandlung dauerhaft fort.
Nach dem Beginn der Behandlung mit Soliris konnte eine Abnahme der terminalen Komplementaktivität beobachtet werden. Soliris reduzierte die Anzeichen einer Komplement-vermittelten TMA, was der Anstieg der medianen Thrombozytenzahl zwischen Studieneinschluss und Woche 26 zeigt. In der Studie stieg die mediane Thrombozytenzahl von 88±42x109/l bei Studieneinschluss auf 281±123x109/l nach Woche 1; diese Entwicklung konnte 26 Wochen lang aufrechterhalten werden (mediane Thrombozytenzahl in Woche 26: 293±106 x109/l).
Die Nierenfunktion, die anhand der eGFR bewertet wurde, konnte unter Soliris verbessert werden. 9 der 11 Patienten, die vor Studienbeginn dialysepflichtig waren, waren nach Tag 15 der Behandlung mit Soliris nicht mehr auf eine Dialyse angewiesen. Das Ansprechen war bei allen Altersstufen zwischen 5 Monaten und 17 Jahren vergleichbar. In der Studie C10-003 zeigten Patienten mit und ohne identifizierte Mutationen in Genen, die Proteine für Komplement-regulierende Faktoren oder gegen den Faktor H gerichtete Autoantikörper kodieren, ein vergleichbares Ansprechen auf Soliris.
Tabelle 18 fasst die Ergebnisse aus der Studie zu aHUS C10-003 hinsichtlich der Wirksamkeit zusammen.
Tabelle 18: Ergebnisse zur Wirksamkeit der prospektiven aHUS Studie C10-003
Wirksamkeitsparameter 1 Monat bis < 12 Alle Patienten(n =
Jahre(n = 18) Bis 22) Bis 26 Wochen
26 Wochen
Vollständige Normalisierung der hämatologischen 14 (78) 35 (13; 78) 18 (82) 35 (13; 78)
Parameter, n (%)Dauer bis zur vollständigen
Normalisierung der hämatologischen Parameter, Median in
Wochen (min, max)
Vollständiges Ansprechen der TMA, n (%)Dauer bis zum 11 (61) 40 (13; 78) 14 (64) 37 (13; 78)
vollständigen Ansprechen der TMA, Median in Wochen
(min, max)1
Abwesenheit von TMA-Ereignissen, n (%)95 % KI 17 (94) n.a. 21 (96) 77; 99
TMA Interventionsrate, Median pro Tag (min, max): n.a. n.a. 0,4 (0; 1,7) 0 (0;
-prä-Eculizumab, -unter Eculizumab 1.01)
Verbesserung der eGFR ≥ 15 ml/min/ 1,73•m2, n (%) 16 (89) 19 (86)
Veränderung der eGFR (≥ 15 ml/min/1.73 m2) in Woche 26, 64 (0; 146) 58 (0; 146)
Median (min, max)
Verbesserung der CKD ≥ 1 Stadium, n (%) 14/16 (88) 17/20 (85)
Keine Plasmainfusion, Plasmaaustausch oder Gabe von 16 (89) 18 (100) 20 (91) 22 (100)
Soliris, in (%)Keine neue Dialyse, n (%) KI 95 % n.a. 85; 100
1Bei cut-off (12. Oktober 2012) mit einer medianen Dauer der Soliris-Therapie von 44 Wochen (Spanne: 1 Dosis bis 88 Wochen).
Eine Langzeitbehandlung mit Soliris (Median 55 Wochen im Bereich von 1 Tag bis 107 Wochen) war mit einem erhöhten Anteil von klinisch bedeutsamen Verbesserungen bei aHUS-Patienten im Kindes- und Jugendalter verbunden. Wenn die Soliris-Therapie länger als 26 Wochen beibehalten wurde, erreichte 1 weiterer Patient (68 % der Patienten insgesamt) ein vollständiges TMA-Ansprechen. Weitere 2 Patienten (91 % der Patienten insgesamt) erreichten eine Normalisierung der hämatologischen Parameter. Bei der letzten Auswertung erreichten 19 der 22 Patienten (86 %) eine Verbesserung der eGFR ≥ 15 ml / min / 1,73 m2, im Vergleich zum Ausgangswert. Kein Patient benötigte unter Soliris eine erneute Dialyse.
Refraktäre generalisierte Myasthenia gravis
Insgesamt 11 pädiatrische Patienten mit refraktärer gMG in der Studie ECU-MG-303 erhielten Soliris. Das mittlere (Spanne) Körpergewicht der behandelten Patienten zu Studienbeginn betrug 59,7 kg (37,2 bis 91,2 kg) und das mittlere (Spanne) Alter beim Screening war 15 Jahre (12 bis 17 Jahre). Bei allen in die Studie einbezogenen Patienten handelte es sich um Patienten mit refraktärer gMG, die eine oder mehrere der folgenden Eigenschaften aufwiesen:
1. Fehlgeschlagene Behandlung ≥ 1 Jahr mit mindestens 1 immunsuppressiven Therapie, definiert als: (i) anhaltende Schwäche mit Beeinträchtigung der Aktivitäten des täglichen Lebens oder (ii) Verschlimmerung und/oder myasthene Krise während der Behandlung oder (iii) Unverträglichkeit gegenüber IST aufgrund von Nebenwirkungen oder Begleiterkrankungen.
2. Benötigen eine Erhaltungstherapie mit PE oder IVIg zur Symptomkontrolle (d. h. Patienten, die regelmässig PE oder IVIg zur Behandlung von Muskelschwäche benötigen, mindestens alle 3 Monate in den letzten 12 Monaten vor dem Screening).
Die Ausgangsparameter der pädiatrischen Patienten mit refraktärer gMG, die in die Studie ECU-MG-303 aufgenommen wurden, sind in Tabelle 19 aufgeführt.
Tabelle 19: Demografische Patientendaten und -parameter
in der Studie ECU-MG-303
Eculizumab (n = 11)
Weiblich n (%) 9 (81,8 %)
Dauer der MG (Zeit von der MG-Diagnosestellung bis zur Mittelwert (SD) 3,99 (2,909) 2,90
ersten Studienmedikation [Jahre]) Median (min, max) (0,1; 8,8)
Ausgangswert MG-ADL-Gesamtscore Mittelwert (SD) 5,0 (5,25) 4,0 (0;
Median (min, max) 19)
Ausgangswert QMG-Gesamtscore Mittelwert (SD) 16,7 (5,64) 15,0
Median (min, max) (10; 28)
MGFA-Klassifizierung beim ScreeningIIa IIb IIIa IIIb n (%) 2 (18,2) 3 (27,3) 3
IVa IVb (27,3) 0 3 (27,3) 0
Anzahl Patienten mit vorangegangenen Exazerbationen der n (%) 4 (36,4) 7 (63,6) 6
MG, einschliesslich MG-Krise, seit der Diagnose Nein Ja (54,5) 3 (27,3)
Exazerbation MG-Krise
IVIg-Langzeittherapie bei Studienbeginn Ja Nein n (%) 6 (54,5) 5 (45,5)
Anzahl der Immunsupressiva-Therapien bei Studienbeginn n (%) 2 (18,2) 4 (36,4) 5
012 (45,5)
Patienten mit Immunsupressiva-Therapiena zu n (%) 8 (72,7) 1 (9,1) 2
Studienbeginn n (%) Kortikosteroide Azathioprin (18,2) 3 (27,3)
Mycophenolat-Mofetil Tacrolimus
aZu den Immunsuppressiva-Therapien gehörten Kortikosteroide, Azathioprin, Cyclophosphamid, Ciclosporin, Methotrexat, Mycophenolat-Mofetil oder Tacrolimus. Zu Studienbeginn erhielt keiner der Patienten Cyclosporin, Cyclophosphamid oder Methotrexat.
Abkürzungen: IVIg = intravenöses Immunglobulin; max = Maxiumum; MG = Myasthenia gravis; MG ADL = Myasthenia Gravis Activities of Daily Living (Myasthenia-Gravis-Profil für Aktivitäten des täglichen Lebens); MGFA = Myasthenia Gravis Foundation of America; min = Minimum; QMG = Quantitative Myasthenia Gravis score for disease severity (Quantitativer Myasthenia Gravis-Score für die Schwere der Erkrankung); SD = Standardabweichung
Der primäre Endpunkt der Studie ECU-MG-303 war die Veränderung des QMG-Gesamtscores gegenüber dem Ausgangswert im Laufe der Zeit, unabhängig von der Notfallbehandlung. Mit Soliris behandelte pädiatrische Patienten zeigten im QMG-Gesamtscore über den gesamten 26wöchigen primären Behandlungszeitraum eine statistisch signifikante Verbesserung gegenüber dem Ausgangswert. Die Ergebnisse für die primären und sekundären Endpunkte der Studie ECU-MG-303 sind in Tabelle 20 aufgeführt.
Die Wirksamkeit der Soliris-Behandlung bei pädiatrischen Patienten mit refraktärer gMG entsprach derjenigen, die bei erwachsenen Patienten mit refraktärer gMG im Rahmen der Zulassungsstudie ECU MG 301 beobachtet wurde (Tabelle 10).
Tabelle 20: Wirksamkeitsergebnisse der Studie ECU-MG-303
Wirksamkeitsendpunkte: Veränderung des Gesamtscores in Differenz der Kleinste-Quadrate-Mitte
Woche 26 gegenüber dem Ausgangswert lwerte (SEM) 95% KI
QMG -5,8 (1,2) (-8,40; -3,13)na = 10
MG-ADL-Gesamtscore -2,3 (0,6) (-3,63; -1,03)na = 10
MGC-Gesamtscore -8,8 (1,9) (-12,93; -4,69)na = 9
na ist die Anzahl der Patienten in Woche 26
Abkürzungen: KI = Konfidenzintervall; LS = kleinste Quadrate; MG-ADL = Myasthenia Gravis Activities of Daily Living (Myasthenia-Gravis-Profil für Aktivitäten des täglichen Lebens); MGC = Myasthenia-Gravis-Komposit; QMG = Quantitative Myasthenia Gravis score for disease severity (Quantitativer Myasthenia Gravis-Score für die Schwere der Erkrankung); SEM = Standardfehler des Mittelwerts;
In der Studie ECU-MG-303 wurde ein klinisches Ansprechen bei den QMG- und MG-ADL-Gesamtscores als eine Verbesserung von mindestens 5 bzw. 3 Punkten gegenüber dem Ausgangswert definiert. Der Anteil der klinischen Responder bei den QMG- und MG-ADL-Gesamtscores betrug in Woche 26, unabhängig von der Notfallbehandlung, 70 % bzw. 50 %. Die 10 Patienten, die den Besuch nach 26 Wochen wahrnahmen, erreichten zu diesem Zeitpunkt einen verbesserten MGFA-postinterventionellen Status (MGFA PIS). Sieben (70 %) Patienten erreichten in Woche 26 eine minimale Manifestation der refraktären gMG.
Bei einem Patienten (9,1 %) wurde während der primären Auswertung des Behandlungszeitraums eine klinische Verschlechterung (myasthene Krise) beobachtet, die eine Notfalltherapie (PE) erforderte, welche zwischen den Studienbesuchen in Woche 22 und Woche 24 verabreicht wurde. Infolgedessen und aufgrund der Entscheidung des Arztes wurden bei diesem Patienten nach Woche 20 keine QMG-, MG-ADL- oder andere Wirksamkeitsbeurteilungen durchgeführt und er wurde nicht in den Verlängerungszeitraum aufgenommen.
Während der primären Auswertung des Behandlungszeitraums bei pädiatrischen Patienten mit refraktärer gMG (Studie ECU-MG-303) reduzierte 1 von 11 Patienten (9,1 %) die tägliche Anticholinesterase-Dosis und 3 von 11 Patienten (27,3 %) reduzierten ihre tägliche Kortikosteroid-Dosis aufgrund einer Verbesserung der MG-Symptome.
Neuromyelitis-optica-Spektrumerkrankungen
Soliris wurde nicht bei Kindern und Jugendlichen mit NMOSD untersucht.
PharmakokinetikAbsorption
Siehe "Pharmakokinetische Parameter"
Distribution
Siehe "Pharmakokinetische Parameter"
Metabolismus
Humane Antikörper werden im retikuloendothelialen System endozytotisch abgebaut. Eculizumab enthält nur natürlich vorkommende Aminosäuren und hat keine bekannten aktiven Metaboliten. Humane Antikörper werden überwiegend durch lysosomale Enzyme zu kleinen Peptiden und Aminosäuren katabolisiert.
Elimination
Es wurden keine speziellen Studien zur Untersuchung der hepatischen, renalen, pulmonalen oder gastrointestinalen Ausscheidungs-/Eliminationswege für Soliris durchgeführt. Von gesunden Nieren werden Antikörper nicht ausgeschieden. Sie sind aufgrund ihrer Grösse von der Filtration ausgeschlossen.
Pharmakokinetische Parameter:
Bei 40 Patienten mit PNH wurden die pharmakokinetischen Parameter nach Mehrfachgabe mittels eines Ein-Kompartiment-Modells untersucht. Die mittlere Clearance betrug 0,31 ± 0,12 ml/h/kg, das mittlere Verteilungsvolumen 110,3 ± 17,9 ml/kg und die mittlere Eliminationshalbwertszeit 11,3 ± 3,4 Tage. Der Steady-State wird bei Anwendung des Dosierungsschemas für erwachsene PNH-Patienten nach 4 Wochen erreicht.
Bei PNH-Patienten korreliert die pharmakodynamische Aktivität direkt mit den Eculizumab-Serumkonzentrationen, und die Aufrechterhaltung von Talspiegeln über ³ 35 µg/ml führt bei den meisten PNH-Patienten zur praktisch vollständigen Blockade der hämolytischen Aktivität.
Eine zweite populationspharmakokinetische Studie mit einem Standard Ein-Kompartimentmodell wurde auf Basis der pharmakokinetischen Daten nach Mehrfachdosis bei 37 aHUS-Patienten durchgeführt, die im Rahmen der Studien C08-002A/B und C08-003A/B mit der empfohlenen Soliris-Dosis behandelt wurden. In diesem Modell lagen die Soliris-Clearance bei einem typischen aHUS-Patienten mit einem Körpergewicht von 70 kg bei 0,0139 l/Std. und das Verteilungsvolumen bei 5,6 l. Die Eliminations-Halbwertszeit betrug 297 Stunden (annähernd 12,4 Tage).
Die zweite pharmakokinetische Studie wurde auf Basis der pharmakokinetischen Daten nach Mehrfachdosis bei 22 Kindern mit aHUS durchgeführt, die im Rahmen der Studie C10-003 mit der empfohlenen Soliris-Dosis behandelt wurden. Die Soliris-Clearance und das Verteilungsvolumen waren abhängig vom Gewicht, was das Dosiseinteilungsschema bei Kindern in Gewichtskategorien stützt ( "Dosierung / Anwendung" ). Die Soliris-Clearance lag bei Kindern mit aHUS bei 10,4, 5,3 bzw. 2,2 ml/h bei Patienten mit einem Körpergewicht von 70, 30 bzw. 10 kg; das jeweilige Verteilungsvolumen lag bei 5,23, 2,76 bzw. 1,21 l. Die Eliminations-Halbwertszeit war beinahe unverändert und betrug zwischen 349 und 378 Stunden (annähernd 14,5 bis 15,8 Tage).
Clearance und Halbwertszeit von Soliris wurden darüber hinaus im Rahmen von Plasmaaustausch-Massnahmen untersucht. Plasmaaustausch führte nach einer einstündigen Intervention zu einer annähernd 50 %igen Abnahme der Eculizumab-Konzentrationen. Die Eliminations-Halbwertszeit von Eculizumab war auf 1,3 Stunden verringert.
Bei aHUS-Patienten ist eine zusätzliche Gabe von Soliris erforderlich, wenn sie eine Plasmainfusion oder einen Plasmaaustausch erhalten (siehe "Dosierung / Anwendung" ).
Alle aHUS-Patienten, die nach dem empfohlenen Dosierungsschema mit Soliris behandelt wurden, erreichten eine rasche und anhaltende Abnahme der terminalen Komplementaktivität.
Bei aHUS-Patienten korreliert die pharmakodynamische Wirkung direkt mit den Eculizumab-Serumkonzentrationen. Das Aufrechterhalten von Talspiegeln von etwa 50-100 Mikrogram/ml reicht für eine praktisch vollständige Hemmung der terminalen Komplementaktivität bei allen aHUS-Patienten aus.
Kinetik spezieller Patientengruppen
Es wurden keine spezifischen Studien zur Untersuchung der Pharmakokinetik von Soliris bei speziellen Patientenpopulationen durchgeführt, die nach Geschlecht, ethnischer Abstammung, Alter (geriatrische Patienten) oder Vorliegen einer Nieren- oder Leberfunktionsstörung identifiziert wurden. Die Populations-PK-Analyse von Daten aus Studien mit PNH-, aHUS-, gMG- und NMOSD-Patienten ergab, dass Geschlecht, ethnische Abstammung, Alter (geriatrisch) oder das Vorliegen einer Nieren- oder Leberfunktionsstörung die PK von Eculizumab nicht beeinflussen. Das Körpergewicht war eine signifikante Kovariable, die bei pädiatrischen Patienten zu einer geringeren Eculizumab-Clearance führte und eine auf dem Körpergewicht basierende Dosierung bei pädiatrischen Patienten erforderte.
Die PK-Parameter in den Patientengruppen mit PNH, aHUS, refraktärer gMG und NMOSD stimmen überein.
Die durch freie C5-Konzentrationen von <0,5 µg/ml gemessene pharmakodynamische Aktivität korreliert mit einer im Wesentlichen vollständigen Blockade der terminalen Komplementaktivität bei Patienten mit PNH, aHUS, refraktärer gMG und NMOSD.
Kinder und Jugendliche
Die Pharmakokinetik von Eculizumab wurde in der Studie M07-005 bei pädiatrischen Patienten mit PNH (Alter 11 bis unter 18 Jahre), in den Studien C08-002, C08-003, C09-001r und C10-003 bei pädiatrischen Patienten mit aHUS (Alter 2 Monate bis unter 18 Jahre) und in der Studie ECU-MG-303 bei pädiatrischen Patienten mit refraktärer gMG (Alter 12 bis unter 18 Jahre) unter Anwendung eines auf dem Körpergewicht basierenden Dosierungsschemas untersucht.
Bei Jugendlichen mit PNH war das Körpergewicht ein signifikanter Einflussfaktor (siehe Angaben zu Kindern mit aHUS oben). In der Studie M07-005 lag die Eculizumab-Clearance bei 10.5 ml/Std. Es fehlen pharmakokinetische Daten in der PNH-Patienten <40 kg Körpergewicht. Aufgrund der vergleichbaren pathogenetischen Mechanismus in PNH und aHUS wird ein vergleichbares PK-Profil in beiden Indikationen angenommen, und es sind keine unterschiedlichen Dosierungen angezeigt.
Präklinische DatenDie Spezifität von Eculizumab für C5 in Humanserum wurde in zwei in-vitro-Studien beurteilt.
Die Gewebe-Kreuzreaktivität von Eculizumab wurde durch Bindung an eine Serie von 38 menschlichen Geweben untersucht. Die C5-Expression der untersuchten Gewebe stimmt mit den in der Literatur publizierten Daten überein. Demnach wird C5 in der glatten Muskulatur, der quergestreiften Muskulatur und im Epithel der proximalen Nierentubuli exprimiert. Es war keine unerwartete Gewebe-Kreuzreaktivität zu beobachten.
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung
In einer 26-wöchigen Toxizitätsstudie an Mäusen mit einem analogen Antikörper gegen murines C5 beeinflusste die Behandlung keinen der untersuchten Toxizitätsparameter. Die hämolytische Aktivität wurde im Verlauf der Studie sowohl bei weiblichen als auch bei männlichen Mäusen wirksam blockiert.
Gentoxizität und Kanzerogenität
Es wurden keine Tierstudien zur Bewertung des genotoxischen und karzinogenen Potenzials von Eculizumab durchgeführt.
Reproduktionstoxizität
Es wurden keine Reproduktionsstudien an Tieren mit Eculizumab durchgeführt, weil bei nicht-menschlichen Spezies keine pharmakologische Aktivität vorliegt.
In reproduktionstoxikologischen Studien an Mäusen mit einem analogen Antikörper zur Hemmung der terminalen Komplementaktivierung, der zur Beurteilung der Sicherheit einer C5-Blockade im Hinblick auf die Reproduktion verwendet wurde, wurden keine eindeutigen behandlungsbedingten Wirkungen oder unerwünschten Wirkungen beobachtet. Diese Studien umfassten eine Beurteilung der Fertilität und frühembryonalen Entwicklung, der Entwicklungstoxizität sowie der prä- und postnatalen Entwicklung.
Bei Exposition des Muttertiers gegenüber dem Antikörper in einer höheren Dosierung ((etwa das 4-Fache der maximalen empfohlenen humanen Soliris-Dosis, auf der Basis eines Körpergewichtsvergleichs) während der Organbildung wurden zwei Fälle von Retinadysplasie und ein Fall von Nabelbruch unter 230 Nachkommen beobachtet. Eine Exposition gegenüber dem Antikörper bewirkte jedoch keinen Anstieg fetaler Verluste oder neonataler Todesfälle.
Sonstige HinweiseInkompatibilitäten
Das Arzneimittel darf, ausser mit den unter "Hinweise für die Handhabung" aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit „EXP“ bezeichneten Datum verwendet werden.
Dieses Arzneimittel enthält keine Konservierungsmittel.
Nach Verdünnung sollte das Arzneimittel sofort verwendet werden. Die chemische und physikalische Gebrauchsstabilität wurde jedoch für 24 Stunden bei 2 °C-8 °C nachgewiesen.
Besondere Lagerungshinweise
Im Kühlschrank lagern (+2 °C bis +8 ºC).
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Arzneimittel für Kinder unzugänglich aufbewahren.
Das originalverpackte Arzneimittel kann für eine einmalige Dauer von bis zu 3 Tagen ausserhalb des Kühlschrankes aufbewahrt werden. Nach dieser Zeit kann das originalverpackte Arzneimittel wieder im Kühlschrank gelagert werden.
Hinweise für die Handhabung
Soliris muss von medizinischem Fachpersonal verabreicht werden.
Die Soliris-Lösung soll vor der Verabreichung visuell auf Partikel und Verfärbung überprüft werden. Die Soliris-Lösung muss klar und farblos sein.
Anweisungen
Die Rekonstitution und Verdünnung muss unter aseptischen Bedingungen und den Regeln entsprechend erfolgen.
-Die gesamte Menge Soliris aus der(den) Durchstechflasche(n) in eine sterile Spritze aufziehen.
-Die empfohlene Dosis in einen Infusionsbeutel überführen.
-Soliris durch Zugabe von 0,9 % Natriumchlorid, 0,45 % Natriumchlorid oder 5 % Glucose in Wasser verdünnt im Infusionsbeutel auf eine Endkonzentration von 5 mg/ml verdünnen.
-Das Endvolumen einer verdünnten Lösung mit 5 mg/ml beträgt 120 ml für 600-mg-Dosen oder 180 ml für 900-mg-Dosen. Die Lösung muss klar und farblos sein.
-Den Infusionsbeutel mit der verdünnten Lösung leicht hin und her bewegen, um sicherzustellen, dass das Arzneimittel und das Verdünnungsmittel gut vermischt werden.
-Die verdünnte Lösung vor der Verabreichung an der Umgebungsluft auf Raumtemperatur erwärmen lassen.
-Nicht verbrauchter Inhalt der Durchstechflasche muss verworfen werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den örtlichen Anforderungen zu entsorgen.
Zulassungsnummer59282 (Swissmedic)
Packungen30 ml Konzentrat in einer Durchstechflasche (A)
ZulassungsinhaberinAlexion Pharma GmbH
Neuhofstrasse 34
6340 Baar
Stand der InformationAugust 2025
|