Eigenschaften/Wirkungen
ATC-Code
R03DX09
Wirkungsmechanismus
Mepolizumab ist ein humanisierter monoklonaler Antikörper (IgG1, kappa), der sich hochaffin und spezifisch gegen das humane Interleukin-5 (IL-5) richtet. IL-5 ist das für Wachstum und Differenzierung, Rekrutierung, Aktivierung und Überleben der Eosinophilen wichtigste Zytokin. Mepolizumab hemmt im nanomolaren Konzentrationsbereich die biologischen Wirkungen von IL-5, indem es verhindert, dass IL-5 an die Alpha-Kette des auf der Zelloberfläche von Eosinophilen exprimierten IL-5-Rezeptorkomplexes bindet; auf diese Weise wird der IL-5-Signalweg blockiert und damit die Bildung von Eosinophilen und deren Überleben reduziert.
Pharmakodynamik
In klinischen Studien wurde nach einer Behandlung mit Mepolizumab eine Verminderung der Eosinophilenzahl im Blut beobachtet. Grössenordnung und Dauer dieser Verminderung waren nach subkutaner Gabe von 12,5 mg-125 mg dosisabhängig.
Die Grössenordnung der Reduktion wurde bei den unten angegebenen Populationen innerhalb von 4 Behandlungswochen beobachtet und über den gesamten Behandlungszeitraum aufrechterhalten.
Bei Patienten mit schwerem Asthma ging nach subkutaner Verabreichung von 100 mg alle 4 Wochen über einen Zeitraum von 32 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 40 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 84% (Woche 32) vs Placebo.
Dieses Ausmass der Verringerung der Eosinophilen im Blut blieb bei Patienten mit schwerem Asthma (n=998), die in offenen Verlängerungsstudien über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, erhalten.
Bei Patienten mit CRSwNP ging nach subkutaner Verabreichung von100 mg alle 4 Wochen über einen Zeitraum von 52 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 60 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 83% vs Placebo. Dieser Rückgang wurde innerhalb der ersten 4 Behandlungswochen beobachtet und blieb während der Therapiedauer erhalten.
Bei Patienten mit COPD ging nach subkutaner Verabreichung von 100 mg alle 4 Wochen über einen Zeitraum von 52-104 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 60 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 79% (Woche 52) und 80% (Woche 104) vs Placebo.
Bei Patienten mit EGPA ging nach subkutaner Verabreichung von 300 mg alle 4 Wochen über einen Zeitraum von 52 Wochen die Eosinophilenzahl im Blut auf einen geometrischen Mittelwert von 38 Zellen/µL zurück. Dies entspricht einer geometrisch gemittelten Reduktion um 83% vs Placebo.
Bei Patienten mit HES ging nach subkutaner Verabreichung von 300 mg alle 4 Wochen über einen Zeitraum von 32 Wochen die Zahl der Eosinophilen im Blut auf einen geometrischen Mittelwert von 70 Zellen/µL zurück. Das entspricht einer geometrisch gemittelten Reduktion um 92% vs Placebo. Diese Reduktionsgrössenordnung wurde für weitere 20 Wochen beibehalten in Patienten, die mit der Mepolizumab-Behandlung in der offenen Verlängerung weiterfuhren.
Immunogenität
Aufgrund des immunogenen Potenzials von protein- oder peptidbasierten Therapeutika können die Patienten nach der Behandlung Antikörper gegen Mepolizumab entwickeln.
Insgesamt entwickelten 15/260 (6 %) der mit mindestens einer subkutanen Dosis in Höhe von 100 mg behandelten Patienten mit schwerem Asthma Antikörper gegen Mepolizumab. Das Immunogenitätsprofil von Nucala bei Patienten mit schwerem Asthma (n=998), die in offenen Verlängerungsstudien über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, war ähnlich wie in den placebo-kontrollierten Studien.
Insgesamt entwickelten 6/196 (3%) der mit mindestens einer subkutanen Mepolizumab-Dosis von 100 mg behandelten CRSwNP-Patienten Antikörper gegen Mepolizumab.
Insgesamt entwickelten 9/381 (2%) der mit mindestens einer subkutanen Mepolizumab-Dosis von 100 mg behandelten COPD-Patienten Antikörper gegen Mepolizumab.
Insgesamt entwickelten 1/68 (1%) der mit mindestens einer subkutanen Mepolizumab-Dosis von 300 mg behandelten EGPA-Patienten Antikörper gegen Mepolizumab.
Insgesamt entwickelten 1/53 (2%) der mit mindestens einer subkutanen Mepolizumab-Dosis von 300 mg behandelten HES-Patienten Antikörper gegen Mepolizumab.
Über alle Indikationen wurden bei einer erwachsenen Person (mit schwerem Asthma) neutralisierende Antikörper festgestellt.
Das Vorliegen von Antikörpern gegen Mepolizumab hatte bei der Mehrzahl der Patienten keine merklichen Auswirkungen auf die PK oder PD von Mepolizumab; Hinweise auf einen Zusammenhang zwischen den Antikörpertitern und einer Änderung der Eosinophilenzahl lagen nicht vor.
Klinische Wirksamkeit
Schweres eosinophiles Asthma
Die Wirksamkeit von Mepolizumab bei hochgradigem eosinophilem Asthma wurde im Rahmen von 3 randomisierten, doppelblinden klinischen Parallelgruppenstudien von 24- bis 52-wöchiger Dauer an Patienten ab 12 Jahren untersucht. Diese Studien waren auf die Beurteilung der Wirksamkeit von Mepolizumab bei Verabreichung in vierwöchigen Abständen als subkutane oder intravenöse Injektion bei Patienten angelegt, die unter ihrer laufenden Standardbehandlung (z.B. inhalative Kortikosteroide [ICS], Kombination aus ICS und langwirkenden Beta2-Sympathomimetika [LABA], Leukotrienmodifikatoren und kurzwirkenden Beta2-Sympathomimetika [SABA]) nicht kontrolliert waren. In MEA112997 und MEA115588 sind klinisch relevante Asthma-Exazerbationen wie folgt definiert: Verschlechterung der Asthmasymptome, die die Anwendung von oralen/systemischen Kortikosteroiden und/oder eine Hospitalisierung bzw. eine Notfallbehandlung erfordert.
Placebokontrollierte Studien
Dosisfindungsstudie MEA112997 (DREAM-Studie)
Die Ergebnisse der randomisierten, doppelblinden, placebokontrollierten, multizentrischen, 52-wöchigen Parallelgruppenstudie MEA112997 an 616 Patienten zeigten, dass Mepolizumab (75 mg, 250 mg oder 750 mg) bei intravenöser Verabreichung zu einer signifikanten Reduktion der Asthma-Exazerbationen gegenüber Placebo führte. Hinsichtlich der Wirksamkeit konnte zwischen den 3 untersuchten Dosierungen kein signifikanter Unterschied festgestellt werden (siehe Tabelle 1).
Tabelle 1: Häufigkeit klinisch relevanter Exazerbationen nach 52 Wochen bei der Intent-to-treat-Population
Mepolizumab IV Placebo
75 mg n = 153 250 mg n = 152 750 mg n = 156 n = 155
Exazerbationen/Jahr 1,24 1,46 1,15 2,40
Prozent Senkung 48 % 39 % 52 %
Verhältnisrate (95%-KI) 0,52 (0,39; 0,69) 0,61 (0,46; 0,81) 0,48 (0,36; 0,64)
p-Wert < 0,001 < 0,001 < 0,001 -
Die Ergebnisse dieser Studie weisen darauf hin, dass eine Eosinophilenzahl im Blut von ≥150 Zellen/µL in der Voruntersuchungsphase bzw. von ≥300 Zellen/µL in den vorausgegangenen 12 Monaten als Biomarker herangezogen werden kann, um vorherzusagen, welche Patienten von der Behandlung mit Mepolizumab profitieren können. Allerdings wurden bei einem Drittel der so ausgewählten Patienten auch unter Placebo während der einjährigen Behandlungsphase keine Exazerbationen mehr beobachtet.
Bei Erhöhung des "Cut off" Wertes der Eosinophilenzahl im Blut wurde bei entsprechend selektionierten Patienten eine verstärkte Abnahme der Exazerbationshäufigkeit beobachtet. Es ist aber nicht untersucht, inwieweit bei höheren "Cut off" Werten Patienten ausgeschlossen wurden, welche von der Mepolizumab-Zusatz-Behandlung profitierten.
Tabelle 2: Berechnete Reduktion der Rate klinisch signifikanter Exazerbationen bei Baseline-Grenzwerten der Eosinophilenzahl im Blut
Studie Baseline Eosinophilenzahl im Blut Verhältnisrate (95%-KI)
MEA112997 150 Zellen/µL 0,70 (0,53; 0,93)
300 Zellen/µL 0,52 (0,41; 0,65)
500 Zellen/µL 0,42 (0,32; 0,54)
MEA115588 150 Zellen/µL 0,61 (0,45; 0,82)
300 Zellen/µL 0,49 (0,38; 0,63)
500 Zellen/µL 0,42 (0,31; 0,55)
Auf Grundlage der Ergebnisse dieser Studie wurden die in späteren Studien zur subkutanen Verabreichung von Mepolizumab zu untersuchenden Dosen ausgewählt. Nucala ist ein Präparat, das nicht intravenös angewendet werden darf, sondern ausschliesslich subkutan verabreicht werden muss.
Reduktion von Exazerbationen (MEA115588), MENSA-Studie
MEA115588 (Mepolizumab as adjunctive therapy in patients with Severe Asthma) war eine randomisierte, doppelblinde, placebokontrollierte, multizentrische Parallelgruppenstudie zur Beurteilung der Wirksamkeit und Sicherheit von Mepolizumab als Zusatztherapeutikum an 576 Patienten mit hochgradigem eosinophilem Asthma.
Die Patienten waren überwiegend 18 Jahre oder älter, hatten in den vorausgegangenen 12 Monaten mindestens zwei Asthma-Exazerbationen durchgemacht und waren unter ihrer laufenden Pharmakotherapie gegen Asthma (hochdosierte inhalative Kortikosteroide [ICS] in Kombination mit mindestens einem anderen Kontrollmedikament, z.B. langwirkenden Beta2-Sympathomimetika [LABA] oder Leukotrienmodifikatoren) nicht kontrolliert. Die Patienten durften orale Kortikosteroide verwenden und erhielten während der Studie weiterhin ihre gewohnten Medikamente gegen Asthma.
Schweres eosinophiles Asthma lag definitionsgemäss vor bei einer Eosinophilenzahl im peripheren Blut von ≥150 Zellen/μL innerhalb der 6 Wochen vor der Randomisierung (erste Dosis) oder einer Eosinophilenzahl im Blut von ≥300 Zellen/μL innerhalb des der Randomisierung vorangegangenen Jahres. Die Patienten erhielten über 32 Wochen hinweg jeweils einmal alle vier Wochen entweder Mepolizumab 100 mg subkutan (s.c.), Mepolizumab 75 mg intravenös (i.v.) oder ein Placebo. Die Ergebnisse für den primären Studienendpunkt, die Verminderung der Häufigkeit klinisch relevanter Asthma-Exazerbationen, fielen statistisch signifikant aus (p < 0,001).
Tabelle 3 fasst die Ergebnisse für den primären und die sekundären Endpunkte aus MEA115588 zusammen.
Tabelle 3: Ergebnisse für den primären und die sekundären Endpunkte nach 32 Wochen in der Intent-to-treat-Population (MEA115588)
Mepolizumab (100 mg Placebo n = 191
s.c.) n = 194
Primärer Endpunkt
Häufigkeit klinisch relevanter Exazerbationen
Exazerbationen/Jahr 0,83 1,74
Prozent Senkung Verhältnisrate (95%-KI) 53 % 0,47 (0,35; 0,64) -
p-Wert < 0,001
Sekundäre Endpunkte
Häufigkeit von Exazerbationen mit erforderlicher
Hospitalisierung/Notfallbehandlung
Exazerbationen/Jahr 0,08 0,20
Prozent Senkung Verhältnisrate (95%-KI) 61 % 0,39 (0,18; 0,83) _
p-Wert 0,015
Häufigkeit hospitalisierungsbedürftiger
Exazerbationen
Exazerbationen/Jahr 0,03 0,10
Prozent Senkung Verhältnisrate (95%-KI) 69 % 0,31 (0,11; 0,91) _
p-Wert 0,034
FEV1 (mL) vor Bronchodilatator nach 32 Wochen
Mittlere Änderung gegenüber Ausgangswert 183 (31,1) 86 (31,4)
(Standardfehler)
Unterschied (Mepolizumab vs. Placebo) 98
95%-KI (11, 184)
p-Wert 0,028
St. George's Respiratory Questionnaire (SGRQ) nach
32 Wochen
Mittlere Änderung gegenüber Ausgangswert -16,0 (1,13) -9,0 (1,16)
(Standardfehler)
Unterschied (Mepolizumab vs. Placebo) -7,0
95%-KI (-10,2; -3,8)
p-Wert < 0,001
Studie zur Reduktion oraler Kortikosteroide (SIRIUS)
MEA115575 beurteilte die Wirkung von Mepolizumab 100 mg s.c. in Bezug auf die Verminderung des Bedarfs an oralen Kortikosteroiden (OCS) zur Erhaltungstherapie, unter Aufrechterhaltung der Asthmakontrolle bei auf systemische Kortikosteroide angewiesenen Patienten mit schwerem eosinophilem Asthma. Die Patienten wiesen in den 12 Monaten vor dem Screening eine Eosinophilenzahl im peripheren Blut von ≥300/μL bzw. vor Behandlungsbeginn eine Eosinophilenzahl im peripheren Blut von ≥150/μL auf und erhielten im Behandlungszeitraum in vierwöchigen Abständen Mepolizumab oder Placebo. Die OCS-Dosis wurde während der OCS-Reduktionsphase (Wochen 4–20) alle 4 Wochen vermindert, solange die Asthmakontrolle dabei aufrechterhalten wurde. Während der Studie setzten die Patienten ihre Ausgangstherapie gegen Asthma fort (hochdosierte inhalative Kortikosteroide [ICS] in Kombination mit mindestens einem weiteren Kontrollmedikament, z.B. langwirkenden Beta2-Sympathomimetika [LABA] oder Leukotrienmodifikatoren).
Die Studienpopulation aus insgesamt 135 Patienten war wie folgt charakterisiert: Durchschnittsalter 50 Jahre, 55 % der Teilnehmer waren weiblich, 48 % nahmen seit mindestens 5 Jahren orale Kortikosteroide und erhielten bei Studienbeginn ein mittleres Prednisolonäquivalent von ca. 13 mg pro Tag.
Primärer Studienendpunkt war die Verminderung der OCS-Tagesdosis (Wochen 20–24) unter Aufrechterhaltung der Asthmakontrolle im Vergleich zu Patienten unter Placebo (siehe Tabelle 4).
Tabelle 4: Ergebnisse für den primären und die sekundären Endpunkte in der Intent-to-treat-Population von MEA115575
Mepolizumab (100 mg s.c.) Placebo n = 66
n = 69
Primärer Endpunkt
OCS-Reduktion gegenüber der Ausgangsdosis nach
20–24 Wochen (%)
90 %–100 % 16 (23 %) 7 (11 %)
75 %–< 90 % 12 (17 %) 5 (8 %)
50 %–< 75 % 9 (13 %) 10 (15 %)
> 0 %–< 50 % 7 (10 %) 7 (11 %)
Keine OCS-Reduktion/unzureichende 25 (36 %) 37 (56 %)
Asthmakontrolle/ Behandlungsabbruch
Odds Ratio (95%-KI) 2,39 (1,25; 4,56)
p-Wert 0,008
Sekundäre Endpunkte
Reduktion der OCS-Tagesdosis (%)
Mindestens 50%ige Reduktion 37 (54 %) 22 (33 %)
Odds Ratio (95%-KI) 2,26 (1,10; 4,65)
p-Wert 0,027
Reduktion der OCS-Tagesdosis (%)
Auf ≤5 mg/Tag 37 (54 %) 21 (32 %)
Odds Ratio (95%-KI) 2,45 (1,12; 5,37)
p-Wert 0,025
Reduktion der OCS-Tagesdosis (%)
Auf null 10 (14 %) 5 (8 %)
Odds Ratio (95%-KI) 1,67 (0,49; 5,75)
p-Wert 0,414
Mediane prozentuale Reduktion der OCS-Tagesdosis
Mediane Reduktion (%) gegenüber Ausgangsdosis 50,0 (20,0; 75,0) 0,0 (-20,0; 33,3)
(95%-KI)
Medianer Unterschied (95%-KI) -30,0 (-66,7; 0,0)
p-Wert 0,007
Ausserdem wurde anhand des SGRQ die gesundheitsbezogene Lebensqualität ermittelt. Nach 24 Wochen war in Bezug auf den mittleren SGRQ-Score unter Nucala eine signifikante Verbesserung gegenüber Placebo feststellbar: -5,8 (95%-KI: -10,6; -1,0; p = 0,019). In Woche 24 war der Anteil der Patienten mit klinisch bedeutsamer Abnahme des SGRQ-Scores (definiert als Rückgang um mindestens 4 Einheiten gegenüber dem Ausgangswert) unter Nucala höher (58 %, 40/69) als unter Placebo (41 %, 27/66).
Das langfristige Wirksamkeitsprofil von Nucala bei Patienten mit schwerem Asthma (n=998), die in den offenen Verlängerungsstudien MEA115666, MEA115661 und 201312 über einen Median von 2,8 Jahren (Spanne von 4 Wochen bis 4,5 Jahren) behandelt wurden, stimmte im Allgemeinen mit den drei placebo-kontrollierten Studien überein.
Chronische Rhinosinusitis mit Nasenpolypen (CRSwNP)
Bei der Studie 205687 handelte es sich um eine 52-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie, in der 407 Patienten im Alter über 18 Jahren mit CRSwNP untersucht wurden.
Die in die Studie aufgenommenen Patienten mussten für die nasale Obstruktion einen VAS-Symptom-Score (visuelle Analogskala) von >5 (von max. 10 Punkten), einen Gesamt-VAS-Symptom-Score >7 (von max. 10 Punkten) und einen endoskopischen bilateralen NP-Score von 5 (von max. 8 möglichen Punkten; mit einem Mindest-Scorewert von 2 pro Nasenhöhle) aufweisen. Ferner mussten die Patienten in den vorangegangenen 10 Jahren mindestens eine Operation infolge von Nasenpolypen erhalten haben.
Die Patienten erhielten eine Dosis 100 mg Mepolizumab oder Placebo, die alle 4 Wochen zusätzlich zur Basistherapie mit intranasalen Kortikosteroiden subkutan verabreicht wurde.
Die demografischen Daten und Baseline-Eigenschaften der an Studie 205687 teilnehmenden Patienten sind unten in Tabelle 5 aufgeführt:
Tabelle 5: Demographische Daten und Baseline-Eigenschaften der CRSwNP
N = 407
Alter (Jahre) der Patienten, Mittelwert (SD) 49 (13)
Weiblich, n (%) 143 (35)
Europäischstämmig, n (%) 379 (93)
Dauer (Jahre) der CRSwNP, Mittelwert (SD) 11,4 (8,39)
Patienten mit ≥1 vorangegangener Operation, n (%) 407 (100)
Patienten mit ≥3 vorangegangenen Operationen, n (%) 124 (30)
OCS-Anwendung wegen NP (≥1 Verlauf) in den vergangenen 12 Monaten, n (%) 197 (48)
Gesamter endoskopischer NP-Scorea,b,c, Mittelwert (SD), max. Scorewert = 8 5,5 (1,29)
VAS-Scorea,d der nasalen Obstruktion, Mittelwert (SD), max. Scorewert = 10 9,0 (0,83)
Gesamt-VAS-Symptom-Scorea,d, Mittelwert (SD), max. Scorewert = 10 9,1 (0,74)
SNOT-22-Gesamtscoree, Mittelwert (SD), Bereich 0-110 64,1 (18,32)
Zusammengesetzter VAS-Symptome-Scorea, Mittelwert (SD), max. Scorewert = 10 9,0 (0,82)
VAS-Scorea,d des Geruchsverlusts, Mittelwert (SD), max. Scorewert = 10 9,7 (0,72)
Asthma, n (%) 289 (71)
AERD, n (%) 108 (27)
Geometrisches Mittel der Eosinophilen-Zahl bei Baseline, Zellen/mcl (95%-KI) 390 (360, 420)
CRSwNP = chronische Rhinosinusitis mit Nasenpolypen, SD = Standardabweichung, OCS = orales Kortikosteroid, NP = Nasenpolypen, VAS = visuelle Analogskala, SNOT-22 = Sino-Nasal-Outcome-Test, AERD = Aspirin-exazerbierte Atemwegserkrankung
a Höhere Scorewerte weisen auf eine höheren Schweregrad der Erkrankung hin.
b Wie von unabhängigen verblindeten Bewertern bewertet
c Der NP-Score ist die Summe der Scorewerte beider Nasenlöcher (auf einer Skala 0-8), bei denen jedes Nasenloch bewertet wurde (0=keine Polypen; 1=kleine Polypen im mittleren Gehörgang, die nicht unter den unteren Rand der mittleren Nasenmuschel ragen; 2=Polypen, die bis unter den unteren Rand der mittleren Nasenmuschel ragen; 3=grosse Polypen, die den unteren Rand der unteren Nasenmuschel oder Polypen medial zur mittleren Ohrmuschel überragen; 4=grosse Polypen, die eine fast vollständige Verstopfung/Obstruktion des unteren Gehörgangs bewirken).
d Täglich von den Patienten anhand einer Skala von 0 bis 10 bewertet (0=kein; 10=extrem schlimm).
e SNOT-22 ist ein Test zur Bewertung der gesundheitsbezogenen Lebensqualität und umfasst 22 Punkte in 6 Bereichen von Symptomen und Auswirkungen im Zusammenhang mit CRSwNP (nasal, nicht-nasal, Ohr/Gesicht, Schlaf, Müdigkeit, emotionale Folgen). Höhere Scorewerte deuten auf eine schlechtere gesundheitsbezogene Lebensqualität hin.
Die koprimären Endpunkte waren die Veränderung des gesamten endoskopischen NP-Scores in Woche 52 gegenüber Baseline und die Veränderung des mittleren VAS-Wertes der nasalen Obstruktion in den Wochen 49-52 gegenüber Baseline.
Patienten, die Mepolizumab erhalten hatten, wiesen im Vergleich zu Placebo signifikant grössere Verbesserungen (Reduktionen) beim endoskopischen NP-Gesamtscore in Woche 52 und beim VAS-Score bei nasaler Obstruktion in den Wochen 49-52 auf (siehe Tabelle 6).
Tabelle 6: Analysen der koprimären Endpunkte (Intent-To-Treat-Population)
Placebo (N=201) Mepolizumab 100 mg s.c.
(N=206)
Endoskopischer Gesamt-Score in Woche 52 a
Medianer Score an Baseline (min, max) 6,0 (0, 8) 5,0 (2, 8)
Mediane Änderung gegenüber Baseline 0,0 -1,0
p-Wert b <0,001
Korrigierter Behandlungsunterschied bei den -0,73 (-1,11, -0,34)
Medianen (95%-KI) c
≥1-Punkt-Verbesserung, n (%) 57 (28) 104 (50)
≥2-Punkt-Verbesserung, n (%) 26 (13) 74 (36)
VAS-Score für die nasale Obstruktion (Wochen 49
bis 52) a
Medianer Score an Baseline (min, max) 9,14 (5,31, 10,00) 9,01 (6,54, 10,00)
Mediane Änderung gegenüber Baseline -0,82 -4,41
p-Wert b <0,001
Korrigierter Behandlungsunterschied bei den -3,14 (-4,09, -2,18)
Medianen (95%-KI) c
>1-Punkt-Verbesserung, n (%) 100 (50) 146 (71)
≥3-Punkt-Verbesserung, n (%) 73 (36) 124 (60)
a Studienteilnehmer mit einer Nasenoperation/Sinuplastik vor dem Studientermin wiesen den schlechtesten beobachteten Scorewert der Situation vor der Nasenoperation/Sinuplastik zu. Teilnehmer, die ohne Nasenoperation/Sinuplastik aus der Studie ausschieden, wiesen ihren schlechtesten beobachteten Scorewerte der Situation vor dem Studienabbruch zu.
b Auf Basis des Wilcoxon-Rangsummentests.
c Quantile Regression mit Kovariaten der Behandlungsgruppe, der geografischen Region, des Baseline-Scorewerts und der log(e)-Eosinophilenzahl im Blut an Baseline.
Abbildung 1: Abbildung des gesamten endoskopischen Nasenpolypen-Scores (zentral abgelesen) Responders per Kontrolle
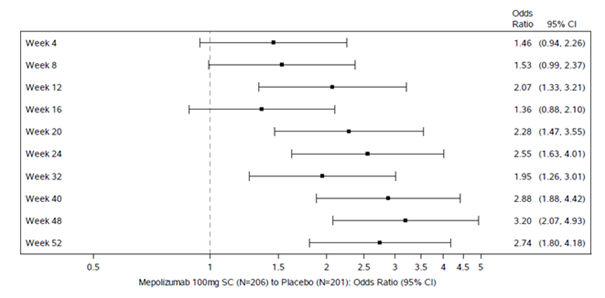
Alle sekundären Endpunkte waren statistisch signifikant und unterstützten die koprimären Endpunkte. Der wichtigste sekundäre Endpunkt war die Zeit bis zur ersten NP-Operation bis Woche 52 (siehe Abbildung 1). Daten zu den anderen sekundären Endpunkten sind in Tabelle 7 aufgeführt.
Zeit bis zur ersten NP-Operation
Über den gesamten Behandlungszeitraum von 52 Wochen war bei den Patienten der Mepolizumab-Gruppe die Wahrscheinlichkeit geringer, sich einer NP-Operation unterziehen zu müssen, als bei den Patienten in der Placebo-Gruppe (als Operation wurde jeder Eingriff definiert, bei dem Instrumente eingesetzt wurden, die zu einer Inzision und Entfernung von Gewebe [Polypektomie] in der Nasenhöhle führten).
Bis Woche 52 hatten sich 18 Patienten (9 %) in der Mepolizumab-Gruppe einer NP-Operation unterzogen, wohingegen dieser Wert in der Placebo-Gruppe bei 46 Patienten (23 %) lag.
Bei Patienten, die Mepolizumab erhielten, war die Zeit bis zur ersten NP-Operation im Vergleich zu Placebo länger. Das Operationsrisiko während des Behandlungszeitraums war bei Patienten, die mit Mepolizumab behandelt wurden, im Vergleich zu Patienten, die mit Placebo behandelt wurden, um 57 % niedriger und somit signifikant geringer (Hazard Ratio: 0,43; 95%-KI 0,25, 0,76; unbereinigt/bereinigt p=0,003), eine Post-hoc-Analyse ergab eine 61%-ige Verringerung der Wahrscheinlichkeit einer Operation (Odds Ratio: 0,39, 95%-KI: 0,21, 0,72; p=0,003.
Abbildung 2: Kaplan-Meier-Kurve für die Zeit bis zur ersten Nasenpolypen-OP

Tabelle 7: Ergebnisse der anderen sekundären Endpunkte in der Intent-To-Treat-Population
Placebo (N=201) Mepolizumab (N=206)
Gesamter VAS-Score (Wochen 49-52) a
Medianer Score an Baseline (min, max) 9,20 (7,21, 10,00) 9,12 (7,17, 10,00)
Mediane Änderung gegenüber Baseline -0,90 -4,48
Unbereinigter/bereinigter p-Wert b,c <0,001/0,003
Korrigierter Behandlungsunterschied bei den Medianen -3,18 (-4,10, -2,26)
(95%-KI) d
≥2,5-Punkte-Verbesserung (%) 40 64
SNOT-22-Gesamtscore in Woche 52 a, g
n 198 205
Medianer Score an Baseline (min, max) 64,0 (19, 110) 64,0 (17, 105)
Mediane Änderung gegenüber Baseline -14,0 -30,0
Unbereinigter/bereinigter p-Wert b,c <0,001/0,003
Korrigierter Behandlungsunterschied bei den Medianen -16,49 (-23,57,
(95%-KI) d -9,42)
≥28-Punkte-Verbesserung (%) g 32 54
Patienten, die wegen Nasenpolypen bis Woche 52
systemische Steroide benötigen
Anzahl der Patienten mit ≥1 Verlauf 74 (37) 52 (25)
Odds Ratio zu Placebo (95%-KI) 0,58 (0,36, 0,92)
Unbereinigter/bereinigter p-Wert c, e 0,020/0,020
Zusammengesetzter VAS-Score – nasale Symptome (Wochen
49-52) a,f
Medianer Score an Baseline (min, max) 9,18 (6,03, 10,00) 9,11 (4,91, 10,00)
Mediane Änderung gegenüber Baseline -0,89 -3,96
Unbereinigter/bereinigter p-Wert b,c <0,001/0,020
Korrigierter Behandlungsunterschied bei den Medianen -2,68 (-3,44, -1,91)
(95%-KI) d
≥2-Punkte-Verbesserung (%) h 40 66
VAS-Score für den Geruchsverlust (Wochen 49-52) a
Medianer Score an Baseline (min, max) 9,97 (6,69, 10,00) 9,97 (0,94, 10,00)
Mediane Änderung gegenüber Baseline 0,00 -0,53
Unbereinigter/bereinigter p-Wert b,c <0,001/0,020
Korrigierter Behandlungsunterschied bei den Medianen -0,37 (-0,65, -0,08)
(95%-KI) d
≥3-Punkte-Verbesserung (%) h 19 36
a Patienten mit einer Nasenoperation/Sinuplastik vor dem Studientermin wiesen den schlechtesten beobachteten Scorewert der Situation vor der Nasenoperation/Sinuplastik zu. Teilnehmer, die ohne Nasenoperation/Sinuplastik aus der Studie ausschieden, wiesen ihren schlechtesten beobachteten Scorewerte der Situation vor dem Studienabbruch zu.
b Auf Basis des Wilcoxon-Rangsummentests.
c Multiplizität kontrolliert mithilfe von Tests der sekundären Endpunkte gemäss einer vordefinierten Hierarchie.
d Quantile Regression mit Kovariaten der Behandlungsgruppe, der geografischen Region, des Baseline-Scorewerts und der log(e)-Eosinophilenzahl im Blut an Baseline.
e Analyse unter Verwendung eines logistischen Regressionsmodells mit Kovariaten der Behandlungsgruppe, der geografischen Region, der Anzahl der OCS-Verläufe für NP in den letzten 12 Monaten (0, 1, >1 als Ordinalzahl), des ENP-Gesamtscores an Baseline (zentral gelesen), des VAS-Scores für nasale Obstruktion und der log(e)-Eosinophilenzahl im Blut an Baseline.
f Zusammengesetzter VAS-Score für nasale Obstruktion, Nasenausfluss, Rachenschleim und Geruchsverlust.
g In allen 6 Bereichen der Symptome und Auswirkungen im Zusammenhang mit CRSwNP wurde eine Verbesserung festgestellt.
h Der Schwellenwert für die Verbesserung jedes einzelnen Endpunktes wurde als sinnvolle intraindividuelle Veränderung bestimmt.
Endpunkte der Subgruppe von Patienten mit komorbidem Asthma
Bei 289 (71 %) Patienten mit komorbidem Asthma ergaben vordefinierte Analysen im Vergleich zu Placebo bei den Patienten, die 100 mg Mepolizumab erhielten, Verbesserungen bei den koprimären Endpunkten, die mit denen der Gesamtpopulation übereinstimmten.
Chronisch-obstruktive Lungenerkrankung (COPD)
Studie 1
Die Wirksamkeit von Mepolizumab (100 mg s.c. alle 4 Wochen) zusätzlich zur Standardtherapie wurde bei 804 erwachsenen Patienten ab 40 Jahren mit COPD vom eosinophilen Phänotyp in einer 52- bis 104-wöchigen randomisierten, placebokontrollierten, multizentrischen pivotalen Studie (208657, MATINEE) untersucht. Die Teilnehmenden mussten eine Eosinophilenzahl im Blut von ≥300 Zellen/µL zum Screeningzeitpunkt und von ≥150 Zellen/µL innerhalb der vorausgegangenen 12 Monate aufweisen. Alle Patienten erhielten für die Dauer der Studie eine inhalative Dreifachtherapie (ICS, langwirksamer Beta-2-Agonist und langwirksamer Muskarinantagonist). Die eingeschlossenen Patienten wiesen trotz regelmässiger Anwendung inhalativer Kortikosteroide plus zweier zusätzlicher Erhaltungstherapien über einen Zeitraum von mindestens 3 Monaten vor dem Screening eine mittelschwere bis sehr schwere Luftstrombehinderung (FEV1 nach Bronchodilatator von 20–80 % des Sollwerts) sowie anamnestisch bekannte Exazerbationen (mindestens 1 schwere, hospitalisierungsbedürftige Exazerbation oder 2 mittelschwere Exazerbationen, die eine Behandlung mit systemischen Kortikosteroiden, mit oder ohne Antibiotika, erforderten) auf.
Primäres Studienziel war die Beurteilung der Wirksamkeit von Mepolizumab auf die annualisierte Rate von mittelschweren (definiert als Verschlechterung der COPD-Symptome, die eine Behandlung mit oralen/systemischen Kortikosteroiden und/oder Antibiotika erforderlich macht) oder schweren Exazerbationen (definiert als hospitalisierungsbedürftig oder zum Tode führend). Symptome und gesundheitsbezogene Lebensqualität wurden anhand einer Responderanalyse des COPD-Assessment-Tests (CAT) (Ansprechen definiert als Verringerung der Punktzahl um 2 oder mehr gegenüber dem Ausgangswert) und einer Responderanalyse des St. George's Respiratory Questionnaire (SGRQ) (Ansprechen definiert als Verringerung der Punktzahl um 4 oder mehr gegenüber dem Ausgangswert) bewertet.
Die demografischen Daten und Baseline-Charakteristika der Patienten sind in Tabelle 8 aufgeführt:
Tabelle 8: Demographische Daten und Baseline-Charakteristika in MATINEE (mITT-Population)
(N = 804)
Alter (Jahre) der Patienten, Mittelwert (SD) 66 (8,0)
Weiblich, n (%) 253 (31)
Weiss, n (%) 673 (84)
Derzeit aktive Raucher, n (%) 222 (28)
Durchschnittliche Raucheranamnese (Packungsjahre), Mittelwert (SD) 43,0 (24,88)
Dauer der COPD (Jahre), Mittelwert (SD) 10,0 (6,28)
mMRC-Score ≥2 (Bereich 0–4), n (%) 611 (76)
Nur Emphysem,a n (%) 252 (31)
Nur chronische Bronchitis,a n (%) 338 (42)
Emphysem und chronische Bronchitis,a n (%) 143 (18)
Leichte Luftstrombehinderung: > 80 % des FEV1-Sollwerts, n (%) 5 (1)
Mässige Luftstrombehinderung: ≥50 % bis < 80 % des FEV1-Sollwerts, n (%) 349 (43)
Schwere Luftstrombehinderung: ≥30 % bis < 50 % des FEV1-Sollwerts, n (%) 340 (42)
Sehr schwere Luftstrombehinderung: < 30 % des FEV1-Sollwerts, n (%) 110 (14)
% des FEV1-Sollwerts nach Bronchodilatator, Mittelwert (SD) 48,2 (15,77)
FEV1/FVC-Verhältnis nach Bronchodilatator, Mittelwert (SD) 0,49 (0,124)
Anzahl der mittelschweren oder schweren Exazerbationen im letzten Jahr, 2,3 (0,94)
Mittelwert (SD)
Eine oder mehrere schwere Exazerbationen im letzten Jahr, n (%) 165 (21)
CAT-Score, Mittelwert (SD) 19,2 (6,85)
SGRQ-Score, Mittelwert (SD) 54,6 (17,80)
Geometrisches Mittel der Eosinophilen-Zahl beim Screening, Zellen/μL (95%-KI) 480 (470; 490)
mITT = modified Intent-to-Treat, mMRC = modified Medical Research Council, FEV1 = forciertes exspiratorisches 1-Sekunden-Volumen, FVC = forcierte Vitalkapazität, CAT = COPD-Assessment-Test, SGRQ = St. George's Respiratory Questionnaire
a COPD-Subtyp gemäss prüfärztlicher Bewertung. 544 (68 %) Patienten gaben im SGRQ Symptome einer chronischen Bronchitis an.
Exazerbationen
Patienten, die Mepolizumab 100 mg erhielten, wiesen im Vergleich zu Placebo eine statistisch signifikante Reduktion der annualisierten Rate von mittelschweren oder schweren Exazerbationen sowie ein geringeres Risiko für mittelschwere/schwere Exazerbationen auf (siehe Tabelle 9).
Tabelle 9: COPD-Exazerbationsendpunkte in der Intent-to-treat-(mITT-)Population
Mepolizumab 100 mg s.c. N Placebo N = 401
= 403
Rate mittelschwerer oder schwerer Exazerbationen
Exazerbationsrate/Jahr 0,80 1,01
Prozent Ratenreduktion Ratenverhältnis vs. 21 % 0,79 (0,66; 0,94)
Placebo (95%-KI)
p-Wert 0,011
Zeit bis zur ersten mittelschweren oder schweren
Exazerbation
Mediane Zeit bis zur ersten Exazerbation a (Tage) 419 321
Prozent Risikoreduktion Hazard Ratio vs. Placebo 23 % 0,77 (0,64; 0,93)
(95%-KI)
p-Wert 0,009
a Kaplan-Meier-Schätzung
In der MATINEE-Studie reduzierte Mepolizumab auch die jährliche Rate der Exazerbationen, die einen Besuch in der Notaufnahme und/oder einen Krankenhausaufenthalt erforderten, um 35% im Vergleich zu Placebo (Rate Ratio [RR] von 0,65; 95 % KI: 0,43, 0,96), Diese Reduktion konnte aber aufgrund der nicht eingehaltenen statistischen Testhierarchie nicht als signifikant gewertet werden.
Bewertung der Symptome und der gesundheitsbezogenen Lebensqualität
Die Ergebnisse der Endpunkte für Symptome und gesundheitsbezogene Lebensqualität (CAT und SGRQ) waren nicht statistisch signifikant. Die SGRQ-Responderrate (Ansprechen definiert als Verringerung der Punktzahl um 4 oder mehr gegenüber dem Ausgangswert) in Woche 52 betrug 50% für Mepolizumab 100 mg gegenüber 46% für Placebo (Odds Ratio: 1,17; 95%-KI: 0,87; 1,57).
Exazerbationen in Patienten-Untergruppen
Analysen nach Raucheranamnese sowie nach Symptomen einer chronischen Bronchitis bei Baseline ergaben in Bezug auf den primären Endpunkt Verbesserungen, die im Einklang standen mit denjenigen in der Gesamtpopulation bei Patienten unter Mepolizumab 100 mg gegenüber Placebo.
Meta-Analyse der Studien 1, 2 und 3
In zwei weiteren 52-wöchigen randomisierten, placebokontrollierten, multizentrischen Studien, Studie 2 (MEA117113 [METREO]) und Studie 3 (MEA117106 [METREX]), wurden 1285 Patienten mit COPD randomisiert und erhielten entweder Mepolizumab 100 mg sc. oder Placebo.
Primäres Studienziel der Studien 2 und 3 war die Beurteilung der Wirksamkeit von Mepolizumab auf die jährliche Rate von mittelschweren (definiert als Verschlechterung der COPD-Symptome, die eine Behandlung mit oralen/systemischen Kortikosteroiden oder Antibiotika erfordert) oder schweren (definiert als hospitalisierungsbedürftig oder zum Tode führend) Exazerbationen.
Eine Metaanalyse integrierte die Daten aller Patienten der Studie 1 sowie einer Untergruppe von Patienten mit vergleichbaren Blut-Eosinophilenzahlen von ≥300 Zellen/µL beim Screening aus den Studien 2 und 3 (kombinierte n = 568 unter Mepolizumab 100 mg und n = 578 unter Placebo). Mepolizumab senkte die jährliche Rate mittelschwerer oder schwerer Exazerbationen im Vergleich zu Placebo um 21 % (RR: 0,79; 95%-KI: 0,68; 0,91). Mepolizumab reduzierte das Risiko einer ersten mittelschweren oder schweren Exazerbation um 28 % im Vergleich zu Placebo (HR: 0,72; 95%-KI: 0,62; 0,85). Mepolizumab reduzierte die jährliche Rate von COPD-Exazerbationen, die einen Notfallstationsbesuch und/oder eine Hospitalisierung erforderten, um 29% im Vergleich zu Placebo (RR: 0,71; 95%-KI: 0,51; 0,98).
Eosinophile Granulomatose mit Polyangiitis (EGPA)
MEA115921 war eine randomisierte, doppelblinde, placebokontrollierte, 52-wöchige Studie, in der 136 Patienten ab 18 Jahren mit rezidivierender oder therapierefraktärer EGPA unter stabil dosierten oralen Kortikosteroiden (OCS; ≥7,5 bis ≤50 mg/Tag Prednisolon/Prednison) untersucht wurden. 53% (n = 72) der Teilnehmer erhielten gleichzeitig Immunsuppressiva in stabiler Dosierung.
Die Patienten erhielten eine Dosis von 300 mg Mepolizumab oder Placebo subkutan einmal alle vier Wochen, ergänzend zu ihrer Prednisolon/Prednison-Basistherapie mit oder ohne Immunsuppressiva. Die OCS-Dosis wurde nach dem Ermessen des Prüfarztes stufenweise reduziert.
Koprimäre Endpunkte waren die kumulative Gesamtremissionsdauer, wobei Remission definiert war als Birmingham Vasculitis Activity Score (BVAS) von 0 (keine aktive Vaskulitis) plus Prednisolon/Prednison-Dosis ≤4 mg/Tag, sowie der Anteil der Teilnehmer in Remission nach 36 sowie nach 48 Behandlungswochen.
Remission
Im Vergleich zu Placebo erreichten die Teilnehmer unter Mepolizumab 300 mg eine signifikant längere Gesamtremissionsdauer. Darüber hinaus war der Anteil der Teilnehmer in Remission sowohl in Woche 36 als auch in Woche 48 unter Mepolizumab 300 mg signifikant höher als unter Placebo (Tabelle 10).
Tabelle 10: Analysen zu den koprimären Endpunkten (ITT-Population)
Anzahl (%) Teilnehmer
Placebo n = 68 Mepolizumab 300 mg n = 68
Gesamtremissionsdauer während 52 Wochen
0 Wochen 55 (81) 32 (47)
> 0 bis < 12 Wochen 8 (12) 8 (12)
12 bis < 24 Wochen 3 (4) 9 (13)
24 bis < 36 Wochen 0 10 (15)
≥36 Wochen 2 (3) 9 (13)
Odds Ratio (Mepolizumab/Placebo) 5,91
95%-KI ---- 2,68; 13,03
p-Wert ---- < 0,001
Teilnehmer in Remission in Woche 36 und 48 2 (3) 22 (32)
Odds Ratio (Mepolizumab/Placebo) 16,74
95%-KI ---- 3,61; 77,56
p-Wert ---- < 0,001
Odds Ratio > 1 spricht zugunsten von Mepolizumab
Teilnehmer unter Mepolizumab 300 mg erreichten eine signifikant längere Gesamtremissionsdauer (p < 0,001), und der Anteil der Teilnehmer in Remission war bei Anwendung der Remissionsdefinition für den sekundären Endpunkt (BVAS = 0 plus Prednisolon/Prednison ≤7,5 mg/Tag) sowohl in Woche 36 als auch in Woche 48 unter Mepolizumab 300 mg höher als unter Placebo (p < 0,001).
Rezidive
Im Vergleich zu Placebo war die Zeit bis zum ersten Rezidiv (definiert als durch Vaskulitis, Asthma oder sinonasale Symptome bedingte Verschlechterung, die eine Dosiserhöhung der Kortikosteroide bzw. der Immunsuppressiva oder eine Hospitalisierung erfordert), bei Teilnehmern unter Mepolizumab 300 mg signifikant länger (p < 0,001). Darüber hinaus war die annualisierte Rezidivrate bei Teilnehmern unter Mepolizumab um 50 % geringer als bei Teilnehmern unter Placebo: 1,14 vs. 2,27.
Dosisreduktion der oralen Kortikosteroide
Im Vergleich zu Teilnehmern unter Placebo erhielten Teilnehmer unter Mepolizumab 300 mg in den Wochen 48 bis 52 eine geringere mittlere Tagesdosis an oralen Kortikosteroiden (p < 0,001). In der Gruppe mit Mepolizumab 300 mg konnten 12 Teilnehmer (18 %) die Behandlung mit oralen Kortikosteroiden gänzlich ausschleichen, gegenüber nur 2 Teilnehmern (3 %) in der Placebogruppe.
Hypereosinophilie-Syndrom (HES)
Studie 200622 war eine randomisierte, doppelblinde, placebokontrollierte, 32-wöchige Studie, in der 108 HES-Patienten im Alter von ≥12 Jahren untersucht wurden. Patienten mit nicht-hämatologischem sekundärem HES (z.B. Medikamentenüberempfindlichkeit, parasitäre Infektion, HIV-Infektion, nicht-hämatologisches Malignom) oder F/P positivem HES wurden von der Studie ausgeschlossen. Die Patienten erhielten subkutan einmal alle 4 Wochen 300 mg Mepolizumab bzw. Placebo, unter Beibehaltung ihrer stabilen HES-Therapie. Einer der 4 eingeschlossenen Jugendlichen erhielt 300 mg Mepolizumab, die anderen 3 erhielten Placebo, alle jeweils 32 Wochen lang. Die Standard-HES-Therapie konnte OCS und Immunsuppressiva oder Zytotoxika umfassen. Die Studienteilnehmer hatten in den letzten12 Monaten mindestens zwei HES-Schübe erlitten und wiesen beim Screening eine Blut-Eosinophilenzahl von ≥1000 Zellen/µL auf.
Primärer Endpunkt der Studie 200622 war der Anteil der Studienteilnehmer, die während des 32-wöchigen Behandlungszeitraums einen HES-Schub erlitten. Ein HES-Schub war definiert als Verschlechterung der klinischen Anzeichen und Symptome des HES oder als Anstieg der Eosinophilen (zu mindestens 2 Zeitpunkten), die eine OCS-Erhöhung oder die Dosiserhöhung bzw. die zusätzliche Verabreichung einer zytotoxischen oder immunsuppressiven HES-Therapie erforderlich machten.
Die Primäranalyse verglich Patienten der Mepolizumab- und der Placebogruppe, die einen HES-Schub erlitten oder aus der Studie ausschieden. Im Vergleich zur Placebogruppe erlitten in der Gruppe mit 300 mg Mepolizumab während des 32-wöchigen Behandlungszeitraums 50 % weniger Patienten einen HES-Schub oder brachen die Studie ab; 28 % versus 56 % (OR 0,28, 95%-KI 0,12–0,64) (siehe Tabelle 11).
Sekundäre Endpunkte waren die Zeit bis zum ersten HES-Schub, der Anteil der Studienteilnehmer, die in Woche 20 bis Woche 32 einen HES-Schub erlitten, die Rate der HES-Schübe und die Änderung des Fatigue-Schweregrads gegenüber dem Ausgangswert. Alle sekundären Endpunkte waren statistisch signifikant und untermauerten den primären Endpunkt (siehe Abbildung 3 und Tabelle 12).
Tabelle 11: Ergebnisse für den primären Endpunkt / Ergebnisse der Primäranalyse in der Intent-to-Treat-Population (Studie 200622)
Mepolizumab N = 54 Placebo N = 54
Anteil der Studienteilnehmer, die einen HES-Schub erlitten
Studienteilnehmer mit ≥1 HES-Schub oder Studienabbruch (%) 15 (28) 30 (56)
Studienteilnehmer mit ≥1 HES-Schub (%) 14 (26) 28 (52)
Studienteilnehmer ohne HES-Schub, die die Studie abbrachen (%) 1 (2) 2 (4)
Odds Ratio (95%-KI) 0,28 (0,12–0,64)
p-Wert im CMH 0,002
CMH = Cochran-Mantel-Haenszel
Zeit bis zum ersten Schub
Bei Studienteilnehmern unter 300 mg Mepolizumab war die Zeit bis zum ersten HES-Schub im Vergleich zu Patienten unter Placebo signifikant länger. Das Risiko eines ersten HES-Schubs während des Behandlungszeitraums war bei Patienten unter Mepolizumab um 66 % niedriger als bei Patienten unter Placebo (Hazard Ratio: 0,34, 95%-KI 0,18–0,67, p = 0,002).
Abbildung 3: Kaplan-Meier-Kurve für die Zeit bis zum ersten HES-Schub

Tabelle 12 Ergebnisse für andere sekundäre Endpunkte in der Intent-to-Treat-Population (Studie 200622)
Mepolizumab N = 54 Placebo N = 54
HES-Schübe in Woche 20 und bis einschliesslich Woche 32
Studienteilnehmer mit ≥1 HES-Schub oder Studienabbruch 9 (17) 19 (35)
(%)
Odds Ratio (95%-KI) 0,33 (0,13–0,85)
CMH-p-Wert (nicht bereinigt/bereinigt)a 0,02/0,02
Rate der HES-Schübe
Geschätzte jährliche Durchschnittsrate 0,50 1,46
Ratenverhältnis (95%-KI) 0,34 (0,19–0,63)
Wilcoxon-p-Wert (nicht bereinigt/bereinigt)a 0,002/0,02
Änderung des Fatigue-Schweregrads gegenüber dem
Ausgangswert, basierend auf Item 3 des Brief Fatigue
Inventory (BFI) (schwerste Fatigue-Ausprägung während
der letzten 24 Stunden) in Woche 32b
Mediane Änderung beim BFI-Item 3 -0,66 0,32
Vergleich (Mepolizumab vs. Placebo) der p-Werte (nicht 0,036/0,036
bereinigt/bereinigt)a
a Bereinigte p-Werte, basierend auf einer vorab festgelegten Hierarchie von Endpunkten.
b Patienten mit fehlenden Daten wurden eingeschlossen unter Verwendung des schlechtesten beobachteten Werts.
CMH = Cochran-Mantel-Haenszel
Offene Verlängerungsphase bei HES
Geeignete Patienten, darunter 4 Jugendliche, fuhren nach Abschluss der Studie 200622 mit der 20-wöchigen offenen Verlängerungsstudie 205203 fort, in der das langfristige Sicherheitsprofil untersucht wurde und zusätzliche Daten zum klinischen Nutzen von Mepolizumab bei HES-Patienten über 32 Wochen hinaus gewonnen wurden.
Die in Studie 200622 beobachtete Wirkung der Mepolizumab-Behandlung auf die Reduktion der HES-Schübe wurde aufrechterhalten bei den Patienten, welche die Mepolizumab-Behandlung in Studie 205203 fortsetzten, in der bei 94 % (47/50) der Patienten keine Schübe auftraten.
In den Wochen 16 bis 20 hatten 28 % aller Patienten, die in den Wochen 0 bis 4 eine durchschnittliche OCS-Dosis von > 0 mg/Tag (Prednison oder Äquivalent) erhalten hatten, eine durchschnittliche tägliche OCS-Dosisreduktion von ≥50 % erreicht. Wirksamkeitsdaten aus dieser Studie deuten darauf hin, dass der klinische Nutzen von Mepolizumab über 52 Wochen anhält und eine Verringerung der OCS-Behandlung bei HES-Patienten ermöglicht.