ZusammensetzungWirkstoffe
Nusinersen (als Nusinersen-Natrium).
Nusinersen ist ein vollständig modifiziertes 2'-O-2-Methoxyethyl Antisense Oligonukleotid, welches an eine spezifische Sequenz im Intron 7 der prä-mRNA von SMN2 bindet.
Hilfsstoffe
Bestandteile der künstlichen Zerebrospinalflüssigkeit:
Natriumdihydrogenphosphat-Dihydrat, Dinatriumhydrogenphosphat, Natriumchlorid, Kaliumchlorid, Calciumchlorid-Dihydrat, Magnesiumchlorid-Hexahydrat, Natriumhydroxid, Salzsäure, Wasser für Injektionszwecke.
Darreichungsform und Wirkstoffmenge pro EinheitInjektionslösung zur intrathekalen Injektion durch Lumbalpunktion
Spinraza 50 mg: Jede Durchstechflasche für den Einmalgebrauch enthält 50 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit.
Spinraza 28 mg: Jede Durchstechflasche für den Einmalgebrauch enthält 28 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit.
Spinraza 12 mg: Jede Durchstechflasche für den Einmalgebrauch enthält 12 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit.
Der pH-Wert beträgt ungefähr 7,2.
Indikationen/AnwendungsmöglichkeitenSpinraza ist für die Behandlung der 5q-assoziierten spinalen Muskelatrophie (SMA) indiziert.
Dosierung/AnwendungSpinraza darf nur in Spital-basierten, spezialisierten neuromuskulären Zentren verabreicht werden. Das behandelnde medizinische Personal muss zwingend Erfahrung mit der Diagnostik und der Behandlung von Patienten mit spinaler Muskelatrophie und in der Durchführung von Lumbalpunktionen haben.
Die Behandlung mit Spinraza ist so früh wie möglich nach der Diagnose einzuleiten. Die Entscheidung zur Behandlung sollte sich auf eine individuelle Einschätzung des zu erwartenden Nutzens der Behandlung für den betroffenen Patienten durch einen Experten stützen und gegen die möglichen Risiken einer Behandlung mit Nusinersen abgewogen werden. Die erwartete Höhe der Verbesserung für den einzelnen Patienten hängt vom klinischen Zustand des Patienten zu Beginn der Behandlung ab. Das Ausmass der Wirkung war zwischen den verschiedenen Phänotypen unterschiedlich aufgrund des Potentials für eine Veränderung der einzelnen Populationen (siehe "Klinische Wirksamkeit" ).
Patienten mit ausgeprägter Hypotonie und Ateminsuffizienz bei der Geburt, bei denen Spinraza nicht untersucht wurde, werden wahrscheinlich aufgrund des schweren SMN-Protein-Mangels keinen klinisch bedeutsamen Nutzen von der Behandlung haben.
Im klinischen Entwicklungsprogramm waren Patienten mit SMA Typ IV, bei denen Symptome erst im Erwachsenenalter auftreten, nicht eingeschlossen.
Die klinischen Daten der Wirksamkeit und Sicherheit sprechen stark dafür, die Therapie mit Nusinersen so schnell wie möglich nach Auftreten der Symptome oder der genetischen Diagnose von SMA zu initiieren. Dies basiert auf dem Hintergrund der raschen progressiven Abnahme, die in Longitudinal-Studien beobachtet wurde und auf den Beobachtungen in den Kontroll-Gruppen der Studien CS3B und CS4.
Empfohlenes Dosierungsschema für nicht mit Spinraza vorbehandelte Patienten:
Eine Aufsättigungsdosis von 50 mg sollte an den Tagen 0 und 14 verabreicht werden. Die Erhaltungsdosis von 28 mg sollte danach einmal alle 4 Monate verabreicht werden.
Empfohlenes Dosierungsschema zur Umstellung von Patienten, die derzeit mit Spinraza 12 mg behandelt werden, auf ein Dosierungsschema von 50/28 mg:
Eine Aufsättigungsdosis von 50 mg sollte mindestens 4 Monate (+/- 14 Tage) nach der letzten Dosis von 12 mg verabreicht werden. Die Erhaltungsdosis von 28 mg sollte danach einmal alle 4 Monate verabreicht werden.
Alternatives Dosierungsschema:
Nach ärztlichem Ermessen kann auch das Dosierungsschema mit 12 mg angewendet werden. Bei diesem Dosierungsschema werden vier Aufsättigungsdosen zu je 12 mg an den Tagen 0, 14, 28 und 63 verabreicht, gefolgt von einer Erhaltungsdosis von 12 mg alle 4 Monate.
Therapiedauer
Der Bedarf für eine Fortsetzung der Therapie sollte in regelmässigen Abständen überprüft und je nach klinischem Erscheinungsbild des Patienten und seinem Ansprechen auf die Behandlung im jeweiligen Einzelfall abgewogen werden.
Spezielle Dosierungsanweisungen
1.Die Lösung ist vor Gebrauch visuell zu prüfen. Es dürfen nur klare und farblose Lösungen ohne Partikel verwendet werden. Die Verwendung von externen Filtern ist nicht notwendig.
2.Spinraza ist unter aseptischen Bedingungen vorzubereiten und zu verabreichen.
3.Je nach klinischer Verfassung des Patienten kann eventuell eine Sedierung zur Verabreichung von Spinraza notwendig sein.
4.Ultraschall (oder eine andere Bildgebungstechnik) kann zur Unterstützung der intrathekalen Verabreichung von Spinraza in Betracht gezogen werden, insbesondere bei jüngeren Patienten und bei Patienten mit Skoliose.
5.Es wird empfohlen, vor der Verabreichung von Spinraza ein Liquorvolumen des Patienten zu entnehmen, dessen Menge dem zu injizierenden Volumen von Spinraza entspricht.
6.Spinraza wird als intrathekaler Bolus über 1 bis 3 Minuten mit einer Spinalanästhesienadel injiziert. Die Injektion darf nicht an infizierten oder entzündeten Hautstellen erfolgen.
7.Nur zur einmaligen Anwendung bei einem Patienten. Nicht verdünnen.
8.Nicht verwendete Reste in der Durchstechflasche sind zu entsorgen.
Patienten mit Leberfunktionsstörungen
Spinraza wurde bei Patienten mit eingeschränkter Leberfunktion nicht untersucht. Spinraza wird nicht über das Zytochrom-P450-Enzymsystem in der Leber metabolisiert; daher sind bei Patienten mit eingeschränkter Leberfunktion voraussichtlich keine Dosierungsanpassungen notwendig (siehe "Interaktionen" ).
Patienten mit Nierenfunktionsstörungen
Spinraza wurde bei Patienten mit eingeschränkter Nierenfunktion nicht untersucht.
Kinder und Jugendliche
Spinraza wurde bei Neugeborenen und Patienten bis zu einem Alter von 15 Jahren (Alter bei Studienbeginn) untersucht (siehe "Klinische Wirksamkeit" ).
Erwachsene
Für Patienten über 18 Jahre liegen Daten von einer kleinen Anzahl von Patienten (n = 7) aus klinischen Studien vor, die als Jugendliche eine Behandlung mit Nusinersen 12 mg begonnen hatten und die bis ins Erwachsenenalter begleitet wurden. In Studie SM203 Teil C gab es 24 Patienten über 18 Jahre. Es liegen Daten von erwachsenen SMA Patienten aus der klinischen Anwendung nach Markteinführung vor (siehe "Klinische Wirksamkeit" ).
Ältere Patienten
Es liegen keine klinischen Studiendaten von Patienten über 66 Jahren vor.
Verspätete oder ausgelassene Dosisgabe
Wenn bei dem Dosierungsschema mit 50/28 mg oder mit 12 mg eine Aufsättigungs- oder Erhaltungsdosis verspätet verabreicht oder ausgelassen wird, sollte Spinraza gemäss dem Schema in Tabelle 1 bzw. Tabelle 2 unten verabreicht werden.
Tabelle 1: Empfehlungen bei einer verspäteten oder ausgelassenen Dosis bei einem Dosierungsschema mit 50/28 mg
Zeitraum der Verspätung / Auslassung der Dosis Anpassung des Zeitpunkts der
Dosisgabe
Zweite Aufsättigungsdosis
-Wenn seit der letzten Dosis weniger als 8,5 Monate vergangen
sind, die verspätete Aufsättigungsdosis (50 mg)
schnellstmöglich verabreichen; die nächste Dosis
(Erhaltungsdosis, 28 mg) am ursprünglich geplanten Datum
verabreichen, wobei ein Mindestabstand von 14 Tagen zwischen
den beiden Dosen nicht unterschritten werden soll* -Wenn seit
der letzten Dosis mehr als 8,5 Monate vergangen sind, das
Dosierungsschema von vorne starten.
Erhaltungsdosis
< 12 Monate seit der letzten Dosis -Die verspätete Dosis
schnellstmöglich verabreichen;
dann -die nächste Erhaltungsdosis
am ursprünglich geplanten Datum
verabreichen, wobei ein
Mindestabstand von 14 Tagen
zwischen diesen beiden Dosen
nicht unterschritten werden soll*
≥12 Monate seit der letzten Dosis -Dosierungsschema von vorne
starten.
* Im Anschluss an die oben genannten Empfehlungen Weiterführung der Erhaltungsdosen alle 4 Monate.
Tabelle 2: Empfehlungen bei einer verspäteten oder ausgelassenen Dosis bei einem Dosierungsschema mit 12 mg
Zeitraum der Verspät Anpassung des Zeitpunkts der Dosisgabe
ung / Auslassung
der Dosis
Aufsättigungsdosis
> 1 bis 3 Monate -Die verspätete oder ausgelassene Dosis schnellstmöglich verabreichen, mit
seit der letzten einem Abstand von mindestens 14 Tagen zwischen den Dosen; Behandlung
Dosis fortsetzen, indem die nachfolgenden Dosen ab der letzten Dosis in den
vorgeschriebenen Abständen verabreicht werden. -Wenn z.B. die dritte
Aufsättigungsdosis mit einer Verspätung von 30 Tagen an Tag 58 verabreicht
wird (anstatt an Tag 28, wie im ursprünglichen Zeitplan), ist die vierte
Aufsättigungsdosis 35 Tage später an Tag 93 (anstatt an Tag 63, wie im
ursprünglichen Zeitplan) zu verabreichen, mit einer Erhaltungsdosis 4 Monate
danach.
Erhaltungsdosis
> 4 bis < 8 Monate -Die verspätete Erhaltungsdosis schnellstmöglich verabreichen, dann -die
seit der letzten nächste Erhaltungsdosis nach Möglichkeit am ursprünglich geplanten Datum
Dosis verabreichen, wobei ein Mindestabstand von 14 Tagen zwischen zwei Dosen nicht
unterschritten werden soll*.
≥8 bis < 16 Monate -Die ausgelassene Dosis schnellstmöglich und die darauffolgende Dosis 14 Tage
seit der letzten später verabreichen*.
Dosis
≥16 bis < 40 Monate -Die ausgelassene Dosis schnellstmöglich und die darauffolgende Dosis 14 Tage
seit der letzten später, gefolgt von einer dritten Dosis 14 Tage später, verabreichen*.
Dosis
≥40 Monate seit der -Das gesamte Aufsättigungsschema in den vorgeschriebenen Abständen (Tage 0,
letzten Dosis 14, 28 und 63) verabreichen*.
* Im Anschluss an die oben genannten Empfehlungen Weiterführung der Erhaltungsdosen alle 4 Monate.
Art der Anwendung
Spinraza ist für die intrathekale Anwendung durch Lumbalpunktion bestimmt.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenEs gibt "bis dato" noch keine Langzeit-Sicherheitsdaten von einer ausreichend grossen Patientenzahl (Stand dieser Fachinformation). Für die Behandlung nach mehr als 3 Jahren Therapie stehen nur begrenzte Informationen für Spinraza zur Verfügung.
Lumbalpunktions-Verfahren
Es besteht ein Risiko für das Auftreten von Nebenwirkungen in Zusammenhang mit der Lumbalpunktion (z.B. Kopfschmerz, Rückenschmerzen, Erbrechen; siehe "Unerwünschte Wirkungen" ). Schwierigkeiten bei dieser Art der Anwendung können unter Umständen bei sehr jungen Patienten sowie bei Patienten mit Skoliose auftreten. Die Verwendung von Ultraschall oder anderen bildgebenden Verfahren kann zur Unterstützung der intrathekalen Anwendung von Spinraza je nach Ermessen des Arztes bzw. der Ärztin in Erwägung gezogen werden.
Thrombozytopenie und Blutgerinnungsstörungen
Nach der Gabe von anderen subkutan oder intravenös angewendeten Antisense-Oligonukleotiden wurden Blutgerinnungsstörungen und Thrombozytopenie, einschliesslich akuter schwerer Thrombozytopenie, beobachtet. Es sollten vor initialer Applikation von Spinraza und regelmässig während der gesamten Spinraza-Therapie Thrombozyten und Gerinnung untersucht werden.
Nierentoxizität
Nach Gabe anderer subkutan oder intravenös angewendeter Antisense-Oligonukleotide wurde eine Nierentoxizität beobachtet. Spinraza ist in der Niere vorhanden und wird über diese ausgeschieden. In einer kombinierten Analyse der Sham-kontrollierten Studien bei Patienten mit early- und later-onset SMA, hatten 71 von 123 (58 %) der Spinraza behandelten Patienten gegenüber 22 von 65 (34 %) der Sham-kontrollierten Patienten erhöhte Urin-Protein-Werte.
Es sollen vor initialer Applikation von Spinraza und regelmässig während der gesamten Spinraza-Therapie die Urin-Protein-Werte überwacht werden (vorzugsweise mittels Morgenurin). Bei einer kontinuierlichen Erhöhung der Urin-Protein-Werte sollte eine weitere Abklärung erwogen werden.
QTc Intervall-Unregelmässigkeiten
In den Sham-kontrollierten Studien bei Patienten mit early- oder later-onset SMA hatten bei den Spinraza-behandelten Patienten mit einem normalen Baseline QTc-Wert 2 Patienten mit early-onset und 2 Patienten mit later-onset einen Post-Baseline QTc-Wert von > 500 msec und eine > 60 msec Erhöhung gegenüber der Baseline. In diesen Studien hatte kein Patient Torsades de Pointes als unerwünschte Wirkung noch wurde eine erhöhte Inzidenz kardialer unerwünschter Wirkungen im Zusammenhang mit einer verspäteten ventrikulären Repolarisierung bei Spinraza-behandelten Patienten im Vergleich zu der Sham-Kontrolle beobachtet.
Ein EKG muss vor Beginn der Therapie und regelmässig während der Therapie mit Spinraza durchgeführt werden. Andere Medikamente mit bekannter QTc-Intervall Verlängerung sollten mit Vorsicht angewendet werden.
Mögliche Neurotoxizität (siehe "Präklinische Daten" )
Präklinische Daten aus Primaten zeigten histologische Veränderungen im Hippocampus, ein Einfluss auf das neurologische Verhalten wurde jedoch nicht gesehen. Die klinische Bedeutung dieser Beobachtungen in Affen ist nicht bekannt.
Aufgrund präklinischer Daten in Primaten kann nicht ganz ausgeschlossen werden, dass bei hohen Dosierungen und / oder Langzeit-Anwendung verhaltensneurologische Defizite auftreten könnten. Jedoch sind bis jetzt (Stand dieser Fachinformation) derartige Veränderungen beim Menschen noch nie festgestellt worden.
Hydrozephalus
Nach Markteinführung wurde über das Auftreten eines kommunizierenden Hydrozephalus bei Patienten unter Behandlung mit Nusinersen 12 mg berichtet, der nicht mit einer Meningitis oder einer Blutung assoziiert war. Einigen Patienten wurde ein ventrikulo-peritonealer Shunt implantiert. Bei Patienten mit Bewusstseinsstörungen ist eine Untersuchung auf einen Hydrozephalus in Betracht zu ziehen. Nutzen und Risiken einer Behandlung mit Nusinersen bei Patienten mit einem ventrikulo-peritonealen Shunt sind derzeit nicht bekannt und die Beibehaltung der Therapie ist sorgfältig abzuwägen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu "natriumfrei" .
Dieses Arzneimittel enthält Kalium, jedoch weniger als 1 mmol (39 mg) Kalium pro Durchstechflasche, d.h. es ist nahezu "kaliumfrei" .
InteraktionenEs sind keine klinischen Studien zu Interaktionen mit anderen Arzneimitteln durchgeführt worden.
Pharmakokinetische Interaktionen
Nusinersen wird über Nukleasen und nicht über das Zytochrom-P450 (CYP450)-System metabolisiert. In vitro-Studien zeigen, dass Nusinersen den CYP450-vermittelten Metabolismus weder induziert noch hemmt.
In vitro-Studien deuten darauf hin, dass die Wahrscheinlichkeit für Wechselwirkungen mit Nusinersen durch kompetitive Plasmaproteinbindung, kompetitive Wirkung auf oder Hemmung von Transportern gering ist.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Bei Frauen im gebärfähigen Alter oder bei Frauen, die während der Therapie schwanger werden, sollte der Nutzen der Behandlung mit Spinraza im Verhältnis zum möglichen Risiko mit der Patientin besprochen werden.
Schwangerschaft
Bisher liegen keine oder nur sehr begrenzte Erfahrungen mit der Anwendung von Nusinersen bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf eine Reproduktionstoxizität (siehe "Präklinische Daten, Reproduktionstoxizität" ). Aus Vorsichtsgründen soll eine Anwendung von Spinraza während der Schwangerschaft vermieden werden.
Geburtsvorgang
Der Einfluss von Spinraza auf die Wehentätigkeit und den Geburtsvorgang ist nicht bekannt.
Stillzeit
Es ist nicht bekannt, ob Nusinersen oder Metabolite in die Muttermilch übergehen.
Ein Risiko für das Neugeborene oder den Säugling kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist, ob auf die Behandlung mit Spinraza verzichtet werden soll oder ob die Behandlung mit Spinraza zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen (siehe auch "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenSpinraza hat keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenKlinische Studien
Erfahrung mit der Verabreichung des 12 mg Dosierungsschemas
Die Sicherheit von Spinraza 12 mg basiert auf:
drei randomisierten, doppel-blinden, Sham-kontrollierten Studien
zwei Phase 3 Studien
bei Kleinkindern (Studie CS3B) mit SMA
bei Kindern (Studie CS4) mit SMA
einer Phase 2 Studie
in Kleinkindern und Kindern (Studie CS7) mit SMA
unverblindeten Phase 1/2 Studien
bei symptomatischen Kleinkindern (Studie CS3A) mit SMA
bei präsymptomatischen Säuglingen mit genetischer diagnostizierter SMA (Studie CS5)
bei Patienten der Altersgruppe 2 - 15 Jahre (bei der ersten Dosis) in einer integrierten Analyse von 4 unverblindeten Studien (Studien CS2, CS12, CS1 und CS10)
in einer Extensionsstudie
-Studie CS11: in Kleinkindern und Patienten mit späterem Krankheitsbeginn, einschliesslich Patienten, die die Studien CS3B, CS4 und CS12 beendet haben.
Teil B der Studie SM203 war eine doppelblinde, randomisierte, kontrollierte Studie mit einer Kohorte von 33 Patienten (25 mit infantiler Krankheitsform und 8 mit späterem Krankheitsbeginn), die das 12 mg Dosierungsschema erhielten.
In Studie CS3B wurden 121 Patienten behandelt, davon erhielten 80 Patienten Spinraza 12 mg (mediane Exposition von 280 Tagen) und 41 Patienten die Sham-Kontrolle (mediane Exposition von 187 Tagen).
In Studie CS4 wurden 126 Patienten behandelt, davon erhielten 84 Patienten Spinraza (mediane Exposition von 451 Tagen) und 42 Patienten die Sham-Kontrolle (mediane Exposition von 450 Tagen).
Insgesamt wurden 385 SMA-Patienten mit 12 mg Spinraza oder einer niedrigeren Dosis behandelt. Die Gesamtdauer der Studienteilnahme lag zwischen 1 Tag bis 10,8 Jahren (Median 6,5 Jahre).
Erfahrung mit der Verabreichung des 50/28 mg Dosierungsschemas
Die Sicherheit von Spinraza in einer Dosierung von 50/28 mg bei Kleinkindern, Kindern und Erwachsenen mit SMA wurde in 2 klinischen Studien bei symptomatischen Patienten (Studien SM203 und SM302) untersucht, die zum Zeitpunkt der ersten Studiendosis zwischen 14 Tage und 65 Jahre alt waren. Studie SM302 ist eine unverblindete Verlängerungsstudie für Patienten, die Studie SM203 abgeschlossen haben.
Insgesamt wurden 128 Patienten mit SMA mit 50 mg oder 28 mg Spinraza behandelt. Die Gesamtdauer der Studienteilnahme lag zwischen 13 Tagen und 4,2 Jahren (median 740,5 Tage).
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention definiert:
"sehr häufig" (≥1/10)
"häufig" (≥1/100, < 1/10)
"gelegentlich" (≥1/1'000, < 1/100)
"selten" (≥1/10'000, < 1/1'000)
"sehr selten" (< 1/10'000)
Klinische Studie der infantilen Form der SMA
In der Sham-kontrollierten Studie CS3B waren die häufigsten Nebenwirkungen, die bei mindestens 20 % der mit Spinraza behandelten Patienten und mindestens 5 % häufiger als in der Kontroll-Gruppe auftraten, untere Atemwegsinfekte, obere Atemwegsinfekte und Verstopfung. Die schwere Nebenwirkung Atelektase war bei Spinraza-behandelten Patienten häufiger (18 %) als in der Kontroll-Gruppe (10 %). Da die Patienten in der Studie Kleinkinder waren, konnten Nebenwirkungen, die verbal gemeldet werden, in dieser Studie nicht bestimmt werden.
Tabelle 3: Unerwünschte Wirkungen, die in der Studie CS3B (Sham-kontrollierte Studie in Kleinkindern mit symptomatischer SMA) bei mindestens 5 % der Spinraza Patienten auftraten und mindestens 5 % häufiger waren oder mindestens zweimal häufiger auftraten als bei Patienten in der Kontrollgruppe
Systemorganklasse Unerwünschte Wirkung Spinraza 12 mg1 N = Sham-Kontrolle N =
en 80 [%] 41 [%]
Infektionen und parasitäre
Erkrankungen
Sehr häufig: Untere Atemwegsinfek 51 37
tionen2
Obere Atemwegsinfektionen 30 22
Nasopharyngitis 19 10
Atemwegsinfektion 11 5
Häufig: Harnwegsinfektion 9 0
Kongestion der oberen Atemwege 8 2
Ohrentzündungen 6 2
Influenza 6 0
Erkrankungen der Atemwege, des
Brustraums und Mediastinums
Sehr häufig: Atelektase 18 10
Erkrankungen des Gastrointestinalt
rakts
Sehr häufig: Verstopfung 35 22
Zahnen 18 7
Häufig: Flatulenz 5 2
Stoffwechsel- und Ernährungsstörun
gen
Häufig: Gewichtsabnahme 5 2
1 Vier Aufsättigungsdosen gefolgt von 12 mg (5 ml) alle 4 Monate
2 Einschliesslich Bronchiolitis, Bronchitis, viraler Bronchitis, untere Atemwegsinfektionen, virale untere Atemwegsinfektionen, Lungeninfektion, Pneumonie, adenovirale Pneumonie, grippale Pneumonie, virale paragrippale Pneumonie, Pneumonie (Moraxella), Pneumokokkale Pneumonie, Pseudomonale Pneumonie, virale syncytiale Atemwegspneumonie, virale Pneumonie, virale syncytiale Atemwegs-Bronchiolitis und virale syncytiale Atemwegs-Bronchitis.
Fälle von Hautausschlag wurden von Spinraza-behandelten Patienten in der Studie CS3B gemeldet. Ein Patient entwickelte 8 Monate nach Beginn der Spinraza Therapie schmerzlose rote Hautläsionen am Vorderarm, Bein und Fuss während einer Zeitspanne von 8 Wochen. Die Läsionen eiterten und verkrusteten innert 4 Wochen und verheilten über mehrere Monate. Ein zweiter Patient entwickelte 10 Monate nach Beginn der Spinraza-Therapie rote Hautläsionen an der Backe und der Hand, welche über 3 Monate abheilten. Beide Patienten erhielten kontinuierlich Spinraza und die Hautauschläge heilten spontan ab.
Klinische Studie der SMA bei späterem Krankheitsbeginn
In der Sham-kontrollierten Studie CS4 waren die häufigsten Nebenwirkungen, die bei mindestens 20 % der mit Spinraza behandelten Patienten auftraten und mindestens 5 % häufiger als in der Kontroll-Gruppe waren Fieber, Kopfschmerzen, Erbrechen und Rückenschmerzen.
Tabelle 4: Unerwünschte Wirkungen, die in der Studie CS4 (Sham-kontrollierte Studie in Kindern mit symptomatischer SMA) bei mindestens 5 % der Spinraza Patienten auftraten und mindestens 5 % häufiger waren oder mindestens zweimal häufiger auftraten als bei Patienten in der Kontrollgruppe
Systemorganklasse Unerwünschte Wirkung Spinraza 12 mg N = Sham-Kontrolle N =
en 84 [%] 42 [%]
Allgemeine Erkrankungen und
Beschwerden am Verabreichungsort
Sehr häufig: Fieber 43 36
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen* 29 7
Erkrankungen des Gastrointestinalt
rakts
Sehr häufig: Erbrechen* 29 12
Erkrankungen der Atemwege, des
Brustraums und Mediastinums
Häufig: Epistaxis 7 0
Kongestion der Atemwege 5 2
Saisonale Allergie 5 2
Skelettmuskulatur-, Bindegewebs-
und Knochenerkrankungen
Sehr häufig: Rückenschmerzen* 25 0
Häufig: Sturz 5 0
*Unerwünschte Ereignisse, die vermutlich mit der Applikation via Lumbalpunktion in Verbindung stehen. Diese Ereignisse können als Manifestationen der Post-Lumbalpunktions-Symptome betrachtet werden.
In Tabelle 5 sind unerwünschte Arzneimittelwirkungen zusammengefasst, die in Studie SM203 Teil B im Kollektiv mit der infantilen Form der Krankheit bei den mit Spinraza 50/28 mg behandelten Patienten mit mindestens 5 % höherer Häufigkeit als bei einer historischen passenden Sham-Kontrolle* und bei > 10 % der Patienten auftraten.
Tabelle 5: Unerwünschte Arzneimittelwirkungen, die in Studie SM203 Teil B (infantile Form) bei den mit Spinraza behandelten Patienten mit mindestens 5 % höherer Häufigkeit als bei einer historischen passenden Sham-Kontrolle* und bei > 10 % der Patienten auftraten
MedDRA-Systemorgankl Bevorzugter MedDRA-B Spinraza n = 50 Sham-Kontrolle n = Spinraza Häufigkeits
asse egriff 20 kategorie
Infektionen und Aspirationspneumonie 7 (14 %) 1 (5 %) Sehr häufig
parasitäre Erkrankun
gen
* Mithilfe eines vorab festgelegten Matching-Algorithmus wurden 20 Sham-Gruppen-Patienten aus Studie CS3B identifiziert, die auf Grundlage des CHOP-INTEND-Scores und der Krankheitsdauer zur Baseline mit der mit 50/28 mg behandelten Gruppe in Studie SM203 übereinstimmten.
Anwendung bei Erwachsenen nach Markteinführung
Es sind keine neuen Sicherheitsaspekte bei der erwachsenen Patientenpopulation bekannt.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Ereignisse im Zusammenhang mit der Lumbalpunktion
Unerwünschte Wirkungen, die auf die Verabreichung per Lumbalpunktion zurückzuführen sind, wie Kopfschmerzen, Rückenschmerzen, Erbrechen und Post-Lumbalpunktions-Syndrom, wurden beobachtet. Die Inzidenz und der Schweregrad dieser Ereignisse stimmten mit den erwarteten Ereignissen bei einer Anwendung via Lumbalpunktion überein. Keine schwerwiegenden Komplikationen der Lumbalpunktion, wie schwerwiegende Infekte, wurden in klinischen Studien beobachtet.
Einige Nebenwirkungen, die häufig in Zusammenhang mit einer Lumbalpunktion auftreten (z.B. Kopfschmerz und Rückenschmerzen), konnten bei dem mit Spinraza behandelten Säuglings-Kollektiv aufgrund der in dieser Altersgruppe begrenzten Möglichkeiten der Kommunikation nicht bewertet werden.
Effekt auf das Wachstum
Die Verabreichung von Spinraza an Kleinkinder könnte eine Reduktion des Wachstums verursachen, wie in einer Sham-kontrollierten Studie bei Messungen der Körpergrösse beobachtet wurde.
Immunogenität
Die immunogene Reaktion auf Spinraza 12 mg oder weniger wurde bei 367 Patienten mit Plasma-Proben nach Behandlungsstart für Anti Drug Antikörper (ADA) bewertet. Insgesamt entwickelten 38 mit Spinraza behandelte Patienten (10,4%) behandlungsbedingte ADA, von denen 16 (4,4%) vorübergehend und 22 (6,0%) dauerhaft waren.
Die immunogene Reaktion auf Spinraza 50/28 mg wurde bei 117 Patienten mit Plasma-Proben nach Behandlungsstart für ADAs bewertet. Elf (11) mit Spinraza behandelte Patienten (9,4 %) entwickelten behandlungsbedingte ADAs, von denen 5 (4,3 %) vorübergehend und 6 (5,1 %) dauerhaft waren.
Das Vorhandensein von ADAs schien die Plasma-Clearance von Spinraza zu verringern. Es wurden keine erkennbaren Auswirkungen von ADAs auf den Plasma-NF-L-Spiegel oder Indikatoren der klinischen Funktion beobachtet. Es wurden keine erkennbaren Auswirkungen von ADAs auf die Sicherheit (Auftreten von UEs, einschliesslich Überempfindlichkeit, anaphylaktischer Reaktion und Angioödem) beobachtet.
Unerwünschte Wirkungen aus der Postmarketingphase
Im Rahmen der Anwendung von Spinraza 12 mg nach Markteinführung wurden Nebenwirkungen festgestellt. Bei Patienten, die Spinraza mittels Lumbalpunktion erhielten, wurden schwerwiegende Infektionen wie Meningitis beobachtet. Ferner wurde über das Auftreten eines kommunizierenden Hydrozephalus, einer Überempfindlichkeit (z.B. Angioödem, Urtikaria, Hautausschlag), aseptischer Meningitis und Arachnoiditis berichtet. Eine wiederholte Lumbalpunktion ist ein Risikofaktor für Arachnoiditis und ein Störfaktor. Die Häufigkeit dieser Nebenwirkungen ist nicht bekannt, da sie erst nach Markteinführung gemeldet wurden.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAus klinischen Studien wurden keine Fälle einer Überdosierung in Zusammenhang mit unerwünschten Reaktionen berichtet.
Anzeichen und Symptome
Den Patienten ist zu raten, im Falle einer Überdosierung von Spinraza einen Arzt oder eine Ärztin hinzuzuziehen, wenn Anzeichen oder Symptome unerwünschter Reaktionen festgestellt werden.
Eigenschaften/WirkungenATC-Code
M09AX07
Wirkungsmechanismus
Spinraza ist ein Antisense-Oligonukleotid (ASO), das speziell für die Behandlung von SMA, einer autosomal-rezessiven progressiven neuromuskulären Krankheit infolge von Mutationen des SMN1-Gens (Survival Motor Neuron 1) auf Chromosom 5q, entwickelt worden ist. Diese Mutationen führen zum Verlust der Funktion des SMN1-Gens und dadurch zu einem Mangel an SMN-Protein. SMN-Protein wird auch vom SMN2-Gen hergestellt, jedoch in viel geringerer Menge. SMA ist der Sammelbegriff für ein klinisches Spektrum von Krankheiten.
Das Alter, in dem sich die Krankheit erstmals äusserst, und die Schwere der Krankheit hängen mit der Anzahl der vorhandenen Kopien des SMN2-Gens zusammen; weniger SMN2-Genkopien sind mit einem altersbezogen früheren Beginn der Krankheit und mit stärkeren Symptomen verbunden.
Spinraza erhöht den Exon-7-Anteil von SMN2 in der transkribierten Messenger-Ribonukleinsäure (mRNA), indem es an eine intronische Splice-Silencing-Stelle (ISS-N1) im Intron 7 der prä-mRNA von SMN2 bindet. Durch diese Hybridisierung verdrängt das ASO Splicing-Faktoren, die das Splicing normalerweise unterdrücken. Die Verdrängung dieser Faktoren führt dazu, dass Exon 7 in der SMN2-mRNA erhalten bleibt. Nachdem die SMN2-mRNA gebildet worden ist, kann sie zu dem funktionsfähigen SMN-Protein in Volllänge translatiert werden.
Pharmakodynamik
Die pharmakodynamischen Wirkungen korrelieren mit den biologischen Wirkungen von Nusinersen.
Autopsie-Proben von behandelten Kleinkindern wiesen höhere Mengen an SMN2-mRNA mit Exon 7 im thorakalen Rückenmark auf als Proben von unbehandelten Kleinkindern mit SMA.
Plasma-NF-L-Biomarker
Plasma-NF-L ist ein blutbasierter Biomarker für axonale Schädigung und Neurodegeneration. In Studie SM203 wurde bei Patienten mit infantiler Form von SMA in der mit 50/28 mg behandelten Gruppe eine Verringerung des Plasma-NF-L-Spiegels zwischen Baseline und Tag 183 um 94 % (Verhältnis der geometrischen Mittelwerte zur Baseline) festgestellt, verglichen mit 30 % in der gematchten Sham-Gruppe (Differenz der Verhältnisse der geometrischen Mittelwerte zwischen der 50/28-mg-Gruppe und der entsprechenden Sham-Gruppe: 92 %; p < 0,0001). Die NF-L-Plasmaspiegel verringerten sich in der 50/28-mg-Gruppe schneller als in der 12-mg-Gruppe, wobei in der 50/28-mg-Gruppe eine Verringerung um 88 % (Verhältnis der geometrischen Mittelwerte zur Baseline) zwischen Baseline und Tag 64 festgestellt wurde, verglichen mit einer Verringerung um 77 % in der 12-mg-Gruppe (Differenz der Verhältnisse der geometrischen Mittelwerte zwischen der 50/28-mg-Gruppe und der 12-mg-Gruppe: 49 %; p = 0,0020).
Entsprechend verringerten sich die NF-L-Plasmaspiegel auch bei Teilnehmern mit späterem Krankheitsbeginn in Teil B in der 50/28-mg-Gruppe schneller, wobei eine Verringerung um 66 % (Verhältnis der geometrischen Mittelwerte zur Baseline) zwischen Baseline und Tag 64 festgestellt wurde, verglichen mit einer Verringerung um 42 % in der 12-mg-Gruppe (Differenz der Verhältnisse der geometrischen Mittelwerte zwischen der 50/28-mg-Gruppe und der 12-mg-Gruppe: 42 %; p = 0,0495) (Abbildung 1).
Abbildung 1: Studie SM203 Teil B, infantile Form von SMA, Plasma-NF-L:LS-Mittelwert-Verhältnis zur Baseline (95 %-KI) nach Visite per ANCOVA-Analyse unter Verwendung von MI und Schätzungen aus der Hauptanalyse der 50/28-mg-Gruppe vs. 12-mg-Gruppe und der Sham-Schätzungen der 50/28-mg-Gruppe vs. Sham-Gruppe: ITT, Gruppe mit gematchter Sham-Kontrolle

Klinische Wirksamkeit
Ergebnisse zur Wirksamkeit
Die Wirksamkeit von Spinraza wurde in 8 klinischen Studien bei symptomatischen Patienten (Studien CS3B, CS3A, CS4, CS2, CS12, CS7, CS11 und SM203) gezeigt, die zum Zeitpunkt der ersten Dosis 15 Tage bis 65 Jahre alt waren sowie in einer klinischen Studie bei präsymptomatischen Patienten (Studie CS5), die zum Zeitpunkt der ersten Dosis 3 bis 42 Tage alt waren. Die Wirksamkeits-Resultate dieser Studien unterstützen einen Therapiebeginn so schnell wie möglich nach der Diagnose.
Symptomatische Patienten unter Behandlung mit 12 mg
Infantile Form
Studie CS3B (ENDEAR)
Studie CS3B war eine Phase-3, randomisierte, doppel-blinde Studie mit Sham-Verfahrenskontrolle, die bei 121 symptomatischen Kleinkindern im Alter von ≤7 Monaten mit diagnostizierter SMA (Symptombeginn vor dem Alter von 6 Monaten) durchgeführt wurde. Die Patienten wurden 2:1 randomisiert und erhielten entweder Spinraza (gemäss dem genehmigten Dosierungsschema) oder die Sham-Kontrolle. Die Behandlungsdauer betrug 6 bis 442 Tage (Medianwert: 258 Tage).
Das mediane Alter beim ersten Auftreten klinischer Anzeichen und Symptome der SMA war 6,5 Wochen (Spanne von 2 - 18 Wochen) bei den Spinraza behandelten Patienten bzw. 8 Wochen (Spanne von 1 - 20 Wochen) bei den Patienten der Sham-Gruppe, wobei 99 % der Patienten 2 Kopien des SMN2-Gens besassen und daher am wahrscheinlichsten von der Entwicklung einer Typ-I-SMA betroffen waren.
Bei der Baseline war der mittlere Gesamtwert der motorischen Meilensteine 1,37 (von 0 - 6), das mediane CHOP INTEND Ergebnis war 28 (von 8 - 50,5) und die mediane CMAP Amplitude war 0,20 (von 0,00 - 0,87) für den Nervus ulnaris und 0,30 (von 0,00 - 1,50) für den Nervus peroneus. Das mediane Alter bei Verabreichung der ersten Dosis betrug bei den Patienten in der Behandlungsgruppe 164,5 Tage (Spanne von 52 - 242 Tagen) und bei den Patienten in der Sham-kontrollierten Gruppe 205 Tage (Spanne von 30 - 262 Tagen).
Bei der Baseline waren die Charakteristika weitgehend dieselben bei den Spinraza behandelten Patienten wie bei der Sham-Kontroll-Gruppe ausser den folgenden Ereignissen, die bei den Spinraza behandelten Patienten häufiger als bei der Sham-Kontrolle auftraten: Paradoxe Atmung (89 % versus 66 %), Pneumonie oder respiratorische Symptome (35 % versus 22 %), Schluck- oder Essbeschwerden (51 % versus 29 %) und der Bedarf respiratorischer Unterstützung (26 % versus 15 %).
Es wurde eine geplante Zwischenanalyse durchgeführt, basierend auf Patienten, welche verstarben, abbrachen, oder eine Behandlung von mindestens 183 Tagen abschlossen. Zum Zeitpunkt der Zwischenanalyse der Studie erhielten 121 Patienten die Behandlung (Spinraza n = 80, Sham-Kontrolle n = 41). Zum Zeitpunkt der Zwischenanalyse erreichten insgesamt 78 Patienten die Behandlungsdauer von mindestens 183 Tagen, verstarben oder brachen die Studie ab und wurden in dem Interim Efficacy Set für die Analyse des primären Endpunktes (Spinraza n = 51, Sham-Kontrolle n = 27) eingeschlossen. Der primäre Endpunkt zum Zeitpunkt der Zwischenanalyse war der Anteil der Responder: Patienten, die einen vordefinierten Bereich der Verbesserung der motorischen Meilensteine gemäss Abschnitt 2 des Hammersmith Infant Neurologic Examination (HINE) erreichten. Ein Behandlungs-Responder war definiert als ein Patient, der eine Erhöhung des Ergebnisses um mindestens 2 Punkte (oder ein maximales Ergebnis von 4) in Bezug auf die Fähigkeit zu Strampeln erreichte oder mindestens eine Erhöhung des Ergebnisses um 1 Punkt in Bezug auf die motorischen Meilensteine Kopfkontrolle, Rollen, Sitzen, Krabbeln, Stehen oder Gehen erreichte. Um als Responder definiert zu werden, mussten sich die Patienten in mehr Kategorien der motorischen Meilensteine verbessern als verschlechtern. Patienten, welche verstarben oder die Studie abbrachen, wurden in dem Interim Efficacy Set eingeschlossen und als Nicht-Responder betrachtet. Von den 78 Patienten, die sich für die Zwischenanalyse qualifizierten, wurde ein statistisch signifikant höherer Prozentsatz als motorische Meilensteine Responder in der Spinraza Gruppe (41 %) als in der Sham-Kontroll-Gruppe (0 %) definiert, bei einem Unterschied (95 % CI) in prozentualen Anteilen von 41,18 % (18,16; 61,20) (Nusinersen – Sham-Kontrolle), p < 0,0001.
Nach der positiven Zwischenanalyse wurde die Studie CS3B gestoppt und die Patienten in einer Open-Label Verlängerungsstudie (CS11) eingeschlossen.
Zum Zeitpunkt der Abschlussanalyse wurde die Zeit bis zum Tod oder bis zur permanenten Beatmung (≥16 Stunden Beatmung pro Tag ununterbrochen während > 21 Tagen ohne ein akutes reversibles Ereignis oder eine Tracheostomie) als primärer Endpunkt ausgewertet. Statistisch signifikante Effekte auf Ereignis-freies Überleben, Gesamt-Überleben, den Anteil der Patienten, die die Definition eines motorischen Meilenstein-Responders erreichten und den Prozentsatz der Patienten mit mindestens 4 Punkten Verbesserung von der Baseline im CHOP INTEND Ergebnis wurden in der Spinraza-Gruppe versus Sham-Kontrolle beobachtet (Tabelle 6).
Eine statistisch signifikante 47%ige Reduktion des Sterberisikos oder einer permanenten Beatmung wurde in der ITT Population (p = 0,0046) beobachtet. Eine mediane Zeit bis zum Tod oder bis zur permanenten Beatmung wurde in der Spinraza Gruppe nicht erreicht, während sie in der Sham-Kontroll-Gruppe 22,6 Wochen betrug. Eine statistisch signifikante 62,8%ige Reduktion des Sterberisikos wurde ebenfalls beobachtet (p = 0,0041).
Im Wirksamkeits-Set benötigten 18 (25 %) Patienten der Spinraza-Gruppe und 12 (32 %) Patienten der Sham-Kontrollgruppe eine permanente Beatmung. Während keiner dieser Patienten der Studie CS3B die permanente Beatmung unterbrach, entsprachen 6 (33 %) Patienten der Spinraza-Gruppe und 0 (0 %) Patienten der Sham-Kontrollgruppe der im Protokoll definierten Kriterien eines motorischen Meilenstein Responders.
11 (61 %) Patienten der Spinraza-Gruppe und 3 (25 %) Patienten der Sham-Kontrollgruppe erreichten mindestens 1 Punkt Verbesserung des Gesamtergebnisses der motorischen Meilensteine. 0 (0 %) Patienten der Spinraza-Gruppe und 3 (25 %) Patienten der Sham-Kontrollgruppe erfuhren mindestens 1 Punkt Verschlechterung im Gesamtergebnis der motorischen Meilensteine.
Ein statistisch signifikant grösserer Prozentsatz (p < 0,0001) der Teilnehmer des Wirksamkeits-Sets, die mit Spinraza behandelt wurden (71 %), erreichte eine Verbesserung von mindestens 4 Punkten von der Baseline im CHOP INTEND Ergebnis gegenüber der Sham-Kontrolle (3 %). Ebenso erfuhren 3 % der Spinraza-Gruppe und 46 % der Sham-Kontroll-Gruppe eine Verschlechterung des CHOP INTEND Ergebnis gegenüber der Baseline.
Tabelle 6: Primäre und sekundäre Endpunkte bei der Abschlussanalyse – Studie CS3B
Wirksamkeits-Parameter Mit Spinraza behande Patienten mit
lte Patienten Sham-Kontrolle
Überleben
Ereignisfreies Überleben2
Anzahl Patienten, die verstarben oder permanent beatmet 31 (39 %) 28 (68 %)
werden mussten
Hazard Ratio (95 % KI) 0,53 (0,32 - 0,89)
p-Wert6 p = 0,0046
Anzahl Patienten, die eine permanente Beatmung 18 (23 %) 13 (32 %)
benötigten2
Hazard Ratio (95 % KI) 0,66 (0,322 – 1,368)
p-Wert6 p = 0,1329
Gesamtüberleben2
Anzahl Patienten, die gestorben sind 13 (16 %) 16 (39 %)
Hazard Ratio (95 % KI) 0,37 (0,18 – 0,77)
p-Wert6 p = 0,0041
Motorische Funktion
Motorische Meilensteine3
Anteil der Patienten, welche die vorab festgelegten 37 (51 %)1 p < 0 (0 %)
Kriterien für Responder mit motorischen Meilensteinen 0,0001
erreichten (HINE Abschnitt 2)4,5
Anteil an Tag 183 30/73 (41 %) 2/37 (5 %)
Anteil an Tag 302 22/49 (45 %) 0/28 (0 %)
Anteil an Tag 394 20/37 (54 %) 0/21 (0 %)
Anteil mit Verbesserung der Gesamtpunktzahl der 49 (67 %) 5 (14 %)
motorischen Meilensteine
Anteil mit Verschlechterung der Gesamtpunktzahl der 1 (1 %) 8 (22 %)
motorischen Meilensteine
CHOP-INTEND3
Anteil mit Verbesserung um 4-Punkte 52 (71 %) p < 0,0001 1 (3 %)
Anteil mit Verschlechterung um 4-Punkte 2 (3 %) 17 (46 %)
Anteil mit jeglicher Verbesserung 53 (73 %) 1 (3 %)
Anteil mit jeglicher Verschlechterung 5 (7 %) 18 (49 %)
1 CS3B wurde nach einer positiven statistischen Analyse des primären Endpunktes bei der Interim-Analyse beendet (ein statistisch signifikant grösserer Prozentsatz von Patienten in der Spinraza-Gruppe (41 %) erfüllte die Definition eines Responders mit motorischem Meilenstein als in der Kontrollgruppe mit Scheinintervention (0 %) (p < 0,0001)).
2 Zum Zeitpunkt der Abschlussanalyse wurden das ereignisfreie Überleben und das Gesamtüberleben anhand des Intent-to-Treat-Kollektivs ausgewertet (ITT: Spinraza n = 80, Kontrolle mit Scheinintervention n = 41).
3 Zum Zeitpunkt der Abschlussanalyse wurden Analysen von CHOP INTEND und der motorischen Meilensteine anhand des Wirksamkeitskollektivs vorgenommen (Spinraza n = 73, Kontrolle mit Scheinintervention n = 37).
4 Bewertung bei Studienvisite am Tag 183, Tag 302 oder Tag 394 der Studie, je nachdem, was der jeweils späteste Erhebungszeitpunkt war.
5 Nach der Hammersmith Infant Neurological Examination (HINE) Abschnitt 2: Verbesserung um ≥2 Punkte [oder Höchstpunktzahl] für die Fähigkeit zu strampeln ODER Verbesserung der motorischen Meilensteine Kopfkontrolle, Rollen, Sitzen, Krabbeln, Stehen oder Gehen um ≥1 Punkt UND Verbesserungen in mehr Kategorien von motorischen Meilensteinen als Verschlechterungen, definiert als Responder in dieser primären Analyse.
6 Basierend auf einem Log Rank Test, welcher stratifiziert wurde mit der Krankheitsdauer.
Von den 121 Patienten (80 mit Spinraza behandelt und 41 mit der Sham-Kontrolle) in der Studie CS3B wurden 89 Patienten (65 mit Spinraza behandelt und 24 Post-Sham) in eine offene Verlängerungsstudie (Studie CS11) eingeschlossen. Bei Patienten, die in der Studie CS3B die Spinraza Behandlung erhielten und in der Verlängerung der Spinraza-Behandlung in der Studie CS11 eingeschlossen wurden, erhielten die Patienten das Medikament für 6 Tage bis 8,3 Jahre (im Median 6,7 Jahre). Bei Patienten, die in der Studie CS3B in der Sham-Gruppe randomisiert wurden und die Spinraza Behandlung in der Studie CS11 begonnen, erhielten die Patienten das Medikament für 65 Tage bis 6,9 Jahre (im Median 5,7 Jahre). Sowohl bei Patienten, die die Behandlung mit Spinraza von der Studie CS3B fortsetzten, als auch bei Patienten, die die Behandlung mit Spinraza in der Studie CS11 begannen, wurden Verbesserungen der motorischen Funktion (Abbildung 3 und 4) beobachtet. Der grösste beobachtete Nutzen war bei Patienten mit einem früheren Behandlungsbeginn assoziiert. Die Mehrheit der Patienten war bei ihrem letzten Besuch am Leben, nachdem sie die Spinraza Behandlung entweder in Studie CS3B oder in Studie CS11 begonnen hatten.
Patienten, die in der Studie CS3B mit Spinraza begannen, hatten ein Medianalter von 5,4 Monaten (Bereich 1,7 bis 14,9 Monate). Ab Beginn der Spinraza-Behandlung und einschliesslich der Verlängerung der Behandlung in der Studie CS11 betrug die mediane Zeit bis zum Tod oder bis zu einer permanenten Beatmung 1,4 Jahre. Am Ende der Studie CS11 waren 60 der 81 (74 %) Patienten am Leben und 41 der 81 (51 %) Patienten waren ohne permanente Beatmung gemäss der Definition der Studie CS11 (≥16 Stunden Beatmung pro Tag ununterbrochen während > 21 Tagen ohne ein akutes reversibles Ereignis oder eine Tracheostomie).
Der durchschnittliche HINE-2-Gesamtwert für motorische Meilensteine stieg um 5,3 (SD 4,6; n = 53) und der CHOP INTEND-Wert stieg um 18,4 (SD 14,7; n = 38) Punkte vom Beginn der Spinraza-Behandlung bis zur Nachbeobachtungszeit 394 Tage bzw. 6 Jahre.
Patienten, die in der Studie CS3B in der Sham-Gruppe randomisiert und erst in der Studie CS11 eine Behandlung mit Spinraza begonnen hatten, hatten ein medianes Alter von 17,8 Monaten (10,1 - 23,0 Monate). Vor der Behandlung mit Spinraza hatten 12 von 24 Patienten (50 %) den Wirksamkeitsendpunkt der Studie CS11 für permanente Beatmung erreicht. Die mediane Zeit bis zum Tod oder zur permanenten Beatmung betrug nach Behandlungsbeginn mit Spinraza in Studie CS11 2,76 Jahre. Am Ende der Studie CS11 waren 19 von 24 (79 %) Patienten am Leben, und 6 von 12 (50 %) waren ohne dauerhafte Beatmung am Leben. Vom Studienbeginn bis zur Nachbeobachtungszeit 394 Tage bzw. 6 Jahre wurden in Studie CS11 Verbesserungen der durchschnittlichen Gesamtpunktzahlen für motorische Meilensteine um 1,4 (SD 1,8; n = 12) und des CHOP INTEND Scores um 11,5 (SD 12,2; n = 10) beobachtet.
Studie CS3A
Studie CS3A war eine offene Phase-2-Studie in symptomatischen Patienten mit SMA-Diagnose. Das mediane Lebensalter bei Beginn der klinischen Anzeichen und Symptome betrug 56 Tage (21 - 254 Tage) und die Patienten wiesen entweder 2 Kopien des SMN2-Gens (n = 17) oder 3 Kopien dieses Gens (n = 2) auf (zu einem Patienten liegen keine Angaben über die Anzahl der SMN2-Gen-Kopien vor). Bei den Patienten in dieser Studie wurde die Möglichkeit, dass sie eine SMA Typ I entwickeln, als sehr wahrscheinlich eingestuft. Das mediane Lebensalter bei der ersten Dosis betrug 162 Tage (37 - 223 Tage). Die Patienten waren für eine mediane Dauer von 3 Jahren (62 Tage - 3,9 Jahre) in der Studie.
Der primäre Endpunkt war der Anteil an Patienten mit Verbesserungen in einer oder mehreren Kategorien von motorischen Meilensteinen (gemäss HINE Abschnitt 2: Verbesserung um ≥2 Punkte [oder Höchstpunktzahl] für die Fähigkeit zu strampeln oder spontanes Greifen oder Verbesserung der motorischen Meilensteine Kopfkontrolle, Rollen, Sitzen, Krabbeln, Stehen oder Gehen um ≥1 Punkt). 12 von 20 Patienten (60 %) erreichten den primären Endpunkt mit einer anhaltenden Verbesserung beim Mittelwert der im Zeitverlauf erzielten motorischen Meilensteine (Abbildung 3 und 4).
Eine anhaltende Verbesserung der mittleren CHOP-INTEND-Punktzahl wurde vom Ausgangswert bis Tag 1072 beobachtet (mittlere Veränderung 21,30). Insgesamt erreichten 11 von 20 Patienten (55 %) den Endpunkt einer Verbesserung der CHOP-INTEND Gesamtpunktzahl um ≥4 Punkte bis zur letzten Studienvisite für die Studie vor dem Stichtag für die Einstellung der Datenerhebung.
Bei der letzten Visite waren von den 20 eingeschlossenen Studienteilnehmern 11 Patienten (55 %) am Leben und benötigten keine permanente Beatmung. Vier Patienten erfüllten die Kriterien der permanenten Beatmung und fünf Patienten verstarben während der Studie.
Späterer Krankheitsbeginn
Studie CS4 (CHERISH)
Studie CS4 ist eine Phase 3, randomisierte, doppel-blinde, Sham-kontrollierte Studie bei 126 symptomatischen Kindern mit late onset SMA (symptomatischer Start nach einem Alter von 6 Monaten). Die Patienten wurden 2:1 zu Spinraza (mit 3 Aufsättigungsdosen sowie Erhaltungsdosen alle 6 Monate) und Sham-Kontrolle randomisiert, mit einer Therapiedauer von 324 bis 482 Tagen (Median 450 Tage). Nach der positiven Zwischenanalyse wurde die Studie CS4 gestoppt und die Patienten in die Open-Label Extensionsstudie (CS11) miteingeschlossen.
Das mediane Alter zum Zeitpunkt des Screenings war 3 Jahre (im Bereich von 2 – 9 Jahren) und das mediane Alter zum Zeitpunkt des Ausbruchs der klinischen Zeichen und Symptome von SMA war 11 Monate (im Bereich von 6 - 20 Monaten). Die Mehrheit der Patienten (88 %) hatte 3 Kopien des SMN2-Gens (8 % haben 2 Kopien, 2 % haben 4 Kopien und 2 % haben eine unbekannte Anzahl an Kopien). Bei der Baseline erreichten die Patienten einen durchschnittlichen HFMSE Score von 21,6 und einen durchschnittlichen RULM von 19,1. Alle Patienten erreichten unabhängig zu sitzen, jedoch erreichte keiner der Patienten unabhängig zu laufen. Die Patienten in dieser Studie wurden als Patienten erachtet, die höchstwahrscheinlich Typ II oder Typ III SMA entwickeln.
Die Baseline Krankheits-Charakteristika waren generell gleich mit der Ausnahme eines Ungleichgewichts beim Anteil der Patienten, die jemals die Fähigkeit erreichten, ohne Hilfe zu stehen (13 % der Spinraza-Gruppe gegenüber 29 % der Sham-Kontrolle) oder mit Hilfe zu gehen (24 % der Spinraza-Patienten gegenüber 33 % der Sham-Kontrolle).
Eine Interim-Analyse erfolgte als bei allen Patienten die Bewertung in Monat 6 abgeschlossen wurde und mindestens 39 Patienten die Bewertung in Monat 15 abgeschlossen hatten. Der zum Zeitpunkt der Interim–Analyse bestimmte primäre Endpunkt war die Veränderung der Punktzahl des HFMSE in Monat 15 gegenüber der Baseline. Die primäre Analyse wurde mit dem ITT-Kollektiv durchgeführt, welches alle einschliesst, die randomisiert und mindestens 1 Dosis Spinraza oder 1 Sham-Kontrolle erhalten hatten (Spinraza n = 84; Sham-Kontrolle n = 42). Post-Baseline HFMSE Daten für Patienten ohne Besuch in Monat 15 wurden mittels Multiple Imputation Method berechnet. Eine statistisch signifikante Verbesserung des Ergebnisses von Baseline HFMSE wurde bei Spinraza-behandelten Patienten im Vergleich zu Sham-kontrollierten Patienten beobachtet (Tabelle 7).
Die Resultate der Abschluss-Analyse stimmen mit den Resultaten der Interim-Analyse überein und zeigen in der Spinraza-Gruppe eine statistisch signifikante Verbesserung der HFMSE-Punktzahl im Monat 15 gegenüber Baseline im Vergleich zur Sham-Kontroll-Gruppe (Tabelle 7, Abbildung 2).
Eine Analyse einer Patienten-Subgruppe aus der ITT-Population, von der Werte vom Monat 15 dokumentiert wurden, zeigte konstante, statistisch signifikante Resultate (p = 0,0000002). Die statistische Analyse wurde mittels eines ANCOVA Modells sowie einer Regression mit Anpassung für das Alter beim Screening und des Baseline HFMSE Score durchgeführt. Von den Patienten mit beobachteten Werten im Monat 15 hatte ein höherer Anteil der Spinraza behandelten Patienten eine Verbesserung (73 % vs. 41 %) und ein kleinerer Anteil eine Verschlechterung (23 % vs. 44 %) der totalen HFMSE-Punktzahl gegenüber der Sham-Kontroll-Gruppe.
Bei der Abschluss-Analyse wurden alle sekundären Endpunkte inklusive funktionale Messwerte und die WHO motorischen Meilenstein-Werte formal statistisch getestet und sind in Tabelle 7 dargestellt.
Ein Therapie-Beginn so früh wie möglich nach der Diagnose zeigte eine frühere und grössere Verbesserung der motorischen Funktion als bei späterem Behandlungs-Beginn. Jedoch zeigte sich bei beiden Gruppen ein Nutzen gegenüber der Sham-Kontrolle.
Abbildung 2: Durchschnittliche Änderung von der Baseline des HFMSE-Ergebnisses mit der Zeit (ITT1) – Studie CS41,2

1Anzahl Patienten mit einem beobachteten Wert zu jedem auf der x-Achse aufgeführten Zeitpunkt. Die aufgeführten Durchschnittswerte wurden für das ITT Set (Spinraza n = 84, Sham-Kontrolle: n = 42) mittels Multiple Imputation Method berechnet.
2Die Fehlerbalken zeigen die +/- Standardabweichungen
Tabelle 7: Primäre und sekundäre Endpunkte bei der Abschluss-Analyse – Studie CS41
Spinraza-behandelte Sham-kontrollierte
Patienten Patienten
HFMSE Punktzahl
Veränderung der HFMSE-Gesamtpunktzahl gegenüber 3,9 (95 % CI: 3,0; 4,9) -1,0 (95 % CI:
dem Ausgangswert nach 15 Monaten1,2,3 p = 0,0000001 -2,5; 0,5)
Anteil der Patienten, die eine Verbesserung um 56,8 % (95 % CI: 45,6; 26,3 % (95 % CI:
mindestens 3 Punkte gegenüber dem Ausgangswert 68,1) p = 0,00065, 6 12,4; 40,2)
erreichten1,4
RULM
Mittlere Veränderung gegenüber dem Ausgangswert 4,2 (95 % CI: 3,4; 5,0) 0,5 (95 % CI: -0,6;
bis Monat 15 bei der RULM-Gesamtpunktzahl1,2,3 p = 0,00000016 1,6)
WHO motorische Meilensteine
Anteil der Patienten, die bis Monat 15 einen 19.7 % (95 % CI: 10,9; 5.9 % (95 % CI:
beliebigen neuen motorischen Meilenstein 31,3) p = 0,0811 0,7; 19,7)
erreichten,4
Durchschnittliche erreichte Zahl neuer motorischer 0,2 (range -1 to 2, 95 -0,2 (range -1 to
Meilensteine2,3,4 % CI: 0,1; 0,3) p = 1, 95 % CI: -0,4;
0,00016 0,0)3
1Bestimmt anhand der Intent to Treat Population (Spinraza, n = 84; Shamcontrol, n = 42); die Daten von Patienten ohne Studienvisite in Monat 15 wurden nach dem Verfahren der multiplen Imputation berechnet.
2Mittelwert der kleinsten Quadrate des ANCOVA Modells, wobei die Behandlung als Effekt fixiert wurde und eine Anpassung bei jedem Patienten für das Alter beim Screening und den Baseline Score vorgenommen wurde.
3 Negative Werte zeigen eine Verschlechterung, positive Werte eine Verbesserung.
4 Bestimmt anhand des Monat 15 Wirksamkeits-Sets (Spinraza n = 66; Sham-Kontrolle n = 34; Analyse basiert auf kalkulierten Werten, wenn die Daten fehlen).
5 Basiert auf logistischer Regression mit Behandlungseffekt und Anpassung um das Alter jedes Patienten beim Screening und den HFMSE-Score bei Baseline
6 Nominaler p-Wert.
Hinweis: Anzahl Patienten in der ITT, welche jeden Besuch wahrgenommen haben: Monate 3, 6, 9 (Spinraza n = 84, Sham-Kontrolle n = 42), Monat 12 (Spinraza n = 77, Sham-Kontrolle n = 41), Monat 15 (Spinraza n = 66, Sham-Kontrolle n = 34).
125 Patienten (83 mit Spinraza behandelt und 42 mit der Sham-Kontrolle) nahmen an der Studie CS4 teil und setzten die Nachbeobachtung in der offenen Extensionsstudie (Studie CS11) fort. Bei Patienten, die in der Studie CS4 zu Spinraza randomisiert und in der Verlängerung der Behandlung mit Spinraza in der Studie CS11 eingeschlossen wurden, erhielten die Patienten das Medikament für eine mediane Dauer von 7,2 Jahren (1,3 - 8,4 Jahre). Bei Patienten, die in der Studie CS4 in der Sham-Gruppe randomisiert und in der Studie CS11 die Spinraza Behandlung begonnen, erhielten die Patienten das Medikament für eine mediane Dauer von 5,8 Jahren (2,7 - 6,7 Jahre). Viele der mit Spinraza behandelten Patienten erfuhren eine Verbesserung der motorischen Funktionen in mindestens einem der beiden motorischen Funktionstests bis zum Nachbeobachtungszeitpunkt von 5,7 Jahren, wobei der grösste Nutzen bei denjenigen beobachtet wurde, die die Behandlung mit Spinraza im jüngsten Alter begonnen hatten.
Patienten, die die Behandlung mit Spinraza in der Studie CS4 begannen, hatten ein medianes Alter von 4,1 Jahren (2,1 - 9,2 Jahre). Vom Beginn der Spinraza-Behandlung und einschliesslich der Verlängerung der Behandlung in der Studie CS11 zeigte der HFMSE-Wert zunächst einen Anstieg auf einen maximalen Mittelwert von 4,5 Punkten (SD 6,1; n = 83) nach einer Nachbeobachtungszeit von 1,9 Jahren, gefolgt von einem Rückgang mit weiterer Nachbeobachtung, sodass ein Anstieg von 1,3 Punkten gegenüber der Baseline (SD 9,4; n = 54) nach einer Nachbeobachtungszeit von 5,7 Jahren verzeichnet wurde. Einige Patienten wurden bis zu 7,6 Jahre nachbeobachtet, mit einer Veränderung von -2.3 Punkten gegenüber der Baseline (SD 9,6; n = 39). RULM-Werte zeigten eine anfängliche Steigerung auf einen maximalen Mittelwert von 6,4 Punkten (SD 5,6; n = 74) zum Nachbeobachtungs-zeitpunkt von 3,9 Jahren, welcher bis zum Nachbeobachtungszeitpunkt von 5,7 Jahren mit 6,4 Punkten (SD 6,5; n = 54) stabil blieb. Einige Patienten wurden bis zu 7,6 Jahre nachbeobachtet, mit einer mittleren Veränderung von 6,0 Punkten gegenüber der Baseline (SD 6.8. n = 40).
Patienten, die in der Studie CS4 in der Sham-Gruppe randomisiert und in der Studie CS11 die Behandlung mit Spinraza begannen, hatten ein medianes Alter von 4,9 Jahren (3,3 - 9,0 Jahre). Die Veränderung des HFMSE-Werts betrug eine durchschnittliche Abnahme von 1,3 (SD 9,3; n = 22) Punkten und der RULM-Wert eine durchschnittliche Steigerung von 4,2 (SD 4,4; n = 23) Punkten zum Nachbeobachtungszeitpunkt von 5,7 Jahren.
Der Krankheitsverlauf unbehandelter Patienten mit vergleichbarem Alter und klinischem Bild ist von einem fortschreitenden Verlust der motorischen Funktion im Laufe der Zeit gekennzeichnet, mit einer geschätzten durchschnittlichen Abnahme des HFMSE-Werts von 6,6 Punkten über einen Zeitraum von 5 Jahren. Im weiteren Langzeitverlauf nahm der HFMSE-Wert weiter ab, mit einer geschätzten durchschnittlichen Abnahme von 8,3 Punkten nach 7 Jahren.
Studien CS2 und CS12
Diese Ergebnisse werden durch 2 offene Studien (Studie CS2 und Studie CS12) unterstützt. Die Analyse schloss 28 Patienten ein, die ihre erste Dosis in Studie CS2 erhielten und dann in die Verlängerungsphase, Studie CS12, übernommen wurden. In die Studien wurden Patienten eingeschlossen, die bei der ersten Dosis zwischen 2 und 15 Jahre alt waren. Von den 28 Patienten waren 3 bei ihrer letzten Studienvisite im Rahmen der Studie mindestens 18 Jahre alt. Von den 28 Patienten hatte einer 2 Kopien des SMN2-Gens, 21 hatten 3 Kopien und 6 hatten 4 Kopien. Die Patienten in dieser Studie wurden mit Typ II oder Typ III SMA diagnostiziert. Der durchschnittliche HFMSE Baseline Wert bei den Patienten mit Typ II SMA (n = 11) war 21,3 (im Bereich von 6 - 35) und bei Patienten mit Typ III SMA (n = 17) 48,9 (im Bereich von 20 - 63).
Der durchschnittliche Baseline Wert des Upper Limb Module (ULM) Tests bei Patienten mit Typ II SMA war 11,9 (im Bereich von 7 - 17) und der durchschnittliche 6-Minuten-Gehtest (6MWT, six-minute walk test) bei Baseline bei den Typ III ambulanten Patienten (n = 13) war 253,3 Meter (Bereich von 0 - 563 Meter).
Die Patienten wurden über einen Behandlungszeitraum von 3 Jahren beurteilt. Eine anhaltende Verbesserung wurde bei Patienten mit Typ II SMA beobachtet, wobei die mittlere Verbesserung der HFMSE-Punktzahl gegenüber dem Ausgangswert nach 253 Tagen bei 5,1 (SD 4,05; n = 11) und nach 2,9 Jahren bei 9,1 (SD 6,61; n = 9) lag. Die mittlere Punktzahl nach 253 Tagen war 26,4 (SD 11,91) und nach 2,9 Jahren 31,3 (SD 13,02), wobei kein Plateau beobachtet wurde. Dies steht im Vergleich zu der Abnahme der Punktzahl, die normalerweise bei Patienten mit later-onset SMA mit der Zeit beobachtet wird.
Patienten mit Typ III SMA zeigten eine mittlere Verbesserung der HFMSE-Punktzahl gegenüber der Baseline von 1,3 Punkten (SD 1,87; n = 16) nach 253 Tagen und von 1,2 (SD 4,64; n = 11) nach 2,9 Jahren.
Bei Patienten mit Typ II SMA war die durchschnittliche Verbesserung von der Baseline des Upper Limb Module 1,9 (SD 2,68; n = 11) nach 253 Tagen und 3.5 (SD 3,32; n = 9) nach 2,9 Jahren. Die mittlere Gesamtpunktzahl war 13,8 (SD 3,09) nach 253 Tagen und 15,7 (SD 1,92) nach 2,9 Jahren.
Der 6-Minuten-Gehtest (6MWT, six-minute-walk test) wurde nur bei gehfähigen Patienten durchgeführt. Bei diesen Patienten wurde nach 253 Tagen eine Verbesserung von 28,6 Meter (SD 47,22; n = 12) und nach 2,9 Jahren von 86,5 Meter (SD 40,58; n = 8) beobachtet. Die mittlere 6MWT-Gehstrecke betrug 278,5 Meter (SD 206,46) nach 253 Tagen und 333,6 Meter (SD 176,47) nach 2,9 Jahren. Zwei Patienten, die zuvor nicht ohne Hilfe gehfähig waren (SMA Typ III) sowie ein nicht gehfähiger Patient (SMA Typ II) erlangten die Fähigkeit, unabhängig zu gehen.
Patienten mit infantiler Form der SMA oder späterem Krankheitsbeginn
Studie CS7 (EMBRACE)
Studie CS7 ist eine zweiteilige Phase 2 Studie, bei der Teil 1 als randomisierte, doppelblinde und Sham kontrollierte und Teil 2 als Open-Label-Erweiterung durchgeführt wurde. In die Studie wurden symptomatische Patienten mit einer infantilen Form der SMA bei Säuglingen (≤6 Monate) oder später auftretender SMA (> 6 Monate) mit 2 oder 3 Kopien von SMN2, die aufgrund des Screening-Alters, des Screening-Verfahrens oder der SMN2-Kopienzahl nicht für eine Teilnahme an den Studien CS3B oder CS4 geeignet waren, eingeschlossen. Die Patienten wurden im Teil 1 der Studie über einen medianen Zeitraum von 302 Tagen beobachtet.
Alle mit Spinraza behandelten Patienten waren am Ende von Teil 1 der Studie am Leben, jedoch starb ein Patient im Kontrollarm am Studientag 289. Darüber hinaus benötigte kein Patient der Spinraza- oder Sham-Kontroll-Gruppe eine permanente Beatmung. Von den 13 Patienten mit infantiler SMA erreichten 7/9 (78 %; 95 % CI: 45, 94) der Spinraza-Gruppe und 0/4 (0 %; 95 % CI: 0, 60) mit Sham-Kontrolle den Wirksamkeitsendpunkt motorische Meilensteinentwicklung (HINE-Abschnitt 2). Von den 8 Patienten mit SMA mit späterem Krankheitsbeginn haben 4/5 (80 %; 95 % CI: 38, 96) der Spinraza-Gruppe und 2/3 (67 %; 95 % CI: 21, 94) mit Sham-Kontrolle diesen Endpunkt erreicht.
Präsymptomatische Säuglinge
Studie CS5 (NURTURE)
Studie CS5 ist eine unverblindete Studie bei präsymptomatischen Kleinkindern mit genetischer SMA-Diagnose, die im Alter von 6 Wochen oder davor in die Studie aufgenommen wurden.
Die Patienten in dieser Studie wurden aufgrund ihres genetischen Hintergrundes so eingeschätzt, dass sie höchstwahrscheinlich eine SMA des Typs I oder II entwickeln werden. Das mediane Alter bei der ersten Dosisgabe war 22 Tage.
Bei der Baseline betrug die mediane Anzahl der erreichten motorischen Meilensteine 3 (zwischen 0 und 7), das mediane Gesamtergebnis in CHOP INTEND betrug 50,0 (zwischen 25 und 60) und die mediane CMAP-Amplitude des Nervus ulnaris zum Baseline-Zeitpunkt betrug 2,65 mV (1,0 - 6,7).
Die Interim-Analyse (Stichtag 19. Februar 2020) wurde durchgeführt, als die Patienten im Median 48,3 Monate (36,6 – 57,1 Monate) in die Studie eingeschlossen waren und beim letzten Studienbesuch ein medianes Alter von 46,0 Monaten hatten (34,0 – 57,0 Monate). Zum Zeitpunkt der Interim-Analyse lebten alle 25 Patienten ohne permanente Beatmung (2 SMN2-Gen-Kopien, n = 15; 3 SMN2-Gen-Kopien, n = 10). Der primäre Endpunkt war die Dauer des Zeitraums bis zum Tod oder bis zu einer respiratorischen Intervention (definiert als invasive oder nicht-invasive Beatmung für ≥6 Stunden/Tag kontinuierlich an ≥7 aufeinander folgenden Tagen ODER Tracheostomie) und konnte aufgrund zu weniger Ereignisse nicht bestimmt werden. Vier Patienten (2 SMN2-Kopien) benötigten aufgrund einer akuten reversiblen Erkrankung als unterstützende Massnahme für > 6 Stunden/Tag über einen Zeitraum von ≥7 Tagen Beatmung.
Die Patienten erreichten Meilensteine, die man bei einer SMA des Typs I oder II nicht erwarten würde und die eher denen einer normalen Entwicklung entsprachen. Bei der Zwischenanalyse hatten alle 25 (100 %) Patienten den motorischen WHO-Meilenstein freies Sitzen, sowie 23 (92 %) Patienten den Meilenstein Laufen mit Unterstützung erreicht und 22 von 25 (88 %) konnten ohne Unterstützung laufen. 21 (84 %) Patienten erreichten den maximalen CHOP INTEND-Wert von 64. Alle Patienten konnten bei dem letzten Besuch (Tag 778) saugen und schlucken; 22 der 25 Säuglinge (88 %) erzielten im HINE Abschnitt 1 die maximale Punktzahl.
Von den Patienten, die bis zur Interim-Analyse den Studienvisitentag 700 (n = 25) erreicht hatten, wurde der Anteil bewertet, der eine klinisch manifeste SMA entwickelte. Die im Prüfplan definierten Kriterien für eine klinisch manifeste SMA waren ein altersangepasstes Körpergewicht unterhalb der fünften Perzentile gemäss WHO, eine Abnahme um 2 oder mehr entscheidenden Kurvenperzentilen der Gewichtszunahme, das Legen einer perkutanen Magensonde und/oder die Unfähigkeit, einen der erwarteten altersentsprechenden WHO-Meilensteine (freies Sitzen, Stehen mit Hilfe und auf Händen und Knien krabbeln, Laufen mit Unterstützung, freies Stehen, Laufen ohne Unterstützung) zu erreichen. Am Tag 700 erreichten 7 der 15 Patienten (47 %) mit 2 SMN2-Kopien und keiner der 10 Patienten mit 3 SMN2-Kopien die im Prüfplan definierten Kriterien für eine klinisch manifestierte SMA. Diese Patienten zeigten jedoch eine Gewichtszunahme und erreichten Meilensteine, die bei einer SMA Typ I normalerweise nicht erreicht werden. Ein Vergleich der motorischen Funktionsentwicklung bei Patienten mit symptomatischer infantiler SMA und präsymptomatischer SMA ist in Abbildung 3 und Abbildung 4 dargestellt.
Abbildung 3: Veränderung der motorischen Meilensteine gemäss HINE versus Studientage für die Studien CS3B (Spinraza- und Sham-Kontrolle), CS3A, CS5 und CS11 (ITT-Population)

Abbildung 4: CHOP INTEND Score versus Studientage für die Studien CS3B (Spinraza und Sham-Kontrolle), CS3A, CS5 und CS11 (ITT-Population)

Symptomatische Patienten unter Behandlung mit dem 50/28-mg-Dosierungsschema
Studie SM203 Teil B war eine randomisierte, doppelblinde Untersuchung der Sicherheit und Wirksamkeit von 50/28 mg Nusinersen bei nicht vorbehandelten Patienten mit der infantilen Form von SMA oder mit SMA mit späterem Krankheitsbeginn. Teil B war darauf ausgelegt, die Wirksamkeit bei Patienten mit infantiler Form der Erkrankung in der 50/28-mg-Gruppe im Vergleich zu einer vorab festgelegten, gematchten Sham-Gruppe aus Studie CS3B zu untersuchen. Das 12-mg-Dosierungsschema in Teil B der Studie SM203 lieferte zwar Belegdaten, aber die Teststärke der Studie war nicht ausreichend, um statistisch signifikante Unterschiede zwischen den zum Erhalt von 50/28 mg und den zum Erhalt von 12 mg Nusinersen randomisierten Patienten festzustellen. Teil C war eine unverblindete Untersuchung der Sicherheit und Wirksamkeit bei Kindern und Erwachsenen mit der infantilen Form von SMA oder mit SMA mit späterem Krankheitsbeginn, die von dem 12-mg- auf das 50/28-mg-Dosierungsschema umgestellt wurden.
Studie SM203, Teil B, zulassungsentscheidende Kohorte mit infantiler Form der Erkrankung
Patienten mit der infantilen Form von SMA (2 SMN2-Kopien; Symptombeginn vor dem Alter von 6 Monaten) wurden im Verhältnis 2:1 randomisiert und im Rahmen des 50/28-mg- oder des 12-mg-Dosierungsschemas behandelt. Anhand vorab festgelegter Analysen wurden 20 von 37 Sham-Patienten aus Studie CS3B aufgrund von Ähnlichkeiten sowohl bei der Krankheitsdauer als auch beim CHOP-INTEND-Score zur Baseline gematcht. Der primäre Endpunkt war die Veränderung des CHOP-INTEND-Scores an Tag 183 bei Patienten mit infantilem Beginn der SMA in der 50/28-mg-Gruppe im Vergleich zu dieser gematchten Sham-Gruppe aus Studie 1. Im Vergleich zu dem Kollektiv mit der infantilen Form von SMA in Studie CS3B hatten die Patienten in Studie SM203 eine kürzere Krankheitsdauer (Zeit vom Symptombeginn bis zum Screening) und niedrigere CHOP-INTEND-Scores zur Baseline, was darauf hindeutet, dass ihre Krankheit schneller und weiter fortgeschritten war. Mithilfe einer vorab festgelegten Zuordnung zu einer Teilgruppe der Sham-Kontroll-Gruppe der Studie CS3B konnte dieses Ungleichgewicht teilweise minimiert werden. Dennoch war in der 50/28-mg- und in der 12-mg-Gruppe der Studie SM203 die mittlere Krankheitsdauer (SD) zur Baseline nach wie vor kürzer und der CHOP-INTEND-Score zur Baseline niedriger als in der entsprechenden Sham-Gruppe (Tabelle 8). Andere wichtige demografische Baseline-Merkmale (Alter bei der ersten Dosis, Alter beim Screening, Alter bei Symptombeginn, SMN2-Kopienzahl und motorische Funktion zur Baseline) waren zwischen der 50/28-mg-Gruppe, der 12-mg-Gruppe und der entsprechenden Sham-Gruppe ausgewogen.
Tabelle 8: Baseline-Merkmale der Patienten in Studie SM203 Teil B
Patienten-Merkmale Spinraza 50/28 mg Spinraza 12 mg (n = CS3B-gematchte
(n = 50) 25) Sham-Kontrolle (n =
20)
Medianes Alter (Bereich) bei der 18,4 (2 bis 33) 15,9 (3 bis 31) 22,2 (4 bis 34)
ersten Dosis (Wochen), Baseline
Mittleres (SD) Alter bei 7,5 (5,26) 5,8 (4,44) 8,8 (5,11)
Symptombeginn (Wochen), Baseline
Mittlere Krankheitsdauer (SD), 9,6 (5,29) 9,2 (6,11) 11,1 (4,92)
Baseline (Zeit vom Symptombeginn
bis zum Screening) (Wochen)
Mittlerer (SD) CHOP-INTEND-Score 20,9 (10,23) 19,9 (9,63) 23,6 (5,84)
(Punkte), Baseline
Mittlere (SD) NF-L-Plasmakonzentra 304,7 (283,9) 329,4 (175,9) 287,3 (140,4)
tion, Baseline (pg/ml)
Die mittlere Veränderung des CHOP-INTEND-Scores zwischen Baseline und Tag 183 war in der 50/28-mg-Gruppe statistisch signifikant grösser (Verbesserung um 15,1 Punkte) als in der entsprechenden Sham-Gruppe (Verschlechterung um 11,1 Punkte) (Differenz der LS-Mittelwerte: 26,19 Punkte [95 %-KI: 20,7; 31,7] p < 0,0001; Abbildung 5).
Die Veränderung des CHOP-INTEND-Scores zwischen Baseline und Tag 302 war in der 50/28-mg-Gruppe numerisch höher als in der 12-mg-Gruppe, basierend auf dem Unterschied in Bezug auf die Ränge, aber dieser Unterschied war basierend auf dem JRT nicht statistisch signifikant (Differenz der LS-Mittelwerte in Bezug auf die Ränge (1,00 [95 %-KI: -9,290; 11,299]; JRT p = 0,8484). Die Veränderung der LS-Mittelwerte zwischen Baseline und Tag 302 basierend auf ANCOVA mit MI war in der 12-mg-Gruppe numerisch höher; 50/28 mg-Gruppe (Verbesserung um 19,6 Punkte), 12 mg-Gruppe (Verbesserung um 21,6 Punkte; 95 %-KI: 16,5; 22,8) (Differenz der LS-Mittelwerte -1,94 [7,77; 3,88]). Ein statistisch signifikant grösserer Anteil der Patienten in der 50/28 mg-Gruppe erfüllte an Tag 183 die Responder-Definition nach HINE-Abschnitt 2 (HINE-2) im Vergleich zur entsprechenden Sham-Gruppe (58 % vs. 0 %; p < 0,0001; Tabelle 9). In einer ergänzenden Analyse erfüllten 60 % der Patienten in der 50/28-mg-Gruppe und 44 % der Patienten in der 12-mg-Gruppe an Tag 302 die HINE-2-Responder-Definition. Die mittlere Veränderung des HINE-2-Werts zwischen Baseline und Tag 183 war in der 50/28-mg-Gruppe (Verbesserung um 3,7 Punkte) signifikant grösser als in der entsprechenden Sham-Gruppe (Verschlechterung um 0,2 Punkte) (Differenz der LS-Mittelwerte: 3,9 (95 %-KI: 2,5; 5,4; p < 0,0001). Die Veränderung des HINE-2-Werts für motorische Meilensteine zwischen Baseline und Tag 302 war in der 50/28-mg-Gruppe (Verbesserung um 5,9 Punkte) numerisch grösser als in der 12-mg-Gruppe (Verbesserung um 5,3 Punkte) (Differenz der LS-Mittelwerte: 0,58 (1,89; 3,04)), aber diese Unterschiede waren nicht statistisch signifikant.
Tabelle 9: Studie SM203 Teil B Infantile Form: Motorische Ergebnisse in der 50/28-mg-Gruppe vs. 12-mg-Gruppe und in der 50/28-mg-Gruppe vs. gematchte Sham-Gruppe
Wirksamkeitsparamete Spinraza50/28-mg-Gru Spinraza12-mg-Gruppe Gematchte Sham-Kontr Unterschiede zwische
r ppe (n = 50) (n = 25) olle aus Studie n den Armen (95
CS3B (n = 20) %-KI)
50/28-mg-Gruppe vs.
gematchte Sham-Kontr
olle
CHOP-INTEND
LS-Mittelwert (95 42,9 (38,7; 47,2) 16,9 (10,1; 23,7) 26,06 (17,94;
%-KI) für den 34,17) p < 0,00013
Rangwert der Verände
rung zwischen
Baseline und Tag 183
Veränderung der 15,1 (12,4; 17,8) -11,1 (-15,9; -6,2) 26,2 (20,7; 31,7)2
LS-Mittelwerte (95
%-KI) zwischen
Baseline und Tag
1831,2
HINE-2, Responder5
Anteil derer, die 29 (58 %) 0 (0 %) 58 % (39,5; 71,8)4
an Tag 183 die p < 0,0001
Kriterien in Bezug
auf motorische
Meilensteine erreich
ten
HINE-2, Gesamtwert
LS-Mittelwert (95 43,1 (39,0; 47,2) 16,5 (9,9; 23,0) 26,67 (18,81;
%-KI) für den 34,53) p < 0,00013
Rangwert der Verände
rung zwischen
Baseline und Tag 183
Veränderung der 3,7 (3,0; 4,4) -0,2 (-1,5; 1,0) 3,94 (2,46; 5,42)2
LS-Mittelwerte (95
%-KI) zwischen
Baseline und Tag
183 des HINE-2-Gesam
twerts1,2
50/28-mg-Gruppe vs.
12-mg-Gruppe
CHOP-INTEND
LS-Mittelwert (95 38,3 (32,7; 44,0) 37,3 (29,1; 45,5) 1,0 (-9,29; 11,30)
%-KI) für den p = 0,8483
Rangwert der Verände
rung zwischen
Baseline und Tag 302
Veränderung der 19,6 (16,5; 22,8) 21,6 (16,6; 26,6) -1,9 (7,8; 3,9)2
LS-Mittelwerte (95
%-KI) zwischen
Baseline und Tag
3021,2
HINE-2, Responder5
Anteil derer, die 30 (60 %) 11 (44 %) 16 % (-7,73; 39,73)
an Tag 302 die p = 0,225
Kriterien in Bezug
auf motorische
Meilensteine erreich
ten
HINE-2, Gesamtwert
LS-Mittelwert (95 40,0 (35,1; 44,9) 33,9 (26,9; 41,0) 6,12 (-2,69; 14,94)
%-KI) für den p = 0,1733
Rangwert der Verände
rung zwischen
Baseline und Tag 302
Veränderung der 5,9 (4,6; 7,2) 5,3 (3,3; 7,4) 0,58 (-1,89; 3,04)
LS-Mittelwerte (95
%-KI) zwischen
Baseline und Tag
3021,2
1 Anwendung von ANCOVA und multipler Imputation (MI)
2 Differenz der Kleinstquadrat-Mittelwerte
3 Joint Rank Test
4 Fisher-Exakt-Test
5 Definition von Responder in dieser primären Analyse: Erhöhung des Ergebnisses um ≥2 Punkte [oder Maximalpunktzahl] in Bezug auf die Fähigkeit zu strampeln ODER Erhöhung des Ergebnisses um ≥1 Punkt in Bezug auf die motorischen Meilensteine Kopfkontrolle, Rollen, Sitzen, Krabbeln, Stehen oder Gehen UND Verbesserung in mehr Kategorien der motorischen Meilensteine als Verschlechterung).
Abbildung 5: Teil B: Infantile Form von SMA: CHOP-INTEND: Veränderung der LS-Mittelwerte (SE) gegenüber Baseline nach Visite, ANCOVA-Analyse mit MI: 50/28-mg-Gruppe und entsprechende Sham-Gruppe.

Als wichtige sekundäre Endpunkte in der Kohorte mit infantiler Krankheitsform in Teil B von Studie SM203 wurden die Veränderungen des NF-L-Plasmaspiegels im Vergleich zur entsprechenden Sham-Gruppe (an Tag 183) und zu der zugelassenen 12-mg-Gruppe (an Tag 64) untersucht. Bei Patienten mit infantiler Form von SMA waren an Tag 183 die mittleren NF-L-Plasmaspiegel in der 50/28-mg-Gruppe um 94 % reduziert, verglichen mit 30 % in der entsprechenden Sham-Gruppe; Differenz zwischen der 50/28-mg-Gruppe und der entsprechenden Sham-Gruppe: 92 %; p < 0,0001. An Tag 64 waren die mittleren NF-L-Plasmaspiegel in der 50/28-mg-Gruppe um 88 % reduziert, verglichen mit 77 % in der 12-mg-Gruppe; Differenz zwischen 50/28-mg-Gruppe und der 12-mg-Gruppe: 49 %; nominaler p-Wert 0,0020.
Tabelle 10: Studie SM203 – Teil B Infantile Form: Verhältnis der geometrischen NF-L-Mittelwerte: Tag 183: 50/28-mg-Gruppe vs. entsprechende Sham-Gruppe, und Tag 64: 50/28-mg-Gruppe vs. 12-mg-Gruppe
Plasma NF-L Spinraza 50/28 mg Spinraza 12 mg (n = Gematchte Sham-Kontr Verhältnisse der
(n = 50) 25) olle, Patienten aus Kleinstquadrat-Mitte
Studie CS3B (n = 20) lwerte der Studienar
m-Ergebnisse Verglei
chsgruppe: 50/28-mg-
Gruppe (95 %-KI)1
Angepasstes Verhältn 0,06 0,70 0,08 (0,05; 0,14) p
is der geometrischen < 0,00013
Mittelwerte,
Baseline: Tag
183.1,2
Angepasstes Verhältn 0,12 0,23 0,51 (0,33; 0,78) p
is der geometrischen < 0,0050†3
Mittelwerte,
Baseline: Tag 64.1,2
† Nominell statistisch signifikanter p-Wert
1 Anwendung von ANCOVA und multipler Imputation (MI)
2 Differenz der Kleinstquadrat-Mittelwerte
3 Joint Rank Test
In der 50/28-mg-Gruppe war das Sterberisiko oder das Risiko einer permanenten Beatmung im Vergleich zur entsprechenden Sham-Gruppe um 68 % (p = 0,0006) und im Vergleich zu der 12-mg-Gruppe um 29,9 % (p = 0,2775) statistisch signifikant reduziert. Die mediane Zeit bis zum Tod oder bis zu permanenter Beatmung wurde in der 50/28-mg-Gruppe nicht erreicht, betrug 24,7 Wochen in der 12-mg-Gruppe und 19,1 Wochen in der gematchten Sham-Gruppe. In Bezug auf das Gesamtüberleben wurden ähnliche Beobachtungen gemacht (Tabelle 11).
Tabelle 11: Studie SM203 – Teil B Infantile Form: Ereignisfreies Überleben und Gesamtüberleben: 50/28-mg-Gruppe vs. entsprechende Sham-Gruppe und 50/28-mg-Gruppe vs. 12-mg-Gruppe
Wirksamkeitsparamete Spinraza 50/28 mg Spinraza 12 mg (n = Gematchte Sham-Kontr Hazard Ratio (95
r (n = 50) 25) olle, Patienten aus %-KI)
Studie CS3B (n = 20)
Überleben
Ereignisfreies 19 (38 %) 12 (48 %) 17 (85 %) 50/28: MS 0,322
Überleben (0,158; 0,657) p <
Anzahl der Patienten 0,0006†1
, die verstarben
oder eine permanente
Beatmung erhielten
50/28:12 0,701
(0,338; 1,452) p =
0,27751
Gesamtüberleben 10 (20 %) 6 (24 %) 11 (55 %) 50/28: MS 0,279
Anzahl der verstorbe (0,112; 0,696) p <
nen Patienten 0,0012†1
50/28: 12 0,730
(0,264; 2,015) p =
0,48211
† Nominell statistisch signifikanter p-Wert
1 p-Wert per Log Rank Test ermittelt
Studie SM203, Teil B, Kohorte mit späterem Krankheitsbeginn
24 Patienten mit SMA mit späterem Krankheitsbeginn (die meisten mit 3 SMN2-Kopien; Symptombeginn nach dem Alter von 6 Monaten) wurden im Verhältnis 2:1 randomisiert und entweder mit dem 50/28-mg-Schema (n = 16) oder dem 12-mg-Schema (n = 8) behandelt. Die Analysen waren vorab so festgelegt, dass das 50/28-mg-Schema mit passenden Teilgruppen aus Studie CS4 verglichen wurde, einschliesslich einer passenden 12-mg-Gruppe (3 Aufsättigungsdosen, gefolgt von Erhaltungsdosen alle 6 Monate) (n = 32) und einer passenden Sham-Kontrolle (n = 16). Die Teststärke der Analysen war nicht darauf ausgelegt, signifikante Unterschiede zwischen den Behandlungsgruppen festzustellen.
Die demografischen Baseline-Merkmale der 50/28-mg-Gruppe, der gematchten Behandlungsgruppe und der gematchten Sham-Gruppe waren im Allgemeinen ausgewogen, mit Ausnahme des Alters bei der ersten Dosis. Das mittlere (SD) Alter bei der ersten Dosis war wie folgt: In der 50/28-mg-Gruppe betrug es 6,1 (3,0) Jahre, in der 12-mg-Gruppe betrug es 5,7 (3,0) Jahre, in der entsprechenden Behandlungsgruppe betrug es 5,47 (1,8) Jahre und in der entsprechenden Sham-Gruppe betrug es 5,13 (1,8) Jahre.
Die Veränderung des HFMSE-Scores zwischen Baseline und Tag 302 (LS-Mittelwert [95 %-KI]) war in der 50/28-mg-Gruppe numerisch höher (3,3 [1,5, 5,0]) als in der 12 mg-Gruppe (2,6 [0,2, 5,1]). Differenz der LS-Mittelwerte: 0,63 (-2,5, 3,8; p = 0,70). Auch die Veränderung des HFMSE-Scores zwischen Baseline und Tag 279 war in der 50/28-mg-Gruppe numerisch höher als in der entsprechenden 12-mg-Gruppe aus Studie CS4 (Tag 274) (Differenz der LS-Mittelwerte: 1,7 (-0,3, 3,6); p = 0,095) und der entsprechenden Sham-Gruppe aus Studie CS4 (Differenz der LS-Mittelwerte: 3,2 (0,2, 6,2); p = 0,037). Die Veränderung des RULM-Scores zwischen Baseline und Tag 302 (LS-Mittelwert [95 %-KI]) war in der 50/28-mg-Gruppe numerisch höher (2,5 [0,7, 4,2]) als in der 12-mg-Gruppe (1,8 [-0,8, 4,4]), aber der Unterschied war nicht statistisch signifikant (p = 0,66). Auch die Veränderung des RULM-Scores zwischen Baseline und Tag 279 war in der 50/28-mg-Gruppe numerisch höher als in der entsprechenden 12-mg-Gruppe aus Studie CS4 (Differenz der LS-Mittelwerte: 0,5 (-1,0, 1,9; p = 0,55)) und der entsprechenden Sham-Gruppe aus Studie CS4 (Differenz der LS-Mittelwerte: 1,7 (-0,2, 3,5); p = 0,076).
Studie SM203, Teil C
Teil C von Studie SM203 war eine unverblindete Kohorte, in die 40 Patienten im Alter von 4 bis 65 Jahren mit 1 bis 4 SMN2-Kopien aufgenommen wurden, die von der 12-mg- auf die 50/28-mg-Behandlung umgestellt worden waren. Die Patienten erhielten eine 50-mg-Dosis, gefolgt von zwei Erhaltungsdosen zu je 28 mg (im Abstand von 4 Monaten).
Bei zwei Patienten (5 %) lag die infantile Form vor, bei 38 (95 %) begann die Krankheit später. Zum Zeitpunkt ihrer 50-mg-Aufsättigungsdosis waren 16 Patienten jünger als 18 Jahre und 24 Patienten älter als 18 Jahre. Das mediane Alter (Bereich) bei Beginn der SMA-Symptome betrug 24 (4 bis 192) Monate. Die mediane Dauer (Bereich) der Behandlung im Rahmen des Therapieschemas mit 12 mg Spinraza betrug 3,9 Jahre (1, 5). 21 Patienten (53 %) konnten zur Baseline 15 Schritte selbstständig gehen.
Nach der Umstellung von dem 12-mg- auf das 50/28-mg-Dosierungsschema wurden zwischen Baseline und Tag 302 Verbesserungen auf der HFMSE (Verbesserung um 1,8 Punkte [SD 3,99]) und im RULM (Verbesserung um 1,2 Punkte [SD 2,14]) beobachtet. Bei erwachsenen Patienten (über 18 Jahre; n = 24) wurden an Tag 302 Verbesserungen auf der HFMSE (2,3 Punkte [SD 3,95]) und im RULM (0,9 Punkte [1,89]) festgestellt (Abbildung 6 und 7).
Abbildung 6: Studie SM203 Teil C: HFMSE: mittlere Veränderung (SE) des Gesamtscores nach Visite: ITT-Gruppe
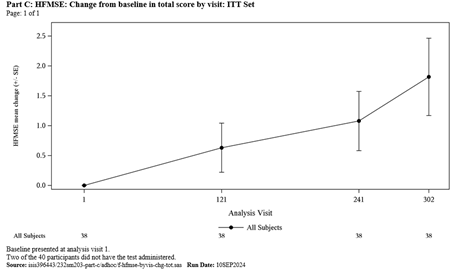
Abbildung 7: Studie SM203 Teil C: RULM: mittlere Veränderung (SE) des Gesamtscores nach Visite: ITT-Gruppe

Therapie bei erwachsenen Patienten mit SMA
Eine Zwischenauswertung liefert klinische Studiendaten von 7 jungen Erwachsenen mit SMA Typ II oder III (im Alter von 18,8 – 22,5 Jahren) mit Behandlungsbeginn als Adoleszente, die 5,3 – 6,8 Jahre in der CS2/CS12/SHINE-Studie beobachtet wurden, und weist auf eine Stabilisierung oder Verbesserung der motorischen Funktionen in diesen Patienten hin, wie der 6-Minuten-Gehtest (6MWT) und HFMSE zeigten.
In Studie SM203 Teil C gab es 24 Patienten über 18 Jahre.
Die Sicherheitsdaten der erwachsenen Patientenpopulation stimmen mit dem bekannten Sicherheitsprofil von Nusinersen und mit Begleiterkrankungen im Zusammenhang mit der SMA-Grunderkrankung überein.
Ergebnisse aus der Anwendung nach Markteinführung bei Erwachsenen
Literaturberichte von Beobachtungsstudien zur Anwendung von Nusinersen 12 mg bei Patienten mit SMA Typ II und III und Therapiebeginn im Erwachsenenalter und einem Follow-Up über 10 bis 14 Monate, weisen auf eine Stabilisierung oder Verbesserung der motorischen Funktionen (HFMSE, 6MWT, RULM) bei Patienten verschiedenen Alters mit unterschiedlich schwerwiegendem Krankheitsverlauf hin, während bei unbehandelten SMA Patienten eine Abnahme der motorischen Funktion im Laufe der Zeit berichtet wurde.
PharmakokinetikDie Pharmakokinetik von Nusinersen nach einmaliger und mehrmaliger Gabe mittels intrathekaler Injektion wurde an pädiatrischen und erwachsenen Patienten mit diagnostizierter SMA untersucht.
Absorption
Durch intrathekale Injektion in die Zerebrospinalflüssigkeit (CSF) kann Nusinersen in vollem Umfang aus dem Liquor in die Zielgewebe des Zentralnervensystems (ZNS) verteilt werden.
Kumulative Daten zur PK im Liquor aus der Langzeitexpositionsstudie (Studie CS11) mit dem zugelassenen 12-mg-Dosierungsschema ergaben eine Akkumulierung der Liquor-Konzentrationen über 9 Jahre bei Patienten mit infantiler Form von SMA und bei Patienten mit SMA mit späterem Krankheitsbeginn.
Ein ähnlicher Trend der Akkumulierung der Liquor-Konzentration wurde auch bei dem 50/28-mg-Dosierungsschema (Studie SM203 und SM302) anhand der über den gesamten Datenzeitraum erhobenen Daten zur PK im Liquor festgestellt.
Die Plasma-Talkonzentrationen von Nusinersen waren nach intrathekaler Verabreichung im Vergleich zur Liquor-Talkonzentration relativ niedrig. Die medianen Tmax-Werte im Plasma lagen im Bereich von 1,7 bis 6,0 Stunden. Die mittleren Cmax- und AUC-Werte im Plasma stiegen im untersuchten Dosisbereich ungefähr dosisproportional an. Nach Mehrfachdosierung fand keine Akkumulierung in Bezug auf Plasmaexpositionsindikatoren (Cmax und AUC) statt.
Distribution
Die Sektionsbefunde bei Patienten (n = 3) zeigen, dass intrathekal verabreichtes Nusinersen im ZNS weit verteilt wird und dass in den Zielgeweben im Rückenmark therapeutische Konzentrationen erreicht werden. Nusinersen wurde auch in Neuronen und in anderen Zelltypen im Rückenmark und im Gehirn sowie in peripheren Geweben, wie etwa im Skelettmuskel, in der Leber und in der Niere, nachgewiesen.
Metabolismus
Nusinersen wird langsam über Exonuklease (3'- und 5')-vermittelte Hydrolyse metabolisiert und ist weder ein Substrat, noch ein Inhibitor oder Induktor von CYP450-Enzymen.
Elimination
Die mittlere terminale Eliminationshalbwertszeit im Liquor beträgt ungefähr 20 Monate. Die Elimination erfolgt vermutlich hauptsächlich durch Ausscheidung von Nusinersen und seinen Metaboliten im Urin.
Interaktionen
In vitro-Studien zeigten, dass Nusinersen keine Induktion oder Inhibition des oxidativen CYP450-vermittelten Stoffwechsels bewirkt, und es sollte daher nicht mit anderen Arzneimitteln um diese Stoffwechselwege konkurrieren. Nusinersen ist kein Substrat oder Inhibitor von humanem BCRP, Pgp, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3 oder BSEP-Transportern.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Pharmakokinetik von Nusinersen wurde bei Patienten mit beeinträchtigter Nieren- oder Leberfunktion nicht untersucht. Die Auswirkung einer Leber- oder Niereninsuffizienz als Kovariablen konnte angesichts der Seltenheit von Patienten, die eine klinisch relevante Leber- oder Niereninsuffizienz aufweisen, im populationspharmakokinetischen Modell nicht umfassend bewertet werden. Populationspharmakokinetische Analysen zeigten keine erkennbare Korrelation zwischen klinischchemischen Leber- und Nierenmarkern und der Variabilität zwischen Subjekten.
Nierenfunktionsstörungen
Siehe unter "Leberfunktionsstörungen"
Geschlecht
Die populationspharmakokinetische Analyse zeigt, dass das Geschlecht keinen Einfluss auf die Pharmakokinetik von Nusinersen hat.
Präklinische DatenMutagenität
Spinraza zeigt keine Anzeichen von Genotoxizität.
Kanzerogenität
In einer Karzinogenitätsstudie wurde Nusinersen männlichen und weiblichen Mäusen über einen Zeitraum von 2 Jahren alle zwei Wochen mittels subkutaner Injektion in Dosen von 0, 5, 15 oder 50 mg/kg verabreicht. Bei der Dosis von 50 mg/kg wurde eine Zunahme der Inzidenz von Gefässtumoren (Hämangiom und Hämangiosarkom kombiniert) beobachtet, die mit einer AUC0-24 assoziiert war, die dem 156-Fachen bzw. 284-Fachen der klinischen Serumexposition bei einer Erhaltungsdosis von 28 mg bzw. 12 mg Nusinersen (auf Basis der jährlichen Dosis) entsprach.
Bei Dosisstufen bis zu 15 mg/kg gab es keinen Hinweis auf eine onkogene Wirkung von Nusinersen. Diese Dosis war mit einer Serum-AUC0-24 assoziiert, die dem 30-Fachen bzw. 55-Fachen der klinischen Serumexposition bei einer Erhaltungsdosis von 28 mg bzw. 12 mg Nusinersen (auf Basis der jährlichen Dosis) entsprach.
Reproduktionstoxizität
Reproduktionstoxikologische Studien wurden an Mäusen und Kaninchen nach subkutaner Verabreichung von Nusinersen durchgeführt. Es wurden keine Auswirkungen auf die Fertilität männlicher oder weiblicher Tiere, auf die embryo-fötale Entwicklung oder die prä- / post-natale Entwicklung festgestellt. Geringe Mengen Nusinersen konnten bei Mäusen in der Milch nachgewiesen werden.
Toxizitätsprüfungen mit juvenilen Tieren
In Studien zur Toxizität nach Mehrfachdosierung (kombiniert über 6 Wochen und 13 Wochen, 14 Wochen und 53 Wochen) an juvenile Cynomolgusaffen führte die intrathekale (i. t.) Gabe von Nusinersen in den untersuchten Dosen nicht zu unerwünschten toxischen Wirkungen. Bei einigen Tieren traten akute, vorübergehende Defizite der unteren Spinalreflexe, eine eingeschränkte Nutzung der Gliedmassen und/oder unkoordinierte Bewegungen auf. Diese Auswirkungen wurden innerhalb weniger Stunden nach Dosisgabe festgestellt, waren innerhalb von 48 Stunden reversibel und wurden nicht als unerwünscht betrachtet. Dosisabhängige Vakuolisierungen von Nervenzellen im Hippocampus wurden bei höheren Dosierungen beobachtet.
Die 53-wöchige Studie zur Toxizität bei juvenilen Affen unterstützt die Sicherheit der Langzeitgabe von Nusinersen, da bei Dosierungen bis zum 14-Fachen und 6,2-Fachen der klinischen Nusinersen-Erhaltungsdosis von 12 mg bzw. 28 mg (drei Erhaltungsdosen pro Jahr) keine unerwünschten Toxizitäten beobachtet wurden. In Lern- und Gedächtnistests wurden keine signifikanten Effekte festgestellt.
Sonstige HinweiseInkompatibilitäten
Keine
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Im Kühlschrank (2-8 °C) lagern.
Nicht einfrieren. Nach versehentlichem Einfrieren verwerfen.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Wenn keine Kühllagerung möglich ist, kann Spinraza vor Licht geschützt bis zu 14 Tage lang bei höchstens 30 °C in der Originalverpackung und Originalkarton aufbewahrt werden.
Ungeöffnete Durchstechflaschen mit Spinraza können, falls erforderlich, vor der Verabreichung aus dem Kühlschrank genommen und wieder in den Kühlschrank gestellt werden. Nach der Entnahme aus der Originalverpackung sollte das Präparat insgesamt nicht länger als 30 Stunden ungekühlt (bei maximal 25 °C) aufbewahrt werden.
Injektionslösung
Spinraza 12 mg
Spinraza 12 mg ist eine klare und farblose sterile Lösung, die 2,4 mg Nusinersen in 1 ml künstlicher Zerebrospinalflüssigkeit enthält.
5 ml Spinraza ist in einer Durchstechflasche aus Typ-I-Glas mit einem Brombutylgummistopfen unter einer Aluminiumversiegelung mit Kunststoffkappe.
Jede Durchstechflasche enthält 12,6 mg Nusinersen-Natrium, was 12 mg Nusinersen als freie Säure entspricht.
Nur zur einmaligen Anwendung.
Spinraza 28 mg
Spinraza 28 mg ist eine klare und farblose sterile Lösung, die 5,6 mg Nusinersen in 1 ml künstlicher Zerebrospinalflüssigkeit enthält.
5 ml Spinraza ist in einer Durchstechflasche aus Typ-I-Glas mit einem Brombutylgummistopfen unter einer Aluminiumversiegelung mit Kunststoffkappe.
Jede Durchstechflasche enthält 29,5 mg Nusinersen-Natrium, was 28 mg Nusinersen als freie Säure entspricht.
Nur zur einmaligen Anwendung.
Spinraza 50 mg
Spinraza 50 mg ist eine klare und farblose sterile Lösung, die 10 mg Nusinersen in 1 ml künstlicher Zerebrospinalflüssigkeit enthält.
5 ml Spinraza ist in einer Durchstechflasche aus Typ-I-Glas mit einem Brombutylgummistopfen unter einer Aluminiumversiegelung mit Kunststoffkappe.
Jede Durchstechflasche enthält 52,6 mg Nusinersen-Natrium, was 50 mg Nusinersen als freie Säure entspricht.
Nur zur einmaligen Anwendung.
Hinweise für die Handhabung
1.Die Durchstechflasche mit Spinraza sollte vor der Verabreichung auf Partikel überprüft werden. Wenn Partikel vorhanden sind und/oder die Flüssigkeit in der Durchstechflasche nicht klar und farblos ist, darf die Durchstechflasche nicht verwendet werden.
2.Die Zubereitung der Spinraza-Lösung für die intrathekale Verabreichung sollte aseptisch erfolgen.
3.Die Durchstechflasche sollte vor der Verabreichung aus dem Kühlschrank genommen werden, um sie Raumtemperatur (25 °C) annehmen zu lassen, ohne dass externe Wärmequellen dazu verwendet werden.
4.Wenn die Durchstechflasche ungeöffnet bleibt und die Lösung nicht verwendet wird, sollte die Durchstechflasche zurück in den Kühlschrank gestellt werden.
5.Direkt vor der Verabreichung die Spritzennadel durch die Mitte der Versiegelung über dem Stopfen in die Durchstechflasche einführen und das erforderliche Volumen entnehmen (siehe "Dosierung/Anwendung" ). Spinraza darf nicht verdünnt werden. Die Verwendung von externen Filtern ist nicht notwendig.
6.Wenn die Lösung nach dem Aufziehen in die Spritze nicht innerhalb von 6 Stunden verwendet wird, ist sie zu verwerfen.
7.Nicht verwendetes Arzneimittel oder Abfallmaterial darf nicht erneut verwendet werden und ist entsprechend den nationalen Anforderungen zu beseitigen.
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer66495 (Swissmedic)
PackungenPackung mit einer Durchstechflasche mit 12 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit. (A)
Packung mit einer Durchstechflasche mit 28 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit. (A)
Packung mit einer Durchstechflasche mit 50 mg Nusinersen in 5 ml künstlicher Zerebrospinalflüssigkeit. (A)
ZulassungsinhaberinBiogen Switzerland AG, 6340 Baar.
Stand der InformationFebruar 2026
|