ZusammensetzungAMGEVITA 80 mg, 40 mg und 20 mg Injektionslösung (100 mg/ml)
Wirkstoffe
Adalimumab (aus gentechnisch veränderten Ovarialzellen des Chinesischen Hamsters hergestellt).
Hilfsstoffe
Milchsäure, Saccharose, Polysorbat 80 (aus gentechnisch verändertem Mais hergestellt), Natriumhydroxid (zur pH-Einstellung), Wasser für Injektionszwecke q.s. ad solutionem.
AMGEVITA 40 mg und 20 mg Injektionslösung (50 mg/ml)
Wirkstoffe
Adalimumab (aus gentechnisch veränderten Ovarialzellen des Chinesischen Hamsters hergestellt).
Hilfsstoffe
Eisessig, Saccharose, Polysorbat 80 (aus gentechnisch verändertem Mais hergestellt), Natriumhydroxid (zur pH-Einstellung), Wasser für Injektionszwecke q.s. ad solutionem.
Darreichungsform und Wirkstoffmenge pro EinheitAMGEVITA 80 mg Injektionslösung in einer Fertigspritze
Jeder Einzeldosis-Fertigspritze enthält 80 mg Adalimumab in 0,8 ml (100 mg/ml).
AMGEVITA 80 mg Injektionslösung im Fertigpen
Jeder Einzeldosis-Fertigpen enthält 80 mg Adalimumab in 0,8 ml (100 mg/ml).
AMGEVITA 40 mg Injektionslösung in einer Fertigspritze
Jede Einzeldosis-Fertigspritze enthält 40 mg Adalimumab in 0,8 ml (50 mg/ml) Lösung bzw. 40 mg Adalimumab in 0,4 ml (100 mg/ml).
AMGEVITA 40 mg Injektionslösung im Fertigpen
Jeder Einzeldosis-Fertigpen enthält 40 mg Adalimumab in 0,8 ml (50 mg/ml) Lösung bzw. 40 mg Adalimumab in 0,4 ml (100 mg/ml).
AMGEVITA 20 mg Injektionslösung in einer Fertigspritze (zur Anwendung bei Kindern und Jugendlichen)
Jede Einzeldosis-Fertigspritze enthält 20 mg Adalimumab in 0,4 ml (50 mg/ml) Lösung bzw. 20 mg Adalimumab in 0,2 ml (100 mg/ml).
Indikationen/AnwendungsmöglichkeitenRheumatoide Arthritis
AMGEVITA ist indiziert zur Reduzierung der Anzeichen und Symptome sowie zur Verlangsamung der Progression struktureller Schäden und zur Verbesserung der körperlichen Funktionsfähigkeit bei erwachsenen Patienten mit einer mässig bis stark ausgeprägten aktiven rheumatoiden Arthritis, die nur unzureichend auf krankheitsmodifizierende Antirheumatika (DMARDs) angesprochen haben.
AMGEVITA kann als Monotherapie oder in Kombination mit Methotrexat bzw. anderen krankheitsmodifizierenden Antirheumatika eingesetzt werden, wobei aber die Kombination von Adalimumab mit Ciclosporin, Azathioprin und anderen anti-TNFα-Therapien nicht untersucht worden ist.
Bei Patienten, bei denen eine mässig bis stark ausgeprägte rheumatoide Arthritis erst kürzlich (<3 Jahre) diagnostiziert wurde und die zuvor nicht mit Methotrexat behandelt worden war, konnte eine Wirksamkeit von Adalimumab in Kombination mit Methotrexat gezeigt werden.
Polyartikuläre juvenile idiopathische Arthritis
AMGEVITA ist in Kombination mit Methotrexat indiziert zur Behandlung der aktiven polyartikulären juvenilen idiopathischen Arthritis bei Kindern und Jugendlichen im Alter von 4 bis 17 Jahren, die nur unzureichend auf ein oder mehrere krankheitsmodifizierende Antirheumatika (DMARDs), einschliesslich Methotrexat, angesprochen haben oder eine solche Therapie nicht tolerieren. AMGEVITA kann im Falle einer Unverträglichkeit gegenüber Methotrexat oder, wenn die weitere Behandlung mit Methotrexat nicht möglich ist, als Monotherapie angewendet werden. Bei Kindern, die jünger als 4 Jahre sind, wurde Adalimumab nicht untersucht.
Psoriasis-Arthritis
AMGEVITA ist indiziert zur Reduzierung der Anzeichen und Symptome der Psoriasis-Arthritis bei Patienten mit ungenügendem Ansprechen auf krankheitsmodifizierende Antirheumatika. AMGEVITA reduziert die Progressionsrate struktureller Schäden und verbessert die körperliche Funktionsfähigkeit bei Patienten mit der polyartikulären, symmetrischen Form der Erkrankung. AMGEVITA kann als Monotherapie oder in Kombination mit krankheitsmodifizierenden Antirheumatika angewendet werden.
Ankylosierende Spondylitis (Morbus Bechterew)
AMGEVITA ist indiziert zur Behandlung von erwachsenen Patienten mit aktiver ankylosierender Spondylitis, die nur unzureichend auf herkömmliche Therapien angesprochen haben.
Morbus Crohn
AMGEVITA ist indiziert zur Behandlung von erwachsenen Patienten mit einem Morbus Crohn mit mässiger bis hoher Krankheitsaktivität, die nur unzureichend auf herkömmliche Therapien angesprochen haben, sowie bei erwachsenen Patienten, die nicht mehr auf Infliximab ansprechen oder dieses nicht vertragen.
Morbus Crohn bei Kindern und Jugendlichen
AMGEVITA ist indiziert zur Reduzierung der Anzeichen und Symptome, einschliesslich Induktion und Erhaltung einer klinischen Remission, von Kindern und Jugendlichen (ab dem Alter von 6 Jahren), mit schwerem aktivem Morbus Crohn, die nur unzureichend auf eine konventionelle Therapie, einschliesslich primärer Ernährungstherapie, einem Glukokortikoid und einem Immunsuppressivum angesprochen haben oder eine Unverträglichkeit gegenüber einer solchen Therapie haben oder bei denen eine solche Therapie kontraindiziert ist.
Colitis ulcerosa
AMGEVITA ist indiziert zur Behandlung der mittelschweren bis schweren aktiven Colitis ulcerosa bei erwachsenen Patienten, die auf die herkömmliche Therapie, einschliesslich Glukokortikoide und/oder 6-Mercaptopurin (6-MP) oder Azathioprin (AZA), unzureichend angesprochen haben oder die eine Unverträglichkeit gegen eine solche Therapie haben oder bei denen eine solche Therapie kontraindiziert ist.
Psoriasis
AMGEVITA ist als Monotherapie indiziert zur Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer, chronischer Plaque-Psoriasis, bei denen eine systemische Therapie oder eine PUVA-Therapie angezeigt ist.
Psoriasis bei Kindern und Jugendlichen
AMGEVITA ist indiziert zur Behandlung der schweren chronischen Plaque-Psoriasis bei Kindern und Jugendlichen (ab dem Alter von 6 Jahren), die nur unzureichend auf Phototherapien oder systemische Vortherapien angesprochen haben oder für die diese Therapien nicht geeignet sind.
Hidradenitis suppurativa (Acne inversa)
AMGEVITA ist indiziert zur Behandlung von erwachsenen Patienten mit aktiver mittelschwerer bis schwerer Hidradenitis suppurativa (Acne inversa), die unzureichend auf eine systemische Antibiotikatherapie angesprochen haben.
Uveitis
AMGEVITA ist indiziert zur Induktion (in Kombination mit Kortikosteroiden) und Erhaltung einer Remission bei erwachsenen Patienten mit Kortikosteroid-abhängiger nicht-infektiöser intermediärer Uveitis, posteriorer Uveitis oder Panuveitis, die nur unzureichend auf Kortikosteroide mit oder ohne Immunmodulatoren angesprochen haben oder die eine Kortikoid sparende Behandlung benötigen. Die Kombination mit Kortikosteroiden und/oder Immunmodulatoren richtet sich nach dem anatomischen und funktionellen Verlauf.
Dosierung/AnwendungEs wird empfohlen, dass die Behandlung mit AMGEVITA durch Fachärzte, die über Erfahrung auf dem Gebiet der Diagnose und Behandlung von Rheumatoider Arthritis (RA), der polyartikulären juvenilen idiopathischen Arthritis (pJIA), Ankylosierender Spondylitis, Psoriasis-Arthritis, Psoriasis, Hidradenitis suppurativa oder der nicht-infektiösen Uveitis verfügen, eingeleitet und überwacht wird. Nach einer entsprechenden Schulung zur subkutanen Injektionsmethode können sich die Patienten AMGEVITA selbst injizieren, wenn ihr Arzt das für angebracht hält und die erforderliche medizinische Nachsorge erfolgt. AMGEVITA ist in verschiedenen Präsentationen erhältlich. Alle Präsentationen sind nur für den einmaligen Gebrauch. AMGEVITA 20 mg Fertigspritze ist ausschliesslich zur Anwendung bei Kindern und Jugendlichen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Erwachsene (18-64 Jahre)
Rheumatoide Arthritis
Die empfohlene AMGEVITA Dosis beträgt für erwachsene Patienten mit rheumatoider Arthritis 40 mg Adalimumab, die alle zwei Wochen als Einmalgabe durch subkutane Injektion zu verabreichen ist. Methotrexat, Glukokortikoide, Salicylate, nicht-steroidale entzündungshemmende Mittel, Analgetika oder andere krankheitsmodifizierende Antirheumatika (ausser Ciclosporin, Azathioprin und andere anti-TNFα-Therapien) können während einer Behandlung mit AMGEVITA weiterhin verabreicht werden.
Bei manchen Patienten kann es unter Monotherapie mit Adalimumab zu einer Verminderung der Wirkung des Arzneimittels kommen. In solchen Fällen kann eine Erhöhung der AMGEVITA Dosis auf 40 mg Adalimumab einmal wöchentlich oder 80 mg Adalimumab alle zwei Wochen von Vorteil sein.
Psoriasis-Arthritis
Die empfohlene AMGEVITA Dosis beträgt für Patienten mit Psoriasis-Arthritis 40 mg Adalimumab, die alle zwei Wochen als Einmalgabe durch subkutane Injektion zu verabreichen ist.
Glukokortikoide, Salicylate, nicht-steroidale entzündungshemmende Arzneimittel, Analgetika oder andere krankheitsmodifizierende Antirheumatika können während einer Behandlung mit AMGEVITA weiterhin verabreicht werden.
Ankylosierende Spondylitis (Morbus Bechterew)
Die empfohlene AMGEVITA Dosis beträgt für Patienten mit ankylosierender Spondylitis 40 mg Adalimumab, die alle zwei Wochen als Einmalgabe durch subkutane Injektion zu verabreichen ist.
Die verfügbaren Daten weisen darauf hin, dass ein klinisches Ansprechen üblicherweise innerhalb von 12 Behandlungswochen erreicht wird. Die Fortsetzung der Therapie sollte bei einem Patienten, der innerhalb dieser Zeitspanne nicht anspricht, nochmals sorgfältig überprüft werden.
Nicht-steroidale entzündungshemmende Arzneimittel können während einer Behandlung mit AMGEVITA weiterhin verabreicht werden.
Morbus Crohn
Das empfohlene Dosierungsschema für AMGEVITA bei erwachsenen Patienten mit Morbus Crohn beträgt 160 mg in Woche 0 (die Dosis kann als 160 mg innerhalb eines Tages oder als 80 mg pro Tag an zwei aufeinanderfolgenden Tagen verabreicht werden), 80 mg in Woche 2 und danach jede zweite Woche 40 mg als subkutane Injektion.
Während der Behandlung mit AMGEVITA können Aminosalicylate, Kortikosteroide bzw. immunomodulatorische Mittel (z.B. 6-Mercaptopurin und Azathioprin) weiterhin gegeben werden.
Colitis ulcerosa
Die empfohlene Induktionsdosis für AMGEVITA beträgt bei erwachsenen Patienten mit mittelschwerer bis schwerer Colitis ulcerosa 160 mg in Woche 0 (die Dosis kann als 160 mg innerhalb eines Tages oder als 80 mg pro Tag an zwei aufeinanderfolgenden Tagen verabreicht werden) und 80 mg in Woche 2. Nach der Induktionsbehandlung beträgt die empfohlene Dosis 40 mg als subkutane Injektion jede zweite Woche.
Während der Erhaltungstherapie können Glukokortikoide gemäss den klinischen Empfehlungen ausgeschlichen werden.
Patienten, bei denen nach primärem Ansprechen ein Wirkverlust auftritt, können von einer Erhöhung der AMGEVITA Dosis auf 40 mg einmal wöchentlich oder 80 mg jede zweite Woche profitieren.
Die Fortführung der Therapie ist regelmässig zu reevaluieren. Die vorhandenen Daten legen nahe, dass ein klinisches Ansprechen gewöhnlich innerhalb von 2 bis 8 Behandlungswochen erreicht wird. Bei Patienten, die in dieser Zeit nicht ansprechen, sollte die Behandlung mit AMGEVITA nicht fortgesetzt werden.
Kontrollierte Daten über 52 Wochen hinaus sind nicht verfügbar.
Psoriasis
Die empfohlene AMGEVITA Dosis für erwachsene Patienten mit Psoriasis ist eine Initialdosis von 80 mg Adalimumab als subkutane Injektion verabreicht, gefolgt von 40 mg Adalimumab alle zwei Wochen als subkutane Injektion verabreicht (beginnend 1 Woche nach der Initialdosis).
Die Fortsetzung der Therapie nach 16 Wochen sollte bei Patienten, die innerhalb dieser Zeitspanne nicht ansprechen, abgebrochen werden.
Nach 16 Wochen kann bei Patienten mit sekundärem Verlust einer initial mindestens partiellen Antwort eine Erhöhung der AMGEVITA Dosis auf 40 mg wöchentlich oder 80 mg jede zweite Woche von Nutzen sein. Wird mit einer höheren Dosis ein ausreichendes Ansprechen erreicht, kann eine erneute Reduktion der Dosis auf 40 mg jede zweite Woche in Betracht gezogen werden. Die Fortsetzung der Therapie sollte bei Patienten, die innerhalb von 16 Wochen auch nach Erhöhung der AMGEVITA Dosis auf 40 mg wöchentlich oder 80 mg jede zweite Woche unzureichend ansprechen, abgebrochen werden.
Bei initial ansprechenden Patienten wurde nach Absetzen der Therapie nach 33 Wochen bis zur 52. Woche nur bei etwas mehr als einem Viertel der Patienten ein Rezidiv nach Therapieabbruch beobachtet, so dass nach einer Therapiedauer von 33 Wochen ein Unterbruch der Behandlung erwogen werden sollte.
Das Nutzen-Risiko dieser Therapie sollte periodisch überwacht werden.
Hidradenitis suppurativa
Die empfohlene AMGEVITA Dosis für erwachsene Patienten mit Hidradenitis suppurativa ist eine Initialdosis von 160 mg in Woche 0 (die Dosis kann als 160 mg innerhalb eines Tages oder als 80 mg pro Tag an zwei aufeinanderfolgenden Tagen verabreicht werden), gefolgt von 80 mg in Woche 2. Beginnend in Woche 4 nach der Initialdosis beträgt die Dosis 40 mg wöchentlich oder 80 mg jede zweite Woche. AMGEVITA wird als subkutane Injektion verabreicht.
Eine Antibiotikatherapie kann während der AMGEVITA Behandlung weitergeführt werden falls notwendig. Es wird empfohlen, dass die Patienten während der Behandlung mit AMGEVITA täglich eine topische antiseptische Waschlösung an ihren HS-Läsionen anwenden.
Bei Patienten, die nach 12 Wochen keine Verbesserung zeigen, sollte ein Therapieabbruch in Erwägung gezogen werden.
Nutzen und Risiko der Behandlung sollten regelmässig beurteilt werden.
In klinischen Studien wurde Adalimumab mit oder ohne Antibiotika verwendet.
Sollte die Behandlung unterbrochen werden müssen, kann AMGEVITA erneut verabreicht werden.
Uveitis
Die empfohlene Dosis AMGEVITA für erwachsene Patienten mit Uveitis ist eine Initialdosis von 80 mg, gefolgt von 40 mg alle zwei Wochen, beginnend 1 Woche nach der Initialdosis.
Die Behandlung mit AMGEVITA kann in Kombination mit Kortikosteroiden und/oder anderen nicht biologischen Immunsuppressiva eingeleitet werden. Eine begleitende Anwendung von Kortikosteroiden kann sukzessive nach der Einleitung der Behandlung mit AMGEVITA reduziert werden (siehe auch "Eigenschaften/Wirkungen" ).
Falls innerhalb von 6 Monaten nicht die angestrebte Wirkung erreicht wird, ist die Fortführung der Therapie mit AMGEVITA unter Abwägung des Nutzens und der Risiken sorgfältig zu re-evaluieren.
Ältere Patienten (älter als 65 Jahre)
Eine Dosisanpassung ist nicht erforderlich.
Kinder und Jugendliche
Polyartikuläre juvenile idiopathische Arthritis (4 bis 17 Jahre)
Bei Patienten mit polyartikulärer juveniler idiopathischer Arthritis im Alter von 4-17 Jahren basiert die empfohlene Dosis auf dem Körpergewicht (Tabelle 1). AMGEVITA wird jede zweite Woche subkutan injiziert.
Tabelle 1: AMGEVITA Dosis für Patienten mit polyartikulärer juveniler idiopathischer Arthritis
Körpergewicht Dosierungsschema
10 kg bis <30 kg 20 mg jede zweite Woche
≥30 kg 40 mg jede zweite Woche
Die verfügbaren Daten weisen darauf hin, dass ein klinisches Ansprechen üblicherweise innerhalb von 12 Behandlungswochen erreicht wird. Die Fortsetzung der Therapie sollte bei einem Patienten, der innerhalb dieser Zeitspanne nicht anspricht, nochmals sorgfältig überdacht werden.
Bei Kindern mit polyartikulärer juveniler idiopathischer Arthritis unter 4 Jahren wurde Adalimumab nicht untersucht. Für die Behandlung mit Adalimumab bei pädiatrischen Patienten mit einem Gewicht unter 15 Kilogramm stehen begrenzt Daten zur Verfügung.
Morbus Crohn bei Kindern und Jugendlichen
Die empfohlene Dosis von AMGEVITA bei Kindern und Jugendlichen mit Morbus Crohn basiert auf dem Körpergewicht (Tabelle 2). AMGEVITA wird subkutan injiziert.
Tabelle 2: AMGEVITA Dosis für Kinder und Jugendliche mit Morbus Crohn
Körpergewicht Induktionsdosis Erhaltungsdosis, beginnend in Woche 4
<40 kg 80 mg in Woche 0 und 40 mg in Woche 2 20 mg jede zweite Woche
≥40 kg 160 mg in Woche 0 und 80 mg in Woche 2 40 mg oder 20 mg jede zweite Woche
Für die Behandlung mit Adalimumab bei pädiatrischen Patienten mit einem Gewicht unter 30 Kilogramm stehen begrenzt Daten zur Verfügung. Alternative Dosierungsschemen sind in internationalen Richtlinien empfohlen.
Einige Patienten, die nur unzureichend ansprechen, können von einer Erhöhung der AMGEVITA Dosis profitieren, wenn ein Wiederaufflammen der Erkrankung oder ein ungenügendes Ansprechen während der Erhaltungsdosis beobachtet wird:
-<40 kg Körpergewicht:
-20 mg wöchentlich
-≥40 kg Körpergewicht:
-Falls bisher 40 mg jede zweite Woche verabreicht wurde: 40 mg wöchentlich respektive 80 mg jede zweite Woche oder
-Falls bisher 20 mg jede zweite Woche verabreicht wurde: 20 mg wöchentlich.
Weitere Bemerkungen
Bei anhaltender Remission sollte eine Reduktion der Dosis erwogen werden.
Die Fortsetzung der Therapie sollte bei einem Patienten, der innerhalb von 12 Wochen nicht anspricht, nochmals sorgfältig überprüft werden. Adalimumab wurde bei Kindern mit Morbus Crohn, die jünger als 6 Jahre sind, nicht untersucht.
Psoriasis bei Kindern und Jugendlichen
Die empfohlene AMGEVITA-Dosis für Patienten ab dem Alter von 6 Jahren mit Plaque-Psoriasis basiert auf dem Körpergewicht (Tabelle 3). AMGEVITA wird subkutan injiziert. AMGEVITA ist in verschiedenen Dosierungsstärken und/oder Präsentationen erhältlich.
Tabelle 3: AMGEVITA -Dosis für Kinder und Jugendliche mit Plaque-Psoriasis
Gewicht des Patiente Dosierungsschema
n
15 kg bis <30 kg Anfangsdosis von 20 mg, gefolgt von einer Dosis von 20 mg jede zweite Woche,
beginnend eine Woche nach der Anfangsdosis
≥30 kg Anfangsdosis von 40 mg, gefolgt von einer Dosis von 40 mg jede zweite Woche,
beginnend eine Woche nach der Anfangsdosis
Die Fortsetzung der Therapie länger als 16 Wochen sollte bei Patienten, die innerhalb dieser Zeitspanne nicht ansprechen, sorgfältig abgewogen werden.
Wenn die Wiederaufnahme der Therapie mit AMGEVITA angezeigt ist, sollte bezüglich Dosis und Behandlungsdauer die oben beschriebene Anleitung befolgt werden.
Adalimumab wurde bei Kindern mit Plaque-Psoriasis, die jünger als 6 Jahre sind, nicht untersucht.
Die Sicherheit und Wirksamkeit von Adalimumab bei pädiatrischen Patienten für andere Indikationen ausser pJIA, pädiatrischem Morbus Crohn und pädiatrischer Psoriasis wurde nicht untersucht.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Aktive Tuberkulose oder andere schwere Infektionen wie Sepsis und opportunistische Infektionen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Mittelschwere bis schwere Herzinsuffizienz (NYHA Klasse III-IV).
Warnhinweise und VorsichtsmassnahmenInfektionen
Wie bei anderen TNF-Antagonisten müssen die Patienten vor, während und nach einer Therapie mit AMGEVITA im Hinblick auf das Auftreten von Infektionen (einschliesslich Tuberkulose) engmaschig überwacht werden.
Es wird empfohlen, eine Behandlung mit AMGEVITA bei Patienten mit aktiven Infektionen (einschliesslich chronischer bzw. Örtlich beschränkter Infektionen) nicht einzuleiten, bis diese Infektionen unter Kontrolle sind.
Patienten, die unter der Therapie mit AMGEVITA eine neue Infektion entwickeln, sind engmaschig zu überwachen und müssen einer vollständigen Untersuchung unterzogen werden.
Wenn sich bei einem Patienten eine neue schwere Infektion entwickelt, wird empfohlen, die Gabe von AMGEVITA abzubrechen, bis die Infektion unter Kontrolle gebracht wurde. Bei Patienten mit rezidivierenden Infektionen in der Anamnese bzw. mit Grundbeschwerden, die diese Patienten für Infektionen prädisponieren könnten, sollte der Arzt eine Anwendung von AMGEVITA sorgfältig abwägen.
In den klinischen Uveitis-Studien wurden unter Adalimumab infektiöse Iridozyklitiden und Herpes simplex-Infektionen des Auges beobachtet.
Eine regelmässige ophthalmologische Kontrolle ist daher angezeigt, und bei entsprechendem klinischem Verdacht ist eine Herpes simplex Infektion auszuschliessen. Bei Auftreten solcher unerwünschter Wirkungen ist die Fortführung von AMGEVITA wiederzuerwägen.
Okulare Tuberkulose- oder Syphilis-Infektionen können eine nicht-infektiöse Uveitis vortäuschen und sind vor Behandlung mit AMGEVITA auszuschliessen.
Schwere Infektionen, aufgrund bakterieller, mykobakterieller, invasiver Pilzinfektionen (disseminierte oder extrapulmonale Histoplasmose, Blastomykose, Aspergillose, Kokzidioidomykose), viraler, parasitärer oder anderer opportunistischer Infektionen, wurden im Zusammenhang mit anti-TNFα-Therapien berichtet.
Sepsis, Candidamykose, Listeriose, Legionellose und Pneumocystis-Infektion sind ebenfalls im Zusammenhang mit anti-TNFα-Therapien, einschliesslich Adalimumab, beschrieben worden. Hospitalisierungen und Todesfälle im Zusammenhang mit Infektionen wurden gemeldet. Viele der schweren Infektionen sind bei Patienten aufgetreten, die gleichzeitig eine immunsuppressive Therapie erhielten, die sie, zusätzlich zu ihrer Grunderkrankung, für Infektionen besonders anfällig gemacht haben könnte.
Tuberkulose
Tuberkulose, einschliesslich Reaktivierung und Neuauftreten von Tuberkulose, ist im Zusammenhang mit Adalimumab beschrieben worden. Die Berichte enthielten Fälle von pulmonaler und extrapulmonaler (z.B. disseminierter) Tuberkulose.
Bevor eine Therapie mit AMGEVITA eingeleitet wird, sind alle Patienten sowohl auf eine aktive als auch auf eine inaktive (latente) Tuberkuloseinfektion zu untersuchen. Es wird empfohlen, dass diese Untersuchung eine genaue medizinische Anamnese enthält, aus der ein eventueller früherer Kontakt mit Personen mit einer aktiven Tuberkulose sowie eine frühere bzw. laufende immunsuppressive Therapie hervorgeht. Die Durchführung geeigneter Screening-Tests (z.B. die Röntgenaufnahme des Thorax und ein Tuberkulintest) im Einklang mit den örtlichen Empfehlungen wird empfohlen. Die Behandlung der aktiven Tuberkulose soll vor Beginn der Therapie mit AMGEVITA eingeleitet werden. Wenn ein Tuberkulin-Hauttest für eine latente Tuberkulose durchgeführt wird, soll eine Induration von 5 mm oder grösser als positiv gewertet werden, selbst dann, wenn vorgängig mit Bacille Calmette Guerin (BCG) geimpft wurde.
Die Möglichkeit einer unentdeckten Tuberkulose soll besonders bei Patienten beachtet werden, welche aus Ländern mit einer hohen Tuberkuloseprävalenz eingewandert sind oder solche bereist haben, wie auch bei Patienten mit engem Kontakt zu Personen mit aktiver Tuberkulose.
Im Falle der Diagnose einer latenten Tuberkulose muss eine Tuberkuloseprophylaxe entsprechend den örtlichen Empfehlungen eingeleitet werden, bevor die Therapie mit AMGEVITA begonnen wird. Die Einleitung einer Tuberkuloseprophylaxe vor einer Therapie mit AMGEVITA soll ebenfalls bei Patienten erwogen werden, welche trotz negativem Tuberkulosetest mehrere oder signifikante Risikofaktoren für eine Tuberkuloseinfektion haben und bei Patienten mit einer latenten oder aktiven Tuberkulose in der medizinischen Anamnese, bei denen eine adäquate Behandlung nicht bestätigt werden kann. Die Entscheidung eine Tuberkulosetherapie bei diesen Patienten einzuleiten soll sowohl vom Risiko für eine latente Tuberkuloseinfektion als auch vom Risiko der Anti-Tuberkulosetherapie abhängen. Falls nötig soll ein Arzt resp. eine Ärztin mit Erfahrung in der Tuberkulosebehandlung konsultiert werden.
Eine Anti-Tuberkulose Therapie bei latenter Tuberkuloseinfektion vermindert das Risiko einer Tuberkulosereaktivierung bei Patienten unter AMGEVITA Therapie. Trotz Tuberkuloseprophylaxe sind Fälle von reaktivierter Tuberkulose bei Patienten aufgetreten, die mit Adalimumab behandelt wurden. Ebenfalls haben Patienten mit einem negativen Screening auf latente Tuberkulose eine aktive Tuberkulose entwickelt und einige Patienten, welche vorgängig erfolgreich gegen eine aktive Tuberkulose behandelt wurden, entwickelten erneut eine Tuberkulose unter der Behandlung mit TNF-Antagonisten.
Patienten, welche AMGEVITA erhalten, müssen auf Zeichen und Symptome einer aktiven Tuberkulose überwacht werden, insbesondere weil der Test auf latente Tuberkulose eventuell falsch negativ ausfallen könnte. Besonders bei Patienten, welche schwer krank oder immunsupprimiert sind, kann der Tuberkulintest falsch negativ ausfallen.
Die Patienten sind darauf hinzuweisen, ärztlichen Rat einzuholen, wenn während oder nach der Behandlung mit AMGEVITA Anzeichen/Symptome auftreten, die auf eine Tuberkulose hindeuten (z.B. anhaltender Husten, Auszehrung/Gewichtsverlust, niedriges Fieber, Apathie).
Andere opportunistische Infektionen
Opportunistische Infektionen, inklusive invasive Pilzinfektionen, wurden unter Adalimumab beobachtet. Diese Infektionen werden bei Patienten, die eine anti-TNFα-Therapie erhalten, nicht durchwegs erkannt, dies hat zu verspäteten Behandlungen manchmal mit Todesfällen geführt.
Patienten, die eine anti-TNFα-Therapie erhalten, sind anfälliger auf schwere Pilzinfektionen, wie Histoplasmose, Kokzidioidomykose, Blastomykose, Aspergillose, Candidamykose und andere opportunistische Infektionen. Patienten, die Fieber oder Unwohlsein, Gewichtsverlust, Schwitzen, Husten, Atemnot und/oder Lungeninfiltrate oder andere systemische Krankheiten entwickeln, mit oder ohne gleichzeitigem Schock, müssen vom Arzt unmittelbar untersucht werden.
Patienten, die in Gebieten wohnen, in denen endemische Mykosen vorkommen, oder solche bereist haben, sollen bei invasiven Pilzinfektionen auf die Entwicklung von Zeichen und Symptomen einer möglichen systemischen Pilzinfektion überwacht werden. Für die Patienten besteht die Gefahr einer Histoplasmose oder einer anderen invasiven Pilzerkrankung, daher soll eine empirische, antimykotische Behandlung erwogen werden bis der/die Krankheitserreger identifizert worden ist/sind.
Antigen- und Antikörptertests auf Histoplasmose können bei einigen Patienten mit aktiver Infektion negativ ausfallen.
Wenn möglich sollte die Entscheidung für eine empirische antimykotische Therapie bei diesen Patienten in Absprache mit einem in Diagnostik und Behandlung von invasiven Pilzerkrankungen erfahrenen Arzt getroffen werden, dabei sind sowohl das Risiko einer schweren Pilzinfektion als auch die Risiken einer antimykotischen Therapie zu berücksichtigen.
Patienten, welche eine schwere Pilzinfektion entwickeln, sind angehalten, die anti-TNFα-Therapie abzubrechen, bis die Infektionen unter Kontrolle gebracht sind.
Hepatitis B Reaktivierung
Die Anwendung von TNF-Antagonisten, einschliesslich Adalimumab, wurde mit Reaktivierung von Hepatitis B Virus (HBV) bei Patienten in Verbindung gebracht, welche chronische Träger dieses Virus sind. In einigen Fällen ist die HBV Reaktivierung unter TNF-Antagonisten tödlich verlaufen. Die meisten dieser Fälle sind bei Patienten aufgetreten, die gleichzeitig andere immunsuppressive Arzneimittel erhalten haben, was ebenfalls zu einer HBV Reaktivierung beitragen kann. Patienten mit einem Risiko für eine HBV Infektion sollten vor Beginn der Therapie mit TNF-Antagonisten auf Hinweise einer HBV Infektion untersucht werden. TNF-Antagonisten sollten mit Vorsicht an Patienten verschrieben werden, die als Träger von HBV identifiziert wurden. Patienten, die Träger von HBV sind und eine Behandlung mit TNF-Antagonisten benötigen, sollen auf Anzeichen und Symptome der aktiven HBV Infektion während und für einige Monate nach Abschluss der Therapie engmaschig überwacht werden. Ausreichende Daten bezüglich Sicherheit und Wirksamkeit bei Patienten, welche Träger von HBV sind, die gleichzeitig antiviral und mit TNF-Antagonisten behandelt werden, um die Reaktivierung einer Hepatitis B zu verhindern, sind nicht vorhanden. Bei Patienten mit einer HBV Reaktivierung sollte AMGEVITA gestoppt werden und eine wirkungsvolle antivirale Therapie sollte eingeleitet werden.
Neurologische Ereignisse
TNF-Antagonisten, u.a. auch Adalimumab, sind in seltenen Fällen mit Neuauftreten oder einer Verschlechterung der klinischen Symptome bzw. dem radiologischen Nachweis demyelinisierender Erkrankungen des ZNS, einschliesslich Multipler Sklerose (wie z.B. Parästhesie und Störungen der Augenfunktion, siehe "Unerwünschte Wirkungen" ), optischer Neuritis und peripherer demyelinisierender Erkrankungen, einschliesslich Guillain-Barré-Syndrom, in Zusammenhang gebracht worden. Der verschreibende Arzt sollte eine Anwendung von AMGEVITA bei Patienten mit bereits bestehenden oder vor kurzem eingetretenen demyelinisierenden Störungen des zentralen oder peripheren Nervensystems mit Sorgfalt abwägen. Bei Auftreten einer solchen Störung ist ein Absetzen von AMGEVITA in Betracht zu ziehen.
Es besteht ein bekannter Zusammenhang zwischen der intermediären Uveitis und demyelinisierenden Erkrankungen des Zentralnervensystems, einschliesslich Multipler Sklerose. Patienten mit nicht-infektiöser intermediärer Uveitis sind vor Beginn einer Behandlung mit AMGEVITA auf das Vorliegen einer demyelinisierenden Erkrankung des Zentralnervensystems zu untersuchen.
Allergische Reaktionen
Allergische Reaktionen (wie z.B. allergischer Hautausschlag, anaphylaktoide Reaktion, fixes Arzneimittelexanthem, unspezifische Arzneimittelreaktion, Urtikaria, angioneurotisches Oedem), die mit Adalimumab in Verbindung gebracht wurden, wurden im Verlaufe der klinischen Prüfungen nur gelegentlich beobachtet. Spontanmeldungen nach Markteinführung über schwerwiegende allergische Reaktionen, einschliesslich Anaphylaxie, wurden nach der Verabreichung von Adalimumab erhalten. Für den Fall, dass eine anaphylaktische Reaktion bzw. eine andere schwere allergische Reaktion auftreten sollte, wird empfohlen, die Gabe von AMGEVITA sofort abzubrechen und eine geeignete Therapie einzuleiten.
Maligne Tumore
In den kontrollierten Phasen der klinischen Studien mit TNF-Antagonisten wurden bei Patienten, die TNF-Antagonisten erhielten mehr Fälle von malignen Tumoren einschliesslich Lymphome beobachtet als in den Kontrollgruppen. Die Grösse der Kontrollgruppe und die begrenzte Dauer der kontrollierten Phasen von Studien lassen jedoch keine sicheren Schlussfolgerungen zu. In Patienten mit hochaktiver, entzündlicher Erkrankung ist zudem das Grundrisiko für Lymphome erhöht, wodurch eine Risikoabschätzung erschwert wird.
Bei Kindern und Jugendlichen, die eine Behandlung mit TNF-Blockern erhielten, wurden maligne Tumore, davon einige mit tödlichem Verlauf, gemeldet. Etwa die Hälfte dieser Fälle waren Lymphome, inklusive Hodgkin's und non-Hodgkin's Lymphome. Die übrigen Fälle umfassten verschiedene andere maligne Tumore, einschliesslich seltener maligner Tumore, die üblicherweise im Zusammenhang mit Immunsuppression stehen. Die malignen Tumore traten median nach 30 Monaten Therapie auf. Die meisten Patienten erhielten gleichzeitig Immunsuppressiva. Bei diesen Fällen handelt es sich um Post-Marketing-Meldungen aus verschiedenen Quellen, inklusive Register und spontane Post-Marketing-Meldungen.
Es liegen sehr seltene Post-Marketing Berichte von hepatosplenischem T-Zell Lymphom (HSTCL) bei Patienten welche mit Adalimumab behandelt wurden, vor. HSTCL ist ein seltenes aggressives Lymphom, welches oft einen tödlichen Verlauf hat. Es handelt sich in einigen Fällen um junge Erwachsene, welche vorgängig mit Infliximab zusammen mit Azathioprin oder 6-Mercaptopurin bei entzündlichen Darmerkrankungen behandelt wurden. Das potenzielle Risiko einer gleichzeitigen Verabreichung von Azathioprin oder 6-Mercaptopurin mit AMGEVITA sollte sorgfältig abgewogen werden. Der kausale Zusammenhang von HSTCL mit Adalimumab ist nicht geklärt, kann aber nicht ausgeschlossen werden.
Aufgrund des aktuellen Wissensstands kann eine mögliche Gefahr für die Entwicklung von Lymphomen oder anderen malignen Tumoren bei Patienten, die mit einem TNF-Antagonisten behandelt werden, nicht ausgeschlossen werden. Alle Patienten, insbesondere Patienten mit einer intensiven immunsuppressiven Therapie in der Vorgeschichte oder Psoriasis-Patienten, die zuvor eine PUVA-Therapie erhalten haben, sollten vor und während der Behandlung mit AMGEVITA auf das Vorliegen von nicht-melanomartigen Hauttumoren untersucht werden.
Im Zusammenhang mit dem Post-Marketing-Einsatz von TNF-Blockern bei rheumatoider Arthritis und anderen Indikationen, wurden Fälle von akuter und chronischer Leukämie berichtet. Patienten mit rheumatoider Arthritis weisen möglicherweise ein höheres Risiko (bis 2mal höher) für die Entwicklung einer Leukämie auf als die übrige Bevölkerung, dies auch ohne Behandlung mit TNF-Blockern.
Nach der aktuellen Datenlage ist nicht bekannt, ob eine Adalimumab-Behandlung das Risiko für die Entwicklung von Dysplasien oder Kolonkrebs beeinflusst. Alle Patienten mit Colitis ulcerosa, die ein erhöhtes Risiko für Dysplasien oder Kolonkarzinom haben (z.B. Patienten mit lange bestehender Colitis ulcerosa oder primär sklerosierender Cholangitis), oder die eine Vorgeschichte für Dysplasie oder Kolonkarzinom hatten, sollten vor der Therapie und während des Krankheitsverlaufs in regelmässigen Intervallen auf Dysplasien untersucht werden. Die Untersuchung sollte Koloskopie und Biopsien entsprechend der nationalen Empfehlungen umfassen.
Intraokulare Lymphome können eine Uveitis vortäuschen und sind vor Behandlung mit AMGEVITA in Uveitis Patienten auszuschliessen.
Immunsuppression
Im Rahmen einer Studie, in der 64 Patienten mit rheumatoider Arthritis mit Adalimumab behandelt wurden, gab es keine Hinweise auf eine Abschwächung der Überempfindlichkeitsreaktion vom Spättyp, eine Senkung der Immunglobulinspiegel oder auf eine Änderung bei der Enumeration der Effektor T- und B-Zellen sowie der NK-Zellen, der Monozyten/Makrophagen und der Neutrophilen.
Impfungen
In einer randomisierten, doppelblinden, Placebo-kontrollierten Studie wurden die Antikörperantworten auf gleichzeitig verabreichte Pneumokokken- und Influenza-Impfungen (H1N1, H3N2, B) bei 226 erwachsenen Patienten mit rheumatoider Arthritis, die mit Adalimumab behandelt wurden, beurteilt. 86% der Patienten in der Adalimumab-Gruppe erreichten schützende Antikörpertiter gegen mindestens 3 von 5 Pneumokokken-Antigene, verglichen mit 82% in der Placebo-Gruppe. Insgesamt 37% der mit Adalimumab behandelten Patienten und 40% der mit Placebo behandelten Patienten erreichten einen mindestens zweifachen Antikörpertiter-Anstieg gegen mindestens 3 von 5 Pneumokokken-Antigene. In der gleichen Studie erreichten 98% der Patienten in der Adalimumab-Gruppe und 95% in der Placebo-Gruppe schützende Antikörpertiter gegen mindestens 2 von 3 Influenza-Antigene. Insgesamt 52% der mit Adalimumab behandelten Patienten und 63% der mit Placebo behandelten Patienten erreichten einen mindestens vierfachen Antikörpertiter-Anstieg gegen mindestens 2 von 3 Influenza-Antigene.
Bei pädiatrischen Patienten wird empfohlen, nach Möglichkeit vor Therapiebeginn mit AMGEVITA alle Immunisierungen in Übereinstimmung mit den gegenwärtigen Richtlinien auf den aktuellen Stand zu bringen.
Mit Ausnahme von Lebendvakzinen können Patienten unter AMGEVITA geimpft werden. Da keine entsprechenden Daten verfügbar sind, wird eine gleichzeitige Gabe von Lebendvakzinen und AMGEVITA nicht empfohlen. Zur Sekundärübertragung einer Infektion bei gleichzeitiger Gabe von Lebendvakzinen und Adalimumab sind keine Daten verfügbar.
Die Verabreichung von Lebendvakzinen (z.B. BCG-Vakzine) an Neugeborene, die in utero gegenüber Adalimumab exponiert waren, wird nicht empfohlen während 5 Monaten nach der letzten Verabreichung von AMGEVITA in der Schwangerschaft.
Herzinsuffizienz
In klinischen Studien mit anderen TNF-Antagonisten wurden eine Verschlechterung der Herzinsuffizienz und eine Erhöhung der Mortalität aufgrund von Herzinsuffizienz beobachtet. Fälle von Verschlechterung einer dekompensierten Herzinsuffizienz wurden auch bei Patienten unter Adalimumab-Therapie berichtet. AMGEVITA sollte bei Patienten mit leichter Herzinsuffizienz (NYHA Klasse I–II) mit Vorsicht angewendet werden. AMGEVITA ist bei Patienten mit mittelschwerer bis schwerer Herzinsuffizienz (NYHA Klasse III-IV) kontraindiziert. Die Therapie mit AMGEVITA ist bei Patienten mit neu auftretenden oder sich verschlimmernden Symptomen der Herzinsuffizienz abzubrechen.
Gleichzeitige Anwendung von biologischen krankheits-modifizierenden Antirheumatika (DMARDS) oder anderen TNF-Antagonisten
Bei der gleichzeitigen Anwendung von Anakinra mit Etanercept, einem anderen anti-TNFα-Therapeutikum, wurden in klinischen Studien schwerwiegende Infektionen beobachtet. Die Kombination brachte kein Vorteil gegenüber der Anwendung von Etanercept alleine.
Aufgrund der Art der unerwünschten Wirkung bei der Kombination von Etanercept und Anakinra besteht die Möglichkeit, dass vergleichbare Effekte auch bei der gleichzeitigen Anwendung von Anakinra mit anderen anti-TNFα-Therapeutika auftreten.
Die Kombination von Adalimumab mit Anakinra wird deshalb nicht empfohlen.
Die gleichzeitige Anwendung von Adalimumab mit anderen biologischen DMARDS (z.B. Anakinra oder Abatacept) oder anderen TNF-Antagonisten wird aufgrund des möglichen Risikos für Infektionen und andere mögliche pharmakologische Interaktionen nicht empfohlen.
Hämatologische Ereignisse
Seltene Fälle von Panzytopenie einschliesslich aplastischer Anämie wurden unter TNF-Antagonisten gemeldet. Unerwünschte Wirkungen bezüglich hämatologischem System einschliesslich medizinisch signifikanter Zytopenie (z.B. Thrombozytopenie, Leukopenie) unter Adalimumab wurden gemeldet. Alle Patienten sollten angewiesen werden, unverzüglich einen Arzt aufzusuchen, falls sie unter AMGEVITA Anzeichen und Symptome, welche auf eine Blut-Dyskrasie hindeuten (z.B. anhaltendes Fieber, Blutergüsse, Blutungen, Blässe), entwickeln. Ein Absetzen der AMGEVITA Therapie sollte bei Patienten mit bestätigten hämatologischen Abnormalitäten in Betracht gezogen werden.
Auto-Antikörper
Die Behandlung mit AMGEVITA kann zur Bildung von Auto-Antikörpern führen. Die Auswirkung einer Langzeitbehandlung mit Adalimumab auf die Entwicklung von Autoimmunerkrankungen ist nicht bekannt. Falls Patienten unter AMGEVITA Behandlung lupusähnliche Symptome entwickeln, sollte die Behandlung mit AMGEVITA gestoppt werden (siehe "Unerwünschte Wirkungen" , "Auto-Antikörper" ).
Antikörper gegen Adalimumab
(Siehe unter "Unerwünschte Wirkungen" ).
Anwendung in der Geriatrie
Die Häufigkeit von schweren Infektionen war bei mit Adalimumab behandelten Patienten über 65 Jahre grösser, als bei Patienten unter 65 Jahre. Von den insgesamt in klinischen Studien mit Adalimumab behandelten Patienten waren 9.6% 65jährig oder älter, etwa 2.0% waren 75jährig oder älter. Da bei älteren Patienten im Allgemeinen eine höhere Inzidenz für Infektionen besteht, ist bei der Behandlung von älteren Patienten Vorsicht geboten.
Warnhinweise für sonstige Bestandteile
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro 0,2 ml / 0,4 ml / 0,8 ml Dosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenDie Wirkung von Adalimumab wurde bei Patienten mit rheumatoider Arthritis, die gleichzeitig Methotrexat einnahmen, untersucht. Die erhaltenen Daten geben keinen Hinweis darauf, dass entweder bei Adalimumab oder bei Methotrexat eine Dosisanpassung angebracht wäre (siehe "Pharmakokinetik" ).
Adalimumab wurde bei Patienten mit rheumatoider Arthritis, polyartikulärer juveniler idiopathischer Arthritis und Psoriasis-Arthritis sowohl als Monotherapie, als auch in der Kombination mit Methotrexat untersucht. Die Bildung von Antikörpern war bei gleichzeitiger Anwendung von Adalimumab und Methotrexat niedriger als unter Monotherapie. Die Anwendung von Adalimumab ohne Methotrexat führte zu einer gesteigerten Bildung von Antikörpern, einer erhöhten Clearance und einer verminderten Wirksamkeit von Adalimumab.
Ausser mit Methotrexat wurden Interaktionen zwischen Adalimumab und anderen Arzneimitteln im Rahmen von pharmakokinetischen Studien nicht untersucht. Im Rahmen der klinischen Prüfungen zur Behandlung der rheumatoiden Arthritis wurden keine Interaktionen beobachtet, wenn Adalimumab zusammen mit häufig eingesetzten krankheitsmodifizierenden Arzneimitteln gegen Rheumatismus (Sulfasalazin, Hydroxychloroquin, Leflunomid und parenterales Gold), Glukokortikoiden, Salicylaten, nicht-steroidalen entzündungshemmenden Mitteln bzw. mit Analgetika verabreicht wurde. Nicht untersucht wurde die Kombination von Adalimumab mit Ciclosporin, Azathioprin und anderen anti-TNFα-Therapien.
Schwangerschaft, StillzeitEs liegt nur eine beschränkte Anzahl klinischer Daten zu einer Exposition mit Adalimumab bei Schwangeren vor.
In einer prospektiven Kohorte des Schwangerschaftsexpositionsregisters wurden 257 Frauen mit rheumatoider Arthritis oder Morbus Crohn, welche mindestens während des ersten Trimesters mit Adalimumab behandelt worden sind und 120 Frauen mit rheumatoider Arthritis oder Morbus Crohn, welche nicht mit Adalimumab behandelt worden sind, eingeschlossen.
Die Rate schwerwiegender Geburtsfehler (primärer Endpunkt) bei allen Schwangerschaften, ausgenommen Lost to follow-up, betrug bei den mit Adalimumab behandelten Frauen 10,1% (25/247) und bei den unbehandelten Frauen 8,1% (9/111). Die limitierten Daten aus dem Schwangerschaftsexpositionsregister weisen auf kein Muster schwerer Geburtsfehler hin. Unterschiede zwischen den Expositionsgruppen hatten möglicherweise Auswirkungen auf das Auftreten von Geburtsfehlern. In Bezug auf die sekundären Endpunkte spontaner Abort, geringfügige Geburtsfehler, Frühgeburt, Körpergrösse bei der Geburt und schwere oder opportunistische Infektionen gab es keine eindeutigen Unterschiede zwischen mit Adalimumab behandelten und unbehandelten Frauen. Es wurden keine Totgeburten oder maligne Erkrankungen berichtet. Die Auswertung der Daten kann durch die methodologischen Limitationen des Registers beeinflusst sein, darunter kleine Stichproben-Grösse und ein nicht-randomisiertes Design.
Aufgrund seiner inhibitorischen Wirkung auf TNFα könnte Adalimumab, bei einer Anwendung während der Schwangerschaft, Auswirkungen auf die normalen Immunreaktionen des Neugeborenen haben.
Adalimumab soll nur während der Schwangerschaft angewendet werden, wenn es klar notwendig ist.
Frauen im gebärfähigen Alter sollten während der Behandlung und fünf Monate nach der Behandlung mit Adalimumab die Anwendung der geeigneten Empfängnisverhütungsmethoden in Betracht ziehen.
Die Verabreichung von Lebendvakzinen (z.B. BCG-Vakzine) an Neugeborene, die in utero gegenüber Adalimumab exponiert waren, wird nicht empfohlen während 5 Monaten nach der letzten Verabreichung von AMGEVITA in der Schwangerschaft.
Anwendung in der Stillzeit
Begrenzte Informationen aus drei Fällen aus publizierter Literatur deuten darauf hin, dass Adalimumab in sehr tiefen Konzentrationen in die Muttermilch ausgeschieden wird und dort in Konzentrationen von 0,1% bis 1% des mütterlichen Serums vorliegt. Die Entwicklungs- und Gesundheitsnutzen des Stillens sollten ebenso in Betracht gezogen werden wie die klinische Notwendigkeit der Adalimumab-Behandlung für die Mutter und jegliche potentielle Nebenwirkungen auf das gestillte Kind durch Adalimumab oder durch die zugrunde liegende mütterliche Erkrankung.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt.
Unerwünschte WirkungenKlinische Studien
Adalimumab wurde bei 9238 Patienten in kontrollierten und offenen Studien über einen Zeitraum von bis zu 60 Monaten oder länger untersucht. Die Daten basieren auf den pivotalen kontrollierten Studien und umfassen 5963 mit Adalimumab behandelte Patienten und 3689 Patienten, die während der kontrollierten Studienphase Placebo oder eine aktive Vergleichssubstanz erhielten.
Der Anteil der Patienten, die die Behandlung während der doppelblinden, kontrollierten Phase der pivotalen Studien aufgrund unerwünschter Ereignisse abbrachen, betrug 6,0% in der Adalimumab-Gruppe und 5,5% in der Kontroll-Gruppe.
Unerwünschte Wirkungen (sowohl klinische als auch Laborwerte betreffend) in klinischen Studien und aus Spontanmeldungen nach der Marktzulassung mit Adalimumab bei Erwachsenen und pädiatrischen Patienten sind unten nach Organsystem und Häufigkeit geordnet (sehr häufig ≥1/10; häufig ≥1/100, <1/10; gelegentlich ≥1/1000, <1/100; selten ≥1/10'000, <1/1000 und Einzelfälle (Häufigkeit kann anhand der verfügbaren Daten nicht geschätzt werden)) aufgelistet. Spezifische Angaben für Kinder sind unten im Anschluss an die generelle Auflistung angefügt. Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Basierend auf der Gruppierung medizinisch ähnlicher Begriffe wurden alle Ereignisse, die zumindest in möglichem Kausalzusammenhang mit der Behandlung mit Adalimumab stehen, für die Berechnung der Häufigkeit eingeschlossen. Die bei den einzelnen Indikationen in den klinischen Studien beobachtete höchste Häufigkeit der unerwünschten Wirkungen wird aufgeführt. Ein * hinter Organsystem weist darauf hin, dass weitere Informationen in den Rubriken "Kontraindikationen" , "Warnhinweise und Vorsichtsmassnahmen" sowie "Unerwünschte Wirkungen" zu finden sind. Ein ** weist darauf hin, dass die unerwünschte Wirkung in Patienten, welche mit einem TNF-Antagonisten (inkl. Adalimumab) behandelt werden, auftritt. Prozentualangaben sind bei sehr häufigen unerwünschten Wirkungen ergänzt.
Unerwünschte Wirkungen in Klinischen Studien und aus Spontanmeldungen
Infektionen und parasitäre Erkrankungen*
Häufig: Infektionen des Respirationstraktes (einschliesslich Infektionen des unteren und oberen Respirationstraktes, Pneumonie, Sinusitis, Pharyngitis, Nasopharyngitis und virale Herpes Pneumonie); Mundinfektionen (einschliesslich Herpes simplex und oraler Herpes), Haut- und Weichteilinfektionen (einschliesslich Paronychie, Impetigo, nektrotisierende Fasciitis, Panniculitis und Herpes zoster), Harnwegsinfektionen (einschliesslich Pyelonephritis), systemische Infektionen (einschliesslich Sepsis und Candidiasis).
Gelegentlich: Ohrinfektionen, intestinale Infektionen (einschliesslich Hepatitis, virale Gastroenteritis), Gelenkinfektionen, Genitaltraktinfektionen (einschliesslich vulvovaginale Pilzinfektionen), Pilzinfektionen, bakterielle Infektionen, Abszess.
Selten: Abdominale Infektionen (einschliesslich Divertikulitis), Augeninfektionen (einschliesslich Herpes simplex), opportunistische Infektionen und Tuberkulose (einschliesslich Histoplasmose und Infektion mit Mycobakterium avium-Komplex), virale Meningitis, schwere parasitäre Infektionen.
Gutartige, bösartige und unspezifische Neubildungen (einschliesslich Zysten und Polypen)*
Gelegentlich: Gutartige Neoplasie.
Selten: Lymphom, malignes Melanom, Hautkrebs, ausgenommen Melanom (einschliesslich Plattenepithelkarzinom), solide Organneoplasien (einschliesslich Brust-, Eierstock- und Hodenkrebs), Leukämie.
Einzelfälle: Merkelzellkarzinom (kutanes neuroendokrines Karzinom), Hepatosplenisches T-Zell Lymphom.
Erkrankungen des Blutes und des Lymphsystems*
Häufig: Leukopenie (einschliesslich Neutropenie und Agranulozytose).
Gelegentlich: Anämie, Thrombozytopenie, Lymphadenopathie, Leukozytose.
Selten: Idiopathische thrombozytopenische Purpura, Panzytopenie.
Erkrankungen des Immunsystems*
Gelegentlich: Überempfindlichkeit, schwerwiegende allergische Reaktionen, einschliesslich Anaphylaxie und angioneurotisches Oedem.
Selten: Sarkoidose, Allergien (einschliesslich saisonale Allergie).
Endokrine Erkrankungen
Selten: Hypothyreose, Struma.
Stoffwechsel und Ernährungsstörungen
Gelegentlich: Erhöhte Blutfettwerte, Hypokaliämie, Anorexie, gesteigerter Appetit, Hyperglykämie.
Selten: Abnorme Blut-Natriumwerte, Hyperkalzämie, Hypokalzämie, Hyperurikämie.
Psychiatrische Erkrankungen
Gelegentlich: Stimmungsschwankungen (einschliesslich Depression), Ängstlichkeit (einschliesslich Nervosität und Agitation), Schlaflosigkeit.
Erkrankungen des Nervensystems*
Häufig: Kopfschmerz, Parästhesien, Benommenheit.
Gelegentlich: Bewusstseinsstörungen, Tremor, zerebrovaskuläre Ereignisse.
Selten: Demyelinisierende Erkrankungen (z.B. Multiple Sklerose, Optikusneuritus, Guillain-Barré-Syndrom), Reflexsstörungen, Lumbago-Ischias-Syndrom, Synkope, Gleichgewichtsstörungen, Aufmerksamkeitsstörungen, Gesichtslähmung.
Augenerkrankungen
Gelegentlich: Konjunctivits, Augenschwellung, Sehbehinderung, Glaukom.
Selten: Blepharitis, Iritis, Panophthalmie, Iridozyklitis.
Erkrankungen des Ohrs und des Labyrinths
Gelegentlich: Ohrbeschwerden (einschliesslich Schmerz und Schwellung), Schwindel, Tinnitus.
Selten: Taubheit.
Herzerkrankungen*
Gelegentlich: Tachykardie, Herzklopfen, Myokardinfarkt.
Selten: Arrhythmie, Vorhofflimmern, Koronarinsuffizienz, Herzgeräusche, Herzstillstand, dekompensierte Herzinsuffizienz, Perikarderguss.
Gefässerkrankungen
Gelegentlich: Hypertonie, Flush.
Selten: Aortenstenose, arterielle Verschlusskrankheit, Vasculitis, Hämatome, Lymphödeme, Thrombophlebitis, Aortenaneurysma.
Erkrankungen der Atemwege, des Brustraums und Mediastinums*
Häufig: Husten.
Gelegentlich: Asthma, Dyspnoe, Dysphonie, Nasenlaufen, Rasselgeräusche, Nasenbluten.
Selten: Pharyngeales Ödem, interstitielle Lungenkrankheit, Pleuritis, Pneumonitis, obere Einflussstauung der Atemwege, nasale Ulcera, Atemversagen, Halsreizung, Lungenembolie, Pleuraerguss.
Erkrankungen des Gastrointestinaltrakts
Häufig: Diarrhoe und Motilitätsstörungen, Abdominalschmerzen, oropharyngeale Schmerzen, Übelkeit.
Gelegentlich: Dyspepsie, Gastrointestinale Blutungen, gastroösophagealer Reflux, Sicca Syndrom, Mundulzeration, entzündliche Darmerkrankung.
Selten: Gastritis, intestinale Obstruktion, Pankreatitis, Divertikel, Dysphagie, Zahnschmerzen, Zahnfleischbluten, oraler Pruritus, Cheilitis, Mundschleimhautverfärbungen, Gesichtsödem, Darmperforation.
Affektionen der Leber und Gallenblase*
Häufig: Erhöhung der Leberenzyme.
Gelegentlich: Hepatoxizität (einschliesslich Lebernekrose, Leberverfettung).
Selten: Cholelithiasis, erhöhtes Bilirubin im Blut, Reaktivierung von Hepatitis B, Autoimmune Hepatitis, Leberversagen.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag (einschliesslich schuppender Hautausschlag), Pruritus, Dermatitis.
Gelegentlich: Haarausfall, Akne, Ekzem, Psoriasis (einschliesslich palmoplantare-pustulöse Psoriasis), Blutergüsse (einschliesslich Purpura und Ekchymose), Hyperhidrosis, Nachtschweiss, Hautpigmentstörungen, Urtikaria.
Selten: Akneförmige Dermatitis, Haar- und Nagelstörungen, Hautverhärtungen, Hautirritation, kutane Vaskulitis, Erythema multiforme, Angioödem, lichenoide Hautreaktion**.
Einzelfälle: Steven Johnson Syndrom, Verschlechterung der Symptome einer Dermatomyositis.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Arthritis, Muskuloskelettale Schmerzen.
Gelegentlich: Muskelkrämpfe (einschliesslich erhöhte Blut-Kreatinphosphokinase).
Selten: Lupus-ähnliches Syndrom, systemischer Lupus erythematodes, Rhabdomyolyse, Tendonitis, Myositis, Schweregefühl.
Erkrankungen der Nieren und Harnwege
Gelegentlich: Hämaturie, Blasen- und Harnröhrenbeschwerden.
Selten: Nierenschmerzen, Nykturie, Proteinurie, eingeschränkte Nierenfunktion.
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Gelegentlich: Vulvo-vaginale Störungen, Störungen des Menstruationszyklus.
Selten: Zysten in der Brust und Empfindlichkeit, erektile Dysfunktion, Uterusstörungen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort*
Sehr häufig: Reaktion an der Injektionsstelle 13% (Kontrolle 7%) (einschliesslich Schmerz, Schwellung, Rötung oder Pruritus).
Häufig: Müdigkeit (einschliesslich Asthenie und Unwohlsein).
Gelegentlich: Brustschmerzen, Fieber, Ödeme, Grippe-ähnliche Symptome, Schmerzen, Schüttelfrost, Gewicht erhöht.
Selten: Entzündung, gesteigerte Energie, abnormales Gefühl, Schleimhautentzündung, Hitzegefühl.
Untersuchungen
Gelegentlich: Verlängerung der aktivierten partiellen Thromboplastinzeit, Nachweis von Autoantikörpern (einschliesslich doppelsträngiger DNA-Antikörper), Lactat-Dehydrogenase Blutwerte erhöht.
Selten: abnorme Urinanalysen.
Verletzungen, Vergiftungen und durch Eingriffe bedingte Komplikationen*
Gelegentlich: Versehentliche Verletzung, beeinträchtigte Wundheilung.
Selten: Anwendungsbedingte Komplikationen.
Hidradenitis suppurativa
Das Sicherheitsprofil einer wöchentlichen Verabreichung von Adalimumab bei Patienten mit Hidradenitis suppurativa war übereinstimmend mit dem bekannten Sicherheitsprofil von Adalimumab.
Uveitis
Das Sicherheitsprofil von Adalimumab bei der Behandlung von Patienten mit nicht-infektiöser Uveitis war übereinstimmend mit dem bekannten Sicherheitsprofil von Adalimumab mit Ausnahme von krankheitsbedingten Ereignissen.
Pädiatrische Population
Bei Kindern waren Häufigkeit und Schweregrad von Infektionen, Überempfindlichkeitsreaktionen und Reaktionen an der Injektionsstelle höher als bei Erwachsenen. Ansonsten waren die bei pädiatrischen Patienten beobachteten unerwünschten Wirkungen bezüglich Häufigkeit und Art ähnlich. Während der kontrollierten 32-Wochen-Phase der pJIA Studie wurden Infektionen in 24% (Kontrolle 15%), Überempfindlichkeitsreaktionen in 1,5% (Kontrolle 0%) und Reaktionen an der Injektionsstelle in 37% (Kontrolle 20%) beobachtet. Über die gesamte Studiendauer, einschliesslich der offenen Extensionsphase über bis 4 Jahre, traten in 80% Infektionen auf, davon 6% schwerwiegend, und in 6% Überempfindlichkeitsreaktionen auf.
Reaktionen an der Einstichstelle
In den pivotalen kontrollierten Studien bei Erwachsenen und Kindern entwickelten total 13% der mit Adalimumab behandelten Patienten Reaktionen an der Einstichstelle (Erytheme und/oder Juckreiz, Hämorrhagie, Schmerzen oder Schwellung) im Vergleich zu total 7% der Patienten unter Placebo oder aktiver Vergleichssubstanz. Die Reaktionen an der Einstichstelle machten im Allgemeinen kein Absetzen des Arzneimittels erforderlich.
Infektionen
In den pivotalen kontrollierten Studien bei Erwachsenen und Kindern betrug die Infektionsrate bei den mit Adalimumab behandelten Patienten 1,51 pro Patientenjahr und bei den Patienten unter Placebo oder aktiver Kontrolle 1,46 pro Patientenjahr. Die Inzidenz der schwerwiegenden Infektionen lag bei 0,04 pro Patientenjahr bei den mit Adalimumab behandelten Patienten sowie bei 0,03 pro Patientenjahr bei den mit Placebo und aktiven Kontrolle behandelten Patienten. Die Infektionen bestanden in der Hauptsache aus Infektionen der oberen Atemwege, Nasopharyngitis und Sinusitis. Der grösste Teil der Patienten führte die Behandlung mit Adalimumab nach Ausheilen der Infektion fort.
In den kontrollierten und offenen Studien bei Erwachsenen und Kindern mit Adalimumab wurden schwere Infektionen (selten auch solche mit tödlichem Ausgang), einschliesslich Tuberkulose (miliar und extrapulmonal) und invasive opportunistische Infektionen (wie z.B. disseminierte oder extrapulmonäre Histoplasmose, Blastomykose, Kokzidioidomykose, Pneumocystis-Infektion, Candidiasis (Soor), Aspergillose und Listeriose) gemeldet. Die meisten Fälle von Tuberkulose traten innerhalb der ersten 8 Monate nach Beginn der Therapie auf und können die Reaktivierung einer latent bestehenden Erkrankung darstellen. Die Inzidenz der Reaktivierung einer Tuberkulose war bei Dosierungen von Adalimumab, die über der empfohlenen Dosis lagen, besonders erhöht.
Maligne Tumore
Im Rahmen der kontrollierten Abschnitte der pivotalen Studien mit Adalimumab bei Erwachsenen, die mindestens 12 Wochen durchgeführt wurden bei Patienten mit mässiger bis schwerer aktiver rheumatoider Arthritis, Psoriasis-Arthritis, ankylosierender Spondylitis (Morbus Bechterew), Morbus Crohn, Colitis ulcerosa, Psoriasis, Hidradenitis suppurativa und Uveitis wurden maligne Erkrankungen (ausgenommen Lymphom und Nicht-Melanom-Hautkrebs) mit einer Inzidenzrate (95%-Konfidenzintervall) von 6,9 (4,4; 10,6) pro 1000 Patientenjahre bei 5196 mit Adalimumab behandelten Patienten beobachtet, verglichen mit einer Rate von 6,4 (3,5; 11,9) pro 1000 Patientenjahre bei 3347 Patienten in den Kontrollgruppen (die mittlere Behandlungsdauer betrug 4,0 Monate im Falle von Adalimumab sowie 3,9 Monate im Falle der mit der Kontrollsubstanz behandelten Patienten). Die Rate (95%-Konfidenzintervall) für Nicht-Melanom-Hautkrebs belief sich auf 8,9 (6,1; 13,1) pro 1000 Patientenjahre bei den mit Adalimumab behandelten Patienten sowie auf 3,2 (1,3; 7,7) pro 1000 Patientenjahre bei den Patienten der Kontrollgruppe. Von diesen Hautkarzinomen trat das Plattenepithelkarzinom mit einer Rate (95%-Konfidenzintervall) von 2,7 (1,4; 5,5) pro 1000 Patientenjahre bei den mit Adalimumab behandelten Patienten sowie mit 0,6 (0,1; 4,6) pro 1000 Patientenjahre bei den Patienten der Kontrollgruppe auf. Die Rate (95%-Konfidenzintervall) der Lymphome betrug 0,7 (0,2; 2,7) pro 1000 Patientenjahre bei den mit Adalimumab behandelten Patienten sowie 0,6 (0,1; 4,6) pro 1000 Patientenjahre bei den Patienten der Kontrollgruppe.
Fasst man die kontrollierten klinischen Studien und die noch laufenden und abgeschlossenen offenen Fortsetzungsstudien zusammen, beläuft sich die beobachtete Inzidenzrate der Malignitäten (Lymphom und Nicht-Melanom-Hautkrebs ausgenommen) auf etwa 8,6 pro 1000 Patientenjahre. Die beobachtete Rate bei Nicht-Melanom-Hautkrebs beträgt etwa 9,8 pro 1000 Patientenjahre und die beobachtete Rate der Lymphome etwa 1,3 pro 1000 Patientenjahre. Die durchschnittliche Dauer dieser Studien beträgt annähernd 3,3 Jahre, umfasste 6279 Patienten, die mindestens während 1 Jahr mit Adalimumab behandelt wurden, oder die innerhalb 1 Jahr nach Therapiestart eine Malignität entwickelten, entsprechend über 26'045 Patientenjahre.
Gemäss Post-Marketing-Erfahrung seit Januar 2003 beläuft sich (vorwiegend bei Patienten mit rheumatoider Arthritis) die gemeldete Inzidenzrate der Malignitäten (Lymphom und Nicht-Melanom-Hautkrebs ausgenommen) auf etwa 1,7 pro 1000 Patientenjahre. Bei Nicht-Melanom-Hautkarzinomen sowie bei Lymphomen betragen die gemeldeten Raten etwa 0,2 bzw. 0,4 pro 1000 Patientenjahre (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Bei 192 pädiatrischen Patienten mit einer Exposition von 258,9 Patientenjahren während einer klinischen Studie mit Adalimumab bei Kindern mit Morbus Crohn wurden keine malignen Erkrankungen beobachtet.
In einer Studie zu Plaque-Psoriasis an pädiatrischen Patienten wurden bei 77 pädiatrischen Patienten mit einer Exposition von 88,2 Patientenjahren keine malignen Erkrankungen beobachtet.
Auto-Antikörper
In den Studien 1-5 zu rheumatoider Arthritis wurden zu verschiedenen Zeitpunkten mehrmals Serumproben der Patienten auf Auto-Antikörper untersucht. In diesen adäquaten und gut kontrollierten Studien wurden bei 11,9% der mit Adalimumab behandelten Patienten, sowie bei 8,1% der mit Placebo und aktiver Vergleichssubstanz behandelten Patienten, die bei der Ausgangsuntersuchung negative Titer bei den antinuklearen Antikörpern aufwiesen, positive Titer in der Woche 24 gefunden. Zwei von den 3989 in allen Studien zu rheumatoider Arthritis, Psoriasis-Arthritis und ankylosierender Spondylitis (Morbus Bechterew) mit Adalimumab behandelten Patienten entwickelten klinische Symptome, die auf den Neuausbruch eines lupusähnlichen Syndroms hinwiesen. Nach Absetzen der Therapie besserte sich der Gesundheitszustand der Patienten. Keiner der Patienten entwickelte eine Lupusnephritis oder Symptome des Zentralnervensystems. Die Auswirkungen einer Langzeitbehandlung mit Adalimumab auf die Entwicklung von Autoimmunkrankheiten sind nicht bekannt.
Psoriasis: Neuauftreten und Verschlechterung
Unter Anwendung von TNF-Blockern, einschliesslich Adalimumab, wurde über Fälle mit neuauftretender Psoriasis, einschliesslich pustulöse Psoriasis und palmoplantare Psoriasis, sowie Fälle mit Verschlechterung einer bereits bestehenden Psoriasis berichtet. Viele dieser Patienten verwendeten gleichzeitig Immunsuppressiva (z.B. Methotrexat, Corticosteroide). Bei einigen dieser Patienten war eine Hospitalisation notwendig. Bei den meisten Patienten verbesserte sich die Psoriasis nach Absetzen des TNF-Blockers. Bei einigen Patienten zeigte sich ein Wiederauflammen der Psoriasis beim Einsatz eines anderen TNF-Blockers. Bei schweren Fällen und wenn sich unter topischer Behandlung keine Besserung oder sogar eine Verschlechterung zeigt, sollte der Abbruch der Behandlung mit AMGEVITA in Betracht gezogen werden.
Leber: Erhöhte ALT-Werte
Rheumatoide Arthritis und Psoriatrische Arthritis:
In kontrollierten Studien der Phase III mit Adalimumab (40 mg s.c. alle zwei Wochen) trat während einer Beobachtungsphase zwischen 4 und 104 Wochen bei 3,7% der mit Adalimumab behandelten Patienten und bei 1,6% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf. Viele Patienten in diesen Studien wendeten auch Medikamente an, die erhöhte Leberenzymwerte hervorrufen (z.B. NSAID, MTX).
Polyartikuläre juvenile idiopathische Arthritis:
In einer kontrollierten Phase III Studie mit Adalimumab trat während einer Beobachtungsphase von 32 Wochen im Methotrexat-Stratum bei 5,3% der mit Adalimumab behandelten Patienten und bei 2,7% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf. Im nicht-Methotrexat-Stratum trat keine Erhöhung der ALT-Werte auf.
Morbus Crohn:
In kontrollierten Studien der Phase III mit Adalimumab (Initialdosis von 160 mg an Tag 1 und 80 mg an Tag 15 bzw. 80 mg an Tag 1 und 40 mg an Tag 15, danach 40 mg alle zwei Wochen) trat während einer Beobachtungsphase zwischen 4 und 52 Wochen bei 0,9% der mit Adalimumab behandelten Patienten und bei 0,9% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf.
Morbus Crohn bei Kindern und Jugendlichen:
In der klinischen Phase-III-Studie zu Adalimumab bei Kindern und Jugendlichen mit Morbus Crohn wurden Wirksamkeit und Sicherheit von zwei körpergewichtsadaptierten Erhaltungstherapieschemata nach erfolgter körpergewichtsadaptierter Induktionstherapie über 52 Behandlungswochen untersucht. Es ergaben sich Erhöhungen der ALT-Werte ≥3x ULN bei 2,6% (5/192) der Patienten, von welchen 4 Patienten zu Therapiebeginn begleitend Immunsuppressiva erhalten hatten.
Ulcerative colitis:
In kontrollierten Studien der Phase III mit Adalimumab (Initialdosis von 160 mg an Tag 1 und 80 mg an Tag 15, danach 40 mg alle zwei Wochen) trat während einer Beobachtungsphase zwischen 1 und 52 Wochen bei 1,5% der mit Adalimumab behandelten Patienten und bei 1% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf.
Plaque-Psoriasis:
In kontrollierten Studien der Phase III mit Adalimumab (Initialdosis von 80 mg, danach 40 mg alle zwei Wochen) trat während einer Beobachtungsphase zwischen 12 und 24 Wochen bei 1,8% der mit Adalimumab behandelten Patienten und bei 1,8% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf.
Plaque Psoriasis bei Kindern und Jugendlichen
In der Phase-III-Studie von Adalimumab bei pädiatrischen Patienten mit Plaque-Psoriasis kam es zu keinen ALT-Erhöhungen ≥3x ULN.
Spondylitis ankylosans:
In kontrollierten Studien der Phase III mit Adalimumab (40 mg alle zwei Wochen) trat während einer Beobachtungsphase zwischen 12 und 24 Wochen bei 2,4% der mit Adalimumab behandelten Patienten und bei 0,66% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf.
Hidradenitis suppurativa:
In kontrollierten Studien mit Adalimumab (Initialdosis von 160 mg in Woche 0 und 80 mg in Woche 2, danach 40 mg wöchentlich, beginnend in Woche 4) trat während einer Beobachtungsphase zwischen 12 und 16 Wochen bei 0,3% der mit Adalimumab behandelten und bei 0,6% der Patienten mit Kontrollbehandlung eine Erhöhung der ALT-Werte (≥3x ULN) auf.
Uveitis:
In kontrollierten Studien zu Adalimumab (Initialdosis von 80 mg in Woche 0, gefolgt von 40 mg alle zwei Wochen, beginnend in Woche 1) bei Patienten mit Uveitis, in denen die Expositionsdauer bei mit Adalimumab behandelten Patienten 165,4 Patientenjahre und bei mit der jeweiligen Kontrollsubstanz behandelten Patienten 119,8 Patientenjahre betrug, kam es bei 2,4% der mit Adalimumab und bei 2,4% der mit der Kontrollsubstanz behandelten Patienten zu einer Erhöhung der ALT-Werte auf ≥3x ULN.
In allen Indikationen waren die Patienten mit erhöhter ALT in den klinischen Studien asymptomatisch. Zudem trat die Erhöhung in den meisten Fällen nur vorübergehend auf und verschwand im Laufe der weiteren Behandlung. Jedoch wurde in Anwendungsbeobachtungen bei Patienten unter TNF-Blockern, darunter Adalimumab, sehr selten von schweren Leberreaktionen (einschl. Leberversagen) berichtet.
Kombinationstherapie mit Azathioprin/6-Mercaptopurin
In den Studien mit erwachsenen Morbus Crohn Patienten war bei Kombination von Adalimumab mit Azathioprin/6-Mercaptopurin die Inzidenz maligner und schwerwiegender infektiöser Nebenwirkungen im Vergleich zu Adalimumab Monotherapie höher.
Immunogenität
Die Bildung von Anti-Adalimumab-Antikörpern ist mit einer erhöhten Clearance und einer verminderten Wirksamkeit von Adalimumab verbunden. Zwischen der Anwesenheit von neutralisierenden Anti-Adalimumab-Antikörpern und dem Auftreten von unerwünschten Ereignissen gibt es keinen offensichtlichen Zusammenhang. Die Langzeitimmunogenität von Adalimumab ist nicht bekannt.
Die Patienten mit rheumatoider Arthritis in den Studien 1, 2 und 3 wurden in den Behandlungszeiträumen von 6 und 12 Monaten zu multiplen Zeitpunkten auf die Bildung von Antikörpern gegen Adalimumab untersucht. In den pivotalen Studien wurden bei 58 von 1053 (5,5%) mit Adalimumab behandelten Patienten und bei 2 von 370 (0,5%) mit Placebo behandelten Patienten Antikörper gegen Adalimumab gefunden. Bei Patienten ohne gleichzeitige Gabe von Methotrexat (Studie 2) war die Häufigkeit 12,4%, verglichen mit 0,6% bei Patienten unter Methotrexat-Komedikation. Patienten unter Adalimumab Monotherapie, welche Adalimumab alle zwei Wochen anwenden, entwickeln möglicherweise häufiger Antikörper als Patienten, welche Adalimumab jede Woche anwenden. Die ACR 20 Antwort war niedriger bei Antikörper-positiven Patienten als bei Antikörper-negativen Patienten, welche die empfohlene Dosis von 40 mg alle zwei Wochen als Monotherapie erhielten.
Bei Patienten mit polyartikulärer juveniler idiopathischer Arthritis wurden Anti-Adalimumab-Antikörper bei 27/171 (15,8%) Patienten, die mit Adalimumab behandelt wurden, festgestellt. Bei Patienten ohne gleichzeitige Methotrexatbehandlung betrug die Häufigkeit 22/86 (25,6%) im Vergleich zu 5/85 (5,9%) bei Kombination von Adalimumab mit Methotrexat.
Bei Patienten mit Psoriasis-Arthritis wurden bei 38 von 376 Patienten (10%) Anti-Adalimumab-Antikörper unter Behandlung mit Adalimumab festgestellt. Bei Patienten ohne gleichzeitige Methotrexatbehandlung betrug die Häufigkeit 13,5% (24 von 178 Patienten) im Vergleich zu 7% (14 von 198 Patienten) bei Kombination von Adalimumab mit Methotrexat.
Bei Patienten mit ankylosierender Spondylitis wurden bei 17 von 204 Patienten (8,3%) Anti-Adalimumab-Antikörper unter Behandlung mit Adalimumab festgestellt. Bei Patienten ohne gleichzeitige Methotrexatbehandlung betrug die Häufigkeit 8,6% (16 von 185 Patienten) im Vergleich zu 5,3% (1 von 19 Patienten) bei Kombination von Adalimumab und Methotrexat.
Bei Patienten mit Morbus Crohn oder Colitis ulcerosa, die mit Adalimumab behandelt wurden, wurden bei 7 von 269 Patienten mit Morbus Crohn (2,6%) und bei 19 von 487 Patienten mit Colitis ulcerosa (3,9%) Anti-Adalimumab-Antikörper identifiziert.
Bei pädiatrischen Patienten mit mässigem bis schwerem aktivem Morbus Crohn, die mit Adalimumab behandelt wurden, betrug die Rate einer Antikörperentwicklung 3,3%.
Bei Patienten mit Plaque-Psoriasis, die Adalimumab langfristig als Monotherapie erhielten und die an einer Studie teilnahmen, in der die Therapie unterbrochen und wieder aufgenommen wurde, war der Anteil an Antikörpern gegen Adalimumab nach Wiederaufnahme der Behandlung ähnlich (11 von 482 Patienten; 2,3%) wie der Anteil, der vor dem Absetzen beobachtet wurde (11 von 590 Patienten; 1,9%).
Bei 5 von 38 Patienten (13%) mit pädiatrischer Psoriasis, die als Monotherapie mit 0,8 mg Adalimumab/kg behandelt wurden, wurden Anti-Adalimumab-Antikörper identifiziert.
Bei Patienten mit mittelschwerer bis schwerer Hidradenitis suppurativa, die mit Adalimumab behandelt wurden, wurden bei 10 von 99 Patienten (10,1%) Anti-Adalimumab Antikörper identifiziert.
Bei Patienten mit nicht-infektiöser Uveitis wurden bei 4,8% (12/249) der mit Adalimumab behandelten Patienten Anti-Adalimumab-Antikörper nachgewiesen.
Da Analysen zur Immunogenität produktspezifisch sind, ist ein Vergleich der Antikörperzahlen mit den unter anderen Arzneimitteln aufgetretenen mit Vorsicht zu interpretieren.
Immunogenität von AMGEVITA in den Vergleichbarkeitsstudien
Bindende und neutralisierende Antikörper wurden mit AMGEVITA und dem Referenzarzneimittel in vergleichbarer Häufigkeit detektiert.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIm Verlaufe der klinischen Studien wurde keine dosis-begrenzende Toxizität festgestellt. Bei der höchsten untersuchten Dosis handelte es sich um eine intravenöse Mehrfachapplikation von 10 mg/kg.
Eigenschaften/WirkungenATC-Code
L04AB04
AMGEVITA ist ein Biosimilar.
Wirkungsmechanismus
Bei Adalimumab handelt es sich um einen unter Einsatz einer rekombinanten DNA-Technologie in CHO-Zellen hergestellten humanen monoklonalen Antikörper. Er wurde unter Verwendung der sogenannten Phage-Display-Methode mit humanen schweren und leichten Ketten entwickelt. Dadurch entsteht ein Antikörper mit variablen Regionen der schweren und leichten Ketten ohne tierische Peptidsequenzen, die zur Spezifität für den humanen Tumor-Nekrose-Faktor (TNF) führen sowie mit humanen konstanten IgG1 (schwere Kette) und kappa (leichte Kette) Regionen. Adalimumab bindet mit einer hohen Affinität und Spezifität den löslichen Tumor-Nekrose-Faktor (TNFα), jedoch nicht Lymphotoxin (TNFβ). Es besteht aus 1330 Aminosäuren und besitzt ein Molekulargewicht von etwa 148 Kilodalton.
Adalimumab bindet spezifisch den TNF und neutralisiert die biologische Funktion des TNF durch die Blockierung seiner Interaktion mit den p55- und p75-TNF-Rezeptoren auf der Zelloberfläche. Bei TNF handelt es sich um ein natürlich vorkommendes Zytokin, das für die normalen Entzündungs- und Immunantworten von Bedeutung ist. Bei Patienten mit rheumatoider Arthritis, Psoriasis-Arthritis oder Ankylosierender Spondylitis (Morbus Bechterew) finden sich erhöhte TNF-Spiegel in der Synovialflüssigkeit, und diese spielen eine wichtige Rolle sowohl für die pathologische Entzündung als auch für die Gelenkdestruktion, beides Kennzeichen für eine rheumatoide Arthritis.
Adalimumab moduliert auch biologische Reaktionen, die durch den TNF induziert oder gesteuert werden, unter anderem Veränderungen bei den Spiegeln von Adhäsionsmolekülen, die für die Leukozytenmigration verantwortlich sind (ELAM-1, VCAM-1 sowie ICAM-1 mit einer IC50 von 1-2 x 10-10 M).
Pharmakodynamik
Nach einer Behandlung mit Adalimumab wurde im Vergleich zu den Basis-Werten bei den Patienten mit rheumatoider Arthritis ein schneller Rückgang bei den Werten für die Reaktanten der akuten Phase einer Entzündung (Creaktives Protein [CRP]), bei der Erythrozytensenkungsrate (ESR) sowie bei den Serumcytokinen (IL-6) beobachtet. Die Serumspiegel der Matrix-Metalloproteinasen (MMP-1 und MMP-3), die den Gewebeumbau herbeiführen, der für die Knorpeldestruktion verantwortlich ist, nahmen nach der Gabe von Adalimumab ebenfalls ab. Bei Patienten mit rheumatoider Arthritis, Psoriasis-Arthritis oder Ankylosierender Spondylitis (Morbus Bechterew) werden oft eine leichte bis mässige Anämie, reduzierte Lymphozytenzahlen sowie erhöhte Neutrophilen- und Thrombozytenzahlen festgestellt. Bei den mit Adalimumab behandelten Patienten zeigte sich im Allgemeinen eine Verbesserung bei diesen hämatologischen Anzeichen einer chronischen Entzündung.
Ein schneller Rückgang der CRP-Werte wurde auch bei Patienten mit Morbus Crohn, Colitis ulcerosa, polyartikulärer juveniler idiopathischer Arthritis und Hidradenitis suppurativa nach Behandlung mit Adalimumab beobachtet. Bei Morbus Crohn-Patienten wurde auch eine Abnahme der Anzahl Entzündungsmarker exprimierenden Zellen im Kolon (nicht statistisch signifikant), einschliesslich einer signifikanten Reduktion der TNFα Expression beobachtet.
Klinische Wirksamkeit
Rheumatoide Arthritis
Im Rahmen sämtlicher klinischer Studien zur rheumatoiden Arthritis wurde Adalimumab bei mehr als 3000 Patienten untersucht. Einige Patienten wurden über einen Zeitraum von mehr als 60 Monaten behandelt. Wirksamkeit und Verträglichkeit von Adalimumab für die Therapie von rheumatoider Arthritis wurden in fünf randomisierten, doppelblinden und gut kontrollierten Studien untersucht.
In Studie 1 evaluierte man 271 Patienten mit einer mässigen bis schweren aktiven rheumatoiden Arthritis, die ≥18 Jahre alt waren, bei denen die Therapie mit mindestens einem, aber mit nicht mehr als vier krankheitsmodifizierenden Antirheumatika versagt und bei denen Methotrexat in einer Dosierung von 12,5 bis 25 mg (10 mg bei Methotrexat-Intoleranz) jede Woche eine ungenügende Wirksamkeit gezeigt hatte, und deren Methotrexat-Dosis während der Studie bei 10 bis 25 mg jede Woche konstant blieb. Die Patienten hatten ≥6 geschwollene Gelenke und ≥9 druckschmerzhafte Gelenke. Die rheumatoide Arthritis hatte man nach den Kriterien des American College of Rheumatology (ACR) diagnostiziert. Über einen Zeitraum von 24 Wochen verabreichte man jede zweite Woche Dosen in Höhe von 20, 40 bzw. 80 mg Adalimumab oder ein Placebo.
Studie 2 evaluierte 544 Patienten mit einer mässigen bis schweren aktiven rheumatoiden Arthritis, die ≥18 Jahre alt waren und bei denen die Therapie mit mindestens einem krankheitsverändernden Antirheumatikum versagt hatte. Die Patienten hatten ≥10 geschwollene Gelenke und ≥12 druckschmerzhafte Gelenke und waren ebenfalls nach den ACR-Kriterien diagnostiziert worden. Über 26 Wochen wurden mittels subkutaner Injektion 20 bzw. 40 mg Adalimumab alle zwei Wochen abwechselnd mit einem Placebo in der darauf folgenden Woche bzw. in jeder Woche verabreicht. Das Placebo wurde jede Woche über den gleichen Zeitraum gegeben. Die Patienten erhielten keine Begleittherapie mit DMARDs.
In Studie 3 bewertete man 619 Patienten mit einer mässigen bis schweren aktiven rheumatoiden Arthritis, die ≥18 Jahre alt waren, bei denen Methotrexat in einer Dosierung von 12,5 bis 25 mg (10 mg bei Methotrexat-Intoleranz) jede Woche eine ungenügende Wirksamkeit gezeigt hatte und deren Methotrexat-Dosis während der Studie bei 12,5 bis 25 mg jede Woche konstant blieb. Anders als in Studie 1 war für die in der Studie 3 eingeschlossenen Patienten das Versagen einer Therapie mit einem krankheitsmodifizierenden Antirheumatikum (Methotrexat ausgenommen) nicht erforderlich. Die Patienten hatten ≥6 geschwollene Gelenke und ≥9 druckschmerzhafte Gelenke. Die rheumatoide Arthritis war nach den ACR-Kriterien diagnostiziert worden. In dieser Studie gab es drei Gruppen. Die Gruppe 1 erhielt über 52 Wochen in jeder Woche eine Injektion mit Placebo. Der zweiten Gruppe verabreichte man über den Zeitraum von 52 Wochen jede Woche 20 mg Adalimumab. Die dritte Gruppe bekam jede zweite Woche 40 mg Adalimumab abwechselnd mit Placebo-Injektionen in der darauf folgenden Woche. Im Anschluss wurden 457 Patienten in eine bis zu 5 Jahren andauernde offene Fortsetzungsperiode überführt und erhielten jede zweite Woche 40 mg Adalimumab.
Studie 4 bewertete 636 Patienten mit einer mässigen bis schweren aktiven rheumatoiden Arthritis, die ≥18 Jahre alt waren. Diese Patienten erfüllten die ACR-Kriterien für die Diagnose der rheumatoiden Arthritis seit mindestens drei Monaten und hatten ≥6 geschwollene Gelenke und ≥9 druckschmerzhafte Gelenke. Die Patienten waren entweder bisher nicht mit krankheitsmodifizierenden Antirheumatika behandelt oder konnten ihre bereits bestehende rheumatologische Behandlung unter der Voraussetzung fortsetzen, dass diese seit mindestens 28 Tagen stabil war. Die Patienten wurden für eine Behandlung in jeder zweiten Woche mit 40 mg Adalimumab bzw. Placebo über den Zeitraum von 24 Wochen randomisiert.
In Studie 5 bei früher rheumatoider Arthritis wurden 525 erwachsene (≥18 Jahre alt) Patienten mit mässiger bis schwerer, aktiver früher Erkrankung (Krankheitsdauer weniger als 3 Jahre) bewertet, welche Methotrexat naiv waren. In dieser Studie wurde die Wirksamkeit von der Adalimumab/Methotrexat Kombinationstherapie gegenüber Methotrexat Monotherapie verglichen in Bezug auf Reduzierung der Anzeichen, Symptome und Progressionsrate von Gelenkschädigung bei rheumatoider Arthritis beurteilt. Die Patienten wurden zu Adalimumab 40 mg/Methotrexat Kombinationstherapie alle zwei Wochen oder Methotrexat Monotherapie alle zwei Wochen randomisiert. Der Behandlungszeitraum betrug 104 Wochen.
Die Ergebnisse aller fünf Studien werden in der Prozentzahl der Patienten mit einer Besserung der rheumatoiden Arthritis unter Verwendung der ACR-Ansprechkriterien ausgedrückt. Als primärer Endpunkt in den Studien 1, 2 und 3 sowie als sekundärer Endpunkt in Studie 4 galt der Prozentsatz an Patienten, die in Woche 24 bzw. 26 eine ACR20-Ansprechrate erreichten. Der primäre Endpunkt in Studie 5 bei früher rheumatoider Arthritis war der Prozentsatz an Patienten, die in Woche 52 eine ACR50-Ansprechrate erreichten. Studien 3 und 5 hatten als zusätzlichen primären Endpunkt in Woche 52 eine Retardierung des Krankheitsverlaufes (festgestellt durch Röntgenuntersuchungen) einbezogen. In Studie 3 wurde ausserdem als primärer Endpunkt Veränderungen bei der Lebensqualität untersucht.
ACR-Ansprechrate
Der Prozentsatz der mit Adalimumab behandelten Patienten, welche ACR20-, ACR50- und ACR70-Ansprechraten erreichten, war konsistent über die Studien 1, 2, 3 und 4. Die Ergebnisse für 40 mg Adalimumab alle zwei Wochen sind in Tabelle 4 zusammengefasst.
Tabelle 4: ACR-Ansprechraten bei Placebo-kontrollierten Prüfungen (in Prozent der Patienten)
Ansprechrate Studie 1a* Studie 2a* Studie 3a* Studie 4
Placebo/ MTXc n=60 Adalimumabb/ MTXc Placebo n=110 Adalimumabb n=113 Placebo/ MTXc n=200 Adalimumabb/ MTXc Standard Behandlung/ Standard Behandlung/
n=63 n=207 Placebo n=318 Adalimumab n=318
ACR 20
6 Monate 13,3% 65,1% 19,1% 46,0% 29,5% 63,3% 34,9% 53,0%
12 Monate NA NA NA NA 24,0% 58,9% NA NA
ACR 50
6 Monate 6,7% 52,4% 8,2% 22,1% 9,5% 39,1% 11,1% 29,2%
12 Monate NA NA NA NA 9,5% 41,5% NA NA
ACR 70
6 Monate 3,3% 23,8% 1,8% 12,4% 2,5% 20,8% 3,2% 14,9%
12 Monate NA NA NA NA 4,5% 23,2% NA NA
a Studie 1 nach 24 Wochen, Studie 2 nach 26 Wochen und Studie 3 nach 24 und 52 Wochen
b 40 mg Adalimumab, jede zweite Woche verabreicht
c MTX=Methotrexat
* p<0,01, Adalimumab vs. Placebo
NA=Nicht zutreffend
Die Patienten, die im Rahmen der Studie 2 jede Woche 40 mg Adalimumab erhielten, erzielten nach sechs Monaten ebenfalls statistisch signifikante ACR20-, ACR50- und ACR70-Ansprechraten in Höhe von 53,4%, 35,0% bzw. 18,4%.
In den Studien 1-4 war bei allen individuellen Komponenten der ACR-Ansprechkriterien (Anzahl der druckschmerzempfindlichen und geschwollenen Gelenke, Einschätzung der Krankheitsaktivität und der Schmerzen durch den Arzt und den Patienten, Bewertung laut HAQ Disability Index sowie CRP-Werte [mg/dl]) im Vergleich mit Placebo nach 24 bzw. 26 Wochen eine Verbesserung zu verzeichnen. In der Studie 3 waren diese Verbesserungen auch über 52 Wochen anhaltend. Darüber hinaus hielten die ACR-Ansprechraten bei der Mehrzahl der Patienten, die an der offenen Fortsetzungsperiode teilnahmen, bis Woche 104 an. Die 2-Jahresergebnisse der Studie zeigen, dass bei 24% der mit Adalimumab behandelten Patienten eine klinische Wirkung, definiert als Erhaltung einer ACR70-Ansprechrate über die Dauer von 6 Monaten, erzielt werden konnte. Eine dauerhaft bis zu 5 Jahren anhaltende klinische Wirkung konnte während der nicht-kontrollierten Phase der Studie 3 gezeigt werden. Die bei Woche 52 beobachtete ACR Ansprechrate konnte aufrecht erhalten werden, wenn Adalimumab ohne Unterbrechung über 5 Jahre verabreicht wurde, mit einer ACR20-Ansprechrate von 75,5% bei der Untergruppe von 220 nach 5 Jahren evaluierten Patienten. Die ACR70-Ansprechrate nach 5 Jahren lag bei 34,7%. Bei 25,7% der Patienten konnte die Dosis des gleichzeitig verabreichten Methotrexats und bei 29,9% derjenigen der Kortikosteroide ohne ein Nachlassen der klinischen Wirkung verringert werden.
Die folgende Abbildung 1 illustriert die Dauerhaftigkeit der in Studie 3 verzeichneten ACR20-Ansprechraten auf Adalimumab. Im Rahmen dieser Studie konnten 84,7% der Patienten, die die ACR20-Ansprechkriterien in Woche 24 erreicht hatten, diese bis in Woche 52 halten.
Abbildung 1: ACR20-Ansprechraten über 52 Wochen in Studie 3

In der Studie 4 zeigten sich die ACR20-Ansprechraten bei den Patienten, die mit Adalimumab plus der Standardtherapie behandelt wurden, als statistisch signifikant besser als bei Patienten, die Placebo plus die Standardtherapie erhielten (p<0,001).
In allen vier Studien erreichten die mit Adalimumab behandelten Patienten die ACR20-, ACR50- und ACR70-Ansprechraten schneller und häufiger als die mit einem Placebo behandelten Patienten. In Studie 1 war ein statistisch signifikanter Unterschied bei den ACR20-Ansprechraten in Woche 1 (erste Untersuchung im Rahmen der Studie) zwischen den mit Adalimumab (26,0%) und den mit Placebo (5,0%) behandelten Patienten zu verzeichnen. Statistisch signifikante Unterschiede bei den ACR20-Ansprechraten wurden auch in den Studien 2, 3 und 4 in Woche 2 (erste Untersuchung im Rahmen der Studie) zwischen den mit Adalimumab (36,4%, 29,1% bzw. 33,7%) und den mit Placebo (7,3%, 13,0% bzw. 8,6%) behandelten Patienten beobachtet. Ein ähnliches Muster stellte man auch in allen vier Studien für die Zeit bis zum Erreichen der ersten ACR50- bzw. ACR70-Ansprache fest.
Für einige Patienten, die nicht gleichzeitig Methotrexat nehmen, könnte eine Erhöhung der Dosierungsfrequenz von Adalimumab auf 40 mg pro Woche einen zusätzlichen Nutzen bringen. Das hat sich im Rahmen einer offenen Langzeitstudie bestätigt, in der man für die Patienten, die nur unvollständig auf das Arzneimittel ansprachen, die Dosisfrequenz von 40 mg jede zweite Woche auf 40 mg pro Woche erhöhte.
In Studie 5 führte die Kombinationstherapie von Adalimumab und Methotrexat bei Patienten mit früher rheumatoider Arthritis, die Methotrexat naiv waren, zu rascher einsetzenden und signifikant höheren ACR-Ansprechraten in Woche 52 als Methotrexat Monotherapie, wobei die Ansprechraten bis Woche 104 aufrechterhalten blieben (siehe Tabelle 5).
Tabelle 5: ACR-Ansprechrate in Studie 5 (in Prozent der Anzahl der Patienten)
Ansprechrate* MTX n=257 Adalimumab/MTX n=268
ACR 20
Woche 52 62,6% 72,8%
Woche 104 56,0% 69,4%
ACR 50
Woche 52 45,9% 61,6%
Woche 104 42,8% 59,0%
ACR 70
Woche 52 27,2% 45,5%
Woche 104 28,4% 46,6%
* p<0,05, Adalimumab/Methotrexat versus Methotrexat für ACR 20
* p<0,001, Adalimumab/Methotrexat versus Methotrexat für ACR 50 und 70
Alle einzelnen Kriterien der ACR-Ansprechrate zeigten unter der Adalimumab/Methotrexat Therapie eine Verbesserung in Woche 52, die bis Woche 104 aufrechterhalten blieb. Im Verlauf der Zweijahres-Studie erreichten 48,5% der Patienten, welche Adalimumab/Methotrexat Kombinationstherapie erhielten, eine bedeutende klinische Ansprechrate (ACR70 für sechs Monate) im Vergleich zu 27,2% der Patienten, welche Methotrexat Monotherapie erhielten (p<0,001).
Tabelle 6: DAS28 Ansprechraten in Studie 5 bei früher rheumatoider Arthritis
DAS28 Ansprechen MTX n=257 Adalimumab/MTX n=268
Mittlere Abweichung von Baseline
Baseline (Mittelwert) 6,3 6,3
Woche 52 (Mittelwert ± SD) -2,8 ± 1,4 -3,6 ± 1,3*
Woche 104 (Mittelwert ± SD) -3,1 ± 1,4 -3,8 ± 1,3*
Remission (DAS28<2,6)
Woche 52 (Prozent von Anzahl Patienten) 20,6% 42,9%*
* p<0,001, Adalimumab/Methotrexat versus Methotrexat
Radiologisches Ansprechen
In Studie 3, in der bei den mit Adalimumab behandelten Patienten die mittlere Dauer der Erkrankung an rheumatoider Arthritis etwa 11 Jahre betrug, wurde der strukturelle Gelenkschaden radiologisch bewertet und als Veränderung im modifizierten Sharp-Gesamtscore (TSS) und seiner Komponenten, dem Erosions-Score und dem Joint Space Narrowing Score = JSN (Score, der die Verengung des Gelenkraums bewertet) ausgedrückt. Ein statistisch signifikanter Unterschied hinsichtlich einer Veränderung im modifizierten Sharp-Gesamtscore sowie beim Erosions-Score wurde nach 6 Monaten beobachtet und blieb bis zu Monat 12 erhalten. Nach 52 Wochen zeigten die mit Adalimumab/Methotrexat behandelten Patienten weniger radiologische Veränderungen als Patienten, die nur Methotrexat erhalten hatten. Diese Wirkung im Sinne einer Verlangsamung der Progression struktureller Schäden konnte über 5 Jahre aufrechterhalten werden.
Von den ursprünglich mit 40 mg Adalimumab alle 2 Wochen behandelten Patienten wurden 55% nach 5 Jahren radiologisch untersucht. Die Verlangsamung der Progression struktureller Schäden konnte aufrecht erhalten werden, und bei 50% dieser verbleibenden Patienten konnte das Fortschreiten der strukturellen Schädigung ganz aufgehalten werden, wie sich durch eine Veränderung im TSS von null oder weniger feststellen liess. Patienten die während der Doppelblindphase der Studie mit Methotrexat behandelt worden waren, zeigten minimale Progression struktureller Schädigung, wenn sie während des offenen Teils der Studie mit Adalimumab behandelt wurden.
Tabelle 7: Radiologische Veränderung über 12 Monate in Studie 3 mit Hintergrundbehandlung Methotrexat
Placebo n=200 Adalimumaba n=207 Unterschied zwischen p-Wert
Adalimumaba und
Placebo
Veränderung beim 2,7 0,1 -2,6 =0,001b
modifizierten
Sharp-Gesamtscore
(Mittel)
Veränderung bei 1,6 0,0 -1,6 =0,001
Erosionen (Mittel)
Keine neuen Erosione 46,2 62,9 16,7 =0,001
n (% der Patienten)
Veränderung beim 1,0 0,1 -0,9 =0,002
JSN-Score (Mittel)
a 40 mg, verabreicht jede zweite Woche
b auf Grundlage der mittleren Werte, gemessen anhand des TSS
In Studie 5 bei früher rheumatoider Arthritis hatten die mit Adalimumab behandelten Patienten eine mittlere Krankheitsdauer der rheumatoiden Arthritis von weniger als 9 Monaten und hatten zuvor kein Methotrexat erhalten. Strukturelle Gelenkschädigung wurde radiologisch ermittelt und als Änderung im modifizierten Total Sharp Score ausgedrückt. Die Resultate nach Woche 52 sind in Tabelle 8 ersichtlich. Eine statistisch signifikante Änderung bezüglich modifiziertem Total Sharp Score und Erosions-Score wurde in Woche 52 beobachtet und blieb bis Woche 104 aufrechterhalten.
Tabelle 8: Radiologische mittlere Abweichungen in Woche 52 in Studie 5
MTX n=257 95% CI Adalimumab/MTX n=268 95% CI p-Wert*
Total Sharp Score 5,7 (4,2-7,3) 1,3 (0,5-2,1) <0,001
Erosion Score 3,7 (2,7-4,7) 0,8 (0,4-1,2) <0,001
JSN Score 2,0 (1,2-2,8) 0,5 (0-0,1) <0,001
* Vergleich zwischen Adalimumab/Methotrexat und Methotrexat mittels Mann-Whitney U test
Der prozentuelle Anteil von Patienten ohne Progression (Zunahme gegenüber Baseline im modifizierten Total Sharp Score ≤0,5) war signifikant höher unter Adalimumab/Methotrexat Kombinationstherapie im Vergleich zu Methotrexat Monotherapie in Woche 52 (63,8% respektive 37,4%, p<0,001) und in Woche 104 (61,2% respektive 33,5%, p<0,001).
Im Rahmen der offenen Fortsetzungsperiode der Studie 3 wurden 77% der ursprünglich mit Adalimumab behandelten Patienten nach 2 Jahren radiologisch bewertet. Die Hemmung des Fortschreitens der strukturellen Schädigung hielt an. 54% der untersuchten Patienten zeigten keine Progression der strukturellen Schädigung entsprechend einer TSS Änderung von 0 oder weniger.
Lebensqualität und körperliche Funktionsfähigkeit
Für die Bewertung der Lebensqualität in Bezug auf den Gesundheitszustand, die in Studie 3 einen vorgeschriebenen Endpunkt nach 52 Wochen darstellte, verwendete man in allen vier adäquaten und gut kontrollierten Prüfungen den im Health Assessment Questionnaire (HAQ = Fragebogen zur Bewertung des Gesundheitszustandes) enthaltenen Behinderungsindex. In allen vier Studien zeigten sich für sämtliche Dosierungen/Therapiepläne von Adalimumab im Vergleich mit Placebo statistisch signifikante, stärkere Verbesserungen beim HAQ Disability Index von den Baseline-Werten bis Monat 6. In Studie 3 belief sich die mittlere Verbesserung (CI) des HAQ zwischen Baseline und Woche 52 auf -0,60 (-0,65, -0,55) bei mit Adalimumab/Methotrexat behandelten Patienten und auf −0,25 (-0,33, -0,17) bei mit Placebo/Methotrexat behandelten Patienten (p<0,001). Bei 82% der mit Adalimumab/Methotrexat behandelten Patienten, bei denen in Woche 52 eine Verbesserung des HAQ von 0,5 oder mehr erzielt werden konnte, blieb diese Verbesserung bis Monat 60 der offenen Fortsetzungsperiode aufrechterhalten.
In Studie 5, der kontrollierten Studie bei früher rheumatoider Arthritis im Vergleich zu Methotrexat, war die Verbesserung im HAQ Disability Index und der physischen Komponente des SF 36 in Woche 52 grösser (p<0,001) unter Adalimumab/Methotrexat Kombinationstherapie als unter Methotrexat Monotherapie und blieb bis Woche 104 aufrechterhalten.
Ausserdem wurde die allgemeine Lebensqualität bezogen auf den Gesundheitszustand in allen vier adäquaten und gut kontrollierten Prüfungen mit Hilfe des Short Form Health Survey (SF 36 = Analyse des Gesundheitszustandes in Kurzform) bewertet. In allen vier Studien zeigten sich für sämtliche Dosierungen/Injektionsfrequenzen von Adalimumab im Vergleich mit Placebo statistisch signifikante, stärkere Verbesserungen bei den summarischen Scores für die physischen Komponenten des SF 36 von den Baseline-Werten bis Monat 6, die in Studie 3 bis zur Woche 52 aufrechterhalten werden konnten. Die summarischen Scores für die mentalen Komponenten des SF 36 in den Studien 2 und 4 lagen ebenfalls, verglichen mit Placebo, im Monat 6 für Adalimumab statistisch signifikant höher. Die auf Schmerzen und Vitalität bezogenen Scores des SF 36 wiesen in allen vier Studien für die Dosierung 40 mg Adalimumab jede zweite Woche im Vergleich mit dem Placebo eine statistisch signifikant stärkere Verbesserung von der Baseline auf Monat 6 aus. Diese Ergebnisse wurden durch die im Rahmen der Functional Assessment of Chronic Illness Therapy (FACIT = funktionelle Bewertung der Therapie von chronischen Erkrankungen) ermittelten Scores gestützt, nach denen sich für alle drei analysierten Studien eine statistisch signifikante Abnahme der Ermüdung in Monat 6 zeigte, die in Studie 3 bis zur Woche 52 aufrechterhalten werden konnte. SF 36 wurde bis zur Woche 156 (3 Jahre) gemessen, und die Verbesserung blieb bei den in der Studie verbleibenden Patienten über diesen Zeitraum erhalten.
Polyartikuläre juvenile idiopathische Arthritis (pJIA)
Die Sicherheit und Wirksamkeit von Adalimumab wurden in einer multizentrischen, randomisierten, doppelblinden Parallel-Gruppenstudie an 171 Kindern (im Alter zwischen 4 bis 17 Jahren) mit polyartikulärer JIA untersucht. Die Analyse erfolgte in den zwei Strata mit Methotrexat (MTX)behandelten und nicht-MTX-behandelten Patienten. Die Patienten erhielten stabile Dosen von NSAR und/oder Prednison (≤0,2 mg/kg/Tag oder maximal 10 mg/Tag). In der offenen Einleitungsphase ( "open-label lead-in" , OL LI) erhielten alle Patienten 16 Wochen lang jede zweite Woche 24 mg/m2 bis zu einer Maximaldosis von 40 mg Adalimumab. Die Patientenverteilung ist in Tabelle 9 dargestellt.
Tabelle 9: Patientenverteilung nach Alter und verabreichter Adalimumab-Dosis während der OL LI-Phase
Altersgruppe Patientenanzahl zu Studienbeginn n (%) Minimale, mittlere und maximale Dosis
4 bis 7 Jahre 31 (18,1) 10, 20 und 25 mg
8 bis 12 Jahre 71 (41,5) 20, 25 und 40 mg
13 bis 17 Jahre 69 (40,4) 25, 40 und 40 mg
Die Patienten mit einem pädiatrischen ACR-30-Ansprechen in Woche 16 wurden in der doppelblinden (DB-)Studienphase randomisiert und erhielten alle 2 Wochen entweder Adalimumab (24 mg/m2 bis zu einer maximalen Einzeldosis von 40 mg) oder Placebo über maximal 32 Wochen oder bis zu einem Wiederaufflammen der Erkrankung. Die Kriterien für ein Wiederaufflammen der Erkrankung waren definiert als eine Verschlechterung von ≥30% im Vergleich zu Studienbeginn bei ≥3 von 6 pädiatrischen ACR-Core-Kriterien, ≥2 aktive Gelenke und eine Verbesserung von >30% in nicht mehr als einem der 6 Kriterien. Nach 32 Wochen oder bei Wiederaufflammen der Erkrankung waren die Patienten für die Überführung in die offene Fortsetzungsphase ( "Open-label extension" , OLE) geeignet.
In der OL LI Phase erreichten 94,1% (80 von 85) der Patienten unter Kombinationstherapie von Adalimumab und MTX und 74,4% (64 von 86) der Patienten unter Adalimumab Monotherapie ein ACR30-Ansprechen in Woche 16. Die Ergebnisse der doppelblinden Periode sind in Tabelle 10 dargestellt.
Tabelle 10: Pädiatrisches ACR30-Ansprechen in der JIA Studie
Ergebnisse zur Wirksamkeit
Doppelblind 32 Adalimumab/MTX Placebo/MTX (n=37) Adalimumab (n=30) Placebo (n=28)
Wochen (n=38)
Wiederaufflammen 36,8% (14/38) 64,9% (24/37)b 43,3% (13/30) 71,4% (20/28)c
der Erkrankung nach
32 Wochena (n/N)
Mittlere Zeit bis >32 Wochen 20 Wochen >32 Wochen 14 Wochen
zum Wiederaufflammen
der Erkrankung
a pädiatrisches ACR 30/50/70-Ansprechen in Woche 48 war signifikant grösser als bei mit Placebo behandelten Patienten
b p=0,015
c p=0,031
Unter den Patienten, die in Woche 16 (n=144) ansprachen, wurde das pädiatrische ACR30/50/70/90-Ansprechen für bis zu sechs Jahre in der OLE-Phase bei denjenigen aufrechterhalten, die Adalimumab während der ganzen Studie über erhielten. Insgesamt wurden 19 Patienten (11 zu Studienbeginn in der Altersgruppe von 4 bis 12 Jahren und 8 zu Studienbeginn in der Altersgruppe von 13 bis 17 Jahren) 6 Jahre oder länger behandelt.
Das Gesamtansprechen bei der Kombinationstherapie von Adalimumab und MTX war besser, und weniger Patienten entwickelten Antikörper im Vergleich zur Adalimumab-Monotherapie. Unter Berücksichtigung dieser Ergebnisse wird der Einsatz von Adalimumab in Kombination mit MTX empfohlen. Nur bei Patienten, bei denen der MTX-Einsatz nicht geeignet ist, wird eine Monotherapie mit Adalimumab empfohlen (siehe "Dosierung/Anwendung" ).
Psoriasis-Arthritis
Die Wirksamkeit von Adalimumab wurde bei 413 Patienten untersucht. In der Hauptstudie wurden 313 erwachsene Patienten mit mässiger bis schwerer Psoriasis-Arthritis behandelt, die nur unzureichend auf nicht-steroidale anti-inflammatorische Therapie angesprochen hatten. 158 (50,5%) der behandelten Patienten nahmen zum Zeitpunkt der Randomisierung Methotrexat. Adalimumab wurde als 40 mg Dosis alle 2 Wochen über einen Zeitraum von 24 Wochen verabreicht. Nach Beenden der Studien wurden 383 Patienten in eine offene Fortsetzungssperiode eingeschlossen, in welcher Adalimumab jede zweite Woche verabreicht wurde. 382 der eingeschlossenen Patienten wurden zumindest anfänglich in dieser Fortsetzungsstudie mit Adalimumab behandelt. Bezüglich der nach 48 und 144 Wochen evaluierbaren Patienten, siehe weiter unten.
ACR und PASI Ansprechrate
In Tabelle 11 ist ersichtlich, dass Adalimumab gegenüber Placebo in allen gemessenen Parametern des Krankheitsverlaufes überlegen war (p<0,001). Bei den Patienten mit Psoriasis-Arthritis, welche Adalimumab erhielten, zeigte sich die klinische Wirkung zum Zeitpunkt der ersten Kontrolle (2 Wochen); sie war nach 12 Wochen signifikant und wurde während der 24 Behandlungswochen aufrechterhalten.
Patienten, bei denen mindestens 3% der Körperoberfläche von Psoriasis betroffen waren, wurden nach Psoriatic Area and Severity Index (PASI) beurteilt. Nach PASI gemessen, verbesserten sich bei diesen Patienten die Psoriasis-bedingten Hautläsionen im Vergleich zu Placebo.
Die Ansprechrate war mit und ohne Methotrexat vergleichbar.
Die ACR Ansprechraten blieben in der offenen Fortsetzungsperiode bis 136 Wochen aufrechterhalten.
Tabelle 11: ACR und PASI Ansprechrate in einer Placebo-kontrollierten Studie bei Patienten mit Psoriasis-Arthritis (in Prozent der Anzahl der Patienten)
Ansprechrate* Placebo Adalimumab
N=162 N=151
ACR 20
Woche 12 14% 58%
Woche 24 15% 57%
ACR 50
Woche 12 4% 36%
Woche 24 6% 39%
ACR 70
Woche 12 1% 20%
Woche 24 1% 23%
N=69 N=69
PASI 50
Woche 12 15% 72%
Woche 24 12% 75%
PASI 75
Woche 12 4% 49%
Woche 24 1% 59%
* p<0,001 für alle Vergleiche von Adalimumab und Placebo
In den Studien zu Psoriasis-Arthritis wurden die radiologischen Veränderungen beurteilt. Röntgenaufnahmen der Hände, Handgelenke und Füsse wurden zu Studienbeginn (Baseline) und in Woche 24 der Doppelblind-Studienperiode aufgenommen, in welcher die Patienten entweder Adalimumab oder Placebo erhielten, sowie in Woche 48, in welcher alle Patienten Adalimumab erhielten. Ein modifizierter Sharp-Gesamtscore (mTSS), welcher distale Interphalangealgelenke einschloss (nicht identisch mit dem Sharp-Gesamtscore für rheumatoide Arthritis), wurde verwendet.
Verglichen mit Placebo reduzierte Adalimumab die Progressionsrate von peripheren Gelenkschäden. Im modifizierten Sharp-Gesamtscore wurde eine Veränderung vom Basiswert von 0,8 ± 2,5 (Mittelwert ± SD) in der Placebo-Gruppe (in Woche 24) im Vergleich zu 0,0 ± 1,9 in der Adalimumab-Gruppe (in Woche 48, n=133); p<0,001) festgestellt.
84% der Patienten, welche mit Adalimumab behandelt wurden und zwischen Baseline und Woche 48 keine radiologische Progression zeigten (n=102), zeigten auch keine radiologische Progression bis Woche 144.
Mittels HAQ Disability Index und Short Form Health Survey (SF 36) bewertet, zeigten die mit Adalimumab behandelten Patienten eine statistisch signifikante Verbesserung der körperlichen Funktionsfähigkeit in Woche 24 im Vergleich zu Patienten, die mit Placebo behandelt wurden.
Die Verbesserung der körperlichen Funktionsfähigkeit blieb in der offenen Fortsetzungsperiode der Studie bis Woche 136 aufrechterhalten.
Morbus Crohn
Sicherheit und Wirksamkeit einer Mehrfachdosis Adalimumab wurden bei mehr als 1500 Patienten mit einer mässigen bis schweren aktiven Crohnschen Erkrankung (Aktivitätsindex des Morbus Crohn [CDAI = Crohn's Disease Activity Index] ≥220 und ≤450) im Rahmen von randomisierten, doppelblinden und placebo-kontrollierten Studien untersucht. Die gleichzeitige Gabe von Aminosalicylaten, Corticosteroiden bzw. immunomodulatorischen Mitteln in gleichbleibenden Dosen war erlaubt, und 80% der Patienten bekamen mindestens eines dieser Arzneimittel verabreicht.
In zwei Studien (CLASSIC I und GAIN) hat man die Einleitung der klinischen Remission (definiert als CDAI<150) bewertet. In der CLASSIC I-Studie wurden 299 TNF-Antagonist-naive Patienten auf vier Behandlungsgruppen randomisiert; die Placebo-Gruppe erhielt Placebo in den Wochen 0 und 2, die 160/80-Gruppe bekam 160 mg Adalimumab in Woche 0 und 80 mg in Woche 2, die 80/40-Gruppe behandelte man mit 80 mg in Woche 0 und 40 mg in Woche 2 und die 40/20-Gruppe mit 40 mg in Woche 0 und mit 20 mg in Woche 2. In der GAIN-Studie wurden 325 Patienten, die nicht mehr auf Infliximab ansprachen bzw. dieses nicht vertrugen, randomisiert und erhielten entweder 160 mg Adalimumab in Woche 0 und 80 mg in Woche 2 oder Placebo in den Wochen 0 und 2.
Die Aufrechterhaltung der klinischen Remission wurde in der CHARM-Studie bewertet. In dieser Studie erhielten 854 Patienten offen 80 mg Adalimumab in Woche 0 und 40 mg Adalimumab in Woche 2. In Woche 4 randomisierte man die Patienten und sie erhielten entweder 40 mg Adalimumab jede zweite Woche, 40 mg Adalimumab jede Woche, oder Placebo. Insgesamt dauerte die Studie 56 Wochen. Patienten mit einer klinischen Reaktion (Abnahme bei CDAI ≥70) in Woche 4 wurden (getrennt von denen, die in Woche 4 keine klinische Reaktion zeigten) stratifiziert und analysiert. Nach Woche 8 war ein Corticosteroid-Taper erlaubt.
Klinische Ergebnisse
Im Vergleich mit Placebo erreichte, ungeachtet der Tatsache, ob die Patienten TNF-Antagonist-naiv waren oder bereits vorher Infliximab ausgesetzt gewesen waren, in den Studien CLASSIC I und GAIN ein statistisch signifikant höherer Prozentsatz der mit 160/80 mg Adalimumab behandelten Gruppen die Einleitung einer klinischen Remission in Woche 4 (siehe Tabelle 12).
Tabelle 12: Einleitung einer klinischen Remission und Reaktion (in Prozent der Patienten)
CLASSIC I: Inflixima GAIN: Infliximab-erf
b-naive Patienten ahrene Patienten
Placebo N=74 Adalimumab 160/80 Placebo N=166 Adalimumab 160/80
mg N=76 mg N=159
Woche 4
Klinische Remission 12% 36%* 7% 21%*
Klinische Reaktion 24% 50%** 25% 38%**
(CR-100)
Klinische Reaktion 34% 58%** 34% 52%**
(CR-70)
Alle p-Werte sind paarweise Vergleiche von Anteilen für Adalimumab vs. Placebo
* p<0,001
** p<0,01
In der CHARM-Studie zeigten 58% (499/854) der Patienten in Woche 4 eine klinische Reaktion und wurden in der Primäranalyse bewertet. 48% der Patienten mit einer klinischen Reaktion in Woche 4 sind bereits vorher einer anderen Anti-TNF-Therapie ausgesetzt gewesen. In den Wochen 26 und 56 erreichte in den Adalimumab-Erhaltungstherapiegruppen im Vergleich mit den Patienten in der Placebo-Erhaltungstherapiegruppe ein statistisch signifikant höherer Anteil der Patienten mit einer klinischen Reaktion in Woche 4 eine klinische Remission. Ausserdem zeigte in den Adalimumab-Erhaltungstherapiegruppen, verglichen mit den Patienten in der Placebo-Erhaltungstherapiegruppe, ein statistisch signifikant höherer Anteil der Patienten, die man an der Baseline gleichzeitig mit Corticosteroiden behandelt hatte, in den Wochen 26 und 56 eine klinische Remission und war in der Lage, die Anwendung von Corticosteroiden mindestens 90 Tage lang abzusetzen (siehe Tabelle 13).
In einer Posthoc-Analyse waren krankheitsbezogene Hospitalisationen und intra-abdominale Operationen in der doppelblinden Studienphase unter Adalimumab verglichen mit Placebo statistisch signifikant vermindert.
Tabelle 13: Erhaltung der klinischen Remission und Reaktion (in Prozent der Patienten)
Placebo 40 mg Adalimumab jede 40 mg Adalimumab
zweite Woche jede Woche
Woche 26 N=170 N=172 N=157
Klinische Remission 17% 40%* 47%*
Klinische Reaktion (CR-100) 27% 52%* 52%*
Klinische Reaktion (CR-70) 28% 54%* 56%*
Patienten mit steroidfreier 3% (2/66) 19% (11/58)** 15% (11/74)**
Remission für ≥90 Tagea
Woche 56 N=170 N=172 N=157
Klinische Remission 12% 36%* 41%*
Klinische Reaktion (CR-100) 17% 41%* 48%*
Klinische Reaktion (CR-70) 18% 43%* 49%*
Patienten mit steroidfreier 5% (3/66) 29% (17/58)* 20% (15/74)**
Remission für ≥90 Tagea
* p<0,001 für Adalimumab vs. Placebo (paarweiser Vergleich der Anteile)
** p<0,002 für Adalimumab vs. Placebo (paarweiser Vergleich der Anteile)
a Von denen, die an der Baseline Corticosteroide erhielten
Die in Tabelle 13 dargestellten Ergebnisse für die klinische Remission blieben, ungeachtet einer vorherigen Exposition gegen TNF-Antagonisten, relativ konstant.
Von den Patienten, die in Woche 4 eine Reaktion zeigten und im Studienverlauf eine Remission erreichten, konnten die Patienten in den Adalimumab-Erhaltungstherapiegruppen diese Remission über einen signifikant längeren Zeitraum aufrechterhalten als die Patienten in der Placebo-Erhaltungstherapiegruppe (siehe Abbildung 2).
Abbildung 2: Tage mit klinischer Remission bei Patienten, die in der CHARM-Studie eine klinische Remission erreichten (Intent-to-Treat-Population)
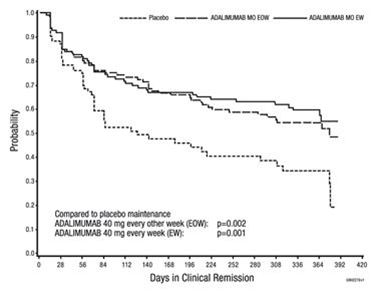
Von den Patienten, die in Woche 4 keine Reaktion zeigten, wurde eine Reaktion bei 43% der Patienten in den Adalimumab-Erhaltungstherapiegruppen bis zur Woche 12 beobachtet, verglichen mit einer Quote von 30% der Patienten in der Placebo-Erhaltungstherapiegruppe. Diese Ergebnisse lassen darauf schliessen, dass eine fortgeführte Erhaltungstherapie bis zur Woche 12 für einige Patienten, die bis zur Woche 4 keine Reaktion gezeigt hatten, von Nutzen sein kann. Eine Fortsetzung der Therapie über 12 Wochen hinaus führte nicht zu signifikant mehr Reaktionen (siehe "Dosierung/Anwendung" ).
117/276 Patienten aus der Studie CLASSIC I und 272/777 Patienten aus den Studien GAIN und CHARM wurden mindestens 3 Jahre in einer offenen Verlängerungs-Studie mit Adalimumab weiterbehandelt. 88 bzw. 189 Patienten blieben nach 3 Jahren weiterhin in klinischer Remission. Die klinische Ansprechrate (CR-100) wurde bei 102 bzw. 233 Patienten erhalten.
135 Patienten wurden in der randomisierten, placebo-kontrollierten endoskopischen Studie M05-769 (EXTEND-Studie), mit Abheilung der Schleimhaut (definiert als Verschwinden von mukosalen Ulzerationen) als primärem Endpunkt, untersucht. Nach einer 4 wöchigen Adalimumab-Induktions-Phase wurden die Patienten randomisiert. In Woche 12 zeigten 27,4% der mit Adalimumab behandelten Patienten eine Abheilung der Schleimhaut verglichen mit 13,1% der mit Placebo behandelten Patienten (p=0,056), in Woche 52 zeigten 24,2% der mit Adalimumab behandelten Patienten eine Abheilung der Schleimhaut verglichen mit 0% der mit Placebo behandelten Patienten (p<0,001).
Ergebnisse aus der Patientenperspektive/Patient-Reported Outcomes
In den Studien CLASSIC I und GAIN wurde im Vergleich mit Placebo eine statistisch signifikante Verbesserung des aufgrund des krankheitsspezifischen "Fragebogens zur entzündlichen Darmerkrankung" (Inflammatory Bowel Disease Questionnaire = IBDQ) ermittelten Gesamtscores in Woche 4 bei Patienten erreicht, die für eine Behandlung mit Adalimumab 160/80 mg randomisiert wurden. Verglichen mit der Placebo-Gruppe hat man in der CHARM-Studie in den mit Adalimumab behandelten Gruppen eine statistisch signifikante Verbesserung beim IBDQ-Gesamtscore gegenüber dem Baseline-Wert in den Wochen 26 und 56 beobachtet.
Lebensqualität und körperliche Funktionsfähigkeit
Gesundheitsbezogene Lebensqualität und körperliche Funktionsfähigkeit wurden in der Studie zur Psoriasis-Arthritis anhand des Health Assessment Questionnaire (HAQ) beurteilt. Die mit Adalimumab behandelten Patienten zeigten im Vergleich zu Patienten, die mit Placebo behandelt wurden, statistisch signifikant stärkere Verbesserungen beim HAQ Disability Index zwischen den Baseline-Werten und Woche 24.
Die Resultate des Short Form Health Survey (SF 36) bekräftigen diese Ergebnisse mit statistisch signifikantem Physical Component Summary (PCS) Score sowie statistisch signifikanten Pain and Vitality Domain Scores.
Morbus Crohn bei Kindern und Jugendlichen
Sicherheit und Wirksamkeit wurden in einer randomisierten, doppelblinden Studie an 192 Kindern und Jugendlichen im Alter zwischen 6 und einschliesslich 17 Jahren mit mässigem bis schwerem Morbus Crohn (MC), definiert als pädiatrischer Morbus-Crohn-Aktivitätsindex (PCDAI)-Score >30, untersucht.
Eingeschlossen wurden Patienten, bei denen eine konventionelle MC-Therapie (einschliesslich eines Glukokortikoids und/oder eines Immunsuppressivums) versagt hatte; es wurden auch Patienten eingeschlossen, die unter Infliximabtherapie einen Verlust des klinischen Ansprechens oder eine Unverträglichkeit entwickelt hatten.
Alle Patienten erhielten eine offene Induktionstherapie mit einer Dosis auf Basis des Körpergewichts zu Studienbeginn: 160 mg zu Woche 0 und 80 mg zu Woche 2 für Patienten ≥40 kg bzw. 80 mg und 40 mg für Patienten <40 kg.
In Woche 4 wurden die Patienten 1:1 auf Basis des derzeitigen Körpergewichts entweder in ein Behandlungsschema mit Standarddosis: 20 mg jede 2. Woche für Patienten <40 kg und 40 mg für Patienten mit ≥40 kg oder mit niedriger Dosis: 10 mg jede 2. Woche für Patienten <40 kg und 20 mg für Patienten mit ≥40 kg, randomisiert.
Ergebnisse zur Wirksamkeit
Der primäre Endpunkt der Studie war die klinische Remission in Woche 26, definiert als PCDAI-Score ≤10.
In Woche 26 waren die Raten für die klinische Remission und das klinische Ansprechen (definiert als Verringerung im PCDAI-Score um mindestens 15 Punkte im Vergleich zur Baseline) 38,7% und 59,1% für die Standarddosierung (N=93) sowie 28,4% und 48,4% für die Patienten mit der niedrigen Dosierung (N=95). Der Unterschied in den Raten der klinischen Remission und des klinischen Ansprechens in Woche 26 war statistisch nicht signifikant (p=0,075 respektive p=0,073).
In Woche 52 betrugen die Raten für die klinische Remission und das klinische Ansprechen 33,3% und 41,9% für die Standarddosierung sowie 23,2% und 28,4% für die Patienten mit der niedrigen Dosierung. Der Unterschied in der Rate des klinischen Ansprechens in Woche 52 war statistisch signifikant (p=0,038).
Bei den Patienten, die die Standarddosierung erhielten, setzten 84,8% in Woche 26 und 69,7% in Woche 52 die Glukokortikoide ab (N=33). Das Absetzen von Immunsuppressiva (nach Ermessen des Prüfarztes zu oder nach Woche 26, wenn der Patient das Kriterium für ein klinisches Ansprechen erfüllte) war 30,0% in Woche 52 (N=60). Fistelremission (definiert als Verschluss aller zum Zeitpunkt des Studienbeginns drainierender Fisteln, nachgewiesen an mindestens zwei aufeinanderfolgenden Visiten im Studienverlauf) war 46,7% in Woche 26 und 40,0% in Woche 52 für Patienten mit Standarddosierung (N=15).
Bei den Patienten mit niedriger Dosierung setzten 65,8% in Woche 26 und 60,5% in Woche 52 die Glukokortikoide ab. Das Absetzen von Immunsuppressiva betrug 29,8% in Woche 52 (N=57). Fistelremission war 38,1% in Woche 26 und 23,8% in Woche 52 für Patienten mit niedriger Dosis (N=21).
Statistisch signifikante Zunahmen (Verbesserungen) im Vergleich zur Baseline wurden im Body Mass Index und der Körpergrösse zu Woche 26 und Woche 52 für beide Behandlungsgruppen beobachtet.
Statistisch und klinisch signifikante Verbesserungen im Vergleich zur Baseline wurden auch in beiden Behandlungsgruppen für die Parameter zur Lebensqualität (einschliesslich IMPACT III) beobachtet.
Colitis ulcerosa
Die Sicherheit und Wirksamkeit wurden bei erwachsenen Patienten mit mittelschwerer bis schwerer aktiver Colitis ulcerosa (Mayo Score 6 bis 12 mit Endoskopie-Subscore 2 bis 3) in zwei randomisierten, doppelblinden, placebo-kontrollierten Studien untersucht. Eine gleichzeitig verabreichte Dauermedikation mit Aminosalicylaten, Glukokortikoiden und/oder immunmodulierenden Substanzen war erlaubt.
Die Induktion einer klinischen Remission (definiert als Mayo Score ≤2 mit keinem Subscore >1) wurde in 390 TNF-Hemmer naiven Patienten untersucht, welche in Woche 0 und 2 jeweils entweder Placebo oder Adalimumab 160 mg/80 mg oder Adalimumab 80 mg/40 mg erhielten, gefolgt von Placebo oder 40 mg Adalimumab in Woche 4 und Woche 6. Während der darauffolgenden Erhaltungsphase erhielten alle Patienten 40 mg Adalimumab alle 2 Wochen.
In Woche 8 wurde mit einer Induktionsdosis von 160 mg/80 mg Adalimumab eine klinische Remission in 18% unter Adalimumab vs. 9% unter Placebo (p=0,031) erreicht. Mit der 80 mg/40 mg Induktionsdosis wurde keine statistisch signifikante Überlegenheit von Adalimumab (10%, p=0,833) beobachtet.
Die Wirksamkeit über die Induktions- und Erhaltungsphase (insgesamt 52 Wochen) wurde in 248 Patienten mit 160 mg/80 mg/40 mg alle 2 Wochen gegen 246 Patienten mit Placebo verglichen. In Woche 8 und Woche 52 waren 16,5% (p=0,019) bzw. 17,3% (p=0,004) unter Adalimumab vs. 9,3% bzw. 8,5% unter Placebo in Remission. Die Raten bezüglich anhaltendem Ansprechen, Remission und Mukosa-Heilung sind in Tabelle 14 zusammengefasst.
Tabelle 14: Anhaltendes Ansprechen, Remission und Mukosa-Heilung in der UC-Studie II, Prozentualer Anteil der Patienten (95%-Konfidenzintervall)
Placebo und 95%-Konfidenzin Adalimumab 40 mg jede zweite Woche und
tervall 95%-Konfidenzintervall
Woche 8 und 52
Anhaltendes Ansprech 12% (CI 8,1-16,3) 24%** (CI 18,5-29,1)
en
Anhaltende Remission 4% (CI 1,6-6,5) 8%* (CI 5,0-11,9)
Anhaltende Mukosa-He 11% (CI 6,7-14,4) 19%* (CI 13,7-23,4)
ilung
a Konfidenzintervall für Proportion basierend auf Normal-Approximation der Binomialverteilung
Klinische Remission bedeutet Mayo Score ≤2 mit keinem Subscore >1;
* p<0,05 für Adalimumab versus Placebo
** p<0,001 für Adalimumab versus Placebo
Mukosa-Heilung bedeutet Endoskopie-Subscore 0 oder 1
Ansprechen bedeutet Reduktion in Mayo Score um ≥3 Punkte und ≥30% gegenüber Baseline sowie einen Rektalblutungs-Subscore von 0 oder 1 oder dessen Reduktion um ≥1 Punkt gegenüber Baseline.
Von den 125 Patienten, die in Woche 8 angesprochen hatten, zeigten in Woche 52 noch 59 (47%) ein Ansprechen, 36 (29%) waren in Remission, 51 (41%) hatten Mukosa-Heilung und 18 (20% der 90 Patienten mit Ansprechen in Woche 8 und Steroid-Behandlung an Baseline) waren in Steroidfreier Remission für ≥90 Tage.
Eine statistisch signifikante Reduktion der durch alle Ursachen sowie durch Colitis ulcerosa bedingten Hospitalisationsrate wurde in der Gesamtanalyse beider UC-Studien beobachtet.
Bei annähernd 40% Patienten der UC-Studie II versagte zuvor die anti-TNF-Behandlung mit Infliximab. Die Wirksamkeit von Adalimumab war bei diesen Patienten im Vergleich zu Patienten, die anti-TNF naïv waren, verringert. In dieser Subgruppe wurde in Woche 52 unter Adalimumab bei 10% vs. 3% unter Placebo eine Remission (p=0,039) erreicht.
Patienten der UC Studien I und II hatten die Möglichkeit, in der offenen Langzeitstudie (UC-III) weiter behandelt zu werden. 3 Jahre nach Behandlung mit Adalimumab waren 75% (301/402) weiterhin in klinischer Remission gemäss partiellem Mayo Score.
Eine Verbesserung der Lebensqualität, gemessen am krankheitsspezifischen Gesamtscore des IBDQ (Inflammatory Bowel Disease Questionnaire) wurde in Woche 52 gegenüber Placebo erreicht (p=0,007).
Ankylosierende Spondylitis (Morbus Bechterew)
Die Wirksamkeit von Adalimumab 40 mg alle 2 Wochen subkutan verabreicht wurde bei 393 Patienten mit aktiver ankylosierender Spondylitis (Krankheitsaktivität [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)]) >4 (mittlere Basiswerte 6,3 sowohl bei der Adalimumab wie auch der Placebo-Gruppe), die nur unzureichend auf herkömmliche Therapien angesprochen haben, in 2 randomisierten doppelblind, placebo-kontrollierten Studien über 24 Wochen untersucht. Als Begleittherapie erhielten 79 Patienten (20,1%) krankheitsmodifizierende Antirheumatika und 37 (9,4%) Glukokortikoide. Der verblindeten Periode folgte eine offene Fortsetzungsperiode, während der die Patienten über bis zu 28 zusätzliche Wochen jede zweite Woche 40 mg Adalimumab subkutan erhielten.
In der grösseren Studie mit 315 Patienten, zeigten die Resultate statistisch signifikante Verbesserungen der Anzeichen und Symptome der ankylosierenden Spondylitis bei denjenigen Patienten, welche Adalimumab erhielten verglichen mit denjenigen, unter Placebo. Das signifikante Ansprechen wurde zuerst nach 2 Wochen gesehen und blieb bis Woche 24 erhalten.
Die Assessment in Ankylosing Spondylitis (ASAS) 20/50/70 Ansprechraten wurden in Woche 12 bei 58%, 38% und 23% der Patienten unter Adalimumab erreicht verglichen mit 21%, 10% und 5% der Patienten unter Placebo (p<0,001 Adalimumab versus Placebo). Ein grob vergleichbares Ansprechen wurde in Woche 24 gesehen.
Wie mit BASDAI festgestellt, führte die Behandlung mit Adalimumab zu Verbesserungen der Anzeichen und Symptome. Bei 45% der mit Adalimumab behandelten Patienten wurde in Woche 12 mindestens eine 50% Reduktion der BASDAI Basiswerte erreicht verglichen mit 16% der mit Placebo behandelten Patienten (p<0,01). Vergleichbare Resultate wurden in Woche 24 gesehen.
Zusätzlich war die mittlere Abweichung der Basiswerte für C-Reaktives-Protein (CRP) in Woche 12 unter Adalimumab-Behandlung (-1,3 mg/dl) grösser verglichen mit Placebo Behandlung (-0,1 mg/dl), (p<0,001).
Vergleichbare Resultate (nicht alle mit statistischer Signifikanz) wurden in der kleineren, randomisierten, doppelblinden, placebo-kontrollierten Studie bei 82 erwachsenen Patienten mit aktiver ankylosierender Spondylitis gesehen.
In den ankylosierenden Spondylitis Studien wurden die von Patienten berichteten Resultate anhand des Generic Health Status Questionnaire Short Form–36 (SF 36) und dem Disease Specific Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) bewertet. Die mit Adalimumab behandelten Patienten hatten signifikant grössere Verbesserungen des ASQoL und des "Physical Components" des SF 36 in Woche 12 als die Patienten in der Placebo-Gruppe, welche bis Woche 24 aufrechterhalten blieben.
Psoriasis
Die Wirksamkeit und Sicherheit von Adalimumab wurde in randomisierten, doppelblinden, kontrollierten Studien bei über 1600 Patienten mit mässiger bis schwerer, chronischer Plaque-Psoriasis untersucht, die ≥18 Jahre alt waren und bei denen eine systemische Therapie oder eine Phototherapie angezeigt war.
In Studie 1 wurden 1212 Patienten mit chronischer Plaque-Psoriasis mit betroffener Körperoberfläche (Body Surface Area, BSA) ≥10% und einem Psoriasis Area and Severity Index (PASI) ≥12 in drei Behandlungsperioden evaluiert. In Periode A erhielten die Patienten subkutan Placebo oder eine Initialdosis 80 mg Adalimumab in Woche 0 gefolgt von 40 mg Adalimumab alle zwei Wochen ab Woche 1. Nach 16 Therapie-Wochen wechselten diejenigen Patienten, welche in Woche 16 mindestens einen PASI 75 erreicht hatten, definiert als PASI Score Verbessung von mindestens 75% relativ zum Basiswert, in die offene Fortsetzungssperiode B. Sie erhielten Adalimumab 40 mg alle zwei Wochen. Nach 17 Wochen unverblindeter Therapie wechselten diejenigen Patienten, welche mindestens PASI 75 in Woche 33 beibehielten und in der Behandlungperiode A aktive Therapie erhielten, in die Behandlungsperiode C. Sie erhielten weitere 19 Wochen lang Adalimumab 40 mg oder Placebo alle 2 Wochen. Über alle Behandlungsgruppen war der mittlere PASI-Basiswert 18,9. Der Physician's Global Assessment (PGA) Basiswert über alle Gruppen reichte von "mässig" (52,6%) über "schwer" (41,3%) bis "sehr schwer" (6,1%).
Studie 2 verglich die Wirksamkeit und Sicherheit von Adalimumab versus Methotrexat und Placebo in 271 Patienten mit chronischer Plaque-Psoriasis mit betroffener BSA 10% und PASI ≥10. Über 16 Wochen erhielten die Patienten Placebo, Methotrexat (7,5–20 mg) oder eine Initialdosis Adalimumab 80 mg subkutan in Woche 0 gefolgt von Adalimumab 40 mg alle zwei Wochen ab Woche 1. Über alle Behandlungsgruppen war der mittlere PASI-Basiswert 19,7. Der PGA Basiswert über alle Gruppen reichte von "mild" (0,4%), "mässig" (47,8%) über "schwer" (45,6%) bis "sehr schwer" (6,3%).
1469 Patienten aus Phase-II- und Phase-III-Studien wurden in einer offenen 3phasigen Fortsetzungsstudie mit einer Weiterbehandlungsphase (104-252 Wochen), einer Behandlungsunterbruchsphase (bis Rückfall oder maximal 52 Wochen) und anschliessenden Wiederbehandlungsphase (16 Wochen) aufgenommen.
In Studie 3 wurden 148 Patienten mit chronischer Plaque-Psoriasis mit betroffener BSA ≥5% über mindestens 1 Jahr evaluiert. Die Patienten erhielten Placebo oder Initialdosis Adalimumab 80 mg in Woche 0 gefolgt von Adalimumab 40 mg alle zwei Wochen ab Woche 1 bzw. eine Initialdosis Adalimumab 80 mg in Woche 0 gefolgt von Adalimumab 40 mg jede Woche ab Woche 1.
Klinische Resultate
Primärer Endpunkt in den Studien 1, 2 und 3 war der Prozentsatz Patienten, die in Woche 16 (Studie 1 und 2) oder in Woche 12 (Studie 3) eine Reduktion im PASI Score von mindestens 75% vom Basiswert (PASI 75) erreichten. Weiter wurden in den Psoriasis Studien 1–3 unter anderem PGA und andere PASI Werte untersucht.
Studie 1: Zusätzlich zum primären Endpunkt oben, hatte Studie 1 als zweiten primären Endpunkt den Verlust an adäquatem Ansprechen nach Woche 33 sowie in oder vor Woche 52. Ein Verlust an adäquater Ansprechrate war definiert als eine <PASI 50 Antwort relativ zum Basiswert mit einem minimalen 6-Punkt erhöhtem PASI Wert relativ zur Woche 33.
Kontrollierte Daten über eine Therapie mit Adalimumab im Vergleich zu Placebo liegen für eine Dauer von 52 Wochen vor. In einer Placebo-kontrollierten Vergleichsstudie mit Patienten, bei welchen unter einer Adalimumab-Therapie eine anhaltende Beschwerdefreiheit bis zur 33. Woche erzielt wurde, blieben unter Weiterführung mit Adalimumab 95,1% der Patienten bis zur 52. Woche rezidivfrei, unter Placebo (d.h. nach Absetzen von Adalimumab in der Woche 33) 71,6%.
Von den Patienten, welche nach der erneuten Randomisierung auf Placebo einen Verlust des adäquaten Ansprechens zeigten und anschliessend in die offene Fortsetzungsperiode eingeschlossen wurden, erzielten 38% (25/66) bzw. 55% (36/66) nach 12 bzw. 24 Wochen aktiver Therapie wieder ein PASI 75-Ansprechen.
Dieses verzögerte Ansprechen nach Rezidiv dürfte im Zusammenhang stehen mit einem schwerwiegenden Verlauf der Psoriasiserkrankung bei dieser Untergruppe von Patienten.
In den Psoriasis Studien 1 und 2 erreichten in Woche 16 mehr der mit Adalimumab behandelten Patienten eine mindestens 75% Reduktion vom PASI Basiswert als die mit Placebo behandelten Patienten. Andere relevante klinische Parameter inklusive PASI 100 (z.B. völliges Verschwinden der Psoriasis Hautanzeichen) und PGA "frei von oder minimal" waren im Vergleich zu Placebo ebenfalls verbessert.
Studie 2: In Psoriasis Studie 2 erreichten die mit Adalimumab behandelten Patienten bessere Resultate für PASI 75, PASI 100 und PGA "frei von oder minimal" als die mit Methotrexat behandelten.
Tabelle 15: Psoriasis Studie 1 - Wirksamkeit in Woche 16 (% der Patienten)
Placebo N=398 Adalimumab 40 mg eow N=814
≥PASI 75 6,5 70,9a
PASI 100 0,8 20,0a
PGA: Clear/minimal 4,3 62,2a
a p<0,001, Adalimumab vs. Placebo
Tabelle 16: Psoriasis Studie 2 - Wirksamkeit in Woche 16 (% der Patienten)
Placebo N=53 MTX N=110 Adalimumab 40 mg eow N=108
≥PASI 75 18,9 35,5 79,6a, b
PASI 100 1,9 7,3 16,7a, b
PGA: Clear/minimal 11,3 30,0 73,1a, b
a p<0,001, Adalimumab vs. Placebo
b p<0,001 Adalimumab vs. Methotrexat
Fortsetzungsstudie: Insgesamt 233 Patienten, die ein PASI-75-Ansprechen in Woche 16 und Woche 33 gezeigt hatten und in der Psoriasis-Studie 1 für 52 Wochen eine Adalimumab Dauertherapie erhalten hatten, wurden mit Adalimumab in der offenen Fortsetzungsstudie weiterbehandelt. Das PASI-75-Ansprechen bzw. das PGA-Ansprechen, definiert als PGA "frei von" oder "minimal" war bei diesen Patienten nach weiteren 108 offenen Behandlungswochen (insgesamt 160 Wochen) 74,7% bzw. 59,0%. In einer Non Responder Imputation- (NRI-) Analyse, in der alle Patienten als Non-Responder betrachtet wurden, die aus der Studie aufgrund von Nebenwirkungen oder mangelnder Wirksamkeit ausschieden oder bei denen die Dosis erhöht wurde, betrug bei diesen Patienten das PASI-75-Ansprechen bzw. das PGA-Ansprechen, definiert als PGA "frei von" oder "minimal" nach weiteren 108 Wochen der offenen Fortsetzungsbehandlung (insgesamt 160 Wochen) 69,6% bzw. 55,7%.
In der Fortsetzungsstudie wurden 347 Patienten, die dauerhaft angesprochen hatten, einer Analyse einer Behandlungsunterbrechung und -wiederaufnahme zugeführt. Während der Phase der Behandlungsunterbrechung kehrten die Psoriasis-Symptome bei 54,2% (188/347) im Verlauf von durchschnittlich etwa 5 Monaten zurück (Verminderung des PGA auf "mittelschwer" oder schlechter). Keiner dieser Patienten erfuhr einen Rebound-Effekt während der Unterbrechungsphase. Insgesamt 76,5% (218/285) der Patienten in der anschliessenden Wiederbehandlungsphase hatten 16 Wochen nach Wiederaufnahme der Behandlung ein PGA-Ansprechen, definiert als PGA "frei von" oder "minimal" , unabhängig davon, ob sie während des Absetzens einen Rückfall hatten (69,1% [123/178] oder nicht 88,8% [95/107]). Es wurde ein ähnliches Sicherheitsprofil in der Phase, in der die Behandlung wiederaufgenommen wurde, beobachtet, wie vor der Behandlungsunterbrechung.
Studie 3: Resultate aus Psoriasis Studie 3 unterstützen die in Studie 1 und 2 gezeigte Wirksamkeit. Patienten in Studie 1, welche PASI 75 Ansprechraten zeigten und welche in Woche 33 in die Adalimumab-Gruppe re-randomisiert wurden, zeigten weniger Verlust in adäquater Ansprechrate in oder vor Woche 52 als in die Placebo-Gruppe re-randomisierte Patienten (4,9% vs. 28,4%, p<0,001).
In einer offenen Fortsetzungsstudie wurde in Patienten, die nach initial mindestens partiellem Ansprechen ein sekundäres Therapieversagen zeigten, d.h. gegenüber der Baseline keine PASI 50-Antwort mehr aufwiesen, und die Dosis von 40 mg jede zweite Woche auf 40 mg wöchentlich gesteigert wurde, bei 28,3% (63/223) bzw. 39,5% (88/223) ein PASI 75-Ansprechen in Woche 12 bzw. 24 beobachtet.
Lebensqualität
Von Patienten berichtete Ergebnisse (Patient Reported Outcomes, PRO) wurden anhand diverser Parameter evaluiert. Lebensqualität wurde anhand des krankheitsspezifischen Dermatology Life Quality Index (DLQI) in Studie 1 und 2 bestimmt. In Studie 1 zeigten die mit Adalimumab behandelten Patienten in Woche 4 und 16 Verbesserungen im DLQI Gesamtscore, Krankheitsgrad, Schmerzen und Pruritus verglichen mit den mit Placebo behandelten Patienten. Die DLQI Resultate blieben bis Woche 52 aufrechterhalten.
In Studie 2 zeigten die mit Adalimumab behandelten Patienten in Woche 16 Verbesserungen im DLQI Gesamtscore, Krankheitsgrad, und Pruritus verglichen mit den mit Placebo oder Methotrexat behandelten Patienten und klinisch aussagekräftige Verbesserungen bezüglich Schmerzen verglichen mit den mit Placebo behandelten Patienten.
Die generelle gesundheitsbezogene Lebenqualität wurde anhand des Short Form Health Survey (SF 36) in Studie 1 bestimmt. Die mit Adalimumab behandelten Patienten zeigten eine signifikant grössere Verbesserung in den SF 36 Physical Component Summary (PCS) and Mental Component Summary (MCS) Scores.
Psoriasis bei Kindern und Jugendlichen
Die Wirksamkeit von Adalimumab wurde in einer randomisierten, doppelblinden, kontrollierten Studie mit 114 pädiatrischen Patienten ab 4 Jahren mit schwerer chronischer Plaque-Psoriasis untersucht, deren topische Therapie und Heliotherapie oder Phototherapie unzureichend war (schwere chronische Plaque-Psoriasis ist definiert durch einen PGA ≥4 oder >20% BSA-Beteiligung oder >10% BSA Beteiligung mit sehr dicken Läsionen oder PASI ≥20 oder ≥10 mit klinisch relevanter Beteiligung von Gesicht, Genitalbereich oder Händen/Füssen).
Patienten erhielten Adalimumab 0,8 mg/kg jede zweite Woche (bis zu 40 mg), 0,4 mg/kg jede zweite Woche (bis zu 20 mg) oder Methotrexat 0,1–0,4 mg/kg per os wöchentlich (bis zu 25 mg). Ein Vergleich mit höher dosiertem subkutan verabreichtem MTX wurde nicht durchgeführt.
Die Altersverteilung der Patienten ist in Tabelle 17 dargestellt.
Tabelle 17: Patientenverteilung nach Alter (Patienten randomisiert zu Adalimumab 0,8 mg/kg, jede zweite Woche)
Altersgruppe (Jahre) Anzahl Patienten zu Beginn der Studie n (%)
>6-9 7 (18,4)
>9-12 8 (21,1)
>12-15 13 (34,2)
>15 10 (26,3)
17 (45%) der Patienten waren vorgängig systemisch behandelt worden (Etanercept und/oder nichtbiologische Wirkstoffe inklusive Acitretin, Ciclosporin, Methotrexat und weitere). Patienten, welche auf vorhergehende Therapien mit Methotrexat nicht ansprachen, wurden ausgeschlossen.
Die primären Endpunkte entsprachen einem PASI 75 und PGA "frei von" / "minimal" . In Woche 16 hatten mehr Patienten, die in die Adalimumab-Gruppe mit 0,8 mg/kg randomisiert waren, höhere Wirksamkeitsnachweise als jene, die in die MTX-Gruppe randomisiert waren.
Tabelle 18: Wirksamkeitsergebnisse bei pädiatrischer Plaque-Psoriasis in Woche 16
MTXa N=37 Adalimumab 0,8 mg/kg jede
zweite Woche N=38
PASI 75b 12 (32,4%) 22 (57,9%)
PGA: frei/minimalc 15 (40,5%) 23 (60,5%)
a MTX=Methotrexat b p=0,027, Adalimumab 0,8 mg/kg
versus MTX c p=0,083, Adalimumab 0,8 mg/kg versus
MTX
Es liegen keine vergleichenden Daten mit Methotrexat über 16 Wochen hinaus vor.
Bei Patienten, die PASI 75 und einen PGA, definiert als "frei von" oder "minimal" , erreichten, wurde die Behandlung bis zu 36 Wochen lang abgesetzt. Diese Patienten wurden dahingehend beobachtet, ob sie einen Rückfall erlitten (Verlust des Ansprechens hinsichtlich PGA). Die Behandlung der Patienten wurde dann wieder mit 0,8 mg Adalimumab/kg jede zweite Woche für weitere 16 Wochen aufgenommen. Die Ansprechraten, die während der erneuten Behandlung beobachtet wurden, waren: PASI 75-Ansprechen von 78,9% (15 von 19 Patienten) und PGA (definiert als "frei von" oder "minimal" ) von 52,6% (10 von 19 Patienten).
In der offenen Fortsetzungsphase der Studie wurden für bis zu weiteren 52 Wochen ein PASI 75- Ansprechen und PGA, definiert als "frei von" oder "minimal" , aufrechterhalten; es gab keine neuen Erkenntnisse zur Sicherheit.
Hidradenitis suppurativa
Die Wirksamkeit und Sicherheit von Adalimumab wurde in randomisierten, doppelblinden, Placebokontrollierten Studien und in einer offenen Fortsetzungsstudie bei 727 erwachsenen Patienten mit mittelschwerer bis schwerer Hidradenitis suppurativa untersucht. Die Patienten hatten entweder eine Kontraindikation, ein ungenügendes Ansprechen oder eine Intoleranz gegenüber einer systemischen Antibiotikatherapie und waren im Hurley Stadium II oder III mit mindestens 3 Abszessen oder entzündlichen Knoten.
Zwei randomisierte, doppelblinde, Placebo-kontrollierte Phase III Studien (HS-I und -II) mit insgesamt 633 erwachsenen Patienten umfassten jeweils eine initiale 12 Wochen doppelblinde Behandlungsperiode (Periode A) und eine nachfolgende 24 Wochen doppelblinde Behandlungsperiode (Periode B). In Periode A erhielten die Patienten Adalimumab (160 mg in Woche 0, 80 mg in Woche 2 und 40 mg jede Woche ab Woche 4 bis 11) oder Placebo. Nach 12 Wochen wurden Patienten, die in Periode A Adalimumab erhalten hatten, in Periode B rerandomisiert auf Adalimumab 40 mg wöchentlich, Adalimumab 40 mg jede 2. Woche oder Placebo bis Woche 35. Patienten, die in Periode A Placebo erhalten hatten, wurden in Periode B auf Adalimumab 40 mg wöchentlich (HS-I) oder Placebo (HS-II) randomisiert.
Eine gleichzeitige Behandlung mit oralen Antibiotika war in Studie HS-II erlaubt.
Die Patienten beider HS Studien konnten an einer offenen Fortsetzungsstudie teilnehmen, in welcher Adalimumab 40 mg wöchentlich verabreicht wurde. Die durchschnittliche Exposition lag in allen Adalimumab Populationen bei 762 Tagen. In allen 3 Studien verwendeten die Patienten täglich topische antiseptische Spülungen.
Klinische Wirksamkeit
Die Verringerung der entzündlichen Läsionen und die Prävention einer Verschlimmerung der Abzesse und dränierenden Fisteln wurde mit Hilfe des Hidradenitis Suppurativa Clinical Response scores (HiSCR; mindestens 50% Reduktion der Anzahl aller Abszesse und entzündlichen Knoten ohne Anstieg der Anzahl an Abszessen und ohne Anstieg der Anzahl an drainierenden Fisteln gegenüber Baseline) untersucht.
In Woche 12 erzielte in beiden Studien (HS-I und –II) ein signifikant grösserer Anteil der Adalimumab behandelten Patienten den HiSCR gegenüber Placebo sowie in der HS-II Studie erzielte ein signifikant höherer Anteil eine relevante Verminderung des mit HS verbundenen Hautschmerzes (siehe Tabelle 19). Patienten, die mit Adalimumab behandelt wurden, hatten zudem ein signifikant reduziertes Risiko des Wiederaufflammens der Erkrankung während der initialen 12 Wochen der Behandlung.
Tabelle 19: HS Studien I und II – Wirksamkeit in Woche 12
HS Studie I HS Studie II
Placebo Adalimumab 40 mg Placebo Adalimumab 40 mg
wöchentlich wöchentlich
Hidradenitis suppura N=154 40 (26,0%) N=153 64 (41,8%)* N=163 45 (27,6%) N=163 96 (58,9%)***
tiva Klinische
Wirksamkeit (HiSCR)a
* p<0,05, ***p<0,001, Adalimumab vs. Placebo
a Innerhalb aller randomisierter Patienten.
Bei Patienten, die Adalimumab wöchentlich erhielten, wurde die Gesamt- HiSCR Rate bis zur Woche 96 aufrechterhalten.
Uveitis
Die Sicherheit und Wirksamkeit von Adalimumab wurde im Rahmen von zwei randomisierten, placebo-kontrollierten, doppelblinden Studien (UV-Studie 1 (M10-877) und UV-Studie 2 (M10-880)) über maximal 80 Wochen bei erwachsenen Patienten mit nicht-infektiöser intermediärer Uveitis, posteriorer Uveitis oder Panuveitis (auch "nicht-infektiöse Uveitis des hinteren Augenabschnitts" ) untersucht. Dabei waren Patienten mit isolierter anteriorer Uveitis ausgeschlossen. Die Patienten erhielten Placebo oder Adalimumab in einer Initialdosis von 80 mg, und beginnend in der darauffolgenden Woche 40 mg alle zwei Wochen.
Eine Begleitmedikation mit einem konventionellen Immunsuppressivum (Ciclosporin A, Methotrexat, Mycophenolat-Mofetil, Azathioprin, Tacrolimus) war in stabiler Dosierung erlaubt. Beide Studien hatten als primären Wirksamkeitsendpunkt die "Zeit bis zu einem Therapieversagen" . Als Therapieversagen war ein kombinierter Endpunkt definiert mit folgenden Komponenten: 1) entzündliche chorioretinale und/oder entzündliche retinale Gefässveränderungen, 2) der Grad der Vorderkammerzellen, 3) der Grad einer Glaskörpertrübung (Vitreous Haze) und 4) die beste korrigierte Sehschärfe (BCVA).
In UV-Studie 1 wurden 217 Patienten untersucht, die trotz Kortikosteroid-Therapie (orales Prednison in einer Dosis von 10 bis 60 mg/Tag) an einer aktiven Uveitis litten. Alle Patienten erhielten bei Einschluss in die Studie eine standardisierte Prednison-Dosis von 60 mg/Tag mit obligatem Dosisreduktionsschema und vollständigem Absetzen der Kortikosteroide bis Woche 15.
In UV-Studie 2 wurden 226 Patienten mit inaktiver Uveitis untersucht, die bei Studienbeginn zur Kontrolle ihrer Erkrankung eine chronische Kortikosteroid-Therapie (orales Prednison 10 bis 35 mg/Tag) benötigten. Die Studienmedikation wurde unter Fortführung der etablierten Kortikosteroiddosis eingeführt, und es folgte ein obligates Dosisreduktionsschema mit vollständigem Absetzen der Kortikosteroide bis Woche 19.
Klinisches Ansprechen
Die Ergebnisse beider Studien zeigten bei den mit Adalimumab behandelten Patienten eine statistisch signifikant längere Zeit bis zum ersten Therapieversagen als bei den mit Placebo behandelten Patienten (siehe Tabelle 20). Ebenso zeigten beide Studien eine frühe und anhaltende Wirkung von Adalimumab in Bezug auf die Verzögerung des ersten Therapieversagens gegenüber Placebo (siehe Abbildung 3).
Tabelle 20: Zeit bis zum ersten Therapieversagen in den UV-Studien 1 und 2
Untersuchte Behandlu N Versagen N (%) Mediane Zeit bis HRa 95% KI für HRa p-Wertb
ng Versagen (Monate)
Zeit bis zu einem
Therapieversagen in
oder nach Woche 6
in UV-Studie 1
Primäre Analyse
(ITT)
Placebo 107 84 (78,5) 3,0 -- -- --
Adalimumab 110 60 (54,5) 5,6 0,50 0,36; 0,70 <0,001
Zeit bis zu einem
Therapieversagen in
oder nach Woche 2
in UV-Studie 2
Primäre Analyse
(ITT)
Placebo 111 61 (55,0) 8,3 -- -- --
Adalimumab 115 45 (39,1) n.a.c 0,57 0,39; 0,84 0,004
Anmerkung: Ein Therapieversagen in oder nach Woche 6 (UV-Studie 1) bzw. in oder nach Woche 2 (UV-Studie 2) wurde als Ereignis gewertet. Abbrüche aus anderen Gründen als Therapieversagen wurden zum Zeitpunkt des Abbruchs zensuriert.
a HR von Adalimumab vs. Placebo aus der Proportional-Hazards-Regression mit Behandlung als Faktor.
b 2-seitiger p-Wert aus dem Log-Rangtest.
c n.a. = Nicht abschätzbar. Weniger als die Hälfte der infrage kommenden Patienten hatte ein Ereignis während der Studiendauer von 80 Wochen.
Abbildung 3: Kaplan-Meier-Kurven für die Zeit bis zum ersten Therapieversagen in oder nach Woche 6 (UV-Studie 1) oder in oder nach Woche 2 (UV-Studie 2)


Eine Sensitivitätsanalyse des primären Endpunktes, in welcher Patienten, die vorzeitig abbrachen und solche, die unerlaubterweise Kortikosteroide als Begleitmedikation nahmen, als Behandlungsversager gezählt wurden, zeigte einen starken Behandlungseffekt zugunsten von Adalimumab (UV I: HR=0,66; 95% CI 0,48–0,88; p=0,005; UV II: HR=0,59; 95% CI 0,42-0,81; p=0,001).
In beiden Studien trugen alle Komponenten des primären Endpunkts kumulativ zur Gesamtdifferenz zwischen der Adalimumab- und Placebogruppe bei.
In UV-Studie 1 wurden statistisch signifikante Differenzen zugunsten von Adalimumab im Hinblick auf die Änderung der Anzahl der Zellen in der Vorderkammer, beim Grad einer Glaskörpertrübung und beim Visus (logMAR BCVA) beobachtet (mittlere Änderung vom besten Wert vor Woche 6, LOCF-Analyse; p-Werte: 0,011; <0,001 bzw. 0,003).
UV-Studie II tendierte in die gleiche Richtung ohne statistische Signifikanz zu erreichen.
In UV-Studie I zeigten der mittlere Verlauf im Grad der Vorderkammerzellen sowie im Grad der Glaskörpertrübung und des Visus (logMAR BCVA) eine vergleichbare Verbesserung in beiden Behandlungsgruppen während der ersten 4-6 Behandlungswochen unter Ausschleichen der Kortikosteroidbegleitmedikation; anschliessend war unter Adalimumab im Vergleich zu Placebo ein geringerer Anstieg im Ausmass der Entzündung und ein geringerer Verlust der Sehstärke zu beobachten. Auch in der UV-Studie II wurde, verglichen mit Placebo, ein geringerer Anstieg im Ausmass der Entzündung und ein geringerer Verlust der Sehstärke beobachtet.
46 von insgesamt 417 Teilnehmern, die in die unkontrollierte langfristige Fortsetzungsstudie der Studien UV I und UV II aufgenommen wurden, galten als ungeeignet (z.B. Entwicklung von sekundären Komplikationen einer diabetischen Retinopathie, wegen einer Kataraktoperation oder Vitrektomie) und wurden von der primären Wirksamkeitsanalyse ausgeschlossen. Von den 371 verbliebenen Patienten absolvierten 276 auswertbare Patienten die offene Adalimumab-Behandlung bis Woche 78. Auf Grundlage der beobachteten Daten erreichten 222 (80,4%) Patienten unter gleichzeitiger Steroidgabe in einer Dosis von ≤7,5 mg täglich eine Krankheitskontrolle (keine aktiven entzündlichen Läsionen, Grad der Vorderkammerzellen ≤0,5+, Grad der Glaskörpertrübung ≤0,5+), während 184 (66,7%) Patienten eine steroidfreie Krankheitskontrolle erzielten. In Woche 78 hatte sich der BCVA bei 88,4% der Augen entweder verbessert oder war gleich geblieben (Verminderung um <5 Buchstaben). Von den Patienten, die die Studienteilnahme vor Woche 78 beendeten, brachen 11% ihre Teilnahme wegen unerwünschter Ereignisse und 5% wegen eines unzureichenden Ansprechens auf die Adalimumab-Behandlung ab.
Lebensqualität
In UV-Studie 1 bewirkte die Behandlung mit Adalimumab einen Erhalt der Visus-abhängigen Funktion und der gesundheitsbezogenen Lebensqualität (beurteilt anhand des NEI VFQ-25).
Vergleichsstudie zur rheumatoiden Arthritis (RA) zwischen AMGEVITA und dem Referenzarzneimittel
In der randomisierten, aktiv kontrollierten doppelblinden Vergleichsstudie (RA-ABP-Studie 1) an 526 Patienten ≥18 Jahren mit mässiger bis schwerer aktiver rheumatoider Arthritis und unzureichendem Ansprechen auf Methotrexat wurden die Wirksamkeit und die Sicherheit von AMGEVITA im Vergleich mit dem Referenzarzneimittel beurteilt. Die Patienten erhielten entweder 40 mg AMGEVITA oder die gleiche Menge des Referenzarzneimittels subkutan alle zwei Wochen über bis zu 22 Wochen.
Der primäre Endpunkt, das ACR20-Ansprechen, lag innerhalb des vorgegebenen Äquivalenzbereichs und zeigte eine klinische Äquivalenz zwischen AMGEVITA und dem Referenzarzneimittel auf.
Das allgemeine Sicherheitsprofil von AMGEVITA ähnelt dem des Referenzarzneimittels.
Vergleichsstudie zur Plaque-Psoriasis (Ps) zwischen AMGEVITA und dem Referenzarzneimittel
In einer randomisierten, aktiv kontrollierten Doppelblindstudie (Ps-ABP-Studie 1) an 350 Patienten ≥18 Jahren mit mässiger bis schwerer Plaque-Psoriasis (Ps), bei denen eine systemische Therapie oder eine Phototherapie in Frage kam, wurden die Wirksamkeit und die Sicherheit von AMGEVITA im Vergleich mit dem Referenzarzneimittel beurteilt. Die Patienten erhielten entweder AMGEVITA oder das Referenzarzneimittel in einer Anfangs- bzw. Sättigungsdosis von 80 mg subkutan in Woche 1/Tag 1, danach 40 mg subkutan alle zwei Wochen, beginnend eine Woche nach Verabreichung der Anfangsdosis. Die Verbesserung des PASI-Scores in Prozent gegenüber dem Ausgangswert in Woche 16 lag innerhalb des vorgegebenen Äquivalenzbereichs und belegte so die klinische Äquivalenz zwischen AMGEVITA und dem Referenzarzneimittel.
PharmakokinetikAbsorption
Nach subkutaner Applikation einer Einmaldosis von 40 mg kam es zu einer langsamen Absorption des Wirkstoffes Adalimumab. Dabei wurden etwa fünf Tage nach der Gabe die maximalen Serumkonzentrationen erreicht. Die aus drei Studien berechnete durchschnittliche absolute Bioverfügbarkeit von Adalimumab nach Gabe einer Einmaldosis von 40 mg subkutan belief sich auf 64%. Die Konzentrationen nach intravenösen Einmaldosierungen, die zwischen 0,25 bis zu 10 mg/kg lagen, waren annähernd dosisproportional.
Nach einer subkutanen Applikation von 40 mg Adalimumab alle zwei Wochen bei Patienten mit rheumatoider Arthritis liess sich die Akkumulation von Adalimumab, ausgehend von der Halbwertszeit, mit mittleren Konzentrationen bei Steady State von etwa 5 µg/ml (ohne gleichzeitige Gabe von Methotrexat) bzw. 8 bis 9 µg/ml (bei gleichzeitiger Gabe von Methotrexat) voraussagen. Im Steady State stiegen die Talspiegel von Adalimumab im Serum nach subkutaner Gabe von 20, 40 und 80 mg jede zweite Woche bzw. jede Woche etwa proportional zur Dosis an.
Bei Patienten mit Morbus Crohn erreicht man während der Induktionsperiode mit einer Aufsättigungsdosis von 160 mg Adalimumab in Woche 0, gefolgt von 80 mg Adalimumab in Woche 2, Serum-Talspiegel für Adalimumab von etwa 12 mcg/ml. Durchschnittliche Talspiegel von etwa 7 mcg/ml wurden bei Patienten mit Morbus Crohn beobachtet, die eine Erhaltungsdosis von 40 mg Adalimumab jede zweite Woche erhielten.
Bei Patienten mit Colitis ulcerosa wurde mit der Induktionsdosis von 160 mg Adalimumab in Woche 0, gefolgt von 80 mg in Woche 2 eine Talkonzentration von Adalimumab im Serum von ca. 12 µg/ml während der Einleitungstherapie erreicht. Die durchschnittliche Steady-State-Talkonzentration in der Erhaltungsphase lag bei 8 µg/ml.
Bei Patienten mit Hidradenitis suppurativa wurde mit der Dosis von 160 mg Adalimumab in Woche 0, gefolgt von 80 mg in Woche 2 eine Talkonzentration von Adalimumab im Serum von ca. 7 bis 8 µg/ml in Woche 2 und 4 erreicht. Die durchschnittliche Steady-State-Talkonzentration von Woche 12 bis 36 lag bei ca. 8 bis 10 µg/ml während der wöchentlichen Verabreichung von 40 mg Adalimumab.
Bei Patienten mit Uveitis bewirkte eine Aufsättigungsdosis in Höhe von 80 mg Adalimumab in Woche 0, gefolgt von 40 mg Adalimumab alle zwei Wochen, beginnend in Woche 1, mittlere Steady-State-Konzentrationen in Höhe von etwa 8 bis 10 µg/ml.
Distribution
Nach subkutaner Applikation einer Einmaldosis von 40 mg kam es zu einer langsamen Distribution des Wirkstoffes Adalimumab.
Nach intravenöser Einmalgabe von Dosierungen, zwischen 0,25 und 10 mg/kg, belief sich das Distributionsvolumen (Vss) auf Werte zwischen 4,7 und 6,0 Liter, was auf eine gleichmässige Verteilung zwischen der vaskularen und der extravaskularen Flüssigkeit hinweist. Die in der Synovialflüssigkeit bei verschiedenen Patienten mit rheumatoider Arthritis festgestellten Konzentrationen von Adalimumab bewegten sich zwischen 31% und 96% der Werte, die im Serum ermittelt worden waren.
Metabolismus
Keine Angaben.
Elimination
Nach intravenös applizierten Einmaldosierungen, die zwischen 0,25 und 10 mg/kg lagen, belief sich die Clearance normalerweise auf Werte unter 12 ml/Stunde. Die mittlere Halbwertszeit betrug etwa zwei Wochen. Die Körpergewicht-normalisierte Clearance zwischen juvenilen und erwachsenen Patienten ist ähnlich.
Im Rahmen von Langzeitstudien, bei denen das Arzneimittel länger als zwei Jahre verabreicht wurde, ergaben sich keine Hinweise auf Veränderungen bei der Clearance.
Kinetik spezieller Patientengruppen
Die pharmakokinetischen Auswertungen der Patientengruppen, die die Daten von mehr als 1300 erwachsenen Patienten mit Rheumatoider Arthritis und 171 Patienten mit juveniler idiopathischer Arthritis (4-17 Jahre) erfassen, zeigten eine höhere scheinbare Clearance mit steigendem Körpergewicht. Auch in der Gegenwart von Antikörpern gegen Adalimumab ist die Clearance erhöht. Die Kinetik von Adalimumab bei Patienten mit einer eingeschränkten Leber- oder Nierenfunktion ist nicht untersucht worden.
Kinder und Jugendliche
Polyartikuläre juvenile idiopathische Arthritis
Nach subkutaner Verabreichung von 24 mg/m2 (bis zu einer Maximaldosis von 40 mg) jede zweite Woche an Patienten mit polyartikulärer juveniler idiopathischer Arthritis waren die mittleren Steady-State-Talkonzentrationen vergleichbar mit der 40 mg Dosis bei Erwachsenen (Adalimumab-Monotherapie 5,6 ± 5,6 µg/ml (102% CV) und 10,9 ± 5,2 µg/ml (47,7% CV) bei Kombinationstherapie mit Methotrexat, die Messwerte wurden von Woche 20 bis 48 erhoben).
Morbus Crohn bei Kindern und Jugendlichen
Bei Kindern und Jugendlichen mit mässigem bis schwerem aktivem Morbus Crohn war die offene Induktionsdosis von Adalimumab 160/80 mg oder 80/40 mg zu Woche 0 bzw. 2, abhängig vom Körpergewicht mit einem Schnitt bei 40 kg. Zu Woche 4 wurden die Patienten auf Basis ihres Körpergewichts 1:1 entweder zur Erhaltungstherapie mit der Standarddosis (40/20 mg jede zweite Woche) oder mit der niedrigen Dosis (20/10 mg jede zweite Woche) randomisiert. Die mittleren (±SD) Serum-Talkonzentrationen von Adalimumab, die zu Woche 4 erreicht wurden, betrugen für Patienten ≥40 kg (160/80 mg) 15,7 ± 6,6 µg/ml und für Patienten <40 kg (80/40 mg) 10,6 ± 6,1 µg/ml.
Für Patienten, die bei der randomisierten Therapie blieben, betrugen zu Woche 52 die mittleren (±SD) Serum-Talkonzentrationen von Adalimumab 9,5 ± 5,6 µg/ml für die Gruppe mit Standarddosis und 3,5 ± 2,2 µg/ml für die Gruppe mit der niedrigen Dosis. Die mittleren Talkonzentrationen blieben bei Patienten, die weiterhin jede zweite Woche eine Adalimumab-Behandlung erhielten, 52 Wochen lang erhalten. Für Patienten mit Dosiseskalation (Verabreichung von jeder zweiten Woche auf wöchentlich) betrugen die mittleren (±SD) Serum-Talkonzentrationen von Adalimumab zu Woche 52 15,3 ± 11,4 µg/ml (40/20 mg, wöchentlich) bzw. 6,7 ± 3,5 µg/ml (20/10 mg, wöchentlich).
Bei pädiatrischen Patienten mit einem Gewicht unter 30 Kilogramm stehen begrenzte Daten zur Verfügung.
Psoriasis bei Kindern und Jugendlichen
Nachdem pädiatrische Patienten mit chronischer Plaque-Psoriasis jede zweite Woche 0,8 mg Adalimumab/kg (bis zu maximal 40 mg) subkutan erhielten, betrugen die mittleren (±SD) Serum- Talkonzentrationen von Adalimumab ungefähr 7,4 ± 5,8 µg/ml (79% CV).
Exposition und Wirksamkeit in Patienten behandelt mit 80 mg Adalimumab jede zweite Woche
Pharmakokinetische/pharmakodynamische Modellierung und Simulationen prognostizierten eine vergleichbare Adalimumab-Exposition und Wirksamkeit bei Patienten, die jede zweite Woche mit 80 mg behandelt wurden, verglichen mit 40 mg jede Woche. Die pharmakokinetische Modellierung zur Vorhersage der Exposition und die pharmakokinetische/pharmakodynamische Modellierung zur Vorhersage der Wirksamkeit schlossen erwachsene Patienten mit rheumatoider Arthritis, Colitis ulcerosa, Psoriasis, Hidradenitis suppurativa oder pädiatrische Patienten ≥40 kg Körpergewicht mit Morbus Crohn ein. Unter den eingeschlossenen Patienten wurden ausschliesslich Patienten mit rheumatoider Arthritis (74/1664) jede zweite Woche mit 80 mg behandelt.
Interaktionen mit anderen Arzneimitteln
Bei 21 Patienten unter einer stabilen Methotrexat-Therapie zeigten sich nach der Gabe von Adalimumab keine statistisch signifikanten Veränderungen bei den Serum-Methotrexat-Konzentrationsprofilen. Im Gegensatz dazu setzte Methotrexat nach Einmal- bzw. Mehrfachapplikation die scheinbare Clearance von Adalimumab um 29% bzw. 44% herab (siehe "Interaktionen" ).
Präklinische DatenAuf der Grundlage von Studien zur Toxizität nach Einmaldosierung bzw. Mehrfachapplikation sowie zur Genotoxizität lassen die präklinischen Daten keine besondere Gefährdung für den Menschen erkennen.
Eine zur Entwicklungs-/perinatalen Toxizität durchgeführte Studie an Cynomolgus-Affen zeigte kaum schädliche Wirkungen auf Schwangerschaft und die Embryonalentwicklung. Verzögerte Verknöcherung trat bei den Föten der mit 30 und 100 mg/kg behandelten Muttertiere häufiger auf als bei der Kontrollgruppe. Die wiederholte Applikation von Adalimumab führte zu einer Akkumulation, und die Exposition der behandelten Muttertiere war wesentlich höher als bei der Therapie zu erwarten ist.
Aufgrund der Tatsache, dass keine geeigneten Modelle für einen Antikörper mit einer eingeschränkten Cross-Reaktivität gegenüber dem TNF von Nagetieren zur Verfügung stehen sowie der Entwicklung von neutralisierenden Antikörpern bei Nagern wurden keine Studien zur Karzinogenität sowie zur Bewertung der Fertilität und der postnatalen Toxizität mit Adalimumab vorgenommen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Beeinflussung diagnostischer Methoden
Ein Einfluss von Adalimumab auf diagnostische Methoden ist nicht bekannt.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Die Zubereitung enthält kein Konservierungsmittel.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern.
Nicht einfrieren.
AMGEVITA in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
AMGEVITA ausser Reichweite von Kindern aufbewahren.
Eine einzelne Fertigspritze und ein einzelner Fertigpen dürfen für bis zu 14 Tage bei Temperaturen bis zu maximal 25°C gelagert werden. Die Fertigspritze und der Fertigpen müssen vor Licht geschützt werden und müssen entsorgt werden, wenn sie nicht innerhalb dieser 14 Tage verwendet werden, auch wenn sie in den Kühlschrank zurückgelegt werden.
Hinweise für die Handhabung
AMGEVITA Lösung zur Injektion ist für eine Anwendung unter der Anleitung sowie unter der Aufsicht eines Arztes/einer Ärztin gedacht. Nach einer entsprechenden Schulung hinsichtlich der Injektionsmethode können sich die Patienten AMGEVITA Lösung zur Injektion selbst injizieren, wenn ihr Arzt/ihre Ärztin das für angebracht hält und die erforderliche medizinische Nachsorge erfolgt.
Die Injektionslösung ist klar und farblos bis leicht gelblich.
Ausführliche Informationen zum Gebrauch finden sich in der Packungsbeilage.
Es wird empfohlen, nicht verwendete Arzneimittel oder Abfallmaterialien gemäss den lokalen Vorschriften zu entsorgen.
Zulassungsnummer66979, 67204 (Swissmedic).
PackungenAMGEVITA 20 mg/0.4 ml (50 mg/ml) und AMGEVITA 20 mg/0.2 ml (100 mg/ml) Injektionslösung in einer Fertigspritze mit einem Nadelschutz: Packungsgrössen mit einer Fertigspritze (B).
AMGEVITA 40 mg/0.8 ml (50 mg/ml) und AMGEVITA 40 mg/0.4 ml (100 mg/ml) Injektionslösung in einer Fertigspritze mit einem Nadelschutz: Packungsgrössen mit einer, zwei oder Bündelpackung mit sechs (3 x 2) Fertigspritze(n) (B).
AMGEVITA 40 mg/0.8 ml (50 mg/ml) oder 40 mg/0.4 ml (100 mg/ml) Injektionslösung im Fertigpen: Packungsgrössen mit einem, zwei oder Bündelpackung mit sechs (3 x 2) Fertigpen(s) (B).
AMGEVITA 80 mg/0.8 ml Injektionslösung in einer Fertigspritze mit einem Nadelschutz: 1 Fertigspritze (B).
AMGEVITA 80 mg/0.8 ml Injektionslösung im Fertigpen: 1 Fertigpen (B).
Möglicherweise sind nicht alle Packungsgrössen auf dem Markt erhältlich.
ZulassungsinhaberinAmgen Switzerland AG, Risch
Domizil: 6343 Rotkreuz
Stand der InformationApril 2025
|