ZusammensetzungWirkstoffe
Ixekizumab (aus gentechnologisch hergestellten CHO (Chinese Hamster Ovary)-Zellen).
Hilfsstoffe
Saccharose, Polysorbat 80, Wasser für Injektionszwecke. q.s. ad solutionem pro 0.5 ml oder 1 ml. Natriumhydroxid kann zur pH-Wert-Einstellung zugesetzt worden sein, corresp. Natrium approx. 0.0505 mg bzw. 0.1010 mg.
Darreichungsform und Wirkstoffmenge pro EinheitTaltz 80 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 80 mg Ixekizumab in 1 ml (80 mg/ml).
Taltz 80 mg Injektionslösung in einem Fertigpen
Jeder Fertigpen enthält 80 mg Ixekizumab in 1 ml (80 mg/ml).
Taltz 40 mg Injektionslösung in einer Fertigspritze
Jede Fertigspritze enthält 40 mg Ixekizumab in 0.5 ml (80 mg/ml).
Indikationen/AnwendungsmöglichkeitenPlaque-Psoriasis
Taltz ist zur Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis indiziert, die auf andere systemische Therapien (einschliesslich Ciclosporin oder Methotrexat oder PUVA) nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Plaque-Psoriasis bei Kindern und Jugendlichen
Taltz ist zur Behandlung der mittelschweren bis schweren Plaque-Psoriasis bei Kindern und Jugendlichen ab 6 Jahren mit einem Körpergewicht von mindestens 25 kg indiziert, die auf andere systemische Therapien (einschliesslich Ciclosporin oder Methotrexat oder PUVA) nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Psoriasis-Arthritis
Taltz, alleine oder in Kombination mit konventionellen krankheitsmodifizierenden Antirheumatika (DMARD, disease-modifying anti-rheumatic drugs), ist zur Behandlung erwachsener Patienten mit aktiver Psoriasis-Arthritis indiziert, die auf eine Behandlung mit einem oder mehreren DMARDs unzureichend angesprochen haben oder diese nicht vertragen haben.
Axiale Spondyloarthritis
• Ankylosierende Spondylitis (Morbus Bechterew)
Taltz ist indiziert zur Behandlung der schweren aktiven ankylosierenden Spondylitis bei erwachsenen Patienten, die auf eine konventionelle Therapie (z.B. nichtsteroidale entzündungshemmende Medikamente [NSAIDs]) unzureichend angesprochen haben oder diese nicht vertragen.
• Nicht-radiographische axiale Spondyloarthritis
Taltz ist indiziert zur Behandlung der schweren aktiven nicht-radiographischen axialen Spondyloarthritis bei erwachsenen Patienten, die auf NSAIDs unzureichend angesprochen haben. Patienten sollten objektive Anzeichen einer Entzündung im Magnetresonanztomographie (MRT) und durch erhöhtes C-reaktives Protein (CRP) zeigen.
Dosierung/AnwendungDie Anwendung von Taltz sollte unter Anleitung und Aufsicht eines in Diagnose und Behandlung der für Taltz indizierten Erkrankungen, erfahrenen Arztes erfolgen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Plaque-Psoriasis bei Erwachsenen
Die empfohlene Dosis beträgt 160 mg als subkutane Injektion (zwei 80 mg Injektionen) in Woche 0, gefolgt von 80 mg (eine Injektion) in den Wochen 2, 4, 6, 8, 10 und 12, und danach 80 mg (eine Injektion) alle 4 Wochen.
Bei Patienten <100 kg kann ein alternatives Dosisschema mit 160 mg in Woche 0 und ab Woche 2 80 mg alle 4 Wochen erwogen werden (siehe "Eigenschaften/Wirkungen" ).
Plaque-Psoriasis bei Kindern und Jugendlichen (ab 6 Jahren)
Die empfohlene Dosis für die subkutane Injektion bei pädiatrischen Patienten ab 6 Jahren mit einem Körpergewicht von mindestens 25 kg basiert auf den folgenden Gewichtsklassen:
Gewicht des pädiatrischen Einmalige Initialdosis Dosis ab Woche 4 und danach alle 4
Patienten (Woche 0) Wochen
Über 50 kg 160 mg (zwei Injektionen zu 80 mg (eine Injektion zu 80 mg)
80 mg)
25 bis 50 kg 80 mg (eine Injektion zu 80 40 mg
mg)
Bei pädiatrischen Patienten, bei denen 80 mg verschrieben werden, kann die komplette Dosis direkt aus der 80 mg Fertigspritze oder aus dem Fertigpen verwendet werden.
Bei pädiatrischen Patienten, bei denen 40 mg verschrieben werden, kann die komplette Dosis direkt aus der 40 mg Fertigspritze verwendet werden, oder eine 40 mg Dosis aus einer 80 mg Fertigspritze hergestellt werden. Anweisungen zur Vorbereitung einer Injektionsspritze mit Taltz 40 mg Dosen aus einer Fertigspritze mit 80 mg finden Sie unter "Sonstige Hinweise / Hinweise für die Handhabung" .
Die Anwendung von Taltz wird bei Kindern mit einem Körpergewicht unter 25 kg nicht empfohlen.
Psoriasis-Arthritis
Die empfohlene Dosis beträgt 160 mg als subkutane Injektion (zwei 80 mg Injektionen) in Woche 0, gefolgt von 80 mg (eine Injektion) alle 4 Wochen.
Für Patienten mit Psoriasis-Arthritis und begleitender mittelschwerer bis schwerer Plaque-Psoriasis sind die Dosierungsempfehlungen dieselben wie für die Plaque-Psoriasis.
Axiale Spondyloarthritis
Die empfohlene Dosis beträgt 80 mg (eine Injektion) als subkutane Injektion alle 4 Wochen.
Bei der axialen Spondyloarthritis können konventionelle DMARDs (z.B. Sulfasalazin), Kortikosteroide, NSAIDs und/oder Analgetika während der Behandlung mit Taltz angewendet werden.
Therapiedauer
In allen Indikationen (Plaque-Psoriasis, Psoriasis-Arthritis, axiale Spondyloarthritis), sollte bei Patienten, die nach 16 bis 20 Wochen Therapie kein Ansprechen gezeigt haben, ein Abbruch der Therapie in Erwägung gezogen werden. Einige Patienten, die initial ein teilweises Ansprechen gezeigt haben, können später, bei Fortsetzung der Therapie über 20 Wochen hinaus, eine Besserung zeigen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Taltz wurde in dieser Patientenpopulation nicht untersucht.
Patienten mit Nierenfunktionsstörungen
Taltz wurde in dieser Patientenpopulation nicht untersucht.
Ältere Patienten
Von den 7088 Patienten mit Plaque Psoriasis, die in klinischen Entwicklungsstudien Taltz erhalten haben, waren insgesamt 493 Patienten 65 Jahre alt oder älter und 74 Patienten 75 Jahre alt oder älter. Von den 1401 Patienten mit Psoriasis-Arthritis, die in klinischen Studien Taltz erhalten haben, waren insgesamt 132 Patienten 65 Jahre alt oder älter und 10 Patienten 75 Jahre alt oder älter. Dosisanpassungen sind nicht erforderlich (siehe "Pharmakokinetik" ). Von den 932 Patienten mit axialer Spondyloarthritis, die in klinischen Studien mit Taltz behandelt wurden, waren insgesamt 40 Patienten 65 Jahre oder älter und 4 Patienten 75 Jahre oder älter.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Taltz bei Kindern unter 6 Jahren bei der Behandlung von Plaque-Psoriasis wurden bisher nicht belegt. Die Erfahrung bei Kindern und Jugendlichen ist limitiert. Die Wirksamkeit und Sicherheit bei der Anwendung über ein Jahr hinaus sind ungenügend belegt. Die Sicherheit und Wirksamkeit von Taltz bei Kindern und Jugendlichen unter 18 Jahren bei der Behandlung von Psoriasis-Arthritis und axiale Spondyloarthritis wurden bisher nicht belegt.
Art der Anwendung
Vor Therapie-Beginn muss der Arzt resp. die Ärztin Folgendes sicherstellen:
Der Patient, bzw. bei Kindern und Jugendlichen die erziehungsberechtigte Person versteht, dass Taltz eine neuartige Therapie mit limitierter Erfahrung und unbekannten Langzeitrisiken darstellt.
Taltz wird subkutan alternierend am Oberarm, Bauch und Oberschenkel injiziert.
Von Psoriasis befallene Hautbereiche sollen als Injektionsstellen vermieden werden.
Nach sachgemässer Schulung zur subkutanen Injektionstechnik können sich erwachsene Patienten Taltz selbst injizieren, wenn der behandelnde Arzt dies für angebracht hält. Umfangreiche Anweisungen zur Anwendung sind in der Bedienungsanleitung der Spritze und des Pens beschrieben.
Plaque-Psoriasis bei Kindern und Jugendlichen:
Bei pädiatrischen Patienten können Pflegepersonen nach der Schulung zur subkutanen Injektionstechnik Injektionen mit den Fertigpens oder Fertigspritzen verabreichen.
Bei pädiatrischen Patienten von 25 kg bis zu 50 kg, wenn 40 mg Fertigspritzen nicht zur Verfügung stehen, müssen Ixekizumab-Dosen von 40 mg von einer qualifizierten medizinischen Fachperson vorbereitet und verabreicht werden. Verwenden Sie nur die im Handel erhältliche Taltz 80 mg/1 ml-Fertigspritze, wenn Sie die vorgeschriebenen pädiatrischen Dosen von 40 mg vorbereiten (siehe "Sonstige Hinweise / Hinweise für die Handhabung" ).
Nach der Verabreichung von Taltz sind Kinder und Jugendliche über einen angemessenen Zeitraum medizinisch nachzubeobachten.
KontraindikationenSchwere Überempfindlichkeit gegen den Wirkstoff oder einen der Hilfsstoffe.
Schwere aktive Infektionen (z.B. aktive Tuberkulose, Sepsis, schwere opportunistische Infektionen).
Warnhinweise und VorsichtsmassnahmenInfektionen
Die Therapie mit Taltz ist mit einer dosisabhängigen erhöhten Infektionsrate wie Infektionen der oberen Atemwege, orale Candidiasis, Konjunktivitis oder Tinea-Infektionen assoziiert (siehe "Unerwünschte Wirkungen" ). Unter immunomodulatorischer Therapie kann es zur Reaktivierung latenter Infektionen kommen.
Bei Patienten mit chronischer oder aktiver Infektion oder rezidivierenden Infektionen in der Vorgeschichte muss Taltz mit Vorsicht angewendet werden. Taltz sollte nicht an Patienten mit aktiver Infektion, insbesondere HIV-, HBV- oder HCV-Infektionen, verabreicht werden. Wenn sich unter Therapie mit Taltz eine solche Infektion entwickelt, ist eine sorgfältige Überwachung erforderlich. Die Patienten sollten angewiesen werden, bei Auftreten von Anzeichen oder Symptomen einer Infektion ärztlichen Rat einzuholen. Die Therapie mit Taltz muss bei Nicht-Ansprechen einer antiinfektiösen Standardtherapie sowie bei schwerwiegenden Infekten beendet werden. Die Therapie mit Taltz ist erst fortzusetzen, wenn die Infektion abgeklungen ist.
Patienten mit aktiver Tuberkulose dürfen Taltz nicht erhalten. Bei Patienten mit latenter Tuberkulose sollte vor dem Beginn der Taltz Therapie eine antituberkulöse Therapie erwogen werden.
Maligne Erkrankungen
Daten aus klinischen Studien über 3 Jahren bei Erwachsenen zeigten kein erhöhtes Risiko für maligne Erkrankungen. Weitere Untersuchungen zur Langzeitsicherheit sind noch nicht abgeschlossen. Da es sich bei Psoriasis-Patienten um eine Risikopopulation handelt, sollten Psoriasis-Patienten vor und unter der Behandlung mit Taltz auf das Vorliegen von Hauttumoren untersucht werden.
Schwerwiegende unerwünschte kardiovaskuläre Ereignisse (MACE)
Bei Patienten mit axialer Spondyloarthritis können MACE auftreten. Zur Vorsicht wird geraten bei Patienten mit axialer Spondyloarthritis und Risiko für MACE. In klinischen Studien wurden bei Patienten mit axialer Spondyloarthritis unter Langzeitbehandlung mit Ixekizumab MACE einschliesslich Myokardinfarkt (IR=0.3 auf 100 Patientenjahre mit Exposition) berichtet. Bei allen Patienten bestand eine relevante medizinische Vorgeschichte mit kardiovaskulären Begleiterkrankungen oder Risikofaktoren.
Überempfindlichkeitsreaktionen
Schwerwiegende Überempfindlichkeitsreaktionen, einschliesslich Fälle von Anaphylaxie, Angioödem und Urtikaria wurden berichtet. Wenn eine schwerwiegende Überempfindlichkeitsreaktion auftritt, muss die Anwendung von Taltz umgehend abgebrochen und eine geeignete Therapie eingeleitet werden.
Schwere ekzematöse Ausschläge
Nach der Markteinführung wurden bei Patienten, die Taltz erhielten, Fälle von schweren ekzematösen Ausschlägen, einschliesslich atopischer Dermatitis-ähnlicher Ausschläge, dyshidrotischem Ekzem und Erythrodermie, gemeldet; einige Fälle führten zur Hospitalisierung. Der Beginn der ekzematösen Ausschläge variierte und lag zwischen Tagen und Monaten nach der ersten Dosis Taltz. Um den ekzematösen Ausschlag zu beheben, muss die Behandlung möglicherweise unterbrochen werden. Einige Patienten mit begrenzten Behandlungsmöglichkeiten für Psoriasis wurden erfolgreich gegen Ekzeme behandelt, während sie Taltz weiter einnahmen.
Depression
Depression ist eine bekannte Komorbidität bei Patienten mit ankylosierender Spondylitis und Plaque-Psoriasis (einschliesslich pädiatrischer Plaque-Psoriasis). Bei Patienten mit einem Risiko für Depressionen ist Vorsicht geboten. Suizidalität und selbstverletzendes Verhalten sind in den kontrollierten klinischen Studien bei Erwachsenen mit Plaque-Psoriasis (IR = 0.4 pro 100 PYE) und ankylosierender Spondylitis (IR = 0.6 pro 100 PYE) aufgetreten. Alle Patienten hatten in der Anamnese eine signifikante Vorgeschichte mit psychiatrischen Komorbiditäten oder Risikofaktoren für diese Ereignisse.
Entzündliche Darmerkrankungen, einschliesslich Morbus Crohn und Colitis ulzerosa
Es wurden Fälle einer Neuerkrankung oder einer Exazerbation von entzündlichen Darmerkrankungen mit Taltz berichtet (siehe "Unerwünschte Wirkungen" ). Taltz wird für Patienten mit einer entzündlichen Darmerkrankung nicht empfohlen. Wenn ein Patient Anzeichen und Symptome einer entzündlichen Darmerkrankung oder eine Exazerbation einer bereits existierenden entzündlichen Darmerkrankung entwickelt, soll Taltz abgesetzt und eine angemessene medizinische Behandlung eingeleitet werden
Impfungen
Taltz darf nicht zusammen mit Lebendimpfstoff verwendet werden, da keine Daten zu Impfungen mit Lebendimpfstoffen unter Taltz verfügbar sind.
Es wird empfohlen, geplante Impfungen vor Beginn der Therapie mit Taltz abzuschliessen. Dies ist insbesondere bei der Anwendung in der pädiatrischen Population zu beachten. Der zeitliche Abstand zwischen Impfungen mit Lebendimpfstoffen und dem Beginn der Therapie gemäss den aktuellen Impfrichtlinien zu immunsuppressiven Wirkstoffen ist einzuhalten.
In einer offenen Studie an 83 gesunden Probanden mit 1:1 Randomisierung nach einer Kontrollgruppe oder Ixekizumab 160 mg in Woche 0 und 80 mg in Woche 2 wurde die Immunantwort auf zwei inaktivierte Impfstoffe gegen Tetanus (Boostrix) bzw. Pneumokokken (Pneumovax-23) untersucht, die in Woche 2 verabreicht wurden. Bis Woche 6 nach Immunisierung wurden in diesem kleinen Kollektiv gesunder Probanden zwar grundsätzlich keine Sicherheitsbedenken festgestellt. Die Extrapolation der Wirksamkeit und Sicherheit inaktivierter Impfungen auf die Zielpopulation ist jedoch nur beschränkt möglich. Die Daten zur Immunantwort waren nicht ausreichend, um auf eine adäquate Immunantwort auf diese Impfstoffe nach der Verabreichung von Taltz in der Zielpopulation schliessen zu können.
Die Impfantwort bei Kindern und Jugendlichen wurde nicht untersucht.
Kombination mit anderen Biologika
Die gleichzeitige Verabreichung von Taltz mit anderen Biologika wurde nicht untersucht und wird nicht empfohlen.
Weitere Hinweise
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 80 mg Dosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenIn Plaque-Psoriasis Studien wurde die Sicherheit von Taltz in Kombination mit anderen Immunmodulatoren oder Phototherapie sowie Impfungen mit Lebendimpfstoffen nicht untersucht.
Cytochrom P450 Substrate
Ergebnisse einer Arzneimittelinteraktionsstudie bei Patienten mit mittelschwerer bis schwerer Psoriasis zeigten, dass die Verabreichung von Ixekizumab mit Arzneimitteln, die durch CYP3A4 (untersucht: Midazolam), CYP2C9 (untersucht: Warfarin), CYP2C19 (untersucht: Omeprazol), CYP1A2 (untersucht: Koffein) oder CYP2D6 (untersucht: Dextromethorphan) metabolisiert werden, keinen klinisch signifikanten Einfluss auf die Pharmakokinetik dieser Arzneimittel hat.
Bei der Verabreichung von Taltz in Kombination mit Methotrexat (MTX), und/oder Kortikosteroiden bei Patienten mit Psoriasis-Arthritis hatten diese Medikamente keinen Einfluss auf die Pharmakokinetik von Taltz.
Die Clearance von Ixekizumab wurde durch die gleichzeitige Gabe von oralen Kortikosteroiden, NSAIDs oder konventionellen DMARDs (z.B. Sulfasalazin und Methotrexat) bei Patienten mit axialer Spondyloarthritis nicht beeinflusst.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt begrenzte Daten zur Anwendung von Ixekizumab bei schwangeren Frauen. Es ist bekannt, dass humane IgGs die Plazentaschranke überwinden, und Ixekizumab ist ein IgG. Daher besteht die Möglichkeit, dass Ixekizumab von der Mutter auf den Fötus übergeht. Während der Schwangerschaft und bei gebärfähigen Frauen, die keine wirksame Empfängnisverhütung verwenden, darf Taltz nicht verabreicht werden, es sei denn, dies ist eindeutig erforderlich. Patientinnen sollen angewiesen werden für mindestens 10 Wochen nach der letzten Gabe von Taltz wirksame Verhütungsmethoden anzuwenden.
Tierexperimentelle Studien lassen nicht eindeutig auf direkte oder indirekte schädigende Wirkungen auf Schwangerschaft, embryonale/fetale Entwicklung, Geburt oder postnatale Entwicklung schliessen (siehe "Präklinische Daten" ). Da sich anhand tierexperimenteller Reproduktionsstudien nicht immer die Reaktion beim Menschen vorhersagen lässt, sollte Taltz nur dann während einer Schwangerschaft angewendet werden, wenn der Nutzen gegenüber den möglichen Risiken eindeutig überwiegt.
Stillzeit
Es ist nicht bekannt, ob Ixekizumab beim Menschen in die Muttermilch übertritt oder nach oraler Aufnahme systemisch resorbiert wird. Allerdings geht Ixekizumab bei Cynomolgus Affen in geringen Mengen in die Muttermilch über. Da viele Medikamente, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Wegen möglichen Schaden für den gestillten Säugling wird es empfohlen während der Behandlung mit Taltz und während mindestens 10 Wochen nach der letzten Dosis nicht zu stillen. Eine Entscheidung, entweder mit dem Stillen aufzuhören oder die Behandlung mit Taltz abzubrechen sollte unter Berücksichtigung der Vorteile des Stillens für das Kind und der Vorteile der Therapie für die Mutter, gefällt werden.
Fertilität
Die Wirkung von Ixekizumab auf die humane Fertilität wurde nicht untersucht. Tierstudien weisen nicht auf direkte oder indirekte schädliche Wirkungen auf die Fertilität hin (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDer Einfluss von Taltz auf die Fahrtüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen wurde nicht untersucht.
Unerwünschte WirkungenInsgesamt wurden 10'117 Patienten in verblindeten und offenen klinischen Studien zu Plaque-Psoriasis, Psoriasis-Arthritis, axialer Spondyloarthritis und anderen Indikationen mit Taltz behandelt. Insgesamt wurden 9421 Patienten in Plaque-Psoriasis (7088 Patienten), Psoriasis-Arthritis (1401 Patienten) und axiale Spondyloarthritis (932 Patienten) Indikationen mit Taltz behandelt, was kumulativ einer Exposition von 22'371.0 erwachsenen Patientenjahren und 342.8 pädiatrischen Patientenjahren (196 pädiatrische Patienten) entspricht. Von diesen 9421 Patienten hatten 6805 Patienten Taltz über mindestens ein Jahr hinweg erhalten.
Erwachsene
Die häufigsten berichteten unerwünschten Wirkungen waren Infektionen der oberen Atemwege (einschliesslich Nasopharyngitis und Pharyngitis) (38.7%)* und Reaktionen an der Injektionsstelle (19.2%)**. Die meisten Reaktionen waren von leichtem oder mittlerem Schweregrad.
*Häufigkeit in Plaque-Psoriasis; integrierte Häufigkeit für alle Indikationen beträgt 36.6 %
**Häufigkeit in axialer Spondyloarthritis; integrierte Häufigkeit für alle Indikationen beträgt 16.3 %
Liste der unerwünschten Wirkungen in placebo-kontrollierten und nicht kontrollierten klinischen Studien (Phase III, integrierte Daten aus den Indikationen Plaque Psoriasis, Psoriasis-Arthritis, und axiale Spondyloarthritis)
Die Häufigkeiten der unerwünschten Wirkungen werden wie folgt angegeben: "sehr häufig" (≥1/10), "häufig" (≥1/100, <1/10), "gelegentlich" (≥1/1000, <1/100), "selten" (≥1/10'000, <1/1000) und "sehr selten" (<1/10'000).
Infektionen und Infestationen
Sehr häufig: Infektionen der oberen Atemwege.
Häufig: Tinea Infektion, Herpes simplex (mukokutan), orale Candidiasis, Rhinitis, Influenza, Konjunktivitis.
Gelegentlich: oesophageale Candidiasis.
Störungen des Blut- und Lymphsystems
Gelegentlich: Neutropenie, Thrombozytopenie.
Störungen des Immunsystems
Gelegentlich: Anaphylaxie (Angioödem, Larynxödem, Urtikaria).
Respiratorische, thorakale und mediastinale Funktionsstörungen
Häufig: oropharyngeale Schmerzen.
Gastrointestinale Störungen
Häufig: Übelkeit, Diarrhö.
Gelegentlich: Morbus Crohn, Colitis ulzerosa (siehe "Warnhinweise und Vorsichtsmassnahmen" )
Leber- und Gallenerkrankungen
Häufig: erhöhte Leberenzyme.
Funktionsstörungen der Haut und des Unterhautzellgewebes
Häufig: Urtikaria.
Gelegentlich: dyshidrotisches Ekzem.
Selten: exfoliative Dermatitis.
Allgemeine Störungen und Reaktionen an der Applikationsstelle
Sehr häufig: Reaktionen an der Injektionsstelle.
Beschreibung ausgewählter Nebenwirkungen bei Erwachsenen
Reaktionen an der Injektionsstelle
Die am häufigsten an der Injektionsstelle beobachteten Reaktionen waren Erythem und Schmerzen. Diese Reaktionen waren vorwiegend von leichtem bis mittlerem Schweregrad und führten nicht zum Abbruch von Taltz.
Die oben beschriebenen Häufigkeiten beziehen sich auf die ursprüngliche Formulierung von Taltz. In einer einfach verblindeten, randomisierten Cross-Over-Studie an 45 gesunden Probanden, in der die ursprüngliche Formulierung mit der weiterentwickelten, citratfreien Formulierung verglichen wurde, wurden während und direkt nach der Injektion statistisch signifikant geringere Schmerzen berichtet.
Infektionen
Die Mehrzahl der Infektionen bestand aus nicht-schwerwiegenden und leichten bis mittelschweren unerwünschten Ereignissen wie Nasopharyngitis und Infektionen der oberen Atemwege, die einen Therapieabbruch nicht erforderlich machten.
In den placebokontrollierten Phasen III klinischen Studien bei Plaque Psoriasis in Erwachsenen (insgesamt wurden 2328 Patienten mit Taltz und 791 Patienten mit Placebo über bis zu 12 Wochen behandelt) wurden Infektionen bei 27.2 % der Patienten unter Taltz im Vergleich zu 22.9 % der Patienten unter Placebo berichtet.
Schwerwiegende Infektionen traten bei 13 (0.6 %) der Patienten unter Taltz und bei 3 (0.4 %) der Patienten unter Placebo auf (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Mit Infektionen zusammenhängende schwerwiegende unerwünschte Ereignisse (SAE), die von mehr als 1 Patient der gesamten Ixekizumab-Gruppe berichtet wurden, waren Cellulitis (n = 3), Appendizitis (n = 2) und Erysipel (n = 2). Der Anteil der Patienten, die die Therapie aufgrund eines mit Infektionen zusammenhängenden unerwünschten Ereignisses abbrach, war in der gesamten Ixekizumab-Gruppe (8 Patienten [0.3 %]) und in der Placebo-Gruppe (2 Patienten [0.3 %]) ähnlich.
Übereinstimmend mit dem Wirkmechanismus zeigte sich in der Ixekizumab-Gruppe in der placebokontrollierten Phase der klinischen Studien Phase III bei Erwachsenen mit Plaque-Psoriasis ein Anstieg oraler Candidiasis und mit einer Ausnahme waren alle Fälle von leichtem oder mittlerem Schweregrad. Es wurde über keine SAE oder Therapieabbrüche aufgrund von Candidiasis berichtet.
Über die gesamte Behandlungsdauer, die die kontrollierten und unkontrollierten Phasen der Studien bei Erwachsenen mit Plaque-Psoriasis umfasste (insgesamt 6892 erwachsene Patienten welche 18'025.7 Patientenjahre Exposition entsprechen), wurden Infektionen bei 62.5 % der Patienten unter Taltz berichtet (23.9 pro 100 Patientenjahre). Schwerwiegende Infektionen wurden von 3.4 % der Patienten unter Taltz berichtet (1.3 pro 100 Patientenjahre).
Die in den placebokontrollierten, klinischen Studien zur Psoriasis-Arthritis und axialen Spondyloarthritis beobachteten Infektionsraten waren ähnlich wie die in den Plaque-Psoriasis-Studien beobachteten Infektionsraten, mit Ausnahme der Häufigkeit der Nebenwirkungen von Influenza und Konjunktivitis, die bei Patienten mit Psoriasis-Arthritis häufig auftraten.
Die Häufigkeiten von Infektionen und schwerwiegenden Infektionen waren in den kontrollierten und unkontrollierten Phasen der klinischen Studien zu Psoriasis-Arthritis und axialer Spondyloarthritis ähnlich wie in Studien zu Psoriasis bei Erwachsenen.
Bei der axialen Spondyloarthritis -Langzeitbehandlung mit Ixekizumab waren die von mehr als 1 Patient berichteteten infektionsbedingten SAE Appendizitis (n=3) und Clostridium difficile Colitis (n=2). Zudem wurden auch die unerwünschten Ereignisse Herpes simplex und Herpes zoster berichtet.
Neutropenie und Thrombozytopenie
In placebokontrollierten Plaque-Psoriasis Studien entwickelten 9 % der mit Taltz behandelten Patienten eine Neutropenie. Ein solcher Grad der Neutropenie kann weiterbestehen, fluktuieren oder vorübergehend sein.
0.1 % der mit Taltz behandelten Patienten entwickelten eine Neutrophilenzahl von < 1000 Zellen/mm³. Im Allgemeinen erforderte die Neutropenie kein Absetzen von Taltz.
3 % der mit Taltz behandelten Patienten mit Plaque-Psoriasis und einem normalen Ausgangswert der Thrombozytenzahl zeigten eine Verminderung dieser Zahl auf Werte zwischen 75'000 und 150'000 Zellen/mm³. Die Thrombozytopenie kann weiterbestehen, fluktuieren oder vorübergehend sein.
Die Häufigkeit von Neutropenie und Thrombozytopenie in kontrollierten und unkontrollierten klinischen Studien zur Psoriasis-Arthritis und axialen Spondyloarthritis ist vergleichbar zu der in Plaque-Psoriasis-Studien beobachteten Häufigkeit
Immunogenität
Etwa 9 – 17 % der mit der empfohlenen Taltz Dosis behandelten Plaque-Psoriasis erwachsenen Patienten entwickelten Antikörper gegen Ixekizumab, bei der Mehrzahl waren die Titer niedrig und in der bis zu 60 Wochen dauernden Therapie nicht mit einem verminderten klinischen Ansprechen verbunden. Etwa 1 % der mit Taltz behandelten Patienten hatten jedoch bestätigte, neutralisierende Antikörper, die mit niedrigen Arzneimittel-Konzentrationen und reduziertem klinischen Ansprechen verbunden waren.
Von den Psoriasis-Arthritis Patienten, die mit Taltz im empfohlenen Dosierschema bis zu 52 Wochen behandelt wurden, entwickelten 11% Antikörper gegen den Wirkstoff, wovon die Mehrheit einen niedrigen Titer aufwies, und 8% hatten nachweislich neutralisierende Antikörper. Die Häufigkeit von Antikörpern gegen den Wirkstoff und neutralisierenden Antikörpern betrug 15.4% bzw. 10.3% in der Gruppe ohne gleichzeitige Gabe von cDMARDs gegenüber 9.0% bzw. 6.0% in der Gruppe, die Ixekizumab in Kombination mit cDMARDs erhielt. Es konnte kein eindeutiger Zusammenhang zwischen der Anwesenheit von neutralisierenden Antikörpern und einem Einfluss auf die Wirkstoffkonzentration oder die Wirksamkeit beobachtet werden.
Bei Patienten mit ankylosierender Spondylitis, die mit Taltz im empfohlenen Dosierschema behandelt wurden, entwickelten bis Woche 16 und Woche 52 10 Patienten (5.2%) bzw. 23 Patienten (11.9 %) Antikörper gegen den Wirkstoff, wovon die Mehrheit einen niedrigen Titer aufwies; 3 Patienten (1.5%) hatten neutralisierende Antikörper bis Woche 16, und keine neuen Patienten entwickelten neutralisierende Antikörper zwischen Woche 16 und Woche 52. Bei einigen Antikörper gegen den Wirkstoff-positiven Patienten wurden tendenziell niedrigere Talspiegel beobachtet.
Bei Patienten mit nicht-radiographischer axialer Spondyloarthritis, die mit Taltz im empfohlenen Dosierschema bis zu 52 Wochen behandelt wurden, entwickelten 5 Patienten (8.9%) Antikörper gegen den Wirkstoff, wovon alle einen niedrigen Titer aufwiesen; kein Patient hatte neutralisierende Antikörper. Es konnte kein offensichtlicher Zusammenhang zwischen der Anwesenheit von Antikörpern gegen den Wirkstoff und einem Einfluss auf die Wirksamkeit oder die Sicherheit beobachtet werden.
In einer Langzeitstudie zu Taltz bei Patienten mit radiographischen und nicht-radiographischen axialen Spondyloarthritis, in der die Patienten die Behandlung über 156 Wochen fortsetzten, entwickelten 25 weitere Patienten (3.3 %) erstmals Anti-Ixekizumab-Antikörper. Auch neutralisierende Antikörper gegen Ixekizumab konnten in diesem Behandlungszeitraum erstmals nachgewiesen werden. Anti-Ixekizumab-Antikörper, die sich während der Langzeitverlängerung entwickelten, hatten überwiegend einen niedrigen Titer. Ein Zusammenhang zwischen Antikörperspiegel und klinischer Wirksamkeit oder Sicherheit wurde in der Langzeitbehandlung nicht beobachtet.
Bei allen Indikationen, wurde kein Zusammenhang zwischen Immunogenität und während der Therapie aufgetretener unerwünschter Ereignissen festgestellt.
Sicherheit im Vergleich zu Etanercept
In den 2 aktiv kontrollierten klinischen Studien UNCOVER-2 und UNCOVER-3 waren die Raten für schwerwiegende unerwünschte Ereignisse sowohl für Taltz als auch für Etanercept 1.9% und die Abbruchraten aufgrund von unerwünschten Ereignissen waren 1.2% für Etanercept und 2% für Taltz. Die Infektionsrate war 21.5% für Etanercept und 26% für Taltz wobei der Schweregrad für die meisten Ereignisse mild bis mässig war. Die Rate für schwere Infektionen lag bei Etanercept bei 0.4% und bei Taltz bei 0.5%.
Pädiatrie
Insgesamt stimmt das Sicherheitsprofil, das bei pädiatrischen Patienten mit Plaque-Psoriasis, die alle 4 Wochen mit Taltz behandelt werden mit dem Sicherheitsprofil bei erwachsenen Patienten mit Plaque-Psoriasis überein, mit Ausnahme folgender unerwünschter Wirkungen, die im Vergleich zu Erwachsenen häufiger beobachtet wurden, oder aber spezifisch bei Kindern und Jugendlichen auftraten:
Störungen des Blut- und Lymphsystems
Häufig: Neutropenie.
Störungen des Immunsystems
Sehr häufig: Überempfindlichkeitsreaktionen (10.2 %)
Häufig: Dermatitis, Rash
Gelegentlich: Bronchospasmus.
Gastrointestinale Störungen
Häufig: Morbus Crohn
Funktionsstörungen der Haut und des Unterhautzellgewebes
Häufig: Ekzem.
Allgemeine Störungen und Reaktionen an der Applikationsstelle
Häufig: Fieber.
Beschreibung ausgewählter Nebenwirkungen bei pädiatrischen Patienten
Neutropenie
In der pädiatrischen Studie in 114 Patienten im Alter von 6-17 mit mittelschwerer oder schwerer Plaque-Psoriasis entwickelten 8.8 % der mit Taltz behandelten Patienten eine Neutropenie, welche weiterbestehen, fluktuieren oder transient auftreten können.
0.9 % der mit Taltz behandelten Patienten entwickelten eine Neutrophilenzahl von < 1000 Zellen/mm³. In der pädiatrischen Studie wurde Taltz bei keinem Patienten wegen Neutropenie abgesetzt.
Überempfindlichkeitsreaktionen
In der pädiatrischen Studie traten während des 12-wöchigen, placebokontrollierten Zeitraums bei 5.2% der Patienten in der Taltz-Gruppe und 1.8% der Patienten in der Placebo-Gruppe, Überempfindlichkeitsreaktionen auf. Alle Überempfindlichkeitsreaktionen waren nicht-anaphylaktischer Natur.
Entzündliche Darmerkrankungen.
In der pädiatrischen Studie trat während des 12-wöchigen, placebokontrollierten Zeitraums ein Morbus Crohn bei 0.9% der Patienten in der Taltz-Gruppe und bei 0% der Patienten in der Placebo-Gruppe auf. Über die Gesamtlaufzeit einschliesslich der langfristigen Verlängerung der pädiatrischen klinischen Studie trat ein Morbus Crohn bei insgesamt 4 mit Taltz behandelten Probanden (2.0%) auf.
Reaktionen an der Injektionsstelle
In der pädiatrischen Plaque-Psoriasis Studie traten, während des 12-wöchigen placebokontrollierten Zeitraums, bei 12.2% der Patienten in der Taltz-Gruppe und 1.8% der Patienten in der Placebo-Gruppe, Reaktionen an der injektionsstelle auf. Die am häufigsten an der Injektionsstelle beobachteten Reaktionen waren Schmerzen und Erythem. Diese Reaktionen waren vorwiegend von leichtem bis mittlerem Schweregrad und führten nicht zum Abbruch von Taltz.
Immunogenität
Von den pädiatrischen Patienten mit Plaque-Psoriasis, die mit Taltz im empfohlenen Dosierschema bis zu 12 Wochen behandelt wurden, entwickelten 21 Patienten (18%) Antikörper gegen den Wirkstoff. Bei ungefähr der Hälfte waren die Titer niedrig, und 5 Patienten (4%) hatten bestätigte neutralisierende Antikörper, die mit niedrigen Arzneimittel-Konzentrationen verbunden waren. Während einer Langzeit-Verlängerungsphase von bis zu 108 Wochen entwickelten 56 Patienten (29 %) Antikörper gegen den Wirkstoff, etwa die Hälfte hatte einen niedrigen Titer und 5 Patienten (2.6 %) hatten neutralisierende Antikörper. Es gab keinen Zusammenhang mit klinischem Ansprechen oder unerwünschten Ereignissen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien wurden Dosierungen bis zu 180 mg subkutan ohne Auftreten dosislimitierender Toxizitäten angewendet. Es wurde über Überdosierungen bis zu 240 mg subkutan berichtet, ohne dass schwerwiegende unerwünschte Ereignisse auftraten Bei Überdosierung wird empfohlen, Patienten auf Anzeichen und Symptome unerwünschter Wirkungen zu überwachen und unverzüglich eine angemessene symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
L04AC13
Wirkungsmechanismus
Ixekizumab ist ein für Interleukin-17A selektiver, rekombinanter, humanisierter, monoklonaler Antikörper. Ixekizumab ist ein aus CHO (Chinese Hamster Ovary) - Zellen hergestellter modifizierter monoklonaler IgG4 Antikörper, der mit hoher Affinität (< 3 pM) und Spezifität an das proinflammatorische Zytokin Interleukin 17A (sowohl an das IL-17A als auch das Heterodimer IL-17A/F) bindet. Erhöhte Werte von IL-17A wurden mit der Pathogenese verschiedener Autoimmunerkrankungen in Verbindung gebracht. Bei Psoriasis spielt der IL-17A-Ligand eine wichtige Rolle bei der überschiessenden Keratinozytenproliferation und -aktivierung. Die Neutralisierung von IL-17A durch Ixekizumab hemmt diese Wirkungen. IL-17A spielt eine Schlüsselrolle in der Pathogenese von Plaque-Psoriasis und Psoriasis-Arthritis und ist hochreguliert in von Läsionen betroffener Haut im Gegensatz zu nicht von Läsionen betroffener Haut von Plaque Psoriasis Patienten.
Bei der axialen Spondyloarthritis spielt der IL-17A-Ligand eine wichtige Rolle durch die Zunahme der Entzündungen, die zu erosiven Knochenschäden und pathologischer Knochenneubildung führt.
Ixekizumab bindet nicht an die Liganden IL-17B, IL-17C, IL-17D, IL-17E oder IL-17F.
Ixekizumab hat eine geringe Fähigkeit zu Bindung an Fcγ Rezeptoren oder Komponenten des Komplementsystems. In-vitro durchgeführte Bindungsversuche bestätigten, dass Ixekizumab nicht an die humanen Fcγ Rezeptoren I, IIa und IIIa oder die Komplementkomponente C1q bindet.
Pharmakodynamik
Auf Grundlage der Daten einer Phase I Studie mit Biopsien, die aus Psoriasis-betroffener Haut entnommen wurden, gab es vom Ausgangswert bis Tag 43 einen dosisabhängigen Trend zur Verringerung von Epidermisdicke, Anzahl proliferierender Keratinozyten, T-Zellen und dendritischer Zellen sowie Verringerung der lokalen Entzündungsmarker. Als direkte Folge reduziert die Behandlung mit Ixekizumab Erythem, Verhärtung und Schuppung in Plaque-Psoriasis-Läsionen.
Taltz hat eine Senkung des C-reaktiven Proteins-Levels (innerhalb von 1 Woche Behandlung) gezeigt.
Klinische Wirksamkeit
Plaque-Psoriasis
Die Wirksamkeit und Sicherheit von Taltz wurde in drei randomisierten, doppelblinden, placebokontrollierten Phase III Studien bei erwachsenen Patienten mit mittelschwerer bis schwerer Plaque Psoriasis untersucht, die Kandidaten für eine Phototherapie oder systemische Therapie waren (UNCOVER-1, UNCOVER-2 und UNCOVER-3). Die Wirksamkeit und Sicherheit von Taltz wurde auch im Vergleich zu Etanercept untersucht (UNCOVER-2 und UNCOVER-3). Patienten, die ursprünglich auf Taltz randomisiert wurden und die in Woche 12 einen static Physician Global Assesment Responder -Wert von 0 oder 1 (sPGA (0,1)) erreichten, wurden erneut auf Placebo oder Taltz für weitere 48 Wochen (UNCOVER-1 und UNCOVER-2) randomisiert. Auf Placebo, Etanercept oder Taltz randomisierte Patienten die sPGA (0,1) non Responder waren, erhielten Taltz für bis zu 48 Wochen. Zusätzlich wurde die Langzeitwirksamkeit und -sicherheit in allen drei Studien für bis zu 5 Jahre bei Patienten untersucht, die über die gesamte Studienzeit teilgenommen haben.
Von den 3866 in diese placebokontrollierten Studien eingeschlossenen Patienten hatten 64 % zuvor eine systemische Therapie erhalten (biologische, konventionelle systemische oder PUVA), 43.5 % hatten zuvor eine Phototherapie erhalten, 49.3 % eine konventionelle systemische Therapie und 26.4 % eine biologische Therapie der Psoriasis. Von allen Patienten hatten 14.9 % mindestens eine anti-TNF-alpha Behandlung bekommen, und 8.7 % eine anti-IL-12/IL-23 Behandlung. Zu Studienbeginn hatten 23.4 % der Patienten eine Psoriasis-Arthritis in der Vorgeschichte.
Ausschlusskriterien: wesentliche Ausschlusskriterien, die einer Teilnahme an UNCOVER-1, UNCOVER-2 und UNCOVER-3 entgegenstanden, waren die begleitende Anwendung von systemischen oder biologischen Psoriasis-Therapien oder Phototherapie, jede instabile schwerwiegende medizinische Störung oder Erkrankung ausgenommen Plaque Psoriasis, und jede derzeit bestehende oder kürzlich bestandene schwerwiegende Infektion. In UNCOVER-2 und UNCOVER-3 war jede vorherige Anwendung von Etanercept ein Ausschlusskriterium.
Die kombinierten primären Endpunkte in allen drei Studien waren der Anteil Patienten im Vergleich zu Placebo, die in Woche 12 ein PASI-75-Ansprechen (Psoriasis Area and Severity Index; PASI 75 = Verbesserung des PASI um mindestens 75 %) und einen sPGA-Wert von 0 ( "frei von" ) oder 1 ( "minimal" ) erreichten. Der Baseline Medianwert des PASI für Patienten aller Therapiegruppen lag zwischen 17.4 und 18.3; 48.3 % bis 51.2 % der Patienten hatten einen Baseline sPGA-Wert von "schwer" oder "sehr schwer" , und der Medianwert der itch Numeric Rating Scale (itch NRS) lag bei Baseline zwischen 6.3 und 7.1.
Tabelle 1. Klinisches Ansprechen nach 12 Wochen (NRI) aus UNCOVER-1, -2 und -3
Placebo Taltz 80 mg Q4W Taltz 80 mg Q2W Etanercept 50 mg 2x
wöchentlich
UNCOVER 1
Anzahl Patienten (N) 431 432 433 NA
sPGA "0" (frei von) 14(3.2%) 330(76.4%)a 354(81.8%)a NA
oder "1" (minimal),
n (%)
sPGA "0" (frei 0 149(34.5%)a 160(37.0%)a NA
von), n (%)
PASI 75, n (%) 17(3.9%) 357(82.6%)a 386(89.1%)a NA
PASI 90, n (%) 2 (0.5%) 279(64.6%)a 307(70.9%)a NA
PASI 100, n (%) 0 145(33.6%)a 153(35.3%)a NA
UNCOVER 2
Anzahl Patienten (N) 168 347 351 358
sPGA "0" (frei von) 4(2.4%) 253(72.9%)a 292(83.2%)a 129(36.0%)a
oder "1" (minimal),
n (%)
sPGA "0" (frei 1(0.6%) 112(32.3%)a,b 147(41.9%)a,b 21(5.9%)c
von), n (%)
PASI 75, n (%) 4 (2.4%) 269(77.5%)a 315(89.7%)a 149(41.6%)a
PASI 90, n (%) 1 (0.6%) 207(59.7%)a,b 248(70.7%)a,b 67(18.7%)a
PASI 100, n (%) 1 (0.6%) 107(30.8%)a,b 142(40.5%)a,b 19(5.3%)c
UNCOVER 3
Anzahl Patienten (N) 193 386 385 382
sPGA "0" (frei von) 13(6.7%) 291(75.4%)a,b 310(80.5%)a,b 159(41.6%)a
oder "1" (minimal),
n (%)
sPGA "0" (frei 0 139(36.0%)a,b 155(40.3%)a,b 33(8.6%)a
von), n (%)
PASI 75, n (%) 14(7.3%) 325(84.2%)a,b 336(87.3%)a,b 204(53.4%)a
PASI 90, n (%) 6(3.1%) 252(65.3%)a,b 262(68.1%)a,b 98(25.7%)a
PASI 100, n (%) 0 135(35.0%)a,b 145(37.7%)a,b 28(7.3%)a
Abkürzungen: n = Anzahl Patienten in dieser Kategorie; N = Anzahl Patienten in der Intent-to-treat Population; NA = nicht zutreffend; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; Q2W = alle zwei Wochen; Q4W = all vier Wochen; sPGA = static Physician Global Assessment.
a p < 0.001 im Vergleich zu Placebo
b p < 0.001 im Vergleich zu Etanercept
c p < 0.01 im Vergleich zu Placebo
UNCOVER-1 schloss 1296 Patienten ein. Die Patienten wurden randomisiert (1:1:1) auf 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg).
UNCOVER-2 schloss 1224 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
UNCOVER-3 schloss 1346 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
Die Dosierung 80 mg Q2W zeigte in allen Studien bei allen Endpunkten eine überlegene Wirksamkeit (siehe Tabellen oben), insbesondere bei hohen Graden der Hautverbesserung (PASI 90, PASI 100, sPGA 0) und bei der Reduktion des Juckreizes. Im Vergleich zu Placebo und Etanercept wurden in Woche 12 signifikant grössere Verbesserungen des Ausgangswerts gezeigt, bei Nagel Psoriasis (gemessen anhand des Nail Psoriasis Severity Index [NAPSI]), bei Kopfhaut Psoriasis (gemessen anhand des Psoriasis Scalp Severity Index [PSSI]) und bei palmoplantarer Psoriasis (gemessen anhand des Psoriasis Palmoplantar Severity Index [PPASI]).
Taltz war mit einem raschen Einsetzen der Wirkung mit einer Reduktion des mittleren PASI in Woche 2 um > 50% verbunden (Abbildung 1). Der Anteil der Patienten, der PASI 75 erreichte, war bereits in Woche 1 in allen drei Studien unter Taltz signifikant grösser als unter Placebo und unter Etanercept. Etwa 25 % der mit Taltz behandelten Patienten erreichte bis Woche 2 einen PASI-Wert< 5, mehr als 55 % erreichten einen PASI-Wert < 5 bis Woche 4 mit einem Anstieg auf 85 % bis Woche 12 (im Vergleich zu 3 %, 14 % und 50 % unter Etanercept). Signifikante Verbesserungen des Juckreiz-Schweregrades wurden in Woche 1 unter Taltz beobachtet.
Taltz zeigte auch einen günstigen Einfluss auf den Pruritus, der Placebo signifikant überlegen war.
Abbildung 1. PASI Werte, prozentuale Verbesserung bei jeder Visite nach der Baseline Visite (mBOCF) in der Intent-to-treat Population während der Anfangsphase der Therapie (UNCOVER-2 und UNCOVER-3)

Die Wirksamkeit und Sicherheit von Taltz wurde unabhängig von Alter, Geschlecht, ethnischer Abstammung, Körpergewicht, PASI Baseline Schweregrad und vorangegangener Therapie mit einem Biologikum, gezeigt. Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q2W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (90.1% bzw. 85.7%) und sPGA 0 oder 1 (84.5% bzw. 75.6%). Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q4W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (85.6% bzw. 74.2%) und sPGA 0 oder 1 (79.0% bzw. 67.4%). Das Ansprechen unterschied sich nicht bei Patienten mit Nagel Psoriasis, Psoriasis im Gesichtsbereich oder Kopfhaut Psoriasis zu Studienbeginn. Taltz war wirksam bei Patientenmit früheren systemischen Therapien, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen. Die Verbesserungen von sPGA und PASI Endpunkten bei Patienten mit begleitender Psoriasis-Arthritis zu Studienbeginn waren ähnlich zu jenen der gesamten Population mit mittelschwerer bis schwerer Plaque Psoriasis. Etwa 45 % der Patienten wiesen bei Studienbeginn Psoriasis im Gesichtsbereich auf. Von diesen Patienten waren 80.4 % der mit Taltz behandelten Patienten in Woche 12 frei von Psoriasis im Gesichtsbereich.
Wirksamkeit bei Etanercept-Non-Respondern: Bei Patienten, die in Woche 12 der UNCOVER-2 als Etanercept-sPGA (0,1) Non-Responder angesehen wurden (N = 200) und die nach einer 4-wöchigen Auswaschphase auf Taltz 80 mg Q4W umgestellt wurden, erreichten nach 12 Wochen 73 % einen sPGA (0,1) und 83.5% einen PASI 75.
Anhalten des Ansprechens bis Woche 60 und bis zu 5 Jahren
Zur Beurteilung des anhaltenden Ansprechens wurden Patienten aus UNCOVER-1 und UNCOVER-2, die ursprünglich auf Taltz randomisiert worden waren und in Woche 12 angesprochen hatten (d.h. sPGA-Werte von 0 oder 1) erneut auf eine der folgenden Therapien für weitere 48 Wochen, randomisiert: Placebo oder Taltz (80 mg alle vier oder zwölf Wochen [Q4W oder Q12W]). Patienten, die in Woche 12 einen sPGA (0,1) nicht erreicht hatten oder während der Erhaltungsphase einen Rückfall (sPGA ≥3) erlitten, erhielten anschliessend Taltz 80 mg Q4W.
Bei den sPGA (0,1) Respondern in Woche 12 (der kombinierten Studien UNCOVER-1 und UNCOVER-2) war der Anteil der Patienten, der dieses Ansprechen in Woche 60 aufrecht erhalten hatte bei Patienten unter Taltz 80 mg Q4W (71 %) signifikant grösser im Vergleich zu Patienten unter Taltz 80 mg Q12W (35.5 %) oder Placebo (7 %).
Tabelle 2 zeigt die Ansprechraten der bei der erneuten Randomisierung auf die empfohlene Erhaltungsdosis von 80 mg Taltz alle 4 Wochen randomisierten Patienten in Abhängigkeit der randomisierten Dosis zu Studienbeginn.
Tabelle 2. Anhaltendes Ansprechen und Wirksamkeit in Woche 60 (kombinierte Ergebnisse aus den Studien UNCOVER-1 und UNCOVER-2; NRI)
Endpunkt 80 mg Q2W (Induktion 80 mg Q4W (Induktion 80 mg Q2W (Induktion 80 mg Q4W (Induktion
)/Placebo (Erhaltung )/ Placebo (Erhaltun )/80 mg Q4W (Erhaltu )/ 80 mg Q4W (Erhalt
) (N=211) g) (N=191) ng) (N=221) ung) (N=195)
Beibehalten eines 7.6 % 6.3 % 78.3 % 68.7 %
sPGA "0" (frei von)
oder "1" (minimal)
sPGA "0" (frei von) 2.8 % 1.6 % 58.8 % 49.2 %
beibehalten oder
erreicht
PASI 75 beibehalten 9.0 % 7.9 % 83.3 % 74.4 %
oder erreicht
PASI 90 beibehalten 4.7 % 4.7 % 76.5 % 66.7 %
oder erreicht
PASI 100 beibehalten 2.8 % 1.6 % 57.5 % 49.7 %
oder erreicht
Abkürzungen: N = Anzahl Patienten in der Analysenpopulation; NRI = Non-Responder Imputation
Von den sPGA (0,1) Respondern in Woche 12, die der Erhaltungstherapie Taltz 80 mg Q4W zugeordnet wurden, hatten, in Woche 60, 76.4 % einen PASI < 5 beibehalten oder erreicht. Die Verbesserung des Juckreiz-Schweregrades hielt bei den Taltz Patienten, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, bis Woche 60 an. Im Hinblick auf das anhaltende Ansprechen bis Woche 60 war Taltz wirksam bei Patienten mit früherer systemischer Therapie, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen.
Von den sPGA (0,1) Respondern in Woche 12, die Placebo rerandomisiert wurden, betrug die Zeit bis zum Rückfall (sPGA ≥3) in den kombinierten UNCOVER-1 und UNCOVER-2 Studien 164 (95% CI [143, 169]) Tage. Von diesen Patienten erreichten 71.5 % innerhalb von 12 Wochen nach erneutem Beginn mit Taltz 80 mg Q4W wieder einen sPGA-Wert von 0 oder 1.
Bei Patienten unter Taltz, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, hielten auch die Verbesserungen bei Nagel Psoriasis, Kopfhaut Psoriasis und palmoplantarer Psoriasis bis Woche 60 an.
Von den 591 Patienten, die Taltz Q2W während der Induktionsphase und Q4W als Erhaltungstherapie in den Studien UNCOVER-1, UNCOVER-2 und UNCOVER-3 erhalten hatten, vollendeten 427 Patienten 5 Jahre der Taltz-Behandlung. Davon erforderten 101 Patienten eine Dosis-Eskalation. Von den Patienten, die die Untersuchung in Woche 264 vollendet haben (N = 427), zeigte sich bei 295 Patienten (69 %) ein sPGA (0 oder 1), bei 289 Patienten (68 %) ein PASI 90 und bei 205 Patienten (48 %) ein PASI 100 Ansprechen in Woche 264. Der DLQI wurde nach der Induktionsphase in UNCOVER-1 und UNCOVER-2 erfasst, und bei 113 Patienten (66 %) zeigte sich ein DLQI (0,1) Ansprechen.
Quality of life/Patientenberichtete Ergebnisse
Für den DLQI (Dermatology Life Quality Index) wurden statistisch signifikante Verbesserungen in Woche 12 (Studien 1-3) gegenüber dem Ausgangswert gezeigt; die Verbesserungen blieben über 60 Wochen bestehen Taltz war mit signifikant grösseren Verbesserungen der Hautschmerzen (gemessen anhand der visuellen Analogskala, VAS) verbunden.
Postmarketing, direkte Vergleichsstudien
Die Wirksamkeit und Sicherheit von Ixekizumab wurden auch in der doppelblinden Studie RHBS (IXORA-S) im Vergleich zu Ustekinumab untersucht. Dabei war Ixekizumab im Hinblick auf den primären Endpunkt (PASI 90-Ansprechen in Woche 12, Tabelle 3) überlegen.). In allen drei Kategorien des PASI-Ansprechens zeigte sich die Überlegenheit versus Ustekinumab schnell. Die Überlegenheit von Ixekizumab versus Ustekinumab wurde ebenfalls in den nach Gewicht stratifizierten Subgruppen gezeigt.
Tabelle 3: PASI-Ansprechraten aus der Vergleichsstudie Ixekizumab versus Ustekinumab
Woche 12 Woche 52
Ixekizumab* Ustekinumab** Ixekizumab* Ustekinumab**
Patienten (n) 136 166 136 166
PASI 75, n (%) 120 (88.2 %) 114 (68.7 %) 120 (88.2 %) 126 (75.9 %)
PASI 90, n (%) 99 (72.8 %)§ 70 (42.2 %) 104 (76.5 %) 98 (59.0 %)
PASI 100, n (%) 49 (36.0 %) 24 (14.5 %) 71 (52.2 %) 59 (35.5 %)
* Ixekizumab 160 mg wurde als Initialdosis verabreicht, gefolgt von 80 mg in den Wochen 2, 4, 6, 8, 10 und 12, und anschliessend 80 mg Q4W
** Gewichtsbasierte Dosierung: Patienten, die mit Ustekinumab behandelt wurden, erhielten 45 mg oder 90 mg in Woche 0 und 4, anschliessend alle 12 Wochen bis Woche 52 (dosiert nach Gewicht gemäss zugelassener Dosierung)
§ p < 0.001 versus Ustekinumab (p-Wert nur für den primären Endpunkt)
Die Wirksamkeit und Sicherheit von Ixekizumab wurden auch in der 24-wöchigen randomisierten, doppelblinden, Parallelgruppen-Studie RHCR (IXORA-R) untersucht, in der Ixekizumab mit Guselkumab verglichen wurde. Hierbei zeigte sich eine überlegene Wirksamkeit von Ixekizumab ab Woche 4 im Erreichen vollständig erscheinungsfreier Haut sowie im Erreichen des primären Endpunktes PASI 100 in Woche 12 [Ixekizumab 41.3% vs Guselkumab 24.9% (p<0.001)] und eine Nicht-Unterlegenheit im PASI 100 Ansprechen in Woche 24 [Ixekizumab 50.0% vs Guselkumab 52.3% (p=0.414)].
Wirksamkeit bei genitaler Psoriasis
Eine randomisierte, doppelblinde, placebokontrollierte Studie (IXORA-Q) wurde an 149 erwachsenen Patienten (24 % Frauen) mit einer mittelschweren bis schweren genitalen Psoriasis (sPGA des Genitalbereichs von ≥3) durchgeführt. Die Patienten hatten eine Hautbeteiligung von mindestens 1 % Body Surface Area (BSA) (60.4 % hatten eine Hautbeteiligung von ≥10% BSA) und haben auf mindestens eine vorangegangene topische Therapie zur Behandlung der genitalen Psoriasis nicht angesprochen oder diese nicht vertragen. Die Patienten hatten zumindest eine mittelschwere Plaque-Psoriasis (definiert als sPGA-score ≥3 und waren geeignet für eine Phototherapie und/oder systemische Therapie) über mindestens 6 Monate.
Studienteilnehmer, die auf Taltz randomisiert wurden, erhielten eine Initialdosis von 160 mg, gefolgt von 80 mg alle 2 Wochen für eine Dauer von 12 Wochen. Der primäre Endpunkt war der Anteil an Patienten, die einen sPGA im Genitalbereich von "0" (frei von) oder "1" (minimal) (sPGA-G 0/1) erreichten. In Woche 12 erreichten, unabhängig von der Hautbeteiligung zu Studienbeginn, signifikant mehr Studienteilnehmer unter Taltz einen sPGA-G 0/1 und einen sPGA 0/1 als Studienteilnehmer unter Placebo (Hautbeteiligung zu Studienbeginn von 1 % bis < 10 % BSA bzw. ≥10 % BSA: sPGA-G von "0" oder "1" : Taltz 71 % bzw. 75 %; Placebo: 0 % bzw. 13 %).
Pädiatrische Plaque-Psoriasis
Eine randomisierte, doppelblinde, multizentrische, placebokontrollierte Studie (IXORA-Peds) umfasste 201 pädiatrische Patienten im Alter von 6 bis unter 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis (definiert durch einen sPGA-Wert ≥3, der ≥10% der Körperoberfläche einbezieht, und einen PASI-Wert ≥12), die Kandidaten für eine Phototherapie oder systemische Therapie waren oder bei einer topischen Therapie unzureichend kontrolliert waren.
Die wesentlichen Ausschlusskriterien waren ähnlich wie bei den oben genannten Studien in Erwachsenen, insbesondere aktive oder kürzlich durchgemachte schwerwiegende Infektionen sowie begleitende systemische konventionelle oder biologische Psoriasis-Therapien oder Phototherapie.
Während der 12-wöchigen doppelblinden Placebo- und aktiv-kontrollierten Phase wurden die Patienten mit Placebo (n=56), Etanercept (n=30) oder Taltz (n=115) behandelt, wobei die Gewichts-adaptierte Dosierung wie folgt angepasst wurde:
Taltz:
<25 kg: 40 mg in Woche 0, danach 20 mg Q4W
25 kg bis 50 kg: 80 mg in Woche 0, danach 40 mg Q4W
>50 kg: 160 mg in Woche 0, danach 80 mg Q4W
Etanercept:
0.8 mg/kg, maximal 50 mg pro Dosis jede Woche.
Das Ansprechen auf die Behandlung wurde nach 12 Wochen Therapie beurteilt und wurde durch den Anteil der Patienten definiert, die den coprimären Endpunkt eines sPGA-Wert von "0" (frei von) oder "1" (minimal) mit einer Verbesserung von mindestens 2 Punkten gegenüber dem Ausgangswert (Baseline) erreichten, und den Anteil der Patienten, die eine Reduktion des PASI-Wertes von mindestens 75% (PASI 75) gegenüber Baseline erreichten.
Andere evaluierte Ergebnisse in Woche 12 umfassten den Anteil der Patienten, die PASI 90, PASI 100, sPGA von "0" erreichten, und eine Verbesserung des Schweregrads des Juckreizes erreichten, gemessen durch eine Reduktion von mindestens 4 Punkten auf einer 11-Punkte-Juckreiz-Bewertungsskala (itch Numeric Rating Scale, itch NRS).
Die Patienten hatten einen mittleren Baseline PASI-Wert von 17, mit Werten zwischen 12 und 49. Der Baseline sPGA-Wert lag bei 49% der Patienten bei schwer oder sehr schwer. Von allen Patienten erhielten 22% eine vorherige Phototherapie, 32% eine vorherige konventionelle systemische Therapie und 4% eine Biologika-Vortherapie zur Behandlung der Psoriasis.
Die Daten des klinischen Ansprechens sind in Tabelle 4 aufgeführt.
Tabelle 4: Ergebnisse der Wirksamkeit nach 12 Wochen bei pädiatrischen Patienten mit Plaque Psoriasis, NRI
Endpunkte Taltza (N=115) n (%) Placebo (N=56) n (%) Unterschied zu Placebo
(95% CI)
sPGA "0" (frei von) oder 93 (81) 6 (11) 70.2 (59.3, 81.0) d
"1" (minimal)b
sPGA "0" (frei von) 60 (52) 1 (2) 50.4 (40.6, 60.2) d
PASI 75c 102 (89) 14 (25) 63.7 (51.0, 76.4)d
PASI 90 90 (78) 3 (5) 72.9 (63.3, 82.5)d
PASI 100 57 (50) 1 (2) 47.8 (38.0, 57.6)d
Itch NRS (≥4 Punkte 59 (71) 8 (20) 51.1 (35.3, 66.9) d
Verbesserung) c
Abkürzungen: N =Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation.
a In Woche 0 erhielten die Patienten 160 mg (Körpergewicht [KG] >50kg), 80 mg (KG 25-50 kg), oder 40 mg (KG < 25 kg) Taltz, danach 80 mg (KG >50kg), 40 mg (KG 25-50 kg) oder 20 mg (KG < 25 kg) alle 4 Wochen, für 12 Wochen.
b Co-primäre Endpunkte.
c Itch NRS (≥4 Verbesserung) bei Patienten mit Baseline itch NRS ≥4. Anzahl der ITT-Patienten mit Baseline itch NRS-Wert ≥4 wie folgt: Taltz n = 83; Placebo n = 40.
d p<0.001
Insgesamt wurden 87 pädiatrische Patienten mit schwerer Plaque-Psoriasis (PASI ≥20 oder sPGA ≥4) zu Ixekizumab Q4W (38 Patienten), Etanercept Q1W (30 Patienten) oder Placebo (19 Patienten) randomisiert.
In Woche 12 wurden Verbesserungen für die Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe beobachtet, gemessen mit PASI 75 (84.2%, 63.3%, 26.3%) und sPGA (0,1) (76.3%, 53.3% und 5.3%).
In den wichtigsten sekundären Endpunkte zeigten sich statistisch signifikante Verbesserungen in der Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe, gemessen an: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) und sPGA (0) (63.2%, 16.7%, 0 ).
Patienten in der Ixekizumab-Behandlungsgruppe hatten ein höheres CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) Ansprechen in Woche 12 (NRI) im Vergleich zu Placebo. Der Unterschied zwischen den Behandlungsgruppen zeigte sich bereits ab Woche 4.
Die doppelblinde Behandlungsphase betrug 12 Wochen. Darauf folgte die 48-wöchige offene Erhaltungsphase (Phase 3) und eine 48-wöchige Verlängerungsphase (Phase 4) für Patienten aus Ländern ausserhalb der EU, unabhängig vom Ansprechen, und Non-Responder aus der EU (definiert als diejenigen, die sPGA 0,1 nicht erreichten).
Von den 115 Patienten, die Taltz in Woche 0 erhielten, erhielten 94 Patienten weiterhin Taltz bis Woche 108 und wurden in die Primärpopulation zur Wirksamkeit von Ixekizumab aufgenommen. Von den Patienten, die die Bewertung in Woche 108 abgeschlossen haben, erreichten 64 Patienten (68.1 %) PASI 90, 72 Patienten (76.6 %)) PASI 75 und 64 Patienten (68.1 %) sPGA (0,1) in Woche 108. Es liegen keine ausreichenden Daten vor, um das mögliche Risiko eines Aufflammens der Erkrankung nach Absetzen von Taltz bei Kindern zu beurteilen.
Psoriasis-Arthritis
Die Sicherheit und Wirksamkeit von Taltz wurden an 780 Patienten in zwei randomisierten, doppelblinden placebo-kontrollierten Phase III Studien bei Patienten mit aktiver Psoriasis-Arthritis (≥3 geschwollene und ≥3 schmerzhafte Gelenke) untersucht. Bei den Patienten in diesen Studien wurde die Diagnose einer Psoriasis Arthritis (CASPAR Klassifizierungskriterien, Classification Criteria for Psoriatic Arthritis) im Median 5.33 Jahren vor Studieneinschluss gestellt. Randomisierte Patienten hatten gleichzeitig auch Plaque-Psoriasis Hautläsionen (94.0 %) oder eine dokumentierte Plaque-Psoriasis in der Anamnese. Bei Studienbeginn litten 12.1 % der Patienten an einer mittelschweren bis schweren Plaque-Psoriasis. Von den Psoriasis-Arthritis Patienten hatten zu Studienbeginn mehr als 58.9 % eine Enthesitis bzw. 22.3 % eine Daktylitis. Für beide Studien war der primäre Endpunkt das American College of Rheumatology (ACR) 20 Ansprechen in Woche 24, gefolgt von einer Langzeit-Verlängerungsphase von Woche 24 bis Woche 156 (3 Jahre).
In der Psoriasis-Arthritis Studie 1 (SPIRIT-P1), wurden Biologika -naive Patienten mit aktiver Psoriasis-Arthritis in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Adalimumab 40 mg alle 2 Wochen (aktive Kontrollgruppe), Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 85.3% der Patienten in dieser Studie hatten eine vorherige Behandlung mit ≥1 cDMARD erhalten. 53% der Patienten haben gleichzeitig MTX mit einer mittleren wöchentlichen Dosis von 15.8 mg angewendet. 67% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichenden Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Adalimumab oder Placebo erhielten, wurden in Woche 16 oder 24 basierend auf den Responderstatus 1:1 auf Taltz Q2W oder Q4W neu randomisiert. 243 Patienten beendeten die Verlängerungsphase der Studie mit Taltz über 3 Jahre.
Psoriasis-Arthritis Studie 2 (SPIRIT-P2) schloss Patienten ein, die zuvor mit einem anti-TNF Wirkstoff behandelt wurden und den anti-TNF Wirkstoff wegen ungenügender Wirksamkeit oder Unverträglichkeit (anti-TNF-IR Patienten) abgebrochen haben. Patienten wurden in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 56% und 35% der Patienten haben unzureichend auf 1 TNF bzw auf ≥2 TNF angesprochen. In SPIRIT-P2 wurden 363 Patienten ausgewertet, von denen 41% gleichzeitig MTX bei einer mittleren wöchentlichen Dosis von 16.1 mg angewendet haben. 73.2% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichendem Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Placebo erhielten, wurden in der Woche 16 oder 24 basierend auf den Responderstatus 1: 1 auf Taltz Q2W oder Q4W neu randomisiert. 168 Patienten beendeten die Verlängerungsphase der Studie mit Taltz über 3 Jahre.
Anzeichen und Symptome
Die Behandlung mit Taltz zeigte in der Woche 24 eine signifikante Verbesserung der Krankheitsaktivität im Vergleich zu Placebo (siehe Tabelle 5).
Tabelle 5. Wirksamkeitsergebnisse in SPIRIT-P1 und SPIRIT-P2 in Woche 24
SPIRIT-P1 SPIRIT-P2
Endpunkte Unterschied von Unterschied von
Placebo in Ansprechr Placebo in Ansprechr
ate (95% CI) ate (95% CI)
PBO (N = 106) Taltz Q4W (N = 107) Taltz Q2W (N = 103) Taltz Q4W Taltz Q2W PBO (N = 118) Taltz Q4W (N = 122) Taltz Q2W (N = 123) Taltz Q4W Taltz Q2W
ACR 20 Ansprechen,
n (%)
Woche 24 32 (30.2) 62 (57.9) 64 (62.1) 27.8 (15.0, 40.6)* 31.9 (19.1, 44.8) * 23 (19.5) 65 (53.3) 59 (48.0) 33.8 (22.4, 45.2) * 28.5 (17.1, 39.8) *
ACR 50 Ansprechen,
n (%)
Woche 24 16 (15.1) 43 (40.2) 48 (46.6) 25.1 (13.6, 36.6) * 31.5 (19.7, 43.3) * 6 (5.1) 43 (35.2) 41 (33.3) 30.2 (20.8, 39.5) * 28.3 (19.0, 37.5) *
ACR 70 Ansprechen,
n (%)
Woche 24 6 (5.7) 25 (23.4) 35 (34.0) 17.7 (8.6, 26.8) * 28.3 (18.2, 38.5) * 0 27 (22.1) 15 (12.2) 22.1 (14.8, 29.5) * 12.2 (6.4, 18.0) *
Abkürzungen: ACR 20/50/70 = American College of Rheumatology 20%/50%/70% Ansprechrate; CI = confidence interval; Q4W = Taltz 80 mg alle 4 Wochen; Q2W = Taltz 80 mg alle 2 Wochen; N = Anzahl der Patienten in der untersuchten Population; n = Anzahl der Patienten in der spezifischen Kategorie; NRI = non-responder imputation; PBO = Placebo.
Hinweis: Patienten die in Woche 16 die Rettungstherapie bekommen haben oder die Therapie abgebrochen hatten, oder bei denen es fehlende Daten gab, wurden als Non-Responder für die Analysen der Woche 24 gezählt.
Gleichzeitige cDMARDs Anwendung schloss MTX, Leflunomid und Sulfasalazin ein.
* p<0.001 im Vergleich zu Placebo.
Bei Patienten mit bereits vorhandener Daktylitis oder Enthesitis führte die Behandlung mit Taltz Q4W zu einer Verbesserung der Daktylitis bzw. der Enthesitis in Woche 24 im Vergleich zu Placebo (p<0.01).
Bei Patienten mit gleichzeitiger Plaque-Psoriasis (≥3% BSA) und Psoriasis-Arthritis führte die Behandlung mit Taltz Q4W zu einer Verbesserung psoriatischer Hautläsionen, wie anhand von PASI 75-, PASI 90- und PASI 100-Ansprechen in Woche 24 gezeigt werden konnte (p < 0.001).
Bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis und Psoriasis-Arthritis zeigte Taltz Q2W Dosierungsschema signifikante höhere Ansprechrate für PASI 75, PASI 90 und PASI 100 im Vergleich zu Placebo (p<0.001) und einen klinisch bedeutsamen Nutzen gegenüber dem Q4W Dosierungsschema.
Das Ansprechen auf die Behandlung mit Taltz war bereits in Woche 1 für ACR 20, in Woche 4 für ACR 50 signifikant höher als das Ansprechen auf Placebo und in Woche 8 für ACR 70 und wurde durchgängig bis Woche 24 beibehalten; die Effekte hielten bei jenen Patienten, die in der Studie verblieben, 3 Jahre lang an.
Abbildung 2. ACR 20 Ansprechen in SPIRIT-P1 über die Zeit bis zur Woche 24

Für Taltz Q2W und Q4W: b p<0.01 und c p<0.001 im Vergleich zu Placebo.
In SPIRIT-P1 und SPIRIT-P2 wurden ähnliche Ansprechen für ACR 20/50/70 in Psoriasis-Arthritis Patienten beobachtet, ungeachtet der Tatsache ob eine Begleittherapie mit cDMARDs, einschliesslich MTX, vorhanden war oder nicht.
In SPIRIT-P1 und SPIRIT-P2 konnten in allen Komponenten der ACR Scores, einschliesslich der Patientenbeurteilung zum Schmerz, Verbesserungen gezeigt werden.
In SPIRIT-P1 wurde anhaltend die Wirksamkeit über 52 Wochen anhand der Ansprechraten auf die Parameter ACR 20/50/70, Besserung der Enthesitis, Besserung der Dactylitis und PASI 75/90 /100, gezeigt.
Die Wirksamkeit und Sicherheit von Taltz wurde ungeachtet von Alter, Geschlecht, Abstammung, Dauer der Erkrankung, Körpergewicht zu Studienbeginn, Psoriasis Beteiligung zu Studienbeginn, CRP Ausgangwert, DAS28-CRP Ausgangwert, Begleittherapie mit Kortikosteroiden, und vorherige Behandlung mit Biologika gezeigt. Taltz war wirksam bei Biologika-naiven, Biologika-exponierten und Biologika Non-Responder Patienten.
Es gab zu wenige Patienten mit Arthritis mutilans, isolierter Arthritis mit Befall der distalen Interphalangealgelenken (DIP) und begleitender Spondylitis in den Zulassungsstudien, um aussagekräftige Resultate in diesen spezifischen Untergruppen zu erhalten.
In SPIRIT-P1 vollendeten 63 Patienten 3 Jahre Therapie mit Q4W Ixekizumab. Unter den 107 Patienten, die auf Ixekizumab Q4W randomisiert wurden (NRI Analyse der ITT Population), zeigten in Woche 156 54 Patienten (50 %) ein ACR20, 41 Patienten (38 %) ein ACR50, 29 Patienten (27 %) ein ACR70 und 36 Patienten (34 %) ein MDA Ansprechen.
In SPIRIT-P2 vollendeten 70 Patienten 3 Jahre Therapie mit Q4W Ixekizumab. Unter den 122 Patienten, die auf Ixekizumab Q4W randomisiert wurden (NRI Analyse der ITT Population), zeigten in Woche 156 56 Patienten (46 %) ein ACR20, 39 Patienten (32 %) ein ACR50, 24 Patienten (20 %) ein ACR70 und 33 Patienten (27 %) ein MDA Ansprechen.
Radiographisches Ansprechen
In SPIRIT-P1 wurde die Hemmung der Progression struktureller Gelenkschäden radiographisch untersucht, und anhand von modifiziertem Total Sharp Score (mTSS) und seinen Komponenten Erosions Score (ES) und dem Joint Space Narrowing Score (JSN) in den Wochen 24 und 52 im Vergleich zu den Ausgangswerten gemessen.
Taltz hemmte die Progression struktureller Gelenkschäden (mTSS) im Vergleich zu Placebo in Woche 24.
Der Prozentanteil der Patienten ohne radiographische Progression struktureller Gelenkschäden (definiert als eine mTSS Veränderung gegen dem Ausgangswert ≤0.5) von der Randomisierung bis zur Woche 24 betrug 94.8% für Taltz Q2W, 89.0 % für Taltz Q4W, und 77.4% für Placebo. Die Hemmung der strukturellen Schäden wurde mit der Taltz-Behandlung bis zur Woche 52 beibehalten.
Körperliche Funktion und gesundheitsrelevante Lebensqualität
In SPIRIT-P1 und SPIRIT-P2 zeigten mit Taltz Q2W (p<0.001) und Q4W (p<0.001) behandelte Patienten eine durch Health Assesment Questionnaire-Disability Index (HAQ-DI) bewertet Verbesserung der körperlichen Funktion in Woche 24. Diese wurde in SPIRIT-P1 bis Woche 52 beibehalten.
Patienten, die mit Taltz behandelt wurden, berichteten von Verbesserungen in der gesundheitsrelevanten Lebensqualität, die mithilfe des Physical Component Summary des Short Form-36 Health Survey (SF-36 PCS) Score gemessen wurden (p < 0.001). Auch eine statistisch signifikante Verbesserung der Müdigkeit konnte im Fatigue Severity NRS gezeigt werden.
Ankylosierende Spondylitis (radiographische axiale Spondyloarthritis, Morbus Bechterew)
Die Sicherheit und Wirksamkeit von Taltz wurden bei 657 Patienten in 2 randomisierten, doppelblinden, placebokontrollierten Studien (COAST-V und COAST-W) bei erwachsenen Patienten ≥18 Jahren mit ankylosierender Spondylitis untersucht. Die Patienten hatten eine aktive Krankheit definiert nach dem Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und trotz einer Therapie mit NSAID Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala. In beiden Studien hatten die Patienten bei Baseline Symptome einer ankylosierenden Spondylitis über einen Zeitraum von durchschnittlich 17 Jahren (Median von 16 Jahren). Bei Baseline erhielten etwa 32% der Patienten begleitend eine cDMARD-Therapie (in COAST-V und in COAST-W: Methotrexat 8.5% und 13.0%, Sulfasalazin 28.5% und 14.6%, Hydroxychloroquin 0.6% und 0).
Primärer Endpunkt in beiden Studien war der Prozentsatz von Patienten, die in Woche 16 ein Assessment of Spondyloarthritis International Society 40 (ASAS40)-Ansprechen erreichten.
COAST-V bewertete 341 biologisch-naive Patienten, die entweder mit Taltz 80 mg oder 160 mg in Woche 0, gefolgt von 80 mg alle 2 Wochen (Q2W) oder 4 Wochen (Q4W), Adalimumab 40 mg alle 2 Wochen oder mit Placebo behandelt wurden. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W). Die mit Adalimumab behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (80 mg Q2W oder Q4W).
COAST-W bewertete 316 Patienten, die Vorerfahrungen mit 1 oder 2 TNF-Hemmern hatten (90% hatten ein unzureichendes Ansprechen, und 10% hatten eine Unverträglichkeit auf TNF-Hemmer). Alle Patienten wurden mit Taltz 80 oder 160 mg in Woche 0 und danach mit 80 mg Q2W oder Q4W oder mit Placebo behandelt. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W).
Klinisches Ansprechen
In beiden Studien wiesen die mit Taltz 80 mg Q2W oder 80 mg Q4W behandelten Patienten in Woche 16 grössere Verbesserungen im ASAS40- und ASAS20-Ansprechen im Vergleich zu Placebo auf (Tabelle 6). Das Ansprechen war bei den Patienten unabhängig von den begleitenden Therapien vergleichbar. Bei COAST-W zeigte sich das Ansprechen unabhängig von der Anzahl der früheren Therapien mit TNF-Hemmern.
Tabelle 6. Wirksamkeitsergebnisse in COAST-V und COAST-W in Woche 16
COAST-V, Biologika-n COAST-W, TNF-Hemmer-
aiv erfahren
Taltz 80 mg Q4Wa Placebo (N=87) Unterschied zu Adalimumab 40 mg Taltz 80 mg Q4Wc Placebo (N=104) Unterschied zu
(N=81) Placebo (95% CI) Q2W (N= 90) (N=114) Placebo (95% CI)
ASAS20-Ansprechenb, 52(64.2%) 35(40.2%) 24.0(9.3, 38.6)** 53(58.9%) 55(48.2%) 31(29.8%) 18.4(5.7, 31.1)**
n (%), NRI
ASAS40-Ansprechenb,c 39(48.1%) 16(18.4%) 29.8(16.2, 43.3)*** 32(35.6%) 29(25.4%) 13(12.5%) 12.9(2.7, 23.2)*
, n (%), NRI
ASDAS Ausgangswert 3.7[-1.4] 3.9[-0.5] -1.0(-1.3, -0.7)*** 3.7[-1.3]*** 4.2[-1.2] 4.1[-0.1] -1.1(-1.3, -0.8)***
[CFB]
BASDAI-Score Ausgang 6.8[-2.9] 6.8[-1.4] -1.5(-2.1, -0.9)*** 6.7[-2.5]*** 7.5[-2.2] 7.3[-0.9] -1.2(-1.8, -0.7)***
swert [CFB]
MRI Spine SPARCCd 14.5[-11.0] 15.8[-1.5] -9.5(-12.6, -6.4)*** 20.0[-11.6]*** 8.3[-3.0] 6.4[3.3] -6.3(-10.0, -2.5)**
Ausgangswert [CFB]
BASDAI50e (%), NRI 42 17 25(11, 38)*** 32* 22 10 12(3, 22)*
ASDAS <2.1 (%) 43 13 31(18, 43)*** 38*** 18 5 13(5, 21)**
(Geringe Krankheitsa
ktivität), NRI
ASAS HI Ausgangswert 7.5[-2.4] 8.1[-1.3] -1.1(-2.0, -0.3)* 8.2[-2.3]* 10.0[-1.9] 9.0[-0.9] -1.0(-1.9, -0.1)*
[CFB]
SF-36 PCS Ausgangswe 34.0[7.7] 32.0[3.6] 4.1(1.9, 6.2)*** 33.5[6.9]** 27.5[6.6]*** 30.6[1.4] 5.2(3.0, 7.4)***
rt [CFB]
Abkürzungen: N = Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation; Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB (change from baseline) = LSM (least square mean)-Änderungen zum Ausgangswert in Woche 16; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 discovertebral unit scale), SF-36 PCS = Short Form health survey physical component summary.
a In Woche 0 erhielten Patienten 80 mg oder 160 mg Taltz.
b ASAS20-Ansprechen: Verbesserung um ≥20% und absolute Verbesserung um ≥1 Einheit (Skala von 0–10) gegenüber Baseline bei ≥3 von 4 Bereichen (Patient Global, Spinal Pain, Function, Inflammation), wobei keine Verschlechterung des verbleibenden Bereichs (definiert als Verschlechterung um ≥20% und durchschnittliche Verschlechterung um ≥1 Einheit, Skala von 0–10) zu verzeichnen ist. ASAS40-Ansprechen: Verbesserung um ≥40% und absolute Verbesserung gegenüber Baseline um ≥2 Einheiten in ≥3 von 4 Bereichen ohne Verschlechterung im verbleibenden Bereich.
c Primärer Endpunkt.
d Die Anzahl der ITT-Patienten mit MRT-Daten zu Studienbeginn lautet: COAST-V: Taltz, n = 81; PBO, n = 82, ADA, n=85. COAST-W: Taltz, n = 58; PBO, n = 51.
e BASDAI50-Ansprechen ist definiert als eine Verbesserung von ≥50% des BASDAI-Score gegenüber dem Ausgangswert.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
In Woche 16 gab es klinisch signifikante Verbesserungen der Hauptkomponenten der ASAS40-Ansprechkriterien (Wirbelsäulenschmerzen, BASFI, globale Beurteilung des Patienten, Steifheit) und anderer Messungen der Krankheitsaktivität, einschliesslich CRP.
Der Prozentsatz der Patienten, die bei Untersuchungen in COAST-V und COAST-W ein ASAS40-Ansprechen erreichten, ist in Abbildung 3 dargestellt.
Abbildung 3. ASAS40-Ansprechen in COAST-V und COAST-W bis Woche 16, NRIa
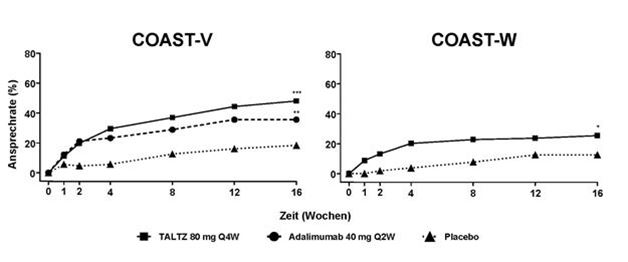
a Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
Vergleichbare ASAS40-Ansprechraten wurden bei Patienten unabhängig von CRP zu Studienbeginn, von ASDAS-Scores zu Studienbeginn und von MRI Spine SPARCC-Scores festgestellt. Der ASAS40 Ansprechen wurde unabhängig von Alter, Geschlecht, Rasse, Krankheitsdauer, Körpergewicht zu Studienbeginn, BASDAI-Score zu Studienbeginn und vorheriger biologischer Behandlung nachgewiesen.
In COAST-V und COAST-W wurde die Wirksamkeit bis zu Woche 52 beibehalten, wie dies durch die oben in Woche 16, dargestellten Endpunkte, einschliesslich ASAS20-, ASAS40-, ASDAS-, BASDAI-, BASFI- und ASAS HI-Ansprechen beurteilt wurde.
Gesundheitsbezogene Ergebnisse
Es zeigten sich bereits ab Woche 1 Verbesserungen bei Wirbelsäulenschmerzen gegenüber Placebo, und diese wurden bis Woche 16 aufrechterhalten (Taltz vs. Placebo: COAST-V -3.2 vs. -1.7; COAST-W -2.4 vs. -1.0); Müdigkeit und Wirbelsäulenbeweglichkeit zeigten in Woche 16 Verbesserungen gegenüber Placebo. Die Verbesserungen von Wirbelsäulenschmerzen, Müdigkeit und Wirbelsäulenbeweglichkeit blieben bis zu Woche 52 erhalten.
Nicht-radiographische axiale Spondyloarthritis
Die Wirksamkeit und Sicherheit von Taltz wurden in einer randomisierten, doppelblinden Studie mit einem 52wöchigen placebokontrollierten Zeitraum (COAST-X) bei 303 Patienten ≥18 Jahren mit aktiver axialer Spondyloarthritis seit mindestens 3 Monaten beurteilt. Die Patienten mussten objektive Anzeichen einer Entzündung, angezeigt durch erhöhte Werte des C-reaktiven Proteins (CRP) und/oder Sakroiliitis bei der Magnetresonanztomographie (MRT), und keinen definitiven radiographischen Nachweis von strukturellen Schäden an den Iliosakralgelenken gehabt haben. Die Patienten hatten eine aktive Krankheit wie durch den Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 definiert, sowie Wirbelsäulenschmerzen ≥4 auf einer Numerical Rating Scale (NRS) von 0 bis 10, obwohl sie mit NSAID behandelt wurden. Die Patienten wurden entweder mit Taltz 80 mg oder 160 mg in Woche 0, gefolgt von 80 mg alle 2 Wochen (Q2W) oder 80 mg alle 4 Wochen (Q4W) oder mit Placebo behandelt. Die Einleitung und/oder Dosisanpassung von Begleitmedikationen (NSAID, cDMARD, Kortikosteroide, Analgetika) war ab Woche 16 erlaubt.
Bei Studienbeginn hatten die Patienten durchschnittlich seit 11 Jahren Symptome einer nicht-radiographischen axialen Spondyloarthritis. Etwa 39 % der Patienten erhielten begleitend eine Behandlung mit cDMARD.
Der primäre Endpunkt war der Prozentsatz von Patienten, die in Woche 16 ein ASAS40-Ansprechen erreichten.
Klinisches Ansprechen
In Woche 16 erreichten im Vergleich zu Placebo höhere Anteile der mit Taltz 80 mg Q4W behandelten Patienten ein ASAS40-Ansprechen (Tabelle 7). Das Ansprechen war unabhängig von Begleittherapien vergleichbar.
Tabelle 7. Wirksamkeitsergebnisse in Woche 16 in COAST-X, NRI
Taltz 80 mg Q4Wa Placebo (N=105) Unterschied zu Placebo
(N=96) (95% CI) f
ASAS20-Ansprechen b, n (%), 52(54.2%) 41(39.0%) 15.1(1.5, 28.8)*
NRI
ASAS40-Ansprechen b,c, n 34(35.4%) 20(19.0%) 16.4(4.2, 28.5)**
(%), NRI
ASDAS Ausgangswert [CFB] 3.8[-1.1] 3.8[-0.6] -0.5(-0.8, -0.3)***
BASDAI Score Ausgangswert 7.0[-2.2] 7.2[-1.5] -0.7(-1.3, -0.1)*
[CFB]
MRI SIJ SPARCCd Ausgangswert 5.1[-3.4] 6.3[-0.3] -3.1(-4.6, -1.6)***
[CFB]
ASDAS <2.1, n (%) (Low 26(27.7%) 13(12.4%) 15.3(4.3, 26.3)**
Disease Activity), NRIe
SF-36 PCS Ausgangswert [CFB] 33.5[8.1] 32.6[5.2] 2.9(0.6, 5.1)*
Abkürzungen: N = Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation, ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB (change from baseline) = LSM (least square mean)-Änderungen zum Ausgangswert in Woche 16; MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint, SF-36 PCS = Short Form health survey physical component summary.
Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
a In Woche 0 erhielten die Patienten 80 mg oder 160 mg Taltz
b ASAS20-Ansprechen: Verbesserung um ≥20% und absolute Verbesserung um ≥1 Einheit (Skala von 0–10) gegenüber Baseline bei ≥3 der folgenden 4 Bereiche ((Patient Global, Spinal Pain, Function, Inflammation), wobei keine Verschlechterung um ≥20% und um ≥1 Einheit (Skala 0–10) im verbleibenden Bereich zu verzeichnen ist. ASAS40-Ansprechen: Verbesserung um ≥40% und absolute Verbesserung gegenüber Baseline um ≥2 Einheiten in ≥3 von 4 Bereichen ohne Verschlechterung im verbleibenden Bereich.
c Primärer Endpunkt in Woche 16.
d Die Anzahl der ITT-Patienten mit MRT-Daten zu Studienbeginn und in Woche 16 lautet: Taltz, n = 85; PBO, n = 90.
e Patienten mit fehlenden Daten wurden als Non-Responder gewertet. Die Prozentsätze basieren auf der Anzahl der Patienten in der ITT-Population mit ASDAS ≥2.1 zu Studienbeginn.
f Die berichteten Werte werden als Unterschied in % (95 % CI) für kategoriale Variablen und Unterschied in LSM (95 % CI) für kontinuierliche Variablen angegeben.
* p<0.05; ** p<0.01, *** p<0.001 im Vergleich zu Placebo.
Die Verbesserung der Hauptkomponenten der ASAS40-Ansprechkriterien (Wirbelsäulenschmerzen, BASFI, globale Beurteilung des Patienten, Steifheit) und anderer Messungen der Krankheitsaktivität zeigte in Woche 16 eine signifikante klinische Verbesserung.
Der Prozentsatz der Patienten, die bei Untersuchungen ein ASAS40-Ansprechen erreichten, ist in Abbildung 4 dargestellt.
Abbildung 4. ASAS40-Ansprechen bis Woche 16 in COAST-X, NRIa
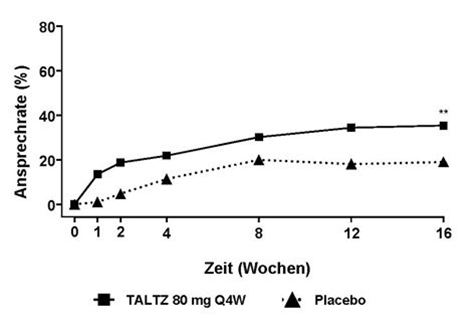
a Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
** p<0.01 im Vergleich zu Placebo.
In COAST-X wurde die Wirksamkeit bis Woche 52 beibehalten.
Langzeit-Ergebnisse bei axialer Spondyloarthritis
Es wurde den Patienten, die eine der drei pivotalen Studien COAST-V/W/X (52 Wochen) beendet hatten, angeboten, an der Langzeit-Verlängerungs- und randomisierten Absetzstudie teilzunehmen (COAST-Y, mit 350 Patienten auf Taltz Q4W und 423 Patienten auf Q2W). Von den 157/773 (20.3 %) mit erreichter Remission (definiert als Ankylosing Spondylitis Disease Activity Score [ASDAS] < 1.3 zumindest einmal, und kein ASDAS Score ≥2.1 in den Wochen 16 und 20) wurden 155 Patienten, die zuvor bis zu 76 Wochen (52 Wochen aus Studie COAST-V/W/X + 24 Wochen aus COAST-Y) mit Taltz behandelt worden waren, in Woche 24 der COAST-Y-Studie randomisiert (randomisierte Absetzpopulation: Placebo, N = 53; Taltz Q4W, N = 48; und Taltz Q2W, N = 54); von diesen schlossen 148 (95.5 %) den Besuch in Woche 64 ab (Placebo, N = 50; Taltz Q4W, N = 47; Taltz Q2W, N = 51). Der primäre Endpunkt war der Anteil an Patienten in der randomisierten Absetzpopulation, die keinen Krankheitsschub während der Wochen 24-64 entwickelten (kombiniert für die Q2W- und Q4W-Gruppen versus Placebo). Ein signifikant grösserer Anteil an Patienten (non responder imputation, NRI) in den kombinierten Taltz-Gruppen (83.3 % (85/102); p < 0.001) und Taltz Q4W (83.3 % (40/48), p = 0.003) zeigte keinen Krankheitsschub während der Wochen 24-64 verglichen mit denen, die von Taltz auf Placebo umgestiegen waren (54.7 % (29/53)). Taltz (in beiden kombinierten Gruppen bzw. In der Taltz Q4W-Gruppe) verzögerte signifikant die Zeit bis zu einem Krankheitsschub (Log-Rank Test p < 0.001 bzw. p < 0.01) im Vergleich zu Placebo.
PharmakokinetikDie pharmakokinetische Eigenschaften von Taltz waren bei den Indikationen Plaque-Psoriasis, Psoriasis-Arthritis, und axiale Spondyloarthritis vergleichbar.
Absorption
Bei Patienten mit Psoriasis wurden nach subkutaner (SC) Gabe einer Ixekizumab Einzeldosis im Dosisbereich von 5 bis 160 mg innerhalb von 4 bis 7 Tagen mittlere maximale Serumkonzentrationen erreicht. Die mittlere (Standardabweichung) maximale Serumkonzentration (Cmax) von Ixekizumab nach einer Startdosis von 160 mg betrug 19.9 (8.15) µg/ml.
Nach einer Startdosis von 160 mg wurde der Steady-State mit dem 80 mg Q2W Dosierungsschema in der Woche 8 erreicht. Die mittleren (Standardabweichung) geschätzten Cmax,ss, und C trough,ss betrugen 21.5 (9.16) µg/ml und 5.23 (3.19) µg/ml.
Nach Umstellung der Dosierungsschema in Woche 12 von 80 mg Q2W auf 80 mg Q4W, würde der Steady-State nach etwa 10 Wochen erreicht werden. Die mittleren (Standardabweichung) geschätzten Cmax,ss, und C trough,ss betrugen 14.6 (6.04) µg/ml und 1.87 (1.30) µg/ml.
Die durchschnittliche absolute Bioverfügbarkeit von Ixekizumab lag über die Analysen hinweg geschätzt in einem Bereich von 54 % bis 90 %. Nach subkutaner Injektion in den Bauch, Oberarm oder Oberschenkel werden ähnliche Ixekizumab Serumkonzentrationen erreicht.
Distribution
Basierend auf den Populations-pharmakokinetischen Analysen betrug das mittlere gesamte Verteilungsvolumen im Steady-State 7.11 Liter.
Metabolismus
Ixekizumab ist ein monoklonaler Antikörper, sodass in gleicher Art wie bei endogenen IgGs ein Abbau über katabole Prozesse in kleine Peptide und Aminosäuren erwartet wird.
Elimination
In der Populations-pharmakokinetischen Auswertung betrug die mittlere Serum-Clearance 0.0161 l/Std. Die Clearance ist von der Dosis unabhängig. Basierend auf Schätzungen der Populations-pharmakokinetischen Auswertung betrug die mittlere Eliminations-Halbwertszeit bei Patienten mit Plaque Psoriasis 13 Tage.
Linearität/Nicht Linearität
Die Exposition (AUC) im Dosisbereich von 5 bis 160 mg als subkutane Injektion stieg proportional zur verabreichten Dosis an.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Spezifische klinische pharmakologische Studien zur Untersuchung der Auswirkungen von Leberfunktionsinsuffizienz auf die Pharmakokinetik von Ixekizumab wurden nicht durchgeführt.
Nierenfunktionsstörungen
Spezifische klinische pharmakologische Studien zur Untersuchung der Auswirkungen von Nierenfunktionsinsuffizienz auf die Pharmakokinetik von Ixekizumab wurden nicht durchgeführt.
Ältere Patienten
Basierend auf einer populationspharmakokinetischen Analyse mit einer begrenzten Anzahl älterer Patienten (n = 94 mit ≥65 Jahre und n = 12 mit ≥75 Jahre) war die Clearance bei älteren Patienten mit der Clearance bei Patienten unter 65 Jahren vergleichbar.
Kinder und Jugendliche
Pädiatrische Psoriasis-Patienten (Alter 6 bis weniger als 18 Jahre) wurden 12 Wochen lang mit Ixekizumab im empfohlenen pädiatrischen Dosierungsschema behandelt. Patienten mit einem Gewicht von >50 kg und 25 bis 50 kg hatten in Woche 12 eine mittlere ±SD Steady-state-Talspiegelkonzentration von 3.8 ± 2.2 µg/ml bzw. 3.9 ± 2.4 µg/ml. Diese Konzentrationen waren vergleichbar mit Konzentrationen bei erwachsenen Patienten, die mit Ixekizumab 80 mg Q4W dosiert wurden, wobei die mittlere ±SD Steady-state-Talspiegelkonzentration 3.5 ± 2.2 μg / ml betrug.
Präklinische DatenNicht-klinische Daten von Cynomolgus Affen zeigten basierend auf Studien zur Toxizität bei wiederholter Gabe, pharmakologischen Auswertungen zur Sicherheit, sowie Studien zur Entwicklungs- und Reproduktionstoxizität keine besonderen Gefahren für den Menschen.
Toxizität bei wiederholter Verabreichung
Bei Cynomolgus Affen rief die subkutane Gabe von Ixekizumab in Dosierungen von bis zu 50 mg/kg/Woche über 39 Wochen keine Organtoxizität oder unerwünschten Wirkungen auf die Immunfunktion hervor (z.B. T-Zell-abhängige Antikörper-Antworten und NK-Zell-Aktivität). Die wöchentliche subkutane Gabe von 50 mg/kg bei Affen entspricht etwa dem 19-Fachen der Taltz 160 mg Startdosis und führt bei Affen zu einer Exposition (AUC) die mindestens 61-fach höher ist als die mittlere vorhergesagte Exposition beim Menschen im Steady-State bei empfohlener Dosierung.
Mutagenität / Karzinogenität
Nicht-klinische Studien zur Untersuchung des karzinogenen oder mutagenen Potentials von Ixekizumab wurden nicht durchgeführt.
Reproduktionstoxizität
Bei geschlechtsreifen Cynomolgus Affen, die über 13 Wochen hinweg wöchentlich 50 mg/kg Ixekizumab subkutan erhalten haben, wurden keine Wirkungen auf die Reproduktionsorgane, den Menstruationszyklus oder die Spermien beobachtet.
In Studien zur Entwicklungstoxizität führte die subkutane Gabe von Ixekizumab in Dosierungen bis zu 50 mg/kg einmal wöchentlich bei Cynomolgus Affen vom Beginn der Organogenese bis entweder kurz vor Ende der Schwangerschaft oder bis zur Geburt nicht zu Embryotoxizität oder Teratogenität und hatte keine Wirkungen auf den Geburtsverlauf oder die morphologische, funktionelle oder immunologische Entwicklung des Nachwuchs von der Geburt bis zu einem Alter von 6 Monaten. Die höhere Inzidenz von postnatalen Todesfällen beim Affen war in erster Linie auf mütterliche Vernachlässigung oder Frühgeburten zurückzuführen. Es handelt sich dabei um Befunde, die beim Affen häufig auftreten und vermutlich nicht auf Ixekizumab zurückzuführen sind. Wegen der geringen Tierzahlen ist der Vorhersagewert der Studie begrenzt. Es wurde gezeigt, dass Ixekizumab plazentagängig ist und im Blut des Nachwuchses bis zu einem Alter von 6 Monaten vorhanden war.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend (einzeldosiertes Arzneimittel zur subkutanen Anwendung)
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren. Taltz, das eingefroren war, darf nicht verwendet werden.
Temporäre Lagerung: Taltz kann bis zu 5 Tagen ungekühlt bei Temperaturen unter 30°C aufbewahrt werden.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Nicht schütteln.
Taltz ist eine sterile, Konservierungsmittel-freie, klare und farblose bis leicht gelbliche Lösung. Nicht verwenden, wenn Partikel auftreten oder wenn die Lösung wolkig und/oder merklich braun ist.
Die Fertigspritze und der Fertigpen sind zum einmaligen Gebrauch bestimmt.
Die der Patienteninformation beigelegte Bedienungsanleitung der Fertigspritze bzw. des Fertigpens muss sorgfältig beachtet werden.
Vorbereitung von Taltz 40 mg für pädiatrische Patienten von 25 kg bis zu 50 kg
Ixekizumab-Dosen von 40 mg müssen von einer qualifizierten medizinischen Fachperson vorbereitet und verabreicht werden. Verwenden Sie nur die im Handel erhältliche Taltz 80 mg/1 ml-Fertigspritze, wenn Sie die vorgeschriebene pädiatrische Dose von 40 mg vorbereiten.
1.den gesamten Inhalt der Fertigspritze in eine sterile Durchstechflasche aus klarem Glas injizieren. Die Durchstechflasche NICHT schütteln oder schwenken.
2.eine 0.5-ml- oder 1-ml-Einwegspritze und eine sterile Nadel verwenden, um 0.5 ml aus der Durchstechflasche zu entnehmen.
3.die Nadel wechseln und eine 27 Gauge sterile Nadel verwenden, um dem Patienten die Injektion zu verabreichen. Unbenutztes Ixekizumab in der Durchstechflasche entsorgen.
Das vorbereitete Ixekizumab muss innerhalb von 4 Stunden nach dem Durchstechen der sterilen Durchstechflasche bei Raumtemperatur verabreicht werden.
Zulassungsnummer65906, 65907 (Swissmedic).
PackungenTaltz 80 mg Injektionslösung in einer Fertigspritze: 1 (B)
Taltz 80 mg Injektionslösung in einem Fertigpen: 1 (B)
Taltz 40 mg Injektionslösung in einer Fertigspritze: 1 (B)
ZulassungsinhaberinEli Lilly (Suisse) SA, 1214 Vernier/GE.
Stand der InformationMai 2025
|