ZusammensetzungWirkstoffe
Liraglutide
Hilfsstoffe
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile.
Das Arzneimittel enthält 0.01596 mmol/ml Natrium.
Darreichungsform und Wirkstoffmenge pro EinheitInjektionslösung im Fertigpen.
1 ml enthält 6 mg Liraglutide (gentechnisch in Saccharomyces cerevisiae hergestellt).
Ein Fertigpen enthält 3 ml (entsprechend 18 mg Liraglutide).
Indikationen/AnwendungsmöglichkeitenVictoza wird zur Behandlung Erwachsener, Jugendlichen und Kindern ab 10 Jahren mit unzureichend kontrolliertem Diabetes mellitus Typ 2 ergänzend zu Diät und Bewegung angewendet:
-Als Monotherapie bei Kontraindikation oder Unverträglichkeit für Metformin.
-In Kombination mit anderen blutzuckersenkenden Arzneimitteln.
(siehe "Klinische Wirksamkeit" für Ergebnisse zu den in klinischen Studien untersuchten Kombinationen von Victoza mit anderen blutzuckersenkenden Mitteln).
Victoza ist indiziert zur Prävention kardiovaskulärer Ereignisse bei Patienten mit Typ 2 Diabetes mellitus und bereits manifester kardiovaskulärer Erkrankung (siehe "Klinische Wirksamkeit" ).
Dosierung/AnwendungVictoza wird einmal täglich zu einem beliebigen Zeitpunkt unabhängig von den Mahlzeiten angewendet.
Übliche Dosierung
Um die gastrointestinale Verträglichkeit zu verbessern, soll die Behandlung mit Victoza bei allen Patienten mit einer Dosis von 0.6 mg begonnen werden. Diese Dosis sollte mindestens eine Woche lang beibehalten und anschliessend auf 1.2 mg erhöht werden. Nach mindestens einer weiteren Woche kann die Dosis je nach klinischem Ansprechen auf 1.8 mg erhöht werden. Höhere Tagesdosen als 1.8 mg werden nicht empfohlen.
Kombinationstherapie
Victoza kann in Kombination mit anderen blutzuckersenkenden Arzneimitteln angewendet werden, wobei die Dosis von Metformin, Thiazolidindion und Natrium-Glukose Co-Transporter 2 Inhibitor (SGLT2i) nicht anzupassen ist.
Victoza kann zusätzlich zu einer bestehenden Behandlung mit einem Sulfonylharnstoff oder Metformin und einem Sulfonylharnstoff angewendet werden. Wird Victoza zusätzlich zu einem Sulfonylharnstoff gegeben, sollte eine Dosisreduktion des Sulfonylharnstoffs erwogen werden, um das Risiko einer Hypoglykämie zu senken.
Wenn Victoza zusätzlich zu einer bestehenden Behandlung mit Basalinsulin angewendet wird, sollte eine Reduktion der Insulindosis erwogen werden, um das Risiko einer Hypoglykämie zu senken.
Eine Messung des Blutzuckerspiegels durch den Patienten zur Anpassung der Victoza-Dosis ist nicht erforderlich. Eine Eigenkontrolle des Blutzuckers durch den Patienten ist erforderlich, um die Dosis des Sulfonylharnstoffs oder des Insulins anzupassen. Dies gilt insbesondere bei Beginn der Behandlung mit Victoza und bei einer Reduktion der lnsulindosis. Es wird empfohlen, die lnsulindosis schrittweise zu senken.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit Leberfunktionsstörungen sind keine Dosisanpassungen erforderlich.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter, mittelschwerer oder schwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit Nierenfunktionsstörungen im Endstadium liegen keine klinischen Erfahrungen vor und die Anwendung von Victoza bei diesen Patienten kann nicht empfohlen werden.
Ältere Patienten
Eine altersabhängige Dosisanpassung ist nicht erforderlich.
Kinder und Jugendliche
Bei Jugendlichen und Kindern ab 10 Jahren ist keine Dosisanpassung erforderlich. Es liegen keine Daten bei Kindern unter 10 Jahren vor (siehe "Eigenschaften/Wirkung" ).
Art der Anwendung
Victoza kann in die Bauchdecke, den Oberschenkel oder den Oberarm subkutan injiziert werden. Die Einstichstelle ist bei jeder Injektion innerhalb derselben Körperregion zu wechseln, um das Risiko einer kutanen Amyloidose zu reduzieren (siehe "Unerwünschte Wirkungen" ). Die Injektionsstelle und der Zeitpunkt der Injektion können ohne Dosisanpassung geändert werden.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenVictoza sollte nicht bei Patienten mit Diabetes mellitus Typ 1 oder zur Behandlung der diabetischen Ketoazidose eingesetzt werden.
Victoza (Liraglutide) ist kein Ersatz für Insulin. Insulin soll nicht abgesetzt werden in Patienten, die auf Insulin angewiesen sind. Es liegen Berichte über diabetische Ketoazidose bei insulinabhängigen Patienten nach raschem Absetzen oder einer schnellen Dosisreduktion von Insulin vor (siehe "Dosierung/Anwendung" ).
Es gibt keine klinischen Erfahrungen bei Patienten mit Herzinsuffizienz des New York Heart Association (NYHA) Stadiums IV, daher wird die Anwendung von Liraglutide bei diesen Patienten nicht empfohlen.
Aspiration im Zusammenhang mit einer Vollnarkose oder tiefer Sedierung
Bei Patienten, die GLP-1-Rezeptor-Agonisten erhielten und die sich einer Vollnarkose oder einer tiefen Sedierung unterzogen, wurden Fälle von pulmonaler Aspiration berichtet, trotz der berichteten Einhaltung der präoperativen Nüchternempfehlungen. Daher sollte vor der Durchführung von Eingriffen unter Vollnarkose oder tiefer Sedierung das erhöhte Risiko für Restmageninhalt aufgrund einer verzögerten Magenentleerung berücksichtigt werden.
Entzündliche Darmerkrankung und diabetische Gastroparese
Bei Patienten mit entzündlichen Darmkrankheiten und diabetischer Gastroparese liegen nur begrenzte Erfahrungen vor. Die Anwendung von Liraglutide wird bei diesen Patienten nicht empfohlen, da sie mit vorübergehenden gastrointestinalen Nebenwirkungen, einschliesslich Übelkeit, Erbrechen und Durchfall, verbunden ist.
Pankreatitis
Akute Pankreatitis wurde bei der Anwendung von GLP-1 Rezeptoragonisten beobachtet. Patienten sollten über die charakteristischen Symptome einer akuten Pankreatitis informiert werden. Wird eine Pankreatitis vermutet, ist Liraglutide abzusetzen; falls eine akute Pankreatitis bestätigt wird, ist die Behandlung mit Liraglutide nicht wieder aufzunehmen (siehe "Unerwünschte Wirkungen" und "Eigenschaften/Wirkungen" ). Eine isolierte Erhöhung der Pankreasenzyme unter der Behandlung mit Victoza (ohne charakteristische Symptomatik) manifestiert nicht zwingend eine akute Pankreatitis (siehe "Unerwünschte Wirkungen" ).
Schilddrüsenerkrankungen
Über unerwünschte Ereignisse in Zusammenhang mit der Schilddrüse wie Struma wurde insbesondere bei Patienten mit bestehender Schilddrüsenerkrankung in klinischen Studien berichtet (siehe "Unerwünschte Wirkungen" ). Victoza sollte somit bei diesen Patienten mit Vorsicht angewendet werden.
Dehydrierung
Bei Patienten, die mit Liraglutide behandelt wurden, wurde über Anzeichen und Symptome von Dehydrierung einschliesslich Beeinträchtigung der Nierenfunktion und akutem Nierenversagen berichtet. Patienten, die mit Liraglutide behandelt werden, müssen auf das potenzielle Dehydrierungs-Risiko im Zusammenhang mit gastrointestinalen Nebenwirkungen hingewiesen werden und Vorkehrungen gegen Flüssigkeitsverluste treffen.
Hypoglykämie
Patienten, die Victoza in Kombination mit einem Sulfonylharnstoff oder einem Basalinsulin erhalten, können ein erhöhtes Risiko für eine Hypoglykämie haben (siehe "Unerwünschte Wirkungen" ). Das Risiko einer Hypoglykämie kann durch Reduktion der Sulfonylharnstoff- oder Basalinsulin-Dosis gesenkt werden.
Sonstige Bestandteile
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenPharmakokinetische Interaktionen
In-vitro-Beurteilung von Arzneimittel-Wechselwirkungen mit Liraglutide
Das Potential von Liraglutide zur Beteiligung an pharmakokinetischen Wechselwirkungen mit anderen Wirkstoffen, die mit Cytochrom P450 (CYP) und der Bindung an Plasmaproteine in Zusammenhang stehen, hat sich als sehr gering erwiesen.
In-vivo-Beurteilung von Arzneimittel-Wechselwirkungen mit Liraglutide
Für die Untersuchung von Arzneimittel-Wechselwirkungen wurden stellvertretend für verschiedene Löslichkeitsgrade und Permeabilitätseigenschaften Paracetamol (Acetaminophen), Digoxin, Lisinopril, Griseofulvin und Atorvastatin herangezogen. Darüber hinaus wurde die Wirkung von Liraglutide auf die Resorption von Ethinylestradiol und Levonorgestrel aus einem Kombinationspräparat zur oralen Kontrazeption untersucht.
Die durch Liraglutide verursachte geringe Verzögerung der Magenentleerung führte zu keiner klinisch relevanten Beeinflussung der Resorption oral verabreichter Arzneimittel, weshalb keine Dosisanpassung erforderlich ist.
Einige Patienten erlitten unter Liraglutide-Behandlung schwere Durchfälle. Diarrhö kann die Resorption gleichzeitig oral gegebener Arzneimittel beeinträchtigen.
Paracetamol (Acetaminophen)
Liraglutide bewirkte keine Veränderung der AUC gegenüber Paracetamol nach Gabe einer Einzeldosis von 1'000 mg. Die Cmax von Paracetamol ging um 31 % zurück; die mittlere tmax stieg um bis zu 15 min. Bei gleichzeitiger Anwendung von Paracetamol ist keine Dosisanpassung erforderlich.
Atorvastatin
Liraglutide bewirkte keine Veränderung der AUC gegenüber Atorvastatin nach Gabe einer Einzeldosis von 40 mg Atorvastatin. Daher ist bei gleichzeitiger Anwendung mit Liraglutide keine Anpassung der Atorvastatin-Dosis erforderlich. Liraglutide führte zu einer Verminderung der Cmax von Atorvastatin um 38 % und einer Zunahme der mittleren tmax von 1 h auf 3 h.
Griseofulvin
Liraglutide bewirkte keine Veränderung der AUC gegenüber Griseofulvin nach Gabe einer Einzeldosis von 500 mg Griseofulvin. Die Griseofulvin-Cmax stieg um 37 %, während die mittlere tmax unverändert blieb.
Anpassungen der Dosis von Griseofulvin und anderen Substanzen mit geringer Löslichkeit und hoher Permeabilität sind nicht erforderlich.
Lisinopril und Digoxin
Nach Verabreichung einer Einzeldosis von 20 mg Lisinopril bzw. 1 mg Digoxin mit Liraglutide kam es zu einer Verminderung der Lisinopril- bzw. Digoxin-AUC um 15 % bzw. 16 % und zu einer Abnahme der jeweiligen Cmax um 27 % bzw. 31 %. Die mittlere tmax stieg mit Liraglutide im Fall von Lisinopril von 6 h auf 8 h und im Fall von Digoxin von 1 h auf 1.5 h.
Aufgrund dieser Ergebnisse ist eine Anpassung der Lisinopril- bzw. Digoxindosis nicht erforderlich.
Orale Kontrazeptiva
Liraglutide senkte die Cmax von Ethinylestradiol und Levonorgestrel nach Einzelgabe eines oralen Kontrazeptivums um 12 % bzw. 13 %. Die tmax für beide Substanzen verlängerte sich mit Liraglutide um 1.5 h. Man beobachtete keinen klinisch relevanten Effekt bezüglich der AUC weder von Ethinylestradiol noch von Levonorgestrel. Daher ist davon auszugehen, dass die kontrazeptive Wirkung durch gleichzeitige Verabreichung mit Liraglutide nicht beeinträchtigt wird.
Antikoagulantien
Es wurden keine Interaktionsstudien durchgeführt. Eine klinisch relevante Wechselwirkung mit Wirkstoffen mit schlechter Löslichkeit oder engem therapeutischem Bereich wie z.B. Warfarin kann nicht ausgeschlossen werden. Bei Patienten, welche Antikoagulantien anwenden, wird zu Beginn der Liraglutide-Behandlung eine häufigere Überwachung des INR empfohlen.
Insulin
Es wurden keine pharmakokinetischen oder pharmakodynamischen Interaktionen zwischen Liraglutide und Insulin Detemir beobachtet, wenn man eine Einzeldosis von 0.5 E/kg Insulin Detemir mit Liraglutide 1.8 mg als Steady State in Patienten mit Typ 2 Diabetes verabreicht hat.
Pädiatrische Population
Interaktionsstudien wurden nur bei Erwachsenen durchgeführt.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine ausreichenden Daten zur Anwendung von Victoza während der Schwangerschaft vor.
In Tierstudien zeigte sich eine Reproduktionstoxizität (siehe "Präklinische Daten" ). Das mögliche Risiko für den Menschen ist nicht bekannt.
Victoza sollte während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich. Möchte eine Patientin schwanger werden oder tritt eine Schwangerschaft ein, sollte die Behandlung mit Victoza abgebrochen werden.
Stillzeit
Es ist nicht bekannt, ob Liraglutide in die Muttermilch übertritt. Tierexperimentelle Studien haben gezeigt, dass der Übergang von Liraglutide und strukturell eng verwandten Metaboliten in die Muttermilch gering ist. Präklinische Studien zeigten in Zusammenhang mit der Behandlung eine Abnahme des neonatalen Wachstums von gesäugten Ratten (siehe "Präklinische Daten" ). Aufgrund mangelnder Erfahrung soll Victoza nicht in der Stillzeit angewendet werden.
Fertilität
Abgesehen von einer leichten Reduktion der Implantationsrate zeigten tierexperimentelle Studien bezüglich Fertilität keine unmittelbar schädlichen Effekte (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Die Patienten sollten angewiesen werden, Massnahmen zur Hypoglykämie-Vermeidung bei der Teilnahme am Strassenverkehr oder während des Bedienens von Maschinen zu ergreifen, vor allem wenn Victoza in Kombination mit einem Sulfonylharnstoff oder einem Basalinsulin angewendet wird.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die in klinischen Studien am häufigsten gemeldeten unerwünschten Wirkungen waren gastrointestinaler Art: Übelkeit und Durchfall waren sehr häufig (von >10 % der Patienten berichtet), während Erbrechen, Verstopfung, Bauchschmerzen und Dyspepsie häufig waren.
Diese gastrointestinalen unerwünschten Ereignisse können zu Beginn der Behandlung mit Victoza häufiger auftreten und lassen in der Regel innerhalb weniger Tage oder Wochen unter fortgesetzter Behandlung nach. Kopfschmerzen und Infektionen der oberen Atemwege waren ebenfalls häufig. Ausserdem waren Hypoglykämien häufig und sehr häufig, wenn Victoza in Kombination mit Sulfonylharnstoffen angewendet wurde (>10 % der Patienten). Schwere Hypoglykämien wurden vor allem bei Anwendung in Kombination mit einem Sulfonylharnstoff beobachtet.
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 1 sind Nebenwirkungen aufgeführt, die in kontrollierten Phase-3a-Langzeitstudien und in der LEADER Studie berichtet wurden. Die Nebenwirkungen sind nach Systemorganklassen und Häufigkeit aufgeführt.
Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1'000, <1/100), selten (≥1/10'000, <1/1'000), sehr selten (<1/10'000), nicht bekannt (Häufigkeit auf Grundlage der bekannten Daten nicht abschätzbar).
Tabelle 1: Unerwünschte Wirkungen aus kontrollierten Langzeitstudien der Phase 3a, der Langzeitstudie zu kardiovaskulären Ergebnissen (LEADER) und spontanen Berichten (nach der Markteinführung)
Systemorganklassen Häufigkeit Unerwünschte Wirkungen
gemäss MedDRA
Infektionen und Häufig Infektionen der oberen
parasitäre Erkrankun Atemwege
gen
Erkrankungen des Selten Anaphylaktische Reaktionen
Immunsystems
Endokrine Erkrankung Gelegentlich Neoplasien der Schilddrüse,
en Struma und Erhöhung der
Calcitonin-Konzentration im
Blut
Stoffwechsel- und Häufig Hypoglykämie Anorexie
Ernährungsstörungen Verminderter Appetit
Nicht bekannt Dehydrierung†
Erkrankungen des Häufig Kopfschmerzen Schwindelgefühl
Nervensystems
Gelegentlich Dysgeusie
Herzerkrankungen Häufig Erhöhte Herzfrequenz
Erkrankungen des Sehr häufig Übelkeit Diarrhö
Gastrointestinaltrak
ts
Häufig Erbrechen Dyspepsie Schmerzen im Oberbauch
Obstipation Gastritis Flatulenz
Aufgetriebener Bauch Gastroösophagealer
Reflux Aufstossen
Gelegentlich Verzögerte Magenentleerung
Selten Darmobstruktion†a
Sehr selten Pankreatitis (inklusive nekrotisierender
Pankreatitis)
Leber- und Gallenerk Gelegentlich Cholelithiasis Cholezystitis
rankungen
Erkrankungen der Nicht bekannt Urtikaria† Hautausschlag†
Haut und des Unterha Pruritus† Kutane Amyloidose†
utgewebes
Erkrankungen der Nicht bekannt Akutes Nierenversagen†
Nieren und Harnwege Nierenfunktionsstörungen†
Allgemeine Erkrankun Häufig Müdigkeit Reaktionen an der
gen und Beschwerden Injektionsstelle
am Verabreichungsort
Nicht bekannt Malaise†
Untersuchungen Häufig Erhöhte Lipase* Erhöhte
Amylase*
*Aus kontrollierten klinischen Studien der Phasen 3b und 4, sofern es gemessen wurde
†Arzneimittelnebenwirkung aus Meldungen nach Markteinführung
aDie zusammengefasste Bezeichnung umfasst die unerwünschten Ereignisse Darmobstruktion, Ileus und Dünndarmobstruktion
Beschreibung ausgewählter Nebenwirkungen
Erkrankungen des Immunsystems
Selten – Anaphylaktische Reaktionen
Bei allen klinischen Langzeitstudien mit Victoza wurden wenige Fälle von Angioödemen berichtet (0.05 %).
Endokrine Erkrankungen
Gelegentlich – Neoplasien der Schilddrüse, Struma und Erhöhung der Calcitonin-Konzentration im Blut
Die Gesamthäufigkeit der unerwünschten Ereignisse im Bereich der Schilddrüse in allen intermediären und Langzeitstudien sind 33.5, 30.0 bzw. 21.7 Ereignisse pro 1'000-Probanden-Jahre Behandlung mit Liraglutide, Placebo bzw. Vergleichspräparaten. Bei 5.4, 2.1 bzw. 0.8 Ereignissen pro 1'000 Probanden handelte es sich jeweils um schwerwiegende unerwünschte Ereignisse im Bereich der Schilddrüse.
Neoplasmen der Schilddrüse, Struma sowie eine Erhöhung der Calcitonin-Konzentration im Blut waren die unerwünschten Ereignisse im Bereich der Schilddrüse, über die am häufigsten berichtet wurde. Die Häufigkeiten pro 1'000-Patienten-Jahre waren 6.8, 10.9 und 5.4 bei Behandlung mit Liraglutide, verglichen mit 6.4, 10.7 und 2.1 bei Behandlung mit Placebo sowie 2.5, 6.0 und 1.8 bei Behandlung mit Vergleichspräparaten.
Stoffwechsel- und Ernährungsstörungen
Häufig – Hypoglykämien
Leichte Hypoglykämie (27.4 %) in Kombination mit einem Sulfonylharnstoff und Metformin: sehr häufig
Leichte Hypoglykämie in Kombination mit einem Sulfonylharnstoff: häufig
Schwere Hypoglykämie in Kombination mit einem Sulfonylharnstoff: selten
Schwere Hypoglykämien können v.a. auftreten, wenn Liraglutide zusammen mit einem Sulfonylharnstoff verabreicht wird (0.02 Ereignisse/Probandenjahr), bei Verabreichung von Liraglutide in Kombination mit einem oralen Antidiabetikum, das nicht zu den Sulfonylharnstoffen gehört, wurden sehr wenige Episoden (0.001 Ereignisse/Probandenjahr) beobachtet.
Die meisten in klinischen Studien aufgetretenen Hypoglykämien waren leicht.
Bei Hinzugabe von Insulin Detemir zu Victoza 1.8 mg und Metformin wurden keine schweren Hypoglykämien beobachtet. Die Häufigkeit leichter Hypoglykämien lag bei einer Behandlung mit Liraglutide 1.8 mg, Metformin und Insulin Detemir bei 0.228 Fällen pro Patientenjahr. In den Vergleichsgruppen, die beide mit Liraglutide 1.8 mg und Metformin behandelt wurden, lag die Rate der leichten Hypoglykämien in der randomisierten Gruppe bei 0.034 und in der nicht-randomisierten Gruppe bei 0.115 Fällen pro Patientenjahr. In der LEADER Studie wurden schwere hypoglykämische Episoden unter Liraglutide seltener berichtet als unter Placebo (1.0 vs. 1.5 Ereignisse pro 100 Patientenjahre; geschätztes Ratenverhältnis 0.69 [0.51 bis 0.93]) (siehe "Eigenschaften/Wirkungen" ).
Erkrankungen des Gastrointestinaltrakts
Sehr häufig – Übelkeit (20.7 %) und Diarrhö (12.6 %) bei Verabreichung in Kombination mit Metformin
Häufig – Übelkeit und Diarrhö bei Verabreichung mit einem Sulfonylharnstoff
Die aufgetretenen Fälle von Übelkeit waren überwiegend leicht bis mittelstark und vorübergehend und führten nur selten zu einem Abbruch der Therapie.
Bei Patienten >70 Jahre können unter der Behandlung mit Liraglutide häufiger gastrointestinale Beschwerden auftreten. Patienten mit leichter und mittelschwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance 60–90 ml/min respektive 30–59 ml/min) können unter der Behandlung mit Liraglutide häufiger gastrointestinale Beschwerden haben.
Sehr selten – Pankreatitis (inklusive nekrotisierender Pankreatitis)
Einige Fälle akuter Pankreatitis (<0.2 %) sind während kontrollierter Phase-3 Langzeitstudien mit Victoza berichtet worden. Pankreatitis wurde auch nach Markeinführung berichtet. In der LEADER Studie lag die Häufigkeit akuter Pankreatitis, die durch Adjudikation bestätigt wurde, bei 0.4 % für Liraglutide und bei 0.5 % für Placebo (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Eigenschaften/Wirkungen" ).
Victoza ist mit einem mittleren Anstieg der Pankreasenzyme Lipase und Amylase von bis zu 38 % bzw. 21 % im Vergleich zur Baseline assoziiert (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Bei den meisten der betroffenen Patienten ist diese Erhöhung der Pankreasenzyme nicht mit einer akuten Pankreatitis assoziiert, wenn keine weiteren Anzeichen und Symptome der Erkrankung vorliegen.
Leber- und Gallenerkrankungen
Gelegentlich – Cholelithiasis und Cholezystitis
In klinischen Phase-3a-Langzeitstudien mit Victoza wurden einige Fälle von Cholelithiasis (0.4 %) und Cholezystitis (0.1 %) berichtet. In der LEADER Studie lag die Häufigkeit von Cholelithiasis und Cholezystitis bei 1.5 % und 1.1 % mit Liraglutide und 1.1 % und 0.7 % mit Placebo (siehe "Eigenschaften/Wirkungen" ).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig – Reaktionen an der Injektionsstelle
Reaktionen an der Injektionsstelle wurden von ca. 2 % der Patienten berichtet, die Victoza in kontrollierten Studien (26 Wochen oder länger) erhielten. Die Mehrheit dieser Reaktionen war mild.
Studienabbruch
Die Häufigkeit von Studienabbrüchen aufgrund von unerwünschten Ereignissen belief sich in den kontrollierten Studien (26 Wochen oder länger) auf 7.8 % bei den mit Victoza bzw. 3.4 % bei den mit einem Vergleichspräparat behandelten Patienten. Bei den mit Victoza behandelten Patienten bestanden die Ereignisse, die am häufigsten zu einem Studienabbruch führten, in Übelkeit (2.8 % der Patienten) und Erbrechen (1.5 %).
Unerwünschte Wirkungen nach Markteinführung
Nach Markteinführung von Victoza sind die folgenden unerwünschten Wirkungen berichtet worden. Da dies Post-Marketing Berichte sind, kann keine Aussage über Häufigkeiten gemacht werden (Häufigkeit unbekannt):
Erkrankungen des Immunsystems: Anaphylaktischen Reaktionen mit zusätzlichen Symptomen wie Hypotension, Palpitationen, Dyspnoe, Ödeme.
Stoffwechsel- und Ernährungsstörungen: Dehydrierung (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Leber- und Gallenerkrankungen: Anstiege von Leberenzymkonzentrationen, Hyperbilirubinämie, Cholestase, Hepatitis.
Erkrankungen der Haut und des Unterhautgewebes: Urtikaria, Hautausschlag, Pruritus, kutane Amyloidose
Erkrankungen der Nieren und Harnwege: Akutes Nierenversagen, Nierenfunktionsstörungen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Malaise.
Kinder und Jugendliche
Insgesamt waren Häufigkeit, Art und Schwere der Nebenwirkungen bei Jugendlichen und Kindern ab 10 Jahren vergleichbar mit denen, die bei Erwachsenen beobachtet wurden. Die Rate bestätigter Hypoglykämien war mit Liraglutide höher (0.58 Ereignisse/Patientenjahr) verglichen mit Placebo (0.29 Ereignisse/Patientenjahr). Bei Patienten, die vor einer bestätigten Hypoglykämie mit Insulin behandelt wurden, war die Rate mit Liraglutide höher (1.82 Ereignisse/Patientenjahr) verglichen mit Placebo (0.91 Ereignisse/Patientenjahr). Es traten keine schweren Hypoglykämien in der Behandlungsgruppe mit Liraglutide auf.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien und bei der Anwendung von Liraglutide nach Markteinführung wurden Überdosierungen von bis zu 40-mal der empfohlenen Erhaltungsdosis (72 mg) berichtet.
Anzeichen und Symptome
Die berichteten Ereignisse schliessen schwere Übelkeit, starkes Erbrechen und Durchfall ein, welches auch die zu erwartenden Symptome einer Überdosierung von Liraglutide sind. Es wurden Fälle schwerer Hypoglykämien berichtet. Alle Patienten erholten sich komplikationslos.
Behandlung
Im Fall einer Überdosierung ist eine angemessene unterstützende Behandlung entsprechend den klinischen Anzeichen und Symptomen des Patienten einzuleiten. Der Patient muss bezüglich klinischer Anzeichen von Dehydrierung beobachtet werden und der Blutzuckerspiegel muss überwacht werden.
Eigenschaften/WirkungenATC-Code
A10BJ02
Wirkungsmechanismus
Liraglutide ist ein Analogon des humanen Glucagon-ähnlichen Peptids 1 (GLP-1) mit 97-prozentiger Homologie zu humanem GLP-1, das an den GLP-1-Rezeptor bindet und diesen aktiviert. Der GLP-1-Rezeptor ist der Zielrezeptor des nativen GLP-1, eines endogenen Inkretinhormons, das die glucoseabhängige Insulinsekretion aus den Betazellen des Pankreas verstärkt. Im Gegensatz zu nativem GLP-1 besitzt Liraglutide beim Menschen ein pharmakokinetisches und pharmakodynamisches Profil, das sich für eine einmal tägliche Verabreichung eignet. Die protrahierte Wirkung nach subkutaner Applikation beruht auf drei Mechanismen: Selbstassoziation, die mit verlangsamter Resorption einhergeht, Albuminbindung und enzymatische Stabilität gegenüber den Enzymen DPP-IV und NEP, die eine lange Halbwertszeit im Plasma zur Folge hat.
Die Wirkung von Liraglutide wird durch eine spezifische Interaktion mit GLP-1-Rezeptoren vermittelt, die zu einem Anstieg der cAMP-Konzentration führt. Liraglutide stimuliert die Insulinsekretion glucoseabhängig. Gleichzeitig dämpft Liraglutide ebenfalls glucoseabhängig eine überhöhte Glucagonsekretion. Bei hohem Blutzuckerspiegel wird demnach die Insulinsekretion stimuliert und die Glucagonsekretion vermindert. Umgekehrt reduziert Liraglutide bei Hypoglykämie die Insulinsekretion und lässt die Glucagonsekretion unbeeinträchtigt. Der Mechanismus der Blutzuckersenkung beinhaltet ausserdem eine geringe Verzögerung der Magenentleerung. Liraglutide führt über Mechanismen, die eine Verminderung des Hungergefühls und der Energieaufnahme umfassen, zu einer Abnahme des Körpergewichts und der Körperfettmasse.
GLP-1 ist ein physiologischer Regulator von Appetit und Nahrungsaufnahme. GLP-1-Rezeptoren (GLP-1R) kommen in verschiedenen Hirnregionen vor, die an der Appetitregulation beteiligt sind. Tierexperimentelle Studien zeigten, dass Liraglutide nach peripherer Verabreichung in Hirnregionen aufgenommen wird, die an der Regulation des Appetits beteiligt sind, wie z.B. den Hypothalamus. Die Aktivierung von GLP-1-Rezeptoren in diesen Hirnarealen verstärkte die Sättigung, verringerte Hungersignale und induzierte letztlich eine Abnahme des Körpergewichts.
GLP-1-Rezeptoren kommen auch an spezifischen Stellen im Herz, im Gefässsystem, im Immunsystem und in den Nieren vor. Humane und tierexperimentelle Studien haben aufgezeigt, dass eine Aktivierung dieser Rezeptoren mit Liraglutide kardiovaskuläre und mikrovaskuläre Wirkungen haben kann, einschliesslich einer Entzündungsminderung. In tierexperimentellen Studien hemmte Liraglutide die Entwicklung der Atherosklerose. Im Zielgewebe (Aorta) konnten keine GLP-1R festgestellt werden. Aus diesem Grund ist von einer indirekten Wirkung von Liraglutide auszugehen.
Pharmakodynamik
Liraglutide verfügt über eine Wirkdauer von 24 Stunden. Es verbessert durch eine Senkung des postprandialen und Nüchternblutzuckers die Blutzuckerkontrolle bei Patienten mit Diabetes mellitus Typ 2.
Klinische Wirksamkeit
Im Rahmen von 5 zur Beurteilung der Wirkungen von Victoza auf die Blutzuckerkontrolle durchgeführten doppelblinden, kontrollierten klinischen Sicherheits- und Wirksamkeitsstudien wurden 3'992 Patienten mit Typ 2 Diabetes mellitus randomisiert. Die Behandlung mit Liraglutide führte zu klinisch und statistisch signifikanten Verbesserungen des HbA1c, der Nüchternplasmaglucose (NPG) und der postprandialen Glucose (PPG) gegenüber Placebo.
An diesen Studien nahmen 3'978 exponierte Probanden teil (davon wurden 2'501 mit Liraglutide behandelt), 53.7 % waren männlich und 46.3 % weiblich. 797 Patienten waren ≥65 Jahre alt (davon wurden 508 mit Victoza behandelt); 113 Patienten waren ≥75 Jahre alt (davon wurden 66 mit Victoza behandelt).
Ausserdem wurde eine grosse Studie zu kardiovaskulären Endpunkten (LEADER Studie) mit Liraglutide bei 9'340 Patienten mit Diabetes mellitus Typ 2 und hohem kardiovaskulärem Risiko durchgeführt.
Blutzuckerkontrolle
Monotherapie
Patienten, die zuvor entweder mit Diät und Bewegung oder OAD-Monotherapie in submaximaler Dosis behandelt wurden, zeigten bei einer 52-wöchigen Monotherapie mit Liraglutide 1.2 mg bzw. 1.8 mg eine stärkere Senkung des HbA1c-Werts als bei Monotherapie mit Glimepiride 8 mg (mittlere Differenz [95 % CI] für 1.2 mg -0.33 [-0.53, -0.13] und für 1.8 mg -0.62 [-0.83, -0.42]).
Tabelle 2: Liraglutide klinische Phase 3a Studien in Monotherapie (52 Wochen) und in Kombination mit oralen Antidiabetika (26 Wochen)
N Mitterer HbA1c (%) Mittlere Veränderung Patienten (%), die Mittleres Gewicht Mittlere Veränderung
bei Baseline des HbA1c seit HbA1c <7 % erreichte bei Baseline (kg) des Gewichts seit
Baseline (%) n Baseline (kg)
Monotherapie
Liraglutide 1.2 251 8.18 -0.84 42.80658.38 92.1 -2.05
Liraglutide 1.8 246 8.19 -1.14 50.9662.08 92.6 -2.45
Glimepiride1 248 8.23 -0.51 27.8630.88 93.3 1.12
Add-on zu Metformin2
Liraglutide 1.2 240 8.3 -0.97 35.3652.87 88.5 -2.58
Liraglutide 1.8 242 8.4 -1.00 42.4666.37 88.0 -2.79
Placebo 121 8.4 0.09 10.8622.57 91.0 -1.51
Glimepiride3 242 8.4 -0.98 36.3656.07 89.0 0.95
Add-on zu Glimepirid
e3
Liraglutide 1.2 228 8.5 -1.08 34.5657.47 80.0 0.32
Liraglutide 1.8 234 8.5 -1.13 41.66 55.97 83.0 -0.23
Placebo 114 8.4 0.23 7.5611.87 81.9 -0.10
Rosiglitazone4 231 8.4 -0.44 21.9636.17 80.6 2.11
Add-on zu Metformin2
+Rosiglitazone5
Liraglutide 1.2 177 8.48 -1.48 57.56 95.3 -1.02
Liraglutide 1.8 178 8.56 -1.48 53.76 94.9 -2.02
Placebo 175 8.42 -0.54 28.16 98.5 0.60
Add-on zu Metformin2
+Glimepiride3
Liraglutide 1.8 230 8.3 -1.33 53.16 85.8 -1.81
Placebo 114 8.3 -0.24 15.36 85.4 -0.42
Insulin Glargine9 232 8.1 -1.09 45.86 85.2 1.62
Add-on zu SGLT2i10
± Metformin (≥1500
mg/Tag)
Liraglutide 1.8 203 8.00 -1.02 54.8 91.0 -2.92
Placebo 100 7.96 -0.28 13.9 91.4 -2.06
1 Glimepiride 8 mg/Tag;
2 Metformin 2'000 mg/Tag;
3 Glimepiride 4 mg/Tag;
4 Rosiglitazone 4 mg/Tag,
5 Rosiglitazone 4 mg zweimal pro Tag
6 alle Patienten;
7 vorangegangene Behandlung mit OAD-Monotherapie;
8Patienten, die vorab mit Diät behandelt wurden
10Victoza wurde als add-on zu SGLT2i in allen genehmigten Dosierungen untersucht
9 die Dosierung von Insulin Glargine erfolgte unverblindet und gemäss der Richtlinie zur Titration von Insulin Glargine. Die Dosistitration von Insulin Glargine wurde vom Patienten nach Anweisung des Prüfarztes durchgeführt:
Richtlinie zur Titration von Insulin Glargine
Selbst gemessene Nüchtern-Plasma-Glucose Erhöhung der Dosis von Insulin Glargine (I.E.)
≤5.5 mmol/l (≤100 mg/dl) Zielwert Keine Anpassung
≥5.5 und <6.7 mmol/l (>100 und <120 mg/dl) 0–2 I.E.a
≥6.7 mmol/l (≥120 mg/dl) 2 I.E.
a Entsprechend der individuellen Empfehlung des Studienarztes beim vorangegangenen Besuch, beispielsweise abhängig davon, ob der Proband eine Hypoglykämie hatte.
Eine 26-wöchige Kombinationstherapie mit Victoza und Metformin, einem Sulfonylharnstoff oder Metformin und einem Thiazolidindion oder SGLT2i ± Metformin bewirkte gegenüber Placebo eine statistisch signifikante und anhaltende HbA1c-Senkung (p <0.0001).
Die Wirksamkeit von Victoza 0.6 mg wurde ausserdem in Kombination mit einem Sulfonylharnstoff oder Metformin geprüft und erwies sich als überlegen im Vergleich zu Placebo, jedoch geringer als die anderen Victoza-Dosisstärken von 1.2 mg und 1.8 mg.
In Patienten, bei denen keine ausreichende Blutzuckerkontrolle mit Victoza und Metformin erreicht wurde, resultierte die Zugabe von Insulin Detemir in einer verbesserten Wirksamkeit verglichen mit Victoza und Metformin alleine nach einer 26-wöchigen Behandlung (geschätzte Behandlungsdifferenz von -0.52 % bezüglich HbA1c (Tabelle 3)).
Tabelle 3: Resultate einer 12 + 26-wöchigen Studie, bei der Insulin Detemir zu Liraglutide und Metformin hinzugegeben wurde in Patienten, die keine ausreichende Blutzuckerkontrolle (HbA1c <7.0 %) nach einer 12-wöchigen Run-in-Phase mit Liraglutide erreichten.
Die Studie schloss anfänglich 988 Patienten in der Run-in-Phase ein. Die Patienten, die zuvor mit Metformin und Sulfonylharnstoff behandelt wurden, wurden angewiesen die Sulfonylharnstoffbehandlung abzubrechen, aber alle Patienten setzten ihre bisherige Metforminbehandlung fort. Nach der Run-in-Phase erreichten 498 Patienten (61 % der Absolventen 988 Patienten) einen HbA1c Zielwert von <7 % mit Liraglutide und Metformin und diese setzten daher die Behandlung in einem "nicht-randomisierten" Arm fort, während die übrigen 323 Patienten (39 %) mit einem HbA1c ≥7 % randomisiert wurden, um entweder die Therapie unverändert mit Liraglutide 1.8 mg und Metformin als Kontrolle (n = 161) fortzusetzen oder eine zusätzliche Intensivierung mit Insulin Detemir als Zusatztherapie (n = 162) zu erhalten.
Insulin Detemir als
Zusatztherapie
Patienten, die einen Patienten, die den HbA1c
HbA1c Zielwert1 nach 12 Zielwert1 nach 12 Wochen
Wochen Therapie mit Therapie mit Liraglutide
Liraglutide und Metformin und Metformin nicht
erreichen erreichen
Nicht-randomisierte Randomisiert + Insulin Randomisierte Kontrollgru
kontinuierliche Therapie Detemir ppe
N (exponiert) 498 162 161
HbA1c (%)
Woche -12 (Mittelwert) 7.72 8.2 8.3
Run-in-Beginn2Woche 0 6.4 7.6 7.6
(Mittelwert)
Veränderung vom HbA1c +0.2 -0.53 -0.13
von Woche 0 zur Woche
26(randomisierte Phase)
Patienten, bei denen
ein HbA1c-Wert <7 %
erreicht wurde
Insulin Dosis (Mittelwer 39 E
t in Woche 38)
Körpergewicht (kg)
Woche -12 (Mittelwert) 99.0 kg 99.5 kg 98.8 kg
Run-in-Beginn2Woche 0 94.65 kg 95.97 kg 95.34 kg
(Mittelwert)
Veränderung vom -0.45 kg -0.5 kg3 -1.24 kg3
Körpergewicht von Woche
0 zur Woche 26(randomisi
erte Phase)
1 HbA1c <7 %
2 (Bei Eintritt in die Run-in-Phase wurden alle Patienten von Metformin + Sulfonylharnstoff + Liraglutide 1.8 mg umgestellt oder erhielten Liraglutide 1.8 mg als Zusatztherapie zu Metformin für 12 Wochen. Bei beiden Szenarien blieb die Metforminbehandlung unverändert ≥1'500 mg/Tag.)
3 Die Schätzungen sind von einem ANCOVA-Modell mit einer Behandlung, früherer OAD und Land als fixer Effekt und den Ausgangswert als Kovariate
Anteil der Patienten, bei denen eine adäquate HbA1c-Senkung erreicht wurde
Unter Liraglutide allein erreichte ein statistisch signifikant höherer Anteil der Patienten (1.8 mg p <0.0001, 1.2 mg p = 0.0007) nach 52 Wochen einen HbA1c-Wert von ≤7 %, als dies bei mit Glimepiride behandelten Patienten der Fall war.
Eine Kombinationstherapie mit Victoza und Metformin, einem Sulfonylharnstoff, einem Thiazolidindion oder SGLT2i ± Metformin führte nach 26 Wochen im Vergleich zur monotherapeutischen Anwendung dieser Arzneimittel bei einem statistisch signifikant höheren Anteil der Patienten zum Erreichen eines HbA1c ≤6.5 % (p ≤0.0001).
Bei Patienten, die keine adäquate Blutzuckerkontrolle mit Victoza + Metformin erreichten, war der Anteil derer, die einen HbA1c Zielwerte von <7 % und ≤6.5 % erreichten, statistisch signifikant höher bei einer Behandlung mit Insulin Detemir + Liraglutide 1.8 mg + Metformin als bei einer Behandlung mit Liraglutide 1.8 mg + Metformin, (p ≤0.0001/p = 0.0016).
Nüchternplasmaglucose
Die Behandlung mit Victoza als Monotherapie und in Kombination mit einem oder zwei oralen Antidiabetika bewirkte eine Senkung der Nüchternplasmaglucose um 0.72–2.42 mmol/l (13–43.5 mg/dl) bereits innerhalb der ersten beiden Behandlungswochen.
Postprandiale Glucose
Victoza reduziert die postprandialen Glucosespiegel nach allen drei Hauptmahlzeiten um 1.68–2.71 mmol/l (31–49 mg/dl).
Betazellfunktion
Klinische Studien mit Victoza weisen auf eine verbesserte Betazellfunktion hin. Dabei wurden Messungen wie das homeostasis model assessment for beta-cell function (HOMA-B) und das Verhältnis von Proinsulin zu Insulin zugrunde gelegt. Nach 52-wöchiger Behandlung mit Victoza wurde bei einer Subgruppe von Patienten mit Diabetes mellitus Typ 2 (n = 29) eine Verbesserung von erster und zweiter Phase der Insulinausschüttung nachgewiesen.
Körpergewicht
Victoza alleine und in Kombination mit Metformin, Metformin und einem Sulfonylharnstoff oder Metformin und einem Thiazolidindion war über die Dauer der Studien mit einer nachhaltigen Gewichtsabnahme von 1.0 bis 2.8 kg verbunden.
Die Gewichtsreduktion fiel umso stärker aus, je höher der Body-Mass-Index (BMI) bei Studienbeginn war.
Der Gewichtsverlust, der in Patienten beobachtet wurde, die mit Liraglutide in Kombination mit Metformin behandelt wurden, blieb nach Hinzugabe von Insulin Detemir erhalten.
Kardiovaskuläres Risiko
Die LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results) Studie ist eine randomisierte, multizentrische, doppelblinde klinische Studie, in welcher 9'340 Patienten mit Liraglutide (4'668) oder Placebo (4'672) zusätzlich zur vorbestehenden Therapie zur Senkung des Blutzuckers und kardiovaskulärer (CV) Risikofaktoren behandelt wurden. Die antihyperglykämische Behandlung und weitere kardiovaskuläre Begleitmedikationen konnten während der gesamten Studie gemäss Behandlungsstandard angepasst werden. Ausgeschlossen davon war der Einsatz alternativer Inkretin-basierter Therapien. Eingeschlossen wurden Patienten mit Typ 2 Diabetes mellitus, die 50 Jahre oder älter waren und eine manifeste kardiovaskuläre Erkrankung aufwiesen sowie Patienten ohne kardiovaskuläre Vorerkrankung, wenn diese 60 Jahre oder älter waren und mindestens einen kardiovaskulären Risikofaktor aufwiesen. Patienten mit einem akuten koronaren oder zerebrovaskulären Ereignis in den letzten 14 Tagen waren von der Studie ausgeschlossen. Die Beobachtungsdauer betrug mindestens 3.5 Jahre (maximal 5 Jahre). Die Studienpopulation umfasste Patienten mit leichter (n=3'907), moderater (n=1'934) oder schwerer (n=224) Nierenfunktionsstörung. Das mittlere Alter betrug 64 Jahre (4'329 Patienten ≥65 Jahren, 836 Patienten ≥75 Jahre), der mittlere BMI 32.5 kg/m² und die mittlere Dauer des Diabetes 12.8 Jahre.
Der primäre Endpunkt war die Zeit von der Randomisierung bis zum ersten Auftreten eines zusammengesetzten kardiovaskulären Endpunkts (3-Punkt MACE) bestehend aus kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt und nicht-tödlichem Schlaganfall. Liraglutide reduzierte im Vergleich zu Placebo das Risiko für das Auftreten des MACE signifikant (Abbildung 1). Diese Risikoreduktion war konsistent für alle Komponenten des Primärendpunkts (Abbildung 2). Liraglutide verlängerte auch die Zeit bis zum Auftreten eines erweiterten MACE, welcher zusätzlich Fälle einer koronaren Revaskularisation, einer Hospitalisierung aufgrund von instabiler Angina pectoris oder Herzinsuffizienz einschloss.
Abbildung 1: Kaplan-Meier-Plot der Zeit bis zum ersten schwerwiegenden unerwünschten kardiovaskulären Ereignis (MACE) – FAS-Population
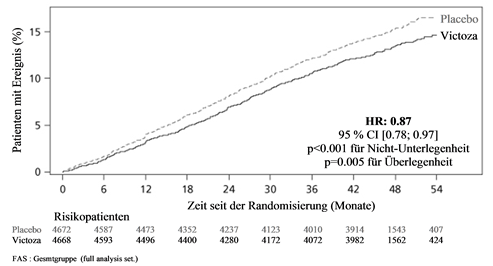
Abbildung 2: Forest Plot der Analysen einzelner Arten kardiovaskulärer Ereignisse – FAS-Population
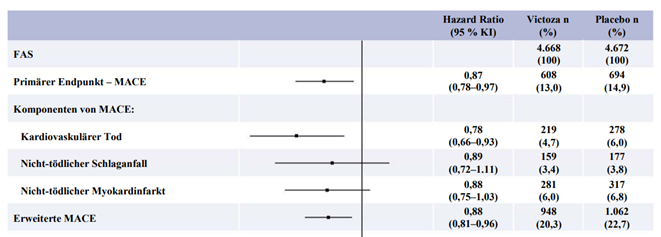

Ungeachtet einer Therapie gemäss Behandlungsstandard in beiden Armen zeigten Patienten im Liraglutide-Arm im Vergleich zu Placebo bis einschliesslich Monat 36 eine überlegene glykämische Kontrolle (geschätzte Therapiedifferenz [ETD] -0.40 % [-0.45; -0.34]) und bessere Reduktion des Körpergewichts (ETD -2.26 [-2.54; -1.99]). Post hoc Cox-Regressionsanalysen weisen darauf hin, dass diese Unterschiede allein nicht für die Reduktion des kardiovaskulären Risikos verantwortlich waren. Die Notwendigkeit einer Initiierung der Insulinbehandlung wurde unter Liraglutide bei Patienten, die zu Studienbeginn Insulin-naiv waren, im Vergleich zu Placebo um 48 % vermindert (HR 0.52 [0.48; 0.57]).
Blutdruck und Herzfrequenz
Im Verlauf der Phase-3a-Studien verminderte Liraglutide den systolischen Blutdruck ab der Baseline im Durchschnitt um 2.3 bis 6.7 mmHg, gegenüber dem Vergleichsprodukt betrug die Verminderung 1.9 bis 4.5 mmHg. In der LEADER Studie wurde nach 36 Monaten der systolische Blutdruck mit Liraglutide im Vergleich zu Placebo reduziert (-1.4 mmHg vs. -0.2 mmHg; ETD: -1.29 mmHg [-1.92; -0.48]), während der diastolische Blutdruck unter Liraglutide im Vergleich zu Placebo weniger stark vermindert wurde (-0.8 mmHg vs. -1.4 mmHg, ETD: 0.59 [0.19; 0.99]). Ein mittlerer Anstieg der Herzfrequenz im Vergleich zur Baseline von 2 bis 3 Schlägen pro Minute wurde mit Liraglutide in langfristigen klinischen Studien, einschliesslich LEADER, beobachtet. In der LEADER Studie wurde keine langfristige Auswirkung der erhöhten Herzfrequenz auf das Risiko kardiovaskulärer Ereignisse beobachtet.
Mikrovaskuläre Beurteilung
In der LEADER Studie umfassten mikrovaskuläre Ereignisse die Nephropathie und Retinopathie. Die Analyse der Zeit bis zum ersten mikrovaskulären Ereignis unter Liraglutide im Vergleich zum Placebo wies eine HR von 0.84 [0.73, 0.97] auf. Die HR für Liraglutide vs. Placebo betrug 0.78 [0.67, 0.92] hinsichtlich der Zeit bis zum ersten Auftreten der Nephropathie und 1.15 [0.87, 1.52] bis zum ersten Auftreten der Retinopathie.
Die geschätzte Therapiedifferenz für die Veränderung der Albumin/Kreatinin-Ausscheidung mit dem Urin von der Baseline bis Monat 36 betrug 0.81 [0.76, 0.86].
Pädiatrische Population
In einer doppelblinden Studie mit Jugendlichen und Kindern ab 10 Jahren mit Typ-2-Diabetes, die die Wirksamkeit und Sicherheit von Victoza 1.8 mg im Vergleich zu Placebo als Zusatz zu Metformin ± Insulin verglich, war Victoza der Behandlung mit Placebo hinsichtlich der Senkung des HbA1c nach 26 Wochen überlegen (-1.06, [-1.65, -0.46]). Die Behandlungsdifferenz hinsichtlich des HbA1c betrug 1.3 % nach einer unverblindeten Verlängerung von zusätzlich 26 Wochen und bestätigte die anhaltende glykämische Kontrolle von Victoza.
Das Wirksamkeits- und Sicherheitsprofil von Victoza war vergleichbar mit dem, welches bei mit Victoza behandelten Erwachsenen beobachtet wurde. Basierend auf einer adäquaten glykämischen Kontrolle oder Verträglichkeit verblieben 30 % der Probanden bei einer Dosis von 0.6 mg, 17 % steigerten die Dosis auf 1.2 mg und 53 % steigerten die Dosis auf 1.8 mg.
Weitere klinische Daten
Victoza verbesserte die mittels HOMA-IR gemessene Insulinsensitivität im Vergleich zu einer Sulfonylharnstoff-Therapie über einen Zeitraum von 52 Wochen.
Entsprechend den potentiell immunogenen Eigenschaften von protein- und peptidhaltigen Arzneimitteln können Patienten während der Behandlung mit Victoza gegen Liraglutide gerichtete Antikörper bilden. Im Durchschnitt bildeten 8.6 % der Patienten Antikörper. Die Antikörperbildung ist nicht mit einer Minderung der Wirksamkeit von Victoza verbunden.
Patienten mit Nierenfunktionsstörungen
In einer doppelblinden Studie, in der die Wirksamkeit und Sicherheit von Liraglutide 1.8 mg, zusätzlich zu Insulin und/oder oralen Antidiabetika (OAD) gegeben, bei Patienten mit Typ 2 Diabetes und moderater Nierenfunktionsstörung gegen Placebo verglichen wurde, war Liraglutide der Placebo-Behandlung hinsichtlich der Reduktion des HbA1c-Wertes nach 26 Wochen (-1.05 % vs. -0.38 %, p <0.0001) überlegen. Das Risiko für hypoglykämische Episoden war in den beiden Behandlungsgruppen vergleichbar. Das Sicherheitsprofil von Liraglutide war im Allgemeinen vergleichbar mit demjenigen, das in anderen Studien mit Liraglutide beobachtet wurde.
PharmakokinetikAbsorption
Liraglutide wird nach subkutaner Verabreichung langsam resorbiert; die Maximalkonzentration wird 8–12 Stunden nach der Gabe erreicht. Die geschätzte Maximalkonzentration von Liraglutide lag nach subkutaner Applikation einer Einzeldosis von 0.6 mg bei 9.4 nmol/l. Nach Verabreichung von 1.8 mg Liraglutide erreichte die durchschnittliche Steady-state-Konzentration (AUCt/24) etwa 34 nmol/l. Mit zunehmendem Körpergewicht sinkt die Liraglutid-Exposition. In einer Studie an gesunden Probanden wurde eine Dosisproportionalität für AUCt und Cmax nachgewiesen im Dosisbereich 0.4–0.9 mg und in einer zweiten Studie für AUCt im Dosisbereich 1.2–1.8 mg. Der intraindividuelle Variationskoeffizient der Liraglutide-AUC belief sich nach Einzelgabe auf 11 %. Die subkutane Verabreichung von Liraglutide kann am Bauch, Oberschenkel oder Oberarm erfolgen.
Die absolute Bioverfügbarkeit von Liraglutide nach subkutaner Verabreichung beträgt etwa 55 %.
Distribution
Das scheinbare Verteilungsvolumen nach subkutaner Verabreichung liegt bei 11–17 l. Das mittlere Verteilungsvolumen nach intravenöser Gabe von Liraglutide beträgt 0.07 l/kg.
Liraglutide wird in grossem Umfang an Plasmaproteine gebunden (>98 %).
Metabolismus
Innerhalb eines Zeitraums von 24 Stunden nach Verabreichung einer Einzeldosis [3H]-Liraglutide bei gesunden Probanden lag im Plasma hauptsächlich intaktes Liraglutide vor. Es wurden zwei Nebenmetaboliten im Plasma nachgewiesen (≤9 % und ≤5 % der Gesamtradioaktivität im Plasma). Liraglutide wird in ähnlicher Weise wie grosse Proteine endogen metabolisiert, ohne dass ein bestimmtes Organ als Hauptweg für die Elimination verantwortlich ist.
Elimination
Nach Verabreichung von [3H]-Liraglutide wurde kein intaktes Liraglutide in Urin oder Faeces nachgewiesen. Nur ein geringer Teil der verabreichten Radioaktivität wurde in Form von Liraglutide-verwandten Metaboliten in Urin und Faeces ausgeschieden (6 % bzw. 5 %). Die Ausscheidung der Radioaktivität über Urin und Faeces fand hauptsächlich innerhalb der ersten 6–8 Tage statt und entsprach derjenigen der drei Nebenmetabolite.
Die mittlere Clearance nach subkutaner Applikation einer Einzeldosis Liraglutide beträgt etwa 1.2 l/h bei einer Eliminationshalbwertszeit von rund 13 Stunden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
In einer Einzeldosis-Studie wurde die Pharmakokinetik von Liraglutide bei Patienten mit unterschiedlichen Graden einer Leberfunktionsstörung beurteilt. Verglichen mit gesunden Probanden war die totale Liraglutide-Exposition bei Patienten mit leichter bis mittelschwerer Leberfunktionsstörung um 13–23 % vermindert.
Bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Score >9) war die totale Exposition deutlich geringer (44 %).
Nierenfunktionsstörungen
Bei Patienten mit Niereninsuffizienz war die Liraglutide-Exposition im Vergleich zu Personen mit normaler Nierenfunktion reduziert. Bei Patienten mit leichter (Creatinin-Clearance ClCr 50–80 ml/min), mittelschwerer (ClCr 30–50 ml/min) und schwerer (ClCr <30 ml/min) Nierenfunktionsstörung und bei dialysepflichtigen Patienten mit einer Nierenerkrankung im Endstadium war die Liraglutide-Exposition um 33 %, 14 %, 27 % bzw. 26 % vermindert.
Ältere Patienten
Die Ergebnisse einer pharmakokinetischen Studie an gesunden Probanden und einer populationspharmakokinetischen Analyse von Patientendaten (18 bis 80 Jahre) zeigen keinen klinisch relevanten Einfluss des Alters auf die Pharmakokinetik von Victoza.
Kinder und Jugendliche
Victoza wurde bei Kindern und Jugendlichen nicht untersucht.
Ethnische Zugehörigkeit
Die Ergebnisse einer populationspharmakokinetischen Analyse unter Einschluss weisser, schwarzer, asiatischer und hispanischer Probanden zeigten keinen klinisch relevanten Einfluss der ethnischen Zugehörigkeit auf die Pharmakokinetik von Victoza.
Fettleibigkeit
Eine populationspharmakokinetische Analyse lässt darauf schliessen, dass der Body-Mass-Index (BMI) keinen signifikanten Einfluss auf die Pharmakokinetik von Liraglutide hat.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe oder Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Karzinogenität
In zweijährigen Karzinogenitätsstudien an Ratten und Mäusen wurden nichtletale C-Zelltumoren der Schilddrüse beobachtet. Bei über einen Zeitraum von 20 Monaten behandelten Affen traten diese Tumoren nicht auf. Die Befunde bei Nagern werden durch einen nicht-genotoxischen, spezifischen, über den GLP-1-Rezeptor vermittelten Mechanismus verursacht, für den Nagetiere im Gegensatz zu Affen und Menschen besonders anfällig sind. Die Relevanz für den Menschen ist wahrscheinlich gering, kann jedoch nicht komplett ausgeschlossen werden. Im Zusammenhang mit der Behandlung wurden keine anderen Tumoren festgestellt.
Reproduktionstoxizität
Tierexperimentelle Studien zeigten keine direkt schädigende Wirkung hinsichtlich Fertilität, aber bei der höchsten Dosis eine leicht erhöhte Embryonensterblichkeit in frühen Stadien. Eine Anwendung von Victoza während des mittleren Abschnitts der Tragzeit führte zu einer Reduktion des mütterlichen Gewichts und des Fötuswachstums mit nicht eindeutigen Auswirkungen auf die Rippen von Ratten und Skelettveränderungen bei Kaninchen. Unter Einwirkung von Victoza war bei Ratten das neonatale Wachstum reduziert. In der Gruppe mit der höchsten Dosis hielt dieser Effekt in der Zeit nach dem Abstillen an. Es ist nicht bekannt, ob das verminderte Wachstum der Jungtiere durch eine geringere Milchaufnahme aufgrund einer direkten GLP-1-Wirkung oder durch geringere Milchproduktion der Muttertiere aufgrund einer verminderten Kalorienaufnahme verursacht wird.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Haltbarkeit der gebrauchsfertigen Lösung nach Anbruch: Nach dem erstmaligen Gebrauch des Victoza-Pens kann das Produkt 1 Monat bei Raumtemperatur (nicht über 30°C) oder im Kühlschrank (2–8°C) aufbewahrt werden.
Besondere Lagerungshinweise
Im Kühlschrank (2–8°C) lagern. Nicht im Gefrierfach oder in unmittelbarer Nähe von Kühlelementen lagern. Victoza nicht einfrieren.
Wenn der Victoza-Pen nicht in Gebrauch ist, sollte die Schutzkappe aufgesetzt werden, um den Inhalt vor Licht zu schützen.
Victoza muss vor übermässiger Hitze und Sonnenlicht geschützt werden.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Die Injektionsnadel sollte nach jeder Injektion entfernt und der Victoza-Pen ohne Injektionsnadel aufbewahrt werden. Dies verhindert Kontamination, Infektion und Auslaufen. Ausserdem wird eine korrekte Dosierung sichergestellt.
Der Patient ist anzuweisen, die Nadel nach jeder Injektion zu entsorgen.
Der Victoza-Pen darf nur von einer Person verwendet werden.
Victoza darf nicht verwendet werden, wenn es nicht klar und farblos oder fast farblos aussieht.
Victoza darf nicht mehr verwendet werden, wenn es gefroren war.
Zulassungsnummer59329 (Swissmedic)
PackungenPackungen zu 2 Fertigpens. [B]
Jeder Fertigpen enthält 30 Dosen zu 0.6 mg, 15 Dosen zu 1.2 mg oder 10 Dosen zu 1.8 mg.
Injektionsnadeln sind nicht enthalten. Für die Verabreichung von Victoza eignen sich Nadeln mit einer Länge von bis zu 8 mm und einem Kaliber von mindestens 31 G. Der Pen ist für eine Verwendung mit NovoFine Einmalnadeln vorgesehen.
ZulassungsinhaberinNovo Nordisk Pharma AG, Kloten
Domizil: Zürich
Stand der InformationApril 2025
|