ZusammensetzungWirkstoffe
Toxinum botulinicum A (150 kD).
Hilfsstoffe
Albuminum humanum, Saccharum.
Darreichungsform und Wirkstoffmenge pro EinheitPulver zur Herstellung einer Injektionslösung.
Eine Durchstechflasche enthält:
Wirkstoff: 50, 100 oder 200 LD50-Einheiten* Botulinumtoxin Typ A (150 kD), frei von Komplexproteinen*
* Eine Einheit entspricht der mittleren letalen Dosis (LD50) nach der unter definierten Bedingungen erfolgten intraperitonealen Injektion der rekonstituierten Lösung in Mäuse.
Die Einheiten sind spezifisch für Xeomin und daher nicht auf andere Botulinumtoxin-Präparate übertragbar.
Zur intramuskulären und intraglandulären Anwendung.
Indikationen/AnwendungsmöglichkeitenXeomin wird angewendet bei Erwachsenen zur symptomatischen Behandlung von
-Blepharospasmus,
zervikaler Dystonie mit überwiegend rotatorischer Komponente (Torticollis spasmodicus),
fokaler Spastik der oberen und unteren Extremitäten (siehe Rubriken "Dosierung/Anwendung" und "klinische Wirksamkeit" ),
chronischer, beeinträchtigender Sialorrhoe.
Xeomin wird angewendet bei Kindern und Jugendlichen im Alter von 2 bis 17 Jahren zur symptomatischen Behandlung von
chronischer beeinträchtigender Sialorrhoe aufgrund neurologischer Erkrankungen / Entwicklungsstörungen.
Dosierung/AnwendungAllgemeine Hinweise
Xeomin darf nur von Ärzten mit entsprechender Fachkenntnis in der Behandlung mit Botulinumtoxin Typ A angewendet werden.
Die nachfolgend empfohlenen Dosierungen sind aufgrund der unterschiedlichen LD50-Testmethoden spezifisch für Xeomin und nicht auf andere Botulinumtoxin-Präparate Typ A übertragbar.
Rekonstituiertes Xeomin ist zur intramuskulären oder intraglandulären Injektion bestimmt. Zu Hinweisen zur Rekonstitution und Verdünnung der Durchstechflaschen siehe "Sonstige Hinweise" und "Hinweise für die Handhabung" .
Die optimale Dosis, Häufigkeit und die Anzahl an Injektionsstellen sind vom behandelnden Arzt für jeden Patienten individuell festzulegen. Dabei sollte eine Dosistitration durchgeführt werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Blepharospasmus
Dosierung
Die empfohlene Initialdosis beträgt 1,25-2,5 Einheiten pro Injektionsstelle. Initial sollten nicht mehr als 25 Einheiten pro Auge appliziert werden.
Üblicherweise stellt sich der erste Effekt der Injektion innerhalb von vier Tagen ein. Die Wirkung einer Behandlung hält im Allgemeinen etwa 3-4 Monate an, sie kann jedoch erheblich länger oder kürzer andauern. Bei Bedarf kann die Behandlung wiederholt werden.
Bei der Behandlung des Blepharospasmus wird empfohlen, eine Gesamtdosis von 100 Einheiten nicht zu überschreiten. Der Zeitraum zwischen zwei Behandlungen sollte mindestens 8 Wochen betragen. Die Behandlungsintervalle sollten nach dem individuellen Bedarf jedes Patienten festgelegt werden.
Bei Wiederholungsbehandlungen kann die Dosis bis auf das Doppelte erhöht werden, wenn die Reaktion auf die Initialbehandlung als ungenügend erachtet wird – gewöhnlich definiert als Effekt, der nicht länger als zwei Monate anhält. Es scheint jedoch, dass eine Applikation von mehr als 5,0 Einheiten pro Injektionsstelle keinen zusätzlichen Nutzen hat.
Anwendung
Die rekonstituierte Lösung von Xeomin wird mit einer geeigneten sterilen Nadel injiziert (z.B. 27-30 G/0,30-0,40 mm Durchmesser/12,5 mm Länge). Es wird ein Injektionsvolumen von etwa 0,05 bis 0,1 ml pro Injektionsstelle empfohlen.
Xeomin wird in den medialen und lateralen M. orbicularis oculi des Oberlids und den lateralen M. orbicularis oculi des Unterlids injiziert. Weitere Injektionen in die Augenbrauengegend, in den lateralen M. orbicularis und in die obere Gesichtshälfte können erfolgen, wenn dort befindliche Krämpfe das Sehvermögen stören.
Torticollis spasmodicus
Dosierung
Die Dosierung muss für jeden Patienten individuell gewählt werden, basierend auf der Stellung von Kopf und Hals des Patienten, dem Ausmass einer eventuellen Muskelhypertrophie, dem Körpergewicht des Patienten sowie seiner Reaktion auf frühere Injektionen.
In der Praxis beträgt die Gesamtdosis gewöhnlich nicht mehr als 200 Einheiten. Dosen bis zu 300 Einheiten können gegeben werden. Dabei sollten pro Injektionsstelle nicht mehr als 50 Einheiten appliziert werden. Therapienaive Patienten benötigen üblicherweise eine niedrigere Startdosis als Patienten, welche bereits mit Botulinumtoxin Typ A vorbehandelt sind.
In den M. sternocleidomastoideus sollte nicht bilateral injiziert werden, da ein erhöhtes Risiko für das Auftreten unerwünschter Wirkungen (besonders Dysphagie) besteht, wenn bilaterale Injektionen oder Dosen von mehr als 100 Einheiten in diesen Muskel verabreicht werden.
Üblicherweise stellt sich der erste Effekt der Injektion innerhalb von sieben Tagen ein. Die Wirkung einer Behandlung hält im Allgemeinen etwa 3-4 Monate an, sie kann jedoch erheblich länger oder kürzer andauern. Der Zeitraum zwischen zwei Behandlungen sollte mindestens 8 Wochen betragen. Die Behandlungsintervalle sollten nach dem individuellen Bedarf jedes Patienten festgelegt werden.
Anwendung
Für die Injektion in oberflächliche Muskeln werden geeignete sterile Nadeln verwendet (z.B. 25-30 G/0,30-0,50 mm Durchmesser/37 mm Länge), für tiefer liegende Muskeln können beispielsweise Nadeln mit 22 G/0,70 mm Durchmesser/75 mm Länge eingesetzt werden. Es wird ein Injektionsvolumen von etwa 0,1 bis 0,5 ml pro Injektionsstelle empfohlen.
Xeomin wird üblicherweise in den M. sternocleidomastoideus, M. levator scapulae, M. scalenus, M. splenius capitis und/oder M. trapezius injiziert. Diese Liste ist nicht vollständig, da alle Muskeln, die für die Kontrolle der Kopfhaltung verantwortlich sind, beteiligt sein können und möglicherweise ebenfalls behandelt werden müssen. Treten bei der Isolation der einzelnen Muskeln Schwierigkeiten auf, sollten die Injektionen mit Verfahren wie zum Beispiel elektromyographischer Ableitung oder Ultraschall durchgeführt werden.
Die Wahl mehrerer Injektionsstellen ermöglicht einen gleichmässigeren Kontakt des Toxins mit den innervierten Gebieten des dystonen Muskels und ist besonders bei grösseren Muskeln günstig. Die optimale Anzahl der Injektionsstellen hängt von der Grösse des Muskels ab, der chemisch denerviert werden soll.
Spastik der oberen und unteren Extremitäten
Dosierung
Spastik der oberen Extremitäten
Die genaue Dosis, die Häufigkeit der Injektionen und die Anzahl der Injektionsstellen sollten individuell auf den Patienten je nach Grösse, Anzahl und Lage der beteiligten Muskeln, Schweregrad der Spastik und dem Vorliegen einer lokalen Muskelschwäche abgestimmt werden.
Es werden folgende Dosierungen pro Muskel empfohlen:
Klinisches Bild Muskel Einheiten (Dosisbere Anzahl der Injektionsst
ich) ellen pro Muskel
Handgelenkbeugung
Flexor carpi radialis Flexor carpi ulnaris 25-100 20-100 1-2 1-2
Gefaustete Hand
Flexor digitorum superficialis Flexor digitorum 25-100 25-100 2 2
profundus
Ellbogenbeugung
Brachioradialis Biceps Brachialis 25-100 50-200 25-100 1-3 1-4 1-2
Unterarmpronation
Pronator quadratus Pronator teres 10-50 25-75 1 1-2
Daumen-in-Hand-Stellung
Flexor pollicis longus Adductor pollicis Flexor 10-50 5-30 5-30 1 1 1
pollicis brevis / Opponens pollicis
Innenrotierte/retrovertierte/adduzierte Schulter
Deltoideus, pars clavicularis Latissimus dorsi 20-150 25-150 1-3 1-4 1-6 1-4 1-2
Pectoralis major Subscapularis Teres major 20-200 15-100 20-100
Die maximale Gesamtdosis für die Behandlung der Spastik der oberen Extremitäten sollte 500 Einheiten pro Behandlungssitzung nicht überschreiten, wobei in die Schultermuskeln nicht mehr als 250 Einheiten appliziert werden sollten.
Nach Angaben der Patienten setzte die Wirkung 4 Tage nach Behandlungsbeginn ein und erreichte nach etwa 4 Wochen ein Maximum. Der Therapieeffekt hält im Allgemeinen ca. 12 Wochen an, kann jedoch erheblich länger oder kürzer andauern. Es wird empfohlen, bei Wiederholungsinjektionen einen Zeitabstand von 12 Wochen einzuhalten. Die Behandlungsintervalle sollten nach dem individuellen Bedarf jedes Patienten festgelegt werden.
Spastik der unteren Extremitäten
Die genaue Dosis, die Häufigkeit der Injektionen und die Anzahl der Injektionsstellen sollten individuell auf den Patienten je nach Grösse, Anzahl und Lage der beteiligten Muskeln, Schweregrad der Spastik und dem Vorliegen einer lokalen Muskelschwäche abgestimmt werden.
Es werden folgende Dosierungen pro Muskel empfohlen:
Klinisches Bild Muskel Einheiten (Dosisbere Anzahl der Injektionsstellen
ich) pro Muskel
Pes Equinus einschliesslich gebeugter Zehen
Gastrocnemius medial/lateral 50-200 2-6
Soleus 50-200 2-4
Tibialis posterior Flexor digitorum longus 50-150 50-100 25-75 2-3 1-3 1-2
Flexor hallucis longus
Die empfohlene Gesamtdosis für die Behandlung der Spastik der unteren Extremitäten beträgt maximal 400 Einheiten pro Behandlungssitzung. Es wird empfohlen, bei Wiederholungsinjektionen einen Zeitabstand von 12 Wochen, mindestens aber 10 Wochen einzuhalten. Die Behandlungsintervalle sollten nach dem individuellen Bedarf jedes Patienten festgelegt werden.
Spastik der oberen und unteren Extremitäten
Die kombinierte Behandlung der multifokalen Spastik der oberen und unteren Extremitäten sollte auf der Grundlage der oben genannten Dosen und Behandlungsempfehlungen durchgeführt werden.
Dabei sollte bei der ersten Behandlung einer kombinierten Spastik von oberer und unterer Extremität eine maximale Gesamtdosis von 500 Einheiten nicht überschritten werden. Wenn diese Dosierung gut vertragen wurde, kann diese bei weiteren Behandlungen auf bis zu 600 Einheiten gesteigert werden.
Anwendung
Rekonstituiertes Xeomin wird mit geeigneten sterilen Nadeln injiziert (z.B. 26 G/0,45 mm Durchmesser/37 mm Länge bei oberflächlichen Muskeln und längere Nadeln, z.B. 22 G/0,7 mm Durchmesser/75 mm Länge, bei tiefer liegenden Muskeln).
Falls Schwierigkeiten bei der Isolation der individuellen Muskeln auftreten, wird die Lokalisierung der involvierten Muskeln mit Verfahren wie zum Beispiel elektromyographischer Ableitung oder Ultraschall empfohlen. Die Verwendung mehrerer Injektionsstellen ermöglicht einen gleichmässigeren Kontakt des Toxins mit den innervierten Gebieten des Muskels, was insbesondere bei Injektion in grössere Muskeln günstig ist.
Bei der Behandlung der Spastik der unteren Extremitäten wird ein Injektionsvolumen von etwa 0,2 bis 1 ml pro Injektionsstelle empfohlen.
Chronische Sialorrhoe (Erwachsene)
Dosierung
Es sollte eine rekonstituierte Lösung mit einer Konzentration von 5 Einheiten/0,1 ml verwendet werden.
Xeomin wird in die Glandula parotidea und die Glandula submandibularis auf beiden Seiten injiziert (pro Behandlung insgesamt vier Injektionen). Die Gesamtdosis ist im Verhältnis 3:2 wie folgt zwischen der Glandula parotidea und der Glandula submandibularis aufgeteilt:
Drüsen Einheiten Volumen
Glandula parotidea 30 pro Seite 0,6 ml pro Injektion
Glandula submandibularis 20 pro Seite 0,4 ml pro Injektion
Die empfohlene Gesamtdosis pro Behandlung beträgt 100 Einheiten.
Es wird empfohlen bei Wiederholungsinjektionen einen Zeitabstand von 16 Wochen einzuhalten. Die Behandlungsintervalle sollten nach dem individuellen Bedarf jedes Patienten festgelegt werden.
Anwendung
Die Injektion darf nur unter Ultraschallführung und durch Ärzte mit Erfahrung in intraglandulärer Applikation erfolgen. Rekonstituiertes Xeomin wird mit geeigneten sterilen Nadeln intraglandulär injiziert (z.B. 27-30 G/0,30-0,40 mm Durchmesser/12,5 mm Länge).
Chronische Sialorrhoe (Kinder/Jugendliche)
Dosierung
Es sollte eine rekonstituierte Lösung in einer Konzentration von 2,5 Einheiten/0,1 ml angewendet werden.
Xeomin wird in die Glandula parotidea und die Glandula submandibularis beider Seiten injiziert (pro Behandlung insgesamt vier Injektionen). Die an das Körpergewicht angepasste Dosis wird in einem Verhältnis von 3:2 zwischen Glandula parotidea und die Glandula submandibularis entsprechend nachfolgender Tabelle aufgeteilt.
Die Dosierungen sollten an das Körpergewicht angepasst werden und die Gesamtdosis sollte 75 Einheiten pro Behandlungssitzung nicht überschreiten. Für Kinder mit einem Körpergewicht unter 12 kg können keine Dosierungsempfehlungen gegeben werden.
Körpergewicht Glandula parotidea,j Glandula submandibul Gesamtdosis, beide
ede Seite aris,jede Seite Drüsen, beide Seiten
Dosis pro Drüse Volumen pro Injektio Dosis pro Drüse Volumen pro Injektio
n n
kg Einheiten ml Einheiten ml Einheiten
≥12 und < 15 6 0,24 4 0,16 20
≥15 und < 19 9 0,36 6 0,24 30
≥19 und < 23 12 0,48 8 0,32 40
≥23 und < 27 15 0,60 10 0,40 50
≥27 und < 30 18 0,72 12 0,48 60
≥30 22,5 0,90 15 0,60 75
Die Injektionsstelle sollte nahe der Drüsenmitte liegen.
Die Behandlungsintervalle sollten nach dem tatsächlichen klinischen Bedarf jedes Patienten festgelegt werden.
Wiederholungsinjektionen häufiger als alle 16 Wochen werden nicht empfohlen.
Anwendung
Die Injektion darf nur unter Ultraschallführung und durch Ärzte bzw. Ärztinnen mit Erfahrung in intraglandulärer Applikation erfolgen. Nach Rekonstitution wird die Xeomin-Lösung mit einer geeigneten sterilen Nadel (z. B. 27-30 G/0,30-0,40 mm Durchmesser/12,5 mm Länge) intraglandulär injiziert.
Alle Indikationen
Sollte nach durchgeführter Erstapplikation auch nach einem Monat kein therapeutischer Effekt eingetreten sein, sollten folgende Massnahmen durchgeführt werden:
klinische Verifizierung der Neurotoxinwirkung auf den injizierten Muskel: Dies kann z.B. eine elektromyographische Untersuchung in einer hierfür spezialisierten Einrichtung beinhalten.
-Analyse der Gründe für das Therapieversagen, z.B. schlechte Isolierung der Muskeln, die injiziert werden sollten, zu geringe Dosis, schlechte Injektionstechnik, fixe Kontraktur, zu schwacher Gegenmuskel, mögliche Antikörperbildung.
-Überprüfung der Behandlung mit Botulinumtoxin Typ A als angemessene Therapieform.
-Sofern im Rahmen der Initialbehandlung keine unerwünschten Wirkungen aufgetreten sind, kann eine Wiederholungsbehandlung unter folgenden Voraussetzungen vorgenommen werden:
1.Dosisanpassung unter Berücksichtigung der Analyse des vorausgegangenen Therapieversagens,
2.Lokalisierung der involvierten Muskeln mit Verfahren wie zum Beispiel einer elektromyographischen Ableitung,
3.Einhaltung des Mindestintervalls zwischen der Initial- und der Wiederholungsbehandlung.
Bei Therapieversagen sind alternative Behandlungsmethoden in Betracht zu ziehen.
Spezielle Dosierungsanweisungen
Kinder und Jugendliche
Es können keine Dosierungsempfehlungen für Indikationen, ausser für chronische Sialorrhoe bei Kindern und Jugendlichen im Alter von 2 bis 17 Jahren und einem Körpergewicht von ≥12 kg gegeben werden.
Die pädiatrischen klinischen Daten zu Xeomin werden im Abschnitt "Klinische Wirksamkeit" beschrieben.
Kontraindikationen-Überempfindlichkeit gegenüber dem Wirkstoff Botulinumtoxin Typ A oder einem der Inhaltsstoffe.
-Generalisierte Störungen der Muskelaktivität (z.B. Myasthenia gravis, Lambert-Eaton-Syndrom).
-Infektion oder Entzündung an der vorgesehenen Injektionsstelle.
Warnhinweise und VorsichtsmassnahmenAllgemein
Bevor der Arzt Xeomin verabreicht, muss er sich mit der Anatomie des Patienten sowie ggf. mit anatomischen Veränderungen aufgrund chirurgischer Eingriffe vertraut machen. Besondere Vorsicht ist erforderlich, wenn sich die Injektionsstelle nahe an empfindlichen Strukturen wie der Arteria carotis, den Lungenapizes oder der ösophagealen Muskulatur befindet. Es ist sicherzustellen, dass Xeomin nicht intravasal injiziert wird.
Die empfohlenen Einzeldosen sollten nicht überschritten und die angegebenen Dosierungsintervalle nicht verkürzt werden.
Xeomin sollte nur mit Vorsicht angewendet werden:
beim Vorhandensein von Gerinnungsstörungen aller Arten
bei Patienten, die mit Antikoagulantien oder anderen Wirkstoffen mit antikoagulierender Wirkung behandelt werden
bei Patienten mit amyotropher Lateralsklerose
bei Patienten mit anderen Erkrankungen, die zu peripheren neuromuskulären Dysfunktionen führen
bei ausgeprägter Schwäche oder Atrophie des zu injizierenden Muskels
Lokale und entfernte Ausbreitung der Toxinwirkung
Unerwünschte Nebenwirkungen können insbesondere auch durch falsch platzierte Injektionen von Botulinumtoxin Typ A hervorgerufen werden, die vorübergehend auch benachbarte Muskelgruppen lähmen.
Es wurden Nebenwirkungen im Zusammenhang mit der Ausbreitung des Botulinumtoxins Typ A an vom Injektionsort entfernten Stellen mit bisweilen tödlichem Ausgang berichtet (siehe "Unerwünschte Wirkungen" ), die in manchen Fällen mit Dysphagie, Pneumonie und/oder ausgeprägten Schwächezuständen assoziiert waren. Eine Dysphagie kann für zwei bis drei Wochen nach der Injektion anhalten, es wurde jedoch in einem Fall auch über ein Andauern von bis zu fünf Monaten berichtet. Die Dysphagie scheint dosisabhängig zu sein.
Über Dysphagie wurde auch nach Injektionen berichtet, die nicht in die zervikale Muskulatur erfolgten.
Bereits bestehende neuromuskuläre Erkrankungen
Bei mit therapeutischen Dosen behandelten Patienten kann eine übermässige Muskelschwäche auftreten. Fälle von iatrogenem Botulismus wurden nach der Injektion von Botulinumtoxinprodukten berichtet. Bei Patienten mit neurologischen Grunderkrankungen wie Schluckstörungen besteht ein erhöhtes Risiko für diese Nebenwirkungen. Das Arzneimittel sollte bei diesen Patienten nur unter Aufsicht eines Spezialisten angewendet werden und die Anwendung darf nur dann erfolgen, wenn der Nutzen der Behandlung das Risiko überwiegt.
Im Allgemeinen sollten Patienten mit einer Vorgeschichte von Aspiration und Dysphagie mit Vorsicht behandelt werden. Die Behandlung einer zervikalen Dystonie bei diesen Patienten sollte mit äusserster Vorsicht erfolgen.
Patienten bzw. Betreuungspersonen sind darauf hinzuweisen, dass der ärztliche Notdienst sofort zu verständigen ist, wenn sie Anzeichen oder Symptome bemerken, die auf eine Ausbreitung der Botulinumtoxinwirkung hindeuten oder wenn Schluck-, Sprech- oder Atemstörungen auftreten.
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen wurden nach der Gabe von Botulinumtoxin Typ A berichtet. Bei schwerwiegenden Überempfindlichkeitsreaktionen (z.B. Anaphylaxie) und/oder Überempfindlichkeitsreaktionen vom Soforttyp sollte eine angemessene medizinische Therapie erfolgen.
Antikörperbildung
Zu häufige Injektionen mit Botulinumtoxin Typ A können eine Antikörperbildung hervorrufen, so dass es auch bei der Behandlung von anderen Indikationen zu einem Therapieversagen kommen kann.
Kinder und Jugendliche
Spontanmeldungen über eine mögliche Ausbreitung des Toxins an vom Applikationsort entfernte Stellen wurden bei Kindern und Jugendlichen mit Begleiterkrankungen, hauptsächlich mit infantiler Zerebralparese, sehr selten mit anderen Botulinumtoxin Typ A-Präparaten berichtet. Im Allgemeinen lag die Dosierung, die in diesen Fällen verwendet wurde, über der für diese Präparate empfohlenen Dosierung.
Selten wurde bei Kindern mit schwerer Zerebralparese nach einer Behandlung mit Botulinumtoxinpräparaten, inklusive nicht-zugelassener (off-label) Anwendung (z.B. im Nackenbereich), über Todesfälle berichtet, die bisweilen mit Aspirationspneumonien im Zusammenhang stehen. Das Risiko gilt bei Kindern und Jugendlichen mit einem schlechten Gesundheitszustand aufgrund von Vorerkrankungen oder bei Patienten mit ausgeprägten neurologischen Defiziten, Dysphagie oder einer Vorgeschichte einer kürzlich erlittenen Aspirationspneumonie bzw. Lungenerkrankung als besonders hoch.
Warnhinweise nach Indikation
Blepharospasmus
Aufgrund der anticholinergen Wirkung von Botulinumtoxin Typ A sollte Xeomin bei Patienten mit erhöhtem Risiko für ein Engwinkelglaukom nur mit Vorsicht angewendet werden.
Injektionen in die Nähe des M. levator palpebrae superioris sollten vermieden werden, um das Risiko einer Ptosis gering zu halten. Aufgrund der Diffusion von Botulinum Neurotoxin Typ A in den M. obliquus inferior kann sich eine Diplopie entwickeln. Diese unerwünschte Wirkung kann vermieden werden, wenn auf die mediale Injektion am unteren Augenlid verzichtet wird.
Durch reduziertes Blinzeln nach der Injektion von Xeomin in den M. orbicularis kann der Schutz der Hornhaut herabgesetzt werden, was zu andauernden epithelialen Defekten und Hornhautulzerationen führen kann. In den Weichteilen der Augenlider treten leicht Ekchymosen auf. Dieses Risiko kann durch sanfte Druckbehandlung an der Injektionsstelle unmittelbar nach der Injektion reduziert werden.
Torticollis spasmodicus
Patienten sollten darauf hingewiesen werden, dass Injektionen von Xeomin zur Behandlung des Torticollis spasmodicus milde bis schwere Dysphagien hervorrufen können, verbunden mit der Gefahr einer Aspiration und Dyspnoe. Ein medizinisches Eingreifen kann notwendig werden (z.B. in Form künstlicher Ernährung). Die Begrenzung der in den M. sternocleidomastoideus injizierten Dosis auf weniger als 100 Einheiten senkt die Häufigkeit des Auftretens von Dysphagien. Patienten mit geringerer Masse der Halsmuskeln oder Patienten, die bilaterale Injektionen in den M. sternocleidomastoideus benötigen, sind einem grösseren Risiko ausgesetzt. Für das Auftreten von Dysphagien wird die Ausweitung der pharmakologischen Wirkung von Xeomin als Folge der Ausbreitung des Neurotoxins in die ösophageale Muskulatur verantwortlich gemacht.
Spastik der oberen und unteren Extremitäten
Xeomin wurde zur Behandlung der fokalen Spastik zusammen mit üblichen Standard-Behandlungsmethoden untersucht und ist nicht als Ersatz für diese gedacht. Xeomin ist wahrscheinlich nicht geeignet, Bewegungseinschränkungen eines Gelenkes aufgrund einer fixen Kontraktur zu verbessern.
Neu oder wiederholt auftretende epileptische Anfälle wurden typischerweise bei Patienten mit Prädisposition für solche Ereignisse berichtet. Der genaue Zusammenhang dieser Ereignisse mit der Botulinumtoxin-Injektion ist nicht nachgewiesen.
Spastik der unteren Extremitäten
Bei der Behandlung, insbesondere von älteren Patienten, die ein erhöhtes Sturzrisiko haben könnten, ist Vorsicht geboten.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Theoretisch kann die Wirkung von Botulinumtoxin Typ A durch Aminoglykosid-Antibiotika oder andere Arzneimittel, die auf die neuromuskuläre Reizleitung wirken, z.B. Muskelrelaxantien des Tubocurarin-Typs, potenziert werden. Die gleichzeitige Anwendung von Xeomin mit Aminoglykosiden erfordert deshalb besondere Sorgfalt. Periphere Muskelrelaxantien sollten mit Vorsicht eingesetzt werden, gegebenenfalls sollte die Initialdosis des Relaxans verringert oder eine mittellangwirksame Substanz, wie Vercuronium oder Atracurium, anstelle einer langwirksamen Substanz eingesetzt werden.
Wenn für die Behandlung von chronischer Sialorrhoe verwendet, kann die Bestrahlung des Kopfes und Halses und/oder die gleichzeitige Verabreichung von Anticholinergika (z.B. Atropin, Glycopyrronium, Scopolamin) die Wirkung des Toxins erhöhen.
4-Aminochinoline können die Wirkung von Xeomin abschwächen.
Werden unterschiedliche Botulinumtoxin-Präparate gleichzeitig oder innerhalb von mehreren Monaten verabreicht, sind die möglichen Effekte nicht bekannt. Eine stark ausgeprägte neuromuskuläre Schwäche kann sich durch die Verabreichung eines anderen Botulinumtoxins vor vollständigem Abklingen der Wirkungen eines zuvor verabreichten Botulinumtoxins verstärken.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung bei Schwangeren. In tierexperimentellen Studien fand sich eine Reproduktionstoxizität (siehe "Präklinische Daten" ). Das potentielle Risiko für den Menschen ist nicht bekannt. Xeomin darf daher während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich und der potentielle Nutzen rechtfertigt das Risiko.
Stillzeit
Es ist nicht bekannt, ob Botulinumtoxin Typ A in die Muttermilch übergeht. Daher kann die Anwendung von Xeomin in der Stillzeit nicht empfohlen werden.
Fertilität
Daten aus klinischen Studien mit Botulinumtoxin Typ A liegen nicht vor. In einer tierexperimentellen Studie wurden keine nachteiligen Wirkungen auf die männliche oder weibliche Fertilität festgestellt (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt. Patienten sollten darauf hingewiesen werden, dass bei Symptomen wie Müdigkeit, Schwindel, Sehstörungen, Ptosis, Verlust der Muskelkraft oder Muskelschwäche die Verkehrstüchtigkeit und die Fähigkeit zur Durchführung von potentiell gefährlichen Tätigkeiten eingeschränkt sein könnten und solche Tätigkeiten daher vermieden werden sollten.
Unerwünschte WirkungenAllgemein
Gewöhnlich treten Nebenwirkungen innerhalb der ersten Woche nach der Injektion auf und sind vorübergehend. Nebenwirkungen können mit dem Wirkstoff, dem Injektionsverfahren oder beidem zusammenhängen.
Wie bei jeder Injektion können im Zusammenhang mit der Injektion lokale Schmerzen, Entzündungen, Parästhesien, Hypoästhesien, Druckempfindlichkeit, Schwellungen/Ödeme, Erytheme, Juckreiz, lokale Infektionen, Hämatome, Blutungen und/oder Blutergüsse auftreten.
Durch den Injektionsprozess verursachter Schmerz und/oder Angst können zu vasovagalen Reaktionen führen wie z.B. vorübergehende symptomatische Hypotonie, Übelkeit, Tinnitus und Synkopen.
Lokale Muskelschwäche stellt eine erwartete pharmakologische Wirkung von Botulinumtoxin Typ A dar.
In sehr seltenen Fällen wurden unerwünschte Wirkungen im Zusammenhang mit der Ausbreitung des Toxins an vom Injektionsort entfernte Stellen berichtet, bei denen die Symptome der Wirkung von Botulinumtoxin Typ A gleichen (übermässige Muskelschwäche, Dysphagie, Aspirationspneumonie mit bisweilen tödlichem Ausgang).
Die Behandlung des Torticollis spasmodicus kann Dysphagien unterschiedlichen Schweregrades mit der Gefahr der Aspiration hervorrufen, so dass medizinisches Eingreifen notwendig werden kann (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Selten wurden unter Botulinumtoxin-Präparaten schwerwiegende Überempfindlichkeitsreaktionen und/oder Überempfindlichkeitsreaktionen vom Soforttyp berichtet, einschliesslich Anaphylaxie, Serumkrankheit, Dyspnoe, Weichteilödem und Urtikaria. Einige dieser Reaktionen wurden nach alleiniger Anwendung von herkömmlichen Präparaten mit dem Botulinumtoxin-Typ-A-Komplex berichtet, andere nach Anwendung von Botulinumtoxin Typ A in Kombination mit anderen Wirkstoffen, die dafür bekannt sind, ähnliche Reaktionen auszulösen.
Unerwünschte Wirkungen aus klinischen Studien
Die Häufigkeiten sind folgendermassen definiert: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000), Einzelfälle ( basierend auf Spontanmeldungen aus der Marktüberwachung, genaue Häufigkeit kann nicht angegeben werden).
Blepharospasmus
Systemorganklasse Nebenwirkung Häufigkeit
Erkrankungen des Nervensystems Parästhesien Häufig
Kopfschmerzen, Facialisparese Gelegentlich
Augenerkrankungen Blepharoptosis Sehr häufig
Trockene Augen, Sehverschlechterungen, Häufig
Verschwommensehen
Diplopie, vermehrter Tränenfluss Gelegentlich
Erkrankungen des Gastrointestinaltrakts Mundtrockenheit Häufig
Dysphagie Gelegentlich
Erkrankungen der Haut und des Ausschlag Gelegentlich
Unterhautzellgewebes
Skelettmuskulatur-, Bindegewebs- und Muskelschwäche Gelegentlich
Knochenerkrankungen
Allgemeine Erkrankungen und Beschwerden am Schmerzen an der Injektionsste Häufig
Verabreichungsort lle
Müdigkeit Gelegentlich
Torticollis spasmodicus
Systemorganklasse Nebenwirkung Häufigkeit
Infektionen und parasitäre Infektion der oberen Atemwege Häufig
Erkrankungen
Erkrankungen des Nervensystems Kopfschmerzen, Schwindel, Präsynkopen Häufig
Tremor des Kopfes, Sprechstörungen Gelegentlich
Erkrankung der Atemwege, des Dysphonie, Dyspnoe Gelegentlich
Brustraums und Mediastinums
Erkrankungen des Gastrointestinaltrakt Dysphagie (10,4 %) Sehr häufig
s
Mundtrockenheit, Übelkeit Häufig
Erbrechen, Diarrhoe Gelegentlich
Erkrankungen der Haut und des Vermehrtes Schwitzen Häufig
Unterhautzellgewebes
Ausschlag, Erythem, Pruritus Gelegentlich
Skelettmuskulatur-, Bindegewebs- und Nackenschmerzen, Muskelschwäche, Häufig
Knochenerkrankungen Myalgien, Muskelsteifigkeit,
muskuläre Spasmen
Allgemeine Erkrankungen und Schmerzen an der Injektionsstelle, Häufig
Beschwerden am Verabreichungsort Asthenie
Spastik der oberen Extremitäten
Systemorganklasse Nebenwirkung Häufigkeit
Erkrankungen des Nervensystems Kopfschmerzen, Hypoästhesie Gelegentlich
Erkrankungen des Gastrointestinaltrakt Mundtrockenheit Häufig
s
Übelkeit, Dysphagie Gelegentlich
Erkrankungen der Haut und des Hautausschlag Gelegentlich
Unterhautzellgewebes
Skelettmuskulatur-, Bindegewebs- und Schmerzen in den Extremitäten, Gelegentlich
Knochenerkrankungen Gelenkschwellung, Muskelschwäche,
Myalgien
Allgemeine Erkrankungen und Asthenie, periphere Ödeme Gelegentlich
Beschwerden am Verabreichungsort
Spastik der unteren Extremitäten
Systemorganklasse Nebenwirkung Häufigkeit
Skelettmuskulatur-, Bindegewebs- und Muskelschwäche Schmerzen in den Häufig Nicht bekannt
Knochenerkrankungen Extremitäten
Verletzung, Vergiftung und durch Eingriffe Sturz Häufig
bedingte Komplikationen
Chronische Sialorrhoe (Erwachsene)
Systemorganklasse Nebenwirkung Häufigkeit
Erkrankungen des Nervensystems Parästhesie Häufig
Sprechstörungen Gelegentlich
Erkrankungen des Gastrointestinaltrakts Mundtrockenheit, Dysphagie Häufig
Veränderter (verdickter) Speichel, Dysgeusie Gelegentlich
Es wurden Fälle langanhaltender (> 110 Tage) starker Mundtrockenheit berichtet, die zu weiteren Komplikationen wie Gingivitis, Dysphagie und Karies führen kann.
Chronische Sialorrhoe (Kinder/Jugendliche)
Systemorganklasse Nebenwirkung Häufigkeit
Erkrankungen des Gastrointestinal Veränderter (verdickter) Speichel, Gelegentlich
trakts Mundtrockenheit, Dysphagie
Unerwünschte Wirkungen nach Markteinführung
Die folgenden Nebenwirkungen wurden mit unbekannter Häufigkeit bei der Verwendung von Xeomin seit Markteintritt unabhängig von der Indikation berichtet:
Systemorganklasse Nebenwirkung
Erkrankungen des Überempfindlichkeitsreaktionen wie Schwellungen, Ödeme (auch entfernt
Immunsystems von der Injektionsstelle), Erytheme, Pruritus, Hautausschlag (lokal
oder generalisiert) und Atemnot
Skelettmuskulatur-, Muskelatrophie
Bindegewebs- und
Knochenerkrankungen
Allgemeine Erkrankungen Grippeähnliche Symptome
und Beschwerden am
Verabreichungsort
Unerwünschte Wirkungen, welche unter anderen Botulinumtoxin-Präparaten berichtet wurden
Folgende andere unerwünschte Ereignisse wurden nach Verabreichung von herkömmlichen Präparaten mit dem Botulinumtoxin-Typ-A-Komplex berichtet: Anorexie, Schläfrigkeit, Paralyse des Gesichtes, Dysarthrie, Ektropium, Entropium, Photophobie, Keratitis superficialis punctata, Ulcus corneae, Tinnitus, Hypoakusis, Bauchschmerzen, Hyperhidrose, psoriasiformes Exanthem, Erythema multiforme und Radikulopathie.
Selten wurden nach Verabreichung des Botulinumtoxin-Typ-A-Komplexes unerwünschte Wirkungen des kardiovaskulären Systems berichtet, wie Arrhythmie oder Myokardinfarkt, einige davon mit letalem Ausgang. Es ist unklar, ob diese Todesfälle durch den Botulinumtoxin-Typ-A-Komplex oder durch vorbestehende Herz-Kreislauf-Erkrankungen verursacht wurden.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungErhöhte Dosen von Botulinumtoxin Typ A können ausgeprägte und/oder von der Injektionsstelle entfernte neuromuskuläre Lähmungen erzeugen. Mögliche Symptome einer Überdosierung sind allgemeine Schwäche, Ptosis, Diplopie, Atem- und Sprechstörungen, Parese der Atemmuskulatur oder Schluckstörungen. In der Folge kann es auch zu einer Aspirationspneumonie kommen.
Die Symptome treten üblicherweise nicht unmittelbar nach der Injektion auf. Im Falle einer Überdosierung oder einer Ausbreitung des Toxins muss der Patient daher mehrere Tage (bis zu mehreren Wochen) lang medizinisch auf Symptome wie übermässige Muskelschwäche und Muskellähmung überwacht werden. Symptomatische Behandlungen können notwendig sein, ggf. einschliesslich Hospitalisierung. Wenn es zur Paralyse der Atemmuskulatur kommt, können Intubation und assistierte Beatmung erforderlich sein.
Eigenschaften/WirkungenATC-Code
M03AX01
Wirkungsmechanismus
Botulinumtoxin Typ A blockiert die cholinerge Signalübertragung an den neuromuskulären Verbindungen und Speicheldrüsen, indem es die Freisetzung von Acetylcholin hemmt. Die Nervenendigungen der neuromuskulären Verbindungen reagieren nicht länger auf Nervenimpulse, und die Sekretion des Neurotransmitters an den motorischen Endplatten wird verhindert (chemische Denervation). Die vollständige Wiederherstellung der Endplattenfunktion/Impulsübertragung nach einer Injektion erfolgt normalerweise innerhalb von 3-4 Monaten durch neugebildete Nervenendigungen und deren Wiederverbindung mit den motorischen Endplatten.
Der Wirkmechanismus von Botulinumtoxin Typ A an den cholinergen Nervenendigungen kann als vierstufiger, sequenzieller Prozess beschrieben werden, der folgende Schritte umfasst:
-Bindung: Die schwere Kette des Botulinumtoxins Typ A bindet mit aussergewöhnlich hoher Selektivität und Affinität an Rezeptoren, die sich nur an den cholinergen Nervenendigungen befinden.
-Eintritt oder Einschluss (Internalisierung): Einschnürung der Membran der Nervenendigung und Aufnahme des Toxins in die Nervenendigung (Endozytose).
-Translokation: Der aminoterminale Teil der schweren Kette des Neurotoxins bildet eine Pore in der Vesikelmembran, die Disulfidbrücke wird gespalten und die leichte Kette des Neurotoxins gelangt durch die Pore in das Zytosol.
-Wirkung: Nach der Freisetzung der leichten Kette spaltet diese sehr spezifisch ein Zielprotein (SNAP-25), welches für die Freisetzung von Acetylcholin erforderlich ist.
Die inhibitorische Wirkung des Neurotoxins auf die präsynaptische Freisetzung von Acetylcholin ist nachgewiesen. Bei der Speicheldrüse bleibt unklar, ob auch die postsynaptischen Zellen direkt betroffen sind. Nach Neurotoxingabe in die Glandula submandibularis von Ratten wird ein Aquaporin auf der Drüsenzellmembran herunterreguliert, dies kann jedoch auch ein sekundärer Effekt der funktionellen Denervierung sein.
Pharmakodynamik
Siehe Rubrik "Wirkmechanismus" .
Klinische Wirksamkeit
Blepharospasmus
Die Wirksamkeit und Sicherheit von Xeomin (bis zu 50 Einheiten pro Auge) wurden in einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studie an 109 Patienten (Xeomin: n=75; Placebo: n=34) mit bilateralem Blepharospasmus und einem Schweregrad in der Jankovic Rating Scale (JRS) von mindestens 2 Punkten untersucht. 102 Patienten nahmen anschliessend an einer offenen Verlängerungsphase mit bis zu 5 Injektionszyklen teil. Die mittlere Gesamtdosis pro Sitzung betrug 66,9 Einheiten. Während der Verlängerungsphase wurde eine kumulative Dosis von 276,7 Einheiten verabreicht.
Xeomin erzielte einen Behandlungseffekt sowohl nach der ersten Injektion als auch während wiederholter Injektionszyklen. Im Primärendpunkt (Änderung des JRS-Schweregradscores nach 6 Wochen) zeigte Xeomin eine statistisch signifikante Überlegenheit gegenüber Placebo (Xeomin: -0,83 Punkte [1,178]; Placebo: 0,21 Punkte [0,914]; Differenz: -1,0 Punkte; 95 % CI -1,4 bis -0,5; p<0,001). Auch für die Sekundärendpunkte fand sich eine signifikante Überlegenheit von Xeomin gegenüber Placebo zu fast allen Messzeitpunkten. In der Verlängerungsphase resultierte zudem eine signifikante Verbesserung der Symptomatik während wiederholter Injektionszyklen.
In einer doppelblinden Phase-III-Studie wurden die Wirksamkeit und Sicherheit von Xeomin ausserdem im Vergleich zu einem Vergleichsprodukt, welches den herkömmlichen Botulinumtoxin-Typ-A-Komplex Onabotulinumtoxin A (900 kD) enthält, an n=300 Patienten mit Blepharospasmus untersucht. Patienten erhielten einmalig Xeomin oder das Vergleichsprodukt von bis zu 35 Einheiten pro Auge. In beiden Gruppen zeigte sich eine klinisch relevante und statistisch signifikante Reduktion des JRS-Summenscores nach 3 Wochen (Abnahme gegenüber Baseline -2,90 für Xeomin und -2,67 für das Vergleichsprodukt). Die Befunde der übrigen Endpunkte waren hierzu konsistent. Non-Inferiority von Xeomin gegenüber dem Vergleichsprodukt konnte gezeigt werden. Die Wirkung von Xeomin trat im Median 4 Tage post injectionem ein, der Median der Wirkdauer betrug 110 Tage. Die Ergebnisse dieser Vergleichsstudie zeigen bei einem Dosisverhältnis von 1:1 eine Äquipotenz der beiden Präparate bei der Behandlung des Blepharospasmus.
Torticollis spasmodicus
Xeomin wurde in einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studie an 233 Patienten (Xeomin: 240 Einheiten, n=81; 120 Einheiten, n=78; Placebo: n=74) mit zervikaler Dystonie mit überwiegend rotatorischer Komponente (Torticollis spasmodicus) und einem Schweregradscore von mindestens 10 Punkten auf der Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) untersucht; 143 Patienten waren bereits mit herkömmlichem Botulinumtoxin-Typ-A-Komplex vorbehandelt. 214 Patienten nahmen anschliessend an einer doppelblinden Verlängerungsphase mit bis zu 5 Injektionszyklen in 2 Dosisgruppen teil (240 Einheiten, n=111; 120 Einheiten, n=103).
Xeomin erzielte einen Behandlungseffekt sowohl nach der ersten Injektion als auch während der wiederholten Injektionszyklen. Im Primärendpunkt (Änderung des TWSTRS-Gesamtscores 4 Wochen nach Einzelinjektion) zeigten sowohl 240 Einheiten Xeomin (-10,9 Punkte) als auch 120 Einheiten Xeomin (-9,9 Punkte) eine statistisch signifikante Überlegenheit gegenüber Placebo (-2,2 Punkte; Differenz, 240 Einheiten: -9,0 Punkte, 95 % CI -12,0 bis -5,9; 120 Einheiten: -7,5 Punkte, 95 % CI -10,4 bis -4,6; p<0,001). Für vorbehandelte (240 Einheiten: -11,4 Punkte; 120 Einheiten: -8,5 Punkte; Placebo: -2,4 Punkte) und nicht vorbehandelte Patienten (240 Einheiten: -10,0 Punkte; 120 Einheiten: -11,9 Punkte; Placebo: -2,0 Punkte) wurden ähnliche Ergebnisse erzielt. Auch für die meisten Sekundärpunkte fand sich eine signifikante Überlegenheit von Xeomin gegenüber Placebo. In der Verlängerungsphase resultierten beide Dosen zudem in einer deutlichen Verbesserung der Symptomatik während wiederholter Injektionszyklen. Unterschiede zwischen beiden Dosisgruppen waren statistisch nicht signifikant. Die mittleren Gesamtdosen pro Sitzung waren 241,11 Einheiten bzw. 119,58 Einheiten. Während der Verlängerungsphase wurden kumulative Dosen von 917,75 Einheiten (Patienten mit 240 Einheiten) bzw. 423,74 Einheiten (Patienten mit 120 Einheiten) verabreicht.
In einer multizentrischen, prospektiven, randomisierten Phase-III-Studie wurden ausserdem 463 Patienten mit zervikaler Dystonie mit überwiegend rotatorischer Komponente (Torticollis spasmodicus) und einem Schweregradscore von mindestens 10 Punkten auf der TWSTRS entweder mit Xeomin oder einem Vergleichsprodukt, welches den herkömmlichen Botulinumtoxin-Typ-A-Komplex Onabotulinumtoxin A (900 kD) enthält, behandelt. Vier Wochen nach Behandlungsbeginn zeigte sich in beiden Gruppen eine signifikante Abnahme des TWSTRS-Scores gegenüber Baseline (-6,6 für Xeomin und -6,4 für das Vergleichsprodukt). Non-Inferiority von Xeomin gegenüber dem Vergleichsprodukt konnte gezeigt werden. Die Wirkung von Xeomin trat im Median 7 Tage nach der Injektion ein, der Median der Wirkdauer betrug 110 Tage. Die Ergebnisse dieser Vergleichsstudie zeigen bei einem Dosisverhältnis von 1:1 eine Äquipotenz der beiden Präparate bei der Behandlung der zervikalen Dystonie.
Spastik der oberen Extremitäten
Xeomin wurde in einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Phase-III-Studie an 148 Patienten (Xeomin: n=73; Placebo: n=75) mit Spastik der oberen Extremitäten nach Schlaganfall untersucht. 145 Patienten nahmen an einer offenen Verlängerungsphase teil (mit der Möglichkeit einer flexiblen Dosierung). Die kumulative Dosis innerhalb der klinischen Studie betrug nach bis zu 6 Wiederholungsbehandlungen über einen Behandlungszeitraum von bis zu 89 Wochen im Durchschnitt 1333 Einheiten (maximal 2395 Einheiten).
Die Responder-Rate nach der Ashworth-Skala für die Handgelenkbeuger nach 4 Wochen (Primärendpunkt; Response definiert als Verbesserung um mindestens 1 Punkt auf der Ashworth-Skala von 0 bis 4 Punkten) war unter Xeomin (68,5 %) signifikant höher als unter Placebo (37,3 %; p<0,001). Patienten, die mit Xeomin behandelt wurden, hatten eine 3,97-fach höhere Chance, auf die Therapie anzusprechen als Patienten unter Placebo (95 % CI: 1,90 bis 8,30).
Der Wirkungseintritt erfolgte im Median 4 Tage nach der Applikation, die maximale Wirkung auf den Muskeltonus wurde im Median nach 4 Wochen beobachtet, und der Therapieeffekt hielt ungefähr 12 Wochen an.
Eine randomisierte, beobachterblinde, multizentrische Phase-III-Studie untersuchte an n=192 Patienten mit Spastik unterschiedlicher Ätiologie – bei vergleichbarer Dosierung – den Einfluss unterschiedlicher Verdünnungen von Xeomin (20 U/mL, n=97 oder 50 U/mL, n=95) auf die Wirksamkeit.
Im Primärendpunkt (definiert als Verbesserung um mindestens 1 Punkt auf der Disability Assessment Scale 4 Wochen nach Injektion) konnte eine Non-Inferiority der 20 U/mL-Gruppe im Vergleich zur 50 U/mL-Gruppe gezeigt werden. Diese Ergebnisse wurden auch in den meisten anderen Wirksamkeitsparametern bestätigt.
Eine weitere doppelblinde, placebokontrollierte klinische Phase-III-Studie wurde mit 317 vorher unbehandelten Patienten durchgeführt, deren Schlaganfall mindestens 3 Monate zurücklag. In der Hauptphase der Studie wurde eine fixe Gesamtdosis von 400 Einheiten Xeomin intramuskulär in das ausgewählte, primäre, spastische Zielmuster appliziert, darunter die Ellbogenbeugung, Handgelenkbeugung, gefaustete Hand oder andere betroffene Muskelgruppen (n=210). Die konfirmatorische Analyse der primären und co-primären Wirksamkeitsparameter, jeweils 4 Wochen nach Injektion, zeigte statistisch signifikante Verbesserungen in der Responder-Rate der Ashworth-Skala oder in den Veränderungen gegenüber Baseline im Ashworth-Punktwert und dem Gesamteindruck der klinischen Veränderung (Prüfarzturteil).
296 behandelte Patienten beendeten die Hauptstudienphase und nahmen am ersten Injektionszyklus der offenen Verlängerungsphase (OLEX) teil. Während der Verlängerungsphase erhielten die Patienten bis zu drei Injektionen. In jedem Injektionszyklus erhielten die Patienten eine Gesamtdosis von 400 Einheiten Xeomin, flexibel verteilt auf alle betroffenen Muskeln, gefolgt von einer 12-wöchigen Beobachtungsphase. Die Gesamtstudiendauer betrug 48 Wochen.
Spastik der unteren Extremitäten
An der doppelblinden, placebokontrollierten klinischen Phase-III-Studie nahmen insgesamt 219 asiatische (japanische) Patienten mit Spastik der unteren Extremitäten teil, deren Schlaganfall mindestens 6 Monate zurücklag. Die Studie umfasste eine offene Verträglichkeitsphase zu Beginn, eine doppelblinde Hauptphase (MP) und eine offene Verlängerungsphase (OLEX). Während der MP wurde eine fixe Gesamtdosis Xeomin (400 Einheiten) oder Placebo intramuskulär in die Muskeln des Pes equinus verabreicht (M. gastrocnemius (medial und lateral), M. tibialis posterior und falls erforderlich einschliesslich der Zehenmuskel (M. flexor digitorum longus und M. flexor hallucis longus). In der konfirmatorischen Analyse zeigte der primäre Wirksamkeitsparameter (Veränderungen gegenüber Baseline in den Punktwerten auf der Modifizierten Ashworth Skala (MAS) für die Plantarflexoren, berechnet als Fläche unter der Kurve (Area Under the Curve, AUC) bis zum Ende der MP (Woche 12), bei einer Behandlung mit Xeomin eine signifikante Verbesserung der Spastik nach Schlaganfall im Vergleich zur Placebogruppe.
-Primärer Endpunkt: Mittlere Änderung im MAS-Ausgangswert der Plantarflexoren gegenüber Baseline bis zum Ende der MP
NT 201(n=104) Placebo(n=104)
Mittlere Änderung –7.74 –4.76
LS mittlere Änderung –8.40 –5.81
p = 0.0041
LS = Least Square (kleinstes Quadrat)
-Sekundärer Endpunkt: Mittlere Änderung im MAS-Ausgangswert der Plantarflexoren gegenüber Baseline zu den Wochen 4, 6, und 8 der MP
NT 201(N = 104) Placebo(N = 104)
Woche 4 Mittlere Änderung –0.75 –0.48
LS mittlere Änderung –0.81 –0.57
p = 0.0125
Woche 6 Mittlere Änderung –0.85 –0.51
LS mittlere Änderung –0.91 –0.62
p = 0.0042
Woche 8 Mittlere Änderung –0.73 –0.41
LS mittlere Änderung –0.81 –0.52
p = 0.0033
LS = Least Square (kleinstes Quadrat)
Insgesamt 202 Patienten wurden in die OLEX-Phase aufgenommen, in der die Patienten bis zu drei Injektionen erhielten. Jeder OLEX-Zyklus bestand aus einer einzigen Behandlungssitzung (Gesamtdosis von 400 Einheiten Xeomin), gefolgt von einem Beobachtungszeitraum von 10 bis 14 Wochen beim ersten und zweiten OLEX-Zyklus und von 12 Wochen beim dritten OLEX-Zyklus. Die Gesamtdauer der Studie betrug bis zu 52 Wochen. Die Ergebnisse der OLEX-Phase bestätigten die Ergebnisse der MP, indem ein anhaltender Behandlungserfolg gezeigt wurde.
In einer weiteren randomisierten, placebokontrollierten Studie an 290 Patienten mit Spastik der unteren Extremitäten konnte unter der Behandlung mit Xeomin im Vergleich zu Placebo kein statistisch signifikantes Ergebnis in der primären Wirksamkeitsvariable (Änderung gegenüber Baseline in den Punktwerten auf der Ashworth Skala für die Plantarflexoren) gezeigt werden.
Spastik der oberen und unteren Extremitäten
Aus einer unkontrollierten, offenen Phase-III-Studie, an der 155 Patienten mit einer kombinierten Spastik der oberen und unteren Extremitäten teilnahmen, liegen Sicherheitsdaten vor, welche die maximale Gesamtdosis von 600 Einheiten zur kombinierten Behandlung der fokalen Spastik der oberen und unteren Extremität bei Erwachsenen unterstützen.
Chronische Sialorrhoe (Erwachsene)
Die zulassungsrelevante, doppelblinde, Placebo-kontrollierte Phase-III-Studie umfasste insgesamt 184 Patienten, die mindestens drei Monate an Sialorrhoe infolge von Morbus Parkinson, atypischem Parkinson-Syndrom, Schlaganfall oder posttraumatischem Hirnschaden litten. Während der Hauptperiode (MP) wurde eine fixe Gesamtdosis von Xeomin (100 oder 75 Einheiten) oder Placebo intraglandulär bis zu einem definierten Dosisverhältnis von 3:2 in die Glandula parotidea bzw. Glandula submandibularis verabreicht. Die bestätigende Analyse der primären Wirksamkeitsvariablen (nicht stimulierte Speichelflussrate (uSFR) und Skala zur Einschätzung der Veränderung des klinischen Gesamtzustands (GICS) in einer Range von +3 bis -3 in Woche 4 nach der Injektion) zeigte statistisch signifikante Verbesserung der Behandlungsgruppe mit 100 Einheiten im Vergleich zu Placebo (Reduktion der uSFR um 90 Mikrogramm/Minute, sowie Zunahme des GICS um 0.58 Einheiten). Die Behandlungsgruppe mit 75 Einheiten zeigte eine Reduktion der uSFR von 20 Mikrogramm/Minute, sowie eine Zunahme des GICS von 0.35 Einheiten gegenüber Placebo, allerdings ohne statistische Signifikanz. Statistisch signifikante Verbesserungen der Wirksamkeitsparameter der uSFR und des GICS in Woche 8 und 12 nach der Injektion konnten in beiden aktiven Behandlungsgruppen gezeigt werden. Am letzten Beobachtungspunkt der Hauptperiode in Woche 16 war die Wirksamkeit deutlich geringer, respektive statistisch nicht mehr signifikant.
173 behandelte Patienten beendeten die Hauptperiode und nahmen am ersten Zyklus der Erweiterungsperiode (EP) teil. Die EP bestand aus drei verblindeten Dosiszyklen, mit jeweils einer einzigen Behandlungssitzung (100 oder 75 Einheiten der Xeomin Gesamtdosis, mit dem gleichen Dosisverhältnis wie in der MP), gefolgt von einer 16-wöchigen Beobachtungsperiode. 151 Patienten haben die EP abgeschlossen. Die Ergebnisse des EP bestätigten die Ergebnisse der MP, welche einen anhaltenden Behandlungsvorteil zeigte.
Chronische Sialorrhoe (Kinder/Jugendliche)
In einer doppelblinden, Placebo-kontrollierten klinischen Phase-III-Studie wurden insgesamt 255 Kinder und Jugendliche (im Alter von 2 bis 17 Jahren) mit einem Körpergewicht (KG) von mindestens 12 kg, die an chronischer Sialorrhoe im Zusammenhang mit neurologischen Erkrankungen und/oder Intelligenzminderung litten, behandelt. Während der Hauptphase erhielten 220 Patienten im Alter von 6 bis 17 Jahren eine Behandlung mit Xeomin entsprechend der Körpergewichtsklasse und bis zu 75 Einheiten oder Placebo. Die Behandlung erfolgte ultraschallgeführt intraglandulär mit einem definierten Dosisverhältnis von 3:2 in die Glandula parotidea und die Glandula submandibularis.
Die konfirmatorische Analyse der coprimären Wirksamkeitsvariablen (uSFR und GICS in Woche 4 nach der Injektion) zeigte statistisch signifikante und klinisch relevante Verbesserungen in der Xeomin -Gruppe im Vergleich zu Placebo. Für beide Wirksamkeitsparameter wurden bis zum Ende der Hauptphase in Woche 16 statistisch signifikante Unterschiede zwischen den Behandlungsgruppen beobachtet.
Alle 35 Kinder im Alter von 2 bis 5 Jahren wurden entsprechend ihrer Körpergewichtsklasse mit Xeomin behandelt wobei kein Placebo-Arm als Kontrolle verwendet wurde. Die untersuchten Wirksamkeitsvariablen zeigten eine ähnliche Verbesserung wie in der Xeomin-Behandlungsgruppe der 6 bis 17-Jährigen.
Am anschliessenden ersten Zyklus der offenen Verlängerungsphase nahmen 247 Patienten teil. Die Verlängerungsphase bestand aus drei zusätzlichen Zyklen mit jeweils einer einzelnen Behandlungssitzung, gefolgt von einer 16wöchigen Beobachtungsphase. Alle Patienten erhielten Xeomin nach dem gleichen vorgegebenen Dosierungsschema und dem gleichen Dosierungsverhältnis wie in der Hauptphase. Insgesamt 222 Patienten beendeten die Verlängerungsphase. Die Ergebnisse der offenen Verlängerungsphase bestätigten die Ergebnisse der Hauptphase und zeigten einen anhaltenden Behandlungsnutzen. Es wurden keine neuen oder unerwarteten Sicherheitsbedenken festgestellt.
PharmakokinetikAbsorption
Klassische Kinetik- und Verteilungsstudien können mit Botulinumtoxin Typ A nicht durchgeführt werden, da der Wirkstoff in äusserst geringen Dosen (Picogramm pro Injektion) appliziert wird und schnell und irreversibel an die cholinergen Nervenendigungen bindet.
Natives Botulinumtoxin vom Serotyp A stellt einen hochmolekularen Komplex dar (Botulinumtoxin-Komplex Typ A), der zusätzlich zu dem Neurotoxin (150 kD) auch andere nicht-toxische Proteine wie Hämagglutinine und Non-Hämagglutinine enthält. Im Gegensatz zu dem Botulinumtoxin-Typ-A-Komplex enthält Xeomin das reine Neurotoxin und ist frei von Komplexproteinen.
Wie für viele andere Proteine wurde für Botulinumtoxin Typ A gezeigt, dass es nach intramuskulärer Injektion einem retrograden axonalen Transport unterliegt. Eine retrograde transsynaptische Passage des aktiven Botulinum Neurotoxins Typ A in das zentrale Nervensystem wurde dagegen nicht gefunden.
Rezeptor-gebundenes Botulinumtoxin Typ A wird durch Endozytose in die Nervenendigung aufgenommen, bevor es sein Ziel (SNAP-25) erreicht. Es wird schliesslich intrazellulär abgebaut. Frei zirkulierende Botulinumtoxin Typ A-Moleküle, die nicht an präsynaptische Rezeptoren auf den cholinergen Nervenendigungen gebunden haben, werden durch Phagozytose oder Pinocytose aufgenommen und wie andere frei zirkulierende Proteine abgebaut.
Distribution
Siehe Rubrik "Absorption" .
Metabolismus
Siehe Rubrik "Absorption" .
Elimination
Siehe Rubrik "Absorption" .
Präklinische DatenSicherheitspharmakologie
Präklinische, sicherheitspharmakologische Studien zur Untersuchung der Einflüsse von Xeomin auf das kardiovaskuläre System zeigten kein erhöhtes Risiko für den Menschen.
Mutagenität und Karzinogenität
Es wurden keine Studien zum genotoxischen oder kanzerogenen Potential von Xeomin durchgeführt.
Reproduktionstoxizität
Reproduktionstoxikologische Studien mit Xeomin zeigten weder eine Beeinträchtigung der männlichen oder weiblichen Fertilität bei Kaninchen noch eine direkte Beeinflussung der embryofötalen oder der prä- und postnatalen Entwicklung bei Ratten und/oder Kaninchen. Allerdings führte die Gabe maternal-toxischer Dosen von Xeomin, welche eine maternale Körpergewichtsreduktion verursachen, zu einer erhöhten Abortrate in einer Embryotoxizitätsstudie bei Kaninchen und zu leicht reduzierten Geburtsgewichten in einer Embryotoxizitätsstudie in Ratten.
Weitere Daten (Lokale Toxizität, Phototoxizität, Immunotoxizität)
Die Befunde in Studien zur systemischen Toxizität von Xeomin nach wiederholter intramuskulärer Gabe waren überwiegend eine Folge der pharmakodynamischen Eigenschaften.
In einer Untersuchung der chronischen Toxizität an Ratten wurde Xeomin in die Speicheldrüse injiziert. Das Gewicht der injizierten Glandula submandibularis war bei allen Dosierungen reduziert, und eine Atrophie des Speicheldrüsenazinus wurde bei der höchsten Dosis von 40 U/kg bei einigen Tieren beobachtet. Bei den niedrigeren Dosen von 2 und 10 U/kg trat keine Atrophie auf. Dementsprechend konnte ein NOAEL von 10 U/kg bestimmt werden, welcher 6-mal höher als die maximale klinische Dosierung ist (100 U/Patient entsprechen 1,67 U/kg für einen Patienten von 60 kg).
Es wurden keine Hinweise auf lokale Unverträglichkeit beobachtet.
Dementsprechend waren die Sicherheitsmargen in Bezug auf die klinische Therapie im Allgemeinen in Bezug auf hohe klinische Dosen gering.
Sonstige HinweiseInkompatibilitäten
Dieses Arzneimittel darf, ausser mit den unter "Hinweise für die Handhabung" aufgeführten, nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Die chemische und physikalische Stabilität der rekonstituierten/verdünnten Lösung wurde für einen Zeitraum von 24 Stunden bei 2 bis 8°C gezeigt.
Aus mikrobiologischer Sicht ist die verdünnte Lösung sofort zu verwenden, es sei denn, die Rekonstitution/Verdünnung ist unter kontrollierten und validierten aseptischen Bedingungen erfolgt.
Wird die Lösung nicht sofort verwendet, so liegen die Dauer der Lagerung und die Bedingungen in der Verantwortung des Anwenders.
Besondere Lagerungshinweise
Nicht über 25°C lagern. In der Originalverpackung aufbewahren. Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Xeomin wird vor der Anwendung mit steriler, physiologischer Kochsalzlösung (ohne Konservierungsmittel) rekonstituiert. Die Rekonstitution und die Verdünnung sollten unter Einhaltung der Standardbedingungen erfolgen, insbesondere im Hinblick auf die aseptische Handhabung.
Das Rekonstituieren des Flascheninhalts und das Aufziehen der Spritze sollten über plastikbeschichteten Papiertüchern erfolgen, um eventuelle Spritzer aufzufangen.
Eine entsprechende Menge Natriumchlorid-Lösung (siehe Verdünnungstabelle) wird mit einer Spritze aufgezogen. Es wird empfohlen, eine Kurzschliffkanüle (20-27 G) für die Rekonstitution zu verwenden. Der freigelegte Teil des Gummistopfens der Durchstechflasche wird vor dem Einstechen der Nadel mit Alkohol (70 %) gereinigt. Nach dem senkrechten Einstechen der Nadel durch den Gummistopfen sollte die Rekonstitution vorsichtig erfolgen, um eine Schaumbildung zu vermeiden. Die Durchstechflasche ist zu verwerfen, wenn in der Flasche kein Unterdruck vorhanden ist, der das Lösungsmittel aus der Spritze ansaugt. Die Spritze wird von der Flasche entfernt und Xeomin mit dem Lösungsmittel vorsichtig gemischt, indem die Flasche geschwenkt und gedreht wird – nicht stark schütteln. Falls erforderlich, sollte die zur Rekonstitution verwendete Kanüle in der Durchstechflasche verbleiben. Die benötigte Menge an Lösung sollte mit einer neuen sterilen, für die Injektion geeigneten Spritze aufgezogen werden.
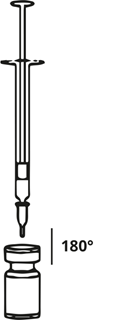

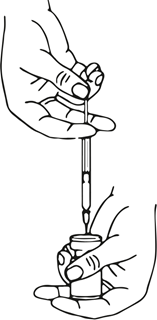
Die rekonstituierte Xeomin-Lösung ist klar, farblos und frei von Partikeln.
Xeomin darf nicht verwendet werden, wenn die rekonstituierte Lösung, die entsprechend der oben genannten Anweisungen hergestellt wurde, eine Trübung zeigt oder Ausflockungen oder Partikel enthält.
Das korrekte Verdünnungsvolumen muss sorgfältig ausgewählt werden, um eine versehentliche Überdosierung zu vermeiden. Wenn verschiedene Grössen von Xeomin-Durchstechflaschen während einer Behandlungssitzung angewendet werden, muss darauf geachtet werden, die korrekte Menge an Lösungsmittel zur Auflösung einer bestimmten Anzahl von Einheiten pro 0,1 ml zu verwenden. Die zugegebene Menge Lösungsmittel ist für Xeomin 50 Einheiten, Xeomin 100 Einheiten und Xeomin 200 Einheiten unterschiedlich und jede Spritze ist entsprechend zu kennzeichnen.
Die folgende Tabelle gibt mögliche Verdünnungen für Xeomin 50 Einheiten, Xeomin 100 Einheiten und Xeomin 200 Einheiten an:
Erhaltene Dosis (in Zugegebene Menge
Einheiten pro 0,1 Lösungsmittel
ml) (Natriumchlorid 9
mg/ml (0,9 %)
Injektionslösung)
Durchstechflasche Durchstechflasche Durchstechflasche
mit 50 Einheiten mit 100 Einheiten mit 200 Einheiten
20,0 Einheiten 0,25 ml 0,5 ml 1,0 ml
10,0 Einheiten 0,5 ml 1,0 ml 2,0 ml
8,0 Einheiten 0,625 ml 1,25 ml 2,5 ml
5,0 Einheiten 1,0 ml 2,0 ml 4,0 ml
4,0 Einheiten 1,25 ml 2,5 ml 5,0 ml
2,5 Einheiten 2,0 ml 4,0 ml Nicht zutreffend
2,0 Einheiten 2,5 ml 5,0 ml Nicht zutreffend
1,25 Einheiten 4,0 ml Nicht zutreffend Nicht zutreffend
Injektionslösung, die länger als 24 Stunden aufbewahrt wurde, sollte verworfen werden.
VORGEHENSWEISE ZUR SICHEREN ENTSORGUNG DER DURCHSTECHFLASCHEN, SPRITZEN UND VERWENDETEN MATERIALIEN
Nicht verwendete Durchstechflaschen sowie in der Durchstechflasche oder Spritze verbliebene rekonstituierte Lösung kann durch Zusatz einer der folgenden Lösungsmittel inaktiviert werden: 70%iges Ethanol, 50%iges Isopropanol, verdünnte Natriumhydroxid-Lösung (0,1 N NaOH) oder verdünnte Natriumhypochlorit-Lösung (mindestens 0,1%ige NaOCl).
Verwendete Durchstechflaschen, Spritzen und Materialien sollten nicht entleert, sondern in geeigneten Behältern und entsprechend den lokalen Vorschriften entsorgt werden.
-Jegliches verschüttetes Arzneimittel muss aufgewischt werden: entweder – im Fall des Pulvers – mit einem saugfähigen Material, das mit einem der oben genannten Lösungsmittel getränkt wurde, oder – im Fall der rekonstituierten Lösung – mit einem trockenen, saugfähigen Material.
-Kontaminierte Oberflächen müssen mit einem saugfähigen Material gereinigt werden, das mit einer der oben genannten Lösungen getränkt wurde. Anschliessend trocknen lassen.
-Wenn eine Durchstechflasche zerbricht, wie oben beschrieben vorgehen: Glassplitter vorsichtig aufsammeln und das Arzneimittel aufwischen, dabei Schnittverletzungen der Haut vermeiden.
-Wenn das Arzneimittel in Kontakt mit der Haut gelangt, den betroffenen Bereich mit reichlich Wasser abspülen.
-Wenn das Arzneimittel in Kontakt mit den Augen gelangt, gründlich mit reichlich Wasser oder einer Augenspüllösung ausspülen.
-Wenn das Arzneimittel in Kontakt mit einer Wunde, Schnittverletzung oder nicht-intakter Haut gelangt, gründlich mit reichlich Wasser spülen und die entsprechenden medizinischen Massnahmen entsprechend der injizierten Dosis ergreifen.
Zulassungsnummer62080 (Swissmedic)
PackungenXeomin Lyophilisat:
Packungen zu 1 Durchstechflasche à 50 Einheiten (A)
Packungen zu 1 Durchstechflasche à 100 Einheiten (A)
Packungen zu 1 Durchstechflasche à 200 Einheiten (A)
ZulassungsinhaberinMerz Pharma (Schweiz) AG,4123 Allschwil
Stand der InformationMärz 2026
|