Eigenschaften/WirkungenATC-Code
L04AC13
Wirkungsmechanismus
Ixekizumab ist ein für Interleukin-17A selektiver, rekombinanter, humanisierter, monoklonaler Antikörper. Ixekizumab ist ein aus CHO (Chinese Hamster Ovary) - Zellen hergestellter modifizierter monoklonaler IgG4 Antikörper, der mit hoher Affinität (< 3 pM) und Spezifität an das proinflammatorische Zytokin Interleukin 17A (sowohl an das IL-17A als auch das Heterodimer IL-17A/F) bindet. Erhöhte Werte von IL-17A wurden mit der Pathogenese verschiedener Autoimmunerkrankungen in Verbindung gebracht. Bei Psoriasis spielt der IL-17A-Ligand eine wichtige Rolle bei der überschiessenden Keratinozytenproliferation und -aktivierung. Die Neutralisierung von IL-17A durch Ixekizumab hemmt diese Wirkungen. IL-17A spielt eine Schlüsselrolle in der Pathogenese von Plaque-Psoriasis und Psoriasis-Arthritis und ist hochreguliert in von Läsionen betroffener Haut im Gegensatz zu nicht von Läsionen betroffener Haut von Plaque Psoriasis Patienten.
Bei der axialen Spondyloarthritis spielt der IL-17A-Ligand eine wichtige Rolle durch die Zunahme der Entzündungen, die zu erosiven Knochenschäden und pathologischer Knochenneubildung führt.
Ixekizumab bindet nicht an die Liganden IL-17B, IL-17C, IL-17D, IL-17E oder IL-17F.
Ixekizumab hat eine geringe Fähigkeit zu Bindung an Fcγ Rezeptoren oder Komponenten des Komplementsystems. In-vitro durchgeführte Bindungsversuche bestätigten, dass Ixekizumab nicht an die humanen Fcγ Rezeptoren I, IIa und IIIa oder die Komplementkomponente C1q bindet.
Pharmakodynamik
Auf Grundlage der Daten einer Phase I Studie mit Biopsien, die aus Psoriasis-betroffener Haut entnommen wurden, gab es vom Ausgangswert bis Tag 43 einen dosisabhängigen Trend zur Verringerung von Epidermisdicke, Anzahl proliferierender Keratinozyten, T-Zellen und dendritischer Zellen sowie Verringerung der lokalen Entzündungsmarker. Als direkte Folge reduziert die Behandlung mit Ixekizumab Erythem, Verhärtung und Schuppung in Plaque-Psoriasis-Läsionen.
Taltz hat eine Senkung des C-reaktiven Proteins-Levels (innerhalb von 1 Woche Behandlung) gezeigt.
Klinische Wirksamkeit
Plaque-Psoriasis
Die Wirksamkeit und Sicherheit von Taltz wurde in drei randomisierten, doppelblinden, placebokontrollierten Phase III Studien bei erwachsenen Patienten mit mittelschwerer bis schwerer Plaque Psoriasis untersucht, die Kandidaten für eine Phototherapie oder systemische Therapie waren (UNCOVER-1, UNCOVER-2 und UNCOVER-3). Die Wirksamkeit und Sicherheit von Taltz wurde auch im Vergleich zu Etanercept untersucht (UNCOVER-2 und UNCOVER-3). Patienten, die ursprünglich auf Taltz randomisiert wurden und die in Woche 12 einen static Physician Global Assesment Responder -Wert von 0 oder 1 (sPGA (0,1)) erreichten, wurden erneut auf Placebo oder Taltz für weitere 48 Wochen (UNCOVER-1 und UNCOVER-2) randomisiert. Auf Placebo, Etanercept oder Taltz randomisierte Patienten die sPGA (0,1) non Responder waren, erhielten Taltz für bis zu 48 Wochen. Zusätzlich wurde die Langzeitwirksamkeit und -sicherheit in allen drei Studien für bis zu 5 Jahre bei Patienten untersucht, die über die gesamte Studienzeit teilgenommen haben.
Von den 3866 in diese placebokontrollierten Studien eingeschlossenen Patienten hatten 64 % zuvor eine systemische Therapie erhalten (biologische, konventionelle systemische oder PUVA), 43.5 % hatten zuvor eine Phototherapie erhalten, 49.3 % eine konventionelle systemische Therapie und 26.4 % eine biologische Therapie der Psoriasis. Von allen Patienten hatten 14.9 % mindestens eine anti-TNF-alpha Behandlung bekommen, und 8.7 % eine anti-IL-12/IL-23 Behandlung. Zu Studienbeginn hatten 23.4 % der Patienten eine Psoriasis-Arthritis in der Vorgeschichte.
Ausschlusskriterien: wesentliche Ausschlusskriterien, die einer Teilnahme an UNCOVER-1, UNCOVER-2 und UNCOVER-3 entgegenstanden, waren die begleitende Anwendung von systemischen oder biologischen Psoriasis-Therapien oder Phototherapie, jede instabile schwerwiegende medizinische Störung oder Erkrankung ausgenommen Plaque Psoriasis, und jede derzeit bestehende oder kürzlich bestandene schwerwiegende Infektion. In UNCOVER-2 und UNCOVER-3 war jede vorherige Anwendung von Etanercept ein Ausschlusskriterium.
Die kombinierten primären Endpunkte in allen drei Studien waren der Anteil Patienten im Vergleich zu Placebo, die in Woche 12 ein PASI-75-Ansprechen (Psoriasis Area and Severity Index; PASI 75 = Verbesserung des PASI um mindestens 75 %) und einen sPGA-Wert von 0 ( "frei von" ) oder 1 ( "minimal" ) erreichten. Der Baseline Medianwert des PASI für Patienten aller Therapiegruppen lag zwischen 17.4 und 18.3; 48.3 % bis 51.2 % der Patienten hatten einen Baseline sPGA-Wert von "schwer" oder "sehr schwer" , und der Medianwert der itch Numeric Rating Scale (itch NRS) lag bei Baseline zwischen 6.3 und 7.1.
Tabelle 1. Klinisches Ansprechen nach 12 Wochen (NRI) aus UNCOVER-1, -2 und -3
Placebo Taltz 80 mg Q4W Taltz 80 mg Q2W Etanercept 50 mg 2x
wöchentlich
UNCOVER 1
Anzahl Patienten (N) 431 432 433 NA
sPGA "0" (frei von) 14(3.2%) 330(76.4%)a 354(81.8%)a NA
oder "1" (minimal),
n (%)
sPGA "0" (frei 0 149(34.5%)a 160(37.0%)a NA
von), n (%)
PASI 75, n (%) 17(3.9%) 357(82.6%)a 386(89.1%)a NA
PASI 90, n (%) 2 (0.5%) 279(64.6%)a 307(70.9%)a NA
PASI 100, n (%) 0 145(33.6%)a 153(35.3%)a NA
UNCOVER 2
Anzahl Patienten (N) 168 347 351 358
sPGA "0" (frei von) 4(2.4%) 253(72.9%)a 292(83.2%)a 129(36.0%)a
oder "1" (minimal),
n (%)
sPGA "0" (frei 1(0.6%) 112(32.3%)a,b 147(41.9%)a,b 21(5.9%)c
von), n (%)
PASI 75, n (%) 4 (2.4%) 269(77.5%)a 315(89.7%)a 149(41.6%)a
PASI 90, n (%) 1 (0.6%) 207(59.7%)a,b 248(70.7%)a,b 67(18.7%)a
PASI 100, n (%) 1 (0.6%) 107(30.8%)a,b 142(40.5%)a,b 19(5.3%)c
UNCOVER 3
Anzahl Patienten (N) 193 386 385 382
sPGA "0" (frei von) 13(6.7%) 291(75.4%)a,b 310(80.5%)a,b 159(41.6%)a
oder "1" (minimal),
n (%)
sPGA "0" (frei 0 139(36.0%)a,b 155(40.3%)a,b 33(8.6%)a
von), n (%)
PASI 75, n (%) 14(7.3%) 325(84.2%)a,b 336(87.3%)a,b 204(53.4%)a
PASI 90, n (%) 6(3.1%) 252(65.3%)a,b 262(68.1%)a,b 98(25.7%)a
PASI 100, n (%) 0 135(35.0%)a,b 145(37.7%)a,b 28(7.3%)a
Abkürzungen: n = Anzahl Patienten in dieser Kategorie; N = Anzahl Patienten in der Intent-to-treat Population; NA = nicht zutreffend; NRI = Non-Responder Imputation; NRS = numeric rating scale; PASI = Psoriasis Area and Severity Index; Q2W = alle zwei Wochen; Q4W = all vier Wochen; sPGA = static Physician Global Assessment.
a p < 0.001 im Vergleich zu Placebo
b p < 0.001 im Vergleich zu Etanercept
c p < 0.01 im Vergleich zu Placebo
UNCOVER-1 schloss 1296 Patienten ein. Die Patienten wurden randomisiert (1:1:1) auf 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg).
UNCOVER-2 schloss 1224 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
UNCOVER-3 schloss 1346 Patienten ein. Die Patienten wurden randomisiert (1:2:2:2) auf entweder 12 Wochen Placebo oder Taltz (80 mg alle zwei oder vier Wochen [Q2W oder Q4W] nach einer Startdosis von 160 mg) oder Etanercept 50 mg 2x wöchentlich.
Die Dosierung 80 mg Q2W zeigte in allen Studien bei allen Endpunkten eine überlegene Wirksamkeit (siehe Tabellen oben), insbesondere bei hohen Graden der Hautverbesserung (PASI 90, PASI 100, sPGA 0) und bei der Reduktion des Juckreizes. Im Vergleich zu Placebo und Etanercept wurden in Woche 12 signifikant grössere Verbesserungen des Ausgangswerts gezeigt, bei Nagel Psoriasis (gemessen anhand des Nail Psoriasis Severity Index [NAPSI]), bei Kopfhaut Psoriasis (gemessen anhand des Psoriasis Scalp Severity Index [PSSI]) und bei palmoplantarer Psoriasis (gemessen anhand des Psoriasis Palmoplantar Severity Index [PPASI]).
Taltz war mit einem raschen Einsetzen der Wirkung mit einer Reduktion des mittleren PASI in Woche 2 um > 50% verbunden (Abbildung 1). Der Anteil der Patienten, der PASI 75 erreichte, war bereits in Woche 1 in allen drei Studien unter Taltz signifikant grösser als unter Placebo und unter Etanercept. Etwa 25 % der mit Taltz behandelten Patienten erreichte bis Woche 2 einen PASI-Wert< 5, mehr als 55 % erreichten einen PASI-Wert < 5 bis Woche 4 mit einem Anstieg auf 85 % bis Woche 12 (im Vergleich zu 3 %, 14 % und 50 % unter Etanercept). Signifikante Verbesserungen des Juckreiz-Schweregrades wurden in Woche 1 unter Taltz beobachtet.
Taltz zeigte auch einen günstigen Einfluss auf den Pruritus, der Placebo signifikant überlegen war.
Abbildung 1. PASI Werte, prozentuale Verbesserung bei jeder Visite nach der Baseline Visite (mBOCF) in der Intent-to-treat Population während der Anfangsphase der Therapie (UNCOVER-2 und UNCOVER-3)

Die Wirksamkeit und Sicherheit von Taltz wurde unabhängig von Alter, Geschlecht, ethnischer Abstammung, Körpergewicht, PASI Baseline Schweregrad und vorangegangener Therapie mit einem Biologikum, gezeigt. Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q2W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (90.1% bzw. 85.7%) und sPGA 0 oder 1 (84.5% bzw. 75.6%). Die Ansprechrate nach 12 Wochen für mit Taltz 80 mg Q4W behandelten Patienten < 100 kg bzw. ≥100kg waren PASI 75 (85.6% bzw. 74.2%) und sPGA 0 oder 1 (79.0% bzw. 67.4%). Das Ansprechen unterschied sich nicht bei Patienten mit Nagel Psoriasis, Psoriasis im Gesichtsbereich oder Kopfhaut Psoriasis zu Studienbeginn. Taltz war wirksam bei Patientenmit früheren systemischen Therapien, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen. Die Verbesserungen von sPGA und PASI Endpunkten bei Patienten mit begleitender Psoriasis-Arthritis zu Studienbeginn waren ähnlich zu jenen der gesamten Population mit mittelschwerer bis schwerer Plaque Psoriasis. Etwa 45 % der Patienten wiesen bei Studienbeginn Psoriasis im Gesichtsbereich auf. Von diesen Patienten waren 80.4 % der mit Taltz behandelten Patienten in Woche 12 frei von Psoriasis im Gesichtsbereich.
Wirksamkeit bei Etanercept-Non-Respondern: Bei Patienten, die in Woche 12 der UNCOVER-2 als Etanercept-sPGA (0,1) Non-Responder angesehen wurden (N = 200) und die nach einer 4-wöchigen Auswaschphase auf Taltz 80 mg Q4W umgestellt wurden, erreichten nach 12 Wochen 73 % einen sPGA (0,1) und 83.5% einen PASI 75.
Anhalten des Ansprechens bis Woche 60 und bis zu 5 Jahren
Zur Beurteilung des anhaltenden Ansprechens wurden Patienten aus UNCOVER-1 und UNCOVER-2, die ursprünglich auf Taltz randomisiert worden waren und in Woche 12 angesprochen hatten (d.h. sPGA-Werte von 0 oder 1) erneut auf eine der folgenden Therapien für weitere 48 Wochen, randomisiert: Placebo oder Taltz (80 mg alle vier oder zwölf Wochen [Q4W oder Q12W]). Patienten, die in Woche 12 einen sPGA (0,1) nicht erreicht hatten oder während der Erhaltungsphase einen Rückfall (sPGA ≥3) erlitten, erhielten anschliessend Taltz 80 mg Q4W.
Bei den sPGA (0,1) Respondern in Woche 12 (der kombinierten Studien UNCOVER-1 und UNCOVER-2) war der Anteil der Patienten, der dieses Ansprechen in Woche 60 aufrecht erhalten hatte bei Patienten unter Taltz 80 mg Q4W (71 %) signifikant grösser im Vergleich zu Patienten unter Taltz 80 mg Q12W (35.5 %) oder Placebo (7 %).
Tabelle 2 zeigt die Ansprechraten der bei der erneuten Randomisierung auf die empfohlene Erhaltungsdosis von 80 mg Taltz alle 4 Wochen randomisierten Patienten in Abhängigkeit der randomisierten Dosis zu Studienbeginn.
Tabelle 2. Anhaltendes Ansprechen und Wirksamkeit in Woche 60 (kombinierte Ergebnisse aus den Studien UNCOVER-1 und UNCOVER-2; NRI)
Endpunkt 80 mg Q2W (Induktion 80 mg Q4W (Induktion 80 mg Q2W (Induktion 80 mg Q4W (Induktion
)/Placebo (Erhaltung )/ Placebo (Erhaltun )/80 mg Q4W (Erhaltu )/ 80 mg Q4W (Erhalt
) (N=211) g) (N=191) ng) (N=221) ung) (N=195)
Beibehalten eines 7.6 % 6.3 % 78.3 % 68.7 %
sPGA "0" (frei von)
oder "1" (minimal)
sPGA "0" (frei von) 2.8 % 1.6 % 58.8 % 49.2 %
beibehalten oder
erreicht
PASI 75 beibehalten 9.0 % 7.9 % 83.3 % 74.4 %
oder erreicht
PASI 90 beibehalten 4.7 % 4.7 % 76.5 % 66.7 %
oder erreicht
PASI 100 beibehalten 2.8 % 1.6 % 57.5 % 49.7 %
oder erreicht
Abkürzungen: N = Anzahl Patienten in der Analysenpopulation; NRI = Non-Responder Imputation
Von den sPGA (0,1) Respondern in Woche 12, die der Erhaltungstherapie Taltz 80 mg Q4W zugeordnet wurden, hatten, in Woche 60, 76.4 % einen PASI < 5 beibehalten oder erreicht. Die Verbesserung des Juckreiz-Schweregrades hielt bei den Taltz Patienten, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, bis Woche 60 an. Im Hinblick auf das anhaltende Ansprechen bis Woche 60 war Taltz wirksam bei Patienten mit früherer systemischer Therapie, mit oder ohne vorherige Biologika/Anti-TNF-Exposition einschliesslich Patienten, die ein Therapieversagen unter einer Biologika/anti-TNF-Behandlung aufwiesen.
Von den sPGA (0,1) Respondern in Woche 12, die Placebo rerandomisiert wurden, betrug die Zeit bis zum Rückfall (sPGA ≥3) in den kombinierten UNCOVER-1 und UNCOVER-2 Studien 164 (95% CI [143, 169]) Tage. Von diesen Patienten erreichten 71.5 % innerhalb von 12 Wochen nach erneutem Beginn mit Taltz 80 mg Q4W wieder einen sPGA-Wert von 0 oder 1.
Bei Patienten unter Taltz, die in Woche 12 zu den sPGA (0,1) Respondern gehörten, hielten auch die Verbesserungen bei Nagel Psoriasis, Kopfhaut Psoriasis und palmoplantarer Psoriasis bis Woche 60 an.
Von den 591 Patienten, die Taltz Q2W während der Induktionsphase und Q4W als Erhaltungstherapie in den Studien UNCOVER-1, UNCOVER-2 und UNCOVER-3 erhalten hatten, vollendeten 427 Patienten 5 Jahre der Taltz-Behandlung. Davon erforderten 101 Patienten eine Dosis-Eskalation. Von den Patienten, die die Untersuchung in Woche 264 vollendet haben (N = 427), zeigte sich bei 295 Patienten (69 %) ein sPGA (0 oder 1), bei 289 Patienten (68 %) ein PASI 90 und bei 205 Patienten (48 %) ein PASI 100 Ansprechen in Woche 264. Der DLQI wurde nach der Induktionsphase in UNCOVER-1 und UNCOVER-2 erfasst, und bei 113 Patienten (66 %) zeigte sich ein DLQI (0,1) Ansprechen.
Quality of life/Patientenberichtete Ergebnisse
Für den DLQI (Dermatology Life Quality Index) wurden statistisch signifikante Verbesserungen in Woche 12 (Studien 1-3) gegenüber dem Ausgangswert gezeigt; die Verbesserungen blieben über 60 Wochen bestehen Taltz war mit signifikant grösseren Verbesserungen der Hautschmerzen (gemessen anhand der visuellen Analogskala, VAS) verbunden.
Postmarketing, direkte Vergleichsstudien
Die Wirksamkeit und Sicherheit von Ixekizumab wurden auch in der doppelblinden Studie RHBS (IXORA-S) im Vergleich zu Ustekinumab untersucht. Dabei war Ixekizumab im Hinblick auf den primären Endpunkt (PASI 90-Ansprechen in Woche 12, Tabelle 3) überlegen.). In allen drei Kategorien des PASI-Ansprechens zeigte sich die Überlegenheit versus Ustekinumab schnell. Die Überlegenheit von Ixekizumab versus Ustekinumab wurde ebenfalls in den nach Gewicht stratifizierten Subgruppen gezeigt.
Tabelle 3: PASI-Ansprechraten aus der Vergleichsstudie Ixekizumab versus Ustekinumab
Woche 12 Woche 52
Ixekizumab* Ustekinumab** Ixekizumab* Ustekinumab**
Patienten (n) 136 166 136 166
PASI 75, n (%) 120 (88.2 %) 114 (68.7 %) 120 (88.2 %) 126 (75.9 %)
PASI 90, n (%) 99 (72.8 %)§ 70 (42.2 %) 104 (76.5 %) 98 (59.0 %)
PASI 100, n (%) 49 (36.0 %) 24 (14.5 %) 71 (52.2 %) 59 (35.5 %)
* Ixekizumab 160 mg wurde als Initialdosis verabreicht, gefolgt von 80 mg in den Wochen 2, 4, 6, 8, 10 und 12, und anschliessend 80 mg Q4W
** Gewichtsbasierte Dosierung: Patienten, die mit Ustekinumab behandelt wurden, erhielten 45 mg oder 90 mg in Woche 0 und 4, anschliessend alle 12 Wochen bis Woche 52 (dosiert nach Gewicht gemäss zugelassener Dosierung)
§ p < 0.001 versus Ustekinumab (p-Wert nur für den primären Endpunkt)
Die Wirksamkeit und Sicherheit von Ixekizumab wurden auch in der 24-wöchigen randomisierten, doppelblinden, Parallelgruppen-Studie RHCR (IXORA-R) untersucht, in der Ixekizumab mit Guselkumab verglichen wurde. Hierbei zeigte sich eine überlegene Wirksamkeit von Ixekizumab ab Woche 4 im Erreichen vollständig erscheinungsfreier Haut sowie im Erreichen des primären Endpunktes PASI 100 in Woche 12 [Ixekizumab 41.3% vs Guselkumab 24.9% (p<0.001)] und eine Nicht-Unterlegenheit im PASI 100 Ansprechen in Woche 24 [Ixekizumab 50.0% vs Guselkumab 52.3% (p=0.414)].
Wirksamkeit bei genitaler Psoriasis
Eine randomisierte, doppelblinde, placebokontrollierte Studie (IXORA-Q) wurde an 149 erwachsenen Patienten (24 % Frauen) mit einer mittelschweren bis schweren genitalen Psoriasis (sPGA des Genitalbereichs von ≥3) durchgeführt. Die Patienten hatten eine Hautbeteiligung von mindestens 1 % Body Surface Area (BSA) (60.4 % hatten eine Hautbeteiligung von ≥10% BSA) und haben auf mindestens eine vorangegangene topische Therapie zur Behandlung der genitalen Psoriasis nicht angesprochen oder diese nicht vertragen. Die Patienten hatten zumindest eine mittelschwere Plaque-Psoriasis (definiert als sPGA-score ≥3 und waren geeignet für eine Phototherapie und/oder systemische Therapie) über mindestens 6 Monate.
Studienteilnehmer, die auf Taltz randomisiert wurden, erhielten eine Initialdosis von 160 mg, gefolgt von 80 mg alle 2 Wochen für eine Dauer von 12 Wochen. Der primäre Endpunkt war der Anteil an Patienten, die einen sPGA im Genitalbereich von "0" (frei von) oder "1" (minimal) (sPGA-G 0/1) erreichten. In Woche 12 erreichten, unabhängig von der Hautbeteiligung zu Studienbeginn, signifikant mehr Studienteilnehmer unter Taltz einen sPGA-G 0/1 und einen sPGA 0/1 als Studienteilnehmer unter Placebo (Hautbeteiligung zu Studienbeginn von 1 % bis < 10 % BSA bzw. ≥10 % BSA: sPGA-G von "0" oder "1" : Taltz 71 % bzw. 75 %; Placebo: 0 % bzw. 13 %).
Pädiatrische Plaque-Psoriasis
Eine randomisierte, doppelblinde, multizentrische, placebokontrollierte Studie (IXORA-Peds) umfasste 201 pädiatrische Patienten im Alter von 6 bis unter 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis (definiert durch einen sPGA-Wert ≥3, der ≥10% der Körperoberfläche einbezieht, und einen PASI-Wert ≥12), die Kandidaten für eine Phototherapie oder systemische Therapie waren oder bei einer topischen Therapie unzureichend kontrolliert waren.
Die wesentlichen Ausschlusskriterien waren ähnlich wie bei den oben genannten Studien in Erwachsenen, insbesondere aktive oder kürzlich durchgemachte schwerwiegende Infektionen sowie begleitende systemische konventionelle oder biologische Psoriasis-Therapien oder Phototherapie.
Während der 12-wöchigen doppelblinden Placebo- und aktiv-kontrollierten Phase wurden die Patienten mit Placebo (n=56), Etanercept (n=30) oder Taltz (n=115) behandelt, wobei die Gewichts-adaptierte Dosierung wie folgt angepasst wurde:
Taltz:
<25 kg: 40 mg in Woche 0, danach 20 mg Q4W
25 kg bis 50 kg: 80 mg in Woche 0, danach 40 mg Q4W
>50 kg: 160 mg in Woche 0, danach 80 mg Q4W
Etanercept:
0.8 mg/kg, maximal 50 mg pro Dosis jede Woche.
Das Ansprechen auf die Behandlung wurde nach 12 Wochen Therapie beurteilt und wurde durch den Anteil der Patienten definiert, die den coprimären Endpunkt eines sPGA-Wert von "0" (frei von) oder "1" (minimal) mit einer Verbesserung von mindestens 2 Punkten gegenüber dem Ausgangswert (Baseline) erreichten, und den Anteil der Patienten, die eine Reduktion des PASI-Wertes von mindestens 75% (PASI 75) gegenüber Baseline erreichten.
Andere evaluierte Ergebnisse in Woche 12 umfassten den Anteil der Patienten, die PASI 90, PASI 100, sPGA von "0" erreichten, und eine Verbesserung des Schweregrads des Juckreizes erreichten, gemessen durch eine Reduktion von mindestens 4 Punkten auf einer 11-Punkte-Juckreiz-Bewertungsskala (itch Numeric Rating Scale, itch NRS).
Die Patienten hatten einen mittleren Baseline PASI-Wert von 17, mit Werten zwischen 12 und 49. Der Baseline sPGA-Wert lag bei 49% der Patienten bei schwer oder sehr schwer. Von allen Patienten erhielten 22% eine vorherige Phototherapie, 32% eine vorherige konventionelle systemische Therapie und 4% eine Biologika-Vortherapie zur Behandlung der Psoriasis.
Die Daten des klinischen Ansprechens sind in Tabelle 4 aufgeführt.
Tabelle 4: Ergebnisse der Wirksamkeit nach 12 Wochen bei pädiatrischen Patienten mit Plaque Psoriasis, NRI
Endpunkte Taltza (N=115) n (%) Placebo (N=56) n (%) Unterschied zu Placebo
(95% CI)
sPGA "0" (frei von) oder 93 (81) 6 (11) 70.2 (59.3, 81.0) d
"1" (minimal)b
sPGA "0" (frei von) 60 (52) 1 (2) 50.4 (40.6, 60.2) d
PASI 75c 102 (89) 14 (25) 63.7 (51.0, 76.4)d
PASI 90 90 (78) 3 (5) 72.9 (63.3, 82.5)d
PASI 100 57 (50) 1 (2) 47.8 (38.0, 57.6)d
Itch NRS (≥4 Punkte 59 (71) 8 (20) 51.1 (35.3, 66.9) d
Verbesserung) c
Abkürzungen: N =Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation.
a In Woche 0 erhielten die Patienten 160 mg (Körpergewicht [KG] >50kg), 80 mg (KG 25-50 kg), oder 40 mg (KG < 25 kg) Taltz, danach 80 mg (KG >50kg), 40 mg (KG 25-50 kg) oder 20 mg (KG < 25 kg) alle 4 Wochen, für 12 Wochen.
b Co-primäre Endpunkte.
c Itch NRS (≥4 Verbesserung) bei Patienten mit Baseline itch NRS ≥4. Anzahl der ITT-Patienten mit Baseline itch NRS-Wert ≥4 wie folgt: Taltz n = 83; Placebo n = 40.
d p<0.001
Insgesamt wurden 87 pädiatrische Patienten mit schwerer Plaque-Psoriasis (PASI ≥20 oder sPGA ≥4) zu Ixekizumab Q4W (38 Patienten), Etanercept Q1W (30 Patienten) oder Placebo (19 Patienten) randomisiert.
In Woche 12 wurden Verbesserungen für die Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe beobachtet, gemessen mit PASI 75 (84.2%, 63.3%, 26.3%) und sPGA (0,1) (76.3%, 53.3% und 5.3%).
In den wichtigsten sekundären Endpunkte zeigten sich statistisch signifikante Verbesserungen in der Ixekizumab Q4W-Gruppe im Vergleich zur Etanercept Q1W-Gruppe und zur Placebo-Gruppe, gemessen an: PASI 90 (76.3%, 40.0%, 0), PASI 100 (60.5%, 16.7%, 0) und sPGA (0) (63.2%, 16.7%, 0 ).
Patienten in der Ixekizumab-Behandlungsgruppe hatten ein höheres CDLQI (children Dermatology Life Quality Index)/DLQI (0,1) Ansprechen in Woche 12 (NRI) im Vergleich zu Placebo. Der Unterschied zwischen den Behandlungsgruppen zeigte sich bereits ab Woche 4.
Die doppelblinde Behandlungsphase betrug 12 Wochen. Darauf folgte die 48-wöchige offene Erhaltungsphase (Phase 3) und eine 48-wöchige Verlängerungsphase (Phase 4) für Patienten aus Ländern ausserhalb der EU, unabhängig vom Ansprechen, und Non-Responder aus der EU (definiert als diejenigen, die sPGA 0,1 nicht erreichten).
Von den 115 Patienten, die Taltz in Woche 0 erhielten, erhielten 94 Patienten weiterhin Taltz bis Woche 108 und wurden in die Primärpopulation zur Wirksamkeit von Ixekizumab aufgenommen. Von den Patienten, die die Bewertung in Woche 108 abgeschlossen haben, erreichten 64 Patienten (68.1 %) PASI 90, 72 Patienten (76.6 %)) PASI 75 und 64 Patienten (68.1 %) sPGA (0,1) in Woche 108. Es liegen keine ausreichenden Daten vor, um das mögliche Risiko eines Aufflammens der Erkrankung nach Absetzen von Taltz bei Kindern zu beurteilen.
Psoriasis-Arthritis
Die Sicherheit und Wirksamkeit von Taltz wurden an 780 Patienten in zwei randomisierten, doppelblinden placebo-kontrollierten Phase III Studien bei Patienten mit aktiver Psoriasis-Arthritis (≥3 geschwollene und ≥3 schmerzhafte Gelenke) untersucht. Bei den Patienten in diesen Studien wurde die Diagnose einer Psoriasis Arthritis (CASPAR Klassifizierungskriterien, Classification Criteria for Psoriatic Arthritis) im Median 5.33 Jahren vor Studieneinschluss gestellt. Randomisierte Patienten hatten gleichzeitig auch Plaque-Psoriasis Hautläsionen (94.0 %) oder eine dokumentierte Plaque-Psoriasis in der Anamnese. Bei Studienbeginn litten 12.1 % der Patienten an einer mittelschweren bis schweren Plaque-Psoriasis. Von den Psoriasis-Arthritis Patienten hatten zu Studienbeginn mehr als 58.9 % eine Enthesitis bzw. 22.3 % eine Daktylitis. Für beide Studien war der primäre Endpunkt das American College of Rheumatology (ACR) 20 Ansprechen in Woche 24, gefolgt von einer Langzeit-Verlängerungsphase von Woche 24 bis Woche 156 (3 Jahre).
In der Psoriasis-Arthritis Studie 1 (SPIRIT-P1), wurden Biologika -naive Patienten mit aktiver Psoriasis-Arthritis in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Adalimumab 40 mg alle 2 Wochen (aktive Kontrollgruppe), Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 85.3% der Patienten in dieser Studie hatten eine vorherige Behandlung mit ≥1 cDMARD erhalten. 53% der Patienten haben gleichzeitig MTX mit einer mittleren wöchentlichen Dosis von 15.8 mg angewendet. 67% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichenden Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Adalimumab oder Placebo erhielten, wurden in Woche 16 oder 24 basierend auf den Responderstatus 1:1 auf Taltz Q2W oder Q4W neu randomisiert. 243 Patienten beendeten die Verlängerungsphase der Studie mit Taltz über 3 Jahre.
Psoriasis-Arthritis Studie 2 (SPIRIT-P2) schloss Patienten ein, die zuvor mit einem anti-TNF Wirkstoff behandelt wurden und den anti-TNF Wirkstoff wegen ungenügender Wirksamkeit oder Unverträglichkeit (anti-TNF-IR Patienten) abgebrochen haben. Patienten wurden in folgende Therapiegruppen randomisiert: subkutane Injektionen mit Placebo, Taltz 80 mg alle 2 Wochen (Q2W), oder 80 mg alle 4 Wochen (Q4W). Beide Taltz-Dosierschema beinhalteten eine Anfangsdosis von 160 mg. 56% und 35% der Patienten haben unzureichend auf 1 TNF bzw auf ≥2 TNF angesprochen. In SPIRIT-P2 wurden 363 Patienten ausgewertet, von denen 41% gleichzeitig MTX bei einer mittleren wöchentlichen Dosis von 16.1 mg angewendet haben. 73.2% der Patienten, die gleichzeitig mit MTX behandelt wurden, hatten eine Dosis von 15 mg oder mehr. Patienten in allen Behandlungsgruppen mit einem unzureichendem Ansprechen in Woche 16 erhielten eine Rettungstherapie (Modifikation der Hintergrundtherapie). Patienten auf Taltz Q2W oder Q4W blieben auf deren initial bestimmter Taltz Dosis. Patienten, die Placebo erhielten, wurden in der Woche 16 oder 24 basierend auf den Responderstatus 1: 1 auf Taltz Q2W oder Q4W neu randomisiert. 168 Patienten beendeten die Verlängerungsphase der Studie mit Taltz über 3 Jahre.
Anzeichen und Symptome
Die Behandlung mit Taltz zeigte in der Woche 24 eine signifikante Verbesserung der Krankheitsaktivität im Vergleich zu Placebo (siehe Tabelle 5).
Tabelle 5. Wirksamkeitsergebnisse in SPIRIT-P1 und SPIRIT-P2 in Woche 24
SPIRIT-P1 SPIRIT-P2
Endpunkte Unterschied von Unterschied von
Placebo in Ansprechr Placebo in Ansprechr
ate (95% CI) ate (95% CI)
PBO (N = 106) Taltz Q4W (N = 107) Taltz Q2W (N = 103) Taltz Q4W Taltz Q2W PBO (N = 118) Taltz Q4W (N = 122) Taltz Q2W (N = 123) Taltz Q4W Taltz Q2W
ACR 20 Ansprechen,
n (%)
Woche 24 32 (30.2) 62 (57.9) 64 (62.1) 27.8 (15.0, 40.6)* 31.9 (19.1, 44.8) * 23 (19.5) 65 (53.3) 59 (48.0) 33.8 (22.4, 45.2) * 28.5 (17.1, 39.8) *
ACR 50 Ansprechen,
n (%)
Woche 24 16 (15.1) 43 (40.2) 48 (46.6) 25.1 (13.6, 36.6) * 31.5 (19.7, 43.3) * 6 (5.1) 43 (35.2) 41 (33.3) 30.2 (20.8, 39.5) * 28.3 (19.0, 37.5) *
ACR 70 Ansprechen,
n (%)
Woche 24 6 (5.7) 25 (23.4) 35 (34.0) 17.7 (8.6, 26.8) * 28.3 (18.2, 38.5) * 0 27 (22.1) 15 (12.2) 22.1 (14.8, 29.5) * 12.2 (6.4, 18.0) *
Abkürzungen: ACR 20/50/70 = American College of Rheumatology 20%/50%/70% Ansprechrate; CI = confidence interval; Q4W = Taltz 80 mg alle 4 Wochen; Q2W = Taltz 80 mg alle 2 Wochen; N = Anzahl der Patienten in der untersuchten Population; n = Anzahl der Patienten in der spezifischen Kategorie; NRI = non-responder imputation; PBO = Placebo.
Hinweis: Patienten die in Woche 16 die Rettungstherapie bekommen haben oder die Therapie abgebrochen hatten, oder bei denen es fehlende Daten gab, wurden als Non-Responder für die Analysen der Woche 24 gezählt.
Gleichzeitige cDMARDs Anwendung schloss MTX, Leflunomid und Sulfasalazin ein.
* p<0.001 im Vergleich zu Placebo.
Bei Patienten mit bereits vorhandener Daktylitis oder Enthesitis führte die Behandlung mit Taltz Q4W zu einer Verbesserung der Daktylitis bzw. der Enthesitis in Woche 24 im Vergleich zu Placebo (p<0.01).
Bei Patienten mit gleichzeitiger Plaque-Psoriasis (≥3% BSA) und Psoriasis-Arthritis führte die Behandlung mit Taltz Q4W zu einer Verbesserung psoriatischer Hautläsionen, wie anhand von PASI 75-, PASI 90- und PASI 100-Ansprechen in Woche 24 gezeigt werden konnte (p < 0.001).
Bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis und Psoriasis-Arthritis zeigte Taltz Q2W Dosierungsschema signifikante höhere Ansprechrate für PASI 75, PASI 90 und PASI 100 im Vergleich zu Placebo (p<0.001) und einen klinisch bedeutsamen Nutzen gegenüber dem Q4W Dosierungsschema.
Das Ansprechen auf die Behandlung mit Taltz war bereits in Woche 1 für ACR 20, in Woche 4 für ACR 50 signifikant höher als das Ansprechen auf Placebo und in Woche 8 für ACR 70 und wurde durchgängig bis Woche 24 beibehalten; die Effekte hielten bei jenen Patienten, die in der Studie verblieben, 3 Jahre lang an.
Abbildung 2. ACR 20 Ansprechen in SPIRIT-P1 über die Zeit bis zur Woche 24

Für Taltz Q2W und Q4W: b p<0.01 und c p<0.001 im Vergleich zu Placebo.
In SPIRIT-P1 und SPIRIT-P2 wurden ähnliche Ansprechen für ACR 20/50/70 in Psoriasis-Arthritis Patienten beobachtet, ungeachtet der Tatsache ob eine Begleittherapie mit cDMARDs, einschliesslich MTX, vorhanden war oder nicht.
In SPIRIT-P1 und SPIRIT-P2 konnten in allen Komponenten der ACR Scores, einschliesslich der Patientenbeurteilung zum Schmerz, Verbesserungen gezeigt werden.
In SPIRIT-P1 wurde anhaltend die Wirksamkeit über 52 Wochen anhand der Ansprechraten auf die Parameter ACR 20/50/70, Besserung der Enthesitis, Besserung der Dactylitis und PASI 75/90 /100, gezeigt.
Die Wirksamkeit und Sicherheit von Taltz wurde ungeachtet von Alter, Geschlecht, Abstammung, Dauer der Erkrankung, Körpergewicht zu Studienbeginn, Psoriasis Beteiligung zu Studienbeginn, CRP Ausgangwert, DAS28-CRP Ausgangwert, Begleittherapie mit Kortikosteroiden, und vorherige Behandlung mit Biologika gezeigt. Taltz war wirksam bei Biologika-naiven, Biologika-exponierten und Biologika Non-Responder Patienten.
Es gab zu wenige Patienten mit Arthritis mutilans, isolierter Arthritis mit Befall der distalen Interphalangealgelenken (DIP) und begleitender Spondylitis in den Zulassungsstudien, um aussagekräftige Resultate in diesen spezifischen Untergruppen zu erhalten.
In SPIRIT-P1 vollendeten 63 Patienten 3 Jahre Therapie mit Q4W Ixekizumab. Unter den 107 Patienten, die auf Ixekizumab Q4W randomisiert wurden (NRI Analyse der ITT Population), zeigten in Woche 156 54 Patienten (50 %) ein ACR20, 41 Patienten (38 %) ein ACR50, 29 Patienten (27 %) ein ACR70 und 36 Patienten (34 %) ein MDA Ansprechen.
In SPIRIT-P2 vollendeten 70 Patienten 3 Jahre Therapie mit Q4W Ixekizumab. Unter den 122 Patienten, die auf Ixekizumab Q4W randomisiert wurden (NRI Analyse der ITT Population), zeigten in Woche 156 56 Patienten (46 %) ein ACR20, 39 Patienten (32 %) ein ACR50, 24 Patienten (20 %) ein ACR70 und 33 Patienten (27 %) ein MDA Ansprechen.
Radiographisches Ansprechen
In SPIRIT-P1 wurde die Hemmung der Progression struktureller Gelenkschäden radiographisch untersucht, und anhand von modifiziertem Total Sharp Score (mTSS) und seinen Komponenten Erosions Score (ES) und dem Joint Space Narrowing Score (JSN) in den Wochen 24 und 52 im Vergleich zu den Ausgangswerten gemessen.
Taltz hemmte die Progression struktureller Gelenkschäden (mTSS) im Vergleich zu Placebo in Woche 24.
Der Prozentanteil der Patienten ohne radiographische Progression struktureller Gelenkschäden (definiert als eine mTSS Veränderung gegen dem Ausgangswert ≤0.5) von der Randomisierung bis zur Woche 24 betrug 94.8% für Taltz Q2W, 89.0 % für Taltz Q4W, und 77.4% für Placebo. Die Hemmung der strukturellen Schäden wurde mit der Taltz-Behandlung bis zur Woche 52 beibehalten.
Körperliche Funktion und gesundheitsrelevante Lebensqualität
In SPIRIT-P1 und SPIRIT-P2 zeigten mit Taltz Q2W (p<0.001) und Q4W (p<0.001) behandelte Patienten eine durch Health Assesment Questionnaire-Disability Index (HAQ-DI) bewertet Verbesserung der körperlichen Funktion in Woche 24. Diese wurde in SPIRIT-P1 bis Woche 52 beibehalten.
Patienten, die mit Taltz behandelt wurden, berichteten von Verbesserungen in der gesundheitsrelevanten Lebensqualität, die mithilfe des Physical Component Summary des Short Form-36 Health Survey (SF-36 PCS) Score gemessen wurden (p < 0.001). Auch eine statistisch signifikante Verbesserung der Müdigkeit konnte im Fatigue Severity NRS gezeigt werden.
Ankylosierende Spondylitis (radiographische axiale Spondyloarthritis, Morbus Bechterew)
Die Sicherheit und Wirksamkeit von Taltz wurden bei 657 Patienten in 2 randomisierten, doppelblinden, placebokontrollierten Studien (COAST-V und COAST-W) bei erwachsenen Patienten ≥18 Jahren mit ankylosierender Spondylitis untersucht. Die Patienten hatten eine aktive Krankheit definiert nach dem Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 und trotz einer Therapie mit NSAID Wirbelsäulenschmerzen ≥4 auf einer numerischen Bewertungsskala. In beiden Studien hatten die Patienten bei Baseline Symptome einer ankylosierenden Spondylitis über einen Zeitraum von durchschnittlich 17 Jahren (Median von 16 Jahren). Bei Baseline erhielten etwa 32% der Patienten begleitend eine cDMARD-Therapie (in COAST-V und in COAST-W: Methotrexat 8.5% und 13.0%, Sulfasalazin 28.5% und 14.6%, Hydroxychloroquin 0.6% und 0).
Primärer Endpunkt in beiden Studien war der Prozentsatz von Patienten, die in Woche 16 ein Assessment of Spondyloarthritis International Society 40 (ASAS40)-Ansprechen erreichten.
COAST-V bewertete 341 biologisch-naive Patienten, die entweder mit Taltz 80 mg oder 160 mg in Woche 0, gefolgt von 80 mg alle 2 Wochen (Q2W) oder 4 Wochen (Q4W), Adalimumab 40 mg alle 2 Wochen oder mit Placebo behandelt wurden. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W). Die mit Adalimumab behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (80 mg Q2W oder Q4W).
COAST-W bewertete 316 Patienten, die Vorerfahrungen mit 1 oder 2 TNF-Hemmern hatten (90% hatten ein unzureichendes Ansprechen, und 10% hatten eine Unverträglichkeit auf TNF-Hemmer). Alle Patienten wurden mit Taltz 80 oder 160 mg in Woche 0 und danach mit 80 mg Q2W oder Q4W oder mit Placebo behandelt. Die mit Placebo behandelten Patienten wurden in Woche 16 neu randomisiert, um Taltz zu erhalten (160 mg Startdosis, gefolgt von 80 mg Q2W oder Q4W).
Klinisches Ansprechen
In beiden Studien wiesen die mit Taltz 80 mg Q2W oder 80 mg Q4W behandelten Patienten in Woche 16 grössere Verbesserungen im ASAS40- und ASAS20-Ansprechen im Vergleich zu Placebo auf (Tabelle 6). Das Ansprechen war bei den Patienten unabhängig von den begleitenden Therapien vergleichbar. Bei COAST-W zeigte sich das Ansprechen unabhängig von der Anzahl der früheren Therapien mit TNF-Hemmern.
Tabelle 6. Wirksamkeitsergebnisse in COAST-V und COAST-W in Woche 16
COAST-V, Biologika-n COAST-W, TNF-Hemmer-
aiv erfahren
Taltz 80 mg Q4Wa Placebo (N=87) Unterschied zu Adalimumab 40 mg Taltz 80 mg Q4Wc Placebo (N=104) Unterschied zu
(N=81) Placebo (95% CI) Q2W (N= 90) (N=114) Placebo (95% CI)
ASAS20-Ansprechenb, 52(64.2%) 35(40.2%) 24.0(9.3, 38.6)** 53(58.9%) 55(48.2%) 31(29.8%) 18.4(5.7, 31.1)**
n (%), NRI
ASAS40-Ansprechenb,c 39(48.1%) 16(18.4%) 29.8(16.2, 43.3)*** 32(35.6%) 29(25.4%) 13(12.5%) 12.9(2.7, 23.2)*
, n (%), NRI
ASDAS Ausgangswert 3.7[-1.4] 3.9[-0.5] -1.0(-1.3, -0.7)*** 3.7[-1.3]*** 4.2[-1.2] 4.1[-0.1] -1.1(-1.3, -0.8)***
[CFB]
BASDAI-Score Ausgang 6.8[-2.9] 6.8[-1.4] -1.5(-2.1, -0.9)*** 6.7[-2.5]*** 7.5[-2.2] 7.3[-0.9] -1.2(-1.8, -0.7)***
swert [CFB]
MRI Spine SPARCCd 14.5[-11.0] 15.8[-1.5] -9.5(-12.6, -6.4)*** 20.0[-11.6]*** 8.3[-3.0] 6.4[3.3] -6.3(-10.0, -2.5)**
Ausgangswert [CFB]
BASDAI50e (%), NRI 42 17 25(11, 38)*** 32* 22 10 12(3, 22)*
ASDAS <2.1 (%) 43 13 31(18, 43)*** 38*** 18 5 13(5, 21)**
(Geringe Krankheitsa
ktivität), NRI
ASAS HI Ausgangswert 7.5[-2.4] 8.1[-1.3] -1.1(-2.0, -0.3)* 8.2[-2.3]* 10.0[-1.9] 9.0[-0.9] -1.0(-1.9, -0.1)*
[CFB]
SF-36 PCS Ausgangswe 34.0[7.7] 32.0[3.6] 4.1(1.9, 6.2)*** 33.5[6.9]** 27.5[6.6]*** 30.6[1.4] 5.2(3.0, 7.4)***
rt [CFB]
Abkürzungen: N = Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation; Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
ASAS HI = Assessment of SpondyloArthritis International Society Health Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB (change from baseline) = LSM (least square mean)-Änderungen zum Ausgangswert in Woche 16; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 discovertebral unit scale), SF-36 PCS = Short Form health survey physical component summary.
a In Woche 0 erhielten Patienten 80 mg oder 160 mg Taltz.
b ASAS20-Ansprechen: Verbesserung um ≥20% und absolute Verbesserung um ≥1 Einheit (Skala von 0–10) gegenüber Baseline bei ≥3 von 4 Bereichen (Patient Global, Spinal Pain, Function, Inflammation), wobei keine Verschlechterung des verbleibenden Bereichs (definiert als Verschlechterung um ≥20% und durchschnittliche Verschlechterung um ≥1 Einheit, Skala von 0–10) zu verzeichnen ist. ASAS40-Ansprechen: Verbesserung um ≥40% und absolute Verbesserung gegenüber Baseline um ≥2 Einheiten in ≥3 von 4 Bereichen ohne Verschlechterung im verbleibenden Bereich.
c Primärer Endpunkt.
d Die Anzahl der ITT-Patienten mit MRT-Daten zu Studienbeginn lautet: COAST-V: Taltz, n = 81; PBO, n = 82, ADA, n=85. COAST-W: Taltz, n = 58; PBO, n = 51.
e BASDAI50-Ansprechen ist definiert als eine Verbesserung von ≥50% des BASDAI-Score gegenüber dem Ausgangswert.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
In Woche 16 gab es klinisch signifikante Verbesserungen der Hauptkomponenten der ASAS40-Ansprechkriterien (Wirbelsäulenschmerzen, BASFI, globale Beurteilung des Patienten, Steifheit) und anderer Messungen der Krankheitsaktivität, einschliesslich CRP.
Der Prozentsatz der Patienten, die bei Untersuchungen in COAST-V und COAST-W ein ASAS40-Ansprechen erreichten, ist in Abbildung 3 dargestellt.
Abbildung 3. ASAS40-Ansprechen in COAST-V und COAST-W bis Woche 16, NRIa
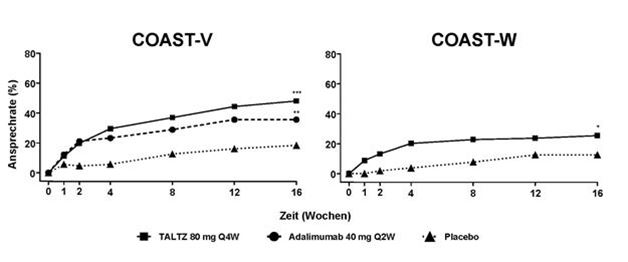
a Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
* p<0.05; ** p<0.01; *** p<0.001 im Vergleich zu Placebo.
Vergleichbare ASAS40-Ansprechraten wurden bei Patienten unabhängig von CRP zu Studienbeginn, von ASDAS-Scores zu Studienbeginn und von MRI Spine SPARCC-Scores festgestellt. Der ASAS40 Ansprechen wurde unabhängig von Alter, Geschlecht, Rasse, Krankheitsdauer, Körpergewicht zu Studienbeginn, BASDAI-Score zu Studienbeginn und vorheriger biologischer Behandlung nachgewiesen.
In COAST-V und COAST-W wurde die Wirksamkeit bis zu Woche 52 beibehalten, wie dies durch die oben in Woche 16, dargestellten Endpunkte, einschliesslich ASAS20-, ASAS40-, ASDAS-, BASDAI-, BASFI- und ASAS HI-Ansprechen beurteilt wurde.
Gesundheitsbezogene Ergebnisse
Es zeigten sich bereits ab Woche 1 Verbesserungen bei Wirbelsäulenschmerzen gegenüber Placebo, und diese wurden bis Woche 16 aufrechterhalten (Taltz vs. Placebo: COAST-V -3.2 vs. -1.7; COAST-W -2.4 vs. -1.0); Müdigkeit und Wirbelsäulenbeweglichkeit zeigten in Woche 16 Verbesserungen gegenüber Placebo. Die Verbesserungen von Wirbelsäulenschmerzen, Müdigkeit und Wirbelsäulenbeweglichkeit blieben bis zu Woche 52 erhalten.
Nicht-radiographische axiale Spondyloarthritis
Die Wirksamkeit und Sicherheit von Taltz wurden in einer randomisierten, doppelblinden Studie mit einem 52wöchigen placebokontrollierten Zeitraum (COAST-X) bei 303 Patienten ≥18 Jahren mit aktiver axialer Spondyloarthritis seit mindestens 3 Monaten beurteilt. Die Patienten mussten objektive Anzeichen einer Entzündung, angezeigt durch erhöhte Werte des C-reaktiven Proteins (CRP) und/oder Sakroiliitis bei der Magnetresonanztomographie (MRT), und keinen definitiven radiographischen Nachweis von strukturellen Schäden an den Iliosakralgelenken gehabt haben. Die Patienten hatten eine aktive Krankheit wie durch den Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 definiert, sowie Wirbelsäulenschmerzen ≥4 auf einer Numerical Rating Scale (NRS) von 0 bis 10, obwohl sie mit NSAID behandelt wurden. Die Patienten wurden entweder mit Taltz 80 mg oder 160 mg in Woche 0, gefolgt von 80 mg alle 2 Wochen (Q2W) oder 80 mg alle 4 Wochen (Q4W) oder mit Placebo behandelt. Die Einleitung und/oder Dosisanpassung von Begleitmedikationen (NSAID, cDMARD, Kortikosteroide, Analgetika) war ab Woche 16 erlaubt.
Bei Studienbeginn hatten die Patienten durchschnittlich seit 11 Jahren Symptome einer nicht-radiographischen axialen Spondyloarthritis. Etwa 39 % der Patienten erhielten begleitend eine Behandlung mit cDMARD.
Der primäre Endpunkt war der Prozentsatz von Patienten, die in Woche 16 ein ASAS40-Ansprechen erreichten.
Klinisches Ansprechen
In Woche 16 erreichten im Vergleich zu Placebo höhere Anteile der mit Taltz 80 mg Q4W behandelten Patienten ein ASAS40-Ansprechen (Tabelle 7). Das Ansprechen war unabhängig von Begleittherapien vergleichbar.
Tabelle 7. Wirksamkeitsergebnisse in Woche 16 in COAST-X, NRI
Taltz 80 mg Q4Wa Placebo (N=105) Unterschied zu Placebo
(N=96) (95% CI) f
ASAS20-Ansprechen b, n (%), 52(54.2%) 41(39.0%) 15.1(1.5, 28.8)*
NRI
ASAS40-Ansprechen b,c, n 34(35.4%) 20(19.0%) 16.4(4.2, 28.5)**
(%), NRI
ASDAS Ausgangswert [CFB] 3.8[-1.1] 3.8[-0.6] -0.5(-0.8, -0.3)***
BASDAI Score Ausgangswert 7.0[-2.2] 7.2[-1.5] -0.7(-1.3, -0.1)*
[CFB]
MRI SIJ SPARCCd Ausgangswert 5.1[-3.4] 6.3[-0.3] -3.1(-4.6, -1.6)***
[CFB]
ASDAS <2.1, n (%) (Low 26(27.7%) 13(12.4%) 15.3(4.3, 26.3)**
Disease Activity), NRIe
SF-36 PCS Ausgangswert [CFB] 33.5[8.1] 32.6[5.2] 2.9(0.6, 5.1)*
Abkürzungen: N = Anzahl Patienten in der Intent-to-treat-Population; NRI = Non-responder Imputation, ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB (change from baseline) = LSM (least square mean)-Änderungen zum Ausgangswert in Woche 16; MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint, SF-36 PCS = Short Form health survey physical component summary.
Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
a In Woche 0 erhielten die Patienten 80 mg oder 160 mg Taltz
b ASAS20-Ansprechen: Verbesserung um ≥20% und absolute Verbesserung um ≥1 Einheit (Skala von 0–10) gegenüber Baseline bei ≥3 der folgenden 4 Bereiche ((Patient Global, Spinal Pain, Function, Inflammation), wobei keine Verschlechterung um ≥20% und um ≥1 Einheit (Skala 0–10) im verbleibenden Bereich zu verzeichnen ist. ASAS40-Ansprechen: Verbesserung um ≥40% und absolute Verbesserung gegenüber Baseline um ≥2 Einheiten in ≥3 von 4 Bereichen ohne Verschlechterung im verbleibenden Bereich.
c Primärer Endpunkt in Woche 16.
d Die Anzahl der ITT-Patienten mit MRT-Daten zu Studienbeginn und in Woche 16 lautet: Taltz, n = 85; PBO, n = 90.
e Patienten mit fehlenden Daten wurden als Non-Responder gewertet. Die Prozentsätze basieren auf der Anzahl der Patienten in der ITT-Population mit ASDAS ≥2.1 zu Studienbeginn.
f Die berichteten Werte werden als Unterschied in % (95 % CI) für kategoriale Variablen und Unterschied in LSM (95 % CI) für kontinuierliche Variablen angegeben.
* p<0.05; ** p<0.01, *** p<0.001 im Vergleich zu Placebo.
Die Verbesserung der Hauptkomponenten der ASAS40-Ansprechkriterien (Wirbelsäulenschmerzen, BASFI, globale Beurteilung des Patienten, Steifheit) und anderer Messungen der Krankheitsaktivität zeigte in Woche 16 eine signifikante klinische Verbesserung.
Der Prozentsatz der Patienten, die bei Untersuchungen ein ASAS40-Ansprechen erreichten, ist in Abbildung 4 dargestellt.
Abbildung 4. ASAS40-Ansprechen bis Woche 16 in COAST-X, NRIa
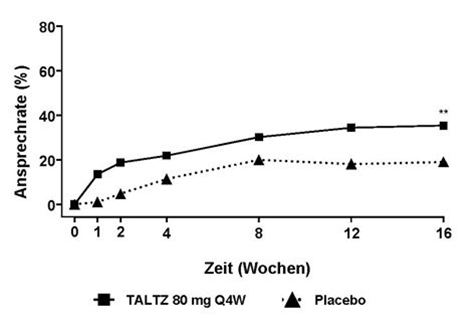
a Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
** p<0.01 im Vergleich zu Placebo.
In COAST-X wurde die Wirksamkeit bis Woche 52 beibehalten.
Langzeit-Ergebnisse bei axialer Spondyloarthritis
Es wurde den Patienten, die eine der drei pivotalen Studien COAST-V/W/X (52 Wochen) beendet hatten, angeboten, an der Langzeit-Verlängerungs- und randomisierten Absetzstudie teilzunehmen (COAST-Y, mit 350 Patienten auf Taltz Q4W und 423 Patienten auf Q2W). Von den 157/773 (20.3 %) mit erreichter Remission (definiert als Ankylosing Spondylitis Disease Activity Score [ASDAS] < 1.3 zumindest einmal, und kein ASDAS Score ≥2.1 in den Wochen 16 und 20) wurden 155 Patienten, die zuvor bis zu 76 Wochen (52 Wochen aus Studie COAST-V/W/X + 24 Wochen aus COAST-Y) mit Taltz behandelt worden waren, in Woche 24 der COAST-Y-Studie randomisiert (randomisierte Absetzpopulation: Placebo, N = 53; Taltz Q4W, N = 48; und Taltz Q2W, N = 54); von diesen schlossen 148 (95.5 %) den Besuch in Woche 64 ab (Placebo, N = 50; Taltz Q4W, N = 47; Taltz Q2W, N = 51). Der primäre Endpunkt war der Anteil an Patienten in der randomisierten Absetzpopulation, die keinen Krankheitsschub während der Wochen 24-64 entwickelten (kombiniert für die Q2W- und Q4W-Gruppen versus Placebo). Ein signifikant grösserer Anteil an Patienten (non responder imputation, NRI) in den kombinierten Taltz-Gruppen (83.3 % (85/102); p < 0.001) und Taltz Q4W (83.3 % (40/48), p = 0.003) zeigte keinen Krankheitsschub während der Wochen 24-64 verglichen mit denen, die von Taltz auf Placebo umgestiegen waren (54.7 % (29/53)). Taltz (in beiden kombinierten Gruppen bzw. In der Taltz Q4W-Gruppe) verzögerte signifikant die Zeit bis zu einem Krankheitsschub (Log-Rank Test p < 0.001 bzw. p < 0.01) im Vergleich zu Placebo.
|