ZusammensetzungWirkstoffe
Lumasiran (als Lumasiran-Natrium).
Hilfsstoffe
Natriumhydroxid (E524), Phosphorsäure 85 % (E338), Wasser für Injektionszwecke.
Enthält 5,5 mg Natrium pro 0,5 ml.
Darreichungsform und Wirkstoffmenge pro EinheitInjektionslösung, subkutan.
Klare, farblose bis gelbe Lösung (pH-Wert ca. 7; Osmolalität: 240–360 mosmol/kg).
Jeder ml Lösung enthält Lumasiran-Natrium, entsprechend 189 mg Lumasiran.
Jede Durchstechflasche enthält 94,5 mg Lumasiran in 0,5 ml.
Indikationen/AnwendungsmöglichkeitenOxlumo wird zur Behandlung der primären Hyperoxalurie Typ 1 (PH1) in allen Altersgruppen angewendet.
Dosierung/AnwendungDie Therapie sollte unter der Aufsicht eines Arztes eingeleitet werden, der Erfahrung mit der Behandlung der Hyperoxalurie hat.
Dosierung
Oxlumo wird als subkutane Injektion verabreicht. Die empfohlene Dosis Oxlumo besteht aus Initialdosen, die während 3 Monaten als monatliche Dosen gegeben werden, gefolgt von Erhaltungsdosen, beginnend einen Monat nach der letzten Initialdosis, gemäss Tabelle 1. Die jeweilige Dosis richtet sich nach dem Körpergewicht.
Die dem Patienten zu verabreichende Menge (in mg) und das Volumen (in ml) sollten wie folgt berechnet werden:
Körpergewicht des Patienten (kg) × Dosis (mg/kg) = zu verabreichende Gesamtmenge (mg) des Arzneimittels.
Gesamtmenge (mg) geteilt durch die Konzentration (189 mg/ml) = zu injizierendes Gesamtvolumen des Arzneimittels (ml).
Tabelle 1: Gewichtsbasierte Dosierung von Oxlumo
Körpergewicht Initialdosis Erhaltungsdosis (beginnend einen Monat nach der
letzten Initialdosis)
unter 10 kg 6 mg/kg einmal monatlich 3 mg/kg einmal monatlich, beginnend einen Monat
während 3 Monaten nach der letzten Initialdosis
10 kg bis unter 20 6 mg/kg einmal monatlich 6 mg/kg einmal alle 3 Monate (vierteljährlich),
kg während 3 Monaten beginnend einen Monat nach der letzten Initialdosis
ab 20 kg 3 mg/kg einmal monatlich 3 mg/kg einmal alle 3 Monate (vierteljährlich),
während 3 Monaten beginnend einen Monat nach der letzten Initialdosis
Patienten unter Hämodialyse
Bei Verabreichung an Dialyse-Tagen sollte Oxlumo nach der Hämodialyse verabreicht werden.
Patienten mit Leberfunktionsstörungen
Oxlumo wurde nicht bei Patienten mit Leberfunktionsstörung untersucht. Es muss keine Dosisanpassung vorgenommen werden bei Patienten mit vorübergehender Erhöhung des Gesamtbilirubins (Gesamtbilirubin > 1 bis 1,5 × ULN). Bei der Behandlung von Patienten mit mittlerer oder schwerer Leberfunktionsstörung ist Vorsicht geboten (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Pharmakokinetik" ).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit Nierenfunktionsstörung (geschätzte glomeruläre Filtrationsrate [eGFR] < 90 ml/min/1,73 m²), einschliesslich terminaler Nierenerkrankung (ESRD) oder Patienten unter Dialyse ist keine Dosisanpassung erforderlich. Zu Patienten mit ESRD oder unter Dialyse liegen nur beschränkt Daten vor. Die Behandlung dieser Patienten muss mit Vorsicht erfolgen (siehe "Warnhinweise und Vorsichtsmassnahmen" und "Pharmakokinetik" ).
Ältere Patienten
Bei Patienten ≥ 65 Jahren ist keine Dosisanpassung erforderlich (siehe "Pharmakokinetik" ).
Kinder und Jugendliche
Von Patienten im ersten Lebensjahr sind nur begrenzte Daten verfügbar. Bei der Behandlung dieser Patienten ist daher Vorsicht geboten (siehe "Pharmakokinetik" ).
Verspätete Dosisgabe
Wenn sich der Erhalt einer Dosis verzögert oder die Dosis ausgelassen wurde, sollte die Dosis so bald wie möglich verabreicht werden. Die verordnete monatliche oder vierteljährliche Dosierung sollte ab der zuletzt verabreichten Dosis wieder aufgenommen werden.
Art der Anwendung
Nur zur subkutanen Anwendung.
Das Arzneimittel wird als gebrauchsfertige Lösung in einer Durchstechflasche zur einmaligen Anwendung bereitgestellt.
-Das benötigte Volumen von Oxlumo wird anhand der empfohlenen gewichtsbasierten Dosierung in Tabelle 1 berechnet.
-Bei einer Dosis über 0,5 ml (94,5 mg) ist mehr als eine Durchstechflasche erforderlich.
-Das maximal zulässige Volumen für eine Einzelinjektion beträgt 1,5 ml. Dosierungen mit einem Volumen über 1,5 ml sollten als Mehrfachinjektionen verabreicht werden, um mögliche Beschwerden an der Injektionsstelle aufgrund der Injektionsvolumina zu vermeiden. Dabei wird die Gesamtdosis gleichmässig auf mehrere Spritzen verteilt, sodass jede Injektion in etwa dasselbe Volumen enthält.
-Achten Sie darauf, dass sich kein Arzneimittel an der Nadelspitze befindet, bevor die Nadel in den Subkutanraum eingestochen wird.
-Das Arzneimittel wird subkutan in den Unterbauch, Oberarm oder Oberschenkel injiziert.
-Für nachfolgende Injektionen oder Dosen sollte die Injektionsstelle gewechselt werden.
-Die Injektion sollte nicht in Narbengewebe oder gerötete, entzündete oder geschwollene Hautbereiche erfolgen.
Oxlumo sollte von einer medizinischen Fachkraft verabreicht werden. Für Anweisungen zur Zubereitung des Arzneimittels vor der Verabreichung siehe "Hinweise zur Handhabung" .
KontraindikationenSchwere Überempfindlichkeit gegen den Wirkstoff oder einen der unter "Zusammensetzung" genannten Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenSchwere oder terminale Niereninsuffizienz
Die Behandlung mit Lumasiran erhöht den Plasmaglykolatspiegel, was bei Patienten mit schwerer oder im Endstadium befindlicher Nierenerkrankung das Risiko einer metabolischen Azidose oder einer Verschlimmerung einer bereits bestehenden metabolischen Azidose erhöhen kann. Diese Patienten sollten daher auf Anzeichen und Symptome einer metabolischen Azidose überwacht werden.
Mässige oder schwere Leberfunktionsstörung
Bei Patienten mit mässiger oder schwerer Leberfunktionsstörung besteht die Möglichkeit einer verminderten Wirksamkeit. Daher sollte die Wirksamkeit bei diesen Patienten überwacht werden (siehe "Pharmakokinetik" ).
Sonstige Bestandteile mit bekannter Wirkung
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro ml, d. h. es ist nahezu „natriumfrei“.
InteraktionenEs wurden keine klinischen Studien zur Erfassung von Wechselwirkungen durchgeführt (siehe "Pharmakokinetik" ).
In-vitro-Studien weisen darauf hin, dass Lumasiran weder ein Substrat noch ein Inhibitor von Cytochrom P450 (CYP)-Enzymen ist. Es wird nicht erwartet, dass Lumasiran CYP-Enzyme hemmt oder induziert oder die Aktivität der Arzneistofftransporter moduliert.
Gleichzeitige Anwendung mit Pyridoxin
Die gleichzeitige Anwendung von Pyridoxin hatte keinen bedeutsamen Einfluss auf die Pharmakodynamik oder Pharmakokinetik von Lumasiran.
Schwangerschaft, StillzeitSchwangerschaft
Bisher liegen keine Erfahrungen mit der Anwendung von Lumasiran bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf die Reproduktionstoxizität (siehe "Präklinische Daten" ). Eine Anwendung von Oxlumo während der Schwangerschaft kann in Betracht gezogen werden, wobei der erwartete Nutzen für die Gesundheit der Frau gegenüber dem potenziellen Risiko für das Ungeborene abgewogen werden muss.
Stillzeit
Es ist nicht bekannt, ob Lumasiran/Metaboliten in die Muttermilch übergehen. Ein Risiko für das Neugeborene/Kind kann nicht ausgeschlossen werden. Es muss eine Entscheidung getroffen werden, ob abgestillt oder die Behandlung mit Oxlumo unterbrochen bzw. auf eine Behandlung verzichtet werden sollte. Dabei ist der Nutzen des Stillens für das Kind gegenüber dem Nutzen der Behandlung für die Frau abzuwägen.
Fertilität
Es liegen keine Daten zu den Auswirkungen von Lumasiran auf die Fertilität beim Menschen vor. In tierexperimentellen Studien wurden keine Auswirkungen auf die männliche oder weibliche Fertilität festgestellt (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOxlumo hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die am häufigsten gemeldeten unerwünschten Wirkungen waren Reaktionen an der Injektionsstelle (35 %) und Abdominalschmerz (16 %).
Liste der unerwünschten Wirkungen
Die folgende Tabelle zeigt die unerwünschten Wirkungen von Lumasiran in klinischen Studien und durch Spontanmeldungen. Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet: „sehr häufig“ (≥1/10); „häufig“ (≥1/100, <1/10); „gelegentlich“ (≥1/1‘000, <1/100); „selten“ (≥1/10‘000, <1/1‘000); „sehr selten“ (<1/10‘000) , nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Tabelle 2: Unerwünschte Wirkungen
Systemorganklasse Nebenwirkung Häufigkeit
Erkrankungen des Immunsystems Überempfindlichkeita Nicht bekannt
Erkrankungen des Gastrointestinaltraktes Abdominalschmerzb Sehr häufig
Allgemeine Erkrankungen und Beschwerden am Reaktionen an der Injektionsste Sehr häufig
Verabreichungsort llec
a Im Rahmen der Anwendung nach der Marktzulassung gemeldete Nebenwirkung
b Umfasst Abdominalschmerz, Schmerzen im Oberbauch, Schmerzen im Unterbauch, abdominale Beschwerden und abdominalen Druckschmerz.
c Umfasst Reaktionen an der Injektionsstelle, Erythem an der Injektionsstelle, Schmerzen an der Injektionsstelle, Juckreiz an der Injektionsstelle, Schwellung an der Injektionsstelle, Beschwerden an der Injektionsstelle, Verfärbung der Injektionsstelle, Raumforderung an der Injektionsstelle, Verhärtung an der Injektionsstelle, Ausschlag an der Injektionsstelle, Blutergüsse an der Injektionsstelle, Hämatome an der Injektionsstelle und Exfoliation der Haut.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Reaktionen an der Injektionsstelle
In placebokontrollierten und offenen klinischen Studien wurde bei 34 von 98 Patienten (34,7 %) Reaktionen an der Injektionsstelle berichtet. Die am häufigsten gemeldeten Symptome waren Erytheme, Schwellungen, Schmerzen, Hämatome, Juckreiz und Verfärbungen. Die meisten Reaktionen an der Injektionsstelle begannen am Tag der Verabreichung, wobei weniger als 2 % der Reaktionen an der Injektionsstelle 5 oder mehr Tage nach der Verabreichung auftraten. Die Reaktionen an der Injektionsstelle waren im Allgemeinen mild, klangen innerhalb von zwei Tagen ab und führten nicht zu einer Unterbrechung oder einem Abbruch der Behandlung.
Abdominalschmerz
In der placebokontrollierten Studie wurde bei 1 von 13 Patienten (7,7 %), die Placebo erhielten, und 4 von 26 Patienten (15,4 %), die Lumasiran erhielten, Abdominalschmerz beobachtet. In den placebokontrollierten, und offenen klinischen Studien berichteten 16 von 98 Patienten (16,3 %) über Abdominalschmerz, darunter Schmerzen im Ober- oder Unterbauch, abdominale Beschwerden oder abdominalen Druckschmerz. Die meisten Ereignisse waren leicht, vorübergehend und bildeten sich ohne Behandlung zurück. Keines der Ereignisse führte zu einem Behandlungsabbruch.
Langzeitsicherheit
Das Sicherheitsprofil von Lumasiran während den offenen Verlängerungsphasen der Studien ILLUMINATE-A und ILLUMINATE-B (mediane Behandlungsdauer 55,0 Monate bzw. 55,5 Monate) stimmte mit dem bekannten Sicherheitsprofil von Lumasiran überein.
Kinder und Jugendliche
Das Sicherheitsprofil von Lumasiran war bei Kindern und Jugendlichen (im Alter von 4 Monaten bis 17 Jahren) ähnlich wie bei Erwachsenen mit PH1.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBei einer Überdosierung empfiehlt es sich, den Patienten bei entsprechender klinischer Indikation auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und gegebenenfalls eine geeignete symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
A16AX18
Wirkungsmechanismus
Lumasiran ist eine doppelsträngige „small interfering RNA“ (siRNA), die den Glykolat-Oxidase (GO)-Enzymspiegel senkt, indem sie in den Hepatozyten mittels RNA-Interferenz auf die Messenger-Ribonukleinsäure (mRNA) des (für Glykolat-Oxidase kodierenden) Gens Hydroxysäureoxidase 1 (HAO1) abzielt. Verringerte GO-Enzymwerte reduzieren die Menge an verfügbarem Glyoxylat, einem Substrat für die Bildung von Oxalat. Dies führt zu einer Senkung der (erhöhten) Oxalatspiegel in Urin und Plasma, der eigentlichen Ursache für Krankheitsmanifestationen bei Patienten mit PH1. Da das GO-Enzym dem defizitären, PH1 verursachenden Alanin-Glyoxylat-Aminotransferase (AGT)-Enzym vorgeschaltet ist, ist der Wirkmechanismus von Lumasiran unabhängig von der zugrundeliegenden AGXT-Genmutation.
Immunogenität
Bei Patienten mit PH1 und gesunden Freiwilligen wurden in klinischen Studien nach Verabreichung von Oxlumo 7 von 120 (5,8 %) Personen positiv auf Anti-Drug-Antikörper (ADA) getestet. Die ADA-Titer waren niedrig, im Allgemeinen vorübergehend und hatten keine Auswirkungen auf die klinische Wirksamkeit, Sicherheit oder das pharmakokinetische oder pharmakodynamische Profil des Arzneimittels.
Pharmakodynamik
Nicht zutreffend.
Klinische Wirksamkeit
Die Wirksamkeit von Lumasiran wurde in einer randomisierten, doppelblinden, placebokontrollierten klinischen Studie an Patienten ab 6 Jahren mit PH1 (ILLUMINATE-A), in einer einarmigen klinischen Studie an Patienten unter 6 Jahren mit PH1 (ILLUMINATE-B) und in einer einarmigen klinischen Studie an pädiatrischen und erwachsenen Patienten mit PH1 und fortgeschrittener Nierenerkrankung, einschliesslich Patienten unter Hämodialyse (ILLUMINATE-C), untersucht.
ILLUMINATE-A
Insgesamt 39 Patienten mit PH1 wurden im Verhältnis 2:1 randomisiert und erhielten während der 6monatigen doppelblinden, placebokontrollierten Studienphase subkutan Lumasiran oder Placebo. Eingeschlossen wurden Patienten ab 6 Jahren mit einer eGFR von ≥ 30 ml/min/1,73 m². Sie erhielten 3 Initialdosen mit 3 mg/kg Lumasiran oder Placebo einmal monatlich und anschliessend vierteljährliche Erhaltungsdosen mit 3 mg/kg Lumasiran oder Placebo (siehe "Dosierung/Anwendung" ). Nach der 6monatigen doppelblinden Behandlungsphase wurden die Patienten, einschliesslich der ursprünglich Placebo zugeordneten Patienten, in eine Verlängerungsphase mit Verabreichung von Lumasiran für bis zu 54 Monate aufgenommen. Die Gesamtexposition gegenüber Lumasiran betrug 165,7 Jahre.
Während der 6-monatigen doppelblinden, placebokontrollierten Phase erhielten 26 Patienten Lumasiran und 13 Placebo. Das mittlere Alter der Patienten bei der ersten Dosis betrug 14,9 Jahre (Bereich von 6,1 bis 61 Jahre). 66,7 % der Patienten waren männlich und 76,9 % waren weiss. Die mediane, nach Körperoberfläche (body surface area, BSA) korrigierte Oxalatausscheidung im 24-Stunden-Sammelurin betrug bei Studienbeginn 1,72 mmol/24 h/1,73 m². Der mediane Oxalat/Kreatinin-Quotient im Spontanurin betrug zu Studienbeginn 0,21 mmol/mmol und der mittlere Plasmaoxalatspiegel zu Studienbeginn 13,1 µmol/l. Insgesamt hatten 33,3 % der Patienten eine normale Nierenfunktion (eGFR ≥ 90 ml/min/1,73 m²), 48,7 % eine leichte Nierenfunktionsstörung (eGFR zwischen 60 und < 90 ml/min/1,73 m²) und 18 % eine mittelschwere Nierenfunktionsstörung (eGFR zwischen 30 und < 60 ml/min/1,73 m²). Von den in die Studie aufgenommenen Patienten berichteten zu Studienbeginn 84,6 % über symptomatische Nierensteine und 53,8 % über Nephrokalzinose in der Vorgeschichte. Die Behandlungsarme waren bei Studienbeginn hinsichtlich Alter, Oxalatspiegel im Urin und eGFR ausgeglichen.
Der primäre Endpunkt war die prozentuale Verringerung der nach Körperoberfläche korrigierten Oxalatausscheidung im 24-Stunden-Sammelurin gegenüber dem Ausgangswert, gemittelt über die Monate 3–6. Lumasiran war mit einer statistisch signifikanten Verringerung des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin von 65,4 % gegenüber 11,8 % in der Placebogruppe assoziiert. Das entspricht einer Differenz von 53,5 % (95-%-KI: 44,8; 62,3; p < 0,0001). In Übereinstimmung mit dem primären Endpunkt wurde in Monat 6 im Lumasiran-Arm eine Verringerung des Oxalat/Kreatinin-Quotienten im Spontanurin um 60,5 % beobachtet, im Vergleich zu einem Anstieg von 8,5 % im Placeboarm. Darüber hinaus kam es bei den mit Lumasiran behandelten Patienten zu einer raschen und anhaltenden Verringerung des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin, wie in Abbildung 1 dargestellt.
Abbildung 1: ILLUMINATE-A: Prozentuale Veränderung des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin gegenüber dem Ausgangswert nach Monaten (6-monatige, doppelblinde, placebokontrollierte Phase)

Abkürzungen: BL = Ausgangswert (Baseline), , M = Monat, SEM = Standardfehler des Mittelwerts (Standard Error of Mean).
Die Ergebnisse werden als Mittelwert (± SEM) der prozentualen Veränderung gegenüber dem Ausgangswert dargestellt.
Bis Monat 6 erreichte ein im Vergleich zu den mit Placebo behandelten Patienten höherer Anteil der mit Lumasiran behandelten Patienten normale oder annähernd normale Werte des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin (≤ 1,5 × ULN), wie in Tabelle 3 dargestellt.
Tabelle 3: ILLUMINATE-A: Ergebnisse zum sekundären Endpunkt der 6monatigen placebokontrollierten Doppelblindphase
Endpunkte Lumasiran (N = 26) Placebo (N = 13) Behandlungsdifferenz p-Wert
(95-%-KI)
Anteil Patienten 0,52 (0,31; 0,72)§ 0 (0; 0,25)§ 0,52 (0,23; 0,70)¶ 0,001#
mit Oxalatspiegeln
im 24-Stunden-Sammel
urin ≤ ULN‡
Anteil Patienten 0,84 (0,64; 0,95)§ 0 (0; 0,25)§ 0,84 (0,55; 0,94)¶ 0,0001#
mit Oxalatspiegeln
im 24-Stunden-Sammel
urin von ≤ 1,5 ×
ULN‡
Prozentuale Verringe 39,8 (2,9)† 0,3 (4,3)† 39,5 (28,9; 50,1) < 0,0001
rung des Oxalats im
Plasma gegenüber
Ausgangswert*Þ
Abkürzungen: ULN = obere Normgrenze (Upper Limit of Normal), SEM = Standardfehler des Mittelwerts (Standard Error of Mean)
Ergebnisse basierend auf Flüssigchromatographie-Tandem-Massenspektrometrie-Test (LC-MS/MS).
* Schätzung basierend auf Kleinste-Quadrate-Mittelwert der prozentualen Verringerung in Monat 3, 4, 5 und 6 unter Verwendung eines gemischten Modells für wiederholte Messungen.
† Kleinste-Quadrate (least squares, LS)-Mittelwert (SEM).
‡ ULN = 0,514 mmol/24 h/1,73 m² für nach Körperoberfläche korrigiertes Oxalat im 24-Stunden-Sammelurin.
§ 95-%-KI basierend auf exaktem Clopper-Pearson-Intervall.
¶ Berechnet mittels Newcombe-Methode basierend auf Wilson-Score-Intervallen.
# p-Wert basierend auf Cochran-Mantel-Haenszel-Test mit Stratifizierung gemäss nach Körperoberfläche korrigiertem Ausgangswert des Oxalats im 24-Stunden-Sammelurin (≤ 1,70 vs. > 1,70 mmol/24 h/1,73 m²).
Þ Auswertung bei 23 Lumasiran- und 10 Placebo-Patienten mit Ausgangswerten, die eine Verringerung zuliessen.
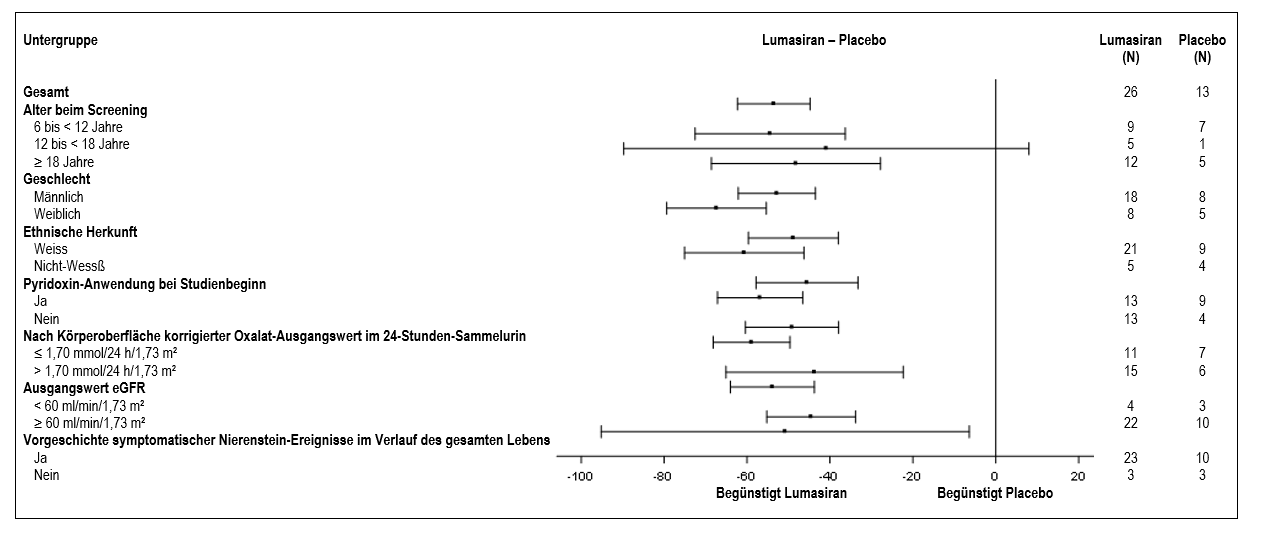
Die Verringerung des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin gegenüber dem Ausgangswert im Vergleich zu Placebo war bei den mit Lumasiran behandelten PH1-Patienten in allen vorspezifizierten Untergruppen ähnlich, einschliesslich Alter, Geschlecht, ethnischer Herkunft, Nierenfunktionsstörung, Anwendung von Pyridoxin (Vitamin B6) bei Studienbeginn und symptomatischer Nierenstein-Ereignisse in der Anamnese (Abbildung 2).
Abbildung 2: ILLUMINATE-A: Prozentuale Veränderung des nach Körperoberfläche korrigierten Oxalats im 24-Stunden-Sammelurin gegenüber dem Ausgangswert, Untergruppenanalyse
Die in der Doppelblindphase beobachtete Verringerung der Oxalatkonzentration blieb bei fortdauernder Behandlung mit Lumasiran während der Verlängerungsphase der Studie über bis zu 60 Monate erhalten. eGFR, Nierenstein-Ereignisse (berichtet nach Ereignissen pro Personenjahr) und medulläre Nephrokalzinose wurden über die 6-monatigen Doppelblind- und Verlängerungsphasen während insgesamt bis zu 60 Monaten beurteilt. Die eGFR blieb bei den mit Lumasiran behandelten Patienten stabil. Die Rate der mittleren jährlichen Veränderung gegenüber dem Ausgangswert während der bis zu 60-monatigen Behandlung mit Lumasiran betrug 0,63 ml/min/1,73 m2/Jahr.
Die Rate der Nierenstein-Ereignisse pro Personenjahr bei Patienten, die zu Lumasiran und Placebo randomisiert wurden in ILLUMINATE A, ist in Tabelle 4 aufgeführt.
Tabelle 4: ILLUMINATE-A: Rate der Nierenstein-Ereignisse pro Personenjahr in der Lumasiran- und Placebo-Gruppe
Zeitraum Lumasiran Rate (95-%-KI) Placebo Rate (95-%-KCI)
12 Monate vor der Einwilligung 3,19 (2,57; 3,96) 0,54 (0,26; 1,13)
6-monatige Doppelblindphase 1,09 (0,63; 1,88) 0,66 (0,25; 1,76)
Während der verlängerten offenen Behandlung mit Lumasiran für bis zu 60 Monate betrug die Raten von Nierensteinereignissen 0,49 pro Personenjahr. 53,8 % der Patienten hatten keine Nierensteinereignisse.
Durch Nieren-Ultraschall beurteilte medulläre Nephrokalzinose-Ergebnisse von Monat 6 im Vergleich zum Ausgangswert sind in Tabelle 5 aufgeführt.
Tabelle 5: ILLUMINATE-A: Patienten mit medullärer Nephrokalzinose in Monat Doppelblinder, placebokontrollierter Zeitraum im Vergleich zum Ausgangswert*
Zeitpunkt Behandlung(n) Verbesserung Unverändert Verschlechterung
Monat 6 Lumasiran(N=22) 3 19 0
Placebo (N=12) 0 11 1
* Patienten mit Nieren-Ultraschall an der Baseline und am entsprechenden Zeitpunkt wurden beurteilt.
Die Beurteilung der medullären Nephrokalzinose wurde nur bei einem Teil der Studienpopulation vorgenommen (17/26 Lumasiran-/Lumasiran-Patienten und 6/13 Placebo-/Lumasiran-Patienten wurden zu Studienbeginn sowie am Ende der 54-monatigen Verlängerungsphase untersucht). In dieser Subpopulation war ein allgemeiner Trend zu einer Besserung im zeitlichen Verlauf nachzuweisen.
ILLUMINATE-B
Insgesamt 18 Patienten wurden in eine laufende, multizentrische, einarmige Studie an PH1-Patienten (ILLUMINATE-B) aufgenommen und mit Lumasiran behandelt. Eingeschlossen wurden Patienten unter 6 Jahren mit einer eGFR > 45 ml/min/1,73 m2 bei Patienten ab einem Alter von 12 Monaten und normalem Serumkreatinin bei Patienten jünger als 12 Monate. In der primären Analyse nach 6 Monaten, bei der ersten Dosis, wogen 3 Patienten weniger als 10 kg, 12 Patienten 10 bis unter 20 kg und 3 Patienten mindestens 20 kg. Das mediane Alter der Patienten bei der ersten Dosis betrug 51,4 Monate (Bereich von 4 bis 74 Monate). 55,6 % der Patienten waren weiblich, und 88,9 % waren weiss. Der mediane Oxalat/Kreatinin-Quotient im Spontanurin betrug zu Studienbeginn 0,47 mmol/mmol. Nach der 6-monatigen primären Analysephase traten die Patienten in eine Verlängerungsphase über, in der Lumasiran bis zu 60 Monate lang verabreicht wurde. Die Gesamtexposition mit Lumasiran betrug 83,2 Patientenjahre.
In Monat 6 erreichten mit Lumasiran behandelte Patienten eine Reduktion von 72 % (95-%-KI: 66,4; 77,5) des Oxalat/Kreatinin-Quotienten im Spontanurin gegenüber dem Ausgangswert (gemittelt über die Monate 3 bis 6), dem primären Endpunkt der Studie. Lumasiran wurde mit einer raschen und anhaltenden Verringerung des Oxalat/Kreatinin-Quotienten im Spontanurin (Abbildung 3) assoziiert, die in allen Gewichtsuntergruppen ähnlich ausfiel. Die prozentuale Verringerung der Oxalatausscheidung im Urin blieb bei fortdauernder Lumasiran-Behandlung bis Monat 30 erhalten, mit einer mittleren (SEM) prozentualen Verringerung von 74,5 % (4,25) gegenüber dem Ausgangswert des Oxalat/Kreatinin-Quotienten im Spontanurin, und dieser Behandlungseffekt stimmte mit den Ergebnissen der Studie ILLUMINATE-A überein.
Abbildung 3: ILLUMINATE-B: Prozentuale Veränderung des Oxalat/Kreatinin-Quotienten im Spontanurin gegenüber dem Ausgangswert nach Monaten

In Monat 6 erreichten neun von 18 Patienten nahezu eine Normalisierung (≤ 1,5 × ULN), darunter 1 Patient, der eine Normalisierung (≤ ULN) des Oxalat/Kreatinin-Quotienten im Spontanurin erreicht hatte. An Monat 12 erreichten zehn von 18 Patienten nahezu eine Normalisierung (≤ 1,5 × ULN), darunter 2 Patienten, die eine Normalisierung (≤ ULN) des Oxalat/Kreatinin-Quotienten im Spontanurin erreicht hatten.
Darüber hinaus wurde von Studienbeginn bis Monat 6 (Durchschnitt aus Monat 3 bis Monat 6) eine mittlere Verringerung des Oxalats im Plasma von 31,7 % (95 %-KI: 23,9; 39,5) beobachtet. Während der primären Analysephase beobachtete verringerte Plasma-Oxalat-Spiegel blieben bei fortgeführter Behandlung mit Lumasiran bis Monat 60 erhalten, mit einer mittleren Reduktion von 24,8 % (95-%-KI: 15,7; 59,5) in Monat 60.
Die eGFR blieb bei allen Patienten bei fortgeführter Behandlung stabil. Die Rate der jährlichen Veränderung der eGFR gegenüber dem Ausgangswert während der bis zu 60-monatigen Behandlung mit Lumasiran betrug 0,26 ml/min/1,73 m2/Jahr.
Die Rate der Nierenstein-Ereignisse pro Personenjahr, die in den 12 Monaten vor der Einwilligung und während der 6monatigen primären Analysephase berichtet wurden, betrug 0,24 (95-%-KI: 0,09; 0,63), respektive 0,24 (95-%-KI: 0,06; 0,96). In Monat 60 betrug die Gesamtrate der Nierenstein-Ereignisse pro Personenjahr 0,11 (95-%-KI: 0,06; 0,21) und 77,8 % der Patienten hatten während der Studie keine Nierenstein-Ereignisse.
Bei der Beurteilung der medullären Nephrokalzinose zeigte sich über einen Zeitraum von 60 Monaten eine Tendenz zur Verbesserung. Von den 18 Patienten, die über 60 Monate behandelt wurden, wiesen 14 Patienten zu Beginn der Studie eine medulläre Nephrokalzinose auf. Von diesen 14 Patienten zeigten 12 eine Verbesserung, wobei 10 eine vollständige Remission der Nephrokalzinose (definiert als Grad 0 beidseitig) erreichten, 1 Patient zeigte keine Veränderung und bei 1 Patient war die Veränderung unbestimmt (eine Niere zeigte eine Verbesserung, die andere eine Verschlechterung). Von den 4 Patienten, die zu Beginn der Studie keine Nephrokalzinose aufwiesen, zeigten alle 4 nach 60 Monaten keine Veränderung.
ILLUMINATE-C
Gesamt wurden 21 Patienten mit PH1 und fortgeschrittener Nierenerkrankung (eGFR ≤ 45 ml/min/1,73 m2 bei Patienten im Alter von mindestens 12 Monaten und mit erhöhtem Kreatinin im Serum bei Patienten unter 12 Monaten), einschliesslich Patienten unter Hämodialyse, in eine laufende multizentrische einarmige Studie aufgenommen und mit Lumasiran behandelt. ILLUMINATE-C umfasste 2 Kohorten: In Kohorte A befanden sich 6 Patienten, die bei Aufnahme in die Studie keine Dialyse benötigten, und in Kohorte B befanden sich 15 Patienten, die dauerhaft mit Hämodialyse behandelt wurden. Die Patienten erhielten die empfohlene Dosierung von Lumasiran basierend auf ihrem Körpergewicht (siehe "Dosierung/Anwendung" ).
Das mittlere Alter der Patienten zum Zeitpunkt der ersten Dosis betrug 8,9 Jahre (Bereich 0 bis 59 Jahre), 57,1 % waren männlich und 76,2 % waren weiss. Bei Patienten in Kohorte A betrug der mittlere Plasma-Oxalat-Spiegel 57,94 µmol/l. Bei Patienten in Kohorte B betrug der mittlere Plasma-Oxalat-Spiegel 103,65 µmol/l.
Primärer Endpunkt der Studie war die prozentuale Veränderung des Plasma-Oxalats von der Baseline zu Monat 6 (Durchschnitt von Monat 3 bis Monat 6) für Kohorte A (N=6) und die prozentuale Veränderung des Plasma-Oxalats vor der Dialyse von der Baseline bis Monat 6 (Durchschnitt von Monat 3 bis Monat 6) für Kohorte B (N=15).
Während der 6monatigen primären Analysephase wurde bei Patienten in beiden Kohorten bereits im ersten Monat eine Verringerung des Plasma-Oxalats beobachtet. Die prozentuale Veränderung des Plasma-Oxalat-Spiegels von der Baseline bis Monat 6 (Durchschnitt von Monat 3 bis Monat 6) war für Kohorte A eine Veränderung des LS-Mittelwerts von -33,3 % (95-%-KI: -81,82, 15,16) und für Kohorte B eine Veränderung des LS-Mittelwerts von 42,4 % (95-%-KI: -50,71, -34,15).
Abbildung 4: ILLUMINATE-C: Prozentuale Veränderung des Plasma-Oxalats (µmol/l) gegenüber dem Ausgangswert zu jedem Termin während der primären Analysephase

Die Ergebnisse werden als Mittelwert (± SEM) der prozentualen Veränderung gegenüber dem Ausgangswert dargestellt.
Abkürzungen: BL = Ausgangswert (Baseline), M = Monat, SEM = Standardfehler des Mittelwerts (Standard Error of Mean).
Für Kohorte A ist der Ausgangswert definiert als Mittelwert aller Plasma-Oxalat-Proben, die vor der ersten Dosis Lumasiran entnommen wurden. Für Kohorte B ist der Ausgangswert definiert als die letzten vier Plasma-Oxalat-Proben vor Dialyse, die vor der ersten Dosis Lumasiran entnommen wurden. In Kohorte B werden nur Proben vor der Dialyse entnommene Proben verwendet.
In Kohorte A betrug die mittlere (SD) eGFR 19,85 (9,6) ml/min/1,73 m2 bei Baseline und 16,43 (9,8) ml/min/1,73 m2 in Monat 6.
Die Rate der Nierenstein-Ereignisse pro Personenjahr, die für Kohorte A in den 12 Monaten vor der Einwilligung und während der 6monatigen primären Analysephase berichtet wurden, betrug 3,20 (95-%-KI: 1,96; 5,22), respektive 1,48 (95-%-KI: 0,55; 3,92).
Pädiatrie
Swissmedic hat für dieses Arzneimittel eine Freistellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in allen pädiatrischen Altersklassen bei der Behandlung von AHP anerkannt.
PharmakokinetikAbsorption
Nach subkutaner Gabe wird Lumasiran rasch absorbiert. Die mediane Zeit bis zum Erreichen der maximalen Plasmakonzentration (tmax) beträgt 4 Stunden (Bereich: 0,5–12 Stunden). Bei Kindern und Erwachsenen mit PH1 und einem Gewicht ≥ 20 kg lagen die maximale Lumasiran-Konzentration im Plasma (Cmax) und die Fläche unter der Konzentrationskurve vom Zeitpunkt Null bis zur letzten messbaren Konzentration nach Verabreichung (AUC0-Endwert) nach einer empfohlenen Lumasiran-Dosis von 3 mg/kg bei jeweils 529 (205–1‘130) ng/ml bzw. 7‘400 (2‘890–10’700) ng·h/ml. Bei Kindern unter 20 kg lagen die Cmax und AUC0-Endwert von Lumasiran nach der empfohlenen Lumasiran-Dosis von 6 mg/kg bei jeweils 912 (523–1‘760) bzw. 7‘960 (5‘920–13‘300). Die Lumasiran-Konzentrationen waren bis zu 24–48 Stunden nach Verabreichung messbar.
Distribution
In Plasmaproben gesunder Erwachsener zeigt Lumasiran in klinisch relevanten Konzentrationen eine mittelstarke bis starke Proteinbindung (77–85 %). Bei einem erwachsenen Patienten mit PH1 beträgt die Populationsschätzung für das scheinbare zentrale Verteilungsvolumen (Vd/F) für Lumasiran 4,9 Liter. Lumasiran wird nach subkutaner Verabreichung primär in die Leber verteilt.
Metabolismus
Lumasiran wird durch Endo- und Exonukleasen zu kürzeren Oligonukleotiden metabolisiert. In-vitro-Studien zeigen, dass Lumasiran nicht über CYP450-Enzyme metabolisiert wird.
Elimination
Lumasiran wird primär über die Leber aus dem Plasma eliminiert. Gemäss gepoolten Daten von gesunden erwachsenen Personen und PH1-Patienten im Alter über 6 Jahren werden nur 7–26 % der verabreichten Dosis als Lumasiran im Urin wiedergefunden. Die mittlere (%CV) terminale Plasmahalbwertszeit von Lumasiran beträgt 5,2 (47 %) Stunden. Die Populationsschätzung für die apparente Plasma-Clearance betrug für einen typischen Erwachsenen mit 70 kg Körpergewicht 26,5 l/h. Lumasiran zeigte bei pädiatrischen und erwachsenen PH1-Patienten eine unbedeutende mittlere renale Clearance zwischen 2 und 3,4 l/h.
Linearität/Nicht Linearität
Nach einmaliger subkutaner Verabreichung von Dosen zwischen 0,3 und 6 mg/kg und Mehrfachdosen von 1 und 3 mg/kg einmal monatlich oder 3 mg/kg vierteljährlich zeigte sich für Lumasiran eine lineare bis leicht nichtlineare, zeitunabhängige Pharmakokinetik im Plasma. Nach wiederholter einmal monatlicher oder vierteljährlicher Verabreichung kam es zu keiner Akkumulation von Lumasiran im Plasma.
Pharmakokinetische/pharmakodynamische Zusammenhänge
Die Plasmakonzentrationen von Lumasiran spiegeln das Ausmass oder die Dauer der pharmakodynamischen Aktivität von Lumasiran nicht wider. Die rasche und gezielte Aufnahme von Lumasiran in der Leber führt zu einer schnellen Verringerung der Plasmakonzentration. In der Leber hat Lumasiran eine lange Halbwertszeit. Dadurch kann die pharmakodynamische Wirkung über das monatliche oder vierteljährliche Dosierungsintervall aufrechterhalten werden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine Studien zu Patienten mit Leberfunktionsstörungen durchgeführt (siehe "Dosierung/Anwendung" ). Begrenzte pharmakokinetische Daten zu Patienten mit leichter und vorübergehender Erhöhung des Gesamtbilirubins (Gesamtbilirubin > 1 bis 1,5 × ULN) zeigten eine vergleichbare Plasmaexposition gegenüber Lumasiran und eine ähnliche Pharmakodynamik wie bei Patienten mit normaler Leberfunktion. Nach Literaturdaten ist die hepatische Expression des Asialoglykoprotein-Rezeptors – also des für die Aufnahme von Lumasiran verantwortlichen Rezeptors – bei Patienten mit Leberfunktionsstörung vermindert. Nichtklinische Daten deuten darauf hin, dass dies möglicherweise keinen Einfluss auf die Leberaufnahme oder die Pharmakodynamik bei therapeutischen Dosen hat. Die klinische Relevanz dieser Daten ist unbekannt.
Nierenfunktionsstörungen
Bei Patienten mit leichter (eGFR 60 bis < 90 ml/min/1,73 m²) Nierenfunktionsstörung waren die Plasmaexposition gegenüber Lumasiran vergleichbar mit der von Patienten mit normaler Nierenfunktion (eGFR ≥ 90 ml/min/1,73 m²). Bei Patienten mit mittelschwerer Nierenfunktionsstörung (eGFR 30 bis < 60 ml/min/1,73 m²) war Cmax ähnlich wie bei Patienten mit normaler Nierenfunktion; die AUC lag, basierend auf begrenzten Daten, 25 % höher. Bei Patienten mit schwerer Nierenfunktionsstörung (eGFR 15 bis < 30 ml/min/1,73 m²), ESRD (eGFR < 15 ml/min/1,73 m²) oder unter Dialyse (siehe "Pharmakokinetik" ) innerhalb derselben Körpergewichtkategorie wurden vorübergehend ein 1,8- bis 3,6fach höheres Cmax und eine 1,6- bis 3,1fach höhere AUC0-Endwert beobachtet (siehe "Pharmakokinetik" ).
Diese Anstiege waren vorübergehend, da die Plasmakonzentrationen, ähnlich wie bei Patienten ohne Nierenfunktionsstörung, innerhalb von 24–48 Stunden unter die Nachweisgrenze abfielen (siehe "Pharmakokinetik" , Pharmakokinetische/pharmakodynamische Zusammenhänge). Die Pharmakodynamik bei Patienten mit Nierenfunktionsstörung (eGFR < 90 ml/min/1,73 m2), einschliesslich ESRD (eGFR < 15 ml/min/1,73 m2) oder unter Dialyse war ähnlich wie bei Patienten mit normaler Nierenfunktion (eGFR ≥ 90 ml/min/1,73 m2) (siehe "Dosierung/Anwendung" ).
Ältere Patienten
Es wurden keine Studien an Patienten im Alter ≥ 65 Jahren durchgeführt. Das Alter war kein signifikantes Kovariat für die Pharmakokinetik von Lumasiran.
Kinder und Jugendliche
Zu unter 1 Jahr alten Kindern liegen nur begrenzte Daten vor. Bei Kindern mit einem Körpergewicht < 20 kg war die Cmax von Lumasiran aufgrund der nominell höheren Dosis von 6 mg/kg und der schnelleren Absorptionsrate um das 2fache höher. Die Pharmakodynamik von Lumasiran war bei pädiatrischen Patienten (im Alter von 4 Monaten bis 17 Jahren) trotz der vorübergehend höheren Plasmakonzentrationen bei Kindern mit einem Gewicht < 20 kg ähnlich wie bei Erwachsenen, was auf die rasche und vorwiegende Verteilung von Lumasiran in die Leber zurückzuführen ist.
Körpergewicht
Die empfohlenen Dosierungsschemata ergaben bei Kindern < 20 kg Körpergewicht ein bis zu 2fach höheres Cmax, während die AUC über die untersuchten Körpergewichte (6,2 bis 110 kg) ähnlich blieb.
Geschlecht und ethnische Herkunft
In klinischen Studien wurden keine Unterschiede in der Plasmaexposition oder Pharmakodynamik von Lumasiran aufgrund des Geschlechts oder der ethnischen Herkunft festgestellt.
Präklinische DatenSicherheitspharmakologie
Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Genotoxizität und zum kanzerogenen Potential lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Bei Ratten, nicht aber bei Affen, wurden mikroskopische Veränderungen in der Leber (z. B. hepatozelluläre Vakuolisierung, Mitose und Karyomegalie) beobachtet, begleitet durch eine Abnahme der Plasmafibrinogenspiegel und anderer Veränderungen von Laborwerten. Der Grund für die offensichtliche Nagetierspezifität ist nicht verstanden und die Relevanz für den Menschen ist unklar.
Reproduktionstoxizität
Lumasiran zeigte keine unerwünschten Wirkungen auf die männliche und weibliche Fertilität und die prä- und postnatale Entwicklung bei Ratten. In embryofötalen Entwicklungsstudien bei Ratten und Kaninchen wurden skelettale Anomalien beobachtet, allerdings verglichen mit der therapeutischen Exposition beim Menschen in hohen Expositionsabständen. Die NOAELs (Dosen ohne beobachtbare schädliche Wirkung) lagen ungefähr 20- bis 70mal höher (basierend auf einer monatlichen Exposition).
Toxizitätsprüfungen mit juvenilen Tieren
Eine Toxizitätsstudie zur Dosisfindung an neugeborenen Ratten ergab keine erhöhte Empfindlichkeit sich entwickelnder Ratten gegenüber der Toxikologie oder Pharmakologie von Lumasiran bei Expositionen, die im Vergleich zu den therapeutischen Expositionen beim Menschen zweimal höher lagen (basierend auf einer monatlichen Exposition).
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit „EXP“ bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach Anbruch der Durchstechflasche ist das Arzneimittel sofort zu verwenden.
Besondere Lagerungshinweise
Nicht über 30°C lagern.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Das Arzneimittel ist gebrauchsfertig und nur für die einmalige Anwendung vorgesehen.
Nur zur subkutanen Anwendung.
Vorbereitung für die Verabreichung
-Die Materialien, die nicht in der Packung enthalten sind, aber für die Verabreichung notwendig sind sollten breitgehalten werden, darunter eine sterile Spritze (0,3 ml, 1 ml oder 3 ml), eine 18-Gauge (G)-Nadel, sowie eine 25-G- bis 31-G-Nadel.
-Das benötigte Volumen von Oxlumo sollte anhand der empfohlenen gewichtsbasierten Dosierung berechnet werden (siehe "Dosierung/Anwendung" ).
-Zur Entnahme von Oxlumo aus der Durchstechflasche sollte eine 18-G-Nadel verwendet werden. Die Durchstechflasche sollte aufrecht oder leicht geneigt gehalten werden und die abgeflachte Seite der Nadel sollte nach unten zeigt.
-Für Volumina unter 0,3 ml wird eine sterile 0,3-ml-Spritze empfohlen.
-Das Arzneimittel sollte mit einer sterilen 25-G- bis 31-G-Nadel mit einer Nadellänge von 13 mm oder 16 mm für die subkutane Injektion verabreicht werden.
-Hinweis: Dieses Arzneimittel sollte nicht in die 25-G- bis 31-G-Nadel gedrückt werden.
-Spritzen, Übertragungsnadeln und Injektionsnadeln dürfen nur einmal verwendet werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den örtlichen Anforderungen zu entsorgen.
Zulassungsnummer68239 (Swissmedic).
PackungenJede Packung enthält eine Durchstechflasche aus Glas mit Fluorpolymer-beschichtetem Gummistopfen und Aluminiumversiegelung mit Flip-off-Knopf. Jede Durchstechflasche enthält 0,5 ml Injektionslösung. [B]
ZulassungsinhaberinAlnylam Switzerland GmbH, Zug.
Stand der InformationDezember 2025
|