ZusammensetzungWirkstoffe
Durvalumab
Hilfsstoffe
L-Histidin, L-Histidin-Hydrochlorid-Monohydrat, α,α-Trehalose-Dihydrat, Polysorbat 80, Wasser für Injektion
Darreichungsform und Wirkstoffmenge pro EinheitKonzentrat zur Herstellung einer Infusionslösung; 50 mg/ml in Einzeldosis-Durchstechflasche zur intravenösen Anwendung.
Eine Durchstechflasche à 2,4 ml enthält 120 mg Durvalumab.
Eine Durchstechflasche à 10 ml enthält 500 mg Durvalumab.
Imfinzi ist eine sterile, klare bis opalisierende, farblose bis leicht gelbliche Lösung, ohne Konservierungsstoffe und ohne sichtbare Partikel.
Imfinzi ist ein humaner monoklonaler Immunglobulin-Antikörper (IgG1κ).
Indikationen/AnwendungsmöglichkeitenBefristet zugelassene Indikationen
Nicht-kleinzelliges Lungenkarzinom (NSCLC)
Imfinzi, in Kombination mit platinbasierter Chemotherapie als neoadjuvante Behandlung, gefolgt von Imfinzi als Monotherapie nach Operation, ist indiziert für die Behandlung von erwachsenen Patienten mit resezierbarem NSCLC (Tumore ≥4 cm und/oder positiver Lymphknotenbefall) ohne bekannte Mutationen des epidermalen Wachstumsfaktor-Rezeptors (EGFR) oder Rearrangements der anaplastischen Lymphomkinase (ALK).
Die Studie AEGEAN war nicht darauf ausgelegt, die Wirksamkeit von Imfinzi in der neoadjuvanten bzw. adjuvanten Therapiephase separat zu analysieren und zu beurteilen (siehe "Eigenschaften/Wirkungen" ). Die zusätzliche adjuvante Therapie mit Imfinzi gegenüber der neoadjuvanten Behandlungsphase allein war mit einer zusätzlichen Toxizität verbunden (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Aufgrund einer zum Zeitpunkt der Begutachtung des Gesuches unvollständigen Dokumentation, wird diese Indikation befristet zugelassen (Art. 9a Heilmittelgesetz). Die befristete Zulassung ist zwingend an die zeitgerechte Erfüllung von Auflagen gebunden. Nach deren Erfüllung kann die befristete Zulassung in eine Zulassung ohne besondere Auflagen überführt werden.
Nicht befristet zugelassene Indikationen
Nicht-kleinzelliges Lungenkarzinom (NSCLC)
Imfinzi ist indiziert für die Behandlung von erwachsenen Patienten mit lokal fortgeschrittenem, nicht resezierbarem nicht-kleinzelligem Lungenkarzinom (NSCLC), deren Erkrankung nach einer definitiven platinbasierten Chemoradiotherapie (CRT) nicht fortgeschritten ist.
Kleinzelliges Lungenkarzinom (SCLC)
Imfinzi als Monotherapie ist indiziert für die Behandlung von erwachsenen Patienten mit inoperablem limited-stage kleinzelligem Lungenkarzinom (LS-SCLC, limited-stage small cell lung cancer), deren Erkrankung nach einer platinbasierten Chemoradiotherapie (CRT) nicht fortgeschritten ist.
Imfinzi, in Kombination mit Etoposid und entweder Carboplatin oder Cisplatin, ist indiziert für die Erstlinienbehandlung von erwachsenen Patienten mit fortgeschrittenem kleinzelligem Lungenkarzinom (ES-SCLC, extensive-stage small cell lung cancer).
Biliäres Karzinom (BTC)
Imfinzi, in Kombination mit Gemcitabin und Cisplatin, ist indiziert für die Erstlinienbehandlung von erwachsenen Patienten mit lokal fortgeschrittenem oder metastasiertem biliärem Karzinom (siehe Rubrik "Klinische Wirksamkeit" ).
Blasenkrebs
Imfinzi in Kombination mit Gemcitabin und Cisplatin als neoadjuvante Behandlung, gefolgt von Imfinzi als adjuvante Monotherapie nach radikaler Zystektomie, ist indiziert für die Behandlung von erwachsenen Patienten mit muskelinvasivem Blasenkrebs (MIBC).
Die Studie NIAGARA war nicht darauf ausgelegt, die Wirksamkeit von Imfinzi in der neoadjuvanten bzw. adjuvanten Therapiephase separat zu analysieren und zu beurteilen (siehe "Eigenschaften/Wirkungen" ). Die zusätzliche adjuvante Therapie mit Imfinzi gegenüber der neoadjuvanten Behandlungsphase allein war mit einer zusätzlichen Toxizität verbunden (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Endometriumkarzinom (EC)
Imfinzi in Kombination mit Carboplatin und Paclitaxel, gefolgt von Imfinzi als Monotherapie, ist indiziert für die Behandlung von erwachsenen Patientinnen mit primärem fortgeschrittenem oder rezidiviertem Endometriumkarzinom mit defizienter Mismatch-Reparatur (dMMR), die ein hohes Rezidivrisiko aufweisen (siehe Rubriken "Warnhinweise und Vorsichtsmassnahmen" und "Klinische Wirksamkeit" ).
Adenokarzinom des Magens oder des gastroösophagealen Übergangs (GC/GEJC)
Imfinzi in Kombination mit FLOT-Chemotherapie als neoadjuvante und adjuvante Behandlung nach radikaler Operation, gefolgt von adjuvanter Imfinzi-Monotherapie, ist indiziert für die Behandlung von erwachsenen Patienten mit resezierbarem Adenokarzinom des Magens oder des gastroösophagealen Übergangs.
Die Studie MATTERHORN war nicht darauf ausgelegt, die Wirksamkeit von Imfinzi in der neoadjuvanten bzw. adjuvanten Therapiephase separat zu analysieren und zu beurteilen (siehe "Eigenschaften/Wirkungen" ). Die zusätzliche adjuvante Therapie mit Imfinzi gegenüber der neoadjuvanten Behandlungsphase allein war mit einer zusätzlichen Toxizität verbunden (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Dosierung/AnwendungDie Behandlung mit Imfinzi sollte von einem erfahrenen Onkologen eingeleitet und überwacht werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Art der Anwendung
Zur intravenösen Anwendung.
Für die Anleitung zur Verdünnung dieses Arzneimittels vor der Anwendung siehe "Sonstige Hinweise" .
Übliche Dosierung
Die empfohlene Dosis von Imfinzi richtet sich nach der Indikation. Imfinzi wird als intravenöse Infusion über 60 Minuten verabreicht.
Lokal fortgeschrittenes NSCLC
Die empfohlene Dosis von Imfinzi ist 10 mg/kg alle 2 Wochen oder 1500 mg alle 4 Wochen, bis zur Krankheitsprogression oder inakzeptablen Toxizität, für maximal 12 Monate.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung erforderlich, entsprechend Imfinzi 10 mg/kg alle 2 Wochen oder 20 mg/kg alle 4 Wochen als Monotherapie bis zum Erreichen eines Körpergewichts von mehr als 30 kg.
Sowohl die Dosierungsempfehlung für die 4-wöchentliche Gabe von 1500 mg als auch die gewichtsbasierte Dosierungsempfehlung für Patienten mit einem Körpergewicht von 30 kg oder weniger werden gestützt durch populationspharmakokinetische Simulationen.
Resezierbares NSCLC
Die empfohlene Dosis von Imfinzi ist 1500 mg in Kombination mit Chemotherapie alle 3 Wochen für bis zu 4 Zyklen vor der Operation, gefolgt von 1500 mg alle 4 Wochen als Monotherapie nach der Operation, bis die Erkrankung als nicht resezierbar gilt, rezidiviert oder eine inakzeptable Toxizität auftritt, oder für maximal 12 Zyklen nach der Operation.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung von 20 mg/kg erforderlich, und zwar mit 20 mg/kg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) vor der Operation, gefolgt von einer Monotherapie mit 20 mg/kg alle 4 Wochen nach der Operation bis zum Erreichen eines Körpergewichts von mehr als 30 kg.
Wenn die Anwendung am selben Tag erfolgt, soll Imfinzi vor der Chemotherapie verabreicht werden. Die Angaben zur Dosierung des Chemotherapeutikums sind der jeweiligen Fachinformation zu entnehmen. Zu den Dosierungsempfehlungen für die Chemotherapie siehe Rubrik "Klinische Wirksamkeit" .
LS-SCLC
Die empfohlene Dosis von Imfinzi ist 1500 mg alle 4 Wochen bis zur Krankheitsprogression, inakzeptablen Toxizität oder für maximal 24 Monate.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung erforderlich, entsprechend Imfinzi 20 mg/kg alle 4 Wochen als Monotherapie, bis zum Erreichen eines Körpergewichts von mehr als 30 kg. Diese Dosierungsempfehlung wird gestützt durch populationspharmakokinetische Simulationen.
Fortgeschrittenes SCLC (ES-SCLC)
Die empfohlene Dosis von Imfinzi ist 1500 mg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) für 4 Zyklen, gefolgt von 1500 mg alle 4 Wochen als Monotherapie, bis zur Krankheitsprogression oder inakzeptablen Toxizität.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung von Imfinzi erforderlich, und zwar mit 20 mg/kg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) für 4 Zyklen, gefolgt von einer Monotherapie mit 20 mg/kg alle 4 Wochen bis zum Erreichen eines Körpergewichts von mehr als 30 kg. Diese Dosierungsempfehlung wird gestützt durch populationspharmakokinetische Simulationen.
Wenn die Anwendung am selben Tag erfolgt, soll Imfinzi vor der Chemotherapie verabreicht werden. Die Angaben zur Dosierung von Etoposid und Carboplatin oder Cisplatin sind der jeweiligen Fachinformation zu entnehmen.
BTC
Die empfohlene Dosis von Imfinzi beträgt 1500 mg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von 1500 mg alle 4 Wochen als Monotherapie, bis zur Krankheitsprogression oder inakzeptablen Toxizität.
Als Chemotherapie wird Gemcitabin 1000 mg/m2 und Cisplatin 25 mg/m2 (jeweils verabreicht an den Tagen 1 und 8) alle 3 Wochen (21 Tage) für bis zu 8 Zyklen verabreicht.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung von Imfinzi erforderlich, und zwar mit 20 mg/kg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von einer Monotherapie mit 20 mg/kg alle 4 Wochen bis zum Erreichen eines Körpergewichts von mehr als 30 kg.
Wenn die Anwendung am selben Tag erfolgt, soll Imfinzi vor der Chemotherapie verabreicht werden.
MIBC
Die empfohlene Dosis von Imfinzi beträgt 1500 mg in Kombination mit Chemotherapie alle 3 Wochen (21 Tage) für 4 Zyklen vor der Operation, gefolgt von 1500 mg alle 4 Wochen als Monotherapie nach der Operation, bis zu einer Krankheitsprogression, die eine endgültige Operation ausschliesst, bis zum Rezidiv der Krankheit, bis zu inakzeptabler Toxizität oder bis zu einem Maximum von 8 Zyklen nach der Operation.
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung von Imfinzi von 20 mg/kg erforderlich. In Kombination mit Chemotherapie ist Imfinzi mit 20 mg/kg alle 3 Wochen (21 Tage) vor der Operation zu verabreichen, gefolgt von einer Monotherapie mit 20 mg/kg alle 4 Wochen nach der Operation, bis das Gewicht auf mehr als 30 kg ansteigt.
Wenn die Anwendung am selben Tag erfolgt, soll Imfinzi vor der Chemotherapie verabreicht werden.
Endometriumkarzinom
Die empfohlene Dosis von Imfinzi beträgt 1120 mg in Kombination mit Carboplatin und Paclitaxel alle 3 Wochen (21 Tage) für mindestens 4 und für bis zu 6 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen als Monotherapie, bis zur Krankheitsprogression oder inakzeptablen Toxizität.
Die Angaben zur Dosierung des entsprechenden Chemotherapeutikums sind der jeweiligen Fachinformation zu entnehmen.
Bei Patientinnen mit einem Körpergewicht von 30 kg oder weniger in der Erhaltungsphase ist eine gewichtsbezogene Dosierung erforderlich, entsprechend Imfinzi 20 mg/kg, bis zum Erreichen eines Körpergewichts von mehr als 30 kg.
GC/GEJC
Die empfohlene Dosis von Imfinzi ist 1500 mg in Kombination mit FLOT-Chemotherapie (5-FU 2600 mg/m2, Leucovorin 200 mg/m2, Oxaliplatin 85 mg/m2, Docetaxel 50 mg/m2) alle 4 Wochen für bis zu 2 Zyklen vor der Operation, gefolgt von 1500 mg kombiniert mit FLOT-Chemotherapie alle 4 Wochen für bis zu 2 Zyklen und anschliessend 1500 mg als Monotherapie alle 4 Wochen für bis zu 10 Zyklen, ingesamt bis zu 12 Zyklen, nach der Operation. In der neoadjuvanten Phase wird Imfinzi bis zur Krankheitsprogression, die eine definitive Operation ausschliesst, oder inakzeptablen Toxizität verabreicht. In der adjuvanten Phase wird Imfinzi bis zur Krankheitsprogression oder einem Rezidiv, inakzeptablen Toxizität oder für maximal 12 Zyklen nach der Operation verabreicht. Die erste Dosis der adjuvanten Therapie sollte nicht früher als 4 Wochen und nicht später als 12 Wochen nach der Operation verabreicht werden. Bei Behandlungsverzögerungen darf die Behandlung mit Imfinzi eine Gesamtdauer von 12 Monaten ab der ersten Dosis der adjuvanten Therapie nicht überschreiten (unabhängig von der Anzahl der verabreichten Behandlungszyklen).
Bei Patienten mit einem Körpergewicht von 30 kg oder weniger ist eine gewichtsbezogene Dosierung von 20 mg/kg In Kombination mit FLOT-Chemotherapie oder als Monotherapie ist eine Dosis von 20 mg/kg alle 4 Wochen bis zum Erreichen eines Körpergewichts von mehr als 30 kg indiziert.
Wenn die Anwendung am selben Tag erfolgt, soll Imfinzi vor der Chemotherapie verabreicht werden.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Es wird keine Dosisreduzierung oder -erhöhung für Imfinzi empfohlen. Im Allgemeinen sollte Imfinzi bei schwerwiegenden (Grad 3) immunvermittelten unerwünschten Wirkungen ausgesetzt werden. Imfinzi sollte permanent abgesetzt werden bei lebensbedrohlichen (Grad 4) immunvermittelten unerwünschten Wirkungen, bei wiederkehrenden schwerwiegenden (Grad 3) immunvermittelten unerwünschten Wirkungen, die eine systemische immunsuppressive Behandlung erfordern, sowie falls die Kortikosteroiddosis nicht innerhalb von 12 Wochen nach Initiierung der Kortikosteroide auf 10 mg oder weniger Prednison oder gleichwertig pro Tag reduziert werden kann.
Immunvermittelte unerwünschte Wirkungen, die ein spezifisches Management erfordern, sind in Tabelle 1 zusammengefasst. Siehe "Warnhinweise und Vorsichtsmassnahmen" für weitere Informationen zur Überwachung und Beurteilung.
Tabelle 1: Empfohlene Behandlungsmodifikationen für Imfinzi
Unerwünschte Wirkungen Schweregrada Imfinzi-Behandlungsmod
ifikationen
Immunvermittelte Pneumonitis / Grad 2 Aussetzen der Dosisb
interstitielle Lungenerkrankung
Grad 3 oder 4 Permanentes Absetzen
Immunvermittelte Hepatitis ALT oder AST > 3 – ≤5 x ULN oder Aussetzen der Dosisb
Gesamt-bilirubin > 1,5 – ≤3 x ULN
AST oder ALT > 5 – ≤10 x ULN Aussetzen der Dosisb
Gleichzeitiger ALT oder AST > 3 x ULN Permanentes Absetzen
und Gesamt-bilirubin > 2 x ULN
ALT oder AST > 10 x ULN oder Permanentes Absetzen
Gesamt-bilirubin > 3 x ULN
Immunvermittelte Colitis oder Diarrhö Grad 2 oder 3 Aussetzen der Dosisb
Grad 4 Permanentes Absetzen
Immunvermittelte Endokrinopathien: Grade 2–4 Aussetzen der Verabrei
Hyperthyreose, Thyreoiditis chung bis klinisch
stabil
Immunvermittelte Endokrinopathien: Grade 2–4 Keine Änderungen
Hypothyreose
Immunvermittelte Endokrinopathien: Grade 2–4 Aussetzen der Verabrei
Nebenniereninsuffizienz, Hypophysitis chung bis klinisch
/ Hypophyseninsuffizienz stabil
Immunvermittelte Endokrinopathien: Grade 2–4 Aussetzen der Verabrei
Typ 1 Diabetes mellitus chung bis klinisch
stabil
Immunvermittelte Nephritis Grad 2 mit Serumkreatinin > 1,5–3 Aussetzen der Dosisb
x (ULN oder Baseline)
Grad 3 mit Serumkreatinin > 3 x Permanentes Absetzen
Baseline oder > 3–6 x ULN; Grad 4 mit
Serumkreatinin > 6 x ULN
Immunvermittelter Ausschlag oder Grad 2> 1 Woche oder Grad 3 Aussetzen der Dosisb
Dermatitis (inkl. Pemphigoid)
Grad 4 Permanentes Absetzen
Immunvermittelte Myokarditis Grade 2–4 Permanentes Absetzen
Immunvermittelte Myositis / Grad 2 oder 3 Aussetzen der Dosisb,c
Polymyositis / Rhabdomyolyse
Grad 4 Permanentes Absetzen
Infusionsbezogene Reaktionen Grad 1 oder 2 Unterbruch oder
Verlangsamen der
Infusionsgeschwindigke
it
Grad 3 oder 4 Permanentes Absetzen
Immunvermittelte Myasthenia gravis Grad 2–4 Permanentes Absetzen
Immunvermittelte Encephalitis Grad 2 – 4 Permanentes Absetzen
Immunvermitteltes Guillain-Barré-Syndr Grad 2 – 4 Permanentes Absetzen
om
Immunvermittelte transverse Myelitis Alle Grade Permanentes Absetzen
Andere immunvermittelte unerwünschte Grad 2 oder 3 Aussetzen der Dosisb
Wirkungend
Grad 4 Permanentes Absetzen
Rezidivierende unerwünschte Wirkung Rezidivierende unerwünschte Permanentes Absetzen
Grad 3 oder 4 Wirkung Grad 3 oder 4
(schwerwiegend oder lebensbedrohen
d)
a Allgemeine Terminologiekriterien für unerwünschte Wirkungen, Version 4.03. ALT: Alanin-Aminotransferase, AST: Aspartat-Aminotransferase, ULN: obere Normgrenze.
b Nach dem Aussetzen kann Imfinzi innerhalb von 12 Wochen wieder verabreicht werden, wenn sich die unerwünschten Wirkungen auf ≤ Grad 1 besserten und die Kortikosteroid-Dosis auf ≤10 mg Prednison oder gleichwertig pro Tag reduziert worden ist. Bei wiederkehrenden unerwünschten Wirkungen des Grades 3 sollte Imfinzi permanent abgesetzt werden.
c Imfinzi ist dauerhaft abzusetzen, wenn die unerwünschte Wirkung sich nicht innerhalb von 30 Tagen auf ≤ Grad 1 bessert oder wenn Anzeichen von respiratorischer Insuffizienz auftreten.
d Beinhaltet Immunthrombozytopenie, Pankreatitis, Arthritis, Uveitis und Polymyalgia rheumatica.
Im Falle von nicht-immunvermittelten Reaktionen ist Imfinzi bei Grad 2 oder 3 der unerwünschten Wirkung auszusetzen und bei Grad 4 abzusetzen. Unerwünschte Wirkungen sind gemäss institutionellen Standards zu behandeln.
Spezielle Dosierungsanweisungen
Eine Dosisanpassung aufgrund von Patientenalter, Körpergewicht, Geschlecht oder ethnischer Gruppe ist nicht erforderlich (siehe "Pharmakokinetik" ).
Patienten mit Nierenfunktionsstörungen
Die Sicherheit und Wirksamkeit von Imfinzi wurde bei Patienten mit eingeschränkter Nierenfunktion nicht untersucht. Basierend auf einer pharmakokinetischen Analyse der Population wird bei Patienten mit leichter oder moderater Nierenfunktionsstörung keine Dosisanpassung von Imfinzi empfohlen (siehe "Pharmakokinetik" ). Die Datenlage bei Patienten mit schwerer Nierenfunktionsstörung ist zu begrenzt, um Schlussfolgerungen bei dieser Population ziehen zu können (siehe "Pharmakokinetik" ).
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Imfinzi wurde bei Patienten mit Leberfunktionsstörung nicht untersucht. Basierend auf einer pharmakokinetischen Analyse der Population wird bei Patienten mit leichter oder mittelschwerer Leberfunktionsstörung keine Dosisanpassung von Imfinzi empfohlen. Imfinzi wurde an Patienten mit schwerer Leberfunktionsstörung nicht untersucht (siehe "Pharmakokinetik" ).
Ältere Patienten
Basierend auf einer pharmakokinetischen Analyse der Population ist für ältere Patienten (≥65 Jahre) keine Dosisanpassung erforderlich (siehe "Eigenschaften / Wirkungen" ).
Von den 476 Patienten mit lokal fortgeschrittenem, inoperablem NSCLC (primäre Wirksamkeitspopulation), die mit Imfinzi behandelt wurden, waren 215 Patienten 65 Jahre oder älter. Es wurden keine gesamthaft klinisch bedeutsamen Unterschiede hinsichtlich der Sicherheit für Patienten ≥65 Jahren verglichen mit jüngeren Patienten berichtet. Die Daten von Patienten ab 75 Jahren aus der PACIFIC-Studie (7,6 %) sind zu begrenzt, um Schlüsse auf diese Population zuzulassen.
Von den 401 Patienten mit resezierbarem NSCLC, die in der AEGEAN-Studie mit Imfinzi in Kombination mit Chemotherapie behandelt wurden, waren 209 Patienten (52 %) 65 Jahre alt oder älter und 49 Patienten (12 %) waren 75 Jahre alt oder älter. Insgesamt wurden keine klinisch bedeutsamen Unterschiede bezüglich der Sicherheit oder Wirksamkeit zwischen ≥65 Jahre alten Patienten und jüngeren Patienten berichtet.
Von den 262 Patienten mit LS-SCLC, die mit Imfinzi behandelt wurden, waren 103 (39,3 %) Patienten 65 Jahre oder älter. Insgesamt wurden keine klinisch bedeutsamen Unterschiede bezüglich der Sicherheit oder Wirksamkeit zwischen ≥65 Jahre alten Patienten und jüngeren Patienten berichtet.
Von den 265 Patienten mit fortgeschrittenem SCLC, die mit Imfinzi in Kombination mit Chemotherapie behandelt wurden, waren 101 (38%) Patienten 65 Jahre oder älter. Insgesamt wurden keine klinisch bedeutsamen Unterschiede bezüglich Sicherheit oder Wirksamkeit bei ≥65 Jahre alten und jüngeren Patienten berichtet.
Von den 338 Patienten mit BTC, die mit Imfinzi in Kombination mit Chemotherapie behandelt wurden, waren 158 (46,7%) Patienten 65 Jahre oder älter. Insgesamt wurden keine klinisch bedeutsamen Unterschiede bezüglich Sicherheit oder Wirksamkeit bei ≥65 Jahre alten und jüngeren Patienten berichtet.
Von den 533 MIBC-Patienten, die mit Imfinzi in Kombination mit Chemotherapie behandelt wurden, waren 275 (51,6 %) Patienten 65 Jahre oder älter. Insgesamt gab es keine klinisch bedeutsamen Unterschiede bezüglich der Sicherheit oder Wirksamkeit zwischen ≥65 Jahre alten Patienten und jüngeren Patienten.
Von den 238 Patientinnen mit Endometriumkarzinom, die randomisiert einer platinbasierten Chemotherapie + Imfinzi zugeteilt wurden, waren 116 (48,7%) Patientinnen 65 Jahre oder älter und 29 (12,2%) waren 75 Jahre oder älter. Es gab keine klinisch bedeutsamen Unterschiede im Hinblick auf die in der Sicherheit und Wirksamkeit zwischen Patientinnen im Alter ab 65 Jahren und jüngeren Patientinnen.
Von den 475 Patienten mit resezierbarem GC/GEJC, die in der MATTERHORN-Studie mit Imfinzi in Kombination mit FLOT-Chemotherapie als neoadjuvante und adjuvante Therapie und anschliessend als Imfinzi-Monotherapie behandelt wurden, waren 184 Patienten (38,7 %) 65 Jahre alt oder älter und 37 Patienten (7,8 %) waren 75 Jahre alt oder älter. Insgesamt wurden keine klinisch bedeutsamen Unterschiede bezüglich der Sicherheit oder Wirksamkeit zwischen ≥65 Jahre alten Patienten und jüngeren Patienten berichtet.
Pädiatrische Population
Imfinzi ist für die Anwendung in der pädiatrischen Population nicht zugelassen. Ausserhalb der zugelassenen Indikationen wurde Imfinzi bei Kindern im Alter von 1 bis 17 Jahren mit Neuroblastom, soliden Tumoren und Sarkomen untersucht.
Die Ergebnisse dieser Studie haben keine Wirksamkeit von Imfinzi in dieser Population nachgewiesen.
Die aktuell verfügbaren Daten werden in der Rubrik "Unerwünschte Wirkungen" beschrieben.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenSiehe Rubrik "Dosierung/Anwendung" Tabelle 1, für empfohlene Behandlungsmodifikationen.
Bei mutmasslichen immunvermittelten unerwünschten Wirkungen sollte eine angemessene Beurteilung durchgeführt werden, um die Ätiologie zu bestätigen oder alternative Ätiologien auszuschliessen. Je nach Schweregrad der unerwünschten Wirkung sollte Imfinzi ausgesetzt oder dauerhaft abgesetzt werden. Eine Behandlung mit Kortikosteroiden oder eine endokrine Therapie sollte eingeleitet werden. Bei Ereignissen, die eine Kortikosteroidtherapie erfordern, oder nach Verbesserung auf ≤ Grad 1 sollte die Kortikosteroidtherapie über mindestens 1 Monat ausgeschlichen werden. Falls eine Verschlechterung oder keine Verbesserung eintritt, ist eine Erhöhung der Kortikosteroiddosis und/oder die Anwendung zusätzlicher systemischer Immunsuppressiva zu erwägen.
Falls die Kortikosteroide nicht ausgeschlichen werden können und innerhalb von 12 Wochen nach der letzten Dosis von Imfinzi keine Reduktion auf ≤10 mg Prednison pro Tag (oder Äquivalent) möglich ist, muss Imfinzi dauerhaft abgesetzt werden.
Immunvermittelte Pneumonitis
Nach Verabreichung von Imfinzi traten bei Patienten immunvermittelte Pneumonitis und interstitielle Lungenerkrankung auf, einschliesslich mit Todesfolge, definiert durch den Bedarf an systemischen Kortikosteroiden und ohne klare alternative Ätiologie (siehe "Unerwünschte Wirkungen" ). Bei Ereignissen des Schweregrades 2 sollte eine Behandlung mit einer Initialdosis von 1 – 2 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung. Bei Ereignissen des Grades 3 oder 4 sollte eine Behandlung mit einer Initialdosis von 2 – 4 mg Methylprednisolon/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung.
Pneumonitis und Strahlenpneumonitis
Strahlenpneumonitis wird häufig bei Patienten mit Bestrahlungstherapie der Lunge beobachtet. Die klinische Präsentation von Pneumonitis und Strahlenpneumonitis ist sehr ähnlich. Patienten sollten hinsichtlich Anzeichen und Symptomen von Pneumonitis oder Strahlenpneumonitis überwacht werden. Ein Verdacht auf Pneumonitis sollte anhand von Röntgenaufnahmen unter Ausschluss anderer infektiöser und krankheitsassoziierter Ursachen bestätigt und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden.
Immunvermittelte Hepatitis
Nach Verabreichung von Imfinzi trat bei Patienten immunvermittelte Hepatitis auf, einschliesslich mit Todesfolge, definiert durch den Bedarf an systemischen Kortikosteroiden und ohne klare alternative Ätiologie (siehe "Unerwünschte Wirkungen" ). Die Patienten sollten vor und während der Behandlung mit Imfinzi regelmässig auf abnorme Leberwerte getestet werden. Immunvermittelte Hepatitis sollte gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei allen Schweregraden sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung.
Immunvermittelte Colitis
Nach Verabreichung von Imfinzi trat bei Patienten immunvermittelte Colitis oder Diarrhö auf, definiert durch den Bedarf an systemischen Kortikosteroiden und ohne klare alternative Ätiologie (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf Anzeichen und Symptome einer Colitis oder Diarrhö überwacht werden und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei Ereignissen der Schweregrade 2-4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung. Bei Verdacht auf Darmperforation jeglichen Grades ist unverzüglich ein Chirurg zu konsultieren.
Immunvermittelte Endokrinopathien
Immunvermittelte Hypothyreose/Hyperthyreose/Thyreoiditis
Immunvermittelte Hypothyreose, Hyperthyreose oder Thyreoiditis ist bei Patienten aufgetreten, die mit Imfinzi behandelt wurden (siehe "Unerwünschte Wirkungen" ). Patienten sollten vor und während der Behandlung regelmässig auf eine abnorme Schilddrüsenfunktion getestet und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei immunvermittelter Hypothyreose der Schweregrade 2-4 ist ein Schilddrüsenhormonersatz einzuleiten, wenn dieser klinisch angezeigt ist. Bei immunvermittelter Hyperthyreose oder Thyreoiditis der Grade 2-4 kann eine symptomatische Behandlung durchgeführt werden.
Immunvermittelte Nebenniereninsuffizienz
Immunvermittelte Nebenniereninsuffizienz ist bei Patienten aufgetreten, die mit Imfinzi behandelt wurden (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf klinische Anzeichen und Symptome einer Nebenniereninsuffizienz untersucht werden. Bei symptomatischer Nebenniereninsuffizienz sollten Patienten gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei immunvermittelter Nebenniereninsuffizienz der Schweregrade 2-4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent, gefolgt von ausschleichender Dosierung, und eine Hormonersatztherapie, sofern klinisch angezeigt, eingeleitet werden.
Immunvermittelter Diabetes mellitus Typ 1
Immunvermittelter Diabetes mellitus Typ 1, der sich mit diabetischer Ketoazidose präsentieren kann, ist bei Patienten aufgetreten, die mit Imfinzi behandelt wurden (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf klinische Anzeichen und Symptome von Diabetes mellitus Typ 1 untersucht werden. Bei symptomatischem Diabetes mellitus Typ 1 sollten Patienten gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei Ereignissen der Schweregrade 2-4 kann eine Behandlung mit Insulin eingeleitet werden, wenn diese klinisch angezeigt ist.
Immunvermittelte Hypophysitis / Hypophyseninsuffizienz
Immunvermittelte Hypophysitis oder Hypophyseninsuffizienz ist bei Patienten aufgetreten, die mit Imfinzi behandelt wurden (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf klinische Anzeichen und Symptome einer Hypophysitis untersucht werden. Bei symptomatischer Hypophysitis oder Hypophyseninsuffizienz sollten Patienten gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei Ereignissen der Schweregrade 2-4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent, gefolgt von ausschleichender Dosierung, und eine Hormonersatztherapie, wenn klinisch angezeigt, eingeleitet werden.
Immunvermittelte Nephritis
Nach Verabreichung von Imfinzi trat bei Patienten immunvermittelte Nephritis auf, definiert durch den Bedarf an systemischen Kortikosteroiden und ohne klare alternative Ätiologie (siehe "Unerwünschte Wirkungen" ). Patienten sollten vor und während der Behandlung mit Imfinzi periodisch auf eine abnorme Nierenfunktion getestet und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei immunvermittelter Nephritis der Schweregrade 2-4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung.
Immunvermitteltes Exanthem
Nach Verabreichung von Imfinzi trat bei Patienten immunvermitteltes Exanthem oder Dermatitis (inkl. Pemphigoid) auf, definiert durch den Bedarf an systemischen Kortikosteroiden und ohne klare alternative Ätiologie (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf Anzeichen und Symptome von Exanthemen oder Dermatitis untersucht und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei immunvermitteltem Exanthem des Grades 2 über > 1 Woche oder der Grade 3 oder 4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 1-2 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung.
Immunvermittelte Myokarditis
Immunvermittelte Myokarditis, die tödlich verlaufen kann, trat bei Patienten auf, die Imfinzi erhielten (siehe "Unerwünschte Wirkungen" ). Patienten sollten auf Anzeichen und Symptome von immunvermittelter Myokarditis überwacht und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei immunvermittelter Myokarditis der Schweregrade 2-4 sollte eine Behandlung mit Kortikosteroiden in einer Initialdosis von 2-4 mg Prednison/kg/Tag oder Äquivalent eingeleitet werden, gefolgt von ausschleichender Dosierung. Wenn trotz der Gabe von Kortikosteroiden innerhalb von 2 bis 3 Tagen keine Besserung eintritt, ist umgehend eine zusätzliche immunsuppressive Therapie einzuleiten. Nach Rekonvaleszenz (Grad 0) sollten die Kortikosteroide über mindestens 1 Monat ausgeschlichen werden.
Immunvermittelte hämophagozytische Lymphohistiozytose (HLH)
HLH ist bei Patienten unter Behandlung mit Imfinzi aufgetreten (siehe "Unerwünschte Wirkungen" ). HLH ist ein potentiell lebensbedrohliches Syndrom mit pathologischer Aktivierung der Immunabwehr. Falls die HLH nicht frühzeitig erkannt und behandelt wird, verläuft sie häufig letal. Die Erkrankung ist gekennzeichnet durch klinische Anzeichen und Symptome einer schweren systemischen Inflammation wie Fieber, Hautausschlag, Hepatosplenomegalie, Zytopenie (v.a. Anämie und Thrombozytopenie), Lymphadenopathie, neurologische Symptome, hohes Serum-Ferritin, Hypertriglyceridämie sowie Störungen der Leberfunktion und der Koagulation. Patienten, bei denen solche Anzeichen und Symptome auftreten, müssen unverzüglich untersucht und im Hinblick auf eine mögliche HLH-Diagnose beurteilt werden. Die Gabe von Imfinzi sollte ausgesetzt werden, solange keine alternative Ätiologie etabliert werden kann.
Andere immunvermittelte Nebenwirkungen
Aufgrund des Wirkmechanismus von Imfinzi könnten andere immunvermittelte unerwünschte Wirkungen auftreten. Patienten sollten auf Anzeichen und Symptome untersucht und gemäss Empfehlungen in "Dosierung/Anwendung" behandelt werden. Bei Patienten, die in klinischen Studien mit Imfinzi als Monotherapie behandelt wurden (n=4045), traten die folgenden klinisch relevanten, immunvermittelten unerwünschte Wirkungen mit einer Häufigkeit von <1 % auf: aseptische Meningitis, hämolytische Anämie, Immunthrombozytopenie, nichtinfektiöse Zystitis, Myositis, Rhabdomyolyse, Encephalitis, Pankreatitis, Guillain-Barré-Syndrom, Arthritis, Uveitis und Polymyalgia rheumatica (siehe "Unerwünschte Wirkungen" ) und entzündliche Augenerkrankungen, einschliesslich Keratitis. Bei einem Patienten, der in einer laufenden klinischen Studie mit Imfinzi behandelt worden war, trat eine Polymyositis mit letalem Ausgang auf. In seltenen Fällen kann Myasthenia gravis als immunvermittelte unerwünschte Wirkung auftreten.
Nach Markteinführung wurden unter Behandlung mit Imfinzi Fälle von transverser Myelitis beobachtet. Die Patienten sollten hinsichtlich Anzeichen und Symptomen überwacht werden, die auf eine Myelitis hindeuten.
Es wurden unter Behandlung mit Imfinzi Fälle von Zöliakie beobachtet.
Die folgenden klinisch relevanten, immunvermittelten unerwünschten Wirkungen wurden für andere Produkte in dieser Klasse gemeldet: Stevens-Johnson-Syndrom (SJS)/toxische epidermale Nekrolyse (TEN), Pankreatitis, exokrine Pankreasinsuffizienz, systemisches inflammatorisches Response-Syndrom, histiozytische nekrotisierende Lymphadenitis, Demyelinisierung, Vaskulitis, hämolytische Anämie, aplastische Anämie, Iritis, Lähmung des Nervus facialis und abducens, Polymyalgia rheumatica, Autoimmun-Neuropathie, Guillain-Barré Syndrom und Vogt-Koyanagi-Harada-Syndrom.
Patienten mit vorbestehender Autoimmunerkrankung
Bei Patienten mit vorbestehender Autoimmunerkrankung (AIE) deuten Daten aus Beobachtungsstudien auf ein erhöhtes Risiko für immunvermittelte unerwünschte Wirkungen nach Therapie mit einem Immun-Checkpoint-Inhibitor im Vergleich zu Patienten ohne vorbestehende AIE hin. Darüber hinaus traten häufig Schübe der zugrundeliegenden AIE auf, die aber überwiegend leicht und gut behandelbar waren.
Infusionsbedingte Reaktionen
Patienten sollten auf Anzeichen und Symptome infusionsbedingter Reaktionen untersucht werden. Bei Patienten unter Imfinzi wurden schwerwiegende infusionsbedingte Reaktionen beobachtet (siehe "Unerwünschte Wirkungen" ). Bei Reaktionen vom Schweregrad 1 oder 2 kann eine Prämedikation zur Prophylaxe nachfolgender Infusionsreaktionen in Betracht gezogen werden. Schwere infusionsbedingte Reaktionen des Grades 3 oder 4 sind entsprechend dem Standardverfahren der Einrichtung, den einschlägigen Leitlinien für die klinische Praxis und/oder den Leitlinien von Fachgesellschaften zu behandeln.
Zerebrovaskuläre Ereignisse
In der TOPAZ-1-Studie wurden bei Patienten mit BTC, die Imfinzi in Kombination mit Chemotherapie erhielten, zerebrovaskuläre Ereignisse, einschliesslich solcher mit Todesfolge, beobachtet. Die meisten dieser Patienten hatten vorbestehende zerebrovaskuläre Risikofaktoren (siehe "Unerwünschte Wirkungen" ).
Unerwünschte Wirkungen bei Transplantationspatienten
Bei mit PD-1 / PD-L1 - Inhibitoren behandelten Patienten wurde im Postmarketing-Umfeld eine Abstossung von soliden Organtransplantaten beobachtet. Bei diesen Patienten sollte der Nutzen der Behandlung mit PD-1 / PD-L1 – Inhibitoren, einschliesslich Durvalumab, gegen das Risiko einer möglichen Organabstossung abgewogen werden.
Krankheitsspezifische Vorsichtsmassnahmen
Cholangitis und Infektionen der Gallengänge bei Patienten mit BTC
Cholangitis und Infektionen der Gallengänge sind bei Patienten mit fortgeschrittenem BTC nicht ungewöhnlich. Fälle von Cholangitis wurden in der TOPAZ-1-Studie in beiden Behandlungsarmen berichtet (14,5 % [Imfinzi + Chemotherapie] vs. 8,2 % [Placebo + Chemotherapie]); diese waren meist mit Gallenstents assoziiert und waren nicht immunvermittelter Ätiologie. Patienten mit BTC (insbesondere solche mit Gallenstents) sollen vor Beginn der Behandlung sorgfältig untersucht und danach regelmässig auf die Entwicklung einer Cholangitis oder einer Infektion der Gallengänge überwacht werden.
NSCLC im Frühstadium
Die abschliessende OS-Analyse der AEGEAN-Studie steht noch aus, wobei ein Überlebensvorteil der perioperativen Durvalumab-Therapie gegenüber Placebo noch nicht nachgewiesen werden konnte. Die Gabe von Durvalumab zusätzlich zur Chemotherapie war mit einer erhöhten Rate immunvermittelter unerwünschter Ereignisse (imAE) im Vergleich zur Chemotherapie allein assoziiert (siehe Rubrik "Unerwünschte Wirkungen" ). Die adjuvante Behandlung mit Durvalumab nach einer neoadjuvanten Therapie war im Vergleich zur adjuvanten Behandlung mit Placebo mit zusätzlicher Toxizität verbunden. In der neoadjuvanten Phase der AEGEAN-Studie (mit DCO vom 14. August 2023) lag die Häufigkeit schwerwiegender unerwünschter Ereignisse (SAEs) bei 20,7% vs. 16,6%, die Häufigkeit von unter der Behandlung auftretenden unerwünschten Ereignissen (TEAEs) des höchsten Grades ≥3 bei 34,4% vs. 37,4% und die Häufigkeit von TEAEs des Grades 5 bei 2,0% vs. 1,0% der Patienten, die Durvalumab und Chemotherapie bzw. Placebo und Chemotherapie erhielten. Über die gesamte Behandlungsphase betrug die Häufigkeit von SAEs 38,9% vs. 31,7%, die Häufigkeit von TEAEs des höchsten Grades ≥3 49,1% vs. 47,0% und die Häufigkeit von TEAEs des Grades 5 5,7% vs. 3,8% bei Patienten, die Durvalumab und Chemotherapie bzw. Placebo und Chemotherapie erhielten. Die vorliegenden Daten erlauben keine Aussage über den Einfluss von Durvalumab bei NSCLC im Frühstadium auf eine nachfolgende anti-PD-(L)1-basierte systemische Therapie im Falle eines fortgeschrittenen/metastasierten Rezidivs, einschliesslich der Möglichkeit einer (erworbenen) Resistenz gegen eine anti-PD-(L)1-basierte systemische Therapie.
Muskelinvasiver Blasenkrebs
Die Gabe von Durvalumab zusätzlich zur Chemotherapie war mit einer erhöhten Rate immunvermittelter unerwünschter Ereignisse (imAE) im Vergleich zur Chemotherapie allein assoziiert (siehe Rubrik "Unerwünschte Wirkungen" ). Die adjuvante Behandlung mit Durvalumab nach einer neoadjuvanten Therapie war im Vergleich zu keiner adjuvanten Behandlung mit zusätzlicher Toxizität verbunden. In der neoadjuvanten Phase der NIAGARA-Studie (DCO vom 29. April 2024) lag die Häufigkeit schwerwiegender unerwünschter Ereignisse (SAEs) bei 23,6% vs. 22,4%, die Häufigkeit von unter der Behandlung auftretenden unerwünschten Ereignissen (TEAEs) des höchsten Grades 3 oder 4 bei 46,8% vs. 50,8% und die Häufigkeit von TEAEs des Grades 5 bei 1,1% vs. 1,9% der Patienten, die Durvalumab und neoadjuvante Chemotherapie bzw. neoadjuvante Chemotherapie erhielten. Über die gesamte Behandlungsphase betrug die Häufigkeit von SAEs 61,5% vs. 54,6%, die Häufigkeit von TEAEs des höchsten Grades 3 oder 4 66,6% vs. 63,9% und die Häufigkeit von TEAEs des Grades 5 5,1% vs. 5,5% bei Patienten, die Durvalumab und Chemotherapie bzw. Chemotherapie erhielten. Die vorliegenden Daten erlauben keine Aussage über den Einfluss von Durvalumab bei MIBC auf eine nachfolgende anti-PD-(L)1-basierte systemische Therapie im Falle eines fortgeschrittenen/metastasierten Rezidivs, einschliesslich der Möglichkeit einer (erworbenen) Resistenz gegen eine anti-PD-(L)1-basierte systemische Therapie.
GC/GEJC
Die Gabe von Durvalumab zusätzlich zur Chemotherapie war mit einer erhöhten Rate immunvermittelter unerwünschter Ereignisse (imAE) im Vergleich zur Chemotherapie allein assoziiert (siehe Rubrik "Unerwünschte Wirkungen" ). In der neoadjuvanten Phase betrug die Häufigkeit von schwerwiegender imAEs 0.8% vs. 1.3%, die Häufigkeit von imAEs des höchsten Grades 3 oder 4 2.1% vs. 1.7%, die Durvalumab und Chemotherapie bzw. Chemotherapie erhielten. In der neoadjuvanten Phase wurden keine imAEs des Grades 5 berichtet. In der adjuvanten Phase betrug die Häufigkeit von schwerwiegender imAEs 4.1% vs. 2.0%, die Häufigkeit von imAEs des höchsten Grades 3 oder 4 5.5% vs. 2.6% und die Häufigkeit von imAEs des Grades 5 0.5% vs. 0% bei Patienten, die Durvalumab und Chemotherapie bzw. Chemotherapie erhielten. Die vorliegenden Daten erlauben keine Aussage über den Einfluss von Durvalumab bei GC/GEJC auf eine nachfolgende anti-PD-(L)1-basierte systemische Therapie im Falle eines fortgeschrittenen/metastasierten Rezidivs, einschliesslich der Möglichkeit einer (erworbenen) Resistenz gegen eine anti-PD-(L)1-basierte systemische Therapie.
Behandlungsspezifische Vorsichtsmassnahmen
Aufgrund der geringen Stichprobengrösse bestätigen die Wirksamkeitsdaten für die ITT Subgruppe der neu diagnostizierten Patientinnen mit Endometriumkarzinom im Stadium III derzeit nicht, dass die Behandlung einen Nutzen im Hinblick auf das progressionsfreie oder das Gesamtüberleben hat (genaue Einschlusskriterien für Patientinnen mit Primärtumor im Stadium III siehe "Klinische Wirksamkeit" ).
Sicherheit in nicht zugelassenen Indikationen
In klinischen Studien zu einer nicht zugelassen Indikation wurden bei Anwendung von Imfinzi in Kombination mit Olaparib nach Behandlung mit Imfinzi in Kombination mit einer platinbasierten Chemotherapie Fälle von Aplasie der roten Blutkörperchen (PRCA, Pure Red Cell Aplasia) und Fälle von autoimmunhämolytischer Anämie (AIHA) (siehe Rubrik "Unerwünschte Wirkungen" ) beschrieben. Wenn eine PRCA oder eine AIHA bestätigt wird, ist die Behandlung mit Imfinzi und Olaparib abzusetzen.
Patientenpopulationen, die nicht in klinischen Studien untersucht wurden
Die folgenden Patientengruppen wurden von den klinischen Studien ausgeschlossen: Patienten mit einem Körpergewicht < 30 kg, aktiven Autoimmun- oder Entzündungskrankheiten, unkontrollierten interkurrenten Erkrankungen einschliesslich interstitieller Lungenkrankheit (Interstitial Lung Disease, ILD), aktiven Infektionen einschliesslich Tuberkulose, Hepatitis C oder HIV, Patienten, die innerhalb von 30 Tagen vor der ersten Dosis Durvalumab einen lebenden, abgeschwächten Impfstoff erhielten, Patienten mit aktueller oder früherer Anwendung hochdosierter systemischer immunsuppressiver Medikamente innerhalb von 14 Tagen vor der ersten Dosis Durvalumab und Patienten mit ECOG Performance Status ≥2.
InteraktionenDurvalumab ist ein humaner Antikörper, der nicht primär über hepatische/renale Wege abgebaut wird, sondern die Elimination erfolgt primär durch Proteinkatabolismus über das retikuloendotheliale System oder durch Elimination des Antikörper-Zielprotein-Komplexes. Da demzufolge keine pharmakokinetischen (PK) Wechselwirkungen mit anderen Arzneimitteln zu erwarten sind, wurden keine formalen Studien zu PK Interaktionen durchgeführt.
PK Wechselwirkungen zwischen Durvalumab und Chemotherapie wurden in der CASPIAN-Studie untersucht und zeigten, dass die gleichzeitige Verabreichung mit Durvalumab keinen Einfluss auf die PK von Etoposid, Carboplatin oder Cisplatin hatte.
Es ist auch nicht zu erwarten, dass Durvalumab die Metabolisierung anderer Arzneimittel durch Cytochrom P450 aktiviert oder hemmt.
Die Anwendung von systemischen Kortikosteroiden oder Immunsuppressiva vor Beginn einer Behandlung mit Durvalumab sollte vermieden werden, da diese die pharmakodynamische Aktivität und Wirksamkeit von Durvalumab beeinträchtigen könnten. Systemische Kortikosteroide oder andere Immunsuppressiva können jedoch nach Beginn der Behandlung mit Durvalumab eingesetzt werden, um immunbedingte unerwünschte Wirkungen zu behandeln (siehe Rubrik "Warnhinweise und Vorsichtsmassnahmen" ).
Schwangerschaft, StillzeitSchwangerschaft
In tierexperimentellen Reproduktionsstudien an trächtigen Javaneraffen war die Verabreichung von Durvalumab von der Bestätigung der Trächtigkeit bis zur Geburt in Expositionen, die etwa 6-20 Mal beziehungsweise 3-11 Mal höher waren als die für die klinische Dosis von 10 mg/kg Durvalumab alle 2 Wochen beziehungsweise 1500 mg Durvalumab alle 3 Wochen (basierend auf der AUC) beobachteten Expositionen, im Vergleich zur gleichzeitigen Kontrollgruppe mit Frühgeburten, Fehlgeburten (Aborte und Totgeburten) und einem Anstieg der neonatalen Todesfälle assoziiert (siehe "Präklinische Daten" ).
Über die Anwendung von Durvalumab bei schwangeren Frauen liegen keine Daten vor. Aufgrund seines Wirkmechanismus könnte sich Durvalumab auf die Aufrechterhaltung der Gravidität auswirken und zu fetalen Schädigungen führen, wenn es schwangeren Frauen verabreicht wird. Das humane IgG1 ist in der Lage, die Plazentaschranke zu überwinden. Imfinzi darf nicht während der Schwangerschaft verabreicht werden, es sei denn, dies ist eindeutig erforderlich. Frauen im gebärfähigen Alter sollten während der Behandlung mit Imfinzi und für mindestens 3 Monate nach Verabreichung der letzten Dosis eine zuverlässige Verhütungsmethode anwenden.
Stillzeit
Über die Präsenz von Durvalumab in der Muttermilch, die Absorption, die Wirkungen auf den gestillten Säugling und die Auswirkungen auf die Milchproduktion liegen keine Informationen vor. Das humane IgG wird mit der menschlichen Muttermilch ausgeschieden. In tierexperimentellen Reproduktionsstudien mit schwangeren Javaneraffen konnte der Zusammenhang zwischen der Verabreichung von Durvalumab mit einer dosisabhängigen, niedrigen Ausscheidungsmenge von Durvalumab in der Muttermilch nachgewiesen werden. Aufgrund der möglichen unerwünschten Nebenwirkungen von Durvalumab bei gestillten Säuglingen sollte stillenden Frauen empfohlen werden, das Stillen während der Behandlung und für mindestens 3 Monate nach der letzten Verabreichung zu unterbrechen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenBasierend auf den pharmakodynamischen Eigenschaften ist es unwahrscheinlich, dass Durvalumab einen Einfluss auf die Fahrtüchtigkeit und das Bedienen von Maschinen hat. Wenn Patienten allerdings unerwünschte Nebenwirkungen feststellen, die ihre Konzentrations- und Reaktionsfähigkeit beeinflussen, ist beim Bedienen von Fahrzeugen oder Maschinen Vorsicht geboten.
Unerwünschte WirkungenDie Anwendung von Imfinzi ist mit immunvermittelten unerwünschten Wirkungen assoziiert. Die meisten davon, einschliesslich schwerer Reaktionen, sind nach Einleitung einer entsprechenden medizinischen Behandlung oder nach Absetzen von Imfinzi wieder abgeklungen (siehe "Beschreibung ausgewählter Nebenwirkungen" unten).
Durvalumab als Monotherapie
Zusammenfassung des Sicherheitsprofils
Zur Sicherheit von Imfinzi als Monotherapie liegen gepoolte Daten von 4642 Patienten aus 15 Studien zu verschiedenen Tumorarten vor (Durvalumab-Pan-Tumor-Pool). Die häufigsten unerwünschten Wirkungen (≥10 %) waren Husten/Husten mit Auswurf (18,1 %), Diarrhö (15,1 %), Ausschlag (15,0 %), Fieber (12,5 %), Abdominalschmerzen (11,8 %), Infektion der oberen Atemwege (11,8 %), Hypothyreose (11,6 %) und Pruritus (11,1 %). Die häufigsten (≥1 %) unerwünschten Wirkungen der Grade 3 oder 4 waren Pneumonie (3,5 %), Aspartataminotransferase erhöht oder Alaninaminotransferase erhöht (2,5 %) und Abdominalschmerzen (1,6 %).
Durvalumab in Kombination mit einer Chemotherapie
Zusammenfassung des Sicherheitsprofils
Die Sicherheit von Imfinzi in Kombination mit einer Chemotherapie basiert auf gepoolten Daten von 2578 Patienten aus 7 Studien (TOPAZ-1, CASPIAN, POSEIDON, AEGEAN, NIAGARA, DUO-E und MATTERHORN). Die häufigsten (≥10 %) unerwünschten Wirkungen waren Neutropenie (43,6 %), Übelkeit (41,6 %), Fatigue (40,1 %), Anämie (38,4 %), Diarrhö (26,3 %), Obstipation (26,1 %), verminderter Appetit (23,7 %), Alopezie (20,5 %), periphere Neuropathie (20,0 %), Thrombozytopenie (19,6 %), Ausschlag (19,4 %), Erbrechen (18,4 %), Abdominalschmerzen (16,8 %), Leukopenie (15,1 %), Fieber (14,4 %), Pruritus (12,1 %), Husten/Husten mit Auswurf (11,3 %), Aspartataminotransferase erhöht oder Alaninaminotransferase erhöht (11,1 %).
Die häufigsten (≥1 %) unerwünschten Wirkungen der Grade 3 oder 4 waren Neutropenie (27,5 %), Anämie (12,6 %), Thrombozytopenie (5,9 %), Leukopenie (5,0 %), Fatigue (3,5 %), Pneumonie (2,6 %), febrile Neutropenie (2,3 %), Diarrhö (2,2 %), akute Nierenschädigung (2,1 %), Hypokaliämie (1,9 %), Aspartataminotransferase erhöht oder Alaninaminotransferase erhöht (1,9 %), Lungenembolie (1,9 %), Hyponatriämie (1,7%), Sepsis (1,4 %), Gamma-Glutamyltransferase erhöht (1,3%), Übelkeit (1,2 %), Erbrechen (1,2 %) und verminderter Appetit (1,2 %).
Unerwünschte Wirkungen (≥10%), die im Arm mit Durvalumab + FLOT der MATTERHORN Studie häufiger auftraten als in den gepoolten Daten, waren Diarrhö (62,3 %), Neutropenie (55,4 %), Übelkeit (50,7 %), periphere Neuropathie (41,1 %), Anämie (25,1 %), Stomatitis (17,7 %) und Leukopenie (15,2 %).
Tabellarische Zusammenfassung der unerwünschten Wirkungen
In Tabelle 2 werden die Inzidenzen von unerwünschten Wirkungen im Monotherapie-Sicherheitsdatensatz aufgeführt sowie unerwünschte Wirkungen, die bekanntermassen unter alleiniger Gabe von Imfinzi oder Chemotherapie auftreten, auch unter Behandlung mit einer Kombination dieser Arzneimittel auftreten, auch wenn diese Wirkungen in klinischen Studien mit der Kombinationstherapie nicht gemeldet wurden. Unerwünschte Wirkungen sind nach den Systemorganklassen gemäss MedDRA aufgeführt. Innerhalb der Systemorganklassen werden die unerwünschten Wirkungen nach abnehmender Häufigkeit und anschliessend nach abnehmendem Schweregrad aufgeführt. Die Häufigkeit der unerwünschten Wirkungen sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1'000 bis < 1/100), selten (≥1/10'000 bis < 1/1'000), sehr selten (< 1/10'000) sowie nicht bekannt.
Tabelle 2: Unerwünschte Wirkungen bei mit Imfinzi behandelten Patienten
Monotherapie In Kombination mit einer Chemotherapie
Infektionen und
parasitäre Erkrankun
gen
Sehr häufig Infektionen der oberen Atemwegea
(11,8 %)
Häufig Pneumonieb,c, Influenza, orale Pneumonieb,c, Infektionen der oberen
Candidose, Infektionen von Zähnen Atemwegea, Influenza, Infektionen von
und oralen Weichteilend Zähnen und oralen Weichteilend, Sepsisc
Gelegentlich Orale Candidose,
Erkrankungen des
Blutes und des
Lymphsystems
Sehr häufig Neutropeniev (43,6 %), Anämie (38,4
%), Thrombozytopeniew (19,6 %),
Leukopeniex (15,1 %)
Häufig Febrile Neutropenie
Gelegentlich Immunthrombozytopeniec Panzytopeniec
Selten Hämophagozytische Lymphohistiozytosec,
Immunthrombozytopenie
Nicht bekannt Hämophagozytische Lymphohistiozytose
Endokrine Erkrankung
en
Sehr häufig Hypothyreose (11,6 %)e
Häufig Hyperthyreosef, TSH erhöht Hyperthyreosef, Hypothyreosee
Gelegentlich TSH erniedrigt, Thyreoiditisg, Nebenniereninsuffizienz, TSH
Nebenniereninsuffizienz, erniedrigt, TSH erhöht, Hypophysitis/Hy
Hypophysitis/Hypophysen-insuffizienz pophysen-insuffizienz8, Thyreoiditisg,
8, Typ-1-Diabetes mellitus Typ 1 Diabetes mellitus
Selten Diabetes insipidus
Augenerkrankungen
Selten Uveitis Uveitis
Stoffwechsel- und
Ernährungsstörungen
Sehr häufig Appetit vermindertc (23,7 %)
Häufig Hypomagnesiämie2, Hypokaliämie,
Hyponatriämie3, Dehydratationc,
Hypokalzämie9
Gelegentlich Hypophosphatämie
Psychiatrische
Erkrankungen
Häufig Schlaflosigkeit
Erkrankungen des
Nervensystems
Sehr häufig Periphere Neuropathie1 (20,0 %)
Häufig Kopfschmerzen
Gelegentlich Enzephalitisi, Myasthenia gravish Zerebrovaskuläre Ereignissec,u,
Myasthenia gravish
Selten Enzephalitisi, Guillain-Barré-Syndrom
Nicht bekannt Transverse Myelitis6, Guillain-Barré
-Syndromc
Erkrankungen des
Ohrs und des Labyrin
ths
Häufig Tinnitus
Herzerkrankungen
Häufig Tachykardie
Gelegentlich Myokarditis Myokarditis
Gefässerkrankungen
Häufig Hypotonie
Erkrankungen der
Atemwege, des
Brustraums und
Mediastinums
Sehr häufig Husten/Husten mit Auswurf (18,1 %) Husten/Husten mit Auswurf (11,3 %)
Häufig Pneumonitisc,10, Dysphonie Pneumonitisc,10, Dysphonie, Dyspnoe,
Lungenemboliec, Schluckauf
Gelegentlich Interstitielle Lungenerkrankung Interstitielle Lungenerkrankungc
Erkrankungen des
Gastrointestinaltrak
ts
Sehr häufig Diarrhö (15,1 %), Abdominalschmerzen Übelkeit (41,6 %), Obstipation (26,1
j (11,8 %) %), Erbrechen (18,4 %), Diarrhö (26,3
%), Abdominalschmerzenj (16,8 %)
Häufig Stomatitisy, Amylase erhöht4, Colitisk
Gelegentlich Colitisk, Pankreatitisl Pankreatitisl
Selten Zöliakie Zöliakie
Leber- und Gallenerk
rankungen
Sehr häufig Aspartataminotransferase erhöht oder
Alaninaminotransferase erhöhtm (11,1 %)
Häufig Hepatitisc,n, Aspartataminotransfera Hepatitisc,n, Bilirubin im Blut
se erhöht oder Alaninaminotransferas erhöht5 Gamma-Glutamyltransferase
e erhöhtc,m erhöht
Erkrankungen der
Haut und des Unterha
utzellgewebes
Sehr häufig Ausschlago (15,0 %), Pruritus (11,1 Alopezie (20,5 %), Ausschlago (19,4
%) %), Pruritus (12,1 %)
Häufig Nächtliche Schweissausbrüche Dermatitis
Gelegentlich Dermatitis, Psoriasis, Pemphigoidp Pemphigoidp, Nächtliche Schweissausbrüc
he, Psoriasis
Skelettmuskulatur-,
Bindegewebs- und
Knochenerkrankungen
Häufig Myalgie Rückenschmerzen, Myalgie, Muskelspasmen
Gelegentlich Myositis7, immunvermittelte Immunvermittelte Arthritis11, Myositis7
Arthritis11
Selten Polymyalgia rheumatica Polymyalgia rheumatica12
Nicht bekannt Polymyositisq
Erkrankungen der
Nieren und Harnwege
Häufig Kreatininwert im Blut erhöht, Kreatininwert im Blut erhöht, Dysurie,
Dysurie Akute Nierenschädigungc, Proteinurie
Gelegentlich Nephritisr, Nichtinfektiöse Zystitis Nephritisr, Nichtinfektiöse Zystitis
Allgemeine Erkrankun
gen und Beschwerden
am Verabreichungsort
Sehr häufig Fieber (12,5 %) Fatiguez (40,1 %), Fieber (14,4 %)
Häufig Peripheres Ödems, Peripheres Ödems, Schüttelfrost, Ödem,
Unwohlsein
Verletzung, Vergiftu
ng und durch Eingrif
fe bedingte Komplika
tionen
Häufig Infusionsbedingte Reaktiont Infusionsbedingte Reaktiont
Die Häufigkeiten der unerwünschten Wirkungen sind möglicherweise nicht vollumfänglich auf Durvalumab allein zurückzuführen, da vielmehr auch die Grunderkrankung oder andere Arzneimittel, die in einer Kombination angewendet werden, dazu beitragen können.
a Umfasst Laryngitis, Nasopharyngitis, Peritonsillarabszess, Pharyngitis, Rhinitis, Sinusitis, Tonsillitis, Tracheobronchitis und Infektionen der oberen Atemwege.
b Umfasst Pneumonie durch Pneumocystis Jirovecii, Pneumonie, Pneumonie durch Candida, Pneumonie durch Legionella, Pneumonie adenoviral, Pneumonie durch Bakterien, Pneumonie durch Zytomegalievirus, Pneumonie durch Hämophilus, Pneumonie durch Pneumo-, Streptokokken und Pneumonie durch Klebsiella.
c Einschliesslich tödlichem Verlauf.
d Umfasst Gingivitis, orale Infektion, Periodontitis, Pulpitis, Zahnabszess und Zahninfektion.
e Umfasst Hypothyreose, autoimmune Hypothyreose und immunvermittelte Hypothyreose.
f Umfasst Hyperthyreose, Morbus Basedow und immunvermittelte Hyperthyreose.
g Umfasst autoimmune Thyreoiditis, immunvermittelte Thyreoiditis, Thyreoiditis und subakute Thyreoiditis.
h Die gemeldete Häufigkeit aus AstraZeneca-gesponserten klinischen Studien ausserhalb des gepoolten Datensatzes ist selten, und es gab keine Fälle Grad > 2.
i Umfasst Enzephalitis, immunvermittelte Enzephalitis, Autoimmun-Enzephalitis und nichtinfektiöse Enzephalitis.
j Umfasst Abdominal-, Unterleibs-, Oberbauch- und Flankenschmerzen.
k Umfasst Colitis, Enteritis, Enterokolitis, immunvermittelte Enterokolitis und Proktitis.
l Umfasst immunvermittelte Pankreatitis, Pankreatitis und akute Pankreatitis.
m Beinhaltet erhöhte Alanin-Aminotransferase, erhöhte Aspartat-Aminotransferase, erhöhte hepatische Enzyme und erhöhte Transaminasen.
n Beinhaltet Hepatitis, autoimmune Hepatitis, toxische Hepatitis, akute Hepatitis, Hepatotoxizität, hepatische Zytolyse und immunvermittelte Hepatitis.
o Umfasst erythematöses Exanthem, makulöses Exanthem, makulopapulöses Exanthem, papulöses Exanthem, pruriginöses Exanthem, pustulöses Exanthem, Erythem, Ekzem und Exanthem.
p Umfasst Pemphigoid, Dermatitis bullös und Pemphigus.
q In einer laufenden Studie ausserhalb der gepoolten Daten wurde Polymyositis mit tödlichem Verlauf bei einem mit Imfinzi behandelten Patienten berichtet. Selten in jeder Grade, selten in Grad 3 und höher nach CTCAE.
r Umfasst autoimmune Nephritis, tubuläre interstitielle Nephritis, immunvermittelte Nephritis, Nephritis, Glomerulonephritis und membranöse Glomerulonephritis.
s Umfasst Ödem peripher und periphere Schwellung.
t Infusionsbedingte Reaktionen beinhalten durch die Infusion ausgelöste Reaktionen und Urtikaria bei Beginn der Medikation oder einen Tag nach der Gabe des Arzneimittels.
u Umfasst zerebralen Infarkt, ischämischen Schlaganfall, zerebrale Blutung und zerebrovaskulären Insult.
v Umfasst Neutropenie und Neutrophilenzahl erniedrigt.
w Umfasst Thrombozytopenie und Thrombozytenzahl vermindert.
x Umfasst Leukopenie und Leukozytenzahl erniedrigt.
y Umfasst Stomatitis und Schleimhautentzündung.
z Umfasst Fatigue und Asthenie.
1 Umfasst periphere Neuropathie, Parästhesie und periphere sensorische Neuropathie.
2 Umfasst Magnesium im Blut erniedrigt und Hypomagnesiämie.
3 Umfasst Natrium im Blut erniedrigt und Hyponatriämie.
4 Umfasst Amylase erhöht und Hyperamylasämie.
5 Umfasst Bilirubin im Blut erhöht und Hyperbilirubinämie.
6 Nach Markteinführung berichtet.
7 Umfasst Myositis und Rhabdomyolyse.
8 Umfasst Hypophysitis und Hypophyseninsuffizienz.
9 Umfasst Kalzium im Blut erniedrigt und Hypokalzämie.
10 Umfasst immunvermittelte Lungenerkrankung und Pneumonitis.
11 Umfasst Autoimmun-Arthritis, immunvermittelte Arthritis, Polyarthritis und rheumatoide Arthritis.
12 Nicht beobachtet im Imfinzi+Chemotherapie-Pool, aber beobachtet in anderen von AstraZeneca gesponserten klinischen Studien.
In klinischen Studien für ein nicht zugelassenes Indikationsgebiet wurde bei der Anwendung von Imfinzi in Kombination mit Olaparib nach einer Behandlung mit Imfinzi in Kombination mit einer platinbasierten Chemotherapie die folgende Nebenwirkung identifiziert.
Tabelle 3: Unerwünschte Wirkungen bei Patientinnen, die mit Imfinzi in Kombination mit einer platinbasierten Chemotherapie, gefolgt von Imfinzi in Kombination mit Olaparib behandelt wurden (nicht zugelassene Indikation)
Erkrankungen des Blutes und des Lymphsystems
Häufig Aplasie der roten Blutkörperchen
Gelegentlich Autoimmunhämolytische Anämie
Beschreibung ausgewählter Nebenwirkungen
Die unten aufgeführten Daten zu signifikanten Nebenwirkungen beziehen sich auf Imfinzi als Monotherapie im gepoolten Sicherheitsdatensatz für verschiedene Tumorarten (n=4642, Durvalumab-Pan-Tumor-Pool) oder auf gepoolte Sicherheitsdaten für Imfinzi + Chemotherapie (n=2578), sofern nichts anderes angegeben ist. Der Imfinzi+Chemotherapie-Pool umfasste Patienten aus 7 Studien: TOPAZ-1 (Durvalumab + Gemcitabin/Cisplatin (Gem/Cis); Erstlinienbehandlung bei fortgeschrittenem BTC), CASPIAN (Durvalumab + Etoposid und entweder Carboplatin oder Cisplatin in Kombination mit platinbasierter Chemotherapie; Erstlinienbehandlung des ES-SCLC), POSEIDON (Durvalumab + platinbasierte Chemotherapie; Erstlinienbehandlung bei metastasiertem NSCLC), AEGEAN (Durvalumab + Chemotherapie vor der Operation, gefolgt von Durvalumab nach der Operation; resezierbares NSCLC), NIAGARA (Durvalumab + Chemotherapie auf Cisplatinbasis als neoadjuvante Behandlung gefolgt von Durvalumab als adjuvante Monotherapie nach radikaler Zystektomie; MIBC) DUO-E (Durvalumab + platinbasierte Chemotherapie; Endometriumkarzinom) und MATTERHORN (Durvalumab + FLOT als neoadjuvante Behandlung, gefolgt von Operation und anschliessend Durvalumab + FLOT nach der Operation; Adenokarzinom des Magens oder des gastroösophagealen Übergangs).
Im Falle von klinisch relevanten Unterschieden bei Anwendung von Imfinzi in Kombination mit einer Chemotherapie sind Details aufgeführt.
Die Richtlinien zur Behandlung dieser Nebenwirkungen sind in den Rubriken "Dosierung/Anwendung" und "Warnhinweise und Vorsichtsmassnahmen" beschrieben.
Immunvermittelte Pneumonitis
Unter Imfinzi-Monotherapie trat immunvermittelte Pneumonitis bei 147 Patienten (3,2 %) auf; dies umfasst 37 Patienten (0,8 %) mit Grad 3, 2 Patienten (<0,1 %) mit Grad 4 und 10 Patienten (0,2 %) mit Grad 5. Die mediane Zeit bis zum Auftreten betrug 56 Tage (Bereich: 1-1308 Tage). 114 der147 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag), 4 Patienten erhielten ausserdem andere Immunsuppressiva einschliesslich Infliximab Ciclosporin. Bei 60 Patienten wurde die Behandlung mit Imfinzi abgebrochen. Rekonvaleszenz trat bei 85 Patienten ein. Immunvermittelte Pneumonitis trat im Vergleich häufiger bei Patienten der PACIFIC-Studie auf, die innerhalb von 1 bis 42 Tagen vor Studienbeginn eine gleichzeitige Radiochemotherapie abgeschlossen hatten (9,9 %), als bei anderen Patienten, die in den kombinierten Sicherheitsdaten erfasst wurden (1,8 %).
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Pneumonitis bei 57 Patienten (2,2 %) auf, einschliesslich 15 Patienten (0,6 %) mit Grad 3, 2 Patienten (0,1 %) mit Grad 4 und 6 Patienten (0,2 %) mit Grad 5. Die mediane Zeit bis zum Auftreten betrug 191 Tage (Bereich: 11-529 Tage). 51 der 57 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag) und 5 Patienten erhielten andere Immunsuppressiva. Bei 19 Patienten wurde Imfinzi und/oder die Chemotherapie abgesetzt. Rekonvaleszenz trat bei 34 Patienten ein.
Bei Patienten in der PACIFIC-Studie mit lokal fortgeschrittenem, inoperablem NSCLC (n=475 in der Imfinzi-Gruppe und n=234 in der Placebo-Gruppe), die eine Behandlung mit gleichzeitiger Radiochemotherapie innerhalb von 1 bis 42 Tagen vor Beginn der Studienbehandlung abgeschlossen hatten, trat eine immunvermittelte Pneumonitis bei 47 Patienten (9,9 %) in der Imfinzi-Gruppe und bei 14 (6,0 %) in der Placebo-Gruppe auf. Dies schliesst Grad 3 bei 9 Patienten (1,9 %) der Imfinzi-Gruppe im Vergleich zu 6 (2,6 %) der Placebo-Gruppe und Grad 5 bei 4 Patienten (0,8 %) der Imfinzi-Gruppe im Vergleich zu 3 (1,3 %) der Placebo-Gruppe mit ein. Die mediane Zeit bis zum Auftreten betrug 46 Tage (Bereich: 2–342 Tage) in der Imfinzi-Gruppe und 57 Tage (Bereich 26-253 Tage) in der Placebo-Gruppe. In der Imfinzi-Gruppe erhielten 30 Patienten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag) und 2 Patienten erhielten zusätzlich Infliximab. In der Placebo-Gruppe wurden 12 Patienten mit systemischen Kortikosteroiden behandelt, und ein Patient wurde zusätzlich mit Cyclophosphamid und Tacrolimus behandelt. Rekonvaleszenz trat bei 29 Patienten in der Imfinzi-Gruppe bzw. bei 6 in der Placebo-Gruppe auf.
Bei Patienten in der ADRIATIC-Studie mit LS-SCLC (n=262 in der Imfinzi-Gruppe und n=265 in der Placebo-Gruppe), die eine Behandlung mit Radiochemotherapie innerhalb von 1 bis 42 Tagen vor Beginn der Studienbehandlung abgeschlossen hatten, trat eine immunvermittelte Pneumonitis bei 31 Patienten (11,8 %) in der Imfinzi-Gruppe und bei 8 (3,0 %) in der Placebo-Gruppe auf. Dies schliesst Grad 3 bei 5 Patienten (1,9 %) der Imfinzi-Gruppe im Vergleich zu 1 (0,4 %) in der Placebo-Gruppe und Grad 5 bei 1 Patienten (0,4 %) in der Imfinzi-Gruppe mit ein. Die mediane Zeit bis zum Auftreten betrug in der Imfinzi-Gruppe 55 Tage (Bereich: 1-375 Tage) und 65,5 Tage (Bereich: 24-124 Tage) in der Placebo-Gruppe. In der Imfinzi-Gruppe erhielten 25 Patienten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag) und 1 Patient erhielt zusätzlich Infliximab. In der Placebo-Gruppe erhielten 7 Patienten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag). Rekonvaleszenz trat bei 18 Patienten in der Imfinzi-Gruppe verglichen mit 3 in der Placebo-Gruppe auf.
Bei Patienten, die eine Radiotherapie der Lunge erhalten, wird häufig eine Strahlenpneumonitis beobachtet; die klinische Präsentation einer Pneumonitis und Strahlenpneumonitis ist sehr ähnlich.
In der PACIFIC-Studie trat bei 161 Patienten (33,9 %) in der Imfinzi-Gruppe und 58 Patienten (24,8 %) in der Placebo-Gruppe Pneumonitis (mit immunvermittelter Pneumonitis und Strahlenpneumonitis) einschliesslich Grad 3 (3,4 % bzw. 3,0 %) und Grad 5 (1,1 % bzw. 1,7 %) auf.
In der ADRIATIC-Studie trat bei 100 Patienten (38,2 %) in der Imfinzi-Gruppe und bei 80 (30,2 %) in der Placebo-Gruppe eine Pneumonitis oder Strahlenpneumonitis auf, einschliesslich Grad 3 bei 8 Patienten (3,1 %) der Imfinzi-Gruppe im Vergleich zu 6 (2,3 %) der Placebo-Gruppe und Grad 5 bei einem Patienten (0,4 %) der Imfinzi-Gruppe im Vergleich zu 0 Patienten in der Placebo-Gruppe.
Immunvermittelte Hepatitis
Unter Imfinzi-Monotherapie trat immunvermittelte Hepatitis bei 120 Patienten (2,6 %) auf, einschliesslich 70 (1,5 %) mit Grad 3, 9 Patienten (0,2 %) mit Grad 4 und 6 Patienten (0,1 %) mit Grad 5. Die mediane Zeit bis zum Auftreten betrug 36 Tage (Bereich 1-644 Tage). 94 der 120 Patienten erhielten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag). 9 Patienten erhielten zusätzlich eine Behandlung mit anderen Immunsuppressiva einschliesslich Mycophenolat. Die Behandlung mit Imfinzi wurde bei 30 Patienten abgesetzt. Rekonvaleszenz trat bei 56 Patienten ein.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Hepatitis bei 59 Patienten (2,3 %) auf, einschliesslich 31 Patienten (1,2 %) mit Grad 3, 9 Patienten (0,3 %) mit Grad 4 und 1 Patient (<0,1 %) mit Grad 5. Die mediane Zeit bis zum Auftreten betrug 80 Tage (Bereich: 6-656 Tage). 53 der 59 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag) und 3 Patienten erhielten andere Immunsuppressiva. Imfinzi und/oder die Chemotherapie wurde bei 24 Patienten abgesetzt. Rekonvaleszenz trat bei 49 Patienten ein.
Immunvermittelte Colitis oder Diarrhö
Unter Imfinzi-Monotherapie trat immunvermittelte Colitis oder Diarrhö bei 79 Patienten (1,7 %) auf, dies beinhaltete 15 Patienten (0,3 %) mit Grad 3, 2 Patienten (< 0,1 %) mit Grad 4 und 1 Patient (< 0,1 %) mit Grad 5. Die mediane Zeit bis zum Auftreten betrug 72 Tage (Bereich 1–920 Tage). 55 der 79 Patienten erhielten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag). 5 erhielten zusätzlich eine Behandlung mit anderen Immunsuppressiva einschliesslich Infliximab und Mycophenolat. Imfinzi wurde bei 15 Patienten abgesetzt. Rekonvaleszenz trat bei 54 Patienten ein.
Unter Imfinzi + Chemotherapie kam es zu immunvermittelter Colitis oder Diarrhö bei 41 Patienten (1,6 %), einschliesslich 13 Patienten (0,5 %) mit Grad 3 und 1 Patient (< 0,1 %) mit Grad 4. Die mediane Zeit bis zum Auftreten betrug 159 Tage (Bereich: 2-657 Tage). 31 der 41 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag) und 3 Patienten erhielten andere Immunsuppressiva. Imfinzi und/oder die Chemotherapie wurde bei 11 Patienten abgesetzt. Rekonvaleszenz trat bei 35 Patienten ein.
Immunvermittelte Endokrinopathien
Immunvermittelte Hypothyreose
Unter Imfinzi-Monotherapie trat immunvermittelte Schilddrüsenunterfunktion bei 384 Patienten (8,3 %) auf, einschliesslich 7 Patienten (0,2 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 91 Tage (Bereich 1–951 Tage). Von den 384 Patienten erhielten 379 eine Hormonersatztherapie, 7 Patienten erhielten hochdosierte Kortikosteroide (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag), gefolgt von einer Hormonersatztherapie. Imfinzi wurde bei einem Patienten aufgrund einer immunvermittelten Schilddrüsenunterfunktion abgesetzt. Der immunvermittelten Hypothyreose war bei 25 Patienten eine immunvermittelte Hyperthyreose und bei 2 Patienten eine immunvermittelte Thyreoiditis vorausgegangen.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Hypothyreose bei 237 Patienten (9,2 %) auf, einschliesslich 2 Patienten (0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 163 Tage (Bereich: 1-659 Tage). 236 Patienten erhielten eine Hormonersatztherapie. Imfinzi und/oder die Chemotherapie wurde bei einem Patienten aufgrund einer immunvermittelten Hypothyreose abgesetzt. Rekonvaleszenz trat bei 50 Patienten ein.
Immunvermittelte Hyperthyreose
Unter Imfinzi-Monotherapie trat immunvermittelte Schilddrüsenüberfunktion bei 76 Patienten (1,6 %) auf und es gab keine Fälle 3. oder 4. Grades. Die mediane Zeit bis zum Auftreten betrug 43 Tage (Bereich 1–253 Tage). 71 der 76 Patienten wurden medikamentös behandelt (Thiamazol, Carbimazol, Propylthiouracil, Perchlorat, Kalziumkanalblocker oder Beta-Blocker), 15 Patienten erhielten systemische Kortikosteroide und 8 der 15 Patienten erhielten eine hochdosierte systemische Kortikosteroidbehandlung (mindestens 40 mg Prednison oder die äquivalente Menge pro Tag). Imfinzi wurde bei einem Patienten aufgrund einer immunvermittelten Schilddrüsenüberfunktion abgesetzt. Rekonvaleszenz trat bei 62 Patienten ein. Bei 31 Patienten kam es nach der Hyperthyreose zu einer Hypothyreose.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Hyperthyreose bei 52 Patienten (2,0 %) auf, einschliesslich 1 Patient (<0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 68 Tage (Bereich: 21-372 Tage). 50 der 52 Patienten erhielten eine Hormonersatztherapie, 5 Patienten erhielten eine hochdosierte systemische Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Bei einem Patienten wurde Imfinzi und/oder die Chemotherapie aufgrund einer immunvermittelten Hyperthyreose abgesetzt. Rekonvaleszenz trat bei 37 Patienten ein.
Immunvermittelte Thyreoiditis
Unter Imfinzi-Monotherapie kam es bei 21 Patienten (0,5 %) zu einer immunvermittelten Thyreoiditis, einschliesslich 2 Patienten (<0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 57 Tage (Bereich: 14-217 Tage). 18 der 21 Patienten erhielten eine Hormonersatztherapie, 3 Patienten erhielten hochdosierte Kortikosteroide (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Imfinzi wurde bei einem Patienten aufgrund einer immunvermittelten Thyreoiditis abgesetzt. Rekonvaleszenz trat bei 8 Patienten ein. Bei fünf Patienten kam es nach der Thyreoiditis zu einer Hypothyreose.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Thyreoiditis bei 11 Patienten (0,4 %) ein. Es gab keine Fälle Grad 3 oder 4. Die mediane Zeit bis zum Auftreten betrug 100 Tage (Bereich: 43-260 Tage). Alle 11 Patienten erhielten eine Hormonersatztherapie und 3 Patienten erhielten hochdosierte Kortikosteroide (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Bei zwei Patienten wurde Imfinzi und/oder die Chemotherapie aufgrund einer immunvermittelten Thyreoiditis abgesetzt. Rekonvaleszenz trat bei 4 Patienten ein.
Immunvermittelte Nebenniereninsuffizienz
Unter Imfinzi-Monotherapie trat immunvermittelte Nebenniereninsuffizienz bei 24 Patienten (0,5 %) auf, einschliesslich 8 Patienten (0,2 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 158 Tage (Bereich: 20-547 Tage). Alle 24 Patienten erhielten systemische Kortikosteroide und 8 der 24 Patienten erhielten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder äquivalente Menge pro Tag). Imfinzi wurde bei einem Patienten aufgrund einer immunvermittelten Nebenniereninsuffizienz abgesetzt. Rekonvaleszenz trat bei 6 Patienten ein.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Nebenniereninsuffizienz bei 23 Patienten (0,9 %) auf, einschliesslich 3 Patienten (0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 234 Tage (Bereich: 29-739 Tage). 22 der 23 Patienten erhielten systemische Kortikosteroide; 2 der 23 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Bei 2 Patienten wurde Imfinzi und/oder die Chemotherapie aufgrund einer Nebenniereninsuffizienz abgesetzt. Rekonvaleszenz trat bei 4 Patienten ein.
Immunvermittelter Typ 1 Diabetes mellitus
Unter Imfinzi-Monotherapie kam es bei 5 Patienten (0,1 %) zu einem immunvermittelten Typ-1-Diabetes mellitus, darunter 3 Patienten (0,1 %) mit Grad 3 und 1 Patient (<0,1 %) mit Grad 4. Die mediane Zeit bis zum Auftreten betrug 43 Tage (Bereich: 29-631 Tage). Alle 5 Patienten erhielten eine Hormonersatztherapie. Bei einem Patienten wurde Imfinzi aufgrund von immunvermitteltem Typ-1-Diabetes mellitus abgesetzt. Rekonvaleszenz trat bei 2 Patienten ein.
Unter Imfinzi + Chemotherapie trat bei 9 Patienten (0,3 %) ein immunvermittelter Typ 1 Diabetes mellitus auf, darunter 4 Patienten (0,2 %) mit Grad 3 und 3 Patienten (0,1 %) mit Grad 4. Die mediane Zeit bis zum Auftreten betrug 134 Tage (Bereich: 14-387 Tage). Ein Patient erhielt eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Alle 9 Patienten erhielten eine Hormonersatztherapie. Imfinzi und/oder Chemotherapie wurde bei 2 Patienten aufgrund des immunvermittelten Typ 1 Diabetes mellitus dauerhaft abgesetzt. Rekonvaleszenz trat bei keinem Patienten ein.
Immunvermittelte Hypophysitis / Hypophyseninsuffizienz
Unter Imfinzi-Monotherapie trat immunvermittelte Hypophysitis/Hypophyseninsuffizienz bei 6 Patienten (0,1 %) auf, darunter 5 Patienten (0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 85 Tage (Bereich: 44-225 Tage). Alle 6 Patienten erhielten systemische Kortikosteroide, und 3 der 6 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Bei 3 Patienten wurde Imfinzi aufgrund einer immunvermittelten Hypophysitis/Hypophyseninsuffizienz abgesetzt.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Hypophysitis/Hypophyseninsuffizienz bei 10 Patienten (0,4 %) auf, einschliesslich 2 Patienten (<0,1 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 243 Tage (Bereich: 53-420 Tage). 9 Patienten erhielten eine systemische Kortikosteroidtherapie und bei 2 Patienten wurde Imfinzi und/oder die Chemotherapie aufgrund einer immunvermittelten Hypophysitis/Hypophyseninsuffizienz abgesetzt. Rekonvaleszenz trat bei 4 Patienten ein.
Immunvermittelte Nephritis
Unter Imfinzi-Monotherapie trat immunvermittelte Nephritis bei 17 Patienten (0,4 %) auf, einschliesslich 4 Patienten (0,1 %) mit Grad 3 und 1 Patient (< 0,1 %) mit Grad 4. Die mediane Zeit bis zum Auftreten betrug 84 Tage (Bereich 4-393 Tage). 12 Patienten (0,3 %) erhielten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder äquivalente Menge pro Tag), und ein Patient erhielt zusätzlich eine Behandlung mit Mycophenolat. Imfinzi wurde bei 7 Patienten abgesetzt. Rekonvaleszenz trat bei 8 Patienten ein.
Unter Imfinzi + Chemotherapie trat eine immunvermittelte Nephritis bei 19 Patienten (0,7 %) auf, einschliesslich 7 Patienten (0,3 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 152 Tage (Bereich: 8-444 Tage). Alle 19 Patienten erhielten systemische Kortikosteroide, und 18 Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag). Die Behandlung mit Imfinzi und/oder Chemotherapie wurde bei 11 Patienten abgesetzt. Rekonvaleszenz trat bei 8 Patienten ein.
Immunvermitteltes Exanthem
Unter Imfinzi-Monotherapie trat immunvermitteltes Exanthem oder Dermatitis (inkl. Pemphigoid) bei 74 Patienten (1,6 %) auf, einschliesslich 20 Patienten (0,4 %) mit Grad 3. Die mediane Zeit bis zum Auftreten betrug 56 Tage (Bereich 4-600 Tage). Alle 74 Patienten erhielten systemische Kortikosteroide, und 37 der 74 Patienten erhielten eine hochdosierte Kortikosteroidbehandlung (mindestens 40 mg Prednison oder äquivalente Menge pro Tag). Die Behandlung mit Imfinzi wurde bei 5 Patienten abgesetzt. Rekonvaleszenz trat bei 46 Patienten ein.
Unter Imfinzi + Chemotherapie trat ein immunvermitteltes Exanthem oder eine Dermatitis (inkl. Pemphigoid) bei 88 Patienten (3,4 %) auf, darunter 20 Patienten (0,8 %) mit Grad 3 und ein Patient (< 0,1 %) mit Grad 4. Die mediane Zeit bis zum Auftreten betrug 90 Tage (Bereich: 1-856 Tage). 87 der 88 Patienten erhielten systemische Kortikosteroide und 37 dieser Patienten erhielten eine hochdosierte Kortikosteroidtherapie (mindestens 40 mg Prednison oder ein entsprechendes Äquivalent pro Tag); ein Patient erhielt andere Immunsuppressiva. Imfinzi und/oder die Chemotherapie wurde bei 15 Patienten abgesetzt. Rekonvaleszenz trat bei 74 Patienten ein.
Infusionsbedingte Reaktionen
Unter Imfinzi-Monotherapie traten infusionsbedingte Reaktionen bei 70 Patienten (1,5 %) auf, darunter maximal Grad 3 oder 4 bei 6 Patienten (0,1 %). Es gab keine Fälle 5. Grades.
Unter Imfinzi + Chemotherapie traten infusionsbedingte Reaktionen bei 86 Patienten (3,3 %) auf, darunter maximal Grad 3 oder 4 bei 6 Patienten (0,2 %). Es gab keine Fälle 5. Grades.
Immunogenität
Die Ergebnisse des Immunogenitätsassays hängen unmittelbar von mehreren Faktoren ab, darunter der Assay-Empfindlichkeit und -spezifität, der Assay-Methodologie, der Handhabung der Proben, dem Zeitpunkt der Probenentnahme, der begleitenden Verabreichung von Arzneimitteln sowie der Grunderkrankung. Aufgrund der eingeschränkten Assay-Leistung wird die Inzidenz der Antikörperentwicklung bei Patienten unter Imfinzi möglicherweise unterschätzt, und ein Vergleich der Inzidenz von Antikörpern gegen Imfinzi mit der Inzidenz von Antikörpern gegen andere Präparate könnte irreführend sein.
Wie andere therapeutische Proteine verfügt auch Durvalumab über ein Immunogenitätspotenzial. Von den 3511 Patienten im Durvalumab-Pan-Tumor-Pool, die hinsichtlich vorhandener therapiebedingter Antikörper (Anti-Drug Antibodies, ADAs) auswertbar waren, wurden 2,6 % (93/3511) positiv auf therapiebedingte ADAs getestet. Neutralisierende Antikörper gegen Durvalumab wurden bei 0,5 % (19/3511) der Patienten nachgewiesen.
In der ADRIATIC-Studie erwiesen sich 7 der 206 Patienten (3,4 %), die mit Imfinzi-Monotherapie behandelt wurden und in Bezug auf das Vorliegen von ADAs auswertbar waren, als positiv für therapiebedingte ADAs. Neutralisierende Antikörper gegen Durvalumab wurden bei 1 % (2/206) der Patienten nachgewiesen. Aufgrund der niedrigen Anzahl an Patienten, die positiv auf während der Behandlung aufgetretene ADAs getestet wurden, liegen nicht genügend Daten vor, um zu beurteilen, ob die Gegenwart von ADAs Auswirkungen auf die Pharmakokinetik oder die Sicherheit von Durvalumab hatte.
In der AEGEAN-Studie wurden von 375 Patienten, die mit Imfinzi 1500 mg in Kombination mit Chemotherapie alle 3 Wochen vor der Operation, gefolgt von Imfinzi 1500 mg alle 4 Wochen nach der Operation, behandelt wurden und hinsichtlich vorhandener therapiebedingter ADAs auswertbar waren, 25 (6,7 %) positiv auf therapiebedingte ADAs getestet. Neutralisierende Antikörper gegen Durvalumab wurden bei 2 Patienten (0,5 %) nachgewiesen. Aufgrund der niedrigen Anzahl an Patienten, die positiv auf während der Behandlung aufgetretene ADAs getestet wurden, liegen nicht genügend Daten vor, um zu beurteilen, ob die Gegenwart von ADAs Auswirkungen auf die Pharmakokinetik oder die Sicherheit von Durvalumab hatte.
In der CASPIAN-Studie wurden 0 (0 %) von 201 auf die Anwesenheit von ADA auswertbaren Patienten, die Imfinzi 1500 mg alle 3 Wochen in Kombination mit Chemotherapie erhalten hatten, positiv auf aufgetretene therapiebedingte ADA getestet. Daher konnten die Auswirkungen auf die Pharmakokinetik und klinische Sicherheit von Durvalumab nicht evaluiert werden.
Die Entwicklung von ADAs war mit niedrigeren Expositionsniveaus von Durvalumab assoziiert, die Reduktion wird jedoch nicht als klinisch relevant angesehen. Es gab keine Hinweise darauf, dass ADAs einen Einfluss auf die Sicherheit von Durvalumab haben. Die möglichen Auswirkungen von ADAs auf die Wirksamkeit von Durvalumab konnten nicht beurteilt werden, da es keine ausreichende Anzahl an therapiebedingten ADA-positiven Patienten gab.
In der TOPAZ-1-Studie wurden von 240 Patienten, die mit Imfinzi 1500 mg alle 3 Wochen in Kombination mit der Chemotherapie, gefolgt von Imfinzi 1500 mg alle 4 Wochen, behandelt wurden und für das Auftreten von ADAs auswertbar waren, 2 (0,8 %) positiv auf während der Behandlung aufgetretene ADAs getestet. Die Anzahl von Patienten mit während der Behandlung auftretenden ADAs oder mit neutralisierenden Antikörpern (jeweils 2 Patienten) war zu gering, um zu beurteilen, ob ADAs die Pharmakokinetik und klinische Sicherheit von Durvalumab beeinflussen.
In der NIAGARA-Studie wurden von den 453 Patienten, die mit Imfinzi 1500 mg in Kombination mit Chemotherapie alle 3 Wochen vor der Operation und anschliessend mit Imfinzi 1500 mg alle 4 Wochen nach der Operation behandelt wurden und für das Auftreten von ADA auswertbar waren, 8 (1,8 %) Patienten positiv auf während der Behandlung aufgetretene ADA getestet. Neutralisierende Antikörper gegen Durvalumab wurden bei 6 (1,3 %) Patienten nachgewiesen. Das Auftreten von ADA hatte keinen erkennbaren Einfluss auf die Pharmakokinetik oder Sicherheit.
In der DUO-E-Studie wurden von den Patientinnen, die mit platinbasierter Chemotherapie + Imfinzi (n=198) behandelt wurden und die im Hinblick auf das Vorliegen von ADA auswertbar waren, 2 (1,0 %) Patientinnen im Arm mit platinbasierter Chemotherapie + Imfinzi positiv auf während der Behandlung auftretende ADA getestet. Neutralisierende Antikörper gegen Durvalumab wurden bei 1 (0,5%) Patienten im Behandlungsarm nachgewiesen. Die Anzahl der Patientinnen mit während der Behandlung auftretenden ADA oder neutralisierenden Antikörpern war zu niedrig, um zu beurteilen, ob ADA die Pharmakokinetik oder Sicherheit von Durvalumab beeinflussen.
In der MATTERHORN-Studie wurden von den 441 Patienten, die mit Imfinzi 1500 mg in Kombination mit FLOT-Chemotherapie als neoadjuvante und adjuvante Therapie behandelt und anschliessend als adjuvante Imfinzi-Monotherapie weiterbehandelt wurden und hinsichtlich vorhandener therapiebedingter ADAs auswertbar waren, 40 (9,1 %) positiv auf therapiebedingte ADAs getestet. Neutralisierende Antikörper gegen Durvalumab wurden bei 2 Patienten (0,5 %) nachgewiesen. Das Vorliegen von ADAs hatte keinen erkennbaren Einfluss auf die Pharmakokinetik oder die Sicherheit von Imfinzi.
Pädiatrische Population
In der klinischen Studie D419EC00001, in der 50 pädiatrische Patienten (< 18 Jahre) untersucht wurden, traten in allen Behandlungsphasen bei mehr als 20 % der Patienten die unerwünschte Ereignisse Erbrechen, Anämie, Kopfschmerzen, Übelkeit, Pyrexie, Bauchschmerzen und eine Erhöhung der Alanin-Aminotransferase auf. In dieser Studie wurden bei 20 von 50 Patienten (40 %) unerwünschte Ereignisse des Grades 3 oder 4 gemeldet. In der pädiatrischen Population wurden keine neuen Sicherheitssignale im Vergleich zu den bekannten Sicherheitsprofilen von Imfinzi bei Erwachsenen beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs gibt keine spezifische Behandlung im Falle einer Durvalumab-Überdosierung und Symptome einer Überdosierung sind nicht bekannt. Bei einer Überdosierung sind allgemeine Hilfsmassnahmen durchzuführen und allfällige Symptome zu behandeln.
Eigenschaften/WirkungenATC-Code
L01FF03
Wirkungsmechanismus / Pharmakodynamik
Die Expression des programmierten Zelltod-Liganden-1 (PD-L1) ist eine adaptive Immunantwort, die Tumore vor dem Immunsystem verbirgt und beschützt. PD-L1 kann durch Entzündungssignale (z.B. IFN-gamma) induziert werden und sowohl auf Tumorzellen als auch auf tumorassoziierten Immunzellen in einer Tumor-Mikroumgebung exprimiert werden. PD-L1 blockiert die Funktion der T-Zellen und deren Aktivierung aufgrund der Interaktion mit PD-1 und CD80 (B7-1). Durch Andocken an ihre Rezeptoren verringert PD-L1 die Aktivität, Vermehrung und Zytokin-Produktion von T-Zellen.
Durvalumab ist ein vollständig humaner, stark bindungsaffiner, monoklonaler Immunglobulin-G1-kappa-Antikörper (IgG1κ), der die Interaktion von PD-L1 mit PD-1 und CD80 (B7-1) selektiv blockiert, dabei jedoch die PD-1/PD-L2-Interaktion intakt lässt. Durvalumab ruft keine antikörperabhängige zellvermittelte Zytotoxizität (ADCC) hervor. Das selektive Blockieren der PD-L1/PD-1- und PD-L1/CD80-Interaktionen steigert die Antitumor-Immunantworten. Diese Antitumor-Antworten können die Eliminierung von Tumoren bewirken.
In präklinischen Studien führte die PD-L1-Blockade zu erhöhter T-Zellen-Aktivierung und verringerten Tumorgrössen.
Klinische Wirksamkeit
Nicht-kleinzelliges Lungenkarzinom (NSCLC) – PACIFIC Studie
Die Wirksamkeit von Imfinzi wurde in der PACIFIC-Studie ermittelt, einer randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studie mit 713 Patienten mit histologisch oder zytologisch nachgewiesenem, lokal fortgeschrittenem, inoperablem NSCLC. Die Patienten hatten innerhalb eines Zeitraums von 1 bis 42 Tagen vor Beginn der Studienbehandlung mindestens zwei Zyklen einer definitiven platinbasierten Radiochemotherapie abgeschlossen und verfügten über einen ECOG-Leistungsstatus von 0 oder 1. 92 % der Patienten hatten eine Gesamtdosis von 54 bis 66 Gy Strahlung erhalten. Nicht für diese Studie infrage kamen Patienten, die nach einer Radiochemotherapie eine Progression aufwiesen, Patienten mit aktiver oder zuvor dokumentierter Autoimmunerkrankung innerhalb eines Zeitraums von 2 Jahren vor Studienbeginn, einer vorbestehenden Immunschwäche, vorbestehenden schweren immunvermittelten unerwünschten Reaktionen, Erkrankungen, die eine systemische Immunsuppression notwendig machten ausgenommen eine physiologische Dosis systemischer Kortikosteroide, Patienten mit aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion oder Patienten, die innerhalb von 30 Tagen vor oder nach dem Beginn der Behandlung mit Imfinzi attenuierten Lebendimpfstoff erhielten. Die Patienten wurden 2:1 randomisiert und erhielten 12 Monate lang oder bis zu inakzeptabler Toxizität oder bis zu bestätigter Krankheitsprogression alle 2 Wochen entweder 10 mg/kg Imfinzi (n=476) oder Placebo (n=237) mittels intravenöser Infusion. Die Randomisierung wurde stratifiziert nach Geschlecht, Alter (< 65 Jahre vs. ≥65 Jahre) und Raucherstatus (Raucher vs. Nichtraucher). Tumorbeurteilungen wurden während den ersten 12 Monaten alle 8 Wochen und danach alle 12 Wochen durchgeführt.
Die Patienten wurden unabhängig von ihrem Tumor-PD-L1-Expressionsstatus in die Studie eingeschlossen. Soweit verfügbar, wurden archivierte Tumorgewebeproben, die vor der Radiochemotherapie entnommen wurden, mit dem VENTANA PD-L1 (SP263) IHC-Assay retrospektiv auf PD-L1-Expression auf Tumorzellen (TC) getestet.
Die demografischen und krankheitsbedingten Eigenschaften bei Studienbeginn waren zwischen den Studienarmen ausgewogen. Die Zusammensetzung der gesamten Studienpopulation zu Studienbeginn war wie folgt: Männer (70 %), Alter ≥65 Jahre (45 %), hellhäutig (69 %), asiatisch (27 %), sonstige (4 %), Raucher (16 %), ehem. Raucher (75 %) und nie geraucht (9 %), WHO/ECOG PS 0 (49 %), WHO/ECOG PS 1 (51 %). Die krankheitsbedingten Eigenschaften waren wie folgt: Stadium IIIA (53 %), Stadium IIIB (45 %), histologische squamöse Untergruppe (46 %), nicht-squamös (54 %).
Die zwei primären Endpunkte der Studie waren Gesamtüberleben (OS) und progressionsfreies Überleben (PFS) von Imfinzi vs. Placebo.
Die Studie zeigte eine statistisch signifikante Verbesserung des OS in der Imfinzi-Gruppe verglichen mit der Placebo-Gruppe [Hazard-Ratio (HR) = 0,68 (95 %-KI: 0,53; 0,87), p = 0,00251]. Zum Zeitpunkt der vorläufigen OS-Analyse (22. März 2018) betrug die mediane Nachbeobachtungszeit 25,2 Monate; 299 Patienten (183 (38,4 %) in der Durvalumab-Gruppe und 116 (48,9 %) in der Placebo-Gruppe) waren verstorben. Das mediane Gesamtüberleben wurde in der Durvalumab-Gruppe noch nicht erreicht; in der Placebo-Gruppe war es gemäss Kaplan-Meier Schätzung 28,7 Monate. Das Gesamtüberleben nach 24 Monaten betrug 66,3 % in der Imfinzi-Gruppe und 55,6 % in der Placebo-Gruppe.
Zum Zeitpunkt der Aktualisierung der Daten nach der 5-jährigen Nachbeobachtung waren insgesamt 23 neue OS-Ereignisse seit der zweiten Analyse der OS-Nachbeobachtung gemeldet worden (insgesamt 419 Todesfälle [264 in der Imfinzi-Gruppe und 155 in der Placebo-Gruppe]). Der Überlebensnutzen von Imfinzi gegenüber Placebo war konsistent mit der primären OS-Analyse, bei einer Reduktion des Mortalitätsrisikos gegenüber Placebo um 28 % [HR = 0,72 (95%-KI: 0,59-0,89)]. Die Kaplan-Meier-Schätzung des medianen OS betrug 29,1 Monate in der Placebo-Gruppe gegenüber 47,5 Monaten in der Imfinzi-Gruppe. Die geschätzten Gesamtüberlebensraten lagen bei 42,9 % für die Imfinzi-Gruppe und 33,4 % für die Placebo-Gruppe.
Zum Zeitpunkt der Interimsanalyse (13. Februar 2017) durch einen verblindeten, unabhängigen, zentralen Review (BICR) gemäss den RECIST 1.1 Kriterien zeigte die Studie eine statistisch signifikante Verbesserung des PFS in der Imfinzi-Gruppe verglichen mit der Placebo-Gruppe [HR = 0,52 (95 %-KI: 0,42; 0,65), p < 0,0001]. Zu diesem Zeitpunkt waren 371 Patienten (214 (45,0 %) in der Durvalumab-Gruppe und 157 (66,2 %) in der Placebo-Gruppe) progredient oder verstorben. Das mediane PFS gemäss Kaplan-Meier Schätzung war 16,8 Monate in der Imfinzi-Gruppe und 5,6 Monate in der Placebo-Gruppe. Das PFS nach 12 Monaten betrug 55,9 % in der Imfinzi-Gruppe und 35,5 % in der Placebo-Gruppe. Das PFS nach 18 Monaten betrug 44,2 % in der Imfinzi-Gruppe und 27,0 % in der Placebo-Gruppe. Die Verbesserungen des OS und des PFS zugunsten der Patienten in der Imfinzi-Gruppe im Vergleich zur Placebo-Gruppe wurde durchgehend in allen analysierten vordefinierten Subgruppen (Ethnizität, Alter, Geschlecht, Raucherstatus, EGFR Mutationsstatus und Histologie) beobachtet.
Zum Zeitpunkt der Analyse der 5-jährigen Nachbeobachtung (11. Januar 2021) basierend auf den BICR-Bewertungen des PFS nach RECIST 1.1 war der in der Imfinzi-Gruppe festgestellte PFS-Nutzen [HR: 0,55 (95%-KI: 0,45-0,68)] konsistent mit der primären Analyse des PFS [HR: 0,52; (95%-KI: 0,42-0,65)]. Die Kaplan-Meier-Schätzung des medianen PFS betrug 16,9 Monate in der Durvalumab-Gruppe (95%-KI: 13,0-23,9) im Vergleich zu 5,6 Monaten in der Placebo-Gruppe (95%-KI: 4,8-7,7).
In der Imfinzi-Gruppe trat bei 21 (4,4 %) Patienten eine unerwünschte Wirkung mit Todesfolge auf, in der Placebo-Gruppe trat bei 14 (6,0 %) Patienten eine unerwünschte Wirkung mit Todesfolge auf.
Post-hoc explorative Subgruppenanalyse entsprechend der PD-L1-Expression
Von den 713 randomisierten Patienten der PACIFIC-Studie waren 451 (63 %) der Gewebeproben von ausreichender Qualität und Quantität zur Bestimmung der PD-L1-Expression. Von den 451 Proben hatten 33 % einen PD-L1 TC < 1 %, 67 % PD-L1 TC ≥1 %, 32 % PD-L1 TC 1 - < 25 %, 35 % PD-L1 TC ≥25 %.
Post hoc Subgruppenanalysen wurden durchgeführt, um die Wirksamkeit bei zusätzlichen, nicht im statistischen Analyseplan vorgesehenen PD-L1-Expressionsleveln des Tumors (< 1 %, ≥1 %, 1 bis < 25 %) zu bewerten.
PFS:
In der Subgruppe mit fehlender PD-L1 Expression (PD-L1 TC < 1 %) war die PFS-HR 0,73; 95 %-KI: 0,48, 1,11.
Für die untersuchten Subgruppen mit positiver PD-L1 Expression (PD-L1 TC ≥1%) und der Subgruppe mit nicht evaluierbarer PD-L1 Expression (PD-L1 TC unbekannt) wurden folgende Resultate ermittelt: PD-L1 TC ≥1 % HR: 0,46; 95 %-KI: 0,33, 0,64; PD-L1 TC 1 bis < 25 % HR: 0,49; 95 %-KI:0,30, 0,80; PD-L1 TC ≥25 % HR: 0,41; 95 %-KI: 0,26, 0,65; PD-L1 TC unbekannt HR: 0,59; 95 %-KI: 0,42, 0,83).
OS:
In der Subgruppe mit fehlender PD-L1 Expression (PD-L1 TC < 1 %) war die OS-HR 1.36; 95 %-KI: 0,79, 2,34.
Für die untersuchten Subgruppen mit positiver PD-L1 Expression (PD-L1 TC ≥1%) und mit nicht evaluierbarer PD-L1 Expression (PD-L1 TC unbekannt) wurden folgende Resultate ermittelt: PD-L1 TC ≥1 % HR: 0,53; 95 %-KI: 0,36, 0,77; PD-L1 TC 1 bis < 25 % HR: 0,60; 95 %-KI: 0,35, 1,03; PD-L1 TC ≥25 % HR: 0,46; 95 %-KI: 0,27, 0,78; PD-L1 TC unbekannt HR: 0,62; 95 %-KI: 0,43, 0,89.
Resezierbares NSCLC – AEGEAN-Studie
AEGEAN ist eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Phase-III-Studie zur Beurteilung der Wirksamkeit von Imfinzi in Kombination mit Chemotherapie als neoadjuvante Behandlung, gefolgt von einer Imfinzi-Monotherapie nach der Operation, bei Patienten mit resezierbarem NSCLC (Stadium IIA bis ausgewählte Patienten mit Stadium IIIB (N2) [AJCC, 8. Auflage]). In die Studie wurden zuvor unbehandelte Patienten mit dokumentiertem plattenepithelialem oder nicht plattenepithelialem NSCLC ohne vorherige Exposition gegenüber einer Immuntherapie, mit einem WHO/ECOG Performance Status von 0 oder 1 und mindestens einer Zielläsion nach RECIST 1.1 aufgenommen. Vor der Randomisierung wurde der PD-L1-Expressionsstatus des Tumors mit dem Ventana PD-L1 (SP263) Test bestätigt.
Patienten mit aktiver oder zuvor dokumentierter Autoimmunerkrankung oder Anwendung einer immunsuppressiven Medikation innerhalb von 14 Tagen vor der ersten Dosis von Durvalumab wurden aus der Studie ausgeschlossen. Aus der Studie ausgeschlossen waren Patienten, deren geplante Operation eine Segmentresektion, eine Keilresektion oder (nach einer Prüfplanänderung) eine Pneumektomie war. Während der Studie durchgeführte Operationen wurden im Rahmen der Standardtherapie durchgeführt und konnten eine Pneumektomie umfassen. Nach einer Prüfplanänderung wurden später keine Patienten mit T4-Tumoren mit Invasion von Zwerchfell, Mediastinum, Herz, grossen Gefässen, Trachea, Nervus laryngeus recurrens, Ösophagus oder Carina mehr in die Studie aufgenommen. Aus der Studienpopulation für die Wirksamkeitsanalyse (modifizierte Intent-to-treat-Analyse [mITT]) wurden Patienten mit bekannten EGFR-Mutationen oder ALK-Rearrangements ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach Krankheitsstadium (Stadium II vs. Stadium III) und PD-L1-Expressionsstatus (TC < 1 % vs. TC ≥1 %).
In der AEGEAN-Studie wurden 802 Patienten im Verhältnis 1:1 für die perioperative Behandlung mit Imfinzi (Arm 1) oder Placebo (Arm 2) in Kombination mit neoadjuvanter Chemotherapie randomisiert. Ein Crossover zwischen den Studienarmen war nicht erlaubt. Die Wirksamkeitsanalyse wurde auf der Basis von 740 Patienten in der mITT-Population durchgeführt.
-Arm 1: Imfinzi 1500 mg + Chemotherapie alle 3 Wochen für bis zu 4 Zyklen vor der Operation, gefolgt von Imfinzi 1500 mg alle 4 Wochen für bis zu 12 Zyklen nach der Operation.
-Arm 2: Placebo + Chemotherapie alle 3 Wochen für bis zu 4 Zyklen vor der Operation, gefolgt von Placebo alle 4 Wochen für bis zu 12 Zyklen nach der Operation.
Die Wahl der Standardchemotherapie lag unter Berücksichtigung der Histologie im Ermessen des Prüfarztes. Bei plattenepithelialen Tumoren: Carboplatin (AUC 6) und Paclitaxel (200 mg/m2) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen oder Cisplatin (75 mg/m2) an Tag 1 und Gemcitabin (1250 mg/m2) an den Tagen 1 und 8 jedes 3-wöchigen Zyklus für maximal 4 Zyklen. Die Anwendung von Carboplatin (AUC 5) kombiniert mit Gemcitabin (anstatt Cisplatin) war bei Patienten erlaubt, die Begleiterkrankungen hatten oder Cisplatin nach Einschätzung des Prüfarztes nicht vertrugen. Bei nicht plattenepithelialen Tumoren: Pemetrexed (500 mg/m2) und Cisplatin (75 mg/m2) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen oder Pemetrexed (500 mg/m2) und Carboplatin (AUC 5) an Tag 1 jedes 3-wöchigen Zyklus für maximal 4 Zyklen. Bei ungünstiger Verträglichkeit konnten die Patienten an jedem Punkt der Studie von der Cisplatin- zur Carboplatin-Therapie wechseln (wenn sie für diese Therapie in Frage kamen).
Patienten, die sich einer Operation mit R0/R1-Resektionsrändern unterzogen hatten, konnten die adjuvante Behandlung fortsetzen.
Eine Tumorbeurteilung nach RECIST 1.1 wurde zur Baseline und nach Abschluss der neoadjuvanten Phase (vor der Operation) durchgeführt. Die erste postoperative CT-/MRT-Untersuchung von Thorax und Abdomen (einschliesslich der gesamten Leber und beider Nebennieren) wurde 5 Wochen ±2 Wochen nach der Operation und vor, aber möglichst nahe am Beginn der adjuvanten Therapie, durchgeführt. Anschliessend erfolgten Tumorbeurteilungen alle 12 Wochen (bezogen auf das Operationsdatum) bis Woche 48, alle 24 Wochen (bezogen auf das Operationsdatum) bis Woche 192 (ca. 4 Jahre) und danach alle 48 Wochen (bezogen auf das Operationsdatum) bis zur radiologisch bestätigten Krankheitsprogression (Progressive disease; PD) gemäss RECIST 1.1, bis zum Widerruf der Einwilligung oder bis zum Tod.
Der Überlebensstatus wurde 2, 3 und 4 Monate nach Beendigung der Behandlung, anschliessend alle 2 Monate bis Monat 12 und danach alle 3 Monate überprüft.
Die primären Endpunkte der Studie waren das vollständige pathologische Ansprechen (pathological complete response; pCR) gemäss verblindeter zentraler pathologischer Untersuchung sowie das ereignisfreie Überleben (event-free survival; EFS) gemäss verblindeter unabhängiger zentraler Beurteilung (BICR). Die wichtigsten sekundären Endpunkte waren das bedeutsame pathologische Ansprechen (major pathological response; MPR) gemäss verblindeter zentraler pathologischer Untersuchung, das krankheitsfreie Überleben (disease free survival, DFS) gemäss BICR und Gesamtüberleben (overall survival; OS). Weitere sekundäre Wirksamkeitsziele waren das EFS (Analysepopulation PD-L1-TC ≥1 %) und pCR (Analysepopulation PD-L1-TC ≥1%).
Bei der geplanten Zwischenanalyse des pCR erreichte die Studie die vorgegebene Grenze für die Erklärung der statistischen Signifikanz für pCR und MPR. In der Folgezeit, bei der ersten geplanten Zwischenanalyse des EFS, erreichte die Studie ihre vorgegebene Grenze für die Erklärung der statistischen Signifikanz für das EFS.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren in den beiden Studienarmen ausgewogen (366 Patienten in Arm 1 und 374 Patienten in Arm 2 der mITT-Population). Die demographischen und krankheitsspezifischen Ausgangsmerkmale der Population für die Wirksamkeitsanalyse (mITT) waren wie folgt: männlich (71,6 %), weiblich (28,4 %), Alter ≥65 Jahre (51,6 %), medianes Alter 65 Jahre (Bereich: 30 bis 88 Jahre), WHO/ECOG PS 0 (68,4 %), WHO/ECOG PS 1 (31,6 %), weiss (53,6 %), asiatisch (41,5 %), schwarz bzw. afroamerikanisch (0,9 %), indigen amerikanisch oder indigen alaskisch (1,4 %), andere Abstammung (2,6 %), hispanisch oder lateinamerikanisch (16,1 %), nicht-hispanisch oder -lateinamerikanisch (83,9 %), aktueller oder ehemaliger Raucher (85,5 %), Nie-Raucher (14,5 %), Plattenepithelhistologie (48,6 %) und Nicht-Plattenepithelhistologie (50,7 %), Stadium II (28,4 %), Stadium III (71,6 %), PD-L1-Expressionsstatus TC ≥1 % (66,6 %), PD-L1-Expressionsstatus TC < 1 % (33,4 %). Die demographischen Daten und Ausgangsmerkmale der mITT-Population waren vergleichbar mit der ITT-Population, mit Ausnahme des Fehlens von Patienten mit bekannten EGFR-Mutationen oder ALK-Rearrangements.
Die Studie zeigte eine statistisch signifikante Verbesserung des EFS [HR = 0,68 (95%-KI: 0,53; 0,88), p = 0,003902] im Imfinzi-Arm verglichen mit dem Placebo-Arm. Ausserdem zeigte die Studie eine statistisch signifikante Verbesserung des pCR [Differenz der Anteile 12,96 % (95%-KI: 8,67, 17,57)] im Imfinzi-Arm verglichen mit dem Placebo-Arm. Siehe Tabelle 4.
Tabelle 4: Wirksamkeitsergebnisse der AEGEAN-Studie (mITT)
Imfinzi + Chemotherapie(n = Placebo + Chemotherapie(n =
366) 374)
EFSa
Anzahl der Ereignisse, n (%) 98 (26,8) 138 (36,9)
Medianes EFS (95%-KI) (Monate) NR (31,9; NR) 25,9 (18,9; NR)
EFS nach 12 Monaten, % (95%-KI) 73,4 (67,9; 78,1) 64,5 (58,8; 69,6)
EFS nach 24 Monaten, % (95%-KI) 63,3 (56,1; 69,6) 52,4 (45,4; 59,0)
Hazard Ratio (95%-KI) 0,68 (0,53; 0,88)
2-seitiger p-Wertd 0,003902
pCRa,b,d
Anzahl der Patienten mit 63 16
Ansprechen
Ansprechrate, % (95%-KI) 17,21 (13,49; 21,48) 4,28 (2,46; 6,85)
Differenz der Anteile, % (95%-KI) 12,96 (8,67; 17,57)
MPRa,c,d
Anzahl der Patienten mit 122 46
Ansprechen
Ansprechrate, % (95%-KI) 33,33 (28,52; 38,42) 12,30 (9,15; 16,06)
Differenz der Anteile, % (95%-KI) 21,03 (15,14; 26,93)
a Die Ergebnisse basieren auf der vorab geplanten EFS-Zwischenanalyse und der abschliessenden pCR-/MPR-Analyse (DCO: 10. November 2022), die 46,3 Monate nach Studienbeginn erfolgte.
b Basierend auf einer vorgegebenen pCR-Zwischenanalyse (DCO: 14. Januar 2022) bei n = 402 war die pCR-Rate statistisch signifikant (p = 0,000036) verglichen mit dem Signifikanzniveau von 0,0082 %.
c Basierend auf einer vorgegebenen MPR-Zwischenanalyse (DCO: 14. Januar 2022) bei n = 402 war die MPR-Rate statistisch signifikant (p = 0,000002) verglichen mit dem Signifikanzniveau von 0,0082 %.
d Der 2-seitige p-Wert für pCR und MPR wurde basierend auf einem stratifizierten CMH-Test berechnet. Der 2-seitige p-Wert für das EFS wurde basierend auf einem stratifizierten Log-Rank-Test berechnet. Die Stratifizierungsfaktoren waren PD-L1 und Krankheitsstadium.
Die Grenze für die Erklärung der statistischen Signifikanz für jeden der Wirksamkeitsendpunkte wurde mit einer Alpha-Spending-Funktion nach Lan-DeMets, die einen O'Brien-Fleming-Ansatz approximiert, bestimmt (EFS = 0,9899 %, pCR = 0,0082 %, MPR = 0,0082 %, 2-seitig).
In der aktualisierten (präspezifizierten) EFS-Analyse (DCO: 10. Mai 2024) betrug die mediane EFS-Nachbeobachtungszeit bei zensierten Patienten 25,9 Monate; HR für EFS war 0,69 (95%-KI: 0,55, 0,88). Bei dieser Analyse war das DFS statistisch nicht signifikant [HR = 0,66 (95%-KI: 0,47, 0,92)]. Das OS wurde nicht auf statistische Signifikanz getestet. Bei dieser Analyse betrug die Reife in Bezug auf das Gesamtüberleben (OS) 35,3 % (261 OS Ereignisse) in der mITT-Population. Die Resultate der deskriptiven OS Analyse zeigten eine OS HR von 0,89 (95%-KI: 0,70, 1,14). In der Subgruppe PD-L1 TC < 1 % der mITT-Population waren 37 Patienten (30,3 %) im Imfinzi + Chemotherapie-Arm und 51 Patienten (40,8 %) im Placebo + Chemotherapie-Arm verstorben. In der Subgruppe PD-L1 TC ≥1 % waren 84 (34,4 %) und 89 (35,7 %) Patienten im jeweiligen Studienarm verstorben.
Kleinzelliges Lungenkarzinom im lokal begrenzten Stadium (LS-SCLC) – ADRIATIC-Studie
In der ADRIATIC-Studie wurde die Wirksamkeit von Imfinzi mit oder ohne Tremelimumab untersucht. ADRIATIC war eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Studie an 730 Patienten mit histologisch oder zytologisch bestätigtem LS-SCLC (Stadium I bis III gemäss AJCC, 8. Ausgabe), deren Erkrankung nach einer gleichzeitigen Chemoradiotherapie nicht fortgeschritten war. Patienten im Stadium I oder II mussten gemäss Feststellung des Prüfarztes internistisch inoperabel sein. Die Patienten schlossen 3-4 Zyklen definitiver platinbasierter Chemoradiotherapie, 60-66 Gy einmal täglich (QD) über 6 Wochen oder 45 Gy zweimal täglich (BID) über 3 Wochen, innerhalb von 1 bis 42 Tagen vor der ersten Dosis der Studienbehandlung ab. Eine prophylaktische Schädelbestrahlung (PCI) konnte im Ermessen des Prüfarztes nach der Chemoradiotherapie innerhalb von 1 bis 42 Tagen vor der ersten Dosis der Studienbehandlung durchgeführt werden.
Von einer Teilnahme ausgeschlossen wurden Patienten mit aktiver oder früherer dokumentierter Autoimmunerkrankung in den letzten 5 Jahren vor Beginn der Studie, anamnestisch bekannter aktiver primärer Immundefizienz, anamnestisch bekannter Pneumonitis Grad ≥2 oder aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion, Patienten mit aktiver interstitieller Lungenerkrankung, Patienten, die innerhalb von 30 Tagen vor Beginn der Behandlung mit Imfinzi attenuierten Lebendimpfstoff erhalten haben sowie Patienten mit Hirnmetastasen, Kompression der Wirbelsäule oder einer Vorgeschichte von leptomeningealer Karzinomatose. Patienten mit gemischter SCLC- und NSCLC-Histologie wurden ebenfalls von einer Teilnahme an der Studie ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach Stadium (I/II oder III) und PCI (ja oder nein). Die Patienten wurden im Verhältnis 1:1:1 randomisiert und den folgenden Behandlungen zugeteilt:
-Arm 1: Imfinzi 1500 mg + Placebo alle 4 Wochen über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen.
-Arm 2: Placebo + zweites Placebo alle 4 Wochen über 4 Zyklen, gefolgt von einem Placebo alle 4 Wochen.
-Arm 3: Imfinzi 1500 mg + Tremelimumab 75 mg alle 4 Wochen über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen.
Nachdem insgesamt 600 Patienten für die drei Studienarme randomisiert worden waren, wurden nachfolgende Patienten im Verhältnis 1:1 randomisiert Arm 1 oder 2 zugeteilt und erhielten entweder Imfinzi 1500 mg alle 4 Wochen oder Placebo alle 4 Wochen.
Die Behandlung wurde bis zur Krankheitsprogression, inakzeptablen Toxizität oder für maximal 24 Monate fortgesetzt. Tumorbeurteilungen wurden während der ersten 72 Wochen alle 8 Wochen, dann bis zur 96. Woche alle 12 Wochen und danach alle 24 Wochen durchgeführt.
Die demografischen und krankheitsbedingten Eigenschaften bei Studienbeginn waren zwischen den Studienarmen gut ausgewogen. Die demografischen Ausgangsdaten und die Krankheitsmerkmale bei Studienbeginn in der Imfinzi- und der Placebo-Gruppe waren: männlich (69,1 %), Alter ≥65 Jahre (39,2 %), weiss (50,4 %), schwarz oder afroamerikanisch (0,8 %), asiatisch (47,5 %), sonstige (1,3 %), hispanisch oder lateinamerikanisch (4,2 %), aktuelle Raucher (22,3 %), frühere Raucher (68,5 %), nie geraucht (9,2 %), WHO/ECOG-PS 0 (48,7 %), WHO/ECOG-PS 1 (51,3 %), Stadium I (3,6 %), Stadium II (9,1 %), Stadium III (87,4 %).
Vor der Randomisierung erhielten alle Patienten eine platinbasierte Chemotherapie (66,2 % Cisplatin-Etoposid, 33,8 % Carboplatin-Etoposid); 72,1 % der Patienten erhielten eine RT QD (darunter 92,4 % mit ≥60 - ≤66 Gy QD); 27,9 % erhielten eine RT BID (darunter 96,6 % mit 45 Gy BID) und 53,8 % der Patiente erhielten eine PCI. Das Ansprechen auf die CRT war wie folgt: vollständiges Ansprechen (12,3 %), partielles Ansprechen (73,8 %), stabile Erkrankung (14,0 %).
Die zwei primären Endpunkte der Studie waren OS und PFS unter Imfinzi vs. Placebo. Sekundäre Endpunkte waren ORR unter Imfinzi vs. Placebo. Die Beurteilung des PFS und der ORR erfolgte durch BICR gemäss RECIST v1.1.
Bei einer geplanten Zwischenanalyse zeigte die Studie eine statistisch signifikante und klinisch bedeutsame Verbesserung des OS für Imfinzi verglichen mit Placebo [HR=0,73 (95%-KI: 0,569, 0,928), p=0,01042]. Die Studie zeigte ausserdem eine statistisch signifikante und klinisch relevante Verbesserung des PFS für Imfinzi verglichen mit Placebo [HR=0,76 (95%-KI: 0,606, 0,950), p=0,01608]. Siehe Tabelle 5.
Tabelle 5: Wirksamkeitsergebnisse der ADRIATIC-Studie
Arm 1: Imfinzi (n = Arm 2: Placebo (n =
264) 266)
OSa
Anzahl der Todesfälle (%) 115 (43,6) 146 (54,9)
Medianes OS (Monate) (95%-KI)b 55,9 (37,3, NR) 33,4 (25,5, 39,9)
HR (95%-KI)c 0,73 (0,569, 0,928)
p-Wertd 0,01042
OS nach 24 Monaten (%) (95%-KI)b 68,0 (61,9, 73,3) 58,5 (52,3, 64,3)
OS nach 36 Monaten (%) (95%-KI)b 56,5 (50,0, 62,5) 47,6 (41,3, 53,7)
PFSe
Anzahl der Ereignisse (%) 139 (52,7) 169 (63,5)
Medianes PFS (Monate) (95%-KI)b 16,6 (10,2, 28,2) 9,2 (7,4, 12,9)
HR (95%-KI)f 0,76 (0,606, 0,950)
p-Wertd 0,01608
PFS nach 18 Monaten (%) (95%-KI)b 48,8 (42,2, 55,0) 36,1 (29,9, 42,2)
PFS nach 24 Monaten (%) (95%-KI)b 46,2 (39,6, 52,5) 34,2 (28,2, 40,3)
ORRe
ORRg n (%) 53/175 (30,3) 54/169 (32,0)
Vollständiges Ansprechen n (%) 5 (2,9) 4 (2,4)
Partielles Ansprechen n (%) 48 (27,4) 50 (29,6)
Odds Ratio (95%-KI) -1,2 (-11,0, 8,5)
Mediane DoRb,e (Monate) (95%-KI) 33,0 (22,4, NR) 27,7 (9,6, NR)
Anteil an Patienten in Remission nach 12 Monatenb,e 73,7 (59,0, 83,8) 60,3 (44,5, 72,9)
(%) (95%-KI)
Anteil an Patienten in Remission nach 18 Monatenb,e 71,5 (56,6, 82,0) 55,2 (39,4, 68,5)
(%) (95%-KI)
a Die mediane Dauer der OS-Nachbeobachtung bei zensierten Patienten betrug 37,19 Monate in der Imfinzi-Gruppe und 37,24 Monate in der Placebo-Gruppe.
b Mittels Kaplan-Meier-Methode berechnet. KI für den Median mit der Brookmeyer-Crowley-Methode generiert.
c Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2-seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für Behandlung mit PCI adjustiert.
d p-Wert basierend auf den Ergebnissen der vorab geplanten Zwischenanalyse. Basierend auf einer Lan-DeMets-Alpha-Spending-Funktion mit O'Brien-Fleming-Grenze und der tatsächlich beobachteten Anzahl der Ereignisse lag die Grenze für die Erklärung der statistischen Signifikanz für OS bei 0,01679 für ein Gesamt-Alpha von 4,5 % und für PFS bei 0,02805 für ein Gesamt-Alpha von 5 % (Lan und DeMets, 1983).
e Beurteilt vom BICR gemäss RECIST v1.1.
f Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2-seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für TNM-Stadium und Behandlung mit PCI adjustiert.
g Basierend auf Subgruppe des vollständigen Analysesets mit messbarer Erkrankung zur Baseline gemäss RECIST v1.1; Imfinzi (n = 175), Placebo (n = 169).
Die Verbesserungen von OS und PFS zugunsten der Patienten, die mit Imfinzi behandelt wurden, verglichen mit den Patienten, die Placebo erhielten, waren allgemein über alle analysierten vordefinierten Subgruppen hinweg konsistent.
Fortgeschrittenes kleinzelliges Lungenkarzinom (ES-SCLC) – CASPIAN Studie
Die Wirksamkeit von Imfinzi in Kombination mit Etoposid und entweder Carboplatin oder Cisplatin in therapienaiven ES-SCLC Patienten wurde in CASPIAN, einer randomisierten, offenen, aktiv-kontrollierten, multizentrischen Studie untersucht. Teilnahmeberechtigte Patienten hatten einem Allgemeinzustand nach WHO/ECOG von 0 oder 1, ein Körpergewicht von > 30 kg, waren für den Erhalt einer platinbasierten Chemotherapie als SCLC-Erstlinientherapie geeignet und hatten eine Lebenserwartung von ≥12 Wochen. Auch Patienten mit asymptomatischen oder behandelten Hirnmetastasen, Patienten mit mindestens einer Zielläsion nach RECIST 1.1 sowie einer ausreichenden Organ- und Knochenmarkfunktion waren teilnahmeberechtigt. Die Wahl des Platin-Wirkstoffs lag unter Berücksichtigung der berechneten Kreatinin-Clearance im Ermessen des Prüfarztes. Von einer Teilnahme ausgeschlossen waren Patienten mit anamnestisch bekannter Thoraxbestrahlung oder einer geplanten Konsolidierungs-Thoraxbestrahlung; einem grösseren chirurgischen Eingriff innerhalb von 28 Tagen vor der ersten Dosis; unkontrollierter zwischenzeitlicher Erkrankung; leptomeningealer Karzinomatose; anamnestisch bekannter aktiver primärer Immundefizienz; Autoimmunerkrankungen einschliesslich des paraneoplastischen Syndroms (PNS); aktiver oder dokumentierter Autoimmunkrankheit oder entzündlicher Erkrankungen; Anwendung systemischer Immunsuppressiva innerhalb von 14 Tagen vor der erstmaligen Gabe der Therapie, physiologische Dosen systemischer Kortikosteroide ausgenommen; aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion sowie Patienten, die innerhalb von 30 Tagen vor oder nach Beginn der Behandlung mit Imfinzi einen attenuierten Lebendimpfstoff erhalten hatten bzw. sollten.
Die Randomisierung erfolgte stratifiziert nach der geplanten platinbasierten Therapie in Zyklus 1 (Carboplatin oder Cisplatin).
Die Auswertung der Wirksamkeit bei ES-SCLC stützte sich auf einen Vergleich zwischen den Gruppen:
-Imfinzi 1500 mg und je nach Wahl des Prüfarztes entweder Carboplatin (AUC 5 oder 6 mg/ml/min) oder Cisplatin (75-80 mg/m2) an Tag 1 und Etoposid (80-100 mg/m2) intravenös an den Tagen 1, 2 und 3 jedes 21-tägigen Zyklus über 4 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen, bis zur Krankheitsprogression oder inakzeptablen Toxizität
-Je nach Wahl des Prüfarztes entweder Carboplatin (AUC 5 oder 6 mg/ml/min) oder Cisplatin (75-80 mg/m2) an Tag 1 und Etoposid (80-100 mg/m2) intravenös an den Tagen 1, 2 und 3 jedes 21tägigen Zyklus über bis zu 6 Zyklen. Nach Abschluss der Chemotherapie prophylaktische Schädelbestrahlung (Prophylactic Cranial Irradiation, PCI), nach Ermessen des Prüfarztes
Die Gabe von Imfinzi als Monotherapie war auch nach einer Progression der Erkrankung gestattet, sofern der Patient klinisch stabil und nach Meinung des Prüfers ein klinischer Nutzen zu erwarten war.
Tumorbeurteilungen fanden 6 und 12 Wochen nach der Randomisierung und anschliessend alle 8 Wochen statt, bis eine gesicherte objektive Krankheitsprogression festgestellt wurde. Der Überlebensstatus wurde nach Beendigung der Behandlung 2 monatlich überprüft.
Die primären Endpunkte der Studie waren das Gesamtüberleben (OS) unter Imfinzi + Chemotherapie vs. Chemotherapie allein. Wichtigster sekundärer Endpunkt war das progressionsfreie Überleben (PFS). Weitere sekundäre Endpunkte waren die Objektive Ansprechrate (ORR), OS- und PFS-Meilensteinen sowie Patient Reported Outcomes (PRO). Die Beurteilung des PFS und der ORR erfolgte durch den Prüfarzt gemäss RECIST v1.1.
Eine geplante (primäre) Interimsanalyse zeigte, dass Imfinzi + Chemotherapie vs. Chemotherapie den primären OS-Endpunkt erreichte. Die Ergebnisse sind untenstehend zusammengefasst.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren ausgewogen in beiden Gruppen (268 Patienten in der Imfinzi + Chemotherapie-Gruppe und 269 Patienten in der Chemotherapie-Gruppe). Zu Studienbeginn wies die Gesamtpopulation der Studie folgende Eigenschaften auf: männlich (69,6 %), Alter ≥65 Jahre (39,6 %), medianes Alter 63 Jahre (Bereich: 28 bis 82 Jahre), weiss (83,8 %), asiatisch (14,5 %), schwarz bzw. afroamerikanisch (0,9 %), andere (0,6 %), nicht-hispanisch oder -lateinamerikanisch (96,1 %), aktueller oder ehemaliger Raucher (93,1 %), Nie-Raucher (6,9 %), WHO/ECOG-PS 0 (35,2 %), WHO/ECOG-PS 1 (64,8 %), Stadium IV 90,3 %, 24,6 % der Patienten erhielten Cisplatin und 74,1 % der Patienten erhielten Carboplatin. In der Chemotherapie-Gruppe erhielten 56,8 % der Patienten 6 Zyklen Chemotherapie und 7,8 % der Patienten erhielten PCI.
In der primären Analyse der Studie zeigte sich eine statistisch signifikante Verbesserung des OS unter Imfinzi + Chemotherapie vs. Chemotherapie allein [HR=0,73 (95 %-KI: 0.591, 0.909), p = 0,0047]. Das mediane Gesamtüberleben betrug in der Imfinzi + Chemotherapie-Gruppe 13,0 Monate (95 %-KI: 11.5, 14.8) und in der Chemotherapie-Gruppe10,3 Monate (95 %-KI: 9.3, 11.2). 336 Patienten (155 (57,8 %) in der Imfinzi + Chemotherapie-Gruppe und 181 (67,3 %) in der Chemotherapie-Gruppe) waren verstorben. Das Gesamtüberleben nach 12 resp. 18 Monaten betrug 53,7 % resp. 33,9 % in der Imfinzi + Chemotherapie-Gruppe und 39,8 % resp. 24,7 % in der Chemotherapie-Gruppe.
In der Analyse der Langzeitnachbeobachtung mit einer medianen Nachbeobachtungszeit von 39,3 Monaten zeigte sich unter Imfinzi + Chemotherapie vs. Chemotherapie allein weiterhin eine anhaltende Verbesserung des OS [HR=0,71 (95%-KI: 0,595; 0,858). Das mediane OS betrug in der Imfinzi + Chemotherapie-Gruppe 12,9 Monate (95%-KI: 11,3; 14,7) und 10,5 Monate (95%-KI: 9,3; 11,2) in der Chemotherapie-Gruppe. 469 Patienten (221 (82,5 %) in der Imfinzi + Chemotherapie-Gruppe und 248 (92,2 %) unter Chemotherapie allein) waren verstorben. Das OS nach 24 bzw. 36 Monaten betrug 22,9 % bzw. 17,6 % in der Imfinzi + Chemotherapie-Gruppe und 13,9 % bzw. 5,8 % in der Chemotherapie-Gruppe.
Unter Imfinzi + Chemotherapie war eine Verbesserung des PFS vs. Chemotherapie allein festzustellen [HR=0,78 (95%-KI: 0.645, 0.936) nominaler p-Wert = 0,0078]. 459 Patienten (226 (84,3 %) in der Imfinzi + Chemotherapie-Gruppe und 233 (86,6 %) in der Chemotherapie-Gruppe) waren progredient oder verstorben. Das mediane PFS war 5,1 Monate in der Imfinzi + Chemotherapie-Gruppe und 5,4 Monate in der Chemotherapie-Gruppe. Das PFS nach 6 Monaten betrug 45,4 % in der Imfinzi + Chemotherapie-Gruppe und 45,6 % in Chemotherapie-Gruppe. Das PFS nach 12 Monaten betrug 17,5 % in der Imfinzi + Chemotherapie-Gruppe und 4,7 % in der Chemotherapie-Gruppe.
Das ORR betrug 67,9 % (182/268) in der Imfinzi + Chemotherapie-Gruppe und 57,6 % (155/269) in der Chemotherapie-Gruppe. Es gab 6 Fälle mit komplettem Ansprechen in der Imfinzi + Chemotherapie-Gruppe und 2 Fälle in der Chemotherapie-Gruppe.
Subgruppenanalyse
Die Verbesserung des OS bei Patienten, die Imfinzi + Chemotherapie erhielten, gegenüber den mit Chemotherapie allein behandelten Patienten war konsistent über die verschiedenen nach Demographie, geographischer Region, Carboplatin- oder Cisplatin-Anwendung und Erkrankungsmerkmalen präspezifizierten Subgruppen hinweg zu beobachten.
Explorative Subgruppenanalyse entsprechend der PD-L1-Expression
Die limitierten explorativen Analysen der PD-L1-Expression wurden retrospektiv im Tumorgewebe mit dem VENTANA PD-L1 (SP263) IHC-Test durchgeführt und umfassten Daten sowohl von Immun- (IC) als auch von Tumorzellen (TC) (52,3% des gesamten Analysesets). Der PD-L1-Status der TC wurde durch Membranfärbung gegenüber Hintergrund bestimmt, während der PD-L1-Status der IC durch Färbung gegenüber Hintergrund von tumorassoziierter IC bestimmt wurde. Die Prävalenz der PD-L1-Expression ist gering, wobei eine PD-L1 positive Expression (≥1%) nur bei 25,8% der IC und bei 5,7% der TC beobachtet wurde.
In der IC-Subgruppe zeigten die Resultate der finalen Analyse eine OS HR von 0,64 (95% CI: 0,473, 0,855) bei PD-L1-negativen Patienten (< 1% IC) und eine OS HR von 0,59 (95% CI: 0,342, 1,019) bei PD-L1-positiven Patienten (≥1% IC). In der TC-Subgruppe zeigten die Resultate der finalen Analyse eine OS HR von 0,62 (95% KI: 0,475, 0,810) bei PD-L1-negativen Patienten (< 1% TC) und eine OS HR von 0,75 (95% KI: 0,238, 2,269) bei PD-L1-positiven Patienten (≥1% TC). Insgesamt zeigen diese Daten keinen klaren Zusammenhang zwischen dem PD-L1-Status und der Wirksamkeit in Bezug auf OS.
Biliäres Karzinom – TOPAZ-1-Studie
In der TOPAZ-1-Studie wurde die Wirksamkeit von Imfinzi in Kombination mit Gemcitabin und Cisplatin untersucht. Die TOPAZ-1-Studie war eine multizentrische, randomisierte, placebokontrollierte Doppelblind-Studie mit 685 Patienten mit histologisch gesichertem lokal fortgeschrittenen oder metastasierten biliären Karzinom und einem ECOG-Performance-Status von 0 oder 1. Die Studie schloss auch Patienten ein, die mehr als 6 Monate nach einem chirurgischen Eingriff und/oder Abschluss einer adjuvanten Therapie ein Rezidiv ausgebildet hatten. Die Patienten mussten mindestens eine Zielläsion gemäss RECIST V1.1 und eine adäquate Organ- und Knochenmarkfunktion aufweisen.
Ausgeschlossen waren Patienten mit Karzinom der Ampulla vateri (Papillenkarzinom), aktiver oder zuvor dokumentierter Autoimmunerkrankung oder entzündlicher Erkrankung, HIV-Infektion oder anderer aktiver Infektion (einschliesslich Tuberkulose und Hepatitis C) und solche, die aktuell oder innerhalb von 14 Tagen vor der ersten Dosis von Imfinzi eine immunsuppressive Medikation erhielten.
Die Randomisierung erfolgte stratifiziert nach Krankheitsstatus und Lokalisation des Primärtumors.
Die Patienten wurden im Verhältnis 1:1 randomisiert und den folgenden Behandlungen zugeteilt:
-Arm 1: Imfinzi 1500 mg intravenös an Tag 1 + Gemcitabin 1000 mg/m2 und Cisplatin 25 mg/m2 (jeweils verabreicht an den Tagen 1 und 8) alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von Imfinzi 1500 mg alle 4 Wochen, solange ein klinischer Nutzen zu beobachten ist oder bis eine inakzeptable Toxizität auftritt, oder
-Arm 2: Placebo intravenös an Tag 1 + Gemcitabin 1000 mg/m2 und Cisplatin 25 mg/m2 (jeweils verabreicht an den Tagen 1 und 8) alle 3 Wochen (21 Tage) für bis zu 8 Zyklen, gefolgt von Placebo alle 4 Wochen, solange ein klinischer Nutzen zu beobachten ist oder bis eine inakzeptable Toxizität auftritt.
Tumorbeurteilungen erfolgten in den ersten 24 Wochen nach dem Tag der Randomisierung alle 6 Wochen und dann alle 8 Wochen bis zu einer bestätigten objektiven Krankheitsprogression.
Der primäre Endpunkt der Studie war OS und der wichtigste sekundäre Endpunkt PFS. Weitere sekundäre Endpunkte waren ORR, Ansprechdauer (DoR) und Daten aus Patientenfragebogen (Patient Reported Outcomes, PRO). Die Beurteilung von PFS, ORR und DoR erfolgte durch den Prüfarzt gemäss RECIST V1.1.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren in den beiden Studienarmen ausgewogen (341 Patienten in Arm 1 und 344 Patienten in Arm 2). Zu Studienbeginn wies die Gesamtpopulation der Studie folgende demographische Merkmale auf: männlich (50,4 %), Alter < 65 Jahre (53,3 %), weiss (37,2 %), asiatisch (56,4 %), schwarz oder afroamerikanisch (2,0 %), andere (4,2 %), nichthispanisch oder -lateinamerikanisch (93,1 %), ECOG PS 0 (49,1 %) vs. PS 1 (50,9 %), Lokalisation des Primärtumors intrahepatisches Cholangiokarzinom (55,9 %), extrahepatisches Cholangiokarzinom (19,1 %) und Gallenblasenkarzinom (25,0 %), Krankheitsstatus rezidiviert (19,1 %) vs. initial nicht resezierbar (80,7 %), metastasiert (86,0 %) vs. lokal fortgeschritten (13,9 %).
Die Studie zeigte eine statistisch signifikante Verbesserung des OS und des PFS bei einer vorab geplanten Zwischenanalyse basierend auf einer Lan-DeMets-Alpha-Spending-Funktion mit O'Brien-Fleming-Grenze und der tatsächlichen Anzahl an beobachteten Ereignissen (Lan und DeMets, 1983) [HR = 0,80 (95%-KI: 0,66, 0,97), p=0,021 für das OS und HR=0,75 (95%-KI: 0,63, 0,89), p=0,001 für das PFS]. Die Reife in Bezug auf das OS betrug 61,9 %, und die Reife in Bezug auf das PFS betrug 83,6 %. Die Ergebnisse dieser Analyse für das PFS sind in Tabelle 6 aufgeführt.
Eine weitere OS-Analyse wurde 6,5 Monate nach der Zwischenanalyse mit einer OS-Reife von 76,9 % durchgeführt. Die HR für das OS betrug 0,76 (95%-KI: 0,64, 0,91) und das mediane Überleben betrug 12,9 Monate (95%-KI: 11,6, 14,1).
Eine explorative OS-Analyse wurde mit rund 3 Jahren Nachbeobachtung ab Randomisierung des letzten Patienten in der globalen Kohorte durchgeführt (3-Jahres-Update; DCO: 23 Okt 2023). Die HR für das OS betrug 0,74 (95 %-KI: 0,63; 0,87) und war konsistent mit der Zwischenanalyse nach 6,5 Monaten. Die Ergebnisse dieser OS-Analyse sind in Tabelle 6 aufgeführt.
Tabelle 6: Wirksamkeitsergebnisse der TOPAZ-1-Studie
Imfinzi + Gemcitabin und Cisplatin Placebo + Gemcitabin und Cisplatin
(n = 341) (n = 344)
OS (DCO: 23 Okt 2023)
Anzahl der Todesfälle (%) 293 (85,5) 319 (92,7)
Medianes OS (Monate) 12, 9(11,6; 14,1) 11, 3(10,1; 12,5)
(95%-KI)a
HR (95%-KI)b 0,74 (0,63; 0,87)
OS nach 24 Monaten (%) 22,9 (18,5; 27,5) 13,1 (9,8; 17,0)
(95%-KI)a
PFS (DCO: 11 Aug 2021)
Anzahl der Ereignisse (%) 276 (80,9) 297 (86,3)
Medianes PFS (Monate)(95% 7,2(6,7; 7,4) 5,7(5,6; 6,7)
-KI)a
HR (95%-KI)b 0,75 (0,63; 0,89)
p-Wertb,c 0,001
a Mit der Kaplan-Meier-Methode berechnet. KI für den Median mit der Brookmeyer-Crowley-Methode generiert.
b Die Analyse für HR erfolgte mit einem stratifizierten Cox-Proportional-Hazards-Modell und der 2seitige p-Wert basiert auf einem stratifizierten Log-Rank-Test; beide sind für Krankheitsstatus und Lokalisation des Primärtumors adjustiert.
c p-Wert basierend auf den Ergebnissen der vorab geplanten Zwischenanalyse. Auf der Basis einer Alpha-Spending-Funktion nach Lan-DeMets mit Pocock-Grenze und der tatsächlich beobachteten Anzahl an Ereignissen.
Für das 3-Jahres-Update wurde eine Subgruppenanalyse nach geographischer Region durchgeführt, die sich auf Patienten stützte, die in Prüfzentren in Asien randomisiert wurden (178 Patienten für Durvalumab + Gem/Cis und 196 Patienten für Placebo + Gem/Cis), sowie auf Patienten, die in weltweit verteilten Prüfzentren randomisiert wurden (163 Patienten bzw. 148 Patienten). Die Hazard Ratio für das OS betrug 0,62 (95% KI: 0,50; 0,78) in Asien und 0.88 (95% KI: 0,69; 1,12) im Rest der Welt (siehe Abbildungen 1 und 2).
Abbildung 1: Kaplan-Meier Kurve für das OS in Asien (DCO: 23 Okt 2023)
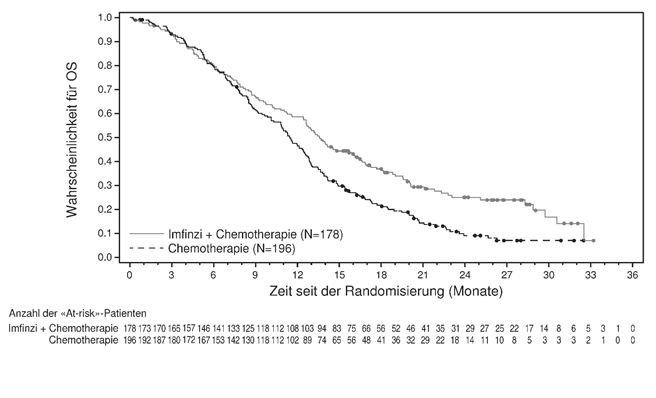
Abbildung 2: Kaplan-Meier-Kurve für das OS im Rest der Welt (DCO: 23 Okt 2023)
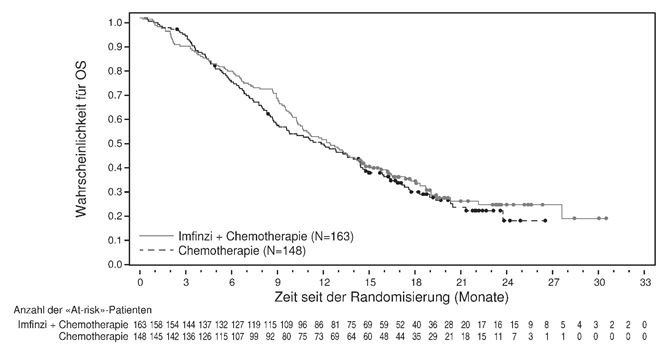
Muskelinvasiver Blasenkrebs (MIBC) – NIAGARA-Studie
NIAGARA war eine randomisierte, offene, multizentrische Phase-III-Studie zur Untersuchung der Wirksamkeit von neoadjuvantem Imfinzi in Kombination mit Gemcitabin und Cisplatin, gefolgt von adjuvanter Imfinzi-Monotherapie bei Patienten mit MIBC. In der Studie wurden 1063 für Cisplatin geeignete Patienten mit klinischem Tumorstadium T2-T4aN0/1M0 randomisiert, die für eine radikale Zystektomie in Frage kamen und zuvor keine systemische Chemotherapie oder immunvermittelte Therapie zur Behandlung von MIBC erhalten hatten. Es konnten Patienten mit angemessener (Kreatinin-Clearance [CrCl] ≥60 ml/min) oder grenzwertiger Nierenfunktion (CrCl ≥40 ml/min bis < 60 ml/min) teilnehmen. Von der Studie ausgeschlossen waren Patienten mit ausschliesslich nichturothelialer Histologie, mit jedweder kleinzelligen Histologie und mit primärem Nicht-Blasen-Krebs (d.h. Harnleiter, Harnröhre oder Nierenbecken) des Urothels, mit aktiver oder früher dokumentierter Autoimmunerkrankung, aktiver Tuberkulose oder Hepatitis B oder C oder HIV-Infektion oder Einnahme von immunsuppressiven Medikamenten innerhalb von 14 Tagen vor der ersten Dosis von Durvalumab mit Ausnahme von systemischen Kortikosteroiden, wenn in physiologischen Dosen oder als Prämedikation verwendet.
Die Randomisierung erfolgte stratifiziert nach klinischem Tumorstadium T2N0 vs. > T2N0 (einschliesslich T2N1, T3 und T4a), Nierenfunktion (angemessene Nierenfunktion: CrCl ≥60 ml/min vs. grenzwertige Nierenfunktion: CrCl ≥40 ml/min und < 60 ml/min), und PD-L1-Expressionsstatus (hoch vs. niedrig/negativ).
Die Patienten wurden im Verhältnis 1:1 auf perioperatives Imfinzi mit neoadjuvanter Chemotherapie (Arm 1) oder neoadjuvante Chemotherapie allein (Arm 2) randomisiert.
-Arm 1 (Imfinzi + Chemotherapie): Imfinzi 1500 mg + Gemcitabin 1000 mg/m² und Cisplatin 70 mg/m² alle 3 Wochen für 4 Zyklen vor der Operation, gefolgt von Imfinzi 1500 mg alle 4 Wochen für bis zu 8 Zyklen nach der Operation, oder
-Arm 2 (Chemotherapie): Gemcitabin 1000 mg/m² und Cisplatin 70 mg/m² alle 3 Wochen für 4 Zyklen vor der Operation, ohne postoperative Behandlung.
Patienten mit grenzwertiger Nierenfunktion erhielten eine geteilte Dosis Cisplatin von 35 mg/m² an den Tagen 1 und 8 eines jeden Zyklus.
Eine RECIST-1.1-Tumorbeurteilung wurde zu Studienbeginn und nach Abschluss der neoadjuvanten Therapie (vor der Operation) durchgeführt. Nach der Operation wurden in den ersten 24 Monaten alle 12 Wochen, dann 36 Monate lang alle 24 Wochen und danach bis zur Krankheitsprogression, zum Ende der Studie oder zum Tod alle 52 Wochen RECIST-1.1-Tumorbeurteilungen durchgeführt.
Die primären Endpunkte waren pathologisch vollständiges Ansprechen (pathological complete response; pCR) durch verblindete zentrale pathologische Überprüfung und ereignisfreies Überleben (event-free survival; EFS), was eine Beurteilung durch verblindete unabhängige zentrale Überprüfung (BICR) einschloss. Der wichtigste sekundäre Endpunkt war das Gesamtüberleben (overall survival; OS).
Die demografischen Daten und die Merkmale der Erkrankung zu Studienbeginn waren zwischen den 533 Patienten in Arm 1 und den 530 Patienten in Arm 2 grundsätzlich gut ausgeglichen. Die demografischen Daten zu Studienbeginn waren wie folgt: männlich (81,8 %), Alter < 65 Jahre (46,9 %), weiss (67 %), asiatisch (27,9 %), hispanisch oder lateinamerikanisch (8,0 %), schwarz oder afroamerikanisch (0,9 %), andere (0,8 %), und ECOG PS 0 (78,4 %) vs. PS 1 (21,6 %). Die Merkmale der Erkrankung waren wie folgt: Tumorstadium T2N0 (40,3 %) und > T2N0a (59,7 %), regionale Lymphknoten N0 (94,5 %) und N1 (5,5 %), angemessene Nierenfunktion (81,1 %) und grenzwertige Nierenfunktion (18,9 %) und PD-L1-Expressionsstatus hoch (73,1 %) und niedrig/negativ (26,9 %). Zu den histologischen Subtypen gehörten Urothelkarzinom (84,5 %), Urothelkarzinom mit Plattenepithel-Differenzierung (8,2 %), Urothelkarzinom mit varianter Histologie (5,0 %) und Urothelkarzinom mit glandulärer Differenzierung (2,4 %).
In der zweiten präspezifizierten Zwischenanalyse zeigte die Studie eine statistisch signifikante Verbesserung des Überlebens im Imfinzi+Chemotherapie-Arm im Vergleich zum Chemotherapie-Arm [HR = 0,68 (95%-KI: 0,56; 0,82), p < 0,0001]. Die Studie zeigte auch eine statistisch signifikante Verbesserung des OS im Imfinzi+Chemotherapie-Arm im Vergleich zum Chemotherapie-Arm [HR = 0,75 (95%-KI: 0,59; 0.93), p = 0,0106]. Eine numerische Verbesserungder pCR-Raten ,welche jedoch nicht statistisch signifikant war, wurde im Imfinzi + Chemotherapie-Arm [Ansprechrate = 37,3 % (95%-KI: 33,2; 41,6)] im Vergleich zum Chemotherapie-Arm [Ansprechrate = 27,5 % (95%-KI: 23.8, 31.6)] beobachtet. Siehe Tabelle 7.
Tabelle 7: Wirksamkeitsergebnisse der NIAGARA-Studie
Imfinzi + Chemotherapie(n = 533) Chemotherapie(n = 530)
EFSa
Anzahl der Ereignisse (%) 187 (35,1) 246 (46,4)
Medianes EFS (Monate) (95% KI)b NR (NR; NR) 46,1 (32,2; NR)
HR (95% KI)c 0,68 (0,56; 0,82)
2-seitiger p-Wertd,e <0,0001
EFS nach 24 Monaten (%) (95% KI)b 67,8 (63,6; 71,7) 59,8 (55,4; 64,0)
pCRf
Anzahl der Patienten mit Ansprechen 199 146
Ansprechrate, % (95% KI)g 37,3 (33,2; 41,6) 27,5 (23,8; 31,6)
Odds Ratio (95% KI)h 1,60 (1,23; 2,09)
OSa
Anzahl Ereignisse (%) 136 (25,5) 169 (31,9)
Medianes OS (Monate) (95% KI)b NR (NR; NR) NR (NR; NR)
HR (95% KI)c 0,75 (0,59; 0,93)
2-seitiger p-Wertd,e 0,0106
OS nach 24 Monaten (%) (95% KI)b 82,2 (78,7; 85,2) 75,2 (71,3; 78.8)
a Die Ergebnisse basieren auf einer vordefinierten Zwischenanalyse (DCO: 29. April 2024), die 68 Monate nach Beginn der Studie durchgeführt wurde.
b Berechnet mittels der Kaplan-Meier-Methode.
c Basierend auf einem stratifizierten Cox-Proportional-Hazard-Modell mit Tumorstadium [T2N0 vs. > T2N0], Nierenfunktion [angemessen vs. grenzwertig] und PD-L1-Status [hoch vs. niedrig/negativ] als Stratifikationsfaktoren.
d Basierend auf dem stratifizierten Log-Rank-Test mit Tumorstadium [T2N0 vs. > T2N0], Nierenfunktion [angemessen vs. grenzwertig] und PD-L1-Status [hoch vs. niedrig/negativ] als Stratifikationsfaktoren.
e Die Grenze für die Erklärung der statistischen Signifikanz für die primären Wirksamkeitsendpunkte pCR-Rate, EFS und den wichtigsten sekundären Endpunkt OS wurde durch ein multiples Testverfahren mit einer alpha-exhaustiven Recycling-Strategie bestimmt. Das Alpha, das bei der Zwischenanalyse dem EFS und OS zugeordnet wurde, basierte auf einer Lan-DeMets Alpha-Ausgabefunktion mit O'Brien-Fleming-Ansatz. (pCR = 0,001, EFS = 0,0412, OS = 0,0154, 2-seitig).
f Basierend auf einer aktualisierten, deskriptiven Analyse des primären Endpunkts. Bei der früheren vorspezifizierten abschliessenden Analyse der pCR (DCO: 14. Jan. 2022) wurde eine numerische Verbesserung der pCR-Raten in dem Imfinzi+Chemotherapie-Arm [Ansprechrate = 33,8 % (95%-KI: 29,8; 38,0)] im Vergleich zum Chemotherapie-Arm [Ansprechrate = 25,8 % (95%-KI: 22,2; 29,8)] [Odds Ratio 1,49 (95%-KI: 1,14; 1,96), p = 0,0038] beobachtet.
g Das KI wurde mit der Clopper-Pearson-Methode berechnet.
h Ermittelt durch logistische Regression, bereinigt um die Stratifikationsfaktoren (Nierenfunktion [angemessen vs. grenzwertig], Tumorstadium [T2N0 vs. > T2N0] und PD-L1-Status [hoch vs. niedrig/negativ] per IVRS).
KI = Konfidenzintervall, HR = Hazard Ratio, NR = Nicht erreicht
Endometriumkarzinom – DUO-E Studie
DUO-E war eine multizentrische, randomisierte, placebokontrollierte, doppelblinde Phase-III-Studie zur Erstlinienbehandlung mittels platinbasierter Chemotherapie in Kombination mit Imfinzi, gefolgt von einer Erhaltungstherapie mit Imfinzi mit oder ohne Olaparib bei Patientinnen mit fortgeschrittenem oder rezidiviertem Endometriumkarzinom. Die Patientinnen mussten ein Endometriumkarzinom einer der folgenden Kategorien aufweisen: neu diagnostizierte Erkrankung im Stadium III (messbare Erkrankung gemäss RECIST 1.1 nach Operation oder diagnostischer Biopsie), neu diagnostizierte Erkrankung im Stadium IV (mit oder ohne Erkrankung nach Operation oder diagnostischer Biopsie) oder rezidivierte Erkrankung (messbare oder nicht messbare Erkrankung gemäss RECIST 1.1), bei der das Heilungspotenzial durch einen chirurgischen Eingriff allein oder in Kombination gering war. Bei Patientinnen mit rezidivierter Erkrankung war eine chemotherapeutische Vorbehandlung nur erlaubt, wenn diese als adjuvante Therapie verabreicht worden war und zwischen dem Datum der letzten Dosis der Chemotherapie und dem Rezidiv mindestens 12 Monate vergangen waren. Die Studie schloss Patientinnen mit epithelialem Endometriumkarzinom aller histologischen Unterformen ein, einschliesslich Karzinosarkomen. Patientinnen mit Endometriumsarkom waren ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach dem MMR(Mismatch-Repair)-Status des Tumorgewebes (kompetent [pMMR] versus defizient [dMMR]), Krankheitsstatus (Rezidiv versus Neudiagnose) und geographischer Region (Asien versus Rest der Welt). Die Patientinnen wurden randomisiert im Verhältnis 1:1:1 einem der folgenden Behandlungsarme zugeteilt:
-Arm 1 (platinbasierte Chemotherapie): Platinbasierte Chemotherapie (Paclitaxel und Carboplatin) alle 3 Wochen für maximal 6 Zyklen mit Durvalumab-Placebo alle 3 Wochen. Nach Abschluss der Chemotherapie erhielten Patientinnen ohne objektive Krankheitsprogression als Erhaltungstherapie Durvalumab-Placebo alle 4 Wochen und Olaparib-Placebo-Tabletten zweimal täglich bis zur Krankheitsprogression.
-Arm 2 (platinbasierte Chemotherapie + Imfinzi): Platinbasierte Chemotherapie (Paclitaxel und Carboplatin) alle 3 Wochen für maximal 6 Zyklen mit 1120 mg Imfinzi alle 3 Wochen. Nach Abschluss der Chemotherapie erhielten Patientinnen ohne objektive Krankheitsprogression als Erhaltungstherapie 1500 mg Durvalumab alle 4 Wochen mit Olaparib-Placebo-Tabletten zweimal täglich bis zur Krankheitsprogression.
-Arm 3 (platinbasierte Chemotherapie + Imfinzi + Olaparib): Platinbasierte Chemotherapie (Paclitaxel und Carboplatin) alle 3 Wochen für maximal 6 Zyklen mit 1120 mg Imfinzi alle 3 Wochen. Nach Abschluss der Chemotherapie erhielten Patientinnen ohne objektive Krankheitsprogression als Erhaltungstherapie 1500 mg Durvalumab alle 4 Wochen mit 300 mg Olaparib-Tabletten zweimal täglich bis zur Krankheitsprogression.
Die Behandlung wurde bis zu einer gemäss RECIST V1.1 definierten Krankheitsprogression oder inakzeptablen Toxizität fortgesetzt. Beurteilungen des Tumorstatus erfolgten in den ersten 18 Wochen nach der Randomisierung alle 9 Wochen und anschliessend alle 12 Wochen.
Der primäre Endpunkt war das PFS, beurteilt durch die Prüfärzte anhand von RECIST 1.1. Die sekundären Wirksamkeitsendpunkte beinhalteten OS.
Die Baseline-Merkmale der Erkrankung waren in den drei Studienarmen im Allgemeinen ausgewogen (241 Patientinnen in Arm 1, 238 Patientinnen in Arm 2 und 239 Patientinnen in Arm 3). Die demographischen Baseline-Merkmale waren: Alter ≥65 Jahre (47%), medianes Alter von 64 Jahren (Spannweite: 22 bis 86 Jahre), weiss (57%), asiatisch (30%), schwarz (5%) und andere (4%). Die Krankheitsmerkmale waren: WHO/ECOG PS 0 (67%) vs. PS 1 (33%), 47% Neudiagnosen und 53% Rezidive;; 67% PD-L1-positiv und 30% PD-L1-negativ; 26% mit HRR-Mutation, 60% ohne HRR-Mutation und 14% mit unbekanntem HRR-Mutationsstatus; 11% mit BRCA-Mutation. Die histologischen Subtypen waren endometrioid (60%), serös (21%), Karzinosarkom (7%), gemischt-epithelial (4%), klarzellig (3%), undifferenziert (2%), muzinös (<1%) und andere (3%).
Die mediane Beobachtungszeit für PFS betrug bei zensierten Patientinnen im Behandlungsarm mit platinbasierter Chemotherapie + Imfinzi 15,4 Monate und im Behandlungsarm mit platinbasierter Chemotherapie 12,6 Monate. Zum Zeitpunkt der primären PFS-Analyse hatten die OS-Daten der Zwischenanalyse eine Reife von 31%, und bei 147 von 479 Patientinnen, die mit platinbasierter Chemotherapie + Imfinzi und platinbasierter Chemotherapie behandelt wurden, waren Ereignisse eingetreten.
Tabelle 8: Wirksamkeitsergebnisse der DUO-E-Studie
Platinbasierte Chemotherapie + Platinbasierte Chemotherapi
Imfinzi n=238 e n=241
PFSa (Primäranalyse, DCO: 12.
April 2023)
Anzahl der Ereignisse (%) 139 (58,4) 173 (71,8)
Medianesb PFS (Monate) (95%-KI) 10,2 (9,7-14,7) 9,6 (9,0-9,9)
HRc (95%-KI) 0,71 (0,57-0,89)
p-Wertd 0,003
a Durch die Prüfärzte beurteilt.
b Mit der Kaplan-Meier-Methode berechnet.
c Eine HR unter 1 spricht zugunsten des Behandlungsarms mit platinbasierter Chemotherapie + Imfinzi vs. platinbasierte Chemotherapie.
d Der p-Wert wurde mit einem Log-Rank-Test berechnet.
Zum Zeitpunkt einer späteren deskriptiven Analyse (DCO 18. Oktober 2023) waren 34,4 % der OS-Ereignisse aufgetreten. Das mediane OS wurde in keinem Arm erreicht. Das OS-HR betrug 0,84 (0,62–1,12) zugunsten der Patienten, die mit platinbasierter Chemotherapie + Imfinzi im Vergleich zu platinbasierter Chemotherapie behandelt wurden.
Der Status der Mismatch-Reparatur (MMR) wurde zentral mithilfe eines MMR-spezifischen immunhistochemischen Panel-Assays bestimmt. Von den insgesamt 718 in der Studie randomisierten Patientinnen wiesen 575 (80%) einen Tumorstatus mit kompetenter MMR (pMMR) und 143 Patientinnen (20%) einen Tumorstatus mit defizienter MMR (dMMR) auf.
Patientinnen mit Endometriumkarzinom mit defizienter MMR (dMMR)
Unter den Patientinnen mit dMMR-Tumorstatus waren die demografischen Merkmale und die Baseline-Merkmale in den Behandlungsarmen im Allgemeinen ausgewogen. Die demografischen Baseline-Merkmale in den drei Studienarmen waren: medianes Alter 62 Jahre (Bereich: 34 bis 85 Jahre), 41% waren 65 Jahre oder älter und 1,5% 75 Jahre oder älter, 62% waren weiss, 29% asiatisch und 2% waren schwarz oder afroamerikanisch. Die Krankheitsmerkmale waren: ECOG PS 0 (58%) oder 1 (42%), 46 % Neudiagnosen und 54% Rezidive, 80% PD-L1-positiv und 18% PD-L1-negativ; 32% mit HRR-Mutation, 43% ohne HRR-Mutation und 26% mit unbekanntem HRR-Mutationsstatus; 13% mit BRCA-Mutation. Die histologischen Subtypen waren endometrioid (83%), gemischt-epithelial (5%), serös (3%), Karzinosarkom (3%), undifferenziert (2%) und andere (3%).
Die Ergebnisse für die Patientinnen mit dMMR-Tumorstatus sind in Tabelle 9 zusammengefasst. Die mediane Nachbeobachtungsdauer für das PFS betrug bei zensierten Patientinnen mit dMMR-Tumorstatus im Behandlungsarm mit platinbasierter Chemotherapie + Imfinzi 15,5 Monate und im Behandlungsarm mit platinbasierter Chemotherapie 10,2 Monate. Zum Zeitpunkt der PFS-Analyse hatten die OS-Daten der Zwischenanalyse eine Reife von 26%, und bei 25 von 95 der mit einer platinbasierten Chemotherapie + Imfinzi bzw. einer platinbasierten Chemotherapie behandelten Patientinnen waren Ereignisse eingetreten.
Tabelle 9: Wirksamkeitsergebnisse der DUO-E-Studie (Patientinnen mit dMMR-Tumorstatus)
Platinbasierte Chemotherapie + Platinbasierte Chemotherapi
Imfinzi N = 46 e N = 49
PFS*,† (Primäranalyse, DCO: 12.
April 2023)
Anzahl an Ereignissen (%) 15 (32,6) 25 (51,0)
Medianes PFS, Monate (95%-KI)‡ n. e. (n. e.; n. e.) 7,0 (6,7; 14,8)
HR (95%-KI) 0,42 (0,22; 0,80) -
* Durch die Prüfärzte beurteilt.
† Ergebnisse der Primäranalyse (PFS) (DCO: 12. April 2023).
‡ Mit der Kaplan-Meier-Methode berechnet.
KI = Konfidenzintervall; HR = Hazard Ratio; n.e. = nicht erreicht
Zum Zeitpunkt einer späteren deskriptiven Analyse (DCO 18. Oktober 2023) waren 25,2 % der OS-Ereignisse aufgetreten. Das mediane OS wurde in keinem Arm erreicht. Das OS-HR betrug 0,40 (0,17–0,86) zugunsten der Patienten, die mit platinbasierter Chemotherapie + Imfinzi im Vergleich zu platinbasierter Chemotherapie behandelt wurden.
Resezierbares Adenokarzinom des Magens oder des gastroösophagealen Übergangs (GC/GEJC) – MATTERHORN-Studie
MATTERHORN war eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Phase-III-Studie zur Beurteilung der Wirksamkeit von Imfinzi in Kombination mit FLOT-Chemotherapie als neoadjuvante und adjuvante Behandlung, gefolgt von adjuvanter Imfinzi-Monotherapie, bei Patienten mit resezierbarem Adenokarzinom des Magens oder des gastroösophagealen Übergangs (klinisches Stadium II bis IVA [cTNM, >T2 N0-3 M0 or T0-4 N1-3 M0, AJCC, 8. Auflage]). In die Studie wurden zuvor unbehandelte Patienten mit resezierbarem GC/GEJC ohne vorherige Exposition gegenüber einer Immuntherapie mit einem WHO/ECOG Performance Status von 0 oder 1 aufgenommen. Die Patienten wurden unabhängig von ihrem Tumor-PD-L1-Expressionsstatus in die Studie aufgenommen, und vor der Randomisierung wurde der PD-L1-Expressionsstatus des Tumors mit dem Ventana PD-L1 (SP263) Test bestätigt. Patienten mit peritonealer Metastasierung, adenosquamösem Karzinom, Plattenepithelkarzinom oder gastrointestinalen Stromatumoren (GIST), aktiver oder zuvor dokumentierter Autoimmunerkrankung oder entzündlicher Erkrankung oder Anwendung einer immunsuppressiven Medikation innerhalb von 14 Tagen vor der ersten Dosis von Imfinzi, wurden aus der Studie ausgeschlossen.
Die Randomisierung erfolgte stratifiziert nach geographischer Region (Asien vs. Nicht-Asien), klinischem Lymphknotenstatus (positiv vs. negativ) und PD-L1-Expressionsstatus (TAP < 1 % vs. TAP ≥1 %).
Die Patienten wurden im Verhältnis 1:1 für einen der folgenden Behandlungsarme randomisiert. Ein Crossover zwischen den Studienarmen war nicht erlaubt.
-Arm 1: Imfinzi 1500 mg an Tag 1 + FLOT-Chemotherapie an den Tagen 1 und 15 alle 4 Wochen für 4 Zyklen (1 Dosis von Imfinzi und 2 Dosen FLOT pro Zyklus; 2 Zyklen in der neoadjuvanten Phase + 2 Zyklen in der adjuvanten Phase), gefolgt von Imfinzi 1500 mg an Tag 1 alle 4 Wochen für bis zu 10 weitere Zyklen nach der Operation, d.h. insgesamt 12 Zyklen (1 Dosis pro Zyklus)
-Arm 2: Placebo an Tag 1 + FLOT-Chemotherapie an den Tagen 1 und 15 alle 4 Wochen für 4 Zyklen (1 Dosis Placebo und 2 Dosen FLOT pro Zyklus; 2 Zyklen in der neoadjuvanten Phase + 2 Zyklen in der adjuvanten Phase), gefolgt von Placebo an Tag 1 alle 4 Wochen für bis zu 10 weitere Zyklen nach der Operation, d.h. insgesamt 12 Zyklen (1 Dosis pro Zyklus)
Patienten in der neoadjuvanten oder adjuvanten Phase, welche die FLOT-Chemotherapie aus anderen Gründen als einer Krankheitsprogression oder einem Rezidiv absetzten, konnten nach Ermessen des Prüfarztes die Behandlung mit einer Imfinzi-Monotherapie wie oben beschrieben fortsetzen.
Eine Baseline-Tumorbeurteilung nach RECIST 1.1 wurde vor Beginn der neoadjuvanten Therapie durchgeführt, und eine Folgeuntersuchung vor der Operation wurde innerhalb von 4 Wochen nach der letzten Chemotherapie-Dosis durchgeführt. Eine adjuvant Baseline-Untersuchung wurde frühestens 4 Wochen nach der Operation und vor Beginn der adjuvanten Behandlung durchgeführt. Tumorbeurteilungen wurden alle 12 Wochen (bezogen auf die adjuvante Baseline-Untersuchung) für 2 Jahre und danach alle 24 Wochen bis zur radiologisch bestätigten Krankheitsprogression gemäss RECIST 1.1, bis zum Widerruf der Einwilligung oder bis zum Tod durchgeführt.
Der primäre Endpunkt der Studie war das EFS gemäss BICR-Beurteilung. Die wichtigsten sekundären Endpunkte waren das OS und die pCR-Rate gemäss verblindeter zentraler pathologischer Untersuchung. Weitere sekundäre Wirksamkeitsziele waren PROs.
Die demographischen Daten und Erkrankungsmerkmale zu Studienbeginn waren generell in den beiden Studienarmen ausgewogen (474 Patienten in Arm 1 und 474 Patienten in Arm 2). Zu Studienbeginn wies die Gesamtpopulation der Studie folgende Eigenschaften auf: männlich (71,9 %), Alter ≥65 Jahre (41,4 %), medianes Alter 62 Jahre (Bereich: 26 bis 84 Jahre), weiss (67,8 %), asiatisch (20,4 %), schwarz bzw. afroamerikanisch (1,1 %), indigen amerikanisch oder alaskisch (4,0 %), andere Ethnie (1,7 %), nicht-hispanisch oder -lateinamerikanisch (80,0 %), WHO/ECOG-PS 0 (74,2 %) versus PS 1 (25,8 %). Die krankheitsbedingten Eigenschaften waren wie folgt: klinisches Stadium II (29,4 %), Stadium III (61,7 %), Stadium IVA (8,8 %), Magen-Adenokarzinom (67,5 %), Adenokarzinom des gastroösophagealen Übergangs (32,5 %), Siewert-Typ 1 (10,4 %), Siewert-Typ 2 (14,8 %), Siewert-Typ 3 (7,3 %), intestinaler Typ (50,9 %), diffuser Typ (26,3 %), unbestimmter Typ (22,8 %), klinischer Lymphknotenstatus positiv (70,4 %), klinischer Lymphknotenstatus negativ (29,2 %), PD-L1-Expressionsstatus TAP ≥1 % (90,0 %), PD-L1-Expressionsstatus TAP < 1 % (10,0 %).
Bei 431 Patienten (90,9 %) in Arm 1 wurde eine Operation mit kurativer Intention versucht, verglichen mit 428 Patienten (90,3 %) in Arm 2. Bei 412 Patienten (86,9 %) in Arm 1 wurde eine Operation mit kurativer Intention durchgeführt, verglichen mit 400 Patienten (84,4 %) in Arm 2.
In einer vorgegebenen Zwischenanalyse zeigte die Studie eine statistisch signifikante und klinisch bedeutsame Verbesserung des EFS [HR = 0,71 (95%-KI: 0,58; 0,86), p = 0,001] im Imfinzi-Arm verglichen mit dem Placebo-Arm. Ausserdem zeigte die Studie eine statistisch signifikante Verbesserung der pCR-Rate für den Imfinzi-Arm [Ansprechrate 19,2 % (95%-KI: 15,7, 23,0)] verglichen mit dem Placebo-Arm [Ansprechrate 7,2 % (95%-KI: 5,0, 9,9)]. In einer abschliessenden OS-Analyse zeigte die Studie eine statistisch signifikante und klinisch bedeutsame Verbesserung des OS [HR = 0,78 (95% CI: 0,63, 0,96), p = 0,021] im Imfinzi-Arm verglichen mit dem Placebo-Arm, siehe Tabelle 10.
Tabelle 10: Wirksamkeitsergebnisse der MATTERHORN-Studie
Imfinzi + FLOT-Chemotherapie(n Placebo + FLOT-Chemotherapie(n
= 474) = 474)
EFSa
Anzahl der Ereignisse, n (%) 167 (35,2 %) 218 (46,0 %)
Medianes EFS (95%-KI) NC (40,7, NR) 32,8 (27,9, NR)
(Monate)b
HR (95 % KI)c 0,71 (0,58, 0,86)
2-seitiger p-Wertd,e < 0,001
EFS nach 24 Monaten, % 67,4 (62,9, 71,6) 58,5 (53,8; 63,0)
(95%-KI)b
OSf
Anzahl der Todesfälle (%) 160 (33,8 %) 192 (40,5 %)
Medianes OS (95%-KI) (Monate)b NR (NR, NR) NR (NR, NR)
HR (95 % KI)c 0,78 (0,63, 0,96)
2-seitiger p-Wertd,g 0,021
OS nach 24 Monaten, % 75,5 (71,3, 79,1) 70,4 (66,0, 74,3)
(95%-KI)b
OS nach 36 Monaten, % 68,6 (64,2, 72,6) 61,9 (57,3, 66,2)
(95%-KI)b
pCRh
Anzahl der Patienten mit 91 34
Ansprechen
Ansprechrate, % (95%-KI)i 19,2 (15,7, 23,0) 7,2 (5,0, 9,9)
Odds Ratio, % (95%-KI) 3,08 (2,0, 4,7)
2-seitiger Wertk,j < 0,001
a Die Ergebnisse basieren auf einer vorgegebenen Zwischenanalyse des EFS (DCO: 20. Dezember 2024).
b Mittels Kaplan-Meier-Methode berechnet.
c Basierend auf einem stratifizierten Cox-Proportional-Hazards-Modell, stratifiziert nach geographischer Region, klinischem Lymphknotenstatus und PD-L1-Expressionsstatus zum Zeitpunkt der Randomisierung. Eine HR <1 spricht zugunsten von Imfinzi. Das KI wurde mithilfe des Profile-Likelihood-Ansatzes berechnet.
d Basierend auf einem stratifizierten Logrank-Test mit Adjustierung um geographische Region, klinischen Lymphknotenstatus und PD-L1-Expressionsstatus zum Zeitpunkt der Randomisierung.
e Basierend auf einer Alpha-Spending-Funktion nach Lan-DeMets mit O'Brien-Fleming-Grenze, berechnet anhand der tatsächlichen Anzahl an Patienten am DCO, betrug die Grenze für die Erklärung der statistischen Signifikanz des EFS 0,0239 für ein Gesamt-Alpha von 5 % (2-seitig).
f Die Ergebnisse basieren auf einer finalen OS-Analyse (DCO: 01. September 2025).
g Ein Alpha von 4,99 % (2-seitig) wurde der abschliessenden Analyse des OS zugeordnet, entsprechend einer Grenze für die Erklärung von statistischer Signifikanz von 0,0499.
h Die Ergebnisse basieren auf der abschliessenden pCR-Analyse (DCO: 01. Februar 2023).
i Das KI wurde mithilfe der Clopper-Pearson-Methode berechnet.
j Ein fixes Alpha von 0,1 % (2-seitig) wurde der Zwischenanalyse der pCR-Rate zugeordnet, entsprechend einer p-Wert-Grenze für die Erklärung von statistischer Signifikanz von 0,001.
k Basierend auf einem stratifizierten Cochran-Mantel-Haenszel-Test mit Adjustierung um geografische Region, klinischen Lymphknotenstatus und PD-L1-Expressionsstatus zum Zeitpunkt der Randomisierung.
KI = Konfidenzintervall, HR = Hazard Ratio, NR = Nicht erreicht
Für die explorative, post-hoc analysierte Subgruppe mit diffusem Adenokarzinom betrug die HR für das EFS 0,93 (95%-KI 0,66-1,32), die HR für das OS 0,98 (95%-KI 0,68-1,40) und pCR von 6.9% im D + FLOT versus 4.2% in Placebo + Arm.
PharmakokinetikDie Pharmakokinetik (PK) von Durvalumab wurde für Imfinzi als Monotherapie und in Kombination mit Chemotherapie beurteilt. Die Pharmakokinetik (PK) von Durvalumab wurde an Patienten mit soliden Tumoren und Dosierungen zwischen 0,1 und 20 mg/kg getestet, die einmal alle zwei, drei oder vier Wochen verabreicht wurden. Es gab keinen klinisch bedeutsamen Unterschied zwischen der PK von Durvalumab als Monotherapie und in Kombination mit Chemotherapie.
Absorption
Die PK-Exposition stieg stärker als dosisproportional (nicht-lineare PK) bei Dosierungen < 3 mg/kg und dosisproportional (lineare PK) bei Dosierungen ≥3 mg/kg.
Distribution
Steady State wurde nach ca. 16 Wochen erreicht. Basierend auf der Analyse der PK-Population, die 1878 Patienten im Dosierungsbereich von 10 mg/kg Q2W, 15 mg/kg Q3W und 20 mg/kg Q4W, umfasste, lag das Verteilungsvolumen (Vd) des geometrisch mittleren Steady State bei 5,64 l.
Metabolismus
Der Metabolismus von Durvalumab wurde nicht ermittelt. Als ein vollständig humaner monoklonaler IgG1-Antikörper wird erwartet, dass Durvalumab wie endogenes IgG via kleinen Peptiden und Aminosäuren katabolisch abgebaut wird.
Elimination
Die Durvalumab-Clearance (CL) nahm mit der Zeit ab und führte zu einer Clearance des geometrischen mittleren Steady States (CLss) von 8,16 ml/h an Tag 365; die CLss-Abnahme wurde nicht als klinisch relevant eingestuft. Die terminale Halbwertszeit (t1/2), basierend auf der Baseline-CL, betrug ungefähr 18 Tage. Die Elimination erfolgt primär durch Proteinkatabolismus über das retikuloendotheliale System oder durch Elimination des Antikörper-Zielprotein-Komplexes.
Kinetik spezieller Patientengruppen
Eine Populations-PK-Analyse ergab, dass Alter (19–96 Jahre), Körpergewicht (31–149 kg), Geschlecht, positiver Status bei therapiebedingten Antikörpern (Anti-Drug Antibodies, ADAs), Albumin-Werte, LDH-Werte, Kreatinin-Werte, löslicher PD-L1, Tumortyp, Ethnizität und ECOG/WHO-Status keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Durvalumab hatten.
Ältere Patienten
Eine Populations-PK-Analyse ergab, dass Alter (19-96 Jahre) keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Durvalumab hatte.
Nierenfunktionsstörungen
Eine Populations-PK-Analyse ergab, dass geringfügige Niereninsuffizienz (creatinin clearance (CRCL) 60 bis 89 ml/min) und mittlere Niereninsuffizienz (CRCL 30 bis 59 ml/min) keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Durvalumab haben. Die Auswirkung schwerer Niereninsuffizienz (CRCL 15 bis 29 ml/min) hinsichtlich der Pharmakokinetik von Durvalumab ist unbekannt.
Leberfunktionsstörungen
Eine Populations-PK-Analyse ergab, dass geringfügige Leberfunktionsstörung (Bilirubin ≤ ULN und AST > ULN oder Bilirubin > 1,0 bis 1,5 x ULN und jeglicher AST) oder mittelschwere Leberfunktionsstörung (Bilirubin > 1,5 bis 3 x ULN und jeglicher AST) keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Durvalumab hatten. Die Auswirkung von schwerer Leberfunktionsstörung (Bilirubin > 3,0 x ULN und jeglicher AST) hinsichtlich der Pharmakokinetik von Durvalumab ist unbekannt.
Pädiatrische Population
Die PK von Durvalumab in Kombination mit Tremelimumab wurde in einer Studie mit 50 pädiatrischen Patienten (von denen 42 in den popPK-Datensatz inkludiert wurden) mit einem Altersbereich von 1 bis 17 Jahren untersucht (Studie D419EC00001). Die Patienten erhielten entweder Durvalumab 20 mg/kg in Kombination mit Tremelimumab 1 mg/kg oder Durvalumab 30 mg/kg in Kombination mit Tremelimumab 1 mg/kg alle 4 Wochen für 4 Zyklen, gefolgt von Durvalumab als Monotherapie alle 4 Wochen. Basierend auf einer populationspharmakokinetischen Analyse war die systemische Exposition gegenüber Durvalumab bei pädiatrischen Patienten ≥35 kg, die Durvalumab 20 mg/kg alle 4 Wochen erhielten, vergleichbar mit der Exposition bei Erwachsenen, die Durvalumab 20 mg/kg alle 4 Wochen erhielten, während bei pädiatrischen Patienten ≥35 kg, die Durvalumab 30 mg/kg alle 4 Wochen erhielten, die Exposition etwa dem 1,5-fachen der Exposition bei Erwachsenen entsprach, die Durvalumab 20 mg/kg alle 4 Wochen erhielten. In pädiatrischen Patienten < 35 kg, die Durvalumab 30 mg/kg alle 4 Wochen erhielten, war die systemische Exposition vergleichbar mit der Exposition bei Erwachsenen, die Durvalumab 20 mg/kg alle 4 Wochen erhielten.
Präklinische DatenKarzinogenität und Mutagenität
Das karzinogene und genotoxische Potenzial von Durvalumab wurde nicht ermittelt.
Reproduktionstoxizität
Es liegen keine Daten über die potenziellen Auswirkungen von Durvalumab auf die Fertilität beim Menschen vor. Wie in Fachtexten berichtet, spielt der PD-1/PD-L1-Pfad eine zentrale Rolle bei der Aufrechterhaltung der Schwangerschaft, indem die Immuntoleranz der Mutter gegenüber dem Fötus aufrechterhalten wird. Anhand allogener Schwangerschaftsmodelle an Mäusen konnte gezeigt werden, dass ein Unterbruch der PD-L1-Signale zu vermehrtem Fetaltod führt. In tierexperimentellen Reproduktionsstudien war die Verabreichung von Durvalumab an trächtige Javaneraffen von der Bestätigung der Trächtigkeit bis zur Geburt in Expositionen, die etwa 6-20 Mal beziehungsweise 3-11 Mal höher waren als die für die klinische Dosis von 10 mg/kg Durvalumab alle 2 Wochen beziehungsweise 1500 mg alle 3 Wochen (basierend auf der AUC) beobachteten Expositionen, im Vergleich zur gleichzeitigen Kontrollgruppe mit Frühgeburten, Fehlgeburten (Aborte und Totgeburten) und einem Anstieg der neonatalen Todesfälle assoziiert (siehe Abschnitt "Schwangerschaft, Stillzeit" ).
Tierische Toxikologie und Pharmakologie
Toxizitätsstudien mit wiederholter Verabreichung von Durvalumab an geschlechtsreifen Javaneraffen mit einer Dauer von bis zu 3 Monaten ergaben keinerlei unerwünschte Wirkungen, die als für den Menschen bedeutsam eingestuft worden wären.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
In der Originalverpackung aufbewahren und vor Licht schützen.
Nicht einfrieren.
Nicht schütteln.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Imfinzi enthält keine Konservierungsmittel. Die zubereitete Infusionslösung unverzüglich verabreichen. Falls die Infusionslösung nicht unverzüglich verabreicht, sondern stattdessen aufbewahrt wird, sind die nachstehenden Empfehlungen zu beachten:
-Die chemische und physikalische Stabilität der zubereiteten Infusionslösung im Infusionsbeutel wurde für bis zu 30 Tage bei 2 bis 8 °C und für bis zu 24 Stunden bei Raumtemperatur (nicht über 25 °C) ab dem Zeitpunkt der Zubereitung nachgewiesen.
Aus mikrobiologischer Sicht sollte die zubereitete Infusionslösung im Infusionsbeutel unverzüglich verwendet werden. Wird sie nicht sofort verwendet, so liegen Lagerungsbedingungen und Dauer der Lagerung in der Verantwortung des Anwenders und dürfen im Normalfall nicht mehr als 24 Stunden bei 2 bis 8 °C oder 12 Stunden bei Raumtemperatur (nicht über 25 °C) betragen, es sei denn, die Verdünnung hat unter kontrollierten und validierten aseptischen Bedingungen stattgefunden.
Vorbereitung der Lösung
Imfinzi wird als Einzeldosis-Durchstechflasche geliefert und enthält keinerlei Konservierungsmittel. Aseptische Methoden sind zu beachten.
-Überprüfen Sie das Arzneimittel optisch auf Partikel und Verfärbungen. Imfinzi ist eine durchsichtige bis opaleszierende und farblose bis gelbliche Lösung. Entsorgen Sie Durchstechflaschen, wenn die enthaltene Lösung trüb, verfärbt oder mit Schwebstoffen verunreinigt ist. Durchstechflasche nicht schütteln.
-Entnehmen Sie die benötigte Menge an Lösung aus der Imfinzi-Durchstechflasche und geben Sie diese in einen Beutel mit 0,9 % Natriumchlorid- oder 5 % Dextrose-Lösung zur intravenösen Anwendung. Mischen Sie die verdünnte Lösung durch sanftes Kippen. Die endgültige Konzentration der verdünnten Lösung sollte zwischen 1 und 15 mg/ml liegen. Lösung nicht einfrieren und nicht schütteln.
-Achten Sie auf Sterilität der zubereiteten Lösungen.
-Benutzen Sie die Durchstechflasche nach der Entnahme des Arzneimittels kein zweites Mal; verabreichen Sie stets nur eine Dosis pro Durchstechflasche.
-Entsorgen Sie die gesamte in der Durchstechflasche verbliebene Restflüssigkeit.
Verabreichung
-Die Infusionslösung wird über einen 60-minütigen Zeitraum und über einen intravenösen Zugang verabreicht, in dem sich ein steriler Einlassfilter mit geringer Proteinbindung in einer Stärke von 0,2 oder 0,22 Mikrometer befindet.
-Keine anderen Arzneimittel über den gleichen intravenösen Zugang verabreichen.
Nicht verwendetes Arzneimittel und Abfallmaterial sind entsprechend den geltenden Anforderungen zu entsorgen.
Zulassungsnummer66548 (Swissmedic)
PackungenImfinzi 120 mg/2,4 ml: 1 Durchstechflasche [A]
Imfinzi 500 mg/10 ml: 1 Durchstechflasche [A]
ZulassungsinhaberinAstraZeneca AG, 6340 Baar
Stand der InformationJuni 2026
|