ZusammensetzungWirkstoffe
Ofatumumab ist ein rekombinanter, vollständig humaner monoklonaler Immunglobulin-G1(IgG1)-Antikörper gegen humanes CD20, das auf B-Zellen exprimiert wird. Ofatumumab wird in einer murinen Zelllinie (NS0) mittels rekombinanter DNA-Technologie hergestellt.
Hilfsstoffe
L-Arginin, Natriumacetat-Trihydrat, Natriumchlorid, Polysorbat 80, Natriumedetat (der Gesamtgehalt an Natrium pro Dosis beträgt 0.92 mg), Salzsäure (zur Einstellung des pH-Werts) und Wasser für Injektionszwecke
Indikationen/AnwendungsmöglichkeitenKesimpta ist für die Behandlung von erwachsenen Patienten mit aktiven, schubförmig verlaufenden Formen der Multiplen Sklerose (MS) indiziert.
Dosierung/AnwendungDie Behandlung sollte von einem Arzt eingeleitet werden, der über Erfahrung in der Behandlung von neurologischen Erkrankungen einschliesslich Multipler Sklerose verfügt.
Übliche Dosierung
Die empfohlene Dosis beträgt 20 mg Kesimpta, verabreicht durch subkutane Injektion in folgenden Abständen:
·initiale Gabe in den Wochen 0, 1 und 2, gefolgt von
·anschliessenden monatlichen Gaben, beginnend in Woche 4.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Ausgelassene Dosen
Wenn eine Injektion von Kesimpta ausgelassen wurde, sollte sie so bald wie möglich verabreicht werden; es sollte nicht bis zur nächsten vorgesehenen Dosis gewartet werden. Die nachfolgenden Dosen sollten in den empfohlenen Abständen verabreicht werden.
Spezielle Dosierungsanweisungen
Ältere Patienten
Bisher wurden keine Studien bei älteren MS-Patienten durchgeführt. Ofatumumab wurde bei Patienten mit schubförmig verlaufender MS (relapsing MS, RMS) im Alter von 18 bis 55 Jahren untersucht. Ergebnisse aus der Populationspharmakokinetik legen nahe, dass bei älteren Patienten keine Dosisanpassung erforderlich ist (siehe «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bisher wurden keine spezifischen Studien zu Ofatumumab bei Patienten mit einer Nierenfunktionsstörung durchgeführt.
Patienten mit leichter Nierenfunktionsstörung wurden in die klinischen Studien eingeschlossen. Es liegen keine Erfahrungen bei Patienten mit mittelschwerer und schwerer Nierenfunktionsstörung vor. Da Ofatumumab jedoch nicht über den Urin ausgeschieden wird, ist nicht zu erwarten, dass bei Patienten mit einer Nierenfunktionsstörung eine Dosisänderung erforderlich ist (siehe «Pharmakokinetik»).
Patienten mit Leberfunktionsstörungen
Bisher wurden keine spezifischen Studien mit Ofatumumab bei Patienten mit einer Leberfunktionsstörung durchgeführt.
Da der hepatische Metabolismus von monoklonalen Antikörpern wie Ofatumumab zu vernachlässigen ist, ist nicht zu erwarten, dass die Pharmakokinetik dieses Arzneimittels durch eine Leberfunktionsstörung beeinflusst wird. Daher ist nicht davon auszugehen, dass bei Patienten mit einer Leberfunktionsstörung eine Dosisänderung erforderlich ist (siehe «Pharmakokinetik».)
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Kesimpta bei Kindern im Alter von 0 bis 18 Jahren wurden bisher nicht untersucht. Es liegen keine Daten vor.
Art der Anwendung
Kesimpta ist für die Selbstverabreichung durch den Patienten als subkutane Injektion vorgesehen.
Die üblichen Stellen für subkutane Injektionen sind der Bauch, der Oberschenkel und die Aussenseite des Oberarms.
Die erste Injektion von Kesimpta sollte unter Anleitung einer medizinischen Fachkraft erfolgen (siehe « Warnhinweise und Vorsichtsmassnahmen»).
Eine ausführliche Anleitung zur Verabreichung der Injektion steht in der Packungsbeilage zur Verfügung.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der in der Rubrik Zusammensetzung aufgeführten Hilfsstoffe.
Stark immungeschwächte Patienten (siehe «Warnhinweise und Vorsichtsmassnahmen», «Interaktionen»).
Vorliegen einer aktiven Infektion (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Bekannte aktive maligne Erkrankungen.
Therapiebeginn während der Schwangerschaft.
Warnhinweise und VorsichtsmassnahmenVor Therapiebeginn muss das medizinische Fachpersonal sicherstellen, dass der Patient die Sicherheitsinformation gelesen und verstanden hat.
Injektionsbedingte Reaktionen
Die Patienten sollten darüber informiert werden, dass injektionsbedingte Reaktionen im Allgemeinen innerhalb von 24 Stunden und überwiegend nach der ersten Injektion auftreten. Etwaige injektionsbedingte Reaktionen können durch eine symptomatische Behandlung bewältigt werden.
Zu den in klinischen Studien festgestellten (lokalen) Symptomen einer Reaktion an der Injektionsstelle gehörten Erythem, Schwellung, Juckreiz und Schmerzen.
Die in klinischen Studien beobachteten systemischen injektionsbedingten Reaktionen traten überwiegend bei der ersten Injektion auf. Zu den beobachteten Symptomen gehörten Fieber, Kopfschmerz, Myalgie, Schüttelfrost und Ermüdung, und diese waren überwiegend (99,7 %) nicht schwerwiegend und von leichter bis mässiger Ausprägung. In den klinischen Studien zur RMS gab es keine lebensbedrohlichen Injektionsreaktionen.
In klinischen Studien zur RMS wurde nur ein begrenzter Nutzen der Prämedikation mit Steroiden, Antihistaminika oder Paracetamol festgestellt. Bei den mit Ofatumumab behandelten Patienten, die eine Prämedikation mit Methylprednisolon (oder einem äquivalenten Steroid) erhielten, traten Symptome wie Fieber, Myalgie, Schüttelfrost und Übelkeit seltener auf. Unter der Anwendung der Steroid-Prämedikation kam es jedoch auch ohne die Behandlung mit Ofatumumab (d.h. bei Patienten in den Teriflunomid-Armen, die Placebo-Injektionen erhielten) zu einem Anstieg der Häufigkeit von Symptomen wie Hitzegefühl, Brustkorbbeschwerden, Hypertonie, Tachykardie und Abdominalschmerz. Die Anwendung einer Prämedikation ist daher nicht notwendig.
Die erste Injektion von Kesimpta sollte unter Anleitung einer entsprechend ausgebildeten medizinischen Fachkraft durchgeführt werden.
Infektionen
Aufgrund seiner Wirkungsweise und entsprechend der verfügbaren klinischen Erfahrung hat Ofatumumab das Potenzial für ein erhöhtes Infektionsrisiko (siehe «Unerwünschte Wirkungen»).
Ofatumumab darf bei Patienten mit einer aktiven Infektion nicht verabreicht werden. Bei Patienten mit einer aktiven Infektion muss mit der Gabe von Ofatumumab zugewartet werden, bis die Infektion abgeheilt ist (siehe «Kontraindikationen»). Bei Patienten, die Anzeichen oder Symptome einer Infektion im Anschluss an eine Behandlung mit Ofatumumab berichten, sollten diese rasch abgeklärt und die Patienten entsprechend behandelt werden. Vor einer weiteren Behandlung sind die Patienten erneut auf ein potenzielles Infektionsrisiko zu untersuchen.
Basierend auf dem Wirkmechanismus von Ofatumumab besteht ein erhöhtes Infektionsrisiko während der Behandlung, einschliesslich schwerwiegenden bakteriellen, fungalen sowie neuen oder reaktivierten viralen Infektionen. In Patienten, welche mit anderen anti-CD20 Antikörpern behandelt wurden, verliefen einige dieser Infektionen tödlich.
In den klinischen Studien zur RMS war der Anteil der Patienten mit Infektionen in der Ofatumumab- und der Teriflunomid-Behandlungsgruppe ähnlich. In den klinischen Zulassungsstudien der Phase III trat bei 51,6 % der mit Ofatumumab behandelten Patienten mindestens eine Infektion auf, verglichen mit 52,7 % der mit Teriflunomid behandelten Patienten.
Progressive multifokale Leukenzephalopathie
PML ist eine opportunistische Infektion, die durch das John-Cunningham-Virus (JCV) verursacht wird und tödlich verlaufen oder zu schweren Behinderungen führen kann. In den klinischen Studien zur RMS wurden für Ofatumumab keine Fälle von progressiver multifokaler Leukenzephalopathie (PML) gemeldet. Tödlich verlaufende Fälle mit PML traten in Patienten auf, die mit Ofatumumab für eine chronisch lymphatische Leukämie behandelt wurden (zwar in deutlich höherer intravenöser Dosierung als für die Behandlung mit MS empfohlen, jedoch für eine kürzere Behandlungsdauer). Zusätzlich wurden bei Patienten, die mit Anti-CD20-Antikörpern und anderen MS-Therapien behandelt wurden, Infektionen mit dem John-Cunningham-Virus (JC-Virus), die zu einer PML führen können, beobachtet. Daher sollten Ärzte bzgl. Frühzeichen und Symptome einer PML, die jede Art von neu auftretenden oder sich verschlechternden neurologischen Zeichen oder Symptomen beinhalten können, wachsam sein, da diese den Symptomen eines MS-Schubs gleichen können.
Die Symptome der PML sind vielfältig, schreiten über Tage bis Wochen fort und können zunehmende Schwäche einer Körperseite oder Ungeschicklichkeit der Gliedmassen, Gleichgewichtsstörungen, Sehstörungen sowie Veränderungen des Denkens, des Gedächtnisses und der Orientierung umfassen, die zu Verwirrung und Persönlichkeitsveränderungen führen. Wird eine PML vermutet, sollte die Behandlung mit Kesimpta so lange ausgesetzt werden, bis eine PML ausgeschlossen wurde.
Bei Verdacht auf eine PML sollte eine Evaluation anhand eines MRTs (vorzugsweise mit Kontrastmittel) im Vergleich zu einem vor der Behandlung angefertigten MRT (vorzugsweise nicht älter als 3 Monate) und eines bestätigenden Liquortests mit Bestimmung der viralen JC-DNA sowie wiederholter neurologischer Untersuchungen erfolgen.
Falls die PML bestätigt ist, muss die Behandlung dauerhaft abgebrochen werden.
Reaktivierung des Hepatitis-B-Virus
In den klinischen Studien mit Kesimpta zur RMS wurde kein Fall einer Reaktivierung des Hepatitis-B-Virus (HBV) festgestellt. Es sind jedoch Fälle der Reaktivierung einer Hepatitis B unter der Behandlung mit Anti-CD-20-Antikörpern aufgetreten, die in einigen Fällen zu einer fulminanten Hepatitis, zum Leberversagen und zum Tod geführt haben.
Patienten mit aktiver Hepatitis-B-Erkrankung dürfen deshalb nicht mit Kesimpta behandelt werden (siehe «Kontraindikationen»). Vor Beginn der Behandlung mit Kesimpta sollte bei allen Patienten ein Screening auf das Hepatitis-B-Virus (HBV) durchgeführt werden. Das Screening sollte als Mindestvorgabe Tests auf das Hepatitis-B-Oberflächenantigen (HBsAg) und den Hepatitis-B-Kernantikörper (Anti-HBc) umfassen. Diese Tests können durch andere geeignete Marker entsprechend den lokalen Leitlinien ergänzt werden. Patienten mit einer positiven Hepatitis-B-Serologie (entweder HBsAg oder Anti-HBc) sollten vor Behandlungsbeginn einen Spezialisten für Lebererkrankungen konsultieren und sie sollten gemäss den lokalen medizinischen Standards überwacht und behandelt werden, um eine Reaktivierung der Hepatitis B zu verhindern.
Im Fall einer HBV-Reaktivierung, sollte Kesimpta pausiert werden bis die aktive Infektion abgeklungen ist.
Behandlung hochgradig immungeschwächter Patienten
Es wird empfohlen, vor Beginn der Therapie mit Kesimpta den Immunstatus des Patienten zu beurteilen. Kesimpta darf nicht an Patienten mit starker Immunsuppression (z.B. einer signifikanten Neutropenie oder Lymphopenie) verabreicht werden (siehe «Kontraindikationen»). Hochgradig immungeschwächte Patienten dürfen so lange nicht behandelt werden, bis die jeweilige Erkrankung abgeklungen ist (siehe «Kontraindikationen»).
Patienten, die aktuell eine immunsuppressive Behandlung erhalten (ausgenommen sind symptomatische Behandlungen mit Kortikosteroiden gegen Rezidive) oder deren Immunsystem durch vorausgehende Therapien geschwächt ist (siehe «Interaktionen») dürfen nicht mit Ofatumumab behandelt werden.
Behandlung mit Immunsuppressiva vor, während oder nach der Behandlung mit Ofatumumab
Bei Einleitung einer Behandlung mit Ofatumumab nach einer immunsuppressiven Therapie bzw. bei Einleitung einer immunsuppressiven Therapie nach einer Behandlung mit Ofatumumab muss das Potenzial für überlappende pharmakodynamische Wirkungen berücksichtigt werden (siehe «Wirkungsmechanismus/Pharmakodynamik»). Ofatumumab wurde nicht in Kombination mit anderen krankheitsmodifizierenden MS-Therapeutika untersucht.
Impfungen
Alle Impfungen sollten gemäss den Impfleitlinien bei Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen mindestens 4 Wochen vor Beginn der Behandlung mit Kesimpta und bei inaktivierten Impfstoffen, wenn möglich, mindestens 2 Wochen vor Beginn der Behandlung mit Kesimpta verabreicht werden.
Kesimpta kann die Wirksamkeit von Totimpfstoffen (inaktivierten Impfstoffen) beeinträchtigen.
Die Sicherheit von Impfungen mit Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen nach einer Therapie mit Kesimpta wurde bisher nicht untersucht. Eine Impfung mit Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen wird während der Behandlung und nach dem Absetzen bis zur B-Zell-Repletion nicht empfohlen (siehe «Eigenschaften/Wirkungen»).
Impfung von Säuglingen, die von Müttern geboren wurden, welche während der Schwangerschaft mit Kesimpta behandelt wurden
Bei Säuglingen von Müttern, die während der Schwangerschaft mit Kesimpta behandelt wurden, sollten so lange keine Lebendimpfstoffe oder abgeschwächten Lebendimpfstoffe verabreicht werden, bis eine Erholung der B-Zellzahl bestätigt wurde. Durch die Depletion der B-Zellen bei diesen Kindern kann das von Lebendimpfstoffen oder abgeschwächten Lebendimpfstoffen ausgehende Risiko ansteigen.
Totimpfstoffe (inaktivierte Impfstoffe) können gemäss der Indikation auch vor der Erholung von der B-Zell-Depletion verabreicht werden, jedoch sollte eine genaue Beurteilung der durch den Impfstoff induzierten Immunreaktionen, ggf. auch unter Hinzuziehung eines qualifizierten Spezialisten, in Betracht gezogen werden, um festzustellen, ob tatsächlich eine schützende Immunantwort ausgelöst wurde (siehe «Schwangerschaft, Stillzeit»).
Malignome
Unter anderen anti-CD20 B-Zell depletierenden Therapien wurde ein erhöhtes Risiko für maligne Erkrankungen, insbesondere Mammakarzinomen, beobachtet. Die Inzidenz lag dabei im Rahmen der bei MS-Patienten zu erwartenden Hintergrundrate.
Patienten mit bestehenden aktiven malignen Erkrankungen (einschliesslich Patienten, die hinsichtlich der Rezidivierung einer malignen Erkrankung aktiv überwacht werden) dürfen nicht mit Ofatumumab behandelt werden (siehe «Kontraindikationen»). Bei Patienten mit bekannten Risikofaktoren für Malignitäten sollte das Nutzen-Risiko-Verhältnis von Ofatumumab sorgfältig abgewogen werden und vor sowie während der Behandlung eine entsprechende Tumorüberwachung durchgeführt werden.
Schwerwiegende unerwünschte Ereignisse während der Behandlung mit anderen anti-CD20 Antikörpern
Unter der Behandlung mit anderen anti-CD20 Antikörpern traten selten schwerwiegende kardiovaskuläre Ereignisse sowie schwerwiegende mukokutane Reaktionen auf. Patienten, welche eine schwerwiegende mukokutane Reaktion während der Behandlung mit Kesimpta zeigen, sollten diese beenden und umgehend medizinisch beurteilen lassen.
Hautreaktionen
Bei anderen anti-CD20-Antikörpern wurden schwere Hautreaktionen wie toxische epidermale Nekrolyse (Lyell-Syndrom), Stevens-Johnson-Syndrom und Pyoderma gangraenosum beobachtet. Für den Fall, dass ein derartiges Ereignis eintritt, ist ein Abbruch der Behandlung in Erwägung zu ziehen.
Natriumgehalt
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Dosis, d.h. es ist nahezu «natriumfrei».
InteraktionenDa keine Interaktionen mit Arzneimitteln über Cytochrom P450-Enzyme, andere metabolisierende Enzyme oder Transportstoffe zu erwarten sind, wurden keine Interaktionsstudien durchgeführt.
Impfungen
Die Sicherheit einer Impfung mit Lebendimpfstoffen, abgeschwächten Lebendimpfstoffen oder Totimpfstoffen (inaktivierten Impfstoffen) während der Behandlung mit Ofatumumab und die Fähigkeit, eine primäre oder anamnestische Immunantwort zu generieren, wurden bisher nicht untersucht. Durch die B-Zell-Depletion kann die Immunantwort auf Impfungen verringert sein. Es wird empfohlen, dass die Patienten die geplanten Impfungen vor Beginn der Therapie mit Kesimpta abschliessen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Andere immunsuppressive oder immunmodulierende Therapien
Bei der Umstellung von Arzneimitteln mit langfristigen Auswirkungen auf das Immunsystem wie Ocrelizumab, Cladribin, Fingolimod, Natalizumab, Teriflunomid, Mitoxantron oder Dimethylfumarat auf Kesimpta muss während der Einleitung der Behandlung mit Kesimpta aufgrund von möglichen additiven immunsuppressiven Wirkungen Dauer und Wirkungsweise dieser Arzneimittel berücksichtigt werden.
Ofatumumab wurde nicht in Kombination mit anderen krankheitsmodifizierenden MS-Therapeutika untersucht.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Während der Schwangerschaft darf keine Therapie mit Kesimpta eingeleitet werden (siehe «Kontraindikationen»). Frauen im gebärfähigen Alter sollten während der Einnahme von Kesimpta und für 6 Monate nach der letzten Anwendung von Kesimpta eine wirksame Empfängnisverhütung anwenden (Methoden, die zu einer Schwangerschaftsrate von weniger als 1 % führen).
Schwangerschaft
Bisher liegen nur begrenzte Erfahrungen mit der Anwendung von Ofatumumab bei Schwangeren vor.
Es wurde über eine vorübergehende periphere B-Zell-Depletion und Lymphozytopenie bei Säuglingen von Müttern berichtet, die während der Schwangerschaft mit anderen Anti-CD20-Antikörpern behandelt worden waren. Die mögliche Dauer der B-Zell-Depletion bei Säuglingen, bei denen in utero eine Exposition gegenüber Ofatumumab bestanden hat, und die Auswirkungen der B-Zell-Depletion auf die Sicherheit und Wirksamkeit von Impfstoffen sind nicht bekannt (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Aufgrund von Erkenntnissen aus tierexperimentellen Studien wird davon ausgegangen, dass Ofatumumab die Plazentaschranke überwinden und eine B-Zell-Depletion beim Föten verursachen kann (siehe «Präklinische Daten»). Nach intravenöser Verabreichung von Ofatumumab an trächtige Affen während der Organogenese wurde keine Teratogenität beobachtet.
Kesimpta sollte während einer Schwangerschaft nicht angewendet werden, es sei denn, der mögliche Nutzen für die Mutter überwiegt gegenüber dem möglichen Risiko für den Fötus.
Stillzeit
Die Anwendung von Ofatumumab bei Frauen während der Stillzeit wurde bisher nicht untersucht. Es ist nicht bekannt, ob Ofatumumab in die Muttermilch übergeht; humanes IgG ist jedoch in der Muttermilch vorhanden. Es liegen keine Daten über die Wirkungen von Kesimpta auf das gestillte Kind oder die Milchproduktion vor. Der Nutzen des Stillens für die Entwicklung und Gesundheit des gestillten Kindes sollte zusammen mit dem klinischen Nutzen von Kesimpta für die Mutter und den möglichen unerwünschten Wirkungen durch Kesimpta auf das gestillte Neugeborene/den gestillten Säugling berücksichtigt werden.
Fertilität
Es liegen keine Erfahrungen im Hinblick auf die Beeinflussung der Fertilität beim Menschen durch Ofatumumab vor. Tierexperimentelle Studien zeigen keine Hinweise auf direkte oder indirekte schädliche Wirkungen in Bezug auf die männliche und weibliche Fertilität (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenKesimpta hat wahrscheinlich keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenUngefähr 1500 Patienten mit RMS haben Ofatumumab in klinischen Studien erhalten. In den beiden Zulassungsstudien der Phase III wurden 1882 Patienten mit RMS randomisiert, 946 davon wurden über eine mediane Dauer von 85 Wochen mit Ofatumumab behandelt; 33 % der Patienten, die Ofatumumab erhielten, wurden über mehr als 96 Wochen behandelt (siehe «Eigenschaften/Wirkungen»).
Der Anteil der Patienten mit unerwünschten Ereignissen (UE) (83,6 % vs. 84,2 %) und die UE, die zum Absetzen des Medikaments führten (5,7 % vs. 5,2 %), waren in der Ofatumumab- und der Teriflunomid-Gruppe ähnlich.
Die unerwünschten Arzneimittelwirkungen (UAW), die im Zusammenhang mit der Anwendung von Ofatumumab in klinischen Studien zur RMS gemeldet wurden, sind untenstehend nach MedDRA-Systemorganklassen aufgeführt. Innerhalb jeder Systemorganklasse sind die unerwünschten Arzneimittelwirkungen nach Häufigkeit aufgelistet, wobei die häufigsten unerwünschten Arzneimittelwirkungen zuerst genannt werden. Innerhalb der einzelnen Häufigkeitsgruppen sind die unerwünschten Arzneimittelwirkungen nach abnehmendem Schweregrad angegeben. Darüber hinaus basiert die entsprechende Häufigkeitskategorie für jede unerwünschte Arzneimittelwirkung auf der folgenden Konvention: sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000); sehr selten (< 1/10'000).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der oberen Atemwege1(39%), Harnwegsinfektion (12%)
Häufig: Oraler Herpes.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (13%)
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Reaktionen an der Injektionsstelle (lokal) (11%)
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Sehr häufig: injektionsbedingte Reaktionen (systemisch) (21%)
Untersuchungen
Häufig: verminderte Immunglobulin M (IgM)-Serumspiegel
1 Bei der Bestimmung der Häufigkeit der UAW wurde die Gruppierung der Vorzugsbenennungen (PTs) berücksichtigt.
Beschreibung ausgewählter unerwünschter Arzneimittelwirkungen
Infektionen
In den klinischen Phase-III-Studien zur RMS war die Gesamthäufigkeit an Infektionen und schweren Infektionen bei den mit Ofatumumab behandelten Patienten vergleichbar mit den mit Teriflunomid behandelten Patienten (51,6% versus 52,7% und 2,5% versus 1,8%).
Infektionen der oberen Atemwege
Infektionen der oberen Atemwege traten bei den mit Ofatumumab behandelten Patienten häufiger auf als bei den mit Teriflunomid behandelten Patienten. In den klinischen Studien zur RMS traten bei 39,4 % der mit Ofatumumab behandelten Patienten Infektionen der oberen Atemwege auf; bei den mit Teriflunomid behandelten Patienten waren es 37,8 %. Die Infektionen waren überwiegend leicht bis mittelschwer und umfassten meistens Nasopharyngitis, Infektionen der oberen Atemwege und Influenza.
Injektionsbedingte Reaktionen und Reaktionen an der Injektionsstelle
In den klinischen Phase-III-Studien zur RMS wurden bei 20,6 % bzw. 10,9 % der mit Ofatumumab behandelten Patienten injektionsbedingte Reaktionen (systemisch) und Reaktionen an der Injektionsstelle (lokal) angegeben.
Die Inzidenz der injektionsbedingten Reaktionen war bei der ersten Injektion am höchsten (14,4 %) und ging bei den nachfolgenden Injektionen deutlich zurück (4,4 % bei der zweiten, < 3 % ab der dritten Injektion). Die injektionsbedingten Reaktionen waren meist (99,8 %) leicht bis mässig ausgeprägt. Nur zwei (0,2 %) der mit Ofatumumab behandelten MS-Patienten berichteten über schwerwiegende injektionsbedingte Reaktionen. Es gab keine lebensbedrohlichen injektionsbedingten Reaktionen. Zu den Symptomen, die am häufigsten angegeben wurden, (≥2 %) gehörten Fieber, Kopfschmerz, Myalgie, Schüttelfrost und Ermüdung.
Lokale Reaktionen am Verabreichungsort waren sehr häufig. Die Reaktionen an der Injektionsstelle waren alle leicht bis mässig ausgeprägt, und es handelte sich dabei nicht um schwerwiegende Ereignisse. Zu den Symptomen, die am häufigsten angegeben wurden (≥2 %) gehörten Erythem, Schmerzen, Juckreiz und Schwellung (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Auffällige Laborwerte
Immunglobuline
Im Verlauf der klinischen Phase-III-Studien zur RMS wurde eine Abnahme des Mittelwerts für das IgM beobachtet, die jedoch nicht mit einem Risiko für Infektionen, einschliesslich schwerer Infektionen, einherging.
Es wurde ein Rückgang des IgM-Wertes um 30,9 % nach 48 Wochen und um 38,8 % nach 96 Wochen festgestellt, während die Mittelwerte für das Serum-IgM insgesamt gut innerhalb der Referenzbereiche blieben. Bei 14,3 % der Patienten führte die Behandlung mit Ofatumumab zu einem Rückgang des IgM-Werts auf einen Wert unter 0,34 g/L.
Die Mittelwerte für das Immunglobulin G (IgG) sanken nach 48 Wochen Behandlung vorübergehend um 4,3 %. Nach 96 Wochen wurde jedoch ein Anstieg um 2,2 % festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung in klinischen Studien zur RMS gemeldet.
In klinischen Studien wurden MS-Patienten Dosen von bis zu 700 mg verabreicht, dabei trat keine dosislimitierende Toxizität auf. Im Falle einer Überdosierung wird eine Überwachung des Patienten auf Anzeichen oder Symptome von unerwünschten Wirkungen empfohlen sowie, falls erforderlich, die Einleitung einer geeigneten symptomatischen Behandlung.
Eigenschaften/WirkungenATC-Code:
L04AA52
Wirkungsmechanismus
B-Zellen spielen aufgrund der Produktion von pro-inflammatorischen Zytokinen, der Freisetzung von autoreaktiven Antikörpern und der Aktivierung von pathogenen T-Zellen eine wichtige Rolle bei der Pathogenese der MS.
Die genauen Mechanismen, durch welche Ofatumumab bei RMS seine therapeutischen klinischen Wirkungen ausübt, sind nicht geklärt, aber es wird eine Immunmodulation durch eine Bindung an CD20-exprimierenden B-Zellen angenommen. Ofatumumab ist ein vollständig humaner monoklonaler Anti-CD20-Antikörper (IgG1). Es bindet an ein bestimmtes Epitop, das sowohl die kleinen als auch die grossen extrazellulären Schleifen des CD20-Moleküls umfasst, was zu einer langsamen Off-Rate und einer hohen Bindungsaffinität führt. Das CD20-Molekül ist ein transmembranäres Phosphoprotein, das von B-Lymphozyten ab dem Prä-B- bis zum reifen B-Lymphozytenstadium exprimiert wird. Das CD20-Molekül wird auch auf einem kleinen Teil der aktivierten T-Zellen exprimiert.
Durch die Bindung von Ofatumumab an CD20 wird die Lyse von CD20-positiven B-Zellen induziert, und zwar in erster Linie durch die komplementabhängige Zytotoxizität (CDC) und in geringerem Masse durch die antikörperabhängige zellvermittelte Zytotoxizität (antibody-dependent cell-mediated cytotoxicity, ADCC). Weiterhin wurde für Ofatumumab gezeigt, dass es eine Lyse sowohl von Zellen mit hoher als auch von solchen mit niedriger CD20-Expression induziert. Auch die CD20-exprimierenden T-Zellen werden durch Ofatumumab depletiert.
Pharmakodynamik
B-Zell-Depletion
In den Phase-III-Studien zur RMS führte die Verabreichung von Ofatumumab 20 mg alle 4 Wochen nach initialer Gabe von 20 mg an den Tagen 1, 7 und 14 bereits zwei Wochen nach Behandlungsbeginn zu einer raschen und anhaltenden Reduktion der B-Zellen unter die untere Grenze des Normbereichs (lower limit of normal, LLN, definiert als 40 Zellen/µl), Der Gesamtanteil der Patienten mit einer B-Zellzahl ≤10 Zellen/μl betrug in Woche 2 81,9 % und in Woche 4 91,8 %. Diese B-Zell-Depletion unter ≤10 Zellen/μl wurde bei 98 % der Patienten in Woche 12 beobachtet, hielt bei etwa 97 % der Patienten bis Woche 96 sowie bis zu 120 Wochen an.
Ähnliche Ergebnisse wurden in einer Studie zur Bioäquivalenz beobachtet, bei der das gleiche Dosierungsschema wie in den Phase-III-Studien angewendet wurde. Vor Beginn der Erhaltungsphase in Woche 4 wurden bei 94 % der Patienten Werte für Gesamtzahl der B-Zellen von < 10 Zellen/µl erreicht, in Woche 12 war dies bei 98 % der Patienten der Fall.
B-Zell-Repletion
Daten aus klinischen RMS-Studien weisen darauf hin, dass die B-Zellen bei mindestens 50 % der Patienten nach 24 bis 36 Wochen nach Absetzen der Behandlung wieder Werte oberhalb des LLN erreichen. In Modellen und Simulationen der B-Zell-Repletion werden diese Daten bestätigt, und es wird eine mittlere Dauer von 40 Wochen bis zur Normalisierung der B-Zell-Zahlen nach dem Absetzen der Behandlung vorausgesagt.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von Kesimpta wurde in zwei zulassungsrelevanten randomisierten, doppelblinden, aktiv-kontrollierten Phase-III-Studien mit identischem Design (G2301 [ASCLEPIOS I] und G2302 [ASCLEPIOS II]) bei Patienten im Alter von 18 bis 55 Jahren untersucht, bei denen eine schubförmige MS (RMS), ein Wert für den MS-bedingten Behinderungsstatus von 0 bis 5,5 auf der Expanded Disability Status Scale (EDSS) beim Screening sowie mindestens ein dokumentierter Schub im Vorjahr oder zwei Schübe in den beiden Vorjahren oder ein positiver MRT-Befund mit Gadolinium-anreichernden Läsionen im Vorjahr vorlag.
In den beiden Studien wurden 927 bzw. 955 Patienten mit RMS im Verhältnis 1:1 randomisiert, um entweder subkutane Injektionen mit 20 mg Ofatumumab alle 4 Wochen, beginnend in Woche 4, nach initialer Verabreichung von drei wöchentlichen 20-mg-Dosen in den ersten 14 Tagen (an den Tagen 1, 7 und 14 ) oder Teriflunomid 14 mg Kapseln oral einmal täglich zu erhalten. Zur Gewährleistung der Verblindung erhielten die Patienten ausserdem ein passendes Placebo, das der Behandlung im jeweils anderen Behandlungsarm entsprach (Double-Dummy-Design).
Die Behandlungsdauer war bei den einzelnen Patienten unterschiedlich und hing davon ab, wann die Kriterien für das Studienende erfüllt waren. Über beide Studien hinweg betrug die mittlere Behandlungsdauer 85 Wochen, wobei 33,0 % der Patienten in der Ofatumumab-Gruppe, verglichen mit 23,2 % der Patienten in der Teriflunomid-Gruppe, länger als 96 Wochen behandelt wurden.
Die Demografie und die Ausgangsmerkmale waren in den Behandlungsarmen und bei beiden Studien ausgewogen (siehe Tabelle 1). Das mittlere Alter betrug 38 Jahre, die mittlere Krankheitsdauer seit dem Auftreten des ersten Symptoms lag bei 8,2 Jahren und der mittlere EDSS-Wert bei 2,9; 40 % der Patienten waren zuvor nicht mit einer krankheitsmodifizierenden Therapie (disease-modifying therapy, DMT), behandelt worden und bei 40 % fanden sich bei Gadolinium- (Gd)-anreichernde T1-Läsionen im Baseline-MRT.
Der primäre Wirksamkeitsendpunkt beider Studien war die jährliche Rate der bestätigten Schübe (annualised rate of confirmed relapses, ARR). Zu den wichtigsten sekundären Wirksamkeitsendpunkten gehörte die Zeit bis zur Verschlimmerung der Behinderung auf der EDSS-Skala (bestätigt nach 3 Monaten und nach 6 Monaten), die definiert war als eine Zunahme des EDSS-Werts von ≥1,5 (bei Baseline-EDSS 0); ≥1 (bei Baseline-EDSS 1 bis 5) oder ≥0,5 (bei Baseline-EDSS≥5,5). Weitere wichtige sekundäre Endpunkte waren die Zeit bis zur Besserung der Behinderung auf der EDSS-Skala (bestätigt nach 6 Monaten), die Anzahl der Gd-anreichernden T1-Läsionen pro MRT-Aufnahme, die annualisierte Rate neuer oder vergrösserter T2-Läsionen, und die Rate der Abnahme des Hirnvolumens (brain volume loss, BVL). Die wichtigsten auf die Behinderung bezogenen sekundären Endpunkte wurden in einer Meta-Analyse der kombinierten Daten aus den Studien G3201 und G2302 ausgewertet, wie es in den Prüfplänen festgelegt war.
Tabelle 1: Demographische Daten und Baseline-Charakteristika
|
Charakteristika
|
Studie G2301
(ASCLEPIOS I)
|
Studie G2302
(ASCLEPIOS II)
| |
|
Ofatumumab
(N = 465)
|
Teriflunomid
(N = 462)
|
Ofatumumab
(N = 481)
|
Teriflunomid
(N = 474)
| |
Mittleres Alter (Jahre)
|
38,9
|
37,8
|
38,0
|
38,2
| |
Altersgruppe (Jahre)
|
19-55
|
18-55
|
18-55
|
18-55
| |
Weiblich (%)
|
68,4
|
68,6
|
66,3
|
67,3
| |
Mittlere / mediane Dauer der MS seit den ersten Symptomen (Jahre)
|
8,36 / 6,41
|
8,18 / 6,69
|
8,20 / 5,70
|
8,19 / 6,30
| |
Mittlere / mediane Dauer der MS seit der Diagnose (Jahre)
|
5,77 / 3,94
|
5,64 / 3,49
|
5,59 / 3,15
|
5,48 / 3,10
| |
Zuvor mit einer krankheitsmodifizierenden Therapie behandelte Patienten (%)
|
58,9
|
60,6
|
59,5
|
61,8
| |
Anzahl der Schübe in den letzten 12 Monaten
|
1,2
|
1,3
|
1,3
|
1,3
| |
Mittlerer/medianer EDSS-Wert
|
2,97 / 3,00
|
2,94 / 3,00
|
2,90 / 3,00
|
2,86 / 2,50
| |
Mittleres Gesamtvolumen der T2-Läsionen (cm3)
|
13,2
|
13,1
|
14,3
|
12,0
| |
Patienten, die frei von Gd-positiven T1-Läsionen sind (%)
|
62,6
|
63,4
|
56,1
|
61,4
| |
Anzahl der Gd-positiven T1-Läsionen (Mittelwert)
|
1,7
|
1,2
|
1,6
|
1,5
|
Die Wirksamkeitsergebnisse für beide Studien sind in Tabelle 2 und Abbildung 1 zusammengefasst.
In beiden Phase-III-Studien (G2301 und G2302) zeigte Kesimpta im Vergleich mit Teriflunomid eine signifikante Verringerung der jährlichen Schubrate von 50,5 % bzw. 58,5 % (beide p < 0,001).
Die vorab festgelegte Meta-Analyse der kombinierten Daten ergab, dass durch Kesimpta das Risiko einer über 3 Monate anhaltenden bestätigten Verschlimmerung der Behinderung (confirmed disability worsening, CDW) (Risikoreduktion = 34,4 %, p = 0,002) und das einer über 6 Monate anhaltenden CDW (Risikoreduktion = 32,5 %, p = 0,012) im Vergleich mit Teriflunomid signifikant reduziert wurde (siehe Abbildung 1).
Durch Kesimpta wurde ausserdem die Anzahl der Gd-anreichernden T1-Läsionen und die Rate der neuen oder sich vergrössernden T2-Läsionen signifikant um 95,9 % bzw. 83,6 % reduziert (in beiden Studien zusammen).
Die Wirksamkeitsergebnisse waren in den beiden Phase-III-Studien (G2301 und G2302) sowie in den exploratorischen Analysen von Untergruppen, die auf der Grundlage von Geschlecht, Alter, Körpergewicht, früherer MS-Therapie, des Ausgangswerts für die Behinderung und Krankheitsaktivität definiert wurden, vergleichbar.
Langzeitwirksamkeits- und Langzeitsicherheitsdaten einer laufenden open label Extensionsstudie der beiden Phase-III-Studien lagen zum Zeitpunkt der Zulassung noch nicht vor.
Tabelle 2: Überblick über die Ergebnisse der Phase-III-Studien zur RMS
|
Endpunkte
|
Studie G2301
(ASCLEPIOS I)
|
Studie G2302
(ASCLEPIOS II)
| |
Ofatumumab 20 mg
(N = 465)
|
Teriflunomid 14 mg
(N = 462)
|
Ofatumumab 20 mg
(N = 481)
|
Teriflunomid 14 mg
(N = 474)
| |
Endpunkte auf Basis von separaten Studien
| |
Jährliche Schubrate (ARR) (primärer Endpunkt)1
|
0,11
|
0,22
|
0,10
|
0,25
| |
Reduzierung der Rate
|
50,5 % (p < 0,001)
|
58,5 % (p < 0,001)
| |
Mittlere Anzahl der Gd-anreichernden T1-Läsionen pro MRT-Aufnahme
|
0,0115
|
0,4523
|
0,0317
|
0,5141
| |
Relative Reduzierung
|
97,5 % (p < 0,001)
|
93,8 % (p<0,001)
| |
Anzahl der neuen oder sich vergrössernden T2-Läsionen pro Jahr
|
0,72
|
4,00
|
0,64
|
4,15
| |
Relative Reduzierung
|
81,9 % (p < 0,001)
|
84,5 % (p < 0,001)
| |
Endpunkte auf der Grundlage von vorab festgelegten Meta-Analysen
| |
Anteil der Patienten mit bestätigter Progression der Behinderung nach 3 Monaten2
|
10,9 % Ofatumumab vs. 15,0 % Teriflunomid
| |
Risikoreduktion
|
34,4 % (p = 0,002)
| |
Anteil der Patienten mit bestätigter Progression der Behinderung nach 6 Monaten2
|
8,1 % Ofatumumab vs. 12,0 % Teriflunomid
| |
Risikoreduktion
|
32,5 % (p = 0,012)
| |
1
Bestätigte Schübe (begleitet von einer klinisch relevanten Veränderung beim EDSS-Wert).
2 Kaplan-Meier in Monat 24. Progression der Behinderung wurde definiert als eine Zunahme im EDSS-Wert um mindestens 1,5, 1, oder 0,5 Punkte bei Patienten mit einem Baseline-EDSS-Wert von 0, 1 bis 5, bzw. 5,5 oder mehr.
|
Abbildung 1: Zeit bis zur ersten CDW im Laufe von 3 Monaten in Abhängigkeit von der Behandlung (G2301 und G2302 kombiniert, vollständiger Analysensatz)
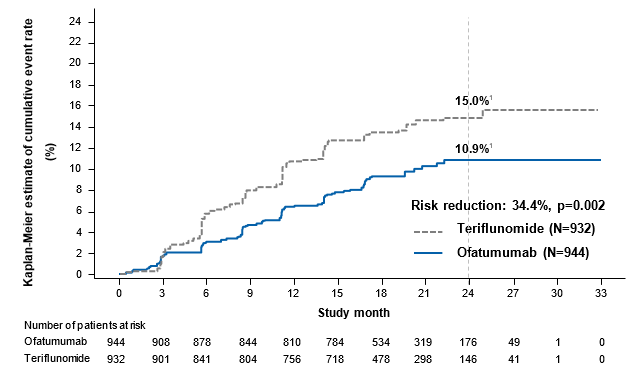
1 The numbers shown on the curves represent Kaplan-Meier estimates of the risk of the event at 24 months (marked by the vertical dashed line).
Immunogenität
Wie bei allen therapeutischen Proteinen besteht das Potenzial für eine Immunogenität. Der Nachweis der Antikörperbildung ist in hohem Masse von der Sensitivität und Spezifität des Assays abhängig. Darüber hinaus kann die beobachtete Inzidenz eines positiven Nachweises von Antikörpern (einschliesslich neutralisierender Antikörper) in einem Assay durch mehrere Faktoren beeinflusst werden. Dazu zählen die Methodik des Assays, die Handhabung der Probe, der Zeitpunkt der Probenentnahme, die Begleitmedikation und die zugrundeliegende Erkrankung. Aus diesen Gründen kann der Vergleich der Inzidenz von Antikörpern in den unten beschriebenen Studien mit der Inzidenz von Antikörpern in anderen Studien oder mit der Inzidenz bei anderen Arzneimitteln, die Ofatumumab enthalten, irreführend sein. Behandlungsinduzierte Antikörper gegen das Arzneimittel («anti-drug antibodies», ADA) in den Phase-III-Studien wurden bei 2 von 923 (0,2 %) der mit KESIMPTA behandelten Patienten nachgewiesen; es wurden keine Patienten mit therapieverstärkenden oder neutralisierenden ADA identifiziert. Bei keinem Patienten wurde eine Auswirkung der positiven ADA-Titer auf die Pharmakokinetik, das Sicherheitsprofil oder die B-Zell-Kinetik festgestellt. Diese Daten reichen jedoch nicht aus, um die Auswirkungen von ADA auf die Sicherheit und Wirksamkeit von KESIMPTA beurteilen zu können.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Es wurden keine Studien zur Untersuchung der Sicherheit und Wirksamkeit bei Kindern und Jugendlichen (< 18 Jahren) durchgeführt.
PharmakokinetikResorption
Basierend auf der 12-wöchigen Bioäquivalenzstudie bei Patienten mit RMS (COMB157G2102) führte eine monatliche subkutane Dosis von 20 mg führt zu einem mittleren AUCtau von 483 µg x h/ml und einer mittleren Cmax von 1,43 µg/ml. Der Tmax wurde in der Regel in der ersten Woche nach der Injektion erreicht.
Es wird angenommen, dass Ofatumumab nach subkutaner Anwendung, ähnlich wie andere therapeutische monoklonale Antikörper, überwiegend über das Lymphsystem resorbiert wird.
Verteilung
Insgesamt waren die Werte für das Verteilungsvolumen im Steady-State (Vss) von Ofatumumab niedrig (3 bis 8 Liter) und konsistent mit den Werten für andere monoklonale Antikörper. Nach zwei intravenösen Infusionen von Ofatumumab (100, 300 oder 700 mg in der ersten oder zweiten Behandlungsphase) bei Patienten mit schubförmig wiederkehrender Multipler Sklerose (RRMS) lag das geometrische Mittel des Vss-Werts nach der zweiten Infusion von Ofatumumab zwischen 2,15 und 2,74 Litern. Auf der Grundlage der pharmakokinetischen Modellierung von Daten aus den Studien mit subkutaner Verabreichung und wiederholter Gabe von 20-mg-Dosen wurde ein zentrales Volumen (Vc) von 2,8 Litern geschätzt.
Metabolismus
Ofatumumab ist ein Protein, bei dem der erwartete Stoffwechselweg im Abbau zu kleinen Peptiden und Aminosäuren durch ubiquitär vorkommende proteolytische Enzyme besteht.
Elimination
Ofatumumab wird auf zweierlei Wegen eliminiert: über einen zielvermittelten Weg, der im Zusammenhang mit der Bindung an B-Zellen steht, und über einen zielunabhängigen Weg wie bei anderen IgG-Molekülen, der durch unspezifische Endozytose, gefolgt von intrazellulärem Katabolismus, vermittelt wird. Bei Baseline vorhandene B-Zellen führen dazu, dass die Komponente der zielvermittelten Clearance von Ofatumumab zu Beginn der Therapie grösser ist. Die Gabe von Ofatumumab hat eine starke B-Zell-Depletion zur Folge, was zu einer reduzierten Gesamt-Clearance führt.
Die Halbwertszeit im Steady-State wurde auf etwa 16 Tage nach wiederholter subkutaner Gabe von 20 mg Ofatumumab geschätzt. Basierend auf den Ergebnissen des Berichts des PopPK-Modells betrug die geschätzte Clearance (CL) von Ofatumumab nach initialer B-Zell-Depletion in einer Patientenpopulation mit schubförmig verlaufender Multipler Sklerose 0,156 l/Tag.
Linearität/Nicht Linearität
Ofatumumab wies eine nicht-lineare Pharmakokinetik auf, die mit der über die Zeit abnehmenden Clearance zusammenhing.
Kinetik spezieller Patientengruppen
Die folgenden Populationsmerkmale haben keinen klinisch bedeutsamen Einfluss auf die Pharmakokinetik von Ofatumumab: Körpergewicht, Geschlecht, Alter, ethnische Herkunft/Hautfarbe oder die Anzahl der B-Zellen bei Baseline.
Leberfunktionsstörungen
Da der hepatische Metabolismus von monoklonalen Antikörpern wie Ofatumumab zu vernachlässigen ist, ist nicht zu erwarten, dass die Pharmakokinetik dieses Arzneimittels durch eine Leberfunktionsstörung beeinflusst wird. Es ist daher nicht davon auszugehen, dass bei Patienten mit einer Leberfunktionsstörung eine Dosisänderung erforderlich ist.
Nierenfunktionsstörungen
Ofatumumab wird nicht über den Urin ausgeschieden; daher ist nicht zu erwarten, dass bei Patienten mit einer Nierenfunktionsstörung eine Dosisänderung nötig ist.
Ältere Patienten
Bisher wurden keine Studien bei älteren MS-Patienten durchgeführt. Ofatumumab wurde bei RMS-Patienten im Alter von 18 bis 55 Jahren untersucht. Aus den Ergebnissen der Populationspharmakokinetik geht hervor, dass eine Dosisanpassung bei älteren Patienten nicht erforderlich ist.
Kinder und Jugendliche
Die Verträglichkeit und Wirksamkeit bei Kindern und Jugendlichen unter 18 Jahren wurde bisher nicht nachgewiesen.
Geschlecht
In einer populationspharmakokinetischen Analyse über mehrere Studien hinweg hatte das Geschlecht einen mässigen (12 %) Einfluss auf das zentrale Verteilungsvolumen von Ofatumumab, wobei bei weiblichen Patienten höhere Werte für die Cmax und die AUC festgestellt wurden (in dieser Analyse waren 48 % der Patienten weiblich und 52 % männlich); diese Effekte wurden als klinisch nicht relevant eingestuft, eine Dosisanpassung wird nicht empfohlen.
Präklinische DatenBasierend auf den konventionellen Studien zur chronischen Toxizität einschliesslich der sicherheitspharmakologischen Endpunkte lassen die präklinischen Daten keine besonderen Risiken für den Menschen erkennen.
Mutagenität und Karzinogenität
Zu Ofatumumab wurden weder Mutagenitäts- noch Karzinogenitätsstudien durchgeführt. Da es sich um einen Antikörper handelt, wird von Ofatumumab keine direkte Interaktion mit der DNA erwartet.
Reproduktionstoxizität
Die Studien zur embryo-fötalen Entwicklung und zur erweiterten prä- und postnatalen Entwicklung (ePPND) an Affen haben gezeigt, dass die intravenöse Verabreichung von Ofatumumab während der Trächtigkeit keine maternale Toxizität und keine Teratogenität verursachte sowie keine Auswirkungen auf die embryo-fötale und die prä-/postnatale Entwicklung hatte. Der NOAEL-Wert für diese Parameter führt zu AUC-basierten Sicherheitsmargen von mindestens dem 160-Fachen im Vergleich zur Exposition des Menschen bei der therapeutischen Monatsdosis von 20 mg. In diesen Studien wurde Ofatumumab im Blut der Föten und Jungtiere nachgewiesen, wodurch die Plazentagängigkeit und die postnatale Exposition der Föten bestätigt wurden. Die Ofatumumab-Behandlung während der Trächtigkeit führte zu der erwarteten Depletion der CD20-positiven B-Zellen bei Muttertieren, Föten und den gesäugten Jungtieren. Bei hohen Dosen (Exposition der Muttertiere mind. das 160-fache im Vergleich zur klinischen Exposition) wurde eine Verminderung des Milzgewichts (ohne histologisches Korrelat) bei Föten und eine Reduzierung der humoralen Immunantwort auf Keyhole-Limpet-Hämocyanin (KLH) bei den gesäugten Jungtieren festgestellt. Alle diese Veränderungen waren während der 6-monatigen behandlungsfreien Periode nach der Geburt reversibel. Bei Jungtieren der Hochdosisgruppe wurde eine frühe postnatale Sterblichkeit festgestellt. Diese ist wahrscheinlich auf opportunistische Infektionen aufgrund der Immunmodulation zurückzuführen. Der NOAEL-Wert in Bezug auf die pharmakologische Aktivität von Ofatumumab für die Jungtiere in der ePPND-Studie führte zu AUC-basierten Sicherheitsmargen von mindestens dem 22-Fachen der Exposition beim Menschen, wenn man die Exposition des Muttertiers beim NOAEL mit der menschlichen Exposition bei der therapeutischen Monatsdosis von 20 mg vergleicht.
In einer Fertilitätsstudie in Affen wurden die Endpunkte der männlichen und weiblichen Fertilität nicht beeinflusst. Die auf den NOAEL-Wert bezogene Exposition (AUC) war mindestens 260-mal höher als die menschliche Exposition bei der therapeutischen Monatsdosis von 20 mg.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8°C) lagern. Nicht einfrieren.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Falls erforderlich, kann Kesimpta ungekühlt einmalig für einen Zeitraum von bis zu 7 Tagen bei Raumtemperatur (nicht über 30 °C) gelagert werden. Wird Kesimpta in diesem Zeitraum nicht verwendet, kann es anschliessend für maximal 7 Tage in den Kühlschrank zurückgelegt werden.
Hinweise für die Handhabung
Anleitung zur Handhabung der Fertigspritze
Die Fertigspritze sollte etwa 15 bis 30 Minuten vor der Injektion aus dem Kühlschrank genommen werden, damit sie Raumtemperatur erreichen kann. Die Fertigspritze sollte bis zur Verwendung im Originalkarton aufbewahrt werden, und die Nadelkappe sollte erst kurz vor der Injektion entfernt werden. Vor der Anwendung sollte die Lösung durch das Sichtfenster visuell überprüft werden. Die Lösung sollte klar bis leicht trüb sein. Wenn die Flüssigkeit sichtbare Partikel enthält oder trüb ist, darf die Fertigspritze nicht verwendet werden.
Eine ausführliche Anleitung zur Verabreichung der Injektion steht in der Packungsbeilage zur Verfügung.
Anleitung zur Handhabung des SensoReady-Fertigpens
Der Fertigpen sollte etwa 15 bis 30 Minuten vor der Injektion aus dem Kühlschrank genommen werden, damit er Raumtemperatur erreichen kann. Der Fertigpen sollte bis zur Verwendung im Originalkarton aufbewahrt werden, und die Nadelkappe sollte erst kurz vor der Injektion entfernt werden. Vor der Anwendung sollte die Lösung durch das Sichtfenster visuell überprüft werden. Die Lösung sollte klar bis leicht trüb sein. Wenn die Flüssigkeit sichtbare Partikel enthält oder trüb ist, darf der Fertigpen nicht verwendet werden.
Eine ausführliche Anleitung zur Verabreichung der Injektion steht in der Packungsbeilage zur Verfügung.
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Anforderungen zu entsorgen.
Zulassungsnummer67757, 67758 (Swissmedic)
Packungen1 Fertigspritze zu 0,4 ml. [B]
1 Fertigpen zu 0,4 ml. [B]
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch; Domizil 6343 Rotkreuz.
Stand der InformationAugust 2023
|