ZusammensetzungWirkstoffe
Morgendosis:
Elexacaftor, Tezacaftor, Ivacaftor
Abenddosis:
Ivacaftor
Hilfsstoffe
Filmtabletten
Morgendosis:
Tablettenkern:
Hypromellose, Hypromelloseacetatsuccinat, Natriumlaurylsulfat, Croscarmellose-Natrium, mikrokristalline Cellulose, Magnesiumstearat
Tabletten-Filmüberzug:
Hypromellose, Hydroxypropylcellulose, Titandioxid, Talkum, gelbes Eisenoxid, rotes Eisenoxid
Jede 50 mg/25 mg/37,5 mg-Tablette enthält 1,34 mg Natrium.
Jede 100 mg/50 mg/75 mg-Tablette enthält 2,68 mg Natrium.
Abenddosis:
Tablettenkern:
Hochdisperses Siliciumdioxid, Croscarmellose-Natrium, Hypromelloseacetatsuccinat, Lactose-Monohydrat, Magnesiumstearat, mikrokristalline Cellulose, Natriumlaurylsulfat
Tabletten-Filmüberzug:
Carnaubawachs, Indigotin, Macrogol 3350, Polyvinylalkohol, Talkum, Titandioxid
Druckfarbe:
Ammoniumhydroxid, schwarzes Eisenoxid, Propylenglycol, Schellack
Jede 75 mg-Tablette enthält 0,90 mg Natrium und 83,6 mg Lactosemonohydrat.
Jede 150 mg-Tablette enthält 1,82 mg Natrium und 167,2 mg Lactosemonohydrat.
Granulat im Beutel
Morgendosis:
Hochdisperses Siliciumdioxid, Croscarmellose-Natrium, Hypromellose, Hypromelloseacetatsuccinat, Lactose-Monohydrat, Magnesiumstearat, Mannitol, Natriumlaurylsulfat, Sucralose
Jeder 80 mg/40 mg/60 mg-Beutel enthält max. 2,75 mg Natrium und 188,6 mg Lactose-Monohydrat.
Jeder 100 mg/50 mg/75 mg-Beutel enthält max. 3,44 mg Natrium und 235,7 mg Lactose-Monohydrat.
Abenddosis:
Hochdisperses Siliciumdioxid, Croscarmellose-Natrium, Hypromelloseacetatsuccinat, Lactose-Monohydrat, Magnesiumstearat, Mannitol, Natriumlaurylsulfat, Sucralose
Jeder 59,5 mg-Beutel enthält max. 1,18 mg Natrium und 87,3 mg Lactose-Monohydrat.
Jeder 75 mg-Beutel enthält max. 1,49 mg Natrium und 109,8 mg Lactose-Monohydrat.
Darreichungsform und Wirkstoffmenge pro EinheitFilmtabletten
Elexacaftor 50 mg/Tezacaftor 25 mg/Ivacaftor 37,5 mg-Tablette und Ivacaftor 75 mg-Tablette
Morgendosis:
Jede 50 mg/25 mg/37,5 mg-Filmtablette enthält 50 mg Elexacaftor, 25 mg Tezacaftor und 37,5 mg Ivacaftor als eine Fixdosis-Kombinationstablette.
Hellorange, kapselförmige Tablette mit der Prägung "T50" auf der einen Seite und auf der anderen Seite ohne Prägung (6,4 mm × 12,2 mm).
Abenddosis:
Jede 75 mg-Filmtablette enthält 75 mg Ivacaftor.
Hellblaue, kapselförmige Tablette, auf der einen Seite mit dem Aufdruck "V 75" in schwarzer Druckfarbe und auf der anderen Seite unbedruckt (12,7 mm × 6,8 mm).
Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg-Tablette und Ivacaftor 150 mg-Tablette
Morgendosis:
Jede 100 mg/50 mg/75 mg-Filmtablette enthält 100 mg Elexacaftor, 50 mg Tezacaftor und 75 mg Ivacaftor als Fixdosis-Kombinationstablette.
Orangefarbene kapselförmige Tablette mit der Prägung "T100" auf der einen Seite und auf der anderen Seite ohne Prägung (7,85 mm × 15,47 mm).
Abenddosis:
Jede 150 mg-Filmtablette enthält 150 mg Ivacaftor.
Hellblaue, kapselförmige Tablette, auf der einen Seite mit dem Aufdruck "V 150" in schwarzer Druckfarbe und auf der anderen Seite unbedruckt (16,5 mm × 8,4 mm).
Granulat im Beutel
Alle Körner des Granulats sind weiss bis gebrochen weiss, gesüsst und nicht aromatisiert und haben einen Korndurchmesser von ca. 2 mm.
Elexacaftor 80 mg/Tezacaftor 40 mg/Ivacaftor 60 mg und Ivacaftor 59,5 mg Granulat im Beutel
Morgendosis:
Jeder Beutel enthält 80 mg Elexacaftor, 40 mg Tezacaftor und 60 mg Ivacaftor.
Abenddosis:
Jeder Beutel enthält 59,5 mg Ivacaftor.
Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg und Ivacaftor 75 mg Granulat im Beutel
Morgendosis:
Jeder Beutel enthält 100 mg Elexacaftor, 50 mg Tezacaftor und 75 mg Ivacaftor.
Abenddosis:
Jeder Beutel enthält 75 mg Ivacaftor.
Indikationen/AnwendungsmöglichkeitenTrikafta ist angezeigt zur Behandlung von Patienten mit zystischer Fibrose (CF) ab 2 Jahren, die mindestens eine F508del-Mutation im CFTR-Gen (Cystic Fibrosis Transmembrane Conductance Regulator) aufweisen oder eine Mutation im CFTR-Gen, bei der klinische Daten und/oder In-vitro-Daten ein Ansprechen zeigen (siehe "Eigenschaften/Wirkungen" , Tabelle 7).
Dosierung/AnwendungTrikafta sollte nur von Ärzten mit Erfahrung in der Behandlung der CF verordnet werden. Wenn der Genotyp des Patienten nicht bekannt ist, muss das Vorliegen von mindestens einer F508del-Mutation oder einer Mutation, bei der klinische Daten und/oder In-vitro-Daten ein Ansprechen zeigen, mithilfe eines Genotypisierungsassays bestätigt werden.
Übliche Dosierung
Erwachsene, Jugendliche und Kinder ab 2 Jahren
Tabelle 1: Dosisempfehlung
für Patienten ab 2 Jahren
Alter Körpergewicht Morgendosis Abenddosis
2 bis <6 Jahre 10-<14 kg Ein Beutel mit Elexacaftor Ein Beutel mit
80 mg/Tezacaftor 40 Ivacaftor 59,5 mg
mg/Ivacaftor 60 mg Granulat Granulat
2 bis <6 Jahre ≥14 kg Ein Beutel mit Elexacaftor Ein Beutel mit
100 mg/Tezacaftor 50 Ivacaftor 75 mg
mg/Ivacaftor 75 mg Granulat Granulat
6 bis <12 Jahre <30 kg Zwei Tabletten mit jeweils Eine Tablette mit
Elexacaftor 50 mg/Tezacaftor Ivacaftor 75 mg
25 mg/Ivacaftor 37,5 mg
6 bis <12 Jahre ≥30 kg Zwei Tabletten mit jeweils Eine Tablette mit
Elexacaftor 100 mg/Tezacafto Ivacaftor 150 mg
r 50 mg/Ivacaftor 75 mg
ab 12 Jahren - Zwei Tabletten mit jeweils Eine Tablette mit
Elexacaftor 100 mg/Tezacafto Ivacaftor 150 mg
r 50 mg/Ivacaftor 75 mg
Die Morgen- und Abenddosis sollen zusammen mit einer fetthaltigen Mahlzeit im Abstand von ungefähr 12 Stunden eingenommen werden (siehe "Art der Anwendung" ).
Verspätete Dosisgabe
Wenn seit der versäumten Morgen- oder Abenddosis höchstens 6 Stunden vergangen sind, soll der Patient die versäumte Dosis baldmöglichst einnehmen und die Einnahme nach dem ursprünglichen Behandlungsplan fortsetzen.
Wenn mehr als 6 Stunden vergangen sind seit:
der versäumten Morgendosis, soll der Patient die versäumte Dosis baldmöglichst einnehmen und die Abenddosis nicht einnehmen. Die nächste geplante Morgendosis soll zur üblichen Zeit eingenommen werden.
der versäumten Abenddosis, soll der Patient die versäumte Dosis nicht einnehmen. Die nächste geplante Morgendosis soll zur üblichen Zeit eingenommen werden.
Die Morgen- und die Abenddosis dürfen nicht zur gleichen Zeit eingenommen werden.
Art der Anwendung
Zum Einnehmen.
Trikafta sollte zusammen mit einer fetthaltigen Mahlzeit eingenommen werden. Beispiele für fetthaltige Mahlzeiten oder Zwischenmahlzeiten sind mit Butter oder Öl zubereitete Mahlzeiten oder Mahlzeiten, die Eier, Erdnussbutter, Käse, Nüsse, Vollmilch oder Fleisch enthalten (siehe "Pharmakokinetik" ).
Auf Speisen oder Getränke, die Grapefruit enthalten, ist während der Behandlung mit Trikafta zu verzichten (siehe "Interaktionen" ).
Filmtabletten
Die Patienten sind anzuweisen, die Tabletten ganz zu schlucken. Die Patienten sollen die Tabletten vor der Einnahme nicht zerkauen, zerbrechen oder auflösen.
Granulat im Beutel
Jeder Beutel ist nur zur einmaligen Anwendung bestimmt.
Der gesamte Inhalt des Beutels sollte mit 5 ml altersgerechter weicher Nahrung oder Flüssigkeit gemischt und die Mischung sofort eingenommen werden. Die Nahrung oder Flüssigkeit sollte Raumtemperatur oder weniger haben. Nach dem Mischen ist das Arzneimittel nachweislich eine Stunde lang stabil und sollte daher in diesem Zeitraum eingenommen werden. Beispiele für weiche Nahrung oder Flüssigkeiten sind püriertes Obst und Gemüse, Joghurt, Apfelmus, Wasser, Milch oder Saft. Unmittelbar vor oder nach der Einnahme sollte eine fetthaltige Mahlzeit oder Zwischenmahlzeit eingenommen werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Die Behandlung von Patienten mit mässig eingeschränkter Leberfunktion (Child-Pugh-Klasse B) wird nicht empfohlen.
Bei Patienten mit mässig eingeschränkter Leberfunktion sollte die Behandlung nur dann in Betracht gezogen werden, wenn ein eindeutiger medizinischer Bedarf vorliegt und der Nutzen der Behandlung die Risiken übersteigt.
Es wurden keine Studien an Patienten mit schwer eingeschränkter Leberfunktion (Child-Pugh-Klasse C) durchgeführt. Patienten mit schwer eingeschränkter Leberfunktion sollten nicht mit Trikafta behandelt werden.
Bei Patienten mit leicht eingeschränkter Leberfunktion (Child-Pugh-Klasse A) wird keine Dosisanpassung empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" , "Unerwünschte Wirkungen" und "Pharmakokinetik" ).
Tabelle 2: Anwendung
sempfehlungen für
Patienten mit
Leberfunktionsstörun
gen
Alter Leicht (Child-Pugh-K Mässig (Child-Pugh-Klasse B) Stark (Child-Pugh
lasse A) Klasse C)
2 bis <6 Jahre Keine Dosisanpassung Anwendung nicht empfohlen. Bei Sollte nicht angewen
Patienten mit mässig eingeschränkt det werden
er Leberfunktion sollte die
Anwendung nur dann in Erwägung
gezogen werden, wenn ein klarer
medizinischer Bedarf vorliegt und
erwartet wird, dass der Nutzen
der Behandlung die Risiken
übersteigt. In solchen Fällen ist
Trikafta mit Vorsicht in einer
niedrigeren Dosis anzuwenden:
-Tag 1-3: ein Beutel Elexacaftor/T
ezacaftor/Ivacaftor Granulat
jeden Tag -Tag 4: Keine Dosis
-Tag 5-6: ein Beutel Elexacaftor/T
ezacaftor/Ivacaftor Granulat
jeden Tag -Tag 7: Keine Dosis
Dieses Dosierungsschema jede
Woche wiederholen. Die
Abenddosis Ivacaftor Granulat
soll nicht eingenommen werden.
ab 6 Jahren Keine Dosisanpassung Anwendung nicht empfohlen. Bei Sollte nicht angewen
Patienten mit mässig eingeschränkt det werden
er Leberfunktion sollte die
Anwendung nur dann in Erwägung
gezogen werden, wenn ein klarer
medizinischer Bedarf vorliegt und
erwartet wird, dass der Nutzen
der Behandlung die Risiken
übersteigt. In solchen Fällen
ist Trikafta mit Vorsicht in
einer niedrigeren Dosis
anzuwenden: -Tag 1: zwei
Elexacaftor/ Tezacaftor/Ivacaftor
Tabletten am Morgen -Tag 2: eine
Elexacaftor/Tezacaftor/ Ivacaftor
Tablette am Morgen Danach
abwechselnd die Dosierung von Tag
1 und Tag 2 wiederholen. Die
Abenddosis Ivacaftor Tabletten
soll nicht eingenommen werden.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leicht und mässig eingeschränkter Nierenfunktion wird keine Dosisanpassung empfohlen. Bei Patienten mit stark eingeschränkter Nierenfunktion oder terminaler Niereninsuffizienz wird zur Vorsicht geraten (siehe "Pharmakokinetik" ).
Gleichzeitige Anwendung von CYP3A-Inhibitoren
Bei gleichzeitiger Gabe mit mässigen CYP3A-Inhibitoren (z.B. Fluconazol, Erythromycin) oder starken CYP3A-Inhibitoren (z.B. Ketoconazol, Itraconazol, Posaconazol, Voriconazol, Telithromycin und Clarithromycin) muss die Dosis entsprechend den Angaben in Tabelle 3 reduziert werden (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Interaktionen" ).
Bei gleichzeitiger Anwendung von Ciprofloxacin wird keine Dosisanpassung empfohlen, da eine klinisch relevante Wirkung auf die Bioverfügbarkeit von Trikafta nicht zu erwarten ist (siehe "Interaktionen" ).
Tabelle 3: Dosierungssch
ema für die gleichzeitig
e Anwendung von
Trikafta mit mässigen
und starken CYP3A-Inhibi
toren
Alter Mässige CYP3A-Inhibitoren Starke CYP3A-Inhibitoren
2 bis <6 Jahre Jeden Tag abwechselnd: -Ein Beutel Ein Beutel Elexacaftor/Tezacafto
Elexacaftor/Tezacaftor/Ivacaftor r/Ivacaftor Granulat zweimal
Granulat am ersten Tag -Ein Beutel wöchentlich, im Abstand von
Ivacaftor Granulat am nächsten Tag etwa 3 bis 4 Tagen. Kein
Kein Abenddosis-Beutel Ivacaftor Abenddosis-Beutel Ivacaftor
Granulat Granulat
ab 6 Jahren Jeden Tag abwechselnd: -Zwei Zwei Elexacaftor/Tezacaftor/Ivac
Elexacaftor/Tezacaftor/Ivacaftor aftor Tabletten zweimal
Tabletten am ersten Tag -Eine wöchentlich, im Abstand von
Ivacaftor Tablette am nächsten Tag etwa 3 bis 4 Tagen. Keine
Keine Abenddosis Ivacaftor Tabletten. Abenddosis Ivacaftor Tabletten.
Kinder
Die Sicherheit und Wirksamkeit von Trikafta bei Kindern unter 2 Jahren sind bisher noch nicht erwiesen (siehe "Unerwünschte Wirkungen" und "Eigenschaften/Wirkungen" ).
Ältere Patienten
Die klinischen Studien mit Trikafta schlossen keine ausreichende Zahl von Patienten ab 65 Jahren ein, um feststellen zu können, ob sie anders auf die Behandlung ansprechen als jüngere Patienten.
KontraindikationenÜberempfindlichkeit gegenüber den Wirkstoffen oder einem der Hilfsstoffe (siehe "Zusammensetzung" ).
Warnhinweise und VorsichtsmassnahmenLeberschädigung
Bei Patienten mit und ohne vorbestehende fortgeschrittene Lebererkrankung wurden innerhalb der ersten 6 Monate der Behandlung Fälle von Leberversagen berichtet, die zu einer Lebertransplantation führten. Bei Patienten mit vorbestehender, fortgeschrittener Lebererkrankung (wie z.B. Leberzirrhose, portale Hypertonie) ist Trikafta mit Vorsicht und unter engmaschiger Überwachung und nur dann anzuwenden, wenn erwartet wird, dass der Nutzen die Risiken übersteigt (siehe "Dosierung/Anwendung" , "Unerwünschte Wirkungen" und "Pharmakokinetik" ).
Erhöhung der Leberenzyme
Erhöhte Transaminasenwerte sind bei CF-Patienten verbreitet und wurden auch bei Patienten mit oder ohne vorbestehender Lebererkrankung festgestellt, die mit Trikafta behandelt wurden. In manchen Fällen waren diese teilweise schwerwiegenden Anstiege mit einem gleichzeitigen Anstieg des Gesamtbilirubins assoziiert. In den Phase-III-Studien traten Transaminasenanstiege in der Trikafta-Gruppe häufiger auf als in der Placebo-Gruppe. Daher werden bei allen Patienten Kontrollen der Transaminasenwerte (ALT und AST) und des Gesamtbilirubins vor Beginn der Trikafta-Behandlung, monatlich während der ersten 6 Monate der Behandlung, alle 3 Monate während der darauffolgenden 6 Monate und danach jährlich empfohlen. Bei Patienten mit anamnestisch bekannten Lebererkrankungen oder Transaminasenanstiegen sind häufigere Kontrollen in Erwägung zu ziehen.
Wenn bei einem Patienten klinische Anzeichen oder Symptome auftreten, die auf eine Leberschädigung hindeuten (z.B. Gelbsucht und/oder dunkler Urin, unerklärliche Übelkeit oder Erbrechen, Schmerzen im rechten Oberbauch oder Anorexie), ist die Einnahme von Trikafta zu unterbrechen und es muss umgehend eine Messung der Serumtransaminasen und des Gesamtbilirubins erfolgen. Im Falle von ALT- oder AST-Werten > 5 × der Obergrenze des Normalbereichs [ULN] resp. ALT oder AST > 3 × ULN mit Bilirubin > 2 × ULN ist die Behandlung zu unterbrechen. Es sind engmaschige Labortests durchzuführen, bis die Abweichungen zurückgegangen sind. Nach deren Normalisierung sind Nutzen und Risiken der Wiederaufnahme der Behandlung abzuwägen (siehe "Dosierung/Anwendung" , "Unerwünschte Wirkungen" und "Pharmakokinetik" ). Patienten, die ihre Behandlung nach einer Unterbrechung wieder aufnehmen, sind engmaschig zu überwachen.
Eingeschränkte Leberfunktion
Die Behandlung wird bei Patienten mit mässig eingeschränkter Leberfunktion nicht empfohlen. Bei Patienten mit mässig eingeschränkter Leberfunktion sollte die Anwendung von Trikafta nur dann in Erwägung gezogen werden, wenn ein klarer medizinischer Bedarf vorliegt und erwartet wird, dass der Nutzen der Behandlung die Risiken übersteigt. In solchen Fällen ist das Arzneimittel mit Vorsicht in einer niedrigeren Dosis anzuwenden (siehe Tabelle 2). Patienten mit stark eingeschränkter Leberfunktion sollen nicht mit Trikafta behandelt werden (siehe "Dosierung/Anwendung" , "Unerwünschte Wirkungen" und "Pharmakokinetik" ).
Depressionen
Bei Patienten, die mit Trikafta behandelt wurden, liegen Berichte über Depressionen (einschliesslich Suizidgedanken und Suizidversuch) vor, die in der Regel innerhalb von drei Monaten nach Behandlungsbeginn und bei Patienten mit psychiatrischen Erkrankungen in der Vorgeschichte auftraten. In einigen Fällen wurde über eine Verbesserung der Symptome nach Dosisreduktion oder nach dem Absetzen der Behandlung berichtet. Patienten (und Betreuer) sind darauf hinzuweisen, dass sie auf depressive Verstimmungen, Suizidgedanken oder ungewöhnliche Verhaltensänderungen achten und bei Auftreten solcher Symptome sofort einen Arzt aufsuchen müssen.
Arzneimittelinteraktionen
CYP3A-Induktoren
Die Bioverfügbarkeit von Ivacaftor wird durch die gleichzeitige Anwendung von CYP3A-Induktoren deutlich reduziert und es ist damit zu rechnen, dass die Bioverfügbarkeit von Elexacaftor und Tezacaftor ebenfalls abnimmt, was zu einem Wirksamkeitsverlust bei Trikafta führen kann. Daher wird die gleichzeitige Anwendung mit starken CYP3A-Induktoren nicht empfohlen (siehe "Interaktionen" ).
CYP3A-Inhibitoren
Die Bioverfügbarkeit von Elexacaftor, Tezacaftor und Ivacaftor wird durch die gleichzeitige Anwendung von starken oder mässigen CYP3A-Inhibitoren erhöht. Daher ist eine Reduktion der Trikafta-Dosis erforderlich, wenn es gleichzeitig mit mässigen oder starken CYP3A-Inhibitoren angewendet wird (siehe "Interaktionen" und Tabelle 3 in "Dosierung/Anwendung" ).
Katarakte
Bei Kindern und Jugendlichen wurde unter einer Behandlung, die Ivacaftor enthielt, über Fälle von nicht kongenitaler Linsentrübung ohne Auswirkungen auf das Sehvermögen berichtet. Obgleich in manchen Fällen andere Risikofaktoren (z.B. die Anwendung von Kortikosteroiden, eine Strahlenexposition) vorhanden waren, kann ein mögliches, auf die Behandlung mit Ivacaftor zurückzuführendes Risiko nicht ausgeschlossen werden. Bei Kindern und Jugendlichen, die eine Therapie mit Trikafta beginnen, werden vor Therapiebeginn sowie zur Verlaufskontrolle Augenuntersuchungen empfohlen (siehe "Präklinische Daten" ).
Patienten nach Organtransplantation
Elexacaftor/Tezacaftor/Ivacaftor wurde nicht bei CF-Patienten nach Organtransplantation untersucht. Daher wird die Anwendung bei Patienten mit Organtransplantaten nicht empfohlen. Siehe "Interaktionen" für Hinweise zu Interaktionen mit Ciclosporin oder Tacrolimus.
Lactose
Dieses Arzneimittel enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tagesdosis, d.h. es ist nahezu "natriumfrei" .
InteraktionenPharmakokinetische Interaktionen
Arzneimittel mit Einfluss auf die Pharmakokinetik von Trikafta
CYP3A-Induktoren
Elexacaftor, Tezacaftor und Ivacaftor sind Substrate von CYP3A (Ivacaftor ist ein sensitives Substrat von CYP3A). Die gleichzeitige Anwendung von CYP3A-Induktoren kann zu einer reduzierten Bioverfügbarkeit und folglich zu einer verminderten Wirksamkeit von Trikafta führen. Bei gleichzeitiger Anwendung von Ivacaftor und Rifampicin, einem starken CYP3A-Induktor, kam es zu einer deutlichen Abnahme der Fläche unter der Kurve (AUC) von Ivacaftor um 89 %. Es ist zu erwarten, dass die Bioverfügbarkeit von Elexacaftor und Tezacaftor bei gleichzeitiger Anwendung mit starken CYP3A-Induktoren herabgesetzt sein wird; die gleichzeitige Anwendung von Trikafta mit starken CYP3A-Induktoren wird daher nicht empfohlen (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Beispiele für starke CYP3A-Induktoren sind:
-Rifampicin, Rifabutin, Phenobarbital, Carbamazepin, Phenytoin und Johanniskraut (Hypericum perforatum)
CYP3A-Inhibitoren
Die gleichzeitige Anwendung mit Itraconazol, einem starken CYP3A-Inhibitor, erhöhte die AUC von Elexacaftor um das 2,8-Fache und die AUC von Tezacaftor um das 4,0- bis 4,5-Fache. Bei gleichzeitiger Anwendung mit Itraconazol und Ketoconazol erhöhte sich die AUC von Ivacaftor um das 15,6- resp. 8,5-Fache. Daher ist eine Reduktion der Trikafta-Dosis erforderlich, wenn es gleichzeitig mit starken CYP3A-Inhibitoren angewendet wird (siehe "Warnhinweise und Vorsichtsmassnahmen" und Tabelle 3 in "Dosierung/Anwendung" ).
Beispiele für starke CYP3A-Inhibitoren sind:
-Ketoconazol, Itraconazol, Posaconazol und Voriconazol
-Telithromycin und Clarithromycin
Simulationen haben gezeigt, dass die gleichzeitige Anwendung mit mässigen CYP3A-Inhibitoren die Elexacaftor- und Tezacaftor-AUC um das etwa 1,9- bis 2,3-Fache erhöhen kann. Die gleichzeitige Anwendung mit Fluconazol erhöhte die Ivacaftor-AUC um das 2,9-Fache. Die Trikafta-Dosis sollte reduziert werden, wenn es gleichzeitig mit mässigen CYP3A-Inhibitoren angewendet wird (siehe "Warnhinweise und Vorsichtsmassnahmen" und Tabelle 3 in "Dosierung/Anwendung" ).
Beispiele für mässige CYP3A-Inhibitoren sind:
-Fluconazol
-Erythromycin
Die gleichzeitige Gabe von Trikafta und Grapefruitsaft, der einen oder mehrere Bestandteile mit mässiger Hemmwirkung auf CYP3A enthält, kann zu einem Anstieg der Bioverfügbarkeit von Elexacaftor, Tezacaftor und Ivacaftor führen. Auf Speisen oder Getränke, die Grapefruit enthalten, ist während der Behandlung mit Trikafta zu verzichten (siehe "Dosierung/Anwendung" ).
Ciprofloxacin
Die gleichzeitige Anwendung von Ciprofloxacin und Trikafta wurde nicht untersucht. Ciprofloxacin zeigte jedoch keine klinisch relevante Wirkung auf die Bioverfügbarkeit von Tezacaftor oder Ivacaftor, und eine klinisch relevante Wirkung auf die Bioverfügbarkeit von Elexacaftor wird nicht erwartet. Bei gleichzeitiger Anwendung von Trikafta und Ciprofloxacin ist eine Dosisanpassung daher nicht erforderlich.
Die Wirkung gleichzeitig angewendeter Arzneimittel auf die Exposition von Elexacaftor, Tezacaftor und/oder Ivacaftor ist in Tabelle 4 gezeigt.
Tabelle 4: Einfluss
anderer Arzneimittel
auf Elexacaftor,
Tezacaftor und/oder
Ivacaftor
Dosis und Behandlung Wirkung auf die PK Geometrischer
sschema von ELX, TEZ und/ode Mittelwertsquotient
r IVA (90 %-KI) von
Elexacaftor, Tezacaf
tor und Ivacaftor
Keine Wirkung = 1,0
AUC Cmax
Itraconazol 200 mg TEZ 25 mg 1 × tgl. ↑ Tezacaftor 4,02(3,71; 4,63) 2,83(2,62; 3,07)
alle 12 Std. an Tag + IVA 50 mg 1 × tgl.
1, gefolgt von 200
mg 1 × tgl.
↑ Ivacaftor 15,6(13,4; 18,1) 8,60(7,41; 9,98)
Itraconazol 200 mg ELX 20 mg + TEZ 50 ↑ Elexacaftor 2,83 (2,59; 3,10) 1,05(0,977; 1,13)
1 × tgl. mg-Einzeldosis
↑ Tezacaftor 4, 51 (3,85; 5,29) 1,48 (1,33; 1,65)
Ketoconazol 400 mg IVA 150 mg-Einzeldos ↑ Ivacaftor 8,45 (7,14; 10,0) 2,65 (2,21; 3,18)
1 × tgl. is
Ciprofloxacin 750 TEZ 50 mg alle 12 ↔ Tezacaftor 1,08(1,03; 1,13) 1,05(0,99; 1,11)
mg alle 12 Std. Std. + IVA 150 mg
alle 12 Std.
↑ Ivacaftor* 1,17(1,06; 1,30) 1,18(1,06; 1,31)
Rifampicin 600 mg 1 IVA 150 mg-Einzeldos ↓ Ivacaftor 0,114(0,097; 0,136) 0,200(0,168; 0,239)
× tgl. is
Fluconazol 400 IVA 150 mg alle 12 ↑ Ivacaftor 2,95(2,27; 3,82) 2,47(1,93; 3,17)
mg-Einzeldosis an Std.
Tag 1, gefolgt von
200 mg 1 × tgl.
↑ = Zunahme, ↓ =
Abnahme, ↔ = keine
Veränderung. KI =
Konfidenzintervall;
ELX = Elexacaftor;
TEZ = Tezacaftor;
IVA = Ivacaftor; PK
= Pharmakokinetik.
* Die Wirkung ist
nicht klinisch
signifikant.
Arzneimittel, die von Trikafta beeinflusst werden
CYP2C9-Substrate
Ivacaftor kann CYP2C9 hemmen; daher wird bei gleichzeitiger Anwendung von Trikafta mit Warfarin eine Überwachung der INR (International Normalized Ratio) empfohlen. Andere Arzneimittel, bei denen es zu einem Anstieg der Bioverfügbarkeit durch Trikafta kommen kann, sind Glimepirid und Glipizid; bei der Anwendung dieser Arzneimittel ist daher Vorsicht geboten.
Interaktionspotenzial mit Transportern
Die gleichzeitige Anwendung von Ivacaftor oder Tezacaftor/Ivacaftor mit Digoxin, einem sensitiven P-Glycoprotein (Pgp)-Substrat, erhöhte die AUC von Digoxin um das 1,3-Fache, was mit einer schwachen Hemmung von Pgp durch Ivacaftor übereinstimmt. Die Anwendung von Trikafta kann die systemische Bioverfügbarkeit von Arzneimitteln, die sensitive Substrate von Pgp sind, erhöhen, wodurch ihre therapeutische Wirkung sowie ihre unerwünschten Wirkungen verstärkt oder länger anhaltend auftreten können. Bei gleichzeitiger Anwendung mit Digoxin oder anderen Pgp-Substraten mit einem engen therapeutischen Index, wie z.B. Ciclosporin, Everolimus, Sirolimus und Tacrolimus, ist Vorsicht geboten und es muss eine angemessene Überwachung durchgeführt werden.
Elexacaftor und M23-ELX hemmen die Aufnahme durch OATP1B1 und OATP1B3 in vitro. Tezacaftor/Ivacaftor vergrösserten die AUC von Pitavastatin, einem OATP1B1-Substrat, um das 1,2-Fache. Die gleichzeitige Anwendung von Trikafta kann die Bioverfügbarkeit von Arzneimitteln erhöhen, die Substrate dieser Transporter sind, wie z.B. Statine, Glyburid, Nateglinid und Repaglinid. Bei gleichzeitiger Anwendung mit Substraten von OATP1B1 oder OATP1B3 ist Vorsicht geboten und es muss eine angemessene Überwachung durchgeführt werden. Bilirubin ist ein OATP1B1- und OATP1B3-Substrat. In Studie 445-102 wurden leichte Anstiege des durchschnittlichen Gesamtbilirubins beobachtet (Veränderung um bis zu 4,0 µmol/l gegenüber dem Ausgangswert). Dieses Ergebnis stimmt überein mit der in vitro beobachteten Hemmung der Bilirubin-Transporter OATP1B1 und OATP1B3 durch Elexacaftor und M23-ELX.
Hormonelle Kontrazeptiva
Trikafta wurde zusammen mit Ethinylestradiol/Levonorgestrel untersucht und hatte keinen klinisch relevanten Einfluss auf die Bioverfügbarkeit des oralen Kontrazeptivums. Es ist nicht zu erwarten, dass Trikafta einen Einfluss auf die Wirksamkeit oraler Kontrazeptiva hat.
Die Wirkung von Elexacaftor, Tezacaftor und/oder Ivacaftor auf die Exposition gegenüber gleichzeitig angewendeten Arzneimitteln ist in Tabelle 5 gezeigt.
Tabelle 5: Einfluss
von Elexacaftor,
Tezacaftor und/oder
Ivacaftor auf
andere Arzneimittel
Dosis und Behandlung Wirkung auf die PK Geometrischer
sschema von anderen Arzneimi Mittelwertsquotient
tteln (90 %-KI) des
anderen Arzneimittel
s Keine Wirkung =
1,0
AUC Cmax
Midazolam 2 mg-Einze TEZ 100 mg 1 × ↔ Midazolam 1,12(1,01; 1,25) 1,13(1,01; 1,25)
ldosis oral tgl./IVA 150 mg
alle 12 Std.
Digoxin 0,5 mg-Einze TEZ 100 mg 1 × ↑ Digoxin 1,30(1,17; 1,45) 1,32(1,07; 1,64)
ldosis tgl./IVA 150 mg
alle 12 Std.
Orales Kontrazeptivu ELX 200 mg 1 × ↑ Ethinylestradiol* 1,33 (1,20; 1,49) 1,26 (1,14; 1,39)
m Ethinylestradiol tgl./TEZ 100 mg 1 ×
30 µg/Levonorgestrel tgl./IVA 150 mg
150 µg 1 × tgl. alle 12 Std.
↑ Levonorgestrel* 1,23 (1,10; 1,37) 1,10 (0,985; 1,23)
Rosiglitazon 4 IVA 150 mg alle 12 ↔ Rosiglitazon 0,975(0,897; 1,06) 0,928(0,858; 1,00)
mg-Einzeldosis oral Std.
Desipramin 50 IVA 150 mg alle 12 ↔ Desipramin 1,04(0,985; 1,10) 1,00(0,939; 1,07)
mg-Einzeldosis Std.
↑ = Zunahme, ↓ =
Abnahme, ↔ = keine
Veränderung. KI =
Konfidenzintervall;
ELX = Elexacaftor;
TEZ = Tezacaftor;
IVA = Ivacaftor; PK
= Pharmakokinetik.
* Die Wirkung ist
nicht klinisch
signifikant.
Schwangerschaft, StillzeitSchwangerschaft
Es wurden keine adäquaten und gut kontrollierten Studien mit Trikafta bei schwangeren Frauen durchgeführt. Tierexperimentelle Studien mit den einzelnen Wirkstoffen zeigten keine direkte Toxizität mit Auswirkung auf Schwangerschaft, embryo-fötale Entwicklung und postnatale Entwicklung (siehe "Präklinische Daten" ). Aus Vorsichtsgründen soll eine Anwendung der Therapie während der Schwangerschaft vermieden werden.
Stillzeit
Einige Daten zeigen, dass Elexacaftor, Tezacaftor und Ivacaftor in die Muttermilch ausgeschieden werden. Ein Risiko für Neugeborene / Kleinkinder kann nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Trikafta verzichtet werden soll / die Behandlung mit Trikafta zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau zu berücksichtigen.
Fertilität
Es liegen keine Daten über den Einfluss von Elexacaftor, Tezacaftor und Ivacaftor auf die Fertilität beim Menschen vor. In tierexperimentellen Studien hatten Elexacaftor und Ivacaftor eine Wirkung auf die Fertilität von Ratten. Tezacaftor zeigte in tierexperimentellen Studien keine Auswirkungen auf das Paarungsverhalten und Fertilitätsparameter (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDer Einfluss von Trikafta auf die Fähigkeit zum Führen eines Fahrzeugs oder zum Bedienen von Maschinen wurde nicht spezifisch untersucht.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Das Sicherheitsprofil von Trikafta basiert auf den Daten von 510 Patienten aus zwei doppelblinden, kontrollierten Phase-3-Studien mit 24 Wochen bzw. 4 Wochen Behandlungsdauer (Studien 445-102 und 445-103). In den zwei kontrollierten Phase-3-Studien erhielten insgesamt 257 Patienten ab 12 Jahren mindestens eine Dosis Trikafta.
In Studie 445-102 betrug der Anteil von Patienten, die die Studienmedikation aufgrund von unerwünschten Ereignissen vorzeitig absetzten, bei den mit Trikafta behandelten Patienten 1 % und bei den mit Placebo behandelten Patienten 0 %.
Schwerwiegende unerwünschte Arzneimittelwirkungen, die bei mit Trikafta behandelten Patienten häufiger auftraten als bei Placebo waren Hautausschlag bei 3 (1,5 %) der mit Trikafta behandelten Patienten vs. 1 (0,5 %) der mit Placebo behandelten Patienten. Die häufigsten (≥10 %) unerwünschten Arzneimittelwirkungen bei mit Trikafta behandelten Patienten waren Kopfschmerz (17,3 %), Diarrhoe (12,9 %) und Infektion der oberen Atemwege (11,9 %).
Das Sicherheitsprofil von Trikafta war in allen Subgruppen von Patienten generell vergleichbar, auch bei einer Analyse nach Alter, Geschlecht und Ausgangswert des forcierten exspiratorischen Volumens in 1 Sekunde in Prozent des Sollwerts (ppFEV1) sowie nach geographischer Region.
Tabellarische Auflistung der unerwünschten Wirkungen
Tabelle 6 zeigt unerwünschte Wirkungen, die unter Elexacaftor/Tezacaftor/Ivacaftor in Kombination mit Ivacaftor, unter Tezacaftor/Ivacaftor in Kombination mit Ivacaftor und unter Ivacaftor-Monotherapie beobachtet wurden. Die unerwünschten Arzneimittelwirkungen von Trikafta sind nach den MedDRA-Häufigkeitsangaben eingestuft: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000); nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 6: Unerwünschte Wirkungen bei
Anwendung von Elexacaftor/Tezacaftor/Iv
acaftor, Tezacaftor/Ivacaftor und
Ivacaftor allein bei Jugendlichen >12
Jahre und Erwachsenen
MedDRA Systemorganklasse Unerwünschte Wirkungen Häufigkeit
Infektionen und parasitäre Erkrankungen Infektion der oberen Atemwege*, Sehr häufig
Nasopharyngitis
Rhinitis*, Influenza* Häufig
Stoffwechsel- und Ernährungsstörungen Hypoglykämie* Häufig
Psychiatrische Erkrankungen Depressionen Nicht bekannt
Erkrankungen des Nervensystems Kopfschmerz*, Schwindelgefühl* Sehr häufig
Erkrankungen des Ohrs und des Ohrenschmerzen, Beschwerden im Ohr, Häufig
Labyrinths Tinnitus, Trommelfellhyperämie,
Gleichgewichtsstörungen (vestibuläre
Störungen)
Verstopfte Ohren Gelegentlich
Erkrankungen der Atemwege, des Oropharyngeale Schmerzen, verstopfte Sehr häufig
Brustraums und Mediastinums Nase*
Rhinorrhoe*, verstopfte Nasennebenhöhle Häufig
n, Rachenrötung, abnormale Atmung*
Giemen* Gelegentlich
Erkrankungen des Gastrointestinaltrakts Diarrhoe*, Bauchschmerzen* Sehr häufig
Übelkeit, Oberbauchschmerzen*, Häufig
Blähungen*
Leber- und Gallenerkrankungen Transaminasenanstiege Sehr häufig
Alaninaminotransferase erhöht*, Häufig
Aspartataminotransferase erhöht*
Erkrankungen der Haut und des Hautausschlag* Sehr häufig
Unterhautzellgewebes
Akne*, Pruritus* Häufig
Erkrankungen der Geschlechtsorgane und Raumforderung in der Brust Häufig
der Brustdrüse
Brustentzündung, Gynäkomastie, Gelegentlich
Affektion der Brustwarzen,
Brustwarzenschmerzen
Untersuchungen Bakterien im Sputum Sehr häufig
Kreatinphosphokinase im Blut erhöht* Häufig
Erhöhter Blutdruck* Gelegentlich
* Unerwünschte Wirkungen, die während
klinischer Studien mit Elexacaftor/Teza
caftor/Ivacaftor in Kombination mit
Ivacaftor beobachtet wurden.
Die Sicherheitsdaten der folgenden Studien stimmten mit den in Studie 445-102 erhobenen Sicherheitsdaten überein.
-Eine 4-wöchige, randomisierte, doppelblinde, aktiv kontrollierte Studie an 107 Patienten (Studie 445-103).
-Eine 192-wöchige offene klinische Studie zur Sicherheit und Wirksamkeit (Studie 445-105) an Patienten, die von Studie 445-102 und 445-103 übernommen wurden.
-Eine 8-wöchige, randomisierte, doppelblinde, aktiv kontrollierte klinische Studie an 258 Patienten (Studie 445-104).
-Eine 24-wöchige, offene Studie (Studie 445-111) an 75 Patienten im Alter von 2 bis unter 6 Jahren.
-Eine 24-wöchige, offene Studie untersuchte 66 Patienten im Alter von 6 bis unter 12 Jahren (Studie 445-106 Teil B). Details zu den die Leber und Haut betreffenden unerwünschten Ereignissen siehe unten.
-Eine 192-wöchige, zweiteilige (Teil A und Teil B), offene Studie zur Sicherheit und Wirksamkeit (Studie 445-107) untersuchte Patienten im Alter ab 6 Jahren, die aus der Studie 445-106 übernommen wurden; eine Interimsanalyse des Teils A wurde mit 64 Patienten in Woche 96 durchgeführt (60 Completers, 93,8 %). Anschliessend wurden 48 (75,0 %) Probanden in Teil B der Studie bis zur Woche 192 übernommen. 39 (60,9 %) Probanden schlossen Teil B ab. Das Sicherheitsprofil war ähnlich wie das in Studie 445-106 beobachtete, jedoch wurden im Langzeitverlauf häufiger Katarakte/Linsenopazität beobachtet (bei 6 Probanden, 9,4 %).
-Eine 24-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie (Studie 445-124) bei 307 Patienten ab 6 Jahren.
Beschreibung ausgewählter Nebenwirkungen
Erhöhte Transaminasenwerte und Leberschädigung
In Studie 445-102 betrug die Inzidenz maximaler Transaminasenwerte (ALT oder AST) > 8, > 5 oder > 3 × ULN bei den mit Trikafta behandelten Patienten 1,5 %, 2,5 % resp. 7,9 % und bei den mit Placebo behandelten Patienten 1,0 %, 1,5 % resp. 5,5 %. Die Inzidenz von Transaminasenanstiegen als unerwünschte Wirkungen betrug 10,9 % bei den mit Trikafta behandelten Patienten und 4,0 % bei den mit Placebo behandelten Patienten. Von den mit Trikafta behandelten Patienten brach keiner die Behandlung wegen Transaminasenanstiegen ab.
In Studie 445-106 Teil B bei 66 Patienten im Alter von 6 bis unter 12 Jahren betrug die Inzidenz maximaler Transaminasenwerte (ALT oder AST) > 8, > 5 und > 3 × ULN 0,0 %, 1,5 % bzw. 10,6 %. Von den mit Trikafta behandelten Patienten hatte kein Patient einen Transaminasenanstieg > 3 × ULN, der mit einem Anstieg des Gesamtbilirubins > 2 × ULN assoziiert war und kein Patient brach die Behandlung wegen Transaminasenanstiegen ab. Bei den unerwünschten Ereignissen mit erhöhten Transaminasen betrug die mittlere (SD) Zeit bis zum Auftreten des ersten Ereignisses 52,1 (62,2) Tage und die mittlere (SD) Dauer betrug 15,3 (9,0) Tage (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
In Studie 445-111 bei Patienten im Alter von 2 bis unter 6 Jahren betrug die Inzidenz maximaler Transaminasenwerte (ALT oder AST) > 8, > 5 und > 3 × ULN 1,3 %, 2,7 % bzw. 8,0 %. Von den mit Trikafta behandelten Patienten hatte kein Patient einen Transaminasenanstieg > 3 × ULN, der mit einem Anstieg des Gesamtbilirubins > 2 × ULN assoziiert war, und kein Patient brach die Behandlung wegen Transaminasenanstiegen ab (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
In Studie 445-124 bei Patienten im Alter ab 6 Jahren betrug die Inzidenz maximaler Transaminasenwerte (ALT oder AST) > 8, > 5 und > 3 × ULN bei den mit Trikafta behandelten Patienten 2,0 %, 2,0 % bzw. 6,3 % und bei den mit Placebo behandelten Patienten 0,0 %. Von den mit Trikafta behandelten Patienten hatte kein Patient einen Transaminasenanstieg > 3 × ULN, der mit einem Anstieg des Gesamtbilirubins > 2 × ULN assoziiert war. Ein Patient brach die Behandlung wegen Transaminasenanstiegen als unerwünschtes Ereignis ab (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Die folgenden unerwünschten Wirkungen wurden im Rahmen der Anwendung von Trikafta nach der Zulassung festgestellt.
-Leberversagen mit daraus resultierender Lebertransplantation bei Patienten mit und ohne vorbestehende fortgeschrittene Lebererkrankung (z.B. Leberzirrhose, portale Hypertonie) (siehe "Dosierung/Anwendung" , "Warnhinweise und Vorsichtsmassnahmen" und "Pharmakokinetik" )
-Leberschädigung, gekennzeichnet durch gleichzeitige Anstiege der Transaminasenwerte (ALT und AST) und des Gesamtbilirubins bei CF Patienten mit oder ohne vorbestehender Lebererkrankung (siehe "Dosierung/Anwendung" , "Warnhinweise und Vorsichtsmassnahmen" und "Pharmakokinetik" ).
Hautausschläge
In Studie 445-102 lag die Inzidenz von Hautausschlägen (z.B. Ausschlag, juckender Ausschlag) bei den mit Trikafta behandelten Patienten bei 10,9 % und bei den mit Placebo behandelten Patienten bei 6,5 %. Die Hautausschläge waren generell leicht bis mittelschwer. Die Inzidenz von Hautausschlägen nach Geschlecht der Patienten ergab einen Anteil von 5,8 % bei den männlichen Patienten und von 16,3 % bei den weiblichen Patienten, die mit Trikafta behandelt wurden, sowie von 4,8 % bei den männlichen Patienten und 8,3 % bei den weiblichen Patienten, die mit Placebo behandelt wurden.
In der Studie 445-106 Teil B an 66 mit Trikafta behandelten Patienten im Alter von 6 bis unter 12 Jahren lag die Inzidenz von Hautausschlägen (z.B. Ausschlag, juckender Ausschlag) bei 24,2 % (n=16). Die spezifischen unerwünschten Ereignisse umfassten Hautausschlag n=8 (12,1 %), erythematöser Hautausschlag n=3 (4,5 %), makulo-papulöser Hautausschlag n=2 (3,0 %), papulärer Hautausschlag n=2 (3,0 %), Hautexfoliation n=1 (1,5 %), Urtikaria n=1 (1,5 %). Ein Patient (1,5 %) hatte einen Ausschlag, der zum Absetzen von Trikafta führte. Die übrigen Patienten hatten Ausschläge, die sich während der Fortsetzung der Behandlung mit Trikafta zurückbildeten.
In Studie 445-124 traten bei 55 (26,8 %) der Patienten in der Trikafta-Gruppe und bei 3 (2,9 %) der Patienten in der Placebo-Gruppe Hautausschläge auf. Die meisten Hautausschläge waren leicht oder mittelschwer. Ein Patient (0,5 %) in der Trikafta-Gruppe hatte einen schwerwiegenden Hautausschlag, der zum Absetzen der Behandlung führte. Kein Patient in der Placebo-Gruppe hatte einen Hautausschlag, der zum Absetzen der Behandlung führte.
Eine Rolle von hormonellen Kontrazeptiva beim Auftreten von Hautausschlag kann nicht ausgeschlossen werden. Bei Patientinnen, die hormonelle Kontrazeptiva einnehmen und einen Hautausschlag entwickeln, ist eine Unterbrechung der Trikafta-Therapie und der hormonellen Kontrazeptiva zu erwägen. Nach dem Abklingen des Hautausschlags ist die Wiederaufnahme der Behandlung mit Trikafta ohne hormonelle Kontrazeptiva zu erwägen. Wenn der Hautausschlag nicht wieder auftritt, kann eine Wiederaufnahme der Behandlung mit hormonellen Kontrazeptiva in Erwägung gezogen werden.
Kreatinphosphokinase erhöht
In Studie 445-102 lag die Inzidenz von maximalen Kreatinphosphokinase-Werten > 5 × des ULN bei den mit Trikafta behandelten Patienten bei 10,4 % und bei den mit Placebo behandelten Patienten bei 5,0 %. Von den mit Trikafta behandelten Patienten brach keiner die Behandlung wegen erhöhter Kreatinphosphokinase-Werte ab.
Erhöhter Blutdruck
In Studie 445-102 betrug der maximale Anstieg des mittleren systolischen Blutdrucks gegenüber dem Ausgangswert 3,5 mmHg und der des mittleren diastolischen Blutdrucks 1,9 mmHg bei den mit Trikafta behandelten Patienten (Ausgangswert: 113 mmHg systolisch und 69 mmHg diastolisch) und 0,9 mmHg resp. 0,5 mmHg bei den mit Placebo behandelten Patienten (Ausgangswert: 114 mmHg systolisch und 70 mmHg diastolisch).
Der Anteil von Patienten, welche bei mindestens zwei Messzeitpunkten einen systolischen Blutdruck > 140 mmHg oder einen diastolischen Blutdruck > 90 mmHg aufwiesen, betrug bei den mit Trikafta behandelten Patienten 5,0 % resp. 3,0 %, im Vergleich zu 3,5 % resp. 3,5 % bei den Patienten, die mit Placebo behandelt wurden.
VX17-445-105 Verlängerungsstudie über 192 Wochen
In der offenen, unkontrollierten Langzeitstudie VX17-445-105 wurden 400 (78,9 %) Patienten mit einer F508del-Mutation/MF-Mutation und 107 (21,1 %) Patienten mit einer homozygoten F508del-Mutation über bis zu 192 Wochen weiterverfolgt. Bis zur Woche 192 erhielt ein Patient keine einzige Dosis, 356 (70,4 %) Patienten schlossen die Behandlung ab und 150 (29,6 %) Patienten brachen aus verschiedenen Gründen vorzeitig ab. Davon brachen 13 (2,6 %) Patienten die Behandlung wegen unerwünschten Ereignissen ab, inklusive 8 (1,6 %) Patienten wegen leberbezogener Ereignisse und 1 (0,2 %) Patient wegen hepatischer Enzephalopathie.
ALT- bzw. AST-Erhöhung > 3, > 5 und > 8 × ULN wurde bei 63 (12,5 %), 36 (7,1 %) und 11 (2,2 %) Patienten festgestellt, davon hatten 2 (0,4 %) Patienten eine ALT- bzw. AST-Erhöhung > 3 × ULN mit gleichzeitiger neu aufgetretener Erhöhung des Gesamtbilirubins > 2 × ULN, wobei ein Patient schon zuvor einen Morbus Meulengracht hatte.
Erhöhungen der CK ≥2,5- ≤5, > 5- ≤10 und > 10 × ULN wurden bei 69 (13,6 %), 38 (7,5 %) und 47 (9,3 %) Patienten festgestellt. Eine CK-Erhöhung wurde als unerwünschtes Ereignis bei 72 (14,2 %) Patienten festgestellt. 3 (0,6 %) Patienten erlitten eine Rhabdomyolyse ohne Nierenbeteiligung oder Myoglobinurie.
Hautausschläge traten bei 89 (17,6 %) Patienten auf. Ein (0,2 %) Patient brach die Behandlung wegen Hautausschlag ab.
Der mittlere systolische Blutdruck stieg zwischen 2,7 und 5,6 mmHg an und der mittlere diastolische Blutdruck stieg zwischen 1,5 und 3,6 mmHg an. Bei 20 (4,0 %) Patienten wurden unerwünschte Ereignisse in Verbindung mit erhöhtem Blutdruck festgestellt.
Bei 5 (1,0 %) Patienten traten unerwünschte Ereignisse in Verbindung mit Katarakten auf, die nicht zu einer Änderung der Dosierung führten.
VX19-445-107 Verlängerungsstudie über 192 Wochen
Eine noch nicht abgeschlossene 192-wöchige unverblindete Studie, VX19-445-107, mit zwei Teilen (Teil A und Teil B) untersuchte Patienten im Alter ab 6 Jahren, die von Studie 445-106 übernommen wurden. Bei 64 Patienten wurde in Woche 96 eine Interimsanalyse durchgeführt. Insgesamt 28 Patienten (43,8 %) waren homozygot für die F508del-Mutation und 36 Patienten (56,3 %) hatten den F508del/MF-Genotyp. In Teil A erhielten 61 der 64 Patienten (95,3 %) mindestens 1 Dosis des Studienmedikaments und beendeten die Behandlung.
Anstiege von ALT oder AST von > 3 ×, > 5 × und > 8 × ULN traten bei 4 (6,3 %), 1 (1,6 %) bzw. 0 Patienten auf. Kein Patient zeigte Anstiege von ALT oder AST > 3 × ULN, die gleichzeitig mit einem neu aufgetretenen Anstieg des Gesamtbilirubins auf >2 × ULN assoziiert waren.
Die meisten Patienten zeigten CK-Anstiege, die innerhalb des Normalbereichs blieben. Vier Patienten (6,3 %) hatten CK-Spiegel von > 2,5 × ULN und kein Patient hatte CK-Spiegel von > 5 × ULN. Kein Patient erlitt eine Rhabdomyolyse.
Ein Hautausschlag trat bei 3 Patienten (4,7 %) auf. Keiner dieser Hautausschläge hatte eine Unterbrechung der Behandlung oder ein Absetzen des Studienmedikaments zur Folge.
Es kam zu keinen klinisch relevanten Blutdruckanstiegen.
Bei 6 Patienten (9,4 %) traten Katarakt-Ereignisse auf, die jedoch zu keiner Dosisänderung führten. Diese Katarakte/Linsentrübungen hatten keine Auswirkungen auf das Sehvermögen und wurden bei 3 dieser Patienten zu Studienende nicht mehr beobachtet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBehandlung
Bei einer Überdosierung mit Trikafta steht kein spezifisches Antidot zur Verfügung. Die Behandlung einer Überdosierung besteht aus allgemeinen supportiven Massnahmen einschliesslich Überwachung der Vitalparameter und Beobachtung des klinischen Zustands des Patienten.
Eigenschaften/WirkungenATC-Code
R07AX32
Wirkungsmechanismus
Elexacaftor und Tezacaftor sind CFTR-Korrektoren, die sich an verschiedene Stellen des CFTR-Proteins binden und eine additive Wirkung bei der Erleichterung der zellulären Verarbeitung und dem Transport von CFTR haben und somit die Menge an CFTR-Protein, das an die Zelloberfläche transportiert wird, im Vergleich zu einem der beiden Moleküle allein erhöhen. Ivacaftor erhöht die Öffnungswahrscheinlichkeit (Gating) des CFTR-Kanals an der Zelloberfläche.
Die kombinierte Wirkung von Elexacaftor, Tezacaftor und Ivacaftor besteht in einer Erhöhung der Menge und Verbesserung der Funktionsfähigkeit des CFTR-Proteins an der Zelloberfläche, was zu einer Zunahme der CFTR-Aktivität führt, wie Messungen des CFTR-vermittelten Chloridtransports zeigten. Die klinischen Resultate stimmten überein mit den Ergebnissen von In-vitro-Untersuchungen und zeigen, dass eine einzige F508del-Mutation ausreicht, um ein signifikantes klinisches Ansprechen zu erzielen (siehe "Klinische Wirksamkeit" ).
CFTR-Chloridtransport-Assay in Fischer-Rat-Thyroid (FRT)-Zellen, die mutiertes CFTR exprimieren
In elektrophysiologischen Studien in der Ussing Kammer wurde das Ansprechen des Chloridtransports von mutiertem CFTR-Protein auf Elexacaftor/Tezacaftor/Ivacaftor untersucht. Dazu wurde ein Panel von FRT-Zelllinien verwendet, die mit einzelnen CFTR-Mutationen transfiziert waren. Elexacaftor/Tezacaftor/Ivacaftor erhöhten den Chloridtransport in FRT-Zellen, die ausgewählte CFTR-Mutationen exprimierten.
Als Schwellenwert für das Ansprechen des In-vitro-CFTR-Chloridtransports wurde eine Nettozunahme von mindestens 10 % des Normalwerts gegenüber Baseline festgelegt, da dies prädiktiv für einen klinischen Nutzen ist oder einen angemessenen klinischen Nutzen erwarten lässt. Bei einzelnen Mutationen korreliert die Grössenordnung der Nettoveränderung gegenüber Baseline des CFTR-vermittelten Chloridtransports in vitro nicht mit dem Ausmass des klinischen Ansprechens.
Tabelle 7 zeigt die ansprechenden CFTR-Mutationen in FRT-Zellen auf der Grundlage des klinischen Ansprechens und/oder von In-vitro-Daten oder Extrapolation, die darauf hinweisen, dass Elexacaftor/Tezacaftor/Ivacaftor den Chloridtransport um mindestens 10 % des Normalwerts gegenüber Baseline erhöhen. Das Vorkommen der in Tabelle 7 aufgeführten CFTR-Mutationen sollte weder anstelle einer Diagnose von zystischer Fibrose noch als alleiniger Faktor für Verschreibungszwecke verwendet werden.
Tabelle 7: Liste
der CFTR-Genmutation
en, die auf Elexacaf
tor/Tezacaftor/Ivaca
ftor ansprechen
Mutationen, die
basierend auf
klinischen Daten
auf Trikafta ansprec
hen
2789+5G→A D1152H L997F P5L R1066H
3272-26A→G F508del L1077P R117C S945L
3849+10kbC→T G85E M1101K R347H T338I
A455E L206W N1303K R347P V232D
Mutationen, die
basierend auf
In-vitro-Daten auf
Trikafta ansprechen
3141del9 E588V G970D L165S R117G S589N
546insCTA E822K G1061R L320V R117H S737F
A46D F191V G1069R L346P R117L S912L
A120T F311del G1244E L453S R117P S977F
A234D F311L G1249R L967S R170H S1159F
A349V F508C G1349D L1324P R258G S1159P
A554E F508C;S1251N † H139R L1335P R334L S1251N
A1006E F575Y H199Y L1480P R334Q S1255P
A1067T F1016S H939R M152V R347L T1036N
D110E F1052V H1054D M265R R352Q T1053I
D110H F1074L H1085P M952I R352W V201M
D192G F1099L H1085R M952T R553Q V456A
D443Y G27R H1375P P67L R668C V456F
D443Y;G576A; R668C † G126D I148T P205S R751L V562I
D579G G178E I175V P574H R792G V754M
D614G G178R I336K Q98R R933G V1153E
D836Y G194R I502T Q237E R1070Q V1240G
D924N G194V I601F Q237H R1070W V1293G
D979V G314E I618T Q359R R1162L W361R
D1270N G463V I807M Q1291R R1283M W1098C
E56K G480C I980K R31L R1283S W1282R
E60K G551D I1027T R74Q S13F Y109N
E92K G551S I1139V R74W S341P Y161D
E116K G576A I1269N R74W; D1270N † S364P Y161S
E193K G576A;R668C † I1366N R74W;V201M † S492F Y563N
E403D G622D K1060T R74W;V201M; D1270N † S549N Y1014C
E474K G628R L15P R75Q S549R Y1032C
Mutationen, die
basierend auf
Extrapolation von
Studie 445-124 auf
Trikafta ansprechen
711+3A→G E831X
† Komplexe/zusammeng
esetzte Mutationen,
bei denen ein
einzelnes Allel des
CFTR-Gens mehrere
Mutationen aufweist.
Sie existieren
unabhängig von
Mutationen auf dem
anderen Allel.
Pharmakodynamik
Pharmakodynamische Wirkungen
Wirkungen auf die Schweisschloridkonzentration
In Studie 445-102 (Patienten mit einer F508del-Mutation in einem Allel und einer Mutation im zweiten Allel, die entweder zum Fehlen von CFTR-Protein oder zu einem CFTR-Protein führt, das nicht auf Ivacaftor und Tezacaftor/Ivacaftor anspricht [Minimalfunktionsmutation]) wurde eine Abnahme der Schweisschloridkonzentration gegenüber Baseline in Woche 4 beobachtet, die während des gesamten 24-wöchigen Behandlungszeitraums anhielt. Der Behandlungsunterschied von Trikafta im Vergleich zu Placebo in Bezug auf die mittlere absolute Veränderung der Schweisschloridkonzentration von Baseline bis einschliesslich Woche 24 betrug -41,8 mmol/l (95 % KI: -44,4; -39,3; p<0,0001).
In Studie 445-103 (Patienten, die homozygot für die F508del-Mutation sind) betrug der Behandlungsunterschied von Trikafta im Vergleich zum Tezacaftor/Ivacaftor und Ivacaftor-Behandlungsschema (Tezacaftor/Ivacaftor) in Bezug auf die mittlere absolute Veränderung der Schweisschloridkonzentration gegenüber Baseline in Woche 4 -45,1 mmol/l (95 % KI: -50,1; -40,1; p<0,0001).
In Studie 445-104 (Patienten, die heterozygot für die F508del-Mutation sind und eine Gating- oder Restfunktionsmutation im zweiten Allel aufweisen) betrug die mittlere absolute Veränderung der Schweisschloridkonzentration von Baseline bis einschliesslich Woche 8 im Anschluss an eine 4-wöchige Ivacaftor- oder Tezacaftor/Ivacaftor-Einlaufphase in der Trikafta-Gruppe -22,3 mmol/l (95 % KI: -24,5; -20,2; p<0,0001). Der Behandlungsunterschied von Trikafta im Vergleich zur Kontrollgruppe (Ivacaftor oder Tezacaftor/Ivacaftor) betrug -23,1 mmol/l (95 % KI: -26,1; -20,1; p<0,0001).
In Studie 445-106 Teil B (Patienten im Alter von 6 Jahren bis unter 12 Jahren, die homozygot für die F508del-Mutation oder heterozygot für die F508del-Mutation und eine Minimalfunktionsmutation sind) betrug die mittlere absolute Veränderung der Schweisschloridkonzentration von Baseline bis einschliesslich Woche 24 -60,9 mmol/l (95 % KI: -63,7; -58,2). Die Messwerte für die Schweisschloridkonzentration wurden an den geplanten Messtagen bei folgender Anzahl Patienten erhoben: Baseline n=62, Tag 15 n=56, Woche 4 n=56, Woche 12 n=50, Woche 24 n=28.
In Studie 445-111 (Patienten im Alter von 2 Jahren bis unter 6 Jahren, die homozygot für die F508del-Mutation oder heterozygot für die F508del-Mutation und eine Minimalfunktionsmutation sind) betrug die mittlere absolute Veränderung der Schweisschloridkonzentration von Baseline bis einschliesslich Woche 24 -57,9 mmol/l (95 % KI: -61,3; -54,6).
In Studie 445-124 (Patienten ab 6 Jahren mit einer auf Elexacaftor/Tezacaftor/Ivacaftor ansprechenden Nicht-F508del-Mutation, durch die sie sich qualifizierten [siehe Tabelle 8]) betrug die mittlere absolute Veränderung der Schweisschloridkonzentration von Baseline bis einschliesslich Woche 24 im Vergleich zu Placebo -28,3 mmol/l (95 %-KI: -32,1, -24,5 mmol/l; p<0,0001).
Kardiovaskuläre Wirkungen
Wirkung auf das QT-Intervall
Nach Dosen vom bis zu 2-Fachen der maximalen empfohlenen Dosis Elexacaftor und dem 3-Fachen der empfohlenen Höchstdosis von Tezacaftor und Ivacaftor war keine klinisch relevante Verlängerung des QT/QTc-Intervalls bei gesunden Probanden festzustellen.
Herzfrequenz
In Studie 445-102 wurden bei mit Trikafta behandelten Patienten mittlere Abnahmen der Herzfrequenz von 3,7 bis 5,8 Schlägen pro Minute (bpm) gegenüber dem Ausgangswert (76 bpm) beobachtet.
Klinische Wirksamkeit
Die Wirksamkeit von Trikafta bei Patienten mit CF wurde in vier doppelblinden, kontrollierten Phase-3-Studien (Studie 445-102, Studie 445-103, 445-104 und 445-124) statistisch nachgewiesen. In diese Studien wurden jeweils CF-Patienten mit mindestens einer F508del-Mutation oder einer der in Tabelle 8 aufgeführten Mutationen, die auf Trikafta ansprechen, aufgenommen. Vier offene, unkontrollierte Phase 3-Studien (Studie 445-105, Studie 445-106 Teil B, Studie 445-111 und Studie 445-107) unterstützen die Wirksamkeit zusätzlich. Trikafta wurde als Kombinationstherapie mit Elexacaftor, Tezacaftor und Ivacaftor entwickelt. Der Nutzen von Elexacaftor allein und von Tezacaftor allein im Vergleich zur Kombinationstherapie wurde in klinischen Studien nicht untersucht und diese Wirkstoffe allein sind nicht als Arzneimittel verfügbar.
Studie 445-102 war eine randomisierte, doppelblinde, placebokontrollierte klinische Studie von 24 Wochen Dauer an Patienten mit einer F508del-Mutation in einem Allel und einer Minimalfunktions (MF)-Mutation im zweiten Allel, die entweder zum Fehlen von CFTR-Protein oder zu einem CFTR-Protein führt, das nicht auf Ivacaftor und Tezacaftor/Ivacaftor anspricht. Insgesamt 403 Patienten ab 12 Jahren (mittleres Alter 26,2 Jahre) wurden randomisiert einer Behandlung mit Trikafta oder Placebo zugewiesen. Die Patienten hatten beim Screening ein ppFEV1 zwischen 40 und 90 %. Der mittlere ppFEV1-Ausgangswert betrug 61,4 % (Bereich: 32,3 %, 97,1 %).
Studie 445-103 war eine 4-wöchige, randomisierte, doppelblinde, aktiv kontrollierte klinische Studie an Patienten, die für die F508del-Mutation homozygot sind. Insgesamt 107 Patienten ab 12 Jahren (mittleres Alter 28,4 Jahre) erhielten während einer 4-wöchigen offenen Einlaufphase ein Behandlungsschema mit Tezacaftor/Ivacaftor und Ivacaftor (Tezacaftor/Ivacaftor) und wurden anschliessend randomisiert einer Behandlung mit Trikafta oder Tezacaftor/Ivacaftor über einen 4wöchigen doppelblinden Behandlungszeitraum zugewiesen. Die Patienten hatten beim Screening ein ppFEV1 zwischen 40 und 90 %. Der mittlere ppFEV1-Ausgangswert nach der Einlaufphase mit Tezacaftor/Ivacaftor betrug 60,9 % (Bereich: 35,0 %; 89,0 %).
Studie 445-104 war eine 8-wöchige, randomisierte, doppelblinde, aktiv kontrollierte klinische Studie an Patienten, die heterozygot für die F508del (F)-Mutation waren und eine Gating (G)- oder Restfunktions (RF)-Mutation im zweiten Allel aufwiesen. Patienten ab 12 Jahren mit einem ppFEV1-Wert zwischen 40 % und 90 % beim Screening erhielten entweder Ivacaftor (Patienten mit F/G-Mutation) oder Tezacaftor/Ivacaftor (Patienten mit F/RF-Mutation) im Rahmen einer 4-wöchigen unverblindeten Einlaufphase. Patienten mit dem F/R117H-Genotyp erhielten Ivacaftor während der Einlaufphase. Anschliessend wurden die Patienten randomisiert der Trikafta-Gruppe zugewiesen oder blieben bei der CFTR-Modulator-Therapie, die sie in der Einlaufphase erhalten hatten. Das mittlere Alter bei Baseline, nach der Einlaufphase, betrug 37,7 Jahre, und der mittlere ppFEV1-Ausgangswert betrug 67,6 % (Bereich: 29,7 %; 113,5 %).
Studie 445-106 war eine zweiteilige 24-wöchige offene, unkontrollierte Studie an 66 Patienten im Alter von 6 Jahren bis unter 12 Jahren (mittleres Alter bei Baseline 9,3 Jahre), die homozygot für die F508del-Mutation oder heterozygot für die F508del-Mutation und eine Minimalfunktionsmutation waren. Teil A evaluierte die Pharmakokinetik und die vorläufigen Ergebnisse zur Sicherheit, Teil B untersuchte die Sicherheit, Verträglichkeit, Wirksamkeit und Pharmakokinetik. Patienten mit einem Körpergewicht <30 kg bei Baseline (36 Patienten, 54,5 %) erhielten zwei Tabletten Elexacaftor 50 mg/Tezacaftor 25 mg/Ivacaftor 37,5 mg am Morgen und eine Tablette Ivacaftor 75 mg am Abend. Patienten mit einem Körpergewicht ≥30 kg bei Baseline (30 Patienten, 45,5 %) erhielten zwei Tabletten Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg am Morgen und eine Tablette Ivacaftor 150 mg am Abend. Beim Screening hatten die Patienten einen ppFEV1-Wert von ≥40 % [mittlerer ppFEV1-Wert bei Baseline 88,8 % (Bereich: 39,0 %, 127,1 %)] und wogen ≥15 kg (verlangtes Einschlusskriterium).
Studie 445-111 war eine 24-wöchige, unverblindete Studie an Patienten im Alter von 2 bis unter 6 Jahren (mittleres Alter bei Baseline 4,1 Jahre). Patienten, die mindestens eine F508del-Mutation oder eine Mutation hatten, die bekanntlich auf Elexacaftor/Tezacaftor/Ivacaftor anspricht, waren für die Studie qualifiziert. Insgesamt 75 Patienten, die homozygot für die F508del-Mutation oder heterozygot für die F508del-Mutation und eine Minimalfunktionsmutation waren, wurden aufgenommen und nach Gewicht behandelt. Patienten mit einem Körpergewicht von 10 kg bis < 14 kg bei Baseline erhielten 1 × täglich morgens Elexacaftor 80 mg /Tezacaftor 40 mg / Ivacaftor 60 mg und 1 × täglich abends Ivacaftor 59,5 mg. Patienten mit einem Körpergewicht von ≥14 kg bei Baseline erhielten 1 × täglich morgens Elexacaftor 100 mg / Tezacaftor 50 mg / Ivacaftor 75 mg und 1 × täglich abends Ivacaftor 75 mg.
Studie 445-124 war eine 24-wöchige, randomisierte, placebokontrollierte, doppelblinde Parallelgruppen-Studie bei Patienten ab 6 Jahren. Patienten mit mindestens einer qualifizierenden Nicht-F508del-Mutation, die auf Elexacaftor/Tezacaftor/Ivacaftor anspricht (siehe Tabelle 8), und ohne Ausschlussmutation (eine andere auf Elexacaftor/Tezacaftor/Ivacaftor ansprechende Mutation) qualifizierten sich für die Studienteilnahme.
Insgesamt wurden 307 Patienten in die Studie aufgenommen und erhielten eine ihrem Alter und Körpergewicht angepasste Dosierung. Patienten im Alter von ≥6 bis < 12 Jahren mit einem Körpergewicht < 30 kg zu Studienbeginn erhielten eine Behandlung mit Elexacaftor 100 mg tgl./Tezacaftor 50 mg tgl./Ivacaftor 75 mg alle 12 Std. Patienten im Alter von ≥6 bis < 12 Jahren mit einem Körpergewicht ≥30 kg zu Studienbeginn erhielten eine Behandlung mit Elexacaftor 200 mg tgl./Tezacaftor 100 mg tgl./Ivacaftor 150 mg alle 12 Std. Patienten im Alter von ≥12 Jahren zu Studienbeginn erhielten Elexacaftor 200 mg tgl./Tezacaftor 100 mg tgl./Ivacaftor 150 mg alle 12 Std. Die Patienten hatten bei der Voruntersuchung einen ppFEV1-Wert ≥40 % und ≤100 % und waren 6 Jahre oder älter. Der mittlere ppFEV1-Wert bei Baseline betrug 67,7 % (Bereich: 34,0 %, 108,7 %)].
Tabelle 8: Geeignete
auf Elexacaftor/Tez
acaftor/Ivacaftor
ansprechende CFTR-Mu
tationen
2789+5G>A D1152H L997F R117C T338I
3272-26A>G G85E M1101K R347H V232D
3849+10kbC>T L1077P P5L R347P
A455E L206W R1066H S945L
Die Patienten in Studie 445-102, 445-103, 445-104, 445-106, 445-111 und 445-124 setzten ihre CF-Therapien (z.B. Bronchodilatatoren, inhalative Antibiotika, Dornase-alfa und hypertone Kochsalzlösung) fort, setzten aber alle vorherigen CFTR-Modulator-Therapien ab, mit Ausnahme der Studienmedikamente. Die Patienten hatten eine bestätigte CF-Diagnose und erfüllten die Eignungskriterien für die Studie.
Von den Studien 445-102, 445-103, 445-104, 445-106, 445-111 und 445-124 waren Patienten, die Lungeninfektionen mit Erregern aufwiesen, die mit einer rascheren Abnahme der Lungenfunktion assoziiert sind, wie u.a. Burkholderia cenocepacia, Burkholderia dolosa oder Mycobacterium abscessus, oder die beim Screening einen abnormalen Leberfunktionstest (ALT, AST, ALP oder GGT ≥3 × ULN oder Gesamtbilirubin ≥2 × ULN) aufwiesen, ausgeschlossen. Von der Studie 445-111 waren Patienten, die ALT- oder AST-Werte ≥2 × ULN aufwiesen, ebenfalls ausgeschlossen.
Patienten aus den Studien 445-102 und 445-103 waren für die Übernahme in eine 192-wöchige unverblindete Verlängerungsstudie (Studie 445-105) qualifiziert. Die Patienten in den Studien 445-104, 445-106 Teil B, 445-111 und 445-124 waren für die Übernahme in unverblindete Verlängerungsstudien qualifiziert.
Studie 445-102
In Studie 445-102 war der primäre Endpunkt die mittlere absolute Veränderung des ppFEV1 von Baseline bis einschliesslich Woche 24. Die Behandlung mit Trikafta führte zu einer statistisch signifikanten Verbesserung des ppFEV1 von 14,3 Prozentpunkten im Vergleich zu Placebo (95 % KI: 12,7; 15,8; p<0,0001) (siehe Tabelle 9). Die durchschnittliche Verbesserung des ppFEV1 setzte schnell ein (Tag 15) und hielt über den gesamten 24wöchigen Behandlungszeitraum an (siehe Abbildung 1). Verbesserungen beim ppFEV1 wurden unabhängig von Alter, Baseline-ppFEV1, Geschlecht und geographischer Region beobachtet. Insgesamt hatten 18 Patienten, die mit Trikafta behandelt wurden, ein ppFEV1 < 40 bei Baseline. Die Sicherheit und Wirksamkeit in dieser Subgruppe war vergleichbar mit den Ergebnissen in der Gesamtstudienpopulation. Eine Zusammenfassung der primären und wichtigsten sekundären Ergebnisse ist Tabelle 9 zu entnehmen.
Tabelle 9: Primäre und wichtigste
sekundäre Wirksamkeitsanalysen,
vollständiges Analyseset (Studie
445-102)
Analyse Statistik Placebo N=203 Trikafta N=200
Primäre Wirksamkeitsanalyse
Absolute Veränderung des ppFEV1 Behandlungsunterschi N.a. 14,3 (12,7; 15,8)
von Baseline bis einschliesslich ed (95 % KI)
Woche 24 (Prozentpunkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -0,4 (0,5) 13,9 (0,6)
(SE)
Wichtigste sekundäre Wirksamkeitsa
nalysen
Absolute Veränderung des ppFEV1 Behandlungsunterschi N.a. 13,7 (12,0; 15,3)
gegenüber Baseline in Woche 4 ed (95 % KI)
(Prozentpunkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -0,2 (0,6) 13,5 (0,6)
(SE)
Anzahl der pulmonalen Anzahl der Ereigniss 113 (0,98) 41 (0,37)
Exazerbationen von Baseline bis e (Ereignisrate pro
einschliesslich Woche 24‡ Jahr††)
Rate Ratio (95 % KI) N.a. 0;37 (0,25; 0,55)
p-Wert N.a. p<0,0001
Absolute Veränderung der Behandlungsunterschi N.a. -41,8 (-44,4; -39,3)
Schweisschloridkonzentra-tion von ed (95 % KI)
Baseline bis einschliesslich
Woche 24 (mmol/l)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -0,4 (0,9) -42,2 (0,9)
(SE)
Absolute Veränderung des Scores Behandlungsunterschi N.a. 20,2 (17,5; 23,0)
der respiratorischen Domäne des ed (95 % KI)
CFQ-R von Baseline bis
einschliesslich Woche 24 (Punkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -2,7 (1,0) 17,5 (1,0)
(SE)
Absolute Veränderung des BMI Behandlungsunterschi N.a. 1,04 (0,85; 1,23)
gegenüber Baseline in Woche 24 ed (95 % KI)
(kg/m²)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe 0,09 (0,07) 1,13 (0,07)
(SE)
Absolute Veränderung der Behandlungsunterschi N.a. -41,2 (-44,0; -38,5)
Schweisschloridkonzentra-tion ed (95 % KI)
gegenüber Baseline in Woche 4
(mmol/l)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe 0,1 (1,0) -41,2 (1,0)
(SE)
Absolute Veränderung des Scores Behandlungsunterschi N.a. 20,1 (16,9; 23,2)
der respiratorischen Domäne des ed (95 % KI)
CFQ-R gegenüber Baseline in Woche
4 (Punkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -1,9 (1,1) 18,1 (1,1)
(SE)
ppFEV1: forciertes exspiratorische
s Volumen in 1 Sekunde in Prozent
des Sollwerts; KI: Konfidenzinterv
all; SE: Standardfehler; N.a.:
nicht anwendbar; CFQ-R: Cystic
Fibrosis Questionnaire-Revised,
überarbeiteter Fragebogen zu
zystischer Fibrose; BMI: Body
Mass Index (Körpermassenindex). ‡
Eine pulmonale Exazerbation war
definiert als eine Veränderung
der antibiotischen Therapie
(i.v., inhalativ oder oral)
aufgrund von mindestens 4 von 12
im Voraus festgelegten
sinopulmonalen Zeichen/Symptomen.
†† Geschätzte Ereignisrate pro
Jahr, berechnet anhand von 48
Wochen pro Jahr.
Abbildung 1: Absolute Veränderung des ppFEV1 gegenüber dem Ausgangswert bei jedem Untersuchungszeitpunkt in Studie 445-102
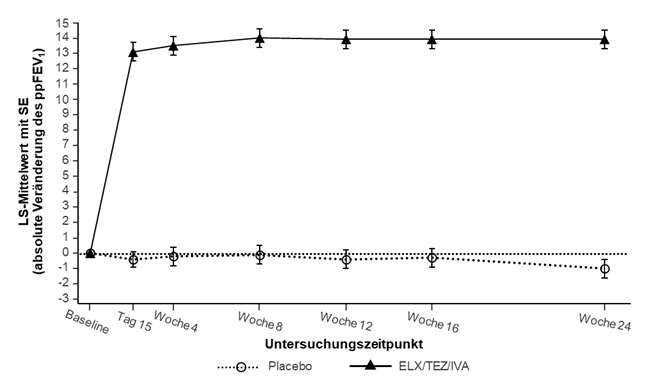
SE: Standardfehler; ELX/TEZ/IVA: Elexacaftor/Tezacaftor/Ivacaftor
Studie 445-103
In Studie 445-103 war der primäre Endpunkt die mittlere absolute Veränderung des ppFEV1 gegenüber Baseline in Woche 4 des doppelblinden Behandlungszeitraums. Die Behandlung mit Trikafta führte zu einer statistisch signifikanten Verbesserung des ppFEV1 von 10,0 Prozentpunkten im Vergleich zum Behandlungsschema mit Tezacaftor/Ivacaftor und Ivacaftor (Tezacaftor/Ivacaftor) (95 % KI: 7,4; 12,6; p<0,0001) (siehe Tabelle 10). Verbesserungen beim ppFEV1 wurden unabhängig von Alter, Geschlecht, Baseline-ppFEV1 und geographischer Region beobachtet. Eine Zusammenfassung der primären und wichtigsten sekundären Ergebnisse ist Tabelle 10 zu entnehmen.
Tabelle 10: Primäre und
wichtigste sekundäre Wirksamkeitsa
nalysen, vollständiges Analyseset
(Studie 445-103)
Analyse* Statistik Tezacaftor/ Ivacafto Trikafta N=55
r# N=52
Primäre Wirksamkeitsanalyse
Absolute Veränderung des ppFEV1 Behandlungsunterschi N.a. 10,0 (7,4; 12,6)
gegenüber Baseline in Woche 4 ed (95 % KI)
(Prozentpunkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe 0,4 (0,9) 10,4 (0,9)
(SE)
Wichtigste sekundäre Wirksamkeitsa
nalysen
Absolute Veränderung der Behandlungsunterschi N.a. -45,1 (-50,1; -40,1)
Schweisschloridkonzentration ed (95 % KI)
gegenüber Baseline in Woche 4
(mmol/l)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe 1,7 (1,8) -43,4 (1,7)
(SE)
Absolute Veränderung des Scores Behandlungsunterschi N.a. 17,4 (11,8; 23,0)
der respiratorischen Domäne des ed (95 % KI)
CFQ-R gegenüber Baseline in Woche
4 (Punkte)
p-Wert N.a. p<0,0001
Veränderung innerhalb der Gruppe -1,4 (2,0) 16,0 (2,0)
(SE)
ppFEV1: forciertes exspiratorische
s Volumen in 1 Sekunde in Prozent
des Sollwerts; KI: Konfidenzinterv
all; SE: Standardfehler; N.a.:
nicht anwendbar; CFQ-R: Cystic
Fibrosis Questionnaire-Revised,
überarbeiteter Fragebogen zu
zystischer Fibrose. * Die
Baseline für den primären und die
wichtigsten sekundären Endpunkte
ist definiert als das Ende der
4-wöchigen Einlaufphase mit
Tezacaftor/Ivacaftor und
Ivacaftor. # Behandlungsschema
von Tezacaftor/Ivacaftor und
Ivacaftor.
Abbildung 2: Absolute Veränderung des ppFEV1 gegenüber dem Ausgangswert bei jedem Untersuchungszeitpunkt in Studie 445-103
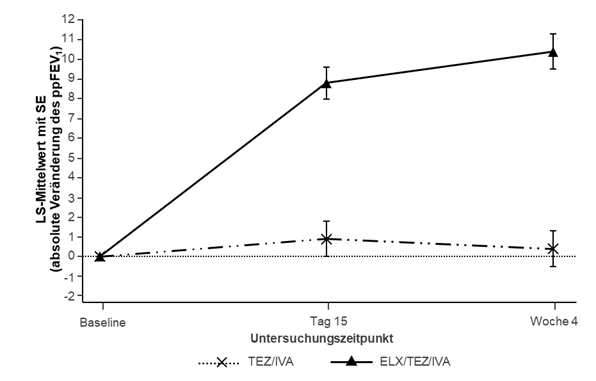
SE: Standardfehler; TEZ/IVA: Tezacaftor/Ivacaftor; ELX/TEZ/IVA: Elexacaftor/Tezacaftor/Ivacaftor
Studie 445-105
Die Studie 445-105 war eine unverblindete Verlängerungsstudie von 192 Wochen Dauer zur Bewertung der Sicherheit und Wirksamkeit einer Langzeitbehandlung mit Trikafta an Patienten, die aus den Studien 445-102 (N=399) und 445-103 (N=107) übernommen wurden. In dieser unverblindeten Verlängerungsstudie haben alle Patienten Trikafta über die gesamte Dauer der Studie erhalten. In Studie 445-105 zeigten Patienten aus den Kontrollarmen der Hauptstudien Verbesserungen der Wirksamkeitsendpunkte, die mit denen von Patienten übereinstimmten, die Trikafta in den Hauptstudien erhielten. Die Patienten aus dem Kontrollarm sowie die Patienten, die in den Hauptstudien Trikafta erhielten, zeigten eine anhaltende Verbesserung des ppFEV1 (siehe Abbildung 3 und Abbildung 4) und anderer Wirksamkeitsendpunkte (siehe Tabelle 11). Der BMI und BMI-z-Score nach 96 Wochen kumulativer Behandlung (Woche 96 in Studie 445-105) waren ähnlich, wie bei Patienten mit den in Studie 445-102 untersuchten Genotypen.
Abbildung 3: Absolute Veränderung des ppFEV1 gegenüber dem Ausgangswert bei jedem Untersuchungszeitpunkt in Studie 445-102 und in Studie 445-105 bei Patienten, die aus Studie 445-102 übernommen wurden

ppFEV1 = forciertes exspiratorisches Volumen in 1 Sekunde in Prozent des Sollwerts; LS-Mittelwert = Mittelwert nach der Methode der kleinsten Quadrate; SE = Standardfehler; OL = unverblindet
Abbildung 4: Absolute Veränderung des ppFEV1 gegenüber dem Ausgangswert bei jedem Untersuchungszeitpunkt in Studie 445-103 und in Studie 445-105 bei Patienten, die aus Studie 445-103 übernommen wurden

ppFEV1 = forciertes exspiratorisches Volumen in 1 Sekunde in Prozent des Sollwerts; LS-Mittelwert = Mittelwert nach der Methode der kleinsten Quadrate; SE = Standardfehler; OL = unverblindet
Tabelle 11: Studie
445-105 sekundäre
unverblindete
Wirksamkeitsanalyse,
vollständiges
Analyseset (F/MF
und F/F Patienten)
Analyse Statistik Studie 445-105
Woche 192
PBO in 445-102 N = ELX/TEZ/IVA in TEZ/IVA in 445-103 ELX/TEZ/IVA in
203 445-102 N = 196 N = 52 445-103 N = 55
Absolute Veränderung n 136 133 32 36
des ppFEV1 gegenübe
r dem Ausgangswert*
(Prozentpunkte)
LS-Mittelwert 15,3 13,8 10,9 10,7
95 %-KI (13,7; 16,8) (12,3; 15,4) (8,2; 13,6) (8,1; 13,3)
Absolute Veränderung n 133 128 31 38
der Schweisschlorid
-konzentration
gegenüber dem
Ausgangswert*
(mmol/l)
LS-Mittelwert -47,0 -45,3 -48,2 -48,2
95 %-KI (-50,1; -43,9) (-48,5; -42,2) (-55,8; -40,7) (-55,1; -41,3)
Anzahl PEx während Anzahl der Ereigniss 385 71
des kumulativen e
TC-Wirksamkeits-zeit
raums†
Geschätzte Ereignisr 0,21 (0,17; 0,25) 0,18 (0,12; 0,25)
ate pro Jahr (95
%-KI)
ppFEV1 = forciertes
exspiratorisches
Volumen in 1 Sekunde
in Prozent des
Sollwerts; SwCl =
Schweisschloridkonze
ntration; PEx =
pulmonale Exazerbati
on; LS-Mittelwert =
Mittelwert nach der
Methode der kleinste
n Quadrate; KI =
Konfidenzintervall;
PBO = Placebo; TC =
Dreifachkombination
* Ausgangswert =
Ausgangswert der
Hauptstudie † Bei
Patienten, die auf
die ELX/TEZ/IVA-Grup
pe randomisiert
wurden, umfasst der
kumulative TC-Wirksa
mkeitszeitraum
Daten aus den
Hauptstudien bis
192 Behandlungswoche
n in Studie 445-105
(N=255, einschliessl
ich 4 Patienten,
die nicht in 445-105
übernommen wurden).
Bei Patienten, die
auf die Placebo-
oder TEZ/IVA-Gruppe
randomisiert wurden,
umfasst der kumulat
ive TC-Wirksamkeitsz
eitraum nur Daten
für einen Behandlung
szeitraum von 192
Wochen in Studie
445-105 (N=255).
Studie 445-104
Im Anschluss an eine 4wöchige Einlaufphase mit Ivacaftor oder Tezacaftor/Ivacaftor zeigte der primäre Endpunkt der mittleren absoluten Veränderung des ppFEV1-Werts innerhalb der Behandlungsgruppe von Baseline bis einschliesslich Woche 8 bei der Trikafta-Gruppe eine statistisch signifikante Verbesserung um 3,7 Prozentpunkte (95 % KI: 2,8; 4,6; p<0,0001) (siehe Tabelle 12). Die mittlere Verbesserung des ppFEV1 wurde bei der ersten Untersuchung an Tag 15 festgestellt. Die Gesamtverbesserungen des ppFEV1 waren unabhängig von Alter, Geschlecht, ppFEV1-Ausgangswert, geographischer Region und Genotyp-Gruppe (F/G oder F/RF) zu beobachten.
Eine Zusammenfassung der primären und sekundären Ergebnisse für die gesamte Studienpopulation ist Tabelle 12 zu entnehmen.
Tabelle 12: Primäre und sekundäre
Wirksamkeitsanalysen,
vollständiges Analyseset (Studie
445-104)
Analyse* Statistik Kontrollgruppe# Trikafta N=132
N=126
Primäre Analyse
Absolute Veränderung des ppFEV1 Veränderung innerhal 0,2 (-0,7; 1,1) 3,7 (2,8; 4,6)
gegenüber dem Ausgangswert bis b der Gruppe (95 %
einschliesslich Woche 8 KI)
(Prozentpunkte)
p-Wert N.a. p<0,0001
Wichtigste und sonstige sekundäre
Analysen
Absolute Veränderung der Veränderung innerhal 0,7 (-1,4; 2,8) -22,3 (-24,5; -20,2)
Schweisschloridkonzentration b der Gruppe (95 %
gegenüber dem Ausgangswert bis KI)
einschliesslich Woche 8 (mmol/l)
p-Wert N.a. p<0,0001
Absolute Veränderung des ppFEV1 Behandlungsunterschi N.a. 3,5 (2,2; 4,7)
gegenüber dem Ausgangswert bis ed (95 % KI)
einschliesslich Woche 8 im
Vergleich zur Kontrollgruppe
(Prozentpunkte)
p-Wert N.a. p<0,0001
Absolute Veränderung der Behandlungsunterschi N.a. -23,1 (-26,1; -20,1)
Schweisschloridkonzentration ed (95 % KI)
gegenüber dem Ausgangswert bis
einschliesslich Woche 8 im
Vergleich zur Kontrollgruppe
(mmol/l)
p-Wert N.a. p<0,0001
Absolute Veränderung des Scores Veränderung innerhal 1,6 (-0,8; 4,1) 10,3 (8,0; 12,7)
der respiratorischen Domäne des b der Gruppe (95 %
CFQ-R gegenüber dem Ausgangswert KI)
bis einschliesslich Woche 8
(Punkte)±
Absolute Veränderung des Scores Behandlungsunterschi N.a. 8,7 (5,3; 12,1)
der respiratorischen Domäne des ed (95 % KI)
CFQ-R gegenüber dem Ausgangswert
bis einschliesslich Woche 8 im
Vergleich zur Kontrollgruppe
(Punkte)±
ppFEV1: forciertes exspiratorische
s Volumen in 1 Sekunde in Prozent
des Sollwerts; KI: Konfidenzinterv
all; N.a.: nicht anwendbar;
CFQ-R: Cystic Fibrosis
Questionnaire-Revised,
überarbeiteter Fragebogen zu
zystischer Fibrose. * Die
Baseline für die primären und
sekundären Endpunkte ist
definiert als das Ende der
4-wöchigen Einlaufphase mit
Ivacaftor oder Tezacaftor/Ivacafto
r. # Ivacaftor-Gruppe oder
Tezacaftor/Ivacaftor-Gruppe. ±
Die CFQ-R-Ergebnisse wurden nicht
nach dem hierarchischen Verfahren
für multiples Testen auf
Multiplizität kontrolliert.
Studie 445-106 Teil B
In Studie 445-106 Teil B wurde der primäre Endpunkt Sicherheit und Verträglichkeit über 24 Wochen bewertet. Sekundäre Endpunkte waren die Bewertung der Wirksamkeit und Pharmakokinetik einschliesslich der absoluten Veränderung des ppFEV1-Werts (1. sekundärer Endpunkt) und der Schweisschloridkonzentration (2. sekundärer Endpunkt, siehe "Pharmakodynamik" ) gegenüber Baseline in Woche 24 und die Anzahl der pulmonalen Exazerbationen von Baseline bis einschliesslich Woche 24. Aufgrund der Durchführung der Studie 445-106 Teil B während der COVID-19 Pandemie konnten nicht alle Messungen wie ursprünglich geplant durchgeführt werden. Die Messungen der sekundären Endpunkte waren in unterschiedlichem Ausmass von nicht durchgeführten Messungen betroffen. Tabelle 13 zeigt die wichtigsten sekundären Wirksamkeitsergebnisse in der Gesamtanalyse über 24 Wochen.
Die Messwerte für die ppFEV1-Werte wurden an den geplanten Messtagen bei folgender Anzahl Patienten erhoben: Baseline n=62, Tag 15 n=51, Woche 4 n=52, Woche 8 n=51, Woche 12 n=43, Woche 16 n=29, Woche 24 n=15.
Die Messwerte für die Schweisschloridkonzentration wurden an den geplanten Messtagen bei folgender Anzahl Patienten erhoben: Baseline n=62, Tag 15 n=56, Woche 4 n=56, Woche 12 n=50, Woche 24 n=28.
Tabelle 13: Sekundäre Wirksamkeitsanalysen, vollständiges Analyseset über 24
Wochen (Studie 445-106, Teil B)
Analyse Veränderung innerhal
b der Gruppe (95 %
KI) für Trikafta
N=66
Absolute Veränderung des ppFEV1-Werts von Baseline bis einschliesslich Woche 10,2 (7,9; 12,6)
24 (Prozentpunkte)
Absolute Veränderung der Schweisschloridkonzentration von Baseline bis -60,9 (-63,7; -58,2)
einschliesslich Woche 24 (mmol/l)
Anzahl der pulmonalen Exazerbationen bis einschliesslich Woche 24‡ 4 (0,12) ††
KI: Konfidenzintervall; ppFEV1: forciertes exspiratorisches Volumen in 1
Sekunde in Prozent des Sollwerts. ‡ Eine pulmonale Exazerbation war definiert
als eine Veränderung der antibiotischen Therapie (i.v., inhalativ oder oral)
aufgrund von mindestens 4 von 12 im Voraus festgelegten sinopulmonalen
Zeichen/Symptomen. †† Anzahl von Ereignissen und geschätzte Ereignisrate pro
Jahr, berechnet anhand von 48 Wochen pro Jahr.
Studie 445-107
Eine 192-wöchige, zweiteilige (Teil A und Teil B), offene Verlängerungsstudie zur Untersuchung der Sicherheit und Wirksamkeit einer Langzeitbehandlung mit Trikafta wurde bei Patienten durchgeführt, welche die Studie 445-106 abgeschlossen hatten. Die Analyse von Teil A (96 Wochen) wurde bei 64 pädiatrischen Patienten im Alter von 6 Jahren und älter durchgeführt und zeigte anhaltende Verbesserungen des ppFEV1-Werts und der Schweisschloridkonzentration, die mit den in der Studie 445-106 beobachteten Ergebnissen übereinstimmen. Anschliessend wurden 48 (75,0 %) Probanden in Teil B der Studie bis zur Woche 192 übernommen. 39 (60,9 %) Probanden schlossen Teil B ab und bestätigten die weiter anhaltende Wirksamkeit. Die sekundären Wirksamkeitsendpunkte der Interims- und der finalen Analyse sind in Tabelle 14 zusammengefasst.
Tabelle 14: Sekundäre
Wirksamkeitsanalyse, vollständiges
Analyseset (N=64) (Studie
445-107)
Analyse Statistik Absolute Veränderung Absolute Veränderung
gegenüber Baseline* gegenüber Baseline*
in Woche 96 in Woche 192
ppFEV1 (Prozentpunkte) n 45 27
LS-Mittelwert 11,2 9,6
95 %-KI (8,3; 14,2) (5,4; 13,7)
Schweisschloridkonzentration n 56 35
(mmol/l)
LS-Mittelwert -62,3 -57,9
95 %-KI (-65,9; -58,8) (-63,3; -52,5)
PEx während des kumulativen Anzahl der Ereigniss 7 11
TC-Wirksamkeitszeitraums† e
Beobachtete Ereignisrate pro Jahr 0,04 0,045
ppFEV1 = forciertes exspiratorisch
es Volumen in 1 Sekunde in
Prozent des Sollwerts; PEx =
pulmonale Exazerbation;
LS-Mittelwert = Mittelwert nach
der Methode der kleinsten
Quadrate; KI = Konfidenzintervall;
TC = Dreifachkombination *
Ausgangswert = Ausgangswert der
Hauptstudie † Der kumulative
TC-Wirksamkeitszeitraum umfasst
die Daten der 66 Patienten, die
in die Hauptstudie (Studie
445-106 Teil B) aufgenommen
wurden und mindestens eine
Behandlungsdosis erhielten
und/oder während der Studie
445-107 mindestens eine Dosis
erhielten.
Studie 445-111
Das pharmakokinetische Profil, die Sicherheit und Wirksamkeit von Trikafta bei CF-Patienten im Alter von 2 bis unter 6 Jahren werden durch Nachweise aus Studien mit Trikafta an Patienten im Alter ab 12 Jahren (Studien 445-102, 445-103 und 445-104) unterstützt. Zusätzliche Daten stammen aus einer 24-wöchigen, offenen Phase-3-Studie mit 75 Patienten im Alter von 2 bis unter 6 Jahren (Studie 445-111).
In Studie 445-111 wurde der primäre Endpunkt Sicherheit und Verträglichkeit über 24 Wochen bewertet. Sekundäre Endpunkte waren die Bewertung der Pharmakokinetik und die Wirksamkeitsendpunkte der absoluten Veränderung der Schweisschloridkonzentration (siehe "Pharmakodynamik" ) und des LCI2.5 von der Baseline bis einschliesslich Woche 24. Tabelle 15 enthält eine Zusammenfassung der sekundären Wirksamkeitsergebnisse.
Tabelle 15: Sekundäre Wirksamkeitsanalysen,
vollständiges Analyseset (Studie 445-111)
Analyse Statistik Veränderung innerhal
b der Gruppe (95 %
KI) für Trikafta
Absolute Veränderung der Schweisschloridkonzentration N* LS-Mittelwert 75 -57,9 (-61,3;
von Baseline bis einschliesslich Woche 24 (mmol/l) (95 % KI) -54,6)
Absolute Veränderung des LCI2.5 von Baseline bis N LS-Mittelwert (95 63‡ -0,83 (-1,01;
einschliesslich Woche 24 % KI) -0,66)
Anzahl der pulmonalen Exazerbationen bis N Anzahl der Ereigni 75 12 (0,32) ††
einschliesslich Woche 24** sse (geschätzte
Ereignisrate pro
Jahr)
KI: Konfidenzintervall; LCI: Lung Clearance Index. * N
ist die Anzahl der Patienten im entsprechenden
vollständigen Analyseset ‡ Der LCI wurde nur bei
Patienten bewertet, die beim Screening 3 Jahre oder
älter waren ** Altersspezifische Definitionen von PEx
werden für Patienten von 2 bis einschliesslich 5 Jahren
und ab 6 Jahren verwendet. †† Anzahl der Ereignisse und
geschätzte Ereignisrate pro Jahr auf der Grundlage von
48 Wochen pro Jahr.
Studie 445-124
Die Wirksamkeit und Sicherheit von Trikafta wurde bei Patienten ab 6 Jahren mit CF ohne F508del-Mutation untersucht (Studie 445-124).
In Studie 445-124 war der primäre Endpunkt für die Wirksamkeit das ppFEV1. Sekundäre Endpunkte waren die absolute Veränderung der Schweisschloridkonzentration, der Score der respiratorischen Domäne des CFQ-R, Wachstumsparameter (BMI, Körpergewicht) und die Anzahl von PEx. Tabelle 16 enthält eine Zusammenfassung der primären und sekundären Wirksamkeitsendpunkte.
Tabelle 16: Primäre und sekundäre
Wirksamkeitsanalysen,
vollständiges Analyseset (Studie
445-124)
Analyse Statistik Placebo N = 102 ELX/TEZ/IVA N = 205
Primäre Analyse
Absolute Veränderung des ppFEV1 Behandlungsunterschi NA 9,2 (7,2; 11,3)
gegenüber Baseline bis ed (95 %-KI)
einschliesslich Woche 24
(Prozentpunkte)
p-Wert NA p<0,0001
Veränderung innerhalb der Gruppe -0,4 (0,8) 8,9 (0,6)
(SE)
Sekundäre Analyse
Absolute Veränderung der Behandlungsunterschi NA -28,3 (-32,1, -24,5)
Schweisschloridkonzentration ed (95 %-KI)
gegenüber Baseline bis
einschliesslich Woche 24 (mmol/l)
p-Wert NA p<0,0001
Veränderung innerhalb der Gruppe 0,5 (1,6) -27,8 (1,1)
(SE)
Absolute Veränderung des Scores Behandlungsunterschi NA 19,5 (15,5; 23,5)
der respiratorischen Domäne des ed (95 %-KI)
CFQ-R von Baseline bis
einschliesslich Woche 24 (Punkte)
p-Wert NA p<0,0001
Veränderung innerhalb der Gruppe -2,0 (1,6) 17,5 (1,2)
(SE)
Absolute Veränderung des BMI Behandlungsunterschi NA 0,47 (0,24; 0,69)
gegenüber Baseline in Woche 24 ed (95 %-KI)
(kg/m2)
p-Wert NA p<0,0001
Veränderung innerhalb der Gruppe 0,35 (0,09) 0,81 (0,07)
(SE)
Absolute Veränderung des Behandlungsunterschi NA 1,3 (0,6; 1,9)
Körpergewichts gegenüber Baseline ed (95 %-KI)
in Woche 24 (kg)
p-Wert NA p<0,0001
Veränderung innerhalb der Gruppe 1,2 (0,3) 2,4 (0,2)
(SE)
Anzahl der PEx bis einschliesslich Rate Ratio (95 %-KI) NA 0,28 (0,15; 0,51)
Woche 24
p-Wert NA p<0,0001
Anzahl der Ereignisse 40 21
Geschätzte Rate pro Jahr 0,63 0,17
BMI: Body Mass Index (Körpermassen
index); CFQ-R RD: Cystic Fibrosis
Questionnaire-Revised Respiratory
Domain (überarbeiteter Fragebogen
zu zystischer Fibrose,
respiratorische Domäne); ELX:
Elexacaftor; IVA: Ivacaftor; N:
Grösse der Gesamtstichprobe; p:
Wahrscheinlichkeit; PEx:
pulmonale Exazerbation; ppFEV1:
forciertes exspiratorisches
Volumen in 1 Sekunde in Prozent
des Sollwerts; SE: Standardfehler;
TEZ: Tezacaftor.
Studie CFD-016
Die Wirksamkeit von Trikafta bei CF-Patienten im Alter ab 6 Jahren wurde auch in einer retrospektiven Studie zur Bewertung der klinischen Ergebnisse anhand von Real World Data bei CF-Patienten ohne F508del-Mutation untersucht. Dazu wurden Daten des Patientenregisters der US Cystic Fibrosis Foundation herangezogen. Der primäre Wirksamkeitsendpunkt in Studie CFD-016 war das ppFEV1. Die mittlere Veränderung des ppFEV1 gegenüber dem Ausgangswert betrug 4,53 Prozentpunkte (n = 422; 95 %-KI: 3,50; 5,56).
PharmakokinetikDie Pharmakokinetik von Elexacaftor, Tezacaftor und Ivacaftor ist bei gesunden erwachsenen Probanden und CF-Patienten vergleichbar. Die pharmakokinetischen Parameter von Elexacaftor, Tezacaftor und Ivacaftor bei CF-Patienten ab 12 Jahren sind in Tabelle 17 gezeigt.
Tabelle 17: Pharmakokinetische
Parameter der Komponenten von
Trikafta
Elexacaftor Tezacaftor Ivacaftor
Allgemeine Angaben
AUC (SD), μg-h/mla 162 (47,5)b 89,3 (23,2)b 11,7 (4,01)c
Cmax, (SD), μg/mla 9,2 (2,1) 7,7 (1,7) 1,2 (0,3)
Zeit bis zum Steady State, Tage Innerhalb von 7 Innerhalb von 8 Innerhalb von 3-5
Tagen Tagen Tagen
Akkumulationsverhältnis 2,2 2,07 2,4
Absorption
Absolute Bioverfügbarkeit 80 % Nicht bestimmt Nicht bestimmt
Mediane Tmax (Bereich), Stunden 6 (4 bis 12) 3 (2 bis 4) 4 (3 bis 6)
Einfluss von Nahrung AUC erhöht sich auf Keine klinisch Exposition erhöht
das 1,9- bis 2,5-Fac signifikante Wirkung sich auf das 2,5-
he (mässig fetthalti bis 4-Fache
ge Mahlzeit)
Distribution
Mittleres (SD) scheinbares 53,7 (17,7) 82,0 (22,3) 293 (89,8)
Verteilungsvolumen, ld
Proteinbindunge > 99 % ungefähr 99 % ungefähr 99 %
Elimination
Mittlere (SD) effektive 27,4 (9,31) 25,1 (4,93) 15,0 (3,92)
Halbwertszeit, Stundenf
Mittlere (SD) scheinbare 1,18 (0,29) 0,79 (0,10) 10,2 (3,13)
Clearance, l/Stunden
Metabolismus
Primärer Stoffwechselweg CYP3A4/5 CYP3A4/5 CYP3A4/5
Aktive Metabolite M23-ELX M1-TEZ M1-IVA
Wirkstärke der Metabolite im Vergleichbar Vergleichbar Etwa 1/6 der Mutters
Vergleich zur Muttersubstanz ubstanz
Ausscheidungg
Primärer Ausscheidungsweg • Fäzes: 87,3 % • Fäzes: 72 % • Fäzes: 87,8 % •
(vorwiegend als (unverändert oder Urin: 6,6 %
Metabolite) • Urin: als M2-TEZ) • Urin:
0,23 % 14 % (0,79 % unverän
dert)
a Ausgehend von Elexacaftor 200
mg und Tezacaftor 100 mg einmal
täglich/Ivacaftor 150 mg alle 12
Stunden im Steady State bei
Patienten mit CF ab 12 Jahren. b
AUC0-24h. c AUC0-12h. d
Elexacaftor, Tezacaftor und
Ivacaftor gehen nicht vorrangig
in rote Blutkörperchen des
Menschen über. e Elexacaftor und
Tezacaftor binden sich vorwiegend
an Albumin. Ivacaftor bindet sich
vorwiegend an Albumin, alpha
1-saures Glycoprotein und an
humanes Gamma-Globulin. f Die
mittleren (SD) terminalen
Halbwertszeiten von Elexacaftor,
Tezacaftor und Ivacaftor betragen
etwa 24,7 (4,87) Stunden, 60,3
(15,7) Stunden bzw. 13,1 (2,98)
Stunden. g Nach radioaktiven
Dosen.
Absorption
Siehe Tabelle 17 "Pharmakokinetische Parameter der Komponenten von Trikafta" .
Distribution
Siehe Tabelle 17 "Pharmakokinetische Parameter der Komponenten von Trikafta" .
Metabolismus
Siehe Tabelle 17 "Pharmakokinetische Parameter der Komponenten von Trikafta" .
Elimination
Siehe Tabelle 17 "Pharmakokinetische Parameter der Komponenten von Trikafta" .
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Elexacaftor allein oder in Kombination mit Tezacaftor und Ivacaftor wurde nicht bei Patienten mit schweren Leberfunktionsstörungen (Child-Pugh-Klasse C, Score 10-15) untersucht. Nach mehreren Dosen von Elexacaftor, Tezacaftor und Ivacaftor über 10 Tage zeigten Patienten mit mässig eingeschränkter Leberfunktion (Child-Pugh-Klasse B, Score 7 bis 9) eine um 25 % höhere AUC und eine um 12 % höhere Cmax für Elexacaftor, eine um 73 % höhere AUC und eine um 70 % höhere Cmax für M23-Elexacaftor, eine um 36 % höhere AUC und eine um 24 % höhere Cmax für die Kombination Elexacaftor und M23-Elexacaftor, eine um 20 % höhere AUC, aber eine vergleichbare Cmax für Tezacaftor sowie eine um 50 % höhere AUC und eine um 10 % höhere Cmax für Ivacaftor im Vergleich zu gesunden Probanden mit vergleichbaren demographischen Merkmalen (siehe" Dosierung/Anwendung" , "Warnhinweise und Vorsichtsmassnahmen" und "Unerwünschte Wirkungen" ).
Tezacaftor und Ivacaftor
Nach wiederholter Gabe von Tezacaftor und Ivacaftor über 10 Tage zeigten Patienten mit mässig eingeschränkter Leberfunktion eine Zunahme der AUC von Tezacaftor um ca. 36 % und eine Zunahme der Cmax von Tezacaftor um 10 % sowie eine Zunahme der AUC von Ivacaftor auf das 1,5-Fache, aber eine vergleichbare Cmax von Ivacaftor, im Vergleich zu gesunden Probanden mit merkmalsgleichen demographischen Daten.
Ivacaftor
In einer Studie mit Ivacaftor allein zeigten Patienten mit mässig eingeschränkter Leberfunktion eine vergleichbare Cmax von Ivacaftor, aber eine um das etwa 2,0-Fache höhere AUC0-∞ von Ivacaftor im Vergleich zu gesunden Probanden mit merkmalsgleichen demographischen Daten.
Nierenfunktionsstörungen
Elexacaftor allein oder in Kombination mit Tezacaftor und Ivacaftor wurde bei Patienten mit stark eingeschränkter Nierenfunktion (eGFR weniger als 30 ml/min/1,73 m2) oder bei Patienten mit terminaler Niereninsuffizienz nicht untersucht.
In humanpharmakokinetischen Studien mit Elexacaftor, Tezacaftor und Ivacaftor wurde eine minimale Elimination von Elexacaftor, Tezacaftor und Ivacaftor mit dem Urin festgestellt (lediglich 0,23 %, 13,7 % [davon 0,79 % als unveränderte Muttersubstanz] resp. 6,6 % der Gesamtradioaktivität).
Auf der Grundlage einer populationspharmakokinetischen (PK) Analyse wurde festgestellt, dass die Bioverfügbarkeit von Elexacaftor bei Patienten mit leicht eingeschränkter Nierenfunktion (N=75; eGFR 60 bis weniger als 90 ml/min/1,73 m2) mit der von Patienten mit normaler Nierenfunktion (N=341; eGFR 90 ml/min/1,73 m2 oder mehr) vergleichbar war.
In einer populationspharmakokinetischen Analyse zeigten Daten von 817 Patienten, die in klinischen Studien der Phase 2 oder 3 mit Tezacaftor allein oder in Kombination mit Ivacaftor behandelt wurden, dass eine leicht eingeschränkte Nierenfunktion (N=172; eGFR 60 bis weniger als 90 ml/min/1,73 m2) und eine mässig eingeschränkte Nierenfunktion (N=8; eGFR 30 bis weniger als 60 ml/min/1,73 m2) keinen bedeutsamen Einfluss auf die Clearance von Tezacaftor haben (siehe "Dosierung/Anwendung" ).
Geschlecht
Auf der Grundlage einer populationspharmakokinetischen Analyse wurde festgestellt, dass die Bioverfügbarkeit von Elexacaftor, Tezacaftor und Ivacaftor bei Männern und Frauen vergleichbar ist.
Pädiatrische Patienten im Alter von 2 Jahren bis unter 18 Jahren
Die Bioverfügbarkeit von Elexacaftor, Tezacaftor und Ivacaftor, und die Bioverfügbarkeit der Metaboliten M1-Tezacaftor und M23-Elexacaftor, die in den Phase-3-Studien mithilfe populationspharmakokinetischer Analysen festgestellt wurde, ist in Tabelle 18 nach Altersgruppen und angewendeter Dosis aufgeführt. Die Bioverfügbarkeit von Elexacaftor, Tezacaftor und Ivacaftor bei Patienten im Alter von 6 bis unter 18 Jahren liegt in dem bei Patienten ab 18 Jahren beobachteten Bereich.
Tabelle 18. Mittlere
(SD) AUC0-24h,ss
von Elexacaftor,
Tezacaftor und
Ivacaftor nach
Altersgruppe
Alters-/ Gewichtsgru Dosis Elexacaftor AUC0-24h M23-Elexacaftor Tezacaftor AUC0-24h, M1-Tezacaftor Ivacaftor AUC0-12h,s
ppe ,ss (μg-h/ml) AUC0-24h,ss (μg-h/ml ss (μg-h/ml) AUC0-24h,ss (μg-h/ml s (μg-h/ml)
) )
Patienten im Alter Elexacaftor 80 mg 128 (24,8) 56,5 (29,4) 87,3 (17,3) 194 (24,8) 11,9 (3,86)
von 2 bis < 6 tgl./Tezacaftor 40
Jahren, < 14 kg mg tgl./Ivacaftor
(N=16) 60 mg morgens und
Ivacaftor 59,5 mg
abends
Patienten im Alter Elexacaftor 100 mg 138 (47,0) 59,0 (32,7) 90,2 (27,9) 197 (43,2) 13,0 (6,11)
von 2 bis < 6 tgl./Tezacaftor 50
Jahren, ≥14 kg mg tgl./Ivacaftor
(N=59) 75 mg alle 12 Std.
Patienten im Alter Elexacaftor 100 mg 116 (39,4) 45,4 (25,2) 67,0 (22,3) 153 (36,5) 9,78 (4,50)
von 6 Jahren bis 1 × tgl./ Tezacaftor
<12 Jahren, <30 kg 50 mg 1 × tgl./Ivac
(N=36) aftor 75 mg alle 12
Std.
Patienten im Alter Elexacaftor 200 mg 195 (59,4) 104 (52) 103 (23,7) 220 (37,5) 17,5 (4,97)
von 6 Jahren bis 1 × tgl./ Tezacaftor
<12 Jahren, ≥30 kg 100 mg 1 × tgl./
(N=30) Ivacaftor 150 mg
alle 12 Std.
Jugendliche Patiente Elexacaftor 200 mg 147 (36,8) 58,5 (25,6) 88,8 (21,8) 148 (333) 10,6 (3,35)
n im Alter von 12 1 × tgl./ Tezacaftor
bis < 18 Jahren 100 mg 1 × tgl./
(N=72) Ivacaftor 150 mg
alle 12 Std.
Erwachsene Patienten Elexacaftor 200 mg 168 (49,9) 64,6 (28,9) 89,5 (23,7) 128 (33,7) 12,1 (4,17)
im Alter von ≥18 1 × tgl./ Tezacaftor
Jahren (N=179) 100 mg 1 × tgl./
Ivacaftor 150 mg
alle 12 Std.
SD: Standardabweichu
ng; AUCss: Fläche
unter der Konzentrat
ions-Zeit-Kurve im
Steady State.
Präklinische DatenElexacaftor/Tezacaftor/Ivacaftor
Studien zur Toxizität nach mehrmaliger Gabe der Wirkstoffkombination bei Ratten und Hunden, in denen Elexacaftor, Tezacaftor und Ivacaftor in Kombination angewendet wurden, um das Potenzial für eine additive und/oder synergistische Toxizität zu bewerten, ergaben keine unerwarteten Toxizitäten oder Interaktionen. Mit Trikafta wurden keine Studien zur Sicherheitspharmakologie, Gentoxizität, Kanzerogenität und Reproduktionstoxizität durchgeführt. Hingegen sind Studien mit den Einzelsubstanzen vorhanden.
Elexacaftor
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Gentoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Toxizität nach wiederholter Verabreichung
In der 6-monatigen Toxizitätsstudie an Ratten waren die primären Zielorgane der Drüsenmagen (Erosion), Hoden und Nebenhoden (Degeneration/Atrophie der Samenkanälchen, Oligospermie/Aspermie) sowie Knochenmark (verminderte hämatopoietische Zellularität). Diese Effekte wurden hauptsächlich unter nicht verträglichen Dosen von ≥40 mg/kg/Tag bei männlichen Tieren bzw. 30 mg/kg/Tag bei weiblichen Tieren beobachtet. Die Plasmaexposition (AUC) der Tiere beim NOAEL (15 mg/kg/Tag) war ca. das 3-Fache (Männchen) bzw. 11-Fache (Weibchen) der für den Menschen empfohlenen Höchstdosis [maximum recommended human dose, MRHD]. In der 9-monatigen Studie zur Toxizität an Hunden lag bei Rüden, denen Elexacaftor in einer Dosis von 14 mg/kg/Tag gegeben wurde (das 15-Fache der MRHD basierend auf den aufsummierten AUCs von Elexacaftor und seinem Metaboliten), eine minimale oder leichte, nicht als schädlich eingestufte, bilaterale Degeneration/Atrophie der Samenkanälchen im Hoden vor, die während der begrenzten Erholungsphase nicht abklang. Sie blieb jedoch ohne weitere Folgeerscheinungen. Die Relevanz dieser Befunde für den Menschen ist nicht bekannt.
Reproduktionstoxizität
Elexacaftor war mit einer geringeren männlichen und weiblichen Fertilität, einer geringeren Paarung bei den männlichen Tieren und geringeren weiblichen Konzeptionsindizes bei einer Dosis von 75 mg/kg/Tag (dem 6-Fachen der MRHD basierend auf den aufsummierten AUCs von Elexacaftor und seinem Metaboliten) bei den männlichen Tieren und bei einer Dosis von 35 mg/kg/Tag (dem 7-Fachen der MRHD basierend auf den aufsummierten AUCs von Elexacaftor und seinem Metaboliten) bei den weiblichen Tieren verbunden.
Elexacaftor war bei Ratten bei einer Dosis von 40 mg/kg/Tag und bei Kaninchen bei einer Dosis von 125 mg/kg/Tag (dem etwa 9- resp. 4-Fachen der MRHD basierend auf den aufsummierten AUCs von Elexacaftor und seinem Metaboliten [bei der Ratte] und der AUC von Elexacaftor [beim Kaninchen]) nicht teratogen. Bei Rattenfeten trat nach Behandlung der Muttertiere mit ≥25 mg/kg/Tag (das ungefähr 4-Fache der MRHD basierend auf AUC) ein geringeres mittleres Körpergewicht auf. In der Studie zur prä- und postnatalen Entwicklung bei der Ratte mit Dosen bis 10 mg/kg/Tag (etwa das 1-Fache der MRHD basierend auf den aufsummierten AUCs von Elexacaftor und seinem Metaboliten) wurden keine unerwünschten Wirkungen festgestellt. Bei trächtigen Ratten wurde ein Übertritt von Elexacaftor in die Plazenta beobachtet.
Juvenile Toxizität
Bei juvenilen Ratten, die ab dem 7. bis zum 70. Tag nach der Geburt mit Dosen behandelt wurden, die zu einer Plasmaexposition von ca. dem 3-Fachen (Männchen) bzw. 5-Fachen (Weibchen) der AUC in pädiatrischen Patienten (ab 12 Jahren) führten, wurden keine unerwünschten Wirkungen festgestellt.
Tezacaftor
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Gentoxizität, zum kanzerogenen Potential und zur Toxizität bei wiederholter Gabe lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Juvenile Toxizität
Studien bei Ratten, die vom 7. bis zum 35. Tag nach der Geburt (postnatal day, PND 7-35) exponiert wurden, zeigten auch bei niedrigen Dosen Mortalität bzw. moribunde Zustände. Die Befunde waren dosisabhängig und im Allgemeinen schwerwiegender, wenn die Gabe von Tezacaftor zu einem früheren Zeitpunkt nach der Geburt begonnen wurde. Bei der Exposition bei Ratten ab PND 21-49 zeigte sich auch bei der höchsten Dosis, die etwa dem Zweifachen der für den Menschen vorgesehenen Dosis entsprach, keine Toxizität. Tezacaftor und sein Metabolit M1-TEZ sind Substrate für P-Glykoprotein. Eine geringere P-Glykoprotein-Aktivität im Gehirn bei jüngeren Ratten führte zu höheren Spiegeln von Tezacaftor und M1-TEZ im Gehirn. Diese Ergebnisse sind für die Anwendung in der pädiatrischen Population im Alter ab 2 Jahren wahrscheinlich nicht relevant, weil ihre P-Glykoprotein-Aktivität der bei Erwachsenen beobachteten entspricht.
Reproduktionstoxizität
Tezacaftor verursachte keine Toxizität im Fortpflanzungssystem von männlichen und weiblichen Ratten bei Anwendung von 100 mg/kg/Tag, der höchsten untersuchten Dosis (etwa das 3-Fache der MRHD basierend auf den aufsummierten AUCs von Tezacaftor und M1-TEZ).
Tezacaftor hatte keine Auswirkungen auf die Fertilitäts- und Reproduktionsleistungsindizes von männlichen und weiblichen Ratten in Dosen von bis zu 100 mg/kg/Tag (etwa dem 3-Fachen der MRHD basierend auf den aufsummierten AUCs von Tezacaftor und M1-TEZ).
Tezacaftor erwies sich bei trächtigen Ratten und Kaninchen in Dosen von etwa dem 3-Fachen bzw. 0,2-Fachen der Tezacaftor-Exposition beim Menschen nach Gabe einer therapeutischen Dosis nicht als teratogen.
In einer prä- und postnatalen Entwicklungsstudie verursachte Tezacaftor keine Entwicklungsstörungen bei den Nachkommen trächtiger Ratten, die orale Dosen von 25 mg/kg/Tag (etwa das 1-Fache der MRHD basierend auf den aufsummierten AUCs für Tezacaftor und M1-TEZ) erhalten hatten. Nach Dosen, die für die Muttertiere toxisch waren (≥50 mg/kg/Tag), führte Tezacaftor zu niedrigeren fetalen Geburtsgewichten, einem niedrigeren Fertilitätsindex und Wirkungen auf die Östrus-Zyklizität (längere Zyklusdauer und Abnahme der Zykluszahl). Nach der höchsten Dosis (100 mg/kg/Tag) traten durch Tezacaftor bedingte Wirkungen bei den Nachkommen auf, wie z.B. schlechtes Überleben der Jungtiere bis zur Entwöhnung, Auswirkungen auf die Entwicklung vor der Entwöhnung und Verzögerungen bei Geschlechtsreife. Bei trächtigen Ratten wurde der Übertritt von Tezacaftor in die Plazenta beobachtet.
Ivacaftor
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Gentoxizität, zum kanzerogenen Potential und zur Toxizität bei wiederholter Gabe lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Reproduktionstoxizität
Ivacaftor beeinträchtigte die Fertilitäts- und Reproduktionsleistungsindizes von männlichen und weiblichen Ratten in Dosen von 200 mg/kg/Tag (dem etwa 7- bzw. 5-Fachen der MRHD basierend auf den aufsummierten AUCs von Ivacaftor und seinen Metaboliten). Bei den weiblichen Tieren war Ivacaftor mit einer Abnahme des Gesamtfertilitätsindex, der Zahl der Trächtigkeiten, der Zahl der Corpora lutea und der Implantationsstellen sowie Änderungen des Östruszyklus assoziiert. Ivacaftor erhöhte auch die Zahl der weiblichen Tiere, bei denen alle Embryos nicht lebensfähig waren, und reduzierte die Zahl der lebensfähigen Embryonen. Bei männlichen Tieren wurde eine leichte Abnahme der Samenblasengewichte beobachtet. Diese Beeinträchtigungen der Fertilität und Reproduktionsleistung bei Ratten unter einer Dosis von 200 mg/kg/Tag wurden einer schwerwiegenden Toxizität zugeschrieben. Nach Dosen von ≤100 mg/kg/Tag (etwa dem 5- bzw. 3-Fachen der MRHD basierend auf den aufsummierten AUCs von Ivacaftor und seinen Metaboliten) wurden keine Wirkungen auf die Fertilitäts- und Reproduktionsleistungsindizes der männlichen oder weiblichen Tiere beobachtet.
Ivacaftor war bei Ratten nach 200 mg/kg/Tag und bei Kaninchen nach 100 mg/kg/Tag (dem etwa 6- bzw. 16-Fachen der MRHD basierend auf den aufsummierten AUCs von Ivacaftor und seinen Metaboliten) nicht teratogen. Bei Ratten wurden bei Dosen, die bei den Muttertieren mit einer erheblichen Toxizität verbunden waren, Effekte auf das fetale Körpergewicht sowie geringfügige Zunahmen von häufigen Variationen bei der Skelettentwicklung festgestellt.
In einer Studie zur prä- und postnatalen Entwicklung bei trächtigen Ratten führten Ivacaftor-Dosen über 100 mg/kg/Tag zu Überlebens- und Laktationsindizes, die 92 % resp. 98 % der Kontrollwerte erreichten, und zu niedrigeren Körpergewichten bei den Nachkommen. Bei trächtigen Ratten und Kaninchen wurde ein Übertritt von Ivacaftor in die Plazenta beobachtet.
Juvenile Toxizität
Bei juvenilen Ratten, die ab dem 7. bis zum 35. Tag nach der Geburt mit Ivacaftor-Dosen von 10 mg/kg/Tag und höher (dem 0,2-Fachen der MRHD basierend auf der systemischen Exposition von Ivacaftor und dessen Metaboliten) behandelt worden waren, wurden Kataraktbefunde festgestellt. Dieser Befund wurde bei Feten von Ratten, die vom 7. bis zum 17. Tag der Trächtigkeit mit Ivacaftor behandelt worden waren, bei Jungtieren von Ratten, die durch Milchaufnahme bis zum 20. Tag nach der Geburt einer gewissen Ivacaftor-Exposition unterlagen, bei 7 Wochen alten Ratten oder bei 3,5 bis 5 Monate alten Hundewelpen, die mit Ivacaftor behandelt wurden, nicht beobachtet. Die mögliche Bedeutung dieser Befunde für den Menschen ist nicht bekannt (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern.
Ausser Sicht- und Reichweite von Kindern aufbewahren.
Zulassungsnummer67773, 69212 (Swissmedic)
PackungenTrikafta-Filmtabletten
-Elexacaftor 50 mg/Tezacaftor 25 mg/Ivacaftor 37,5 mg-Tablette und Ivacaftor 75 mg-Tablette
-Packungsgrösse mit 84 Tabletten (4 Walletpackungen für jeweils eine Woche mit jeweils 14 Elexacaftor 50 mg/Tezacaftor 25 mg/Ivacaftor 37,5 mg-Filmtabletten und mit 7 Ivacaftor 75 mg-Filmtabletten). [A]
-Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg-Tablette und Ivacaftor 150 mg-Tablette
-Packungsgrösse mit 84 Tabletten (4 Walletpackungen für jeweils eine Woche mit jeweils 14 Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg-Filmtabletten und mit 7 Ivacaftor 150 mg-Filmtabletten). [A]
Granulat im Beutel
-Elexacaftor 80 mg/Tezacaftor 40 mg/Ivacaftor 60 mg Granulat im Beutel und Ivacaftor 59,5 mg Granulat im Beutel
-Packungsgrösse mit 56 Beuteln (4 Walletpackungen für jeweils eine Woche mit jeweils 7 Beuteln Elexacaftor 80 mg/Tezacaftor 40 mg/Ivacaftor 60 mg Granulat und 7 Beuteln Ivacaftor 59,5 mg Granulat) [A]
-Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg Granulat im Beutel und Ivacaftor 75 mg Granulat im Beutel
-Packungsgrösse mit 56 Beuteln (4 Walletpackungen für jeweils eine Woche mit jeweils 7 Beuteln Elexacaftor 100 mg/Tezacaftor 50 mg/Ivacaftor 75 mg Granulat und 7 Beuteln Ivacaftor 75 mg Granulat) [A]
ZulassungsinhaberinVertex Pharmaceuticals (CH) GmbH
6300 Zug
Stand der InformationJuli 2025
|