| Patienteninformation zu Mayzent®
Qu'est-ce que Mayzent et quand doit-il être utilisé?
Mayzent fait partie d'un groupe de médicaments qui sont dénommés modulateurs des récepteurs de la sphingosine-1phosphate (S1P). Mayzent contient le principe actif siponimod.
Mayzent est utilisé pour le traitement de patients adultes atteints de sclérose en plaques secondairement progressive (SEP-SP) avec activité inflammatoire de la maladie. Une activité inflammatoire de la maladie est présente en cas de SEP-SP lorsque de nouvelles poussées se manifestent ou lorsque l'IRM (imagerie par résonance magnétique) montre des inflammations.
Mayzent contribue à protéger le système nerveux central (SNC) des attaques du système immunitaire en limitant la capacité de certains globules blancs (lymphocytes) à se déplacer librement dans le corps. Mayzent peut empêcher que les cellules à l'origine de l'inflammation atteignent le cerveau et la moelle épinière. Grâce à cela, l'atteinte nerveuse entraînée par la sclérose en plaques est diminuée. Mayzent contribue ainsi à ralentir les effets de l'activité de la maladie (aggravation du handicap, lésions cérébrales, perte de volume cérébral et poussées). Mayzent a sans doute aussi une influence favorable directe sur certaines cellules cérébrales qui sont impliquées dans la suppression ou le ralentissement de l'atteinte conditionnée par la sclérose en plaques.
Selon prescription du médecin.
De quoi faut-il tenir compte en dehors du traitement?
Veuillez suivre exactement les instructions de votre médecin. Celles-ci peuvent différer des informations générales contenues dans cette notice.
Quand Mayzent ne doit-il pas être pris?
Vous ne devez pas prendre Mayzent
·si vous êtes allergique (hypersensible) au siponimod, aux arachides, au soja ou à l'un des autres composants de Mayzent (voir «Que contient Mayzent»).
·si vous souffrez d'un syndrome d'immunodéficience.
·si vous avez des antécédents d'une infection sévère du cerveau, comme une leucoencéphalopathie multifocale progressive (LEMP) ou une méningite à cryptocoque.
·si vous avez un cancer actif.
·si vous présentez une insuffisance hépatique sévère (classe C de Child-Pugh).
·si vous avez eu, au cours des 6 derniers mois, un infarctus du myocarde, une angine de poitrine instable (une forme de trouble circulatoire du cœur), un accident vasculaire cérébral (y compris un trouble passager de l'irrigation du cerveau appelé AIT) ou une insuffisance cardiaque sévère ayant nécessité un traitement en milieu hospitalier (insuffisance cardiaque de la classe III-IV de la classification NYHA).
·si vous avez des antécédents de rythme cardiaque irrégulier ou anormal sévère (bloc AV du 2e degré de type Mobitz II, bloc AV du 3e degré, blocage sino-auriculaire ou maladie du sinus) et que vous ne portez pas de stimulateur cardiaque (pacemaker) (voir «Quelles sont les précautions à observer lors de la prise de Mayzent?»).
·si le résultat de votre examen sanguin montre que votre corps ne peut pas dégrader suffisamment Mayzent (génotype CYP2C9*3/*3, concerne moins de 0,4–0,5% de la population).
·si vous êtes enceinte ou une femme en âge de procréer qui n'utilise pas de méthode contraceptive fiable.
Quelles sont les précautions à observer lors de la prise de Mayzent?
Veuillez parler à votre médecin avant de prendre Mayzent
·si vous avez une infection. Une infection déjà présente peut s'aggraver. Si vous avez une infection active sévère, votre médecin reportera le traitement par Mayzent jusqu'à la guérison de l'infection.
·si vos défenses immunitaires sont affaiblies, par exemple par une maladie ou par des médicaments qui répriment le système immunitaire (voir «Prise en même temps que d'autres médicaments»).
·si vous n'avez pas encore eu la varicelle ou que vous n'êtes pas vacciné(e) contre cette dernière. S'il vous arrivait de contracter la varicelle pendant le traitement par Mayzent, cela pourrait signifier un risque plus important de complications pour vous. Votre médecin vous demandera peut-être de vous faire vacciner contre la varicelle.
·si vous prévoyez une vaccination. Votre médecin vous conseillera à ce sujet (voir «Prise en même temps que d'autres médicaments»).
·si vous souffrez ou avez souffert de troubles visuels (en particulier une maladie appelée œdème maculaire), d'inflammations ou d'infections des yeux (uvéite). Votre médecin vous demandera, avant de débuter le traitement avec Mayzent, et à des intervalles réguliers pendant votre traitement, de vous soumettre à un examen oculaire. La macula est une petite zone de la rétine sur le fond de l'œil grâce à laquelle vous pouvez voir clairement et nettement les contours, les couleurs et les détails (champ de vision central). Mayzent peut causer un gonflement de la macula. La probabilité qu'un œdème maculaire se développe est plus élevée si vous avez déjà eu un œdème maculaire ou une uvéite ou si vous êtes diabétique.
·si vous avez ou avez eu l'une des maladies suivantes (même si elle a été traitée): une maladie cardiaque sévère, un rythme cardiaque irrégulier ou anormal (arythmie), un accident vasculaire cérébral ou d'autres maladies en rapport avec les vaisseaux sanguins du cerveau, une fréquence cardiaque ralentie, des syncopes, une perturbation du rythme cardiaque (anomalies à l'ECG).
·si vous souffrez d'apnée sévère pendant votre sommeil.
·si vous avez une tension artérielle élevée qui ne peut pas être contrôlée par des médicaments.
·si vous avez ou avez eu des problèmes de foie.
·si vous pourriez ou voudriez tomber enceinte.
Au cours des premiers jours de traitement, Mayzent peut entraîner un ralentissement de la fréquence cardiaque (bradycardie). Peut-être n'allez-vous pas le remarquer du tout. Vous pouvez toutefois éprouver une sensation de vertige ou de fatigue. Au début du traitement, Mayzent peut aussi entraîner un rythme cardiaque irrégulier. Si quelque chose devait indiquer qu'un risque accru d'apparition de ces effets pourrait exister chez vous, votre médecin pourra décider de vous surveiller de plus près au début du traitement, ou de vous adresser d'abord à un cardiologue.
Si l'un des symptômes suivants se manifeste chez vous pendant le traitement par Mayzent, avertissez immédiatement votre médecin, étant donné qu'il pourrait être grave:
·Une infection. Mayzent réduit le nombre de lymphocytes. Vous pouvez donc être plus facilement sujet(te) aux infections pendant votre traitement par Mayzent (et jusqu'à 3 à 4 semaines après la fin de la prise). Ces infections pourraient être graves ou mettre la vie en danger.
·Vous supposez que votre sclérose en plaques s'aggrave (p.ex. faiblesse ou modifications visuelles), ou vous remarquez des symptômes nouveaux ou inhabituels. Il pourrait s'agir de symptômes d'une maladie rare du cerveau provoquée par une infection et appelée leucoencéphalopathie mutlifocale progressive (LEMP).
·Si une LEMP vous est diagnostiquée, le traitement par Mayzent doit être arrêté définitivement. Cependant, en raison de la diminution de la concentration sanguine de Mayzent, une aggravation de la maladie peut se produire chez certains patients, incluant une détérioration de la fonction cérébrale. Ceci peut être dû à une réaction inflammatoire excessive, nommée syndrome inflammatoire de reconstitution immunitaire (en anglais Immune reconstitution inflammatory syndrome, IRIS).
·Vous avez de la fièvre, vous avez l'impression d'avoir la grippe ou des maux de tête qui s'accompagnent d'une raideur de la nuque, d'une sensibilité à la lumière, de nausées et/ou de confusion. Il pourrait s'agir des symptômes d'une méningite et/ou d'une encéphalite causée par une infection virale ou fongique (telle qu'une méningite à cryptocoque).
·Votre vue change, une «tache aveugle» se développe, par exemple, au centre de votre champ visuel ou vous avez des problèmes s'agissant de la perception des couleurs ou de détails fins. Il pourrait s'agir des symptômes d'un œdème maculaire. Celui-ci peut entraîner des perturbations visuelles similaires à celles d'une poussée de SEP (névrite optique). Votre médecin vous conseillera des examens ophtalmologiques supplémentaires.
·Nausées inexpliquées, vomissements, douleurs abdominales, fatigue, jaunissement de la peau ou du blanc des yeux ou urine inhabituellement foncée. Il peut s'agir de problèmes du foie.
·Des nodules cutanés (p.ex. des nodules brillants et nacrés), des taches ou des plaies ouvertes qui ne guérissent pas en l'espace de quelques semaines peuvent compter parmi les symptômes du cancer de la peau. Une croissance anormale ou des modifications du tissu cutané (p.ex. des grains de beauté inhabituels) avec un changement de couleur, de forme ou de taille au cours du temps peuvent être des symptômes d'autres types de cancers de la peau.
·Apparition soudaine de forts maux de tête, de confusion, de convulsions et de troubles visuels. Il pourrait s'agir d'une maladie dénommée syndrome d'encéphalopathie postérieure réversible (SEPR).
Prise en même temps que d'autres médicaments (interactions avec d'autres médicaments, y compris des vaccinations et d'autres thérapies)
Veuillez informer votre médecin avant la prise de Mayzent si vous prenez l'un des médicaments suivants:
·Médicaments contre une perturbation du rythme cardiaque, p.ex. la quinidine, le procaïnamide, l'amiodarone ou le sotalol, étant donné que ceci pourrait augmenter l'effet sur le rythme cardiaque irrégulier.
·Médicaments qui ralentissent la fréquence cardiaque, p.ex. le vérapamil ou le diltiazem (ce que l'on appelle des antagonistes du calcium), l'ivabradine ou la digoxine. Votre médecin peut vous adresser à un cardiologue pour le changement de vos médicaments, car l'effet sur la fréquence cardiaque pourrait être renforcé au cours des premiers jours qui suivent le début du traitement par Mayzent. Si vous prenez un bêtabloquant, votre médecin pourra vous demander éventuellement d'interrompre temporairement votre traitement par bêtabloquant jusqu'à ce que vous ayez atteint votre dose d'entretien de Mayzent.
·Médicaments qui répriment ou modulent le système immunitaire, dont certains agents chimiothérapeutiques et d'autres médicaments pour le traitement de la SEP, étant donné que ceux-ci peuvent renforcer l'action sur le système immunitaire.
·Si vous devez vous faire vacciner, demandez d'abord conseil à votre médecin. Pendant le traitement par Mayzent et pendant 4 semaines après l'arrêt du traitement, vous ne pouvez pas être vacciné(e) avec certains vaccins (vaccins vivants atténués), étant donné que ceux-ci pourraient déclencher l'infection qu'ils sont supposés prévenir. D'autres vaccins peuvent être moins efficaces pendant le traitement par Mayzent et pendant 4 semaines après le traitement. Le traitement par Mayzent doit donc être interrompu si possible 1 semaine avant et jusqu'à 4 semaines après la vaccination (voir «Quelles sont les précautions à observer lors de la prise/de l'utilisation de Mayzent?»).
·Le fluconazole et certains autres médicaments peuvent, chez certains patients, augmenter la concentration de Mayzent dans le sang et il n'est pas recommandé de les utiliser en même temps que Mayzent. Votre médecin vous conseillera à ce sujet.
·La carbamazépine, la rifampicine et certains autres médicaments peuvent diminuer la concentration de Mayzent dans le sang, et donc réduire son efficacité. Votre médecin vous conseillera à ce propos.
·Le modafinil et certains autres médicaments peuvent, chez certains patients, diminuer la concentration de Mayzent dans le sang, et donc diminuer l'efficacité du médicament. Votre médecin vous dira si ceci est important pour vous.
·Photothérapie avec rayonnement UV-B ou photochimiothérapie PUVA. Pendant le traitement par Mayzent, une thérapie aux UV peut augmenter votre risque de cancer de la peau.
Examens avant et pendant le traitement
La vitesse à laquelle Mayzent est dégradé (métabolisé) dans le corps diffère d'un patient à l'autre et est déterminée avant le début du traitement à l'aide d'un examen génétique dans le sang ou la salive afin de déterminer la dose optimale pour vous. Dans de rares cas, le résultat de l'examen pourrait montrer que vous ne pouvez pas prendre Mayzent.
L'effet souhaité du traitement par Mayzent est la diminution du nombre de globules blancs dans votre sang. Cette valeur se normalise généralement en 3 à 4 semaines après l'arrêt du traitement. Si vous devez effectuer des analyses de sang, avertissez le médecin que vous prenez Mayzent, faute de quoi le médecin pourrait ne pas comprendre les résultats du test et devra prélever plus de sang que d'habitude pour certains types d'analyses de sang.
Avant que vous commenciez à prendre Mayzent, votre médecin vérifiera que vous avez suffisamment de globules blancs dans le sang et il ou elle effectuera éventuellement un contrôle régulier. Si vous n'avez pas suffisamment de globules blancs dans le sang, votre médecin devra peut-être diminuer la dose de Mayzent ou interrompre Mayzent.
Avant le début du traitement, votre médecin vérifiera vos paramètres hépatiques. En outre, votre médecin vous interrogera sur les médicaments que vous prenez actuellement de manière à exclure des interactions d'autres médicaments avec Mayzent. Votre médecin vérifiera régulièrement votre tension artérielle et vous demandera peut-être de faire procéder à un examen de l'œil 3 ou 4 mois après le début du traitement et plus tard aussi éventuellement.
Avant le début du traitement et 3 à 4 mois après le début du traitement par Mayzent, un examen ophtalmologique est réalisé.
Un type de cancer de la peau portant le nom de carcinome basocellulaire (CBC) et d'autres types de cancer de la peau, par exemple le carcinome épidermoïde, le mélanome malin, le sarcome de Kaposi et le carcinome à cellules de Merkel ont été rapportés chez des patients traités par des modulateurs des récepteurs de la S1P, dont Mayzent est un représentant. Votre médecin examinera régulièrement votre peau avant et pendant votre traitement par Mayzent. Informez immédiatement votre médecin si vous remarquez une modification suspecte de la peau.
Composants
Les comprimés contiennent du lactose et des phospholipides issus de graines de soja. Veuillez prendre Mayzent uniquement après avoir consulté votre médecin si vous savez que vous souffrez d'une intolérance au sucre. Si vous êtes allergique aux arachides ou au soja, vous ne pouvez pas prendre Mayzent.
Exposition au soleil et protection contre le soleil
Une exposition prolongée au soleil et un système immunitaire faible peuvent influencer le risque de développer un CBC ou d'autres tumeurs cutanées. Vous devez limiter votre exposition au soleil et aux rayons UV en portant des vêtements qui vous protègent de manière adéquate et en appliquant régulièrement des crèmes solaires avec un facteur de protection UV élevé. Une photothérapie avec des rayons UV ou une photochimiothérapie (PUVA) doivent être évitées pendant le traitement par Mayzent (celles-ci peuvent augmenter le risque de développer un cancer de la peau).
Mayzent affaiblit votre système immunitaire et peut donc augmenter votre risque de cancer de la peau. Contrôlez votre peau régulièrement.
Aggravation de la SEP après la fin du traitement par Mayzent
Ne mettez pas fin à la prise de Mayzent ou ne modifiez pas la dose sans en avoir d'abord parlé à votre médecin.
Consultez immédiatement votre médecin si vous pensez que votre SEP s'est aggravée après l'arrêt du traitement par Mayzent.
Veuillez informer votre médecin ou votre pharmacien si
·vous souffrez d'une autre maladie,
·vous êtes allergique ou
·vous prenez déjà d'autres médicaments ou utilisez déjà d'autres médicaments en usage externe (même en automédication!)!
Aptitude à la conduite et capacité à utiliser des machines
Votre médecin vous dira si votre maladie vous permet de conduire des véhicules et d'utiliser des machines en toute sécurité. Mayzent ne devrait pas influencer votre aptitude à la conduite et votre capacité à utiliser des machines si vous conservez votre dose de traitement régulière. Au début du traitement, vous pouvez ressentir occasionnellement des vertiges. Lors de votre première journée de traitement par Mayzent, vous ne devrez donc pas conduire de véhicules ou utiliser de machines.
Mayzent peut-il être pris pendant la grossesse ou l'allaitement?
Si vous êtes enceinte, si vous envisagez une grossesse ou si vous êtes en âge de procréer mais n'utilisez pas de méthode de contraception fiable, vous ne pouvez pas prendre Mayzent. Si vous prenez Mayzent pendant la grossesse, il existe un risque de lésion pour le fœtus.
Si vous êtes en âge de procréer, votre médecin vous demandera, avant le début du traitement par Mayzent, de procéder à un test de grossesse pour vérifier que vous n'êtes pas enceinte. Pendant le traitement et au moins 10 jours après la fin de la prise, une méthode de contraception fiable doit être utilisée pour éviter une grossesse (voir «Quelles sont les précautions à observer lors de la prise/de l'utilisation de Mayzent?»). Discutez des méthodes de contraception fiables avec votre médecin.
En cas de grossesse pendant le traitement par Mayzent, informez-en immédiatement votre médecin. Ce dernier interrompra le traitement.
Vous ne devez pas allaiter pendant le traitement par Mayzent. Mayzent peut passer dans le lait maternel et présente un risque d'effets secondaires graves pour le bébé.
Si Mayzent est arrêté dans le but de planifier une grossesse, un retour éventuel de l'activité de la maladie doit être envisagé.
Comment utiliser Mayzent?
Quelle dose de Mayzent faut-il prendre?
Début du traitement
Avec l'aide d'une boîte de début de traitement, la dose est augmentée progressivement sur 5 jours, ce qui diminue le risque d'effets secondaires cardiaques au début du traitement. Suivez les instructions figurant sur l'emballage (voir aussi tableau ci-après). Pour certains antécédents cardiaques, votre médecin pourra éventuellement ordonner une surveillance étroite au début du traitement.
Boîte de début de traitement
Titration Dose de titration Plan de titration
Jour 1 0,25 mg 1 comprimé
Jour 2 0,25 mg 1 comprimé
Jour 3 0,5 mg 2 comprimés
Jour 4 0,75 mg 3 comprimés
Jour 5 1,25 mg 5 comprimés
Le jour 6, passez à la dose de traitement qui vous est prescrite.
Pendant les 6 premiers jours de traitement, la dose recommandée doit être prise une fois par jour le matin avec ou sans nourriture.
Dose de traitement
La dose journalière normale (dose d'entretien) faisant suite à la boîte de début de traitement est de 2 mg (un comprimé à 2 mg de siponimod) avec ou sans nourriture.
Si la prise de sang réalisée avant le traitement a indiqué que votre corps dégrade Mayzent lentement (voir «Quelles sont les précautions à observer lors de la prise/de l'utilisation de Mayzent?»), votre médecin vous indiquera de ne prendre que 1 mg une fois par jour (un comprimé à 1 mg ou quatre comprimés à 0,25 mg). Si c'est votre cas, le même plan pour l'initiation du traitement (boîte de début de traitement) s'applique et la prise de cinq comprimés à 0,25 mg au jour 5 reste tout de même sûre.
Mayzent doit être pris une fois par jour avec ou sans nourriture. Les comprimés pelliculés de Mayzent doivent être pris entiers avec de l'eau. Si vous prenez Mayzent tous les jours à la même heure, il vous sera plus facile de ne pas oublier la prise.
Pendant combien de temps faut-il prendre Mayzent?
Prenez Mayzent aussi longtemps que votre médecin vous le prescrit. Il s'agit d'un traitement de longue durée qui peut être poursuivi pendant des mois ou des années. Votre médecin surveillera continuellement votre maladie afin de vérifier si le traitement produit l'effet souhaité.
Si vous avez pris plus de Mayzent que vous n'auriez dû
Si vous avez pris trop de comprimés de Mayzent ou si vous avez pris par mégarde le premier comprimé de l'emballage avec la dose de traitement au lieu de la boîte de début de traitement, informez-en immédiatement votre médecin. Celui-ci mettra éventuellement en place une surveillance.
Si vous avez oublié de prendre Mayzent
Si vous avez oublié de prendre une dose un jour pendant les 6 premiers jours de traitement, appelez votre médecin avant de prendre la dose suivante. Celui-ci doit alors vous prescrire une nouvelle boîte de début de traitement. Vous devez recommencer une nouvelle boîte de début de traitement au jour 1.
Si vous avez omis une dose alors que vous recevez déjà la dose de traitement régulière (à partir du jour 7), prenez la dose dès que vous vous apercevez de votre oubli. Si la prise de la prochaine dose est proche, ne prenez pas la dose oubliée et poursuivez votre traitement comme d'habitude. Ne doublez pas la quantité de la prochaine prise.
Si vous suspendez la prise de Mayzent pendant 4 jours consécutifs ou plus, vous devez recommencer le traitement avec une boîte de début de traitement au jour 1.
Si vous arrêtez de prendre Mayzent
Vous ne devez pas mettre fin à la prise de Mayzent ni modifier votre dose sans en avoir d'abord parlé à votre médecin.
Après l'arrêt du traitement, Mayzent peut encore être retrouvé dans le sang pendant au moins 10 jours. Le nombre de vos globules blancs (nombre de lymphocytes) peut rester bas jusqu'à 4 semaines et les effets secondaires décrits dans cette notice peuvent continuer à se manifester.
Si vous reprenez le traitement par Mayzent 4 jours ou plus après cet arrêt, vous devez recommencer le traitement avec une boîte de début de traitement au jour 1.
Contactez immédiatement votre médecin si vous pensez que votre SEP s'est aggravée après l'arrêt du traitement par Mayzent.
Enfants et adolescents (moins de 18 ans)
L'utilisation et la sécurité de Mayzent pour les enfants de moins de 18 ans n'ont pas été établies à ce jour.
Patients âgés
On ne dispose d'aucune expérience concernant l'utilisation de Mayzent chez les patients âgés (plus de 61 ans).
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Mayzent peut-il provoquer?
Comme tous les médicaments, Mayzent peut provoquer des effets secondaires qui ne surviennent toutefois pas chez toutes les personnes.
Les effets secondaires suivants peuvent être graves ou le devenir:
Fréquent (concerne 1 à 10 utilisateurs sur 100)
·Éruption cutanée avec des petites cloques remplies de liquide qui apparaissent sur la peau rougie; signes d'une infection virale qui peut évoluer de manière grave (zona).
·Type de cancer de la peau, nommé carcinome basocellulaire, qui se présente souvent sous forme de nodules nacrés, mais qui peut aussi prendre d'autres formes.
·Fièvre, mal de gorge, ulcères de la muqueuse buccale en raison d'infections (lymphopénie).
·Convulsions, crises convulsives.
·Formation d'une ombre ou «tache aveugle» dans le champ visuel central, vision trouble, problèmes de reconnaissance des couleurs ou des détails (signes d'un gonflement de la zone visuelle centrale de la rétine au fond de l'œil: œdème maculaire).
·Troubles du rythme cardiaque (bloc auriculo-ventriculaire).
·Rythme cardiaque lent (bradycardie).
Occasionnel (concerne 1 à 10 utilisateurs sur 1000)
·Un type de cancer de la peau appelé carcinome épidermoïde pouvant survenir sous la forme d'un nodule rouge solide, d'une plaie avec une croûte ou d'une nouvelle plaie sur une cicatrice déjà existante.
·Un type de cancer de la peau appelé mélanome malin qui se développe en général à partir d'un grain de beauté inhabituel. Des signes possibles de mélanome sont des grains de beauté qui changent de taille, de forme, de hauteur et de couleur au fil du temps, ou des nouveaux grains de beauté. Les grains de beauté peuvent gratter, saigner ou ulcérer.
·Infections à cryptocoque (une sorte d'infection fongique) ou infection virale (causée par le virus de l'herpès ou le virus varicelle-zona), y compris méningite et/ou encéphalite avec symptômes comme des maux de tête associés à une raideur de la nuque, sensibilité à la lumière, nausées et/ou confusion.
Rare (concerne 1 à 10 utilisateurs sur 10 000)
·Une infection cérébrale rare, nommée leucoencéphalite multifocale progressive (LEMP). Les symptômes d'une LEMP peuvent ressembler à ceux de la SEP, comme, par exemple, faiblesse ou troubles de la vision, perte de mémoire, difficultés à penser ou difficultés à la marche.
Si l'un des effets secondaires mentionnés ci-dessus survient, vous devez en informer immédiatement votre médecin.
Autres effets secondaires éventuels
D'autres effets secondaires sont indiqués ci-après. Si ces effets secondaires s'aggravent, parlez-en à votre médecin ou à votre pharmacien.
La plupart des effets secondaires sont d'intensité légère à modérée et disparaissent en règle générale après quelques semaines de traitement.
Très fréquent (concerne plus d'un utilisateur sur 10)
·Maux de tête
·Tension artérielle élevée (hypertension), qui s'accompagne occasionnellement de symptômes comme des maux de tête et des vertiges.
·Valeurs augmentées au test de la fonction hépatique (valeurs hépatiques élevées): taux augmentés des enzymes alanine aminotransférase (ALAT), gammaglutamyltransférase (GGT) et aspartate aminotransférase (ASAT).
Fréquent (concerne 1 à 10 utilisateurs sur 100)
·Taches pigmentées ou grains de beauté (nævi) nouvellement apparus: petites taches (diamètre inférieur à 1 cm), petites papules ou nodules aux limites peu précises, qui peuvent avoir un aspect bleuâtre/noir à brun, rose ou de la couleur de la peau (nævi mélanocytaires).
·Vertiges
·Tremblement involontaire du corps
·Diarrhée
·Nausées
·Douleurs dans les mains ou les pieds
·Gonflements des mains, des chevilles, des jambes ou des pieds (œdème périphérique)
·Faiblesse (asthénie)
·Valeurs abaissées au test de la fonction pulmonaire (baisse des valeurs de la fonction pulmonaire)
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Délai d'utilisation après ouverture
Après l'ouverture, conserver 3 mois à une température ne dépassant pas 25 °C.
Mention concernant l'élimination des médicaments
Ne jetez aucun médicament avec les eaux usées ou les ordures ménagères. Les médicaments périmés ou qui ne sont plus utilisés dans les ménages peuvent être rapportés dans les pharmacies ou dans les centres de collecte. Ces mesures contribueront à protéger l'environnement.
Remarques concernant le stockage
Conserver au réfrigérateur (2–8 °C).
Conserver dans l'emballage d'origine.
Conserver le médicament hors de portée des enfants.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Mayzent?
Principes actifs
Siponimod (sous forme d'acide fumarique de siponimod).
Excipients
Noyau des comprimés:
Lactose monohydraté, cellulose microcristalline, crospovidone, dibéhénate de glycérol et silice colloïdale anhydre.
Chaque comprimé à 0,25 mg contient 62,2 mg de lactose monohydraté.
Chaque comprimé à 1 mg contient 61,4 mg de lactose monohydraté.
Chaque comprimé à 2 mg contient 60,3 mg de lactose monohydraté.
Enrobage des comprimés:
Poly(alcool vinylique), dioxyde de titane (E171), oxyde de fer rouge (E172), talc, lécithine de soja (E322), gomme xanthane, oxyde de fer noir (E172, seulement les comprimés à 0,25 mg et à 1 mg), oxyde de fer jaune (E172, seulement les comprimés à 2 mg).
Où obtenez-vous Mayzent? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Boîte de début de traitement de 12 comprimés pelliculés à 0,25 mg (B)
Boîte de 120 comprimés pelliculés à 0,25 mg (B)
Boîte de 28 comprimés pelliculés à 1 mg (B)
Boîte de 28 comprimés pelliculés à 2 mg (B)
Numéro d'autorisation
67230 (Swissmedic)
Titulaire de l'autorisation
Novartis Pharma Schweiz AG, Risch; domicile: 6343 Rotkreuz.
Cette notice d'emballage a été vérifiée pour la dernière fois en juin 2025 par l'autorité de contrôle des médicaments (Swissmedic).
TravoTim-Vision
Qu’est-ce que le collyre TravoTim-Vision et quand doit-il être utilisé ?
Le collyre TravoTim-Vision est une combinaison du travoprost et du timolol. Le travoprost est un analogue de la prostaglandine qui agit en augmentant l'évacuation d'humeur aqueuse, ce qui diminue la pression dans l'œil. Le timolol est un bêta-bloquant qui agit en réduisant la production d'humeur aqueuse à l'intérieur de l'œil. Les deux substances agissent ensemble et réduisent la pression intra-oculaire.
Le collyre TravoTim-Vision est utilisé pour traiter une pression élevée à l'intérieur de l'œil. Cette pression peut conduire à une maladie appelée glaucome.
Le collyre TravoTim-Vision doit être utilisé uniquement selon la prescription du médecin.
De quoi faut-il tenir compte en dehors du traitement ?
Si vous portez des lentilles de contact souples, retirez vos lentilles avant d'appliquer le collyre. Après avoir instillé le collyre, attendez 15 minutes avant de remettre vos lentilles.
Quand le collyre TravoTim-Vision ne doit-il pas être utilisé ?
N'utilisez pas TravoTim-Vision si vous êtes allergique aux prostaglandines, aux bêta-bloquants ou à l'un des autres composants du médicament.
Le collyre TravoTim-Vision ne doit pas être utilisé:
·par des patients souffrant ou ayant souffert de troubles respiratoires, par ex. d'asthme, de bronchite ou d'autres maladies sévères des voies respiratoires ou de problèmes de bronches, ou d'une bronchopneumopathie chronique obstructive;
·en cas de problèmes cardiaques tels qu'un ralentissement du pouls, un infarctus du myocarde, ou d'autres problèmes de rythme cardiaque;
·par des femmes susceptibles de tomber enceinte, à moins que des mesures de contraception adéquate n'aient été prises;
·si la surface de l'œil (cornée) est trouble;
·lors d'un fort rhume allergique.
Quelles sont les précautions à observer lors de l’utilisation du collyre TravoTim-Vision ?
Veuillez informer votre médecin si vous avez actuellement ou si vous avez eu dans le passé:
·une maladie coronarienne (dont les symptômes peuvent être oppression thoracique, difficulté à respirer);
·des troubles du rythme cardiaque (tels qu'un rythme cardiaque lent ou irrégulier);
·des problèmes respiratoires, de l'asthme ou une bronchite chronique obstructive;
·des problèmes liés à une mauvaise circulation du sang (comme le syndrome de Raynaud);
·un excès d'hormones thyroidiennes;
·du diabète ou d'autres problèmes de glycémie (TravoTim-Vision peut masquer les symptômes d'une hypoglycémie);
·des maladies de la cornée;
·une faiblesse musculaire;
·des réactions allergiques graves.
Veuillez informer votre médecin que vous utilisez TravoTim-Vision avant une intervention chirurgicale car TravoTim-Vision peut modifier les effets de certains médicaments utilisés pendant l'anesthésie.
Le travoprost peut augmenter la longueur, l'épaisseur et/ou le nombre de vos cils et assombrir leur couleur.
Peu à peu, c.-à-d. sur plusieurs mois, voire quelques années, le travoprost peut modifier la couleur de votre iris (partie colorée de votre œil), en particulier si vos yeux sont de couleur mixte. Ce changement peut être permanent. Si vous avez des yeux de couleur mixte, discutez-en avec votre médecin avant de commencer le traitement.
Travoprost peut assombrir la peau autour de l'œil, y compris la paupière.
Si vous avez moins de 18 ans, vous ne devez pas utiliser le collyre TravoTim-Vision car l'efficacité et la sécurité d'emploi de ce médicament n'ont pas encore été examinées chez les patients de moins de 18 ans.
Le travoprost peut être absorbé par la peau et ne devra donc pas être utilisé par les femmes enceintes ou souhaitant de le devenir. En cas de contact du produit avec la peau, lavez immédiatement la zone concernée.
Des interactions sont possibles entre TravoTim-Vision et d'autres médicaments que vous utilisez, y compris les collyres pour le traitement du glaucome.
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie, si vous êtes allergique, ou si vous prenez déjà d'autres médicaments (même en automédication !) ou utilisez d'autres médicaments à usage ophtalmique.
TravoTim-Vision contient de l'huile de ricin hydrogénée PEG-40 qui peut causer des réactions cutanées.
Conduite de véhicule et utilisation de machines
Il est possible que votre vision soit légèrement floue juste après l'application du collyre TravoTim-Vision. Ne conduisez pas de véhicule et n'utilisez pas de machines avant que cet effet ait disparu.
Le collyre TravoTim-Vision peut-il être utilisé pendant la grossesse ou l’allaitement ?
Pendant la grossesse ou l'allaitement, vous ne devez utiliser le collyre TravoTim-Vision que si son emploi vous est expressément autorisé par votre médecin ou votre pharmacien.
Comment utiliser le collyre TravoTim-Vision ?
Posologie usuelle
Adultes: Instiller une fois par jour – le matin ou le soir, mais toujours à la même heure – une goutte dans l'œil ou les yeux à traiter.
Ne traitez les deux yeux avec le collyre TravoTim-Vision que si votre médecin vous a dit de le faire. Utilisez TravoTim-Vision aussi longtemps que votre médecin vous l'a prescrit.
Utilisez le collyre TravoTim-Vision dans les yeux uniquement, à l'exclusion de tout autre emploi.
Comment utiliser le collyre TravoTim-Vision ?
(image)
·Ouvrez le sachet d'emballage juste avant d'utiliser un flacon de TravoTim-Vision pour la première fois (figure 1). Sortez-en le flacon et inscrivez la date d'ouverture sur l'étiquette dans l'espace prévu à cet effet.
·Prenez le flacon de TravoTim-Vision et munissez-vous d'un miroir.
·Lavez-vous les mains.
·Dévissez le capuchon.
·Saisissez le flacon entre le pouce et les autres doigts et maintenez-le en position renversée.
·Penchez la tête en arrière. Avec un doigt propre, tirez doucement votre paupière vers le bas pour créer un sillon entre la paupière et votre œil. Placez l'embout du flacon tout près de votre œil. Utilisez le miroir si cela vous aide.
·Déposez à présent une goutte, comme montré à la figure 2.
·Ne touchez pas votre œil, vos paupières, les surfaces voisines ou d'autres surfaces avec le compte-gouttes, car cela peut infecter le collyre.
·Appuyez légèrement sur le flacon pour libérer une goutte de TravoTim-Vision à la fois (figure 3).
·Après avoir instillé TravoTim-Vision, pressez avec un doigt sur le coin de votre œil près du nez pendant 2 minutes (figure 4). Ceci empêche le passage d'une quantité excessive de TravoTim-Vision dans le reste du corps.
·Si vous devez traiter les deux yeux, répétez ces étapes pour l'autre œil.
·Refermez bien le flacon immédiatement après usage.
Si une goutte tombe à côté de votre œil, recommencez l'instillation.
Si un œil a reçu trop de médicament, rincez-le à l'eau tiède. Ne mettez plus d'autres gouttes avant que l'heure soit venue d'instiller la goutte suivante.
Si vous avez oublié d'appliquer le collyre TravoTim-Vision, continuez le traitement selon le plan habituel. Ne doublez pas la dose pour compenser l'oubli d'une dose précédente.
Si vous utilisez un autre collyre, attendez au moins 5 minutes entre l'application de TravoTim-Vision et celle de l'autre collyre.
L'utilisation et la sécurité d'emploi du collyre travoprost-timolol chez les enfants et les jeunes n'ont pas été vérifiées jusqu'ici. Le médicament n'est donc pas employé dans ce groupe d'âge.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible, ou au contraire trop forte.
Quels effets secondaires TravoTim-Vision collyre peut-il provoquer ?
L'utilisation du collyre TravoTim-Vision peut provoquer les effets secondaires suivants:
Des effets secondaires locaux peuvent apparaître chez certains patients qui utilisent le collyre TravoTim-Vision. Ces effets peuvent être désagréables, mais disparaissent rapidement pour la plupart.
Des modifications cutanées définitives peuvent apparaître dans la région de l'œil, comme un assombrissement de la peau ou des paupières ou une modification de la couleur de l'iris.
En cas d'inquiétude, contactez votre médecin ou votre pharmacien.
Très fréquent (concerne plus d'un utilisateur sur 10)
Effets oculaires: rougeur de l'œil.
Fréquent (concerne 1 à 10 utilisateurs sur 100)
Effets oculaires: inflammations de l'œil, vision floue, sécheresse oculaire, douleurs oculaires, démangeaisons dans l'œil, sensation de corps étranger, irritations oculaires.
Effets sur le corps: nervosité, étourdissement, maux de tête, augmentation ou diminution de la pression artérielle, douleurs dans les mains et les pieds.
Occasionnel (concerne 1 à 10 utilisateurs sur 1’000)
Effets oculaires: inflammations de la surface oculaire, inflammation de l'iris, conjonctivite, inflammation de la paupière, sensibilité à la lumière, vision réduite, démangeaisons sur la paupière, inflammation des glandes lacrymales, fatigue oculaire, saignements dans l'œil, gonflement de l'œil, augmentation du larmoiement, rougeur sur la paupière, encroûtement du bord de la paupière, croissance accrue des cils, modification de la couleur de la peau (paupière et autour de l'œil).
Effets sur le corps: allergie, vertiges, maux de tête, ralentissement du rythme cardiaque, essoufflement, inflammation cutanée, forte croissance capillaire, insuffisance respiratoire, toux.
Rare (concerne 1 à 10 utilisateurs sur 10’000)
Effets oculaires: modifications de la cornée, inflammation des glandes de la paupière, mauvaise position des cils.
Effets sur le corps: modification de la voix, douleurs cervicales, urticaire.
Les effets indésirables supplémentaires de TravoTim-Vision, rapportés après la mise sur le marché et dont la fréquence est cependant inconnue, comprennent:
Effets oculaires: gonflement de la rétine, inflammation des yeux, inflammation de la surface de l’œil, inflammation de la paupière, augmentation du larmoiement, gonflement des yeux, gonflement des paupières, rougeur des yeux, paupière tombante, yeux enfoncés, modifications de la couleur de l'iris (l'anneau coloré autour de la pupille).
Effets secondaires généraux: voir, ressentir ou entendre des choses qui n’existent pas (hallucinations), vertiges, douleurs thoraciques, augmentation du rythme cardiaque, détresse respiratoire, toux, asthme, gonflement des membres, baisse de la tension artérielle, altération du gôut, éruption cutanée, chute des cheveux, dépression.
Les effets indésirables supplémentaires suivants, non rapportés avec TravoTim-Vision, ont été observés chez les patients traités par des collyres contenant du travoprost ou du timolol:
Effets oculaires: yeux rouges douloureux, augmentation du larmoiement, rougeurs, gonflements, cloques sur la paupière, affections de la conjonctive, troubles du cristallin, traumatisme à l'oeil après une intervention chirurgicale sur celui-ci, diminution de la sensibilité cornéenne, vision double.
Effets sur le corps: faible taux de sucre dans le sang, insomnie, cauchemars, perte de mémoire, diminution du débit sanguin vers le cerveau, faiblesse neuromusculaire chronique, palpitations, infarctus, arrêt cardiaque, mains et pieds froids, altération du goût, nausées, douleurs abdominales, desquamation de la peau, douleurs musculaires, troubles sexuels, diminution de la libido, perte de force et d'énergie, douleurs cervicales, sécheresse de la bouche, constipation, réactions allergiques systémiques, gonflement du visage et de la gorge, éruption cutanée qui démange, éruption cutanée locale et sur tout le corps, démangeaisons, difficultés respiratoires, toux, vertiges, douleurs thoraciques, gonflements, rhume, diarrhée, vomissements.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention ?
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention « EXP » sur le récipient.
Délai d’utilisation après ouverture
Vous devez jeter le collyre 4 semaines après avoir ouvert le flacon pour la première fois, car la stérilité du contenu n'est alors plus garantie. Inscrivez la date d'ouverture du flacon sur l'étiquette et sur la boîte.
Remarques concernant le stockage
Conservez le médicament à température ambiante (15–25 °C) et hors de la portée des enfants. Une fois le traitement terminé, rapportez l'emballage avec ce qu'il reste de médicament à la personne qui vous l'a remis (médecin, pharmacien) pour qu'elle l'élimine comme il se doit.
Remarques complémentaires
Pour empêcher une contamination du collyre par des microbes, il est important que vous évitiez tout contact de l'extrémité du compte-gouttes avec les paupières ou le tour des yeux. Vous devez également éviter de toucher l'extrémité du compte-gouttes avec les mains.
Bien fermer le flacon après emploi.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient TravoTim-Vision ?
TravoTim-Vision est un collyre stérile contenant comme principes actifs 40 µg de travoprost et 5 mg de timolol (sous forme de maléate de timolol) par 1 ml.
TravoTim-Vision contient l'agent conservateur chlorure de benzalkonium ainsi que d'autres excipients.
Où obtenez-vous TravoTim-Vision ? Quels sont les emballages à disposition sur le marché ?
Les gouttes oculaires TravoTim-Vision ne vous sont remises en pharmacie que sur présentation d'une ordonnance médicale.
Des emballages de 2,5 ml et de 3 x 2,5 ml sont disponibles.
Numéro d’autorisation
67232 (Swissmedic)
Titulaire de l’autorisation
OmniVision AG, 8212 Neuhausen
Cette notice d’emballage a été vérifiée pour la dernière fois en mars 2021 par l’autorité de contrôle des médicaments (Swissmedic).
Was sollte dazu beachtet werden?
Es ist wichtig, dass der Augeninnendruck durch Ihren Arzt oder Ihre Ärztin regelmässig überprüft wird.
Wann darf GANFORT Unit Dose nicht angewendet werden?
·Wenn Sie eine Atemwegserkrankung haben wie Asthma und/oder schwere chronisch obstruktive Lungenerkrankung (Lungenerkrankung, die krankhafte Atemgeräusche, Atemprobleme und/oder chronischen Husten verursacht) oder anderweitige Atemprobleme oder früher daran gelitten haben.
·Wenn Sie Herzprobleme haben wie niedrige Herzfrequenz, Herzblock (Erregungsleitungsstörung) oder Herzinsuffizienz.
·Wenn Sie auf einen der Wirkstoffe (Bimatoprost oder Timolol) oder Hilfsstoffe von GANFORT Unit Dose oder auf Betablocker allergisch reagieren.
GANFORT Unit Dose soll bei Personen unter 18 Jahren nicht angewendet werden, es sei denn, Ihr Arzt oder Ihre Ärztin hat dies trotzdem empfohlen.
XTANDI™ Filmtabletten
Was ist XTANDI und wann wird es angewendet?
XTANDI enthält den Wirkstoff Enzalutamid. XTANDI ist ein Arzneimittel, welches die Aktivität androgener Hormone (wie z.B. Testosteron) blockiert, wodurch das Wachstum von Prostatakrebs verlangsamt werden kann.
XTANDI wird bei der Behandlung erwachsener Männer
·mit nicht metastasiertem hormonsensitivem Prostatakarzinom und biochemischem Rezidiv die ein hohes Risiko der Metastasierung haben, in Kombination mit einem LHRH-Agonist
·mit metastasiertem Prostatakrebs, in Kombination mit einer Androgenentzugstherapie
·mit nicht metastasiertem Prostatakarzinom, welche auf eine Androgenentzugstherapie oder chirurgische Behandlung zur Senkung des Testosteronspiegels nicht mehr ansprechen, in Kombination mit einem LHRH-Agonist
·mit metastasiertem Prostatakarzinom (CRPC), welche auf eine Androgenentzugstherapie zur Senkung des Testosteronspiegels nicht mehr ansprechen, in Kombination mit einem LHRH-Agonist
·mit metastasiertem Prostatakarzinom (CRPC), welche auf eine Androgenentzugstherapie oder chirurgische Behandlung zur Senkung des Testosteronspiegels nicht mehr ansprechen, nach Versagen einer Chemotherapie, in Kombination mit einem LHRH-Agonist eingesetzt.
XTANDI erhalten Sie auf Verschreibung Ihres Arztes bzw. Ihrer Ärztin.
Wann darf XTANDI nicht eingenommen / angewendet werden?
·Wenn Sie allergisch (überempfindlich) auf Enzalutamid oder einen der anderen Inhaltsstoffe dieses Arzneimittels sind.
·Wenn Sie schwanger sind oder schwanger werden können.
Wann ist bei der Einnahme / Anwendung von XTANDI Vorsicht geboten?
Sprechen Sie vor der Einnahme von XTANDI mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin:
·wenn Sie nach Einnahme von XTANDI oder anderen Arzneimitteln jemals einen schweren Hautausschlag oder Hautabschälung, Blasenbildung und/oder wunde Stellen im Mund entwickelt haben
·wenn bei Ihnen eine Erkrankung der Leber besteht
·wenn bei Ihnen eine Erkrankung der Nieren besteht
·wenn bei Ihnen eine Erkrankung des Herzens besteht
·wenn Sie mit Chemotherapie, wie Docetaxel, behandelt werden
Krampfanfälle
XTANDI kann Krampfanfälle auslösen. (Siehe auch die Abschnitte zu «andere Arzneimittel und XTANDI» bzw. «Welche Nebenwirkungen kann XTANDI haben?»).
Ihr Risiko für einen Krampfanfall könnte unter anderem erhöht sein:
·wenn bei Ihnen in der Vergangenheit Krampfanfälle aufgetreten sind
·wenn Sie entweder regelmässig oder von Zeit zu Zeit grosse Mengen an Alkohol trinken
·wenn Sie in der Vergangenheit eine schwere Kopfverletzung oder einen Unfall mit Beteiligung des Kopfes hatten
·wenn Sie in der Vergangenheit bestimmte Arten eines Schlaganfalls hatten
·wenn Sie in der Vergangenheit einen Hirntumor oder Krebsmetastasen im Gehirn hatten oder aktuell haben
·wenn Sie Arzneimittel einnehmen, die Krampfanfälle verursachen oder welche die Krampfschwelle senken (Siehe auch Abschnitt: «Andere Arzneimittel und XTANDI» weiter unten)
Wenn bei Ihnen während der Behandlung mit XTANDI ein Krampfanfall auftritt:
Informieren Sie so schnell wie möglich Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Ihr Arzt bzw. Ihre Ärztin kann entscheiden, ob Sie die Einnahme von XTANDI beenden müssen.
Wenn Sie allergisch reagieren auf Enzalutamid kann dies zu Hautausschlag oder Schwellung im Gesicht, der Zunge, der Lippen oder des Rachens führen. Wenn Sie allergisch reagieren auf Enzalutamid oder einen der anderen Inhaltsstoffe dieses Arzneimittels sollten Sie XTANDI nicht nehmen.
Risiko für neue Krebsarten (sekundäre Primärtumore)
Es gab Berichte über neue (zweite) Krebsarten, einschliesslich Blasen- und Dickdarmkrebs bei Patienten, die mit XTANDI behandelt wurden.
Suchen Sie so schnell wie möglich Ihren Arzt oder Ihre Ärztin auf, wenn Sie Anzeichen von Magen-Darm-Blutungen, Blut im Urin oder häufig das dringende Bedürfnis Wasser zu lassen bemerken, wenn Sie XTANDI einnehmen.
Schwere Hautreaktionen
Im Zusammenhang mit einer XTANDI Behandlung wurde über schwerwiegenden Hautausschlag oder Hautabschälung, Blasenbildung und/oder wunde Stellen im Mund berichtet, einschliesslich Stevens-Johnson-Syndrom. Beenden Sie die Einnahme von XTANDI und kontaktieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie eines der Symptome in Zusammenhang mit diesen schwerwiegenden Hautreaktionen bemerken (siehe «Welche Nebenwirkungen kann XTANDI haben?»).
Schwierigkeiten beim Schlucken aufgrund der Produktgrösse
Es gab Berichte über Patienten, die aufgrund der Produktgrösse, Schwierigkeiten beim Schlucken dieses Arzneimittels hatten, einschliesslich Berichten über Erstickungsanfälle. Schlucken Sie die Tabletten ganz, mit einer ausreichenden Menge Wasser.
Wenn einer der oben genannten Umstände auf Sie zutrifft oder Sie sich nicht sicher sind, sprechen Sie mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin, bevor Sie dieses Arzneimittel einnehmen.
Kinder und Jugendliche
XTANDI darf nicht von Kindern unter 18 Jahren eingenommen werden.
Andere Arzneimittel und XTANDI
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie andere Arzneimittel einnehmen, vor Kurzem eingenommen haben oder möglicherweise in naher Zukunft einnehmen werden. Bei gleichzeitiger Einnahme mit XTANDI können diese Medikamente das Risiko für einen Krampfanfall erhöhen.
Sie sollten nicht mit der Einnahme eines neuen Arzneimittels beginnen oder mit der Einnahme eines Arzneimittels aufhören, ohne Ihren Arzt bzw. Ihre Ärztin, die Ihnen XTANDI verschrieben hat, davon in Kenntnis zu setzen.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie eines der folgenden Arzneimittel einnehmen. Diese Arzneimittel und XTANDI können sich gegenseitig in ihrer Wirksamkeit beeinflussen:
Dies beinhaltet gewisse Arzneimittel mit folgendem Gebrauch:
·Behandlung bakterieller Infektionen (z.B. Clarithromycin, Doxycyclin, Rifampicin)
·Behandlung von Depressionen oder bestimmten psychiatrischen Erkrankungen (z.B. Diazepam, Midazolam, Haloperidol)
·Senkung des Cholesterinspiegels (z.B. Gemfibrozil, Atorvastatin, Simvastatin)
·Behandlung von Herzerkrankungen oder Senkung des Blutdrucks (z.B. Bisoprolol, Digoxin, Dilitazem, Felodipin, Nicardipin, Nifedipin, Propranolol, Verapamil)
·Verhinderung der Abstossung von transplantierten Organen (z.B. Cyclosporin, Tacrolimus)
·Behandlung einer HIV-Infektion (z.B. Indinavir, Ritonavir)
·Behandlung einer Epilepsie (z.B. Carbamazepin, Clonazepam, Phenobarbiton, Phenytoin, Primidon, Valproinsäure)
·Behandlung von Schlafstörungen (z.B. Zolpidem)
·Behandlung schwerwiegender Erkrankungen infolge Entzündungen (z.B. Dexamethason, Prednisolon)
·Behandlung von Schilddrüsenerkrankungen (z.B. Levothyroxin)
·Vermeidung von Blutgerinnseln (z.B. Warfarin, Acenocumarol, Clopidogrel)
·Behandlung von Krebserkrankungen (z.B. Docetaxel, Cabazitaxel, Irinotecan, Sunitinib, Methotrexat)
·Behandlung eines Ulcus oder einer Magenerkrankung (z.B. Omeprazol)
·Behandlung von Gicht (z.B. Cholchizin)
·Vorbeugung von Herzerkrankungen oder Schlaganfall (z.B. Dabigatran etexilat)
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie eines der oben aufgeführten Arzneimittel einnehmen. Die Dosen anderer Arzneimittel, die Sie einnehmen, müssen eventuell angepasst werden.
XTANDI kann Auswirkungen auf Ihre Fahrtüchtigkeit oder Ihre Fähigkeit, Werkzeuge oder Maschinen zu bedienen, haben. Unter der Behandlung mit XTANDI wurden verminderte Konzentrationsfähigkeit, Schwindel und Krampfanfälle beobachtet. Falls Sie eines dieser Symptome haben oder wenn bei Ihnen das Risiko für Krampfanfälle erhöht ist (siehe auch «Wann ist bei der Einnahme von XTANDI Vorsicht geboten?»), sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin.
Während der Behandlung und bis drei Monate nach Ende der Behandlung ist die Anwendung von Kondomen nötig, wenn der Patient sexuellen Kontakt hat.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d.h. es ist nahezu «natriumfrei».
Informieren Sie Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin, wenn Sie:
·an anderen Krankheiten leiden,
·Allergien haben oder
·andere Arzneimittel (auch selbst gekaufte!) einnehmen
Darf XTANDI während einer Schwangerschaft oder in der Stillzeit eingenommen / angewendet werden?
·XTANDI ist nicht zur Anwendung bei Frauen vorgesehen. Dieses Arzneimittel kann bei Einnahme in der Schwangerschaft das ungeborene Kind schädigen oder möglicherweise zum Abbruch der Schwangerschaft führen und darf daher nicht von Frauen eingenommen werden, die schwanger sind, schwanger werden können oder die ein Kind stillen.
·Dieses Arzneimittel kann möglicherweise Auswirkungen auf die männliche Fruchtbarkeit haben.
·Wenden Sie bei Geschlechtsverkehr mit einer Frau, die schwanger ist oder schwanger werden könnte, während der Behandlung und bis drei Monate nach deren Ende sowohl ein Kondom als auch eine weitere Verhütungsmethode an. Wenden Sie bei Geschlechtsverkehr mit einer schwangeren Frau ein Kondom an, um das ungeborene Kind zu schützen.
·Betreuerinnen/Pflegerinnen (siehe «Wie verwenden Sie XTANDI?»)
Wie verwenden Sie XTANDI?
Nehmen Sie dieses Arzneimittel immer genau nach Anweisung Ihres Arztes bzw. Ihrer Ärztin ein. Bitte fragen Sie bei Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin nach, wenn Sie sich nicht sicher sind. Die übliche Dosis beträgt 160 mg (vier Tabletten zu 40 mg oder zwei Tabletten zu 80 mg), die einmal am Tag jeweils zur selben Zeit eingenommen werden.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Einnahme von XTANDI
·Schlucken Sie die Tabletten ganz mit einer ausreichenden Menge Wasser.
·Die Tabletten nicht kauen, zerschneiden, oder zerkleinern.
·Die Tabletten können mit oder ohne Nahrung eingenommen werden.
·XTANDI Tabletten sollten soweit möglich nur vom Patienten und den Pflegern oder Pflegerinnen angefasst werden. Frauen die schwanger sind oder es werden könnten, sollten gebrochene oder beschädigte XTANDI Tabletten nicht ohne Schutz wie etwa Handschuhe handhaben.
Anwendung bei Kindern und Jugendlichen
Die Anwendung und Sicherheit von XTANDI bei Kindern unter 18 Jahren ist bisher nicht geprüft worden. XTANDI darf von Kindern unter 18 Jahren nicht eingenommen werden.
Wenn Sie eine grössere Menge von XTANDI eingenommen haben, als Sie sollten
Wenn Sie mehr Tabletten einnehmen, als Ihnen verschrieben wurde, beenden Sie die Einnahme von XTANDI und kontaktieren Sie Ihren Arzt bzw. Ihre Ärztin. Ihr Risiko für einen Krampfanfall oder andere Nebenwirkungen ist möglicherweise erhöht.
Wenn Sie die Einnahme von XTANDI vergessen haben
·Wenn Sie vergessen, XTANDI zur üblichen Zeit einzunehmen, nehmen Sie Ihre übliche Dosis ein, sobald Sie sich daran erinnern.
·Wenn Sie vergessen, XTANDI während eines ganzen Tages einzunehmen, nehmen Sie Ihre übliche Dosis am folgenden Tag zur gewohnten Zeit ein.
·Wenn Sie länger als einen Tag vergessen, XTANDI einzunehmen, informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin.
Nehmen Sie nicht die doppelte Dosis ein, um die vergessene Dosis auszugleichen.
Wenn Sie die Einnahme von XTANDI abbrechen
Beenden Sie die Einnahme dieses Arzneimittels nicht, ohne dass Ihr Arzt bzw. Ihre Ärztin Sie dazu auffordert.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, fragen Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin.
Welche Nebenwirkungen kann XTANDI haben?
Wie alle Arzneimittel kann XTANDI Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Krampfanfälle
Krampfanfälle treten mit einer höheren Wahrscheinlichkeit auf, wenn Sie mehr als die empfohlene Dosis dieses Arzneimittels einnehmen, wenn Sie gleichzeitig gewisse andere Arzneimittel einnehmen, oder wenn bei Ihnen unabhängig von den Arzneimitteln ein erhöhtes Risiko für Krampfanfälle vorliegt (siehe «Wann ist bei der Einnahme von XTANDI Vorsicht geboten?»).
Wenn Sie einen Krampfanfall erleiden, informieren Sie Ihren Arzt bzw. Ihre Ärztin. Nehmen Sie XTANDI nicht weiter ein.
Andere mögliche Nebenwirkungen sind:
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
·Infektionen
·Ermüdung
·Hitzewallungen
·Knochenbrüche
·Hoher Blutdruck
·Schwäche
·Stürze
Häufig (betrifft 1 bis 10 von 100 Anwendern)
·Kreislaufkollaps
·Angstgefühle
·Trockene Haut
·Juckreiz
·Gedächtnisschwierigkeiten
·Verminderte Konzentrationsfähigkeit
·Brustvergrösserung bei Männern
·Symptome des Restless-legs-syndrom (unkontrollierter Drang Körperteile zu bewegen, gewöhnlich der Beine)
·Vergesslichkeit
·Geschmacksveränderung
·Schmerzen im Rücken oder den Unterschenkeln
·Erschwerte Blasen- oder Darmkontrolle
·Schlaflosigkeit
·Schwindel
·Kopfschmerzen
·Kribbeln der Haut
·Ischämische Herzkrankheit (verminderte Blutversorgung der Herzkranzgefässe)
·Muskelsteifigkeit
·Blut im Urin
·Durchfall
·Beeinträchtigung der Denkleistung
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern)
·Krampfanfälle
·Halluzinationen
Häufigkeit nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden)
·Schwierigkeiten beim Schlucken dieses Medikaments, einschliesslich Erstickungsanfälle
·Übelkeit
·Erbrechen
·Hautausschlag
·Muskelschmerzen
·Muskelspasmen
·Muskelschwäche
·Rückenschmerzen
·Schwellung des Gesichts, der Zunge, der Lippen und/oder des Rachens
·Schwere Hautreaktionen, die sich als rötliche, nicht erhöhte, zielscheibenähnliche oder kreisrunde Flecken auf dem Rumpf zeigen, häufig mit Blasenbildung in der Mitte, Hautabschälung, Geschwüren in Mund, Rachen, Nase, Genitalien oder Augen, denen Fieber und grippeähnliche Symptome vorausgehen können (Stevens-Johnson-Syndrom)
·Verminderter Appetit
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Nicht über 30 °C lagern.
In der Originalverpackung aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Weitere Hinweise
Nehmen Sie keine Tablette ein die beschädigt ist oder Anzeichen von Manipulation aufweist.
Arzneimittel sollten nicht im Abwasser oder Haushaltsabfall entsorgt werden. Fragen Sie Ihren Apotheker bzw. Ihre Apothekerin, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr benötigen.
Weiter Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in XTANDI enthalten?
Wirkstoffe
XTANDI 40 mg: 1 Filmtablette enthält 40 mg Enzalutamid als Wirkstoff.
XTANDI 80 mg: 1 Filmtablette enthält 80 mg Enzalutamid als Wirkstoff.
Hilfsstoffe
Hypromellose-Acetat-Succinat, Mikrokristalline Cellulose (E460), Siliciumdioxid (E551), Croscarmellose-Natrium (E468) Magnesiumstearat (E572), Hypromellose (E464), Talkum, Macrogol 8000, Titandioxid (E171), gelbes Eisenoxid (E172).
Wo erhalten Sie XTANDI? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
XTANDI 40 mg: Packungen zu 112 Filmtabletten
XTANDI 80 mg: Packungen zu 56 Filmtabletten
Zulassungsnummer
67236 (Swissmedic)
Zulassungsinhaberin
Astellas Pharma AG, Wallisellen.
Diese Packungsbeilage wurde im Dezember 2024 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
XTANDI™ Comprimés filmés
Qu'est-ce que XTANDI et quand doit-il être utilisé?
XTANDI contient comme principe actif de l'enzalutamide. XTANDI est un médicament qui bloque l'activité des hormones androgènes (comme p.ex. la testostérone), ce qui peut ralentir la croissance du cancer de la prostate.
XTANDI est utilisé pour le traitement des hommes adultes
·atteints de cancer de la prostate hormonosensible non métastatique et de récidive biochimique à haut risque de métastases, en association avec un agoniste de la LHRH
·atteints de cancer de la prostate métastatique, en association avec une thérapie de privation androgénique
·atteints de carcinomes de la prostate non métastatiques qui ne répondent plus au traitement par privation androgénique ou au traitement chirurgical visant à abaisser les niveaux de testostérone, en association avec un agoniste de la LHRH
·atteints de cancer de la prostate métastatique (CRPC) qui ne répond plus au traitement de privation androgénique pour abaisser les niveaux de testostérone, en combinaison avec un agoniste de la LHRH
·atteints de carcinome de la prostate métastatique (CRPC) qui ne répond plus au traitement de privation androgénique ou au traitement chirurgical visant à abaisser les niveaux de testostérone, après l'échec de la chimiothérapie, en association avec un agoniste de la LHRH.
XTANDI vous est remis sur prescription médicale.
Quand XTANDI ne doit-il pas être pris/utilisé?
·Si vous êtes allergique (hypersensible) à l'enzalutamide ou à l'un des autres composants de ce médicament.
·Si vous êtes enceinte ou si vous êtes susceptible de l'être.
Quelles sont les précautions à observer lors de la prise/de l'utilisation de XTANDI?
Avant de prendre XTANDI, veuillez informer votre médecin ou votre pharmacien:
·si vous avez déjà développé des éruptions cutanées graves ou une desquamation de la peau, des cloques et/ou des plaies buccales après avoir pris XTANDI ou d'autres médicaments
·si vous souffrez d'une maladie du foie
·si vous souffrez d'une maladie des reins
·si vous souffrez d'une maladie du cœur
·si vous êtes traité par chimiothérapie par docétaxel
Convulsions
XTANDI peut déclencher des crises convulsives (voir aussi les paragraphes « Utilisation de XTANDI et d'autres médicaments » et « Quels effets secondaires XTANDI peut-il provoquer ? »).
Le risque d'apparition d'une crise convulsive pourrait entre autres être accru dans les situations suivantes:
·si vous avez souffert par le passé de crises convulsives
·si vous consommez régulièrement ou sporadiquement de grandes quantités d'alcool
·si, par le passé, vous avez été grièvement blessé à la tête ou si vous avez eu un accident ayant impliqué la tête
·si vous avez souffert par le passé de certains types d'accident vasculaire cérébral
·si vous souffrez actuellement ou avez souffert par le passé d'une tumeur cérébrale ou de métastases dans le cerveau
·si vous prenez des médicaments qui provoquent des crises convulsives ou qui abaissent le seuil épileptogène (voir aussi le paragraphe « Utilisation de XTANDI et d'autres médicaments »)
Si une crise convulsive se déclare pendant le traitement par XTANDI:
Informez votre médecin ou votre pharmacien aussi rapidement que possible de la situation. Votre médecin pourra décider si vous devez interrompre la prise de XTANDI.
Si vous présentez une réaction allergique à l'enzalutamide, celle-ci peut se manifester sous la forme d'éruption cutanée ou de gonflement du visage, de la langue, des lèvres ou dans la gorge. Si vous présentez une réaction allergique à l'enzalutamide ou à l'un des autres ingrédients du présent médicament, vous ne devez pas prendre XTANDI.
Risque de nouveaux cancers (seconds cancers primitifs)
Des cas de nouveaux (seconds) cancers, dont des cancers de la vessie et du côlon, ont été rapportés chez des patients traités par XTANDI.
Consultez votre médecin dès que possible si vous remarquez des signes de saignement gastro-intestinal, de sang dans les urines ou si vous ressentez souvent un besoin urgent d'uriner pendant le traitement par XTANDI.
Réactions cutanées graves
Des éruptions cutanées graves ou une desquamation de la peau, des cloques et/ou des plaies buccales, y compris un syndrome de Stevens-Johnson, ont été rapportées en association avec le traitement XTANDI. Si vous remarquez l'un des symptômes liés à ces réactions cutanées graves, arrêtez d'utiliser XTANDI et consultez immédiatement un médecin (voir « Quels effets secondaires XTANDI peut-il provoquer ? »).
Difficultés de déglutition en raison de la taille du produit
Des cas de patients ayant des difficultés à avaler ce médicament en raison de la taille du produit ont été rapportés, y compris des cas d'étouffement. Il est conseillé d'avaler les comprimés entiers avec une quantité suffisante d'eau.
Si vous êtes concerné par l'une des circonstances mentionnées ci-dessus ou si vous avez des doutes, informez-en votre médecin ou votre pharmacien avant de prendre ce médicament.
Enfants et adolescents
XTANDI ne doit pas être pris par les enfants de moins de 18 ans.
Utilisation de XTANDI et d'autres médicaments
Veuillez informer votre médecin ou votre pharmacien si vous prenez actuellement d'autres médicaments, si vous en avez pris récemment ou si vous allez en prendre prochainement. Si l'un de ces médicaments est pris en même temps que XTANDI, le risque de crise convulsive peut être augmenté.
Ne débutez pas la prise d'un nouveau médicament et ne stoppez pas la prise d'un ancien médicament sans en informer le médecin qui vous a prescrit XTANDI.
Veuillez informer votre médecin ou votre pharmacien si vous prenez l'un des médicaments suivants. Ces médicaments et XTANDI peuvent avoir une influence réciproque sur leur efficacité:
Il s'agit de médicaments utilisés dans les cas suivants:
·traitement des infections bactériennes (p.ex. clarithromycine, doxycycline, rifampicine)
·traitement des dépressions ou de certaines maladies psychiatriques (p.ex. diazépam, midazolam, halopéridol)
·pour abaisser le taux de cholestérol (p.ex. gemfibrozil, atorvastatine, simvastatine)
·traitement de maladies cardiaques ou baisse de la pression artérielle (p.ex. bisoprolol, digoxine, diltiazem, félodipine, nicardipine, nifédipine, propranolol, vérapamil)
·pour empêcher le rejet d'organes transplantés (p.ex. ciclosporine, tacrolimus)
·traitement d'une infection par le VIH (p.ex. indinavir, ritonavir)
·traitement d'une épilepsie (p.ex. carbamazépine, clonazépam, phénobarbitone, phénytoïne, primidone, acide valproïque)
·traitement des troubles du sommeil (p.ex. zolpidem)
·traitement de maladies plus sévères dues à des inflammations (p.ex. dexaméthasone, prednisolone)
·traitement des maladies thyroïdiennes (p.ex. lévothyroxine)
·pour éviter les caillots sanguins (p.ex. warfarine, acénocoumarol, clopidogrel)
·traitement d'affections cancéreuses (p.ex. docétaxel, cabazitaxel, irinotécan, sunitinib, méthotrexate)
·traitement d'un ulcère ou d'une maladie gastrique (p.ex. oméprazole)
·traitement de la goutte (p.ex. colchicine)
·prévention de maladies cardiaques ou d'un accident vasculaire cérébral (p.ex. étexilate de dabigatran)
Informez votre médecin ou votre pharmacien si vous prenez l'un des médicaments mentionnés ci-dessus. Il faudra éventuellement ajuster la posologie d'autres médicaments que vous prenez.
XTANDI peut avoir une influence sur votre aptitude à conduire des véhicules ou à utiliser des outils ou des machines. Une diminution de la capacité de concentration, des vertiges et des convulsions ont été observés sous traitement par XTANDI. Si vous présentez l'un de ces symptômes ou un risque élevé de convulsions (voir aussi « Quelles sont les précautions à observer lors de la prise de XTANDI ? »), parlez-en à votre médecin.
L'utilisation de préservatifs est nécessaire pendant le traitement et jusqu'à trois mois après la fin du traitement lorsque le patient a des rapports sexuels.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu'il est essentiellement « sans sodium ».
Veuillez informer votre médecin ou votre pharmacien si
·vous souffrez d'une autre maladie,
·vous êtes allergique ou
·vous prenez déjà d'autres médicaments (même en automédication!).
XTANDI peut-il être pris/utilisé pendant la grossesse ou l'allaitement?
·XTANDI n'est pas prévu pour une utilisation chez la femme. S'il est pris pendant la grossesse, ce médicament peut porter atteinte à l'enfant à naître ou éventuellement conduire à une interruption de grossesse. C'est pourquoi il ne doit pas être pris par les femmes enceintes, par celles qui pourraient le devenir ou par celles qui allaitent.
·Il est possible que ce médicament ait des répercussions sur la fertilité masculine.
·Lors de rapports sexuels avec une femme enceinte ou qui pourrait tomber enceinte, utilisez un préservatif ainsi qu'une autre méthode contraceptive pendant le traitement et jusqu'à trois mois après la fin de celui-ci. Lors de rapports sexuels avec une femme enceinte, utilisez un préservatif pour protéger l'enfant à naître.
·Tutrices/infirmières (voir « Comment utiliser XTANDI ? »)
Comment utiliser XTANDI?
Prenez toujours ce médicament exactement comme vous l'a indiqué votre médecin. En cas de doute, veuillez vous adresser à votre médecin ou à votre pharmacien. La dose habituelle est de 160 mg (quatre comprimés de 40 mg ou deux comprimés de 80 mg), à prendre une fois par jour toujours au même moment.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Prise de XTANDI
·Avalez les comprimés entiers avec une quantité suffisante d'eau.
·Ne pas mâcher, casser ou écraser les comprimés.
·Les comprimés peuvent être pris avec ou sans nourriture.
·Les comprimés XTANDI doivent, autant que possible, être touchés uniquement par les patients et les infirmiers ou infirmières. Les femmes qui sont enceintes ou en âge de procréer ne doivent pas manipuler les comprimés de XTANDI cassés ou endommagés sans protection, par exemple des gants.
Utilisation chez les enfants et les adolescents
L'utilisation et la sécurité de XTANDI chez les enfants de moins de 18 ans n'ont jusqu'ici pas été examinées. XTANDI ne doit pas être utilisé chez les enfants de moins de 18 ans.
Si vous avez pris plus de XTANDI que nécessaire
Si vous avez pris plus de comprimés que le nombre prescrit, interrompez la prise de XTANDI et contactez votre médecin. Votre risque de crise convulsive ou d'autres effets secondaires est probablement élevé.
Si vous avez oublié de prendre XTANDI
·Si vous oubliez de prendre XTANDI à l'heure habituelle, prenez la dose habituelle dès que vous remarquez votre oubli.
·Si vous oubliez de prendre XTANDI pendant tout un jour, prenez la dose habituelle le jour suivant, à l'heure habituelle.
·Si vous oubliez de prendre XTANDI pendant plus qu'un jour, informez-en immédiatement votre médecin.
Ne doublez pas la dose pour rattraper une dose oubliée.
Si vous interrompez la prise de XTANDI
N'interrompez pas la prise du médicament sans que votre médecin vous l'ait prescrit.
Si vous avez d'autres questions en lien avec l'utilisation du médicament, adressez-vous à votre médecin ou à votre pharmacien.
Quels effets secondaires XTANDI peut-il provoquer?
Comme tous les médicaments, XTANDI peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Crises convulsives
La probabilité de crises convulsives est accrue si vous prenez ce médicament à une dose supérieure à celle qui vous a été recommandée, si vous prenez simultanément certains autres médicaments ou si vous présentez un risque accru de crises convulsives, indépendamment des médicaments que vous prenez (voir « Quelles sont les précautions à observer lors de la prise de XTANDI ? »).
Si vous souffrez d'une crise convulsive, informez-en votre médecin. Interrompez la prise de XTANDI.
D'autres effets indésirables possibles sont:
Très fréquent (concerne plus d'un utilisateur sur 10)
·infections
·fatigue
·bouffées de chaleur
·fractures osseuses
·hypertension artérielle
·asthénie
·chutes
Fréquent (concerne 1 à 10 utilisateurs sur 100)
·collapsus cardiovasculaire
·sentiments d'anxiété
·sécheresse de la peau
·démangeaisons
·troubles de la mémoire
·difficultés de concentration
·développement excessif des glandes mammaires chez l'homme
·symptômes du syndrome des jambes sans repos (besoin incontrôlé de bouger certaines parties du corps, habituellement les jambes)
·pertes de mémoire
·changement de la perception du goût
·douleurs dans le dos ou les jambes
·difficultés de contrôle de la vessie ou de l'intestin
·insomnie
·vertige
·maux de tête
·fourmillements au niveau de la peau
·cardiopathie ischémique (diminution de l'apport de sang des vaisseaux coronaires)
·raideur musculaire
·présence de sang dans l'urine
·diarrhée
·troubles cognitifs
Occasionnel (concerne 1 à 10 utilisateurs sur 1000)
·crise convulsive
·hallucinations
Fréquence inconnue (ne peut être estimée sur la base des données disponibles)
·Difficultés à avaler ce médicament, y compris des cas d'étouffement
·nausée
·vomissements
·éruption cutanée
·douleurs musculaires
·spasmes musculaires
·faiblesses musculaires
·douleurs dorsales
·gonflement du visage, de la langue, des lèvres et/ou dans la gorge
·réactions cutanées sévères, se manifestant par des taches rougeâtres non surélevées en forme de cibles ou de cercles sur le tronc, souvent accompagnées de cloques centrales, de desquamation de la peau, d'ulcères de la bouche, de la gorge, du nez, des organes génitaux et des yeux pouvant être précédées de fièvre et de symptômes pseudo-grippaux (syndrome de Stevens-Johnson)
·diminution de l'appétit
Si vous remarquez des effets secondaires, veuillez en informer votre médecin, votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Ne pas conservez au-dessus de 30 °C.
Conserver dans l'emballage d'origine.
Conserver hors de portée des enfants.
Remarques complémentaires
Si un comprimé est endommagé ou s'il porte des traces de manipulation, ne l'avalez pas.
Les médicaments ne doivent pas être éliminés avec les eaux usées ou les déchets ménagers. Demandez à votre pharmacien comme éliminer le médicament une fois le traitement terminé.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient XTANDI?
Principes actifs
XTANDI 40 mg: 1 comprimé filmé contient 40 mg d'enzalutamide comme principe actif.
XTANDI 80 mg: 1 comprimé filmé contient 80 mg d'enzalutamide comme principe actif.
Excipients
Succinate d'acétate d'hypromellose, cellulose microcristalline (E460), silice colloïdale anhydre (E551), croscarmellose sodique (E468), stéarate de magnésium (E572), hypromellose (E464), talc, macrogol 8000, dioxyde de titane (E171), oxyde de fer jaune (E172).
Où obtenez-vous XTANDI? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
XTANDI 40 mg: emballages de 112 comprimés filmés.
XTANDI 80 mg: emballages de 56 comprimés filmés.
Numéro d'autorisation
67236 (Swissmedic)
Titulaire de l'autorisation
Astellas Pharma SA, Wallisellen.
Cette notice d'emballage a été vérifiée pour la dernière fois en décembre 2024 par l'autorité de contrôle des médicaments (Swissmedic).
Quofenix®
Was ist Quofenix und wann wird es angewendet?
Quofenix ist ein Antibiotikum, das den Wirkstoff Delafloxacin enthält. Delafloxacin gehört zur Arzneimittelgruppe der «Fluorchinolone».
Es wirkt, indem Enzyme des Bakteriums gehemmt werden, die es zur Vervielfältigung und Reparatur seiner DNS braucht. Durch diese Enzymhemmung tötet Quofenix die Bakterien ab, die die Infektion hervorrufen.
Quofenix wird zur Behandlung der folgenden schweren bakteriellen Infektionen bei Erwachsenen eingesetzt, die durch andere Antibiotika nicht behandelt werden können:
·Akute Infektionen der Haut und des Gewebes unter der Haut
·Ausserhalb des Spitals erworbene Lungenentzündung (Pneumonie) Quofenix darf nur auf Verschreibung des Arztes oder der Ärztin eingenommen werden.
Was sollte dazu beachtet werden?
Dieses Arzneimittel wurde Ihnen von Ihrem Arzt bzw. Ihrer Ärztin zur Behandlung Ihrer gegenwärtigen Erkrankung verschrieben.
Das Antibiotikum Quofenix ist nicht gegen alle Mikroorganismen wirksam, welche Infektionskrankheiten verursachen. Die Anwendung eines falsch gewählten oder nicht richtig dosierten Antibiotikums kann Komplikationen verursachen. Wenden Sie es deshalb nie von sich aus für die Behandlung anderer Erkrankungen oder anderer Personen an. Auch bei einem Rückfall Ihrer gegenwärtigen Infektion dürfen Sie Quofenix nicht ohne erneute ärztliche Konsultation anwenden.
Wann darf Quofenix nicht eingenommen werden?
Sie sollten Fluorchinolon-/Chinolon-Antibiotika, einschliesslich Quofenix, nicht anwenden, wenn bei Ihnen in der Vergangenheit bei der Anwendung von Chinolonen oder Fluorchinolonen eine schwerwiegende unerwünschte Wirkung aufgetreten ist. In diesem Fall sollten Sie sich so schnell wie möglich an Ihren Arzt bzw. Ihre Ärztin wenden.
Quofenix darf nicht eingenommen werden,
·wenn Sie allergisch gegen Delafloxacin oder einen der sonstigen Bestandteile dieses Arzneimittels sind.
·wenn Sie allergisch gegen ein anderes Fluorchinolon- oder Chinolon-Antibiotikum (Gyrasehemmer) sind.
·wenn bei Ihnen bereits früher im Zusammenhang mit einer Behandlung mit ähnlichen Wirkstoffen (Chinolone oder Fluorchinolone) Sehnenerkrankungen/-schäden aufgetreten sind.
·wenn Sie schwanger sind, schwanger werden könnten oder vermuten, schwanger zu sein.
·wenn Sie stillen.
·wenn Sie ein Kind oder ein(e) heranwachsende(r) Jugendliche(r) unter 18 Jahren sind.
Wann ist bei der Einnahme von Quofenix Vorsicht geboten?
Fluorchinolon-/Chinolon-Antibiotika, einschliesslich Quofenix, wurden mit sehr seltenen, aber schwerwiegenden Nebenwirkungen in Verbindung gebracht, von denen einige lang anhaltend (über Monate oder Jahre andauernd), die Lebensqualität beeinträchtigend oder möglicherweise bleibend sind. Dazu gehören Sehnen-, Muskel- und Gelenkschmerzen der oberen und unteren Gliedmassen, Schwierigkeiten beim Gehen, ungewöhnliche Empfindungen wie Kribbeln, Prickeln, Kitzeln, Taubheitsgefühl oder Brennen (Parästhesie), sensorische Störungen einschliesslich Beeinträchtigung des Seh-, Geschmacks-, Riech- und Hörvermögens, Depression, eingeschränktes Erinnerungsvermögen, starke Ermüdung und starke Schlafstörungen.
Wenn Sie bei Anwendung von Quofenix eine dieser unerwünschten Wirkungen bemerken, wenden Sie sich sofort an Ihren Arzt bzw Ihre Ärztin, bevor Sie mit der Behandlung fortfahren. Sie und Ihr Arzt bzw Ihre Ärztin werden entscheiden, ob die Behandlung fortgesetzt werden soll, möglicherweise auch mit einem Antibiotikum aus einer anderen Wirkstoffgruppe.
Delafloxacin kann zu schwerwiegenden unerwünschten Wirkungen verschiedener Organsysteme führen, die zusammen auftreten können. Diese unerwünschten Wirkungen sind Sehnenentzündungen und –risse, Gelenkschmerzen, neuropathische Symptome und psychiatrische Reaktionen. Diese unerwünschten Wirkungen können innerhalb von Stunden bis Wochen nach Anwendung von Quofenix und bei Patienten jeden Alters sowie bei Patienten ohne bereits bestehende Risikofaktoren auftreten.
Sehnenentzündungen und -risse können bereits während der ersten 48 Stunden nach Behandlungsbeginn unter der Behandlung mit Chinolonen und Floroquinolonen auftreten, unter Umständen auch erst mehrere Monate nach Ende der Einnahme. Das Risiko hierfür ist bei Ihnen erhöht, wenn Sie älter sind (über 60 Jahre), ein Organtransplantat erhalten haben, unter Nierenproblemen leiden, wenn Sie gleichzeitig mit Kortikosteroiden behandelt werden oder bei anstrengender körperlicher Aktivität. Beim ersten Anzeichen von Schmerz oder Entzündung (z.B. schmerzvolle Schwellung von Fussknöchel, Handgelenk, Ellenbogen, Schulter oder Knie) sollten Sie die Behandlung abbrechen, die betroffenen Gliedmassen ruhigstellen und unverzüglich Ihren Arzt bzw. Ihre Ärztin informieren. Vermeiden Sie jede unnötige Bewegung, da dies das Risiko eines Sehnenrisses erhöhen kann.
Bei Patienten, die Chinolone wie z.B. Delafloxacin erhielten, wurde über neuropathische Symptome wie Schmerzen, Brennen, Kribbeln, Taubheitsgefühl oder Schwächegefühl berichtet; solche Nervenerkrankungen können rasch auftreten. Sollten Sie nach Einnahme von Quofenix solche Symptome bei sich feststellen, sollten Sie die Behandlung abbrechen und sich sofort mit einem Arzt oder einer Ärztin in Verbindung setzen. Dadurch kann das mögliche Risiko für die Herausbildung eines irreversiblen Nervenschadens verringert werden.
Auch können bereits nach der ersten Gabe von Quofenix psychiatrische Reaktionen auftreten (z.B. Nervosität, Agitation Schlaflosigkeit, Angstzustände, Albträume, paranoide Gedanken, Verwirrtheit, Tremor (Zittern), Halluzinationen und Depressionen). In sehr seltenen Fällen wurde beobachtet, dass sich eine Depression oder psychotische Reaktionen gesteigert haben, so dass es zu Suizidgedanken oder selbstgefährdendem Verhalten wie Suizidversuchen kam. Sollten Sie nach Einnahme von Quofenix solche Symptome und Gedanken bei sich feststellen, müssen Sie die Behandlung abbrechen und sich mit einem Arzt oder einer Ärztin in Verbindung setzen!
Quofenix kann wie andere Chinolon-Antibiotika Krampfanfälle auslösen oder die Krampfschwelle herabsetzen. Falls Krampfanfälle auftreten sollte die Behandlung mit Quofenix unterbrochen werden. Sprechen Sie mit Ihrem Arzt/Ihrer Ärztin, wenn Sie an Anfallsleiden (Epilepsie) oder anderen Störungen des Zentralnervensystems (z.B. erniedrigte Krampfschwelle, Krampfanfälle in der Vorgeschichte, verringerte Hirndurchblutung, Veränderung in der Gehirnstruktur oder Schlaganfall) leiden. In diesen Fällen sollte Quofenix, wie andere Arzneimittel dieser Klasse, mit Vorsicht angewendet werden.
Bitte sprechen Sie mit Ihrem Arzt, Apotheker bzw. mit Ihrer Ärztin, Apothekerin, bevor Sie Quofenix einnehmen, wenn
·bei Ihnen eine Vergrösserung oder „Ausbuchtung” eines grossen Blutgefässes (Aortenaneurysma oder Aneurysma eines grossen peripheren Gefässes) diagnostiziert wurde.
·Sie in der Vergangenheit eine Aortendissektion (Riss in der Wand der Hauptschlagader) erlitten haben.
·bei Ihnen undichte Herzklappen (Herzklappeninsuffizienz) diagnostiziert wurden.
·in Ihrer Familie Fälle von Aortenaneurysma oder Aortendissektion aufgetreten sind oder angeborene Herzklappenfehler, oder andere Risikofaktoren oder prädisponierende (begünstigende) Bedingungen vorliegen (z.B. Bindegewebserkrankungen wie das Marfan-Syndrom oder das Ehlers-Danlos-Syndrom, Turner-Syndrom, Sjögren-Syndrom [eine entzündliche Autoimmunkrankheit], oder Erkrankungen von Blutgefässen wie Takayasu-Arteriitis, Riesenzellarteriitis, Morbus Behçet, Bluthochdruck oder bekannte Atherosklerose [Verengung der Blutgefässe aufgrund von Fettablagerungen], rheumatoide Arthritis [Erkrankung der Gelenke] oder Endokarditis [Herzinnenhautentzündung]).
Wenn Sie plötzlich starke Schmerzen im Bauch, im Brustbereich oder im Rücken verspüren, die die Symptome eines Aortenaneurysmas (Ausbeulung der Aortenwand) und einer Aortendissektion (Aufspaltung der Schichten der Aortenwand) sein können, begeben Sie sich sofort in eine Notaufnahme. Ihr Risiko kann bei gleichzeitiger Behandlung mit systemischen Kortikosteroiden erhöht sein.
Sollten Sie plötzlich unter Atemnot leiden, besonders, wenn Sie flach in Ihrem Bett liegen, oder eine Schwellung Ihrer Fußgelenke, Füße oder des Bauchs bemerken, oder neu auftretendes Herzklopfen verspüren (Gefühl von schnellem oder unregelmäßigem Herzschlag), sollten Sie unverzüglich einen Arzt benachrichtigen.
Wenn Sie unter Myasthenia gravis (eine Muskelkrankheit) leiden, sollte Quofenix mit Vorsicht angewendet werden, da sich die Symptome verschlimmern können.
Bei der Anwendung von Antibiotika sind Fälle von schweren Durchfällen (pseudomembranöse Kolitis) beschrieben worden. Verständigen Sie sofort Ihren Arzt bzw. Ihre Ärztin, wenn bei Ihnen während oder bis zu 2 Monaten nach der Behandlung schwerer Durchfall auftritt. Nehmen Sie erst nach Rücksprache mit Ihrem Arzt bzw. Ihrer Ärztin Mittel gegen Durchfall ein. Beim Auftreten solcher Durchfälle dürfen keine Arzneimittel, die die Darmperistaltik (Darmbewegung) hemmen, eingenommen werden.
Nach Einnahme von Quofenix sind einzelne Fälle besonders schwerwiegender allergischer Reaktionen, sog. «bullöse Hautreaktionen», wie das Stevens-Johnson-Syndrom oder die (möglicherweise lebensbedrohliche) toxische epidermale Nekrolyse beschrieben worden. Anzeichen solcher Hautreaktionen sind:
·schwere Störungen des Allgemeinbefindens und hohe Temperaturen,
·Reaktionen der Schleimhäute: es bilden sich schmerzhafte Blasen im Mund-, Rachen- und Genitalbereich,
·schwere Augenbindehautentzündung.
Während der Behandlung mit Fluorchinolonen wurden schwere Hautreaktionen, einschliesslich Arzneimittelreaktionen mit Eosinophilie und systemischen Symptomen (DRESS) beobachtet. DRESS äussert sich zunächst durch grippeähnliche Symptome und Hautausschlag im Gesicht, dann durch einen ausgedehnten Hautausschlag und hohe Körpertemperatur, erhöhte Leberenzymwerte in Bluttests, Zunahme einer bestimmten Art weisser Blutkörperchen (Eosinophilie) und vergrösserte Lymphknoten.
Sollten Sie nach Einnahme von Quofenix solche Symptome bei sich feststellen, müssen Sie die Behandlung abbrechen und sich sofort mit einem Arzt oder einer Ärztin in Verbindung setzen!
Überempfindlichkeit und allergische Reaktionen (z.B. Hautausschlag) können schon nach der ersten Anwendung von Chinolonen auftreten. In sehr seltenen Fällen können sich anaphylaktische Reaktionen bis hin zum lebensbedrohlichen Schock entwickeln. In diesen Fällen ist Quofenix sofort abzusetzen und eine ärztliche Behandlung (z.B. Schocktherapie) ist einzuleiten.
Chinolon-Antibiotika können bewirken, dass Ihre Haut empfindlicher gegenüber Sonnenlicht oder UV-Bestrahlung reagiert. Während der Behandlung mit Quofenix sollten Sie intensives Sonnenbaden sowie künstliche Ultraviolettbestrahlung möglichst vermeiden, da dies zu Hautreaktionen führen kann.
Da bei der Anwendung von Chinolonen wie Quofenix Blutzuckerstörungen (zu hohe oder zu niedrige Blutzuckerspiegel) auftreten können, wird bei Diabetikern eine sorgfältige Überwachung der Blutzuckerwerte empfohlen. Informieren Sie bitte Ihren Arzt bzw. Ihre Ärztin falls Sie mit Insulin oder mit einem oralen Antidiabetikum behandelt werden.
Informieren Sie Ihren Arzt bzw. Ihre Ärztin vor der Einnahme von Quofenix,
·wenn Sie an einer Störung der Nierenfunktion leiden. Er bzw. sie wird entscheiden, ob Sie mit Quofenix behandelt werden sollen.
·wenn Sie oder ein Familienangehöriger einen Mangel an dem Enzym Glucose-6-Phosphat-Dehydrogenase haben.
Dieses Arzneimittel enthält 39 mg Natrium (Hauptbestandteil von Kochsalz/Speisesalz) pro Tablette. Dies entspricht 2% der für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung.
Kinder und Jugendliche
Quofenix darf bei Kindern und Jugendlichen nicht angewendet werden, da die Anwendung in diesen Altersgruppen nicht ausreichend untersucht worden ist.
Einnahme von Quofenix zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen.
Quofenix Tabletten sollten mindestens 2 Stunden vor und 6 Stunden nach folgenden Arzneimitteln eingenommen werden:
·Mittel zur Neutralisierung der Magensäure (Antazida), Multivitaminpräparate oder andere Produkte, die Magnesium, Aluminium, Eisen oder Zink enthalten
·Sucralfat
·Didanosin als gepufferte Tablette zur Herstellung einer Suspension zum Einnehmen oder als Pulver zur Herstellung einer Lösung zum Einnehmen
Informieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie Vitamin-K-Gegenspieler wie Warfarin (zur Vorbeugung von Blutgerinnseln) einnehmen. Die Einnahme dieser Arzneimittel mit Antibiotika kann zu einem erhöhten Blutungsrisiko führen. Ihr Arzt bzw. Ihre Ärztin muss bei Ihnen möglicherweise regelmässige Blutuntersuchungen durchführen, um die Blutgerinnungsaktivität zu überwachen.
Wirkung auf die Fahrtüchtigkeit und das Bedienen von Maschinen
Quofenix kann Schwindel, Benommenheit und/oder Sehstörungen verursachen. Solange Sie nicht wissen, wie Quofenix bei Ihnen wirkt, dürfen Sie kein Fahrzeug lenken, keine Maschinen bedienen und keine anderen Tätigkeiten ausführen, die Reaktionsfähigkeit und Koordination erfordern.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie an anderen Krankheiten leiden, Allergien haben oder andere Arzneimittel (auch selbstgekaufte!) einnehmen oder äusserlich anwenden!
Darf Quofenix während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Quofenix darf nicht während der Schwangerschaft oder Stillzeit verwendet werden. Quofenix darf nicht angewendet werden bei Frauen im gebärfähigen Alter, die nicht verhüten. Wenn Sie schwanger sind oder stillen, oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, fragen Sie vor der Einnahme dieses Arzneimittels Ihren Arzt bzw. Ihre Ärztin um Rat.
Welche Nebenwirkungen kann Quofenix haben?
Die folgenden Nebenwirkungen können bei der Einnahme von Quofenix auftreten:
Häufige Nebenwirkungen (kann bis zu 1 von 10 Behandelten betreffen):
Pilzinfektion, Kopfschmerzen, Durchfall, Erbrechen, Übelkeit, erhöhte Werte von Leberenzymen (durch Bluttests nachgewiesen), Juckreiz.
Gelegentliche Nebenwirkungen (kann bis zu 1 von 100 Behandelten betreffen):
Schwerwiegende Nebenwirkungen
Wenn bei Ihnen eines oder mehrere der folgenden Symptome auftreten, nehmen Sie dieses Arzneimittel nicht mehr ein und wenden Sie sich umgehend an einen Arzt bzw. eine Ärztin:
·Schluck- und/oder Atembeschwerden und Husten; Anschwellen der Lippen, des Gesichts, der Zunge oder des Rachens; Halstrockenheit oder Engegefühl im Hals und starker Hautausschlag.
Alle diese Beschwerden sind möglicherweise Anzeichen einer schweren Überempfindlichkeitsreaktion und können lebensbedrohend sein (siehe auch «Wann ist bei der Einnahme von Quofenix Vorsicht geboten?»).
·Blutdruckabfall; Verschwommensehen; Schwindelgefühl.
·Bauchschmerzen, möglicherweise mit starkem Durchfall; Fieber und Übelkeit.
Diese Beschwerden sind möglicherweise Anzeichen einer Darminfektion, die sich verschlechtern kann. In diesem Fall ist es wichtig, dass Sie keine Arzneimittel einnehmen, welche die Darmbewegungen beeinträchtigen (siehe auch «Wann ist bei der Einnahme von Quofenix Vorsicht geboten?»).
Verringerung der Anzahl weisser Blutkörperchen (Leukopenie), niedriger Hämoglobin-Wert (Anämie), allergische Reaktion, Blutzuckerspiegel zu hoch, verminderter Appetit, Schlaflosigkeit, Muskelschwäche in den Gliedern, Taubheitsgefühl, Kribbeln, «Ameisenkribbeln», verminderter Tastsinn, Geschmacksstörung, Verschwommensehen, Schwindelgefühl oder Verlust des Gleichgewichts, Störung des Gleichgewichtsgefühls, Klingeln oder Summen/Brummen in den Ohren (Tinnitus), wahrnehmbarer Herzschlag (Palpitationen), hoher Blutdruck, tiefer Blutdruck, Hitzegefühl, Atembeschwerden, Entzündung der Mundschleimhaut (Stomatitis), Bauchschmerzen, Magenbeschwerden/-schmerzen oder Verdauungsstörung, Mundtrockenheit, Blähungen, Verstopfung, veränderte Blutwerte in Bezug auf Leberfunktion (alkalische Phosphatase im Blut erhöht), abnormales Schwitzen, allergische Hautreaktionen, Juckreiz, roter Hautausschlag, Gelenkschmerz, Schmerz und Schwellung von Sehnen, Muskelschmerzen und Schmerzen im Muskel- und Skelettsystem (z.B. Schmerzen in einer Extremität, Rückenschmerzen, Nackenschmerzen), erhöhter Blutspiegel der Kreatinphosphokinase (ein Anzeichen für Muskelschädigung), Nierenfunktionsstörung, Ermüdung, erhöhte Körpertemperatur (Fieber), lokale Schwellung.
Seltene Nebenwirkungen (kann bis zu 1 von 1'000 Behandelten betreffen)
Harnwegsinfektion, Nebenhöhlenentzündung (Sinusitis), niedrige Anzahl gewisser weisser Blutkörperchen (Neutropenie), Abnahme der für die Blutgerinnung notwendigen speziellen Blutzellen, Veränderungen bei Tests zur Messung Ihrer Blutgerinnung, jahreszeitbedingte Allergie, Blutzuckerspiegel zu niedrig, hohe Harnsäurespiegel, hohe Kalium-Blutspiegel, niedrige Kalium-Blutspiegel, Hören von Dingen, die nicht existieren (akustische Halluzination), Angst, abnorme Träume, Verwirrtheit, Schläfrigkeit, Gefühl von Benommenheit oder Schwäche, in der Regel wegen eines Blutdruckabfalls, trockenes Auge, unregelmässiger oder schneller Herzschlag, Abnahme des Herzschlags, geschwollene, rote, gereizte Venen, Blutgerinnsel in einer tiefen Vene, Husten, Halstrockenheit, Entzündung der Magenschleimhaut, Sodbrennen/Säurerückfluss, Verlust des Berührungsempfindens im Bereich des Mundes, Verringerung des Berührungsempfindens im Bereich des Mundes, Zungenbrennen, Verfärbung des Stuhls, Veränderung bei einem Bluttest zur Leberfunktion (Albumin im Blut erniedrigt und Gamma-Glutamyltransferase erhöht), kalter Schweiss, Nachtschweiss, abnormaler Haarausfall, Muskelkrämpfe, Muskelentzündung/-schmerz, Entzündung der Gelenke, Blut im Urin, trüber Urin aufgrund der Gegenwart von festen Bestandteilen (Kristall im Urin), Schüttelfrost, peripheres Ödem, Komplikation durch Medizinprodukt, Verschlechterung des Wundzustandes (Wundkomplikation).
Sehr seltene Fälle von lang (bis zu Monaten oder Jahren) andauernden oder dauerhaften Nebenwirkungen, wie Sehnenentzündungen, Sehnenrisse, Gelenkschmerzen, Schmerzen in den Gliedmassen, Gehstörungen, abnormen Empfindungen wie Stechen, Kribbeln, Brennen, Taubheitsgefühl oder Schmerzen (Neuropathie), Müdigkeit, Gedächtnisstörungen und Konzentrationsbeeinträchtigung, Auswirkungen auf die psychische Gesundheit (darunter Schlafstörungen, Angst, Panikanfälle, Depressionen und Suizidgedanken), sowie Beeinträchtigungen des Gehörs, des Sehvermögens und des Geschmacks- und Geruchssinns wurden mit der Verabreichung von Chinolon- und Fluorchinolon-Antibiotika in Verbindung gebracht, in einigen Fällen unabhängig von bestehenden Risikofaktoren (siehe auch «Wann ist bei der Einnahme von Quofenix Vorsicht geboten?»).
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden)
Grossflächiger Ausschlag, hohe Körpertemperatur, erhöhte Leberenzymwerte, Blutanomalien (Eosinophilie), vergrösserte Lymphknoten und Beteiligung anderer Körperorgane (Arzneimittelreaktion mit Eosinophilie und systemischen Symptomen, auch bekannt als DRESS oder Arzneimittel-Hypersensitivitätssyndrom).
Scharf abgegrenzte, rötliche (erythematöse) Flecken mit/ohne Blasenbildung, die sich innerhalb von Stunden nach der Anwendung von Fluorchinolonen entwickeln und nach der Entzündungsphase mit verbleibender Überpigmentierung abheilen; nach erneuter Anwendung von Fluorchinolonen treten sie in der Regel wieder an der gleichen Stelle der Haut oder Schleimhaut auf.
Höhere Empfindlichkeit der Haut gegenüber Sonnenlicht oder UV-Bestrahlung.
Fälle der Erweiterung und Schwächung der Aortenwand oder Einrisse der Aortenwand (Aneurysmen und Aortendissektionen), die reissen können und tödlich sein können, sowie Fälle undichter Herzklappen wurden bei Patienten, die Fluorchinolone anwenden, berichtet. (Siehe auch «Wann ist bei der Einnahme von Quofenix Vorsicht geboten?»).
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Das Arzneimittel nicht über 30°C, in der Originalverpackung und ausser Reichweite von Kindern aufbewahren.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Quofenix enthalten?
Quofenix ist eine beige bis beige gesprenkelte Tablette.
Wirkstoffe
Der Wirkstoff ist Delafloxacin. Jede Tablette enthält 450 mg Delafloxacin (als Delafloxacin-Meglumin).
Hilfsstoffe
Die sonstigen Bestandteile sind mikrokristalline Cellulose, Povidon K30, Crospovidon, Natriumhydrogencarbonat, Natriumdihydrogenphosphat-Monohydrat, Zitronensäure, Magnesiumstearat.
Wo erhalten Sie Quofenix? Welche Packungen sind erhältlich?
In Apotheken gegen ärztliche Verschreibung, die nur zum einmaligen Bezug berechtigt.
Packungen zu 10, 20 oder 30 Tabletten.
Zulassungsnummer
67238 (Swissmedic).
Zulassungsinhaberin
A. Menarini GmbH, Zürich
Diese Packungsbeilage wurde im Mai 2025 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Quofenix®
Qu'est-ce que Quofenix et quand doit-il être utilisé?
Quofenix est un antibiotique dont le principe actif est la délafloxacine. La délafloxacine appartient au groupe des médicaments appelés «fluoroquinolones».
Il agit en bloquant des enzymes dont les bactéries ont besoin pour copier et réparer leur ADN. En bloquant ces enzymes, Quofenix tue les bactéries responsables de l'infection.
Quofenix est utilisé chez l'adulte pour traiter les infections bactériennes suivantes, lorsque les antibiotiques habituels ne sont pas appropriés:
·Infections aiguës de la peau et des tissus situés sous la peau;
·Pneumonie contractée en dehors de l'hôpital.
Quofenix ne doit être pris que sur prescription du médecin.
De quoi faut-il tenir compte en dehors du traitement?
Ce médicament vous a été prescrit par le médecin pour traiter votre affection actuelle.
L'antibiotique Quofenix n'est pas efficace contre tous les microorganismes qui provoquent des maladies infectieuses. L'utilisation d'un antibiotique mal choisi ou mal dosé peut entraîner des complications. Par conséquent, ne l'utilisez jamais de votre propre initiative pour le traitement d'autres maladies ou chez d'autres personnes. Même si votre infection actuelle récidive, vous ne devez pas utiliser Quofenix sans consulter une nouvelle fois votre médecin.
Quand Quofenix ne doit-il pas être pris?
Vous ne devez pas prendre d'antibiotiques de la famille des quinolones/fluoroquinolones, y compris Quofenix, si vous avez déjà présenté un effet indésirable grave par le passé lors de la prise de quinolones ou de fluoroquinolones. Si tel est le cas, avertissez votre médecin au plus vite avant de prendre ce médicament.
Quofenix ne doit pas être pris
·si vous êtes allergique à la délafloxacine ou à l'un des autres composants de ce médicament;
·si vous êtes allergique à un autre antibiotique de la famille des fluoroquinolones ou des quinolones (inhibiteurs de la gyrase);
·si vous avez déjà souffert de maladies/lésions des tendons en rapport avec un traitement avec des principes actifs similaires (quinolones ou fluoroquinolones);
·si vous êtes enceinte, pourriez le devenir ou pensez que vous pourriez être enceinte;
·si vous allaitez;
·si vous êtes un(e) enfant ou un(e) adolescent(e) de moins de 18 ans.
Quelles sont les précautions à observer lors de la prise de Quofenix?
Les antibiotiques de la famille des quinolones/fluoroquinolones, y compris Quofenix, ont été associés à des effets indésirables très rares, mais graves, dont certains peuvent être persistants (durant des mois ou des années), invalidants ou potentiellement irréversibles. Il s'agit notamment de douleurs dans les tendons, les muscles et les articulations des membres supérieurs et inférieurs, de difficultés à marcher, de sensations anormales telles que des picotements, fourmillements, chatouillements, un engourdissement ou une sensation de brûlure (paresthésies), de troubles sensoriels incluant des troubles de la vue, du goût, de l'odorat et de l'audition, d'une dépression, de troubles de la mémoire, d'une fatigue intense et de troubles sévères du sommeil.
Si vous présentez l'un de ces effets indésirables après avoir pris Quofenix, contactez immédiatement votre médecin avant de poursuivre le traitement. Vous déciderez avec votre médecin de la poursuite du traitement en envisageant éventuellement aussi de recourir à un antibiotique d'une autre famille.
La délafloxacine peut provoquer des effets indésirables graves touchant différents systèmes d'organes et pouvant survenir ensemble. Ces effets indésirables comprennent l'inflammation et la déchirure des tendons, les douleurs articulaires, les symptômes neuropathiques et les réactions psychiatriques. Ces effets indésirables peuvent survenir dans les heures ou les semaines qui suivent l'utilisation de Quofenix, chez les patients de tous âges et sans facteurs de risque préexistants.
L'inflammation et les déchirures de tendons peuvent se produire dès les premières 48 heures du traitement aux quinolones et aux fluoroquinolones, mais aussi éventuellement plusieurs mois après la fin du traitement. Votre risque est plus important si vous avez plus de 60 ans, si vous avez reçu une greffe d'organe, si vous avez des problèmes rénaux, si vous prenez actuellement un traitement à base de corticoïdes ou si vous pratiquez une activité physique intense. Vous devez donc interrompre le traitement dès les premiers signes de douleurs ou d'inflammation (par exemple, gonflement douloureux des chevilles, poignets, coudes, épaules ou genoux), mettre les membres atteints au repos et informer immédiatement votre médecin. Vous devez éviter tout effort inutile, car cela pourrait augmenter le risque de rupture d'un tendon.
Des symptômes neuropathiques tels que douleur, brûlure, picotement, engourdissement ou faiblesse ont été rapportés chez des patients ayant reçu des quinolones, telles que la délafloxacine; ces troubles nerveux peuvent survenir rapidement. Si vous ressentez de tels symptômes après avoir pris Quofenix, vous devez arrêter le traitement et contacter un médecin immédiatement. Cela peut réduire le risque potentiel de développer des lésions nerveuses irréversibles.
Des réactions d'ordre psychiatrique peuvent également se produire dès la première administration de Quofenix (par exemple, nervosité, agitation, insomnie, anxiété, cauchemars, pensées paranoïaques, confusion, tremblements, hallucinations et dépression). Dans de très rares cas, on a observé une augmentation des cas de dépression ou de réactions psychotiques, conduisant à des pensées suicidaires ou à des comportements autodestructeurs tels que des tentatives de suicide. Si vous ressentez de tels symptômes et avez de telles pensées après avoir pris Quofenix, vous devez arrêter le traitement et contacter un médecin immédiatement!
Quofenix, comme d'autres antibiotiques de la famille des quinolones, peut déclencher des crises convulsives ou abaisser le seuil de leur déclenchement. Le traitement par Quofenix doit être interrompu en cas de crises convulsives. Consultez votre médecin si vous souffrez d'épilepsie ou d'autres troubles du système nerveux central (par exemple, abaissement du seuil convulsif, antécédents de crises épileptiques, diminution de la circulation cérébrale, modifications de la structure cérébrale ou AVC). Dans ces cas, Quofenix, comme les autres médicaments de cette classe, doit être utilisé avec prudence.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre Quofenix, si
·une dilatation ou une «poche» formée sur un gros vaisseau sanguin (anévrisme de l'aorte ou anévrisme d'un gros vaisseau périphérique) vous a été diagnostiquée;
·vous avez déjà connu une dissection aortique (déchirure de la paroi de l'aorte) dans le passé;
·vous avez reçu un diagnostic de fuite des valves cardiaques (régurgitation des valves cardiaques);
·vous avez des antécédents familiaux d'anévrisme aortique, de dissection aortique ou de valvulopathie cardiaque congénitale, ou présentez d'autres facteurs de risque ou affections prédisposantes (par exemple, des troubles du tissu conjonctif tels que syndrome de Marfan ou syndrome vasculaire d'Ehlers-Danlos syndrome de Turner, syndrome de Sjögren [une maladie auto-immune inflammatoire], ou des troubles vasculaires tels que l'artérite de Takayasu, l'artérite à cellules géantes, la maladie de Behçet, l'hypertension artérielle ou une athérosclérose connue [rétrécissement des vaisseaux sanguins dû à des dépôts de graisse], la polyarthrite rhumatoïde [une maladie des articulations] ou l'endocardite [une infection du coeur]).
Si vous ressentez une douleur soudaine et intensedans l'abdomen, la poitrine ou le dos, qui peuvent être des symptômes d'un anévrisme et d'une dissection aortiques, rendez-vous immédiatement au service des urgences. Votre risque peut être augmenté si vous êtes traité avec des corticostéroïdes administrés par voie générale.
Si vous ressentez soudainement un essoufflement, en particulier lorsque vous vous allongez sur votre lit, ou si vous remarquez un gonflement de vos chevilles, de vos pieds ou de votre abdomen, ou une nouvelle apparition de palpitations cardiaques (sensation de battements de coeur rapides ou irréguliers), vous devez en informer un médecin immédiatement.
Quofenix doit être utilisé avec prudence si vous souffrez de myasthénie, car les symptômes peuvent s'aggraver.
Des cas de diarrhée sévère (colite pseudo-membraneuse) ont été décrits lors de l'utilisation d'antibiotiques. Informez immédiatement votre médecin si vous souffrez d'une diarrhée grave pendant le traitement ou jusqu'à 2 mois après. Ne prenez pas de médicaments contre la diarrhée avant d'avoir consulté votre médecin. En cas de diarrhée de ce type, il est important de ne pas prendre de médicaments qui empêchent le péristaltisme intestinal (mouvement des intestins).
Des cas isolés de réactions allergiques particulièrement graves, dites «réactions cutanées bulleuses», telles que le syndrome de Stevens-Johnson ou la nécrolyse épidermique toxique (pouvant mettre la vie en danger), ont été décrits après la prise de Quofenix. Ces réactions cutanées se manifestent par les signes suivants:
·grave détérioration de l'état général et température élevée,
·réactions des muqueuses: des cloques douloureuses apparaissent dans la bouche, la gorge et la région génitale,
·grave conjonctivite.
Des réactions cutanées graves incluant la réaction médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS) ont été rapportées lors de l'utilisation des fluoroquinolones. Le syndrome DRESS se manifeste initialement par des symptômes pseudo-grippaux et une éruption cutanée sur le visage, qui se propage ensuite, accompagnée d'une température corporelle élevée, d'une augmentation des taux d'enzymes du foie révélée par les analyses de sang et d'une augmentation d'un certain type de globules blancs (éosinophilie) ainsi que d'un gonflement des ganglions lymphatiques.
Si vous ressentez de tels symptômes après avoir pris Quofenix, vous devez arrêter le traitement et contacter un médecin immédiatement!
Une hypersensibilité et des réactions allergiques (par exemple, une éruption cutanée) peuvent survenir après la première utilisation des quinolones. Dans de très rares cas, des réactions anaphylactiques peuvent aller jusqu'au choc anaphylactique, potentiellement mortel. Dans de tels cas, il est indispensable d'arrêter immédiatement le traitement et d'instaurer un traitement médical (traitement du choc, par exemple).
Les antibiotiques de la famille des quinolones peuvent rendre votre peau plus sensible au soleil ou aux rayons ultra-violets (UV). Vous devez éviter une exposition prolongée au soleil ainsi que les rayons ultraviolets artificiels pendant que vous prenez Quofenix, car ils peuvent entraîner des réactions cutanées.
L'utilisation de quinolones, dont Quofenix, pouvant provoquer des troubles de la glycémie (taux de sucre trop élevé ou trop bas), il est recommandé aux diabétiques de surveiller attentivement leur taux de sucre dans le sang. Si vous prenez de l'insuline ou un traitement oral contre le diabète, informez-en votre médecin.
Avant de prendre Quofenix, veuillez informer votre médecin des points suivants s'ils vous concernent:
·Si vous souffrez de troubles rénaux. Votre médecin décidera alors si vous devez prendre Quofenix ou non;
·Si vous ou un membre de votre famille présentez un déficit de l'enzyme glucose-6-phosphate déshydrogénase.
Ce médicament contient 39 mg de sodium (composant principal du sel de cuisine/table) par comprimé, ce qui équivaut à 2% de l'apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
Enfants et adolescents
Quofenix ne doit pas être utilisé chez les enfants et les adolescents, car son utilisation n'a pas été suffisamment étudiée dans ces groupes de patients.
Prise de Quofenix avec d'autres médicaments
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Les comprimés de Quofenix doivent être pris au moins 2 heures avant ou 6 heures après les médicaments suivants:
·préparations destinées à neutraliser l'acide gastrique (antiacides), préparations multivitaminiques ou autres produits contenant du magnésium, de l'aluminium, du fer ou du zinc
·sucralfate
·didanosine sous forme de comprimé tamponné pour préparation d'une suspension orale ou sous forme de poudre pour préparation d'une solution orale
Informez votre médecin si vous prenez des antagonistes de la vitamine K, tels que la warfarine (pour prévenir la formation de caillots sanguins). La prise de ces médicaments avec des antibiotiques peut entraîner un risque accru de saignement. Votre médecin devra peut-être vous demander de faire régulièrement des analyses de sang pour surveiller votre coagulation sanguine.
Effet sur l'aptitude à la conduite et l'utilisation de machines
Quofenix peut provoquer des vertiges, des étourdissements et/ou des troubles visuels. Tant que vous ne savez pas comment Quofenix agit sur vous, vous ne devez pas conduire de véhicule, utiliser une machine ou effectuer toute autre activité qui nécessite de la réactivité et de la coordination.
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie, si vous êtes allergique ou si vous prenez déjà d'autres médicaments (même en automédication!) ou utilisez déjà d'autres médicaments en usage externe!
Quofenix peut-il être pris pendant la grossesse ou l'allaitement?
Quofenix ne doit pas être utilisé pendant la grossesse et l'allaitement. Quofenix ne doit pas être utilisé chez les femmes en âge de procréer qui n'utilisent pas de contraception. Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, prévenez votre médecin avant de prendre ce médicament.
Comment utiliser Quofenix?
Prenez ce médicament toujours exactement comme prescrit par votre médecin. En cas de doute, adressez-vous à votre médecin.
Après un traitement initial par Quofenix sous forme de perfusion, les comprimés Quofenix vous ont été prescrits pour poursuivre le traitement.
C'est votre médecin qui détermine la dose et la durée de traitement. Le meilleur bénéfice possible de Quofenix ne peut être atteint qu'en respectant strictement les prescriptions.
La posologie recommandée pour Quofenix est de 1 comprimé (450 mg) par voie orale toutes les 12 heures pour une durée totale de 2 à 11 jours en cas d'infections de la peau et de 2 à 7 jours en cas de pneumonie, selon l'avis de votre médecin. Avalez les comprimés sans les croquer avec une quantité suffisante d'eau. Les comprimés peuvent être pris indépendamment des repas.
Si vous avez pris une quantité plus grande de Quofenix que vous n'auriez dû
Si vous prenez accidentellement plus de comprimés que vous ne le devriez, contactez un médecin ou rendez-vous dans un hôpital. Prenez l'emballage du médicament avec vous.
Si vous avez oublié de prendre Quofenix
Si vous avez oublié une dose et qu'il reste au moins 8 heures avant la prochaine dose prévue, prenez-la dès que possible. S'il reste moins de 8 heures avant votre prochaine dose, attendez et prenez la dose suivante à l'heure habituelle.
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre Quofenix
Si vous arrêtez de prendre Quofenix sans l'avis de votre médecin, vos symptômes peuvent s'aggraver. Consultez votre médecin ou votre pharmacien avant d'arrêter de prendre le médicament.
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou à votre pharmacien.
Ne changez pas de votre propre chef le dosage prescrit. Si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte, veuillez vous adresser à votre médecin ou à votre pharmacien.
Quels effets secondaires Quofenix peut-il provoquer?
La prise de Quofenix peut provoquer les effets secondaires suivants:
Fréquent (concerne 1 à 10 utilisateurs sur 100):
Infection fongique, maux de tête, diarrhée, vomissements, nausées, taux élevé d'enzymes hépatiques (mis en évidence par des analyses de sang), démangeaisons.
Occasionnel (concerne 1 à 10 utilisateurs sur 1000):
Effets secondaires graves
Si vous présentez un ou plusieurs des symptômes suivants, cessez de prendre ce médicament et contactez immédiatement un médecin:
·Difficulté à avaler et/ou à respirer et toux; gonflement des lèvres, du visage, de la langue ou de la gorge; sécheresse ou serrement de la gorge et éruption cutanée grave.
Tous ces symptômes peuvent être des signes d'une réaction d'hypersensibilité grave et peuvent mettre la vie en danger (voir également «Quelles sont les précautions à observer lors de la prise de Quofenix?»).
·Chute de la pression artérielle; vision trouble; vertiges.
·Douleurs abdominales (maux de ventre) parfois accompagnées d'une forte diarrhée; fièvre et nausées.
Ces symptômes peuvent être des signes d'une infection intestinale qui peut s'aggraver. Dans ce cas, il est important que vous ne preniez aucun médicament qui perturbe le mouvement de l'intestin (voir également «Quelles sont les précautions à observer lors de la prise de Quofenix?»).
Diminution du nombre de globules blancs (leucopénie), faible taux d'hémoglobine (anémie), réaction allergique, taux de sucre dans le sang trop élevé, diminution de l'appétit, insomnie, faiblesse musculaire dans les membres, sensation d'engourdissement, picotements, «fourmillements», sens du toucher réduit, troubles du goût, vision floue, sensations vertigineuses ou perte d'équilibre, trouble du sens de l'équilibre, tintements ou bourdonnements dans les oreilles (acouphènes), perception des battements cardiaques (palpitations), tension artérielle élevée, tension artérielle basse, bouffées de chaleur, troubles respiratoires, inflammation de la muqueuse buccale [stomatite], maux de ventre, troubles/douleurs gastriques ou troubles digestifs, sécheresse de la bouche, flatulences, constipation, modification des valeurs sanguines liées à la fonction hépatique (phosphatase alcaline sanguine augmentée), transpiration anormale, réactions cutanées allergiques, démangeaisons, éruptions cutanées rouges, douleurs articulaires, douleurs et gonflement des tendons, douleurs musculaires et douleurs musculosquelettiques (par exemple douleurs dans un membre, douleurs du dos, douleurs au cou), créatine phosphokinase sanguine augmentée (signe d'une lésion musculaire), trouble de fonctionnement des reins, fatigue, température corporelle élevée (fièvre), gonflement local.
Effets indésirables rares (pouvant affecter 1 personne sur 1000)
Infection des voies urinaires, sinusite, faible taux de certains globules blancs (neutropénie), faible taux d'hémoglobine (anémie), diminution de cellules sanguines spécifiques nécessaires à la coagulation sanguine, modifications lors des tests pour la mesure de la coagulation, allergie saisonnière, taux de sucre dans le sang trop faible, taux élevé d'acide urique, taux élevé de potassium dans le sang, faible taux de potassium dans le sang, perception auditive de choses qui n'existent pas (hallucinations auditives), anxiété, rêves anormaux, confusion, somnolence, sensation d'étourdissement ou de faiblesse, généralement en raison d'une baisse de tension, sécheresse des yeux, battements cardiaques irréguliers ou rapides, ralentissement des battements cardiaques, veines gonflées, rouges, irritées, caillot sanguin dans une veine profonde, toux, gorge sèche, inflammation de la muqueuse gastrique, brûlures d'estomac/remontées acides, perte des sensations tactiles au niveau de la bouche, diminution des sensations tactiles au niveau de la bouche, sensation de brûlure sur la langue, coloration anormale des selles, modification des analyses sanguines en lien avec le fonctionnement du foie (baisse de l'albumine dans le sang, élévation de la gamma-glutamyltransférase dans le sang), sueurs froides, sueurs nocturnes, chute anormale des cheveux, crampes musculaires, inflammation/douleurs au niveau des muscles, inflammation des articulations, sang dans les urines, urines troubles en raison de la présence de particules solides (cristaux dans les urines), frissons, œdème périphérique, complication liée à un médicament, aggravation de la plaie (complication).
Très rares cas d'effets secondaires persistants (jusqu'à plusieurs mois ou années), tels que: inflammations de tendons, ruptures de tendons, douleurs articulaires, douleurs dans les membres, difficultés à marcher, sensations anormales telles que des picotements, fourmillements, une sensation de brûlure, un engourdissement ou des douleurs (neuropathie), une fatigue, des troubles de la mémoire et de la concentration, des troubles mentaux (incluant troubles du sommeil, anxiété, attaques de panique, dépression et idées suicidaires), ainsi que des troubles de l'audition, de la vision, du goût ou de l'odorat, ont été rapportés en lien avec l'administration d'antibiotiques de la famille des quinolones et des fluoroquinolones, dans certains cas indépendamment des facteurs de risque préexistants (voir également «Quelles sont les précautions à observer lors de la prise de Quofenix?»).
Indéterminée (ne peut être estimée sur la base des données disponibles)
Une éruption cutanée généralisée, une température corporelle élevée, une augmentation des enzymes du foie, des anomalies sanguines (éosinophilie), un gonflement des ganglions lymphatiques et une atteinte d'autres organes dans le corps (réaction médicamenteuse avec éosinophilie et symptômes systématiques, également appelée syndrome DRESS ou syndrome d'hypersenibilité médicamenteuse).
Plaques érythémateuses nettement délimitées, avec/sans cloques, apparaissant dans les heures suivant l'administration de fluoroquinolones et laissant une hyperpigmentation résiduelle post-inflammatoire après cicatrisation; la réaction réapparaît habituellement au même endroit sur la peau ou la muqueuse en cas de nouvelle exposition aux fluoroquinolones.
Augmentation de la sensibilité au soleil ou aux rayons UV.
Des cas d'élargissement et d'affaiblissement de la paroi aortique ou de déchirure de la paroi aortique (anévrismes et dissections), qui peuvent se rompre et mettre en jeu le pronostic vital, ainsi que de fuite de valves cardiaques ont été signalés chez des patients recevant des fluoroquinolones. (Voir «Quelles sont les précautions à observer lors de la prise de Quofenix? ».
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Ne pas conserver au-dessus de 30 °C. Conserver dans l'emballage d'origine et hors de portée des enfants.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Quofenix?
Quofenix est un comprimé beige à beige moucheté.
Principes actifs
Le principe actif est la délafloxacine. Chaque comprimé contient 450 mg de délafloxacine (sous forme de sel de méglumine).
Excipients
Les autres composants sont les suivants: cellulose microcristalline, povidone K30, crospovidone, bicarbonate de sodium, dihydrogénophosphate de sodium monohydraté, acide citrique, stéarate de magnésium.
Où obtenez-vous Quofenix? Quels sont les emballages à disposition sur le marché?
En pharmacie, seulement sur ordonnance médicale non renouvelable.
Emballages de 10, 20 ou 30 comprimés.
Numéro d'autorisation
67238 (Swissmedic).
Titulaire de l'autorisation
A. Menarini GmbH, Zurich
Cette notice d'emballage a été vérifiée pour la dernière fois en mai 2025 par l'autorité de contrôle des médicaments (Swissmedic).
Co-Amoxicillin Spirig HC®, Filmtabletten
Was ist Co-Amoxicillin Spirig HC und wann wird es angewendet?
Co-Amoxicillin Spirig HC ist ein Antibiotikum aus der Gruppe der Penicilline. Es besteht aus zwei Wirkstoffen: Clavulansäure und Amoxicillin.
Clavulansäure beherrscht den Hauptabwehr- oder Resistenzmechanismus zahlreicher resistenter Bakterien gegenüber Penicillinen und schützt auf diese Weise Amoxicillin, das damit die Bakterien zerstören kann. Diese Wirkungsweise macht Co-Amoxicillin Spirig HC gegen zahlreiche bakterielle Infektionen wirksam.
Co-Amoxicillin Spirig HC darf nur auf Verschreibung des Arztes oder der Ärztin zur ausschliesslichen Behandlung der folgenden bakteriellen Infektionen angewendet werden:
·Nasen-, Hals-, Mandeln-, Stirn-/Kieferhöhlen- und Ohreninfektionen;
·Infektionen der Atemwege (Bronchien und Lungen);
·Infektionen der Nieren, Blase und Harnwege;
·Infektionen der Geschlechtsorgane (Gonorrhoe, Schleimausscheidung);
·Gynäkologische Infektionen;
·Infektionen der Haut und Weichteile (Furunkel, Abszesse, etc.).
Was sollte dazu beachtet werden?
Dieses Arzneimittel wurde Ihnen von Ihrem Arzt bzw. Ihrer Ärztin zur Behandlung Ihrer gegenwärtigen Erkrankung verschrieben.
Das Antibiotikum in Co-Amoxicillin Spirig HC ist nicht gegen alle Mikroorganismen, welche Infektionskrankheiten verursachen, wirksam. Die Anwendung eines falsch gewählten oder nicht richtig dosierten Antibiotikums kann Komplikationen verursachen. Wenden Sie es deshalb nie von sich aus für die Behandlung anderer Erkrankungen oder anderer Personen an. Auch bei späteren neuen Infektionen dürfen Sie Co-Amoxicillin Spirig HC nicht ohne erneute ärztliche Konsultation anwenden.
Häufig verschwinden die Krankheitssymptome, bevor die Infektion vollständig ausgeheilt ist. Die Behandlung darf deshalb nicht vorzeitig abgebrochen werden, auch wenn Sie sich besser fühlen.
Je nach Umständen und gemäss Vorschrift des Arztes bzw. der Ärztin kann die Behandlung bis zu zwei Wochen oder länger dauern.
Wann darf Co-Amoxicillin Spirig HC nicht eingenommen werden?
Sie sollten Co-Amoxicillin Spirig HC nicht einnehmen, wenn Sie früher auf Co-Amoxicillin Spirig HC, auf Penicilline oder Cephalosporine allergisch reagiert haben. Eine Allergie oder eine Überempfindlichkeit zeigt sich z.B. in Symptomen wie rote Hautflecken, Fieber, Asthma, Atemnot, Kreislaufbeschwerden, Schwellungen der Haut (z.B. Nesselfieber) und der Schleimhäute, Hautausschlägen oder einer schmerzhaften Zunge.
Bei bekannter oder vermuteter Überempfindlichkeit auf einen der anderen Bestandteile des Arzneimittels darf Co-Amoxicillin Spirig HC nicht angewendet werden.
Sie dürfen Co-Amoxicillin Spirig HC nicht einnehmen, wenn Sie an Pfeiffer'schem Drüsenfieber oder an einer lymphatischen Leukämie leiden.
Darf Co-Amoxicillin Spirig HC während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Schwangerschaft
Über die Einnahme von Arzneimitteln jeglicher Art während einer Schwangerschaft ist mit grösster Vorsicht und nur nach Rücksprache mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin zu entscheiden. In Studien bei schwangeren Frauen mit vorzeitigem Blasensprung wurde berichtet, dass die vorbeugende Behandlung mit einer Amoxicillin-Clavulansäure-Kombination ein erhöhtes Risiko von teilweise schwerwiegenden gewebsschädigenden Darmentzündungen beim Neugeborenen verursachen kann.
Stillzeit
Da Co-Amoxicillin Spirig HC in geringer Menge in die Muttermilch übertritt, ist bei Säuglingen mit der Möglichkeit einer Überempfindlichkeitsreaktion (mit Symptomen wie Hautrötung und Fieber) oder Durchfall zu rechnen. Deshalb sollte Co-Amoxicillin Spirig HC während der Stillzeit nicht eingenommen oder abgestillt werden.
Orientieren Sie auf jeden Fall Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie schwanger sind, es werden möchten oder stillen. Es sind die einzigen, die entscheiden können, ob Sie während diesen Zeiten Co-Amoxicillin Spirig HC einnehmen dürfen.
Wie verwenden Sie Co-Amoxicillin Spirig HC?
Übliche Dosierung
Co-Amoxicillin Spirig HC ist vorzugsweise zu Beginn der Mahlzeiten mit mindestens einem halben Glas Wasser einzunehmen. Damit wird eine optimale Wirkung und Verträglichkeit erzielt. Sofern vom Arzt bzw. von der Ärztin nicht anders verordnet, gilt folgende Dosierung:
Erwachsene:
Leichte, mittelschwere bis schwere Infektionen:
·3 × tägl. Co-Amoxicillin Spirig HC 625 mg (500/125) oder in bestimmten Fällen
·2 × tägl. Co-Amoxicillin Spirig HC 1000 mg (875/125)
Die Bruchrille der 1000 mg Filmtablette ist lediglich dazu gedacht, die Tabletteneinnahme zu vereinfachen. Die Tabletten sind nicht zum Halbieren der Dosierung bestimmt. Beide Hälften müssen gleichzeitig eingenommen werden.
Eine begonnene Antibiotika-Therapie sollte so lange wie vom Arzt bzw. von der Ärztin verordnet durchgeführt werden.
Die Krankheitssymptome verschwinden oft, bevor die Infektion vollständig ausgeheilt ist. Brechen Sie aus diesem Grund die Therapie nicht vorzeitig ab, selbst wenn Sie sich wohler fühlen.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Co-Amoxicillin Spirig HC haben?
Gastrointestinale Störungen wie Magenbeschwerden oder Übelkeit. Ebenfalls vorkommen können Reaktionen wie Brechreiz, Appetitlosigkeit, Blähungen, Erbrechen, Durchfall, weicher Stuhl, Dyspepsie, Bauchschmerzen und Entzündung der Zunge oder der Mundschleimhaut.
Wenn Co-Amoxicillin Spirig HC zu Beginn der Mahlzeiten eingenommen wird, sind Magen-Darm-Beschwerden weniger häufig.
Allergische Reaktionen sind mit Co-Amoxicillin Spirig HC häufig, wie bei allen Arzneimitteln der Gruppe der Penicilline.
Hautausschläge, Hautrötungen, Juckreiz und Urtikaria (Nesselsucht) können auftreten.
Ebenfalls können Pilzinfektionen der Haut/Schleimhäute auftreten.
Gelegentlich können Schwindelgefühl und Kopfschmerzen auftreten.
Sehr selten können Hyperaktivität, Entzündung der Hirnhaut (aseptische Meningitis), Erregung, Angst, Schlaflosigkeit, Verwirrung, Verhaltensänderungen, Benommenheit, Krämpfe und Empfindungsstörungen auftreten.
Selten stellte man oberflächliche Zahnverfärbungen fest. Diese Erscheinung verschwindet gewöhnlich mit dem Zähneputzen wieder.
Sehr selten wurde eine dunkel belegte Zunge, Hyperkinese (übermässige Bewegungsaktivität), Blutbildveränderungen, Verlängerung der Blutungsdauer und Prothrombinzeit, Leberentzündung (Hepatitis), Nierenentzündung und Nierenfunktionsstörungen beobachtet.
Sehr selten wurden grippeähnliche Symptome mit Hautausschlag, Fieber, geschwollenen Drüsen und abnormalen Blutwerten (einschliesslich weisse Blutkörperchen (Eosinophilie) und Leberenzyme) (Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS) beobachtet (siehe «Wann ist bei der Einnahme von Co-Amoxicillin Spirig HC Vorsicht geboten»).
Es wurden Fälle von schwerwiegenden Hautreaktionen berichtet.
Sehr selten wurden Schmerzen in der Brust beobachtet, die ein Anzeichen für eine potenziell schwerwiegende allergische Reaktion, das so genannte Kounis-Syndrom, sein können.
Bei der Verabreichung von Amoxicillin im Alter von 0-9 Monate können Zahnschmelzschäden (z.B. weisse Streifung, Verfärbung) der definitiven Schneidezähne nicht ausgeschlossen werden.
Über Gelbsucht wurde selten berichtet.
Konsultieren Sie sofort Ihren Arzt bzw. Ihre Ärztin beim Auftreten von:
·Nesselfieber, grossflächigem Hautausschlag, Hautrötungen;
·gelblicher Farbe der Haut oder des weissen Teils der Augen;
·plötzlich einsetzenden Bauchschmerzen oder Erbrechen;
·schweren, blutigen oder anhaltenden Durchfällen;
·Atemproblemen in Form von Asthmaanfällen und Heuschnupfen.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Nicht über 25°C, in der Originalverpackung vor Feuchtigkeit geschützt und für Kinder unerreichbar aufbewahren.
Wenn Sie eine Verfärbung der Co-Amoxicillin Spirig HC Filmtabletten feststellen, könnte es sich um eine Veränderung des Präparates handeln. Falls dies eintritt, wenden Sie sich sofort an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin.
Nach Beendigung der Behandlung ist das Arzneimittel mit dem restlichen Inhalt Ihrer Abgabestelle (Arzt oder Apotheker bzw. Ärztin oder Apothekerin) zum fachgerechten Entsorgen zu bringen.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Co-Amoxicillin Spirig HC enthalten?
Wirkstoffe
Co-Amoxicillin Spirig HC enthält die Wirkstoffe Amoxicillin (als Amoxicillin-trihydrat) und Clavulansäure (als Kaliumsalz). Nachstehend die ausführliche Zusammensetzung der verschiedenen im Handel erhältlichen Filmtabletten:
1 Filmtablette enthält: Amoxicillin Clavulansäure Verhältnis Amoxicillin / Clavulansäure
Co-Amoxicillin Spirig HC 625 mg (500/125) Filmtabletten 500 mg 125 mg 4:1
Co-Amoxicillin Spirig HC 1000 mg (875/125) Filmtabletten (mit Bruchrille) 875 mg 125 mg 7:1
Hilfsstoffe
Filmtabletten 625 mg: Mikrokristalline Cellulose, Carboxymethylstärke-Natrium (Typ A), Magnesiumstearat, Titandioxid, hochdisperses Siliciumdioxid, Talkum, Ethylcellulose, Cetylalkohol, Natriumdodecylsulfat, Hypromellose, Triethylcitrat.
Filmtabletten 1000 mg: Crospovidon, Mikrokristalline Cellulose, Povidon K 25, Talkum, Ethylcellulose, Cetylalkohol, Natriumdodecylsulfat, Titandioxid, Magnesiumstearat, Hypromellose, hochdisperses Siliciumdioxid, Triethylcitrat.
Wo erhalten Sie Co-Amoxicillin Spirig HC? Welche Packungen sind erhältlich?
In Apotheken gegen ärztliche Verschreibung, die nur zum einmaligen Bezug berechtigt.
Co-Amoxicillin Spirig HC 625 mg (500/125) Filmtabletten: Packungen zu 10 und 20 Filmtabletten.
Co-Amoxicillin Spirig HC 1000 mg (875/125) Filmtabletten (mit Bruchrille): Packungen zu 12 und 20 Filmtabletten.
Zulassungsnummer
67240 (Co-Amoxicillin Spirig HC 625 mg), 67241 (Co-Amoxicillin Spirig HC 1000 mg) (Swissmedic)
Zulassungsinhaberin
Spirig HealthCare AG, 4622 Egerkingen
Diese Packungsbeilage wurde im Juni 2023 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Co-Amoxicilline Spirig HC®, Comprimés pelliculés
Qu'est-ce que Co-Amoxicilline Spirig HC et quand doit-il être utilisé?
Co-Amoxicilline Spirig HC est un antibiotique du groupe des pénicillines. Il se compose de deux principes actifs, à savoir l'acide clavulanique et l'amoxicilline.
L'acide clavulanique contrôle le principal mécanisme de défense ou de résistance de nombreuses bactéries résistantes aux pénicillines et protège de cette manière l'amoxicilline, lui permettant ainsi de détruire ces bactéries. Grâce à ce mode d'action, Co-Amoxicilline Spirig HC agit sur un grand nombre d'infections bactériennes.
Co-Amoxicilline Spirig HC ne doit être utilisé que sur prescription médicale et uniquement dans le traitement des infections bactériennes suivantes:
·infections du nez, de la gorge, des amygdales, des sinus frontaux/maxillaires et infections de l'oreille;
·infections des voies respiratoires (bronches et poumons);
·infections des reins, de la vessie et des voies urinaires;
·infections des organes génitaux (gonorrhée, sécrétion de mucus);
·infections gynécologiques;
·infections de la peau et des parties molles (furoncles, abcès, etc.).
De quoi faut-il tenir compte en dehors du traitement?
Ce médicament vous a été prescrit par votre médecin pour le traitement de votre affection actuelle.
L'antibiotique contenu dans Co-Amoxicilline Spirig HC n'agit pas sur l'ensemble des micro-organismes qui provoquent des maladies infectieuses. L'utilisation d'un antibiotique mal choisi ou incorrectement dosé risque d'entraîner des complications. Ne l'utilisez donc jamais de votre propre initiative pour le traitement d'autres affections ou d'autres personnes. Même lors de l'apparition ultérieure de nouvelles infections, il ne faut pas utiliser Co-Amoxicilline Spirig HC sans consultation médicale préalable.
Les symptômes de la maladie disparaissent fréquemment avant la guérison complète de l'infection. Pour cette raison, il ne faut pas arrêter le traitement avant terme, même si vous vous sentez mieux.
Selon les circonstances et d'après la prescription médicale, le traitement peut durer jusqu'à deux semaines, voire plus longtemps.
Quand Co-Amoxicilline Spirig HC ne doit-il pas être pris?
Si vous avez présenté par le passé une réaction allergique à Co-Amoxicilline Spirig HC, aux pénicillines ou aux céphalosporines, vous ne devez pas prendre Co-Amoxicilline Spirig HC. Une allergie ou une hypersensibilité peut se manifester, par exemple, sous forme de symptômes comme taches rouges sur la peau, fièvre, asthme, dyspnée, troubles circulatoires, gonflements au niveau de la peau (urticaire, par exemple) et des muqueuses, éruptions cutanées ou langue douloureuse.
Co-Amoxicilline Spirig HC ne doit pas être utilisé lors d'une hypersensibilité connue ou présumée à l'un de ses autres composants.
Si vous souffrez d'une mononucléose infectieuse ou d'une leucémie lymphoïde, vous ne devez pas prendre Co-Amoxicilline Spirig HC.
Quelles sont les précautions à observer lors de la prise de Co-Amoxicilline Spirig HC?
Ce médicament peut affecter les réactions, l'aptitude à conduire et la capacité à utiliser des outils ou des machines.
Si vous prenez un contraceptif oral (la pilule), il est possible que son efficacité soit réduite au cours d'un traitement par antibiotiques. Cette remarque s'applique également à Co-Amoxicilline Spirig HC. Votre médecin ou votre pharmacien pourra donc vous proposer d'autres méthodes contraceptives.
Des troubles digestifs peuvent survenir au cours de la prise de Co-Amoxicilline Spirig HC. Lors de troubles gastro-intestinaux graves, avec vomissements et diarrhées, il faut stopper la prise du médicament et en informer immédiatement le médecin. Celui-ci doit également être informé en cas d'apparition d'une éruption cutanée ou de démangeaisons.
À l'apparition de diarrhées, il ne faut prendre aucun médicament ralentissant le péristaltisme intestinal (mouvements de l'intestin).
Après la prise de Co-Amxicilline Spirig HC, des cas de réactions cutanées particulièrement graves et potentiellement mortelles ont été rapportés. Les signes de telles réactions cutanées sont:
·symptômes semblables à ceux de la grippe et fièvre;
·éruption cutanée;
·réactions des muqueuses (comme p.ex. gonflement de la muqueuse buccale ou pharyngée, formation de cloques, saignements);
·gonflements du visage ou d'autres parties du corps;
·douleurs dans la poitrine (syndrome de Kounis).
Si vous présentez de tels symptômes après la prise de Co-Amoxicilline Spirig HC, vous devez arrêter le traitement et contacter immédiatement un médecin.
Si vous souffrez d'allergies, d'asthme allergique, de rhume des foins ou d'urticaire, une prudence particulière est de rigueur lors de la prise de Co-Amoxicilline Spirig HC en raison d'une éventuelle hypersensibilité.
Les patients qui doivent en même temps prendre un médicament contenant de l'allopurinol (p.ex. Zyloric®) sont plus sujets aux éruptions cutanées.
Informez votre médecin si vous souffrez de troubles de la fonction rénale; il vous prescrira alors une posologie adaptée à vos besoins, qui pourra éventuellement s'écarter du schéma indiqué ci-après (voir paragraphe «Posologie usuelle»).
Informez votre médecin si vous prenez des médicaments visant à bloquer la coagulation du sang (anticoagulants).
Informez votre médecin si vous prenez des médicaments contenant du mycophénolate mofétil, utilisés à la suite d'une transplantation d'organe pour prévenir les réactions de rejet du greffon.
Si vous prenez un médicament contenant de la digoxine, il faut en informer votre médecin ou votre pharmacien.
Co-Amoxicilline Spirig HC 625 mg et 1000 mg comprimés pelliculés contiennent moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c.-à-d. qu'ils sont essentiellement « sans sodium ».
En cas de fonction rénale ou hépatique insuffisante, une prudence toute particulière est indiquée.
Les pénicillines peuvent réduire l'excrétion du méthotrexate (utilisé dans le traitement des maladies articulaires inflammatoires, du cancer et du psoriasis sévère), ce qui peut entraîner une éventuelle augmentation des effets indésirables.
Veuillez informer votre médecin ou votre pharmacien si
·vous souffrez d'une autre maladie,
·vous êtes allergique ou
·vous prenez déjà d'autres médicaments ou utilisez déjà d'autres médicaments en usage externe (même en automédication!).
Co-Amoxicilline Spirig HC peut-il être pris pendant la grossesse ou l'allaitement?
Grossesse
La décision de prendre un médicament, de quelque nature qu'il soit, au cours d'une grossesse impose une prudence extrême et ne doit pas être prise sans l'avis préalable de votre médecin ou de votre pharmacien. Dans des études effectuées chez des femmes enceintes présentant une rupture prématurée de la poche des eaux, il a été rapporté que le traitement préventif par une combination d'amoxicilline et d'acide clavulanique serait susceptible de provoquer chez le nouveau-né un risque accru d'inflammations intestinales, parfois graves, entraînant des lésions tissulaires.
Allaitement
Étant donné que Co-Amoxicilline Spirig HC passe en faibles quantités dans le lait maternel, il faut compter avec le risque d'une réaction d'hypersensibilité (avec des symptômes comme rougeurs de la peau ou fièvre) ou de diarrhées chez le nourrisson. C'est pourquoi, Co-Amoxicilline Spirig HC ne devrait pas être pris durant l'allaitement, ou le nourrisson devrait être sevré.
Informez en tout cas votre médecin ou votre pharmacien, si vous êtes enceinte, si vous envisagez une grossesse ou si vous allaitez votre enfant. Ils sont les seuls à pouvoir décider si vous pouvez utiliser Co-Amoxicilline Spirig HC pendant ces périodes.
Comment utiliser Co-Amxicilline Spirig HC?
Posologie usuelle
Prendre Co-Amoxicilline Spirig HC de préférence en début de repas avec un demi-verre d'eau au moins. Cela permet d'assurer une efficacité et une tolérance optimales. Sauf prescription médicale contraire, la posologie suivante est applicable:
Adulte
Infections légères, modérées à sévères:
·625 mg de Co-Amoxicilline Spirig HC (500/125) 3 fois par jour ou, dans certains cas,
·1000 mg de Co-Amoxicilline Spirig HC (875/125) 2 fois par jour.
Le sillon de sécabilité des comprimés pelliculés à 1000 mg est uniquement destiné à faciliter la prise du comprimé. Les comprimés ne sont pas destinés à être divisés en deux. Les deux moitiés doivent être prises simultanément.
Une fois commencé, tout traitement par antibiotiques doit être poursuivi pendant la période prescrite par le médecin.
Les symptômes de la maladie disparaissent fréquemment avant la guérison complète de l'infection. Pour cette raison, il ne faut pas arrêter le traitement avant terme, même si vous vous sentez mieux.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou votre pharmacien, si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Co-Amoxicilline Spirig HC peut-il provoquer?
Troubles gastro-intestinaux tels que maux d'estomac ou nausées. Des réactions telles qu'envie de vomir, manque d'appétit, ballonnements, vomissements, diarrhées, selles molles, dyspepsie, douleurs au ventre et inflammations au niveau de la langue ou des muqueuses sont également possibles.
Les troubles gastro-intestinaux sont moins fréquents si Co-Amoxicilline Spirig HC est pris en début de repas.
Des réactions allergiques sous Co-Amoxicilline Spirig HC sont fréquentes, comme avec tous les médicaments du groupe des pénicillines.
Des éruptions cutanées, rougeurs de la peau, démangeaisons et urticaires peuvent survenir.
Des mycoses au niveau de la peau ou des muqueuses sont également possibles.
Occasionnellement, une sensation de vertige et des maux de tête peuvent apparaître.
Très rarement, hyperactivité, inflammation des méninges (méningite aseptique), agitation, anxiété, insomnie, confusion, troubles du comportement, torpeur, convulsions et troubles de la sensibilité sont susceptibles d'apparaître.
Rarement, une coloration superficielle des dents a été observée. Cette manifestation disparaît en général avec le nettoyage des dents.
Très rarement, on a observé une langue chargée d'une couche foncée, des hyperkinésies (activité motrice excessive), des altérations de la formule sanguine, un allongement du temps de saignement et de prothrombine, une inflammation du foie (hépatite), une inflammation des reins et des troubles de la fonction rénale.
Très rarement, on a observé des symptômes semblables à ceux de la grippe avec une éruption cutanée, de la fièvre, un gonflement des ganglions et des valeurs sanguines anormales (y compris globules blancs (éosinophilie) et enzymes hépatiques) (exanthème médicamenteux avec éosinophilie et symptômes systémiques (DRESS)) (voir «Quelles sont les précautions à observer lors de la prise de Co-Amoxicilline Spirig HC»).
Des cas de réactions cutanées graves ont été rapportés.
Très rarement, des douleurs dans la poitrine ont été observées. Celles-ci peuvent être le signe d'une réaction allergique potentiellement grave, appelée syndrome de Kounis.
Lors de l'administration d'amoxicilline entre 0 et 9 mois, des dommages au niveau de l'émail dentaire (p.ex. rayures blanches, coloration) au niveau des incisives maxillaires définitives ne peuvent être exclus.
Une jaunisse a rarement été signalée.
Consultez immédiatement votre médecin lors de l'apparition:
·d'une urticaire, d'une éruption cutanée à grande surface, de rougeurs de la peau;
·d'une coloration jaune de la peau ou du blanc de l'œil;
·de douleurs au ventre ou de vomissements apparaissant soudainement;
·de diarrhées sévères, sanglantes ou persistantes;
·de troubles respiratoires, survenant sous forme de crises d'asthme, ou d'un rhume des foins.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Conserver dans l'emballage d'origine à une température ne dépassant pas 25 °C et protéger de l'humidité. Conserver hors de portée des enfants.
Si vous constatez une coloration des comprimés pelliculés de Co-Amoxicilline Spirig HC, il pourrait s'agir d'une altération du produit. Si tel est le cas, adressez-vous d'emblée à votre médecin ou votre pharmacien.
Après l'arrêt du traitement, le médicament avec le reste de son contenu est à ramener à son point de vente (médecin ou pharmacien) en vue d'une élimination dans les règles.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée desintée aux professionnels.
Que contient Co-Amoxicilline Spirig HC?
Principes actifs
Co-Amoxicilline Spirig HC contient les principes actifs amoxicilline (sous forme d'amoxicilline trihydratée) et acide clavulanique (sous forme de clavulanate de potassium). Vous trouverez ci-après la composition détaillée des différents comprimés pelliculés commercialisés:
1 comprimé pelliculé contient: Amoxicilline Acide clavulanique Rapport amoxicilline/acide clavulanique
Co-Amoxicilline Spirig HC 625 mg (500/125) Comprimés pelliculés 500 mg 125 mg 4: 1
Co-Amoxicilline Spirig HC 1000 mg (875/125) Comprimés pelliculés (avec rainure de sécabilité) 875 mg 125 mg 7: 1
Excipients
Comprimés pelliculés 625 mg: cellulose microcristalline, carboxymethylamidon sodique (type A)-, stéarate de magnésium, dioxyde de titane, silice colloidale anhydre, talc, ethylcellulose, alcool cétylique, sodium de laurilsulfate, hypromellose, citrate de triéthyle.
Comprimés pelliculés 1000 mg: crospovidonum, cellulose microcristalline, povidone K 25, talc, ethylcellulose, alcool cétylique, laurilsulfate de sodium, dioxyde de titane, stéarate de magnésium, hypromellose, silice colloidale anhydre, citrate de triéthyle.
Où obtenez-vous Co-Amoxicilline Spirig HC? Quels sont les emballages à disposition sur le marché?
En pharmacie, seulement sur ordonnance médicale non renouvelable.
Co-Amoxicilline Spirig HC 625 mg (500/125) Comprimés pelliculés: emballages à 10 et 20 comprimés pelliculés.
Co-Amoxicilline Spirig HC 1000 mg (875/125) Comprimés pelliculés (avec rainure de sécabilité): emballages à 12 et 20 comprimés pelliculés.
Numéro d'autorisation
67240 (Co-Amoxicilline Spirig HC 625 mg), 67241 (Co-Amoxicilline Spirig HC 1000 mg) (Swissmedic).
Titulaire de l'autorisation
Spirig HealthCare SA, 4622 Egerkingen.
Cette notice d'emballage a été vérifiée pour la dernière fois en juin 2023 par l'autorité de contrôle des médicaments (Swissmedic).
Ezetimib Simvastatin Zentiva®
Was ist Ezetimib Simvastatin Zentiva und wann wird es angewendet?
Ezetimib Simvastatin Zentiva enthält die beiden Wirkstoffe Simvastatin und Ezetimib. Es senkt erhöhtes Gesamtcholesterin, LDL-Cholesterin (schlechtes Cholesterin) und Triglyceride (fettartige Substanzen) und erhöht HDL-Cholesterin (gutes Cholesterin) im Blut. Es ist für Patienten geeignet, deren Cholesterinwerte zu hoch sind und bei denen eine Diät allein diese Werte nicht genügend senken konnte.
In Ezetimib Simvastatin Zentiva ergänzen sich die Wirkmechanismen der beiden Wirkstoffe in ihrer cholesterinsenkenden Wirkung. Ezetimib Simvastatin Zentiva vermindert die Aufnahme von Cholesterin aus dem Dünndarm und hemmt die körpereigene Cholesterinproduktion in der Leber.
Ihr Arzt oder Ihre Ärztin hat Ihnen Ezetimib Simvastatin Zentiva zur Senkung erhöhter Cholesterinwerte und Triglyceridwerte im Blut verschrieben. Cholesterin ist eine von mehreren Fettsubstanzen, welche im Blutkreislauf vorkommen. Ihr Gesamtcholesterin besteht hauptsächlich aus LDL- und HDL-Cholesterin.
Ezetimib Simvastatin Zentiva soll nur auf Verschreibung des Arztes oder der Ärztin angewendet werden.
Was sollte dazu beachtet werden?
Vor der Behandlung mit Ezetimib Simvastatin Zentiva soll eine cholesterinsenkende Diät durchgeführt und das vorhandene Übergewicht abgebaut werden. Die fett- und cholesterinarme Diät ist auch während der Behandlung mit Ezetimib Simvastatin Zentiva weiterzuführen.
Wann darf Ezetimib Simvastatin Zentiva nicht eingenommen werden?
Nehmen Sie Ezetimib Simvastatin Zentiva nicht ein, wenn Sie auf Ezetimib, Simvastatin oder irgendeinen der Inhaltsstoffe überempfindlich (allergisch) reagieren. Patienten und Patientinnen, die an einer Lebererkrankung leiden oder bei denen aus nicht bekannten Gründen die Blutwerte der Leberenzyme erhöht sind, dürfen Ezetimib Simvastatin Zentiva nicht anwenden.
Ferner darf Ezetimib Simvastatin Zentiva während der Schwangerschaft oder Stillzeit nicht eingenommen werden. Ezetimib Simvastatin Zentiva darf bei Kindern und Jugendlichen unter 18 Jahren nicht angewendet werden, da in dieser Altersgruppe noch keine Erfahrungen mit dem Arzneimittel vorliegen.
Ezetimib Simvastatin Zentiva darf nicht eingenommen werden, wenn Sie eines oder mehrere der nachfolgenden Arzneimittel einnehmen: Arzneimittel gegen Pilze, z.B. Itraconazol (Sporanox®), Ketoconazol (Nizoral®), Posaconazol (Noxafil®) oder Voriconazol (Vfend®), HIV-Proteasehemmer, z.B. Indinavir (Crixivan®), Nelfinavir (Viracept®), Ritonavir (Norvir®), Saquinavir (Invirase®, Fortovase®), Arzneimittel zur Behandlung von Hepatitis C Virusinfektionen, z.B. Boceprevir (Victrelis®) oder Telaprevir (Incivo®), Antibiotika, z.B. Erythromycin (Erythrocin®), Clarithromycin (Klacid®), Telithromycin, systemische Fusidinsäure (Fucidin® Filmtabletten) und bestimmte Antidepressiva, z.B. Nefazodon (Nefadar®), Arzneimittel, die Cobicistat enthalten (Stribild®), Gemfibrozil (Gevilon®) eine cholesterinsenkende Fibrinsäure, Cyclosporin (Sandimmun®) und Danazol (Danatrol®).
Fragen Sie Ihren Arzt oder Ihre Ärztin, wenn Sie nicht sicher sind, ob Ihr Arzneimittel, das Sie einnehmen, oben aufgelistet ist.
Wann ist bei der Einnahme von Ezetimib Simvastatin Zentiva Vorsicht geboten?
Informieren Sie Ihren Arzt oder Ihre Ärztin über alle medizinischen Probleme, die Sie haben oder früher gehabt haben, insbesondere über Leberprobleme und –krankheiten oder Allergien.
Bei der Einnahme von Ezetimib/Simvastatin sind selten erhöhte Werte bestimmter Lebertests (ohne Krankheitszeichen) aufgetreten. Ihr Arzt bzw. Ihre Ärztin wird deshalb diese Laborwerte periodisch kontrollieren. Bei Patienten und Patientinnen, die übermässig Alkohol konsumieren oder Lebererkrankungen in der Vorgeschichte aufweisen, wird der Arzt oder die Ärztin dieses Arzneimittel nur mit besonderen Vorsichtsmassnahmen verordnen. Informieren Sie deshalb Ihren Arzt bzw. Ihre Ärztin wenn Sie grössere Mengen Alkohol konsumieren oder früher eine Lebererkrankung gehabt haben.
Ihr Arzt bzw. Ihre Ärztin wird Sie während der Einnahme dieses Arzneimittels engmaschig auf Diabetes, respektive auf das Risiko zur Entwicklung von Diabetes untersuchen. Wenn Sie hohe Zucker- und Fettwerte im Blut haben, übergewichtig sind und einen hohen Blutdruck haben, besteht bei Ihnen wahrscheinlich ein Risiko zur Entwicklung von Diabetes.
Konsultieren Sie Ihren Arzt bzw. Ihre Ärztin sofort, wenn Sie unerklärliche Muskelschmerzen, -empfindlichkeit oder –schwäche verspüren. In seltenen Fällen können Muskelprobleme schwerwiegend sein, einschliesslich Muskelabbau, der Nierenschäden und im Weiteren den Tod zur Folge haben kann. Das Risiko eines Muskelabbaus ist grösser bei Patienten und Patientinnen, die höhere Ezetimib Simvastatin Zentiva-Dosierungen einnehmen, besonders bei einer Dosierung von 10/80 mg, bei älteren Patienten und Patientinnen (65 Jahre und älter), beim weiblichen Geschlecht und im Falle von vorbestehenden Nierenschäden oder Schilddrüsenproblemen.
Da sich das Risiko von Muskelproblemen erhöhen kann, wenn Sie gleichzeitig mit Ezetimib Simvastatin Zentiva eines der folgenden Arzneimittel oder Substanzen einnehmen (siehe Welche Nebenwirkungen kann Ezetimib Simvastatin Zentiva haben?), ist es besonders wichtig, dass Sie Ihren Arzt bzw. Ihre Ärztin informieren, wenn Sie eines der folgenden Arzneimittel oder Substanzen einnehmen: das Immunsupressivum Cyclosporin (Sandimmun Neoral®); Danazol (Danatrol®); Mittel gegen Pilzinfektionen, wie Itraconazol (Sporanox®) oder Ketoconazol (Nizoral®), Posaconazol (Noxafil®) oder Voriconazol (Vfend®); Fibrate wie z.B. Gemfibrozil (Gevilon®), Ciprofibrat (Hyperlipen®), Bezafibrat (Cedur®) oder Fenofibrat (Lipanthyl®); die Antibiotika Erythromycin (Erythrocin®), Clarithromycin (Klacid®), Telithromycin und Fusidinsäure (Fucidin®); HIV-Proteasehemmer wie z.B. Indinavir (Crixivan®), Nelfinavir (Viracept®), Ritonavir (Norvir®) und Saquinavir (Invirase®, Fortovase®); antivirale Arzneimittel gegen Hepatitis C wie z.B. Boceprevir (Victrelis®), Telaprevir (Incivo®), Elbasvir oder Grazoprevir (die Wirkstoffe von Zepatier®); das Antidepressivum Nefazodon (Nefadar®); Arzneimittel, die Cobicistat enthalten (Stribild®); Amiodaron (Cordarone®), ein Arzneimittel zur Behandlung des unregelmässigen Herzschlags; Arzneimittel zur Behandlung von Bluthochdruck oder Herzengegefühl oder anderen Herzerkrankungen wie z.B. Verapamil (Isoptin®), Diltiazem (Dilzem®) oder Amlodipin (Norvasc®); Lomitapid (ein Arzneimittel zur Behandlung einer schwerwiegenden und seltenen genetischen Cholesterinerkrankung); Daptomycin (ein Arzneimittel zur Behandlung komplizierter Haut- und Hautstrukturinfektionen und Bakteriämie); Ticagrelor (Brilique®, Thrombozytenaggregationshemmer); Grapefruitsaft (sollte während der Einnahme von Ezetimib Simvastatin Zentiva vermieden werden).
Es ist wichtig, dass Sie Ihren Arzt bzw. Ihre Ärztin informieren, wenn Sie Blutverdünnungsmittel wie Phenprocoumon (Marcoumar®) oder Acenocoumarol (Sintrom®) einnehmen, sowie Colchicin (Arzneimittel gegen Gicht), Niacin oder Digoxin.
Einige dieser Arzneimittel sind bereits oben im Kapitel Wann darf Ezetimib Simvastatin Zentiva nicht eingenommen werden? erwähnt.
Bitte nehmen Sie Ezetimib Simvastatin Zentiva erst nach Rücksprache mit Ihrem Arzt ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit (z.B. gegenüber Lactose) leiden.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Tablette, d.h. es ist nahezu «natriumfrei».
Aufgrund der möglichen unerwünschten Wirkungen kann dieses Arzneimittel die Reaktionsfähigkeit, die Fahrtüchtigkeit und die Fähigkeit, Werkzeuge oder Maschinen zu bedienen, beeinträchtigen.
Bitte sprechen Sie mit Ihrem Arzt oder Apotheker, bevor Sie Ezetimib Simvastatin Zentiva einnehmen, wenn Sie Myasthenie (eine Erkrankung mit allgemeiner Muskelschwäche, einschliesslich in einigen Fällen einer Schwäche der Atemmuskulatur) oder okuläre Myasthenie (eine Erkrankung, die eine Muskelschwäche der Augen verursacht) haben oder hatten, da Statine diese Erkrankung manchmal verschlimmern oder zum Auftreten von Myasthenie führen können (siehe «Welche Nebenwirkungen kann Ezetimib Simvastatin Zentiva haben?»).
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie an anderen Krankheiten leiden, Allergien haben, andere Arzneimittel (auch selbstgekaufte!) einnehmen oder äusserlich anwenden, oder asiatischer Abstammung sind.
Informieren Sie auch Ihren Arzt bzw. Ihre Ärztin, der/die Ihnen ein neues Arzneimittel verschreibt, dass Sie Ezetimib Simvastatin Zentiva einnehmen.
Darf Ezetimib Simvastatin Zentiva während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Ezetimib Simvastatin Zentiva darf während einer Schwangerschaft oder in der Stillzeit nicht eingenommen werden. Nehmen Sie Ezetimib Simvastatin Zentiva auch nicht ein, wenn Sie eine Schwangerschaft planen oder vermuten, dass Sie schwanger sein könnten. Wenn Sie während der Behandlung mit Ezetimib Simvastatin Zentiva schwanger werden, beenden Sie die Behandlung und setzen Sie sich sofort mit Ihrem Arzt bzw. Ihrer Ärztin in Verbindung.
Wie verwenden Sie Ezetimib Simvastatin Zentiva?
Vor Beginn der Behandlung mit Ezetimib Simvastatin Zentiva sollte eine geeignete cholesterinsenkende Diät begonnen werden, die auch während der Behandlung fortgesetzt werden sollte.
·Die übliche Dosis zu Therapiebeginn beträgt 1 Tablette Ezetimib Simvastatin Zentiva 10/10 einmal täglich am Abend. Je nach Situation und Ansprechen wird der Arzt oder die Ärztin die Dosis langsam auf 1 Tablette Ezetimib Simvastatin Zentiva 10/20, Ezetimib Simvastatin Zentiva 10/40 oder Ezetimib Simvastatin Zentiva 10/80 erhöhen.
·Wegen des erhöhten Risikos von Muskelproblemen, ist die 10/80 mg Dosis von Ezetimib Simvastatin Zentiva nur für Patienten und Patientinnen, welche chronisch die 10/80 mg Dosis ohne schwerwiegende Muskelprobleme eingenommen haben. Nebenwirkungen treten bei einer Dosierung von 10/80 mg häufiger auf.
·Ezetimib Simvastatin Zentiva kann unabhängig von den Mahlzeiten eingenommen werden.
·Wenn Ihnen Ihr Arzt oder Ihre Ärztin Ezetimib Simvastatin Zentiva zusammen mit einem Anionenaustauscher wie z.B. Colestyramin (Quantalan®) oder Colestipol (Colestid®) verordnet hat, nehmen Sie Ezetimib Simvastatin Zentiva mindestens 2 Stunden vor oder mindestens 4 Stunden nach der Einnahme des Anionentauschers ein.
·Ezetimib Simvastatin Zentiva sollte wie von Ihrem Arzt bzw. Ihrer Ärztin verschrieben eingenommen werden. Fahren Sie mit der Einnahme der anderen cholesterinsenkenden Arzneimittel fort, bis Ihr Arzt oder Ihre Ärztin Ihnen mitteilt, diese abzusetzen.
Es ist wichtig, dass Sie Ezetimib Simvastatin Zentiva täglich, genau wie von Ihrem Arzt oder Ihrer Ärztin verschrieben, einnehmen.
Selbst wenn Sie Arzneimittel zur Senkung der Cholesterinwerte einnehmen, ist es wichtig, Ihre Cholesterinwerte regelmässig bestimmen zu lassen. Sie sollten Ihre Cholesterinwerte und auch die Zielwerte kennen, die Sie erreichen und beibehalten wollen.
Was ist zu tun, wenn Sie zu viele Tabletten eingenommen haben?
Wenn Sie mehr Ezetimib Simvastatin Zentiva Tabletten eingenommen haben als verordnet, wenden Sie sich bitte an Ihren Arzt oder Apotheker bzw. an Ihre Ärztin oder Apothekerin.
Was ist zu tun, wenn Sie eine Dosis vergessen haben?
Versuchen Sie, Ezetimib Simvastatin Zentiva wie verordnet einzunehmen. Sollten Sie jedoch einmal die Einnahme vergessen, so holen Sie die Einnahme nicht mit einer Extradosis nach, sondern fahren Sie am folgenden Tag wie gewohnt mit der Einnahme fort.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Ezetimib Simvastatin Zentiva haben?
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Muskelschmerzen (siehe Wann ist bei der Einnahme von Ezetimib Simvastatin Zentiva Vorsicht geboten?).
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern)
Gewichtsabnahme, Schwindel, Kopfschmerzen, Kribbeln, Bauchschmerzen, Verdauungsstörungen, Blähungen, Übelkeit, Erbrechen, geblähter Bauch, Durchfall, Mundtrockenheit, Sodbrennen, Ausschlag, Juckreiz, Nesselsucht, Gelenkschmerzen, Muskelschmerzen, Empfindlichkeit und Schwäche der Muskulatur, Muskelverkrampfungen, Nackenschmerzen, Schmerzen in den Armen und Beinen, Rückenschmerzen, ungewöhnliche Müdigkeit oder Schwäche, Kraftlosigkeit, Brustschmerzen, Schwellungen, besonders der Hände, Füsse und Gesicht, Schlafstörungen, Schlaflosigkeit.
Selten (betrifft 1 bis 10 von 10'000 Anwendern)
Entzündung des Nasen-Rachen-Raumes, Überempfindlichkeit, Appetitlosigkeit, verminderter Appetit, Angst, Depression, verminderte Libido, Energielosigkeit, anfallsweises Erröten mit Hitzegefühl, erhöhter Blutdruck, Atembeschwerden einschliesslich hartnäckigem Husten und/oder Atemnot oder Fieber, Nasenbluten, Magenschleimhautentzündung, Akne, Haarverlust im Bereich der Kopfhaut, übermässiges Schwitzen, Schuppenflechte, Flankenschmerz, Gelenkschwellung, Muskelschmerzen, Empfindlichkeit und Schwäche der Muskulatur, häufiger Harndrang, Erektionsstörung, Durst.
Zusätzlich wurde über die folgenden unerwünschten Wirkungen bei der Anwendung von Ezetimib (Ezetrol® Tabletten) oder Simvastatin (Zocor® Filmtabletten) berichtet (Präparate, die einen der Wirkstoffe von Ezetimib Simvastatin Zentiva enthalten): Leberprobleme (manchmal schwerwiegend), Verstopfung, saures Aufstossen, allergische Reaktionen, einschliesslich Schwellung des Gesichts, Lippen, Zunge und/oder Rachens, die Atem- oder Schluckbeschwerden verursachen können (die eine sofortige Behandlung erfordern), Hautausschlag und Nesselausschlag, Hautausschlag mit roten Erhebungen, manchmal scheibenförmig mit zentral gelegenen Läsionen, Muskelschmerzen, Muskelempfindlichkeit oder -schwäche (die in sehr seltenen Fällen auch nach Absetzen von Ezetimib Simvastatin Zentiva nicht verschwinden), Entzündung der Bauchspeicheldrüse, Gallensteine, Gallenblasenentzündung, Gedächtnisprobleme, Gedächtnisverlust, Verwirrtheit. Es wurde über einzelne Beschwerden der Achillessehne, selten verbunden mit Achillessehnenriss, berichtet.
Diabetes kann auftreten – dies ist wahrscheinlicher, wenn Sie hohe Zucker- und Fettwerte im Blut haben, übergewichtig sind und einen hohen Blutdruck haben. Ihr Arzt bzw. Ihre Ärztin wird Sie während der Einnahme dieses Arzneimittels diesbezüglich überwachen.
Wenn Ihnen Ezetimib Simvastatin Zentiva verschrieben wurde, möchte Ihr Arzt oder Ihre Ärztin allenfalls Bluttests zur Überprüfung Ihrer Leber durchführen bevor Sie Ezetimib Simvastatin Zentiva zum ersten Mal einnehmen und falls während der Behandlung mit Ezetimib Simvastatin Zentiva Symptome von Leberproblemen auftreten. Kontaktieren Sie Ihren Arzt oder Ihre Ärztin sofort, falls bei Ihnen die folgenden Symptome von Leberproblemen auftreten: Müdigkeit oder Schwäche, Appetitlosigkeit, Schmerzen im Oberbauch, dunkler Urin, Gelbfärbung der Haut oder des Augenweiss.
Nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): Myasthenia gravis (eine Erkrankung, die zu allgemeiner Muskelschwäche führt, einschliesslich in einigen Fällen einer Schwäche der Atemmuskulatur). Okuläre Myasthenie (eine Erkrankung, die eine Muskelschwäche der Augen verursacht). Sprechen Sie mit Ihrem Arzt, wenn Sie in Ihren Armen oder Beinen ein Schwächegefühl verspüren, das sich nach Phasen der Aktivitätverschlimmert bei Doppeltsehen oder Hängen Ihrer Augenlider, Schluckbeschwerden oder Kurzatmigkeit.
Schwerwiegende Erkrankungen mit schwerem Schälen und Anschwellen der Haut, Blasenbildung auf Haut, Mund, Augen oder Genitalien und Fieber; Hautausschlag mit pink-roten Flecken vor allem auf den Handinnenflächen oder Fusssohlen, der Blasen bilden kann; Möglicherweise treten gleichzeitig auch grippeähnliche Symptome wie Fieber, Schüttelfrost oder Muskelschmerzen auf; grippeähnliche Symptome mit Hautausschlag, Fieber, geschwollenen Drüsen und abnormalen Bluttestergebnissen (einschliesslich erhöhter weisser Blutkörperchen (Eosinophilie) und Leberenzyme).
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, falls Sie irgendeines dieser Symptome oder andere Beschwerden im Zusammenhang mit der Einnahme von Ezetimib Simvastatin Zentiva bemerken.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
In der Originalverpackung, nicht über 25 °C und ausserhalb der Reichweite von Kindern aufbewahren.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Ezetimib Simvastatin Zentiva enthalten?
Jede Tablette Ezetimib Simvastatin Zentiva enthält
Wirkstoffe
10 mg Ezetimib sowie jeweils 10 mg (Ezetimib Simvastatin Zentiva 10/10), 20 mg (Ezetimib Simvastatin Zentiva 10/20), 40 mg (Ezetimib Simvastatin Zentiva 10/40) oder 80 mg (Ezetimib Simvastatin Zentiva 10/80) Simvastatin
Hilfsstoffe
Lactose-Monohydrat, Hypromellose, Croscarmellose-Natrium, mikrokristalline Cellulose, Ascorbinsäure, Citronensäure, wasserfrei, Butylhydroxyanisol (E 320), Propylgallat (E 310), Magnesiumstearat, Eisenoxid gelb (E 172), Eisenoxid rot (E 172), Eisenoxid schwarz (E 172)
Wo erhalten Sie Ezetimib Simvastatin Zentiva? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Ezetimib Simvastatin Zentiva 10/10: Blisterpackungen zu 28 und 98 Tabletten.
Ezetimib Simvastatin Zentiva 10/20: Blisterpackungen zu 28 und 98 Tabletten.
Ezetimib Simvastatin Zentiva 10/40: Blisterpackungen zu 28 und 98 Tabletten.
Ezetimib Simvastatin Zentiva 10/80: Blisterpackungen zu 28 Tabletten.
Zulassungsnummer
67242 (Swissmedic).
Zulassungsinhaberin
Helvepharm AG, Frauenfeld.
Diese Packungsbeilage wurde im März 2024 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Mayzent®
Was ist Mayzent und wann wird es angewendet?
Mayzent gehört zu einer Gruppe von Arzneimitteln, die als Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulatoren bezeichnet werden. Mayzent enthält den Wirkstoff Siponimod.
Mayzent wird angewendet zur Behandlung von Erwachsenen mit sekundär progredienter Multipler Sklerose (SPMS) mit entzündlicher Krankheitsaktivität. Eine entzündliche Krankheitsaktivität bei SPMS liegt dann vor, wenn noch Schübe auftreten, oder wenn die MRT (Magnetresonanztomographie)-Bildgebung Entzündungen zeigt.
Mayzent hilft, das Zentralnervensystem (ZNS) vor Angriffen des körpereigenen Immunsystems zu schützen, indem es die Fähigkeit bestimmter weisser Blutkörperchen (Lymphozyten), sich ungehindert im Körper zu bewegen, einschränkt. Mayzent kann verhindern, dass entzündungsverursachende Zellen das Gehirn und das Rückenmark erreichen. Damit wird die von der MS verursachte Nervenschädigung verringert. Dadurch hilft Mayzent, die Auswirkungen der Krankheitsaktivität (Verschlechterung der Behinderung, Hirnläsionen, Verlust von Hirnvolumen und Schübe) zu verlangsamen. Mayzent hat möglicherweise auch einen direkten günstigen Einfluss auf bestimmte Hirnzellen, welche an der Behebung oder Verlangsamung der MS-bedingten Schädigung beteiligt sind.
Auf Verschreibung des Arztes oder der Ärztin.
Was sollte dazu beachtet werden?
Bitte halten Sie sich genau an die Anweisungen Ihres Arztes oder Ihrer Ärztin. Sie können von den allgemeinen Informationen in dieser Packungsbeilage abweichen.
Wann darf Mayzent nicht eingenommen werden?
Sie dürfen Mayzent nicht einnehmen,
·wenn Sie allergisch (überempfindlich) gegen Siponimod, Erdnüsse, Soja oder einen der sonstigen Inhaltsstoffe von Mayzent sind (siehe «Was ist in Mayzent enthalten»).
·wenn Sie unter einem Immunschwächesyndrom leiden.
·wenn Sie eine Vorgeschichte einer schweren Infektion des Gehirns haben, wie eine progressive multifokale Leukoenzephalopathie (PML) oder Kryptokokkenmeningitis.
·wenn Sie eine aktive Krebserkrankung haben.
·wenn Sie eine schwere Leberfunktionsstörung haben (Child-Pugh-Klasse C).
·wenn Sie in den letzten 6 Monaten einen Herzinfarkt, instabile Angina pectoris (eine Form der Durchblutungsstörung des Herzens), einen Schlaganfall (einschliesslich einer TIA genannten, vorübergehenden Durchblutungsstörung des Gehirns) oder, eine schwere Herzmuskelschwäche mit stationärer Behandlung (Herzmuskelschwäche der Klasse NYHA Klasse III-IV) hatten.
·wenn Sie eine Vorgeschichte von schwerem unregelmässigem oder anomalem Herzrhythmus (AV-Block 2. Grades Mobitz Typ II, AV-Block 3. Grades, sinusatriale Blockierung oder Sick-Sinus-Syndrom) haben und keinen Herzschrittmacher tragen (siehe «Wann ist bei der Einnahme/Anwendung von Mayzent Vorsicht geboten?»).
·wenn das Ergebnis Ihrer Blutuntersuchung zeigt, dass Ihr Körper Mayzent nicht ausreichend abbauen kann (Genotyp CYP2C9*3/*3, betrifft weniger als 0.4-0.5% der Bevölkerung).
·wenn Sie schwanger sind oder eine Frau im gebärfähigen Alter sind, die keine zuverlässige Verhütungsmethode anwendet.
Wann ist bei der Einnahme von Mayzent Vorsicht geboten?
Bitte sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, bevor Sie Mayzent einnehmen,
·wenn Sie eine Infektion haben. Eine bereits bestehende Infektion kann sich verschlechtern. Sollten Sie eine aktive schwere Infektion haben, wird Ihr Arzt bzw. Ihre Ärztin die Behandlung mit Mayzent verschieben, bis die Infektion abgeklungen ist.
·wenn Ihre Immunabwehr geschwächt ist beispielsweise durch eine Erkrankung oder durch Arzneimittel, die das Immunsystem unterdrücken (s. «Einnahme zusammen mit anderen Arzneimitteln»).
·wenn Sie noch nicht an Windpocken erkrankt waren oder nicht dagegen geimpft sind. Sollten Sie während der Behandlung mit Mayzent Windpocken bekommen, könnte bei Ihnen ein grösseres Risiko für Komplikationen bestehen. Ihr Arzt bzw. Ihre Ärztin wird Sie möglicherweise bitten, sich gegen Windpocken impfen zu lassen.
·wenn Sie eine Impfung planen. Ihr Arzt bzw. Ihre Ärztin wird Sie hierzu beraten (s. «Einnahme zusammen mit anderen Arzneimitteln»).
·wenn Sie an Sehstörungen (insbesondere einer als Makulaödem bezeichnete Erkrankung), an Augenentzündungen oder -infektionen (Uveitis) leiden oder gelitten haben. Ihr Arzt oder Ihre Ärztin bittet Sie, vor dem Beginn der Behandlung mit Mayzent sowie in regelmässigen Abständen während Ihrer Behandlung eine Augenuntersuchung vornehmen zu lassen. Die Makula ist ein kleiner Bereich der Netzhaut am Augenhintergrund, mit dem Sie Umrisse, Farben und Details klar und scharf sehen können (zentrales Blickfeld). Mayzent kann eine Schwellung in der Makula verursachen. Die Wahrscheinlichkeit, dass sich ein Makulaödem entwickelt, ist höher, wenn Sie schon einmal ein Makulaödem oder eine Uveitis hatten oder Diabetes haben.
·wenn Sie eine der folgenden Erkrankungen haben oder hatten (auch wenn sie behandelt werden): eine schwere Herzerkrankung, unregelmässigen oder anomalen Herzschlag (Arrhythmie), Schlaganfall oder andere mit den Blutgefässen des Gehirns in Zusammenhang stehende Erkrankungen, eine verlangsamte Herzfrequenz, Ohnmachtsanfälle, Störung des Herzrhythmus, (auffällige Befunde im EKG).
·wenn Sie eine schwere Schlafapnoe haben.
·wenn Sie einen hohen Blutdruck haben, der nicht durch Arzneimittel kontrolliert werden kann.
·wenn Sie Leberprobleme haben oder hatten.
·wenn Sie schwanger werden könnten oder möchten.
In den ersten Tagen der Behandlung kann Mayzent eine Verlangsamung der Herzfrequenz (Bradykardie) hervorrufen. Möglicherweise werden Sie überhaupt nichts davon bemerken. Sie könnten sich aber auch schwindelig oder müde fühlen. Mayzent kann zu Beginn der Behandlung auch einen unregelmässigen Herzschlag verursachen. Sollte etwas darauf hindeuten, dass bei Ihnen ein erhöhtes Risiko für das Auftreten dieser Wirkungen bestehen könnte, wird Ihr Arzt bzw. Ihre Ärztin möglicherweise entscheiden, Sie am Anfang der Behandlung engmaschiger zu überwachen, oder Sie zunächst an einen Kardiologen zu überweisen.
Wenn eines der folgenden Symptome während der Behandlung mit Mayzent bei Ihnen auftritt, benachrichtigen Sie sofort Ihren Arzt oder Ihre Ärztin, weil es schwerwiegend sein könnte:
·Eine Infektion. Mayzent senkt die Zahl der Lymphozyten. Sie können daher anfälliger für Infektionen sein, während Sie Mayzent einnehmen (und bis zu 3 bis 4 Wochen nach Beendigung der Einnahme). Infektionen könnten schwerwiegend oder möglicherweise lebensbedrohlich sein.
·Sie vermuten, dass sich Ihre MS-Erkrankung verschlechtert (z.B. Schwäche oder visuelle Veränderungen), oder Sie bemerken irgendwelche neuen oder ungewöhnlichen Symptome. Dies könnten Symptome einer seltenen Erkrankung des Gehirns sein, die durch eine Infektion verursacht wird und Progressive Multifokale Leukenzephalopathie (PML) genannt wird.
·Wenn solch eine PML bei Ihnen festgestellt wird, sollte die Behandlung mit Mayzent dauerhaft beendet werden. Aufgrund der Konzentrationsabnahme von Mayzent im Blut, kann es allerdings bei manchen Patienten zu einer Verschlechterung der Krankheit, einschliesslich der Gehirnfunktion, kommen. Dies kann durch eine überschiessende Entzündungsreaktion, das sogenannte entzündliche Immunrekonstitutionssyndrom, hervorgerufen werden (engl.: Immune reconstitition inflammatory syndrome, IRIS).
·Sie haben Fieber, fühlen sich, als ob Sie Grippe hätten oder Kopfschmerzen haben, die mit einem steifen Nacken, Lichtempfindlichkeit, Übelkeit und/oder Verwirrtheit einhergehen. Dies könnten Symptome einer Meningitis und/oder Enzephalitis sein, verursacht durch eine Virus- oder Pilzinfektion (wie Kryptokokkenmeningitis).
·Ihre Sehkraft verändert sich, es entwickelt sich beispielsweise ein «blinder Fleck» im Zentrum Ihres Blickfeldes oder Sie haben Probleme mit der Wahrnehmung von Farben oder feinen Details. Es könnte sich um Symptome eines Makulaödems handeln. Es kann ähnliche Sehstörungen verursachen wie ein MS-Schub (Optikusneuritis). Ihr Arzt bzw. Ihre Ärztin wird zusätzliche Augenuntersuchungen veranlassen.
·Unerklärliche Übelkeit, Erbrechen, Bauchschmerzen, Müdigkeit, Gelbfärbung der Haut oder des Weissen in den Augen oder ungewöhnlich dunklen Urin. Es könnte sich um Leberprobleme handeln.
·Zu den Symptomen von Hautkrebs können Hautknötchen (z.B. glänzende perlenartige Knötchen), Flecken oder offene Wunden gehören, die nicht innerhalb von Wochen abheilen. Symptome anderer Hautkrebsarten können abnormales Wachstum oder Veränderungen des Hautgewebes (z.B. ungewöhnliche Muttermale) mit einer Änderung der Farbe, Form oder Grösse im Laufe der Zeit sein.
·Plötzliches Auftreten von starken Kopfschmerzen, Verwirrung, Krampfanfällen und Sehstörungen. Es könnte sich um eine als posteriores reversibles Enzephalopathiesyndrom (PRES) bezeichnete Erkrankung handeln.
Einnahme zusammen mit anderen Arzneimitteln (Wechselwirkungen mit anderen Arzneimitteln einschliesslich Impfungen und anderen Therapien)
Bitte informieren Sie Ihren Arzt bzw. Ihre Ärztin vor der Einnahme von Mayzent, wenn Sie eines der folgenden Arzneimittel einnehmen:
·Arzneimittel gegen Herzrhythmusstörungen, z.B. Chinidin, Procainamid, Amiodaron oder Sotalol, da es die Wirkung auf den unregelmässigen Herzschlag verstärken könnte.
·Arzneimittel, die die Herzfrequenz verlangsamen, z.B. Verapamil oder Diltiazem (sogenannte Calciumantagonisten), Ivabradin oder Digoxin. Ihr Arzt bzw. Ihre Ärztin kann Sie zwecks Wechsel Ihrer Medikamente an einen Kardiologen überweisen, denn der Effekt auf die Herzfrequenz könnte in den ersten Tagen nach Beginn der Behandlung mit Mayzent verstärkt werden. Falls Sie einen Betablocker einnehmen, wird Ihr Arzt bzw. Ihre Ärztin Sie möglicherweise bitten, Ihre Betablockerbehandlung vorübergehend abzusetzen, bis Sie Ihre Erhaltungsdosis von Mayzent erreicht haben.
·Arzneimittel, die das Immunsystem unterdrücken oder modulieren, darunter einige Chemotherapeutika und andere Arzneimittel zur Behandlung von MS, da diese die Wirkung auf das Immunsystem verstärken könnten.
·Sollten Sie eine Impfung benötigen, fragen Sie erst Ihren Arzt oder Ihre Ärztin um Rat. Während und für 4 Wochen nach Abbruch der Behandlung mit Mayzent dürfen Sie nicht mit bestimmten Impfstoffen (abgeschwächten Lebendimpfstoffen) geimpft werden, da diese die Infektion auslösen könnten, die sie eigentlich verhindern sollen. Andere Impfstoffe können während und für 4 Wochen nach der Behandlung mit Mayzent weniger wirksam sein. Die Behandlung mit Mayzent muss deshalb möglicherweise 1 Woche vor und bis zu 4 Wochen nach der Impfung unterbrochen werden (s. «Wann ist bei der Einnahme/Anwendung von Mayzent Vorsicht geboten?»).
·Fluconazol und bestimmte andere Arzneimittel können bei einigen Patienten bzw. Patientinnen die Konzentration von Mayzent im Blut erhöhen und es wird nicht empfohlen, sie gemeinsam mit Mayzent anzuwenden. Ihr Arzt bzw. Ihre Ärztin wird Sie dazu beraten.
·Carbamazepin, Rifampicin und bestimmte andere Arzneimittel können die Konzentration von Mayzent im Blut verringern und daher dessen Wirksamkeit herabsetzen. Ihr Arzt bzw. Ihre Ärztin wird Sie diesbezüglich beraten.
·Modafinil und bestimmte andere Arzneimittel können bei einigen Patienten oder Patientinnen die Konzentration von Mayzent im Blut verringern und daher die Wirksamkeit des Arzneimittels herabsetzen. Ihr Arzt bzw. Ihre Ärztin wird Sie dahingehend beraten, ob dies für Sie von Bedeutung ist.
·Phototherapie mit UV-B-Strahlung oder PUVA-Photochemotherapie. Während der Mayzent-Behandlung kann eine UV-Therapie Ihr Hautkrebsrisiko erhöhen.
Untersuchungen vor und während der Behandlung
Wie schnell Mayzent im Körper abgebaut (metabolisiert) wird, ist von Patient zu Patient bzw. Patientin zu Patientin unterschiedlich und wird vor Beginn der Behandlung mithilfe einer genetischen Untersuchung im Blut oder Speichel ermittelt, um die optimale Dosis für Sie zu bestimmen. In seltenen Fällen könnte das Ergebnis der Untersuchung zeigen, dass Sie Mayzent nicht einnehmen dürfen.
Die erwünschte Wirkung der Behandlung von Mayzent ist die Verringerung der Anzahl der weissen Blutkörperchen in Ihrem Blut. Dieser Wert normalisiert sich in der Regel innerhalb von 3 bis 4 Wochen nach Beendigung der Behandlung. Wenn bei Ihnen Blutuntersuchungen durchgeführt werden müssen, sagen Sie dem Arzt oder der Ärztin, dass Sie Mayzent einnehmen. Andernfalls kann es sein, dass der Arzt oder die Ärztin die Ergebnisse des Tests nicht verstehen kann und für bestimmte Arten von Bluttests muss Ihr Arzt oder Ihre Ärztin möglicherweise mehr Blut als üblich abnehmen.
Bevor Sie mit der Einnahme von Mayzent beginnen, wird sich Ihr Arzt bzw. Ihre Ärztin vergewissern, ob Sie genügend weisse Blutkörperchen im Blut haben, und eventuell eine regelmässige Kontrolle durchführen. Falls Sie nicht genügend weisse Blutkörperchen haben, muss Ihr Arzt bzw. Ihre Ärztin möglicherweise die Dosis von Mayzent verringern oder Mayzent absetzen.
Ihr Arzt bzw. Ihre Ärztin wird vor Beginn der Behandlung Ihre Leberwerte überprüfen. Darüber hinaus wird Ihr Arzt bzw. Ihre Ärztin Sie nach Ihren derzeitigen Begleitmedikationen befragen, um Wechselwirkungen anderer Arzneimittel mit Mayzent auszuschliessen. Ihr Arzt bzw. Ihre Ärztin wird Ihren Blutdruck regelmässig überprüfen und Sie möglicherweise 3 oder 4 Monate nach Behandlungsbeginn und eventuell auch später bitten, eine Augenuntersuchung vornehmen zu lassen. Vor Beginn der Behandlung und 3 bis 4 Monate nach Behandlungsbeginn mit Mayzent wird eine augenärztliche Untersuchung durchgeführt.
Eine Art von Hautkrebs, der Basalzellkarzinom (BCC) genannt wird, und andere Hautkrebsarten, wie z.B. das Plattenepithelzellkarzinom, das maligne Melanom, das Kaposi-Sarkom und das Merkelzellkarzinom sind bei Patienten bzw. Patientinnen, die mit (S1P)-Rezeptor-Modulatoren, zu denen auch Mayzent gehört, behandelt werden, berichtet worden. Ihr Arzt bzw. Ihre Ärztin wird vor und während Ihrer Behandlung mit Mayzent regelmässige Hautuntersuchungen durchführen.
Informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin, wenn Sie eine verdächtige Hautveränderung bemerken.
Inhaltsstoffe
Die Tabletten enthalten Lactose und Phospholipide aus Sojabohnen. Bitte nehmen Sie Mayzent erst nach Rücksprache mit Ihrem Arzt bzw. Ihrer Ärztin ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit leiden. Wenn Sie allergisch gegen Erdnüsse oder Soja sind, dürfen Sie Mayzent nicht einnehmen.
Exposition gegenüber der Sonne und Schutz vor der Sonne
Eine länger dauernde Exposition gegenüber der Sonne und ein schwaches Immunsystem können das Risiko für die Entwicklung von BCC oder anderen Hauttumoren beeinflussen. Sie sollten Ihre Exposition gegenüber der Sonne und UV-Strahlen begrenzen, indem Sie angemessen schützende Kleidung tragen und regelmässig Sonnenschutzmittel mit hohem UV-Schutzfaktor auftragen. Eine Phototherapie mit UV-Strahlung oder eine Photochemotherapie (PUVA) sollte während der Behandlung mit Mayzent vermieden werden (sie kann Ihr Risiko, Hautkrebs zu entwickeln, erhöhen).
Mayzent schwächt Ihr Immunsystem und kann damit Ihr Hautkrebsrisiko erhöhen. Kontrollieren Sie Ihre Haut regelmässig.
Verschlechterung der MS nach Beendigung der Behandlung mit Mayzent
Beenden Sie die Einnahme von Mayzent oder ändern Sie nicht die Dosis, ohne zuvor mit Ihrem Arzt oder Ihrer Ärztin gesprochen zu haben.
Benachrichtigen Sie sofort Ihren Arzt bzw. Ihre Ärztin, wenn Sie glauben, dass sich Ihre MS verschlechtert hat, nachdem Sie die Behandlung mit Mayzent beendet haben.
Informieren Sie Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin, wenn Sie
·an anderen Krankheiten leiden,
·Allergien haben oder
·andere Arzneimittel (auch selbst gekaufte!) einnehmen oder äusserlich anwenden!
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen
Ihr Arzt bzw. Ihre Ärztin wird Ihnen sagen, ob Ihre Erkrankung das sichere Führen von Fahrzeugen und das Bedienen von Maschinen erlaubt. Es ist nicht zu erwarten, dass Mayzent Ihre Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinflusst, wenn Sie Ihre reguläre Behandlungsdosis erhalten. Zu Beginn der Behandlung könnten Sie sich gelegentlich schwindelig fühlen. An Ihrem ersten Behandlungstag mit Mayzent sollten Sie daher kein Fahrzeug führen oder Maschinen bedienen.
Darf Mayzent während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Während der Schwangerschaft, wenn Sie versuchen, schwanger zu werden, oder wenn Sie schwanger werden könnten und keine zuverlässige Verhütungsmethode anwenden, dürfen Sie Mayzent nicht einnehmen. Wenn Mayzent während der Schwangerschaft eingenommen wird, besteht das Risiko, dass das ungeborene Baby geschädigt wird.
Wenn Sie im gebärfähigen Alter sind, wird Ihr Arzt bzw. Ihre Ärztin Sie vor Behandlungsbeginn mit Mayzent bitten, einen Schwangerschaftstest durchzuführen, um sicherzustellen, dass Sie nicht schwanger sind. Während der Behandlung und mindestens 10 Tage nach Beendigung der Einnahme muss eine zuverlässige Verhütungsmethode angewendet werden, um eine Schwangerschaft zu vermeiden (siehe «Wann ist bei der Einnahme/Anwendung von Mayzent Vorsicht geboten?»). Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin über zuverlässige Empfängnisverhütungsmethoden.
Wenn Sie während der Behandlung mit Mayzent schwanger werden, informieren Sie sofort Ihren Arzt oder Ihre Ärztin. Ihr Arzt oder Ihre Ärztin wird die Behandlung abbrechen.
Während der Behandlung mit Mayzent dürfen Sie nicht stillen. Mayzent kann in die Muttermilch übertreten und es besteht das Risiko schwerwiegender Nebenwirkungen für das Baby.
Wird Mayzent zum Zweck einer Schwangerschaftsplanung abgesetzt, sollte eine mögliche Rückkehr der Krankheitsaktivität in Betracht gezogen werden.
Wie verwenden Sie Mayzent?
Wie viel Mayzent ist einzunehmen?
Behandlungsbeginn
Mit Hilfe eines Starter-Sets wird die Dosis über 5 Tage allmählich erhöht und damit das Risiko für kardiale Nebenwirkungen zu Beginn der Behandlung verringert. Befolgen Sie die Anweisungen auf der Packung (s. auch nachfolgende Tabelle). Bei bestimmten Vorerkrankungen des Herzens wird Ihr Arzt bzw. Ihre Ärztin möglicherweise eine engmaschige Überwachung zu Behandlungsbeginn anordnen.
Starter-Set
Titration Titrationsdosis Titrationsplan
Tag 1 0.25 mg 1 Tablette
Tag 2 0.25 mg 1 Tablette
Tag 3 0.5 mg 2 Tabletten
Tag 4 0.75 mg 3 Tabletten
Tag 5 1.25 mg 5 Tabletten
An Tag 6 wechseln Sie zu Ihrer verordneten Behandlungsdosis.
Während der ersten 6 Behandlungstage sollte die empfohlene Dosis einmal täglich morgens mit oder ohne Nahrung eingenommen werden.
Behandlungsdosis
Die normale tägliche Dosis (Erhaltungsdosis) im Anschluss an die Starterpackung beträgt 2 mg (eine Tablette zu 2 mg Siponimod) mit oder ohne Mahlzeit.
Falls die vor Behandlungsbeginn durchgeführte Blutuntersuchung ergeben hat, dass Ihr Körper Mayzent langsam abbaut (s. «Wann ist bei der Einnahme/Anwendung von Mayzent Vorsicht geboten?»), wird Ihr Arzt bzw. Ihre Ärztin Sie anweisen, nur 1 mg einmal täglich (eine Tablette zu 1 mg oder vier Tabletten zu 0.25 mg) einzunehmen. Wenn dies auf Sie zutrifft, gilt derselbe Plan zur Behandlungseinleitung (Starterpackung) und die Einnahme von fünf Tabletten zu 0.25 mg am Tag 5 ist trotzdem sicher.
Mayzent wird einmal täglich mit oder ohne Mahlzeit eingenommen. Mayzent Filmtabletten sollten ganz mit Wasser eingenommen werden. Wenn Sie Mayzent jeden Tag zur selben Zeit einnehmen, fällt es Ihnen leichter, an die Einnahme zu denken.
Wie lange ist Mayzent einzunehmen?
Nehmen Sie Mayzent so lange ein, wie Ihr Arzt bzw. Ihre Ärztin es Ihnen verordnet. Es handelt sich um eine Langzeitbehandlung, die möglicherweise über Monate oder Jahre fortgeführt wird. Ihr Arzt bzw. Ihre Ärztin wird Ihre Erkrankung kontinuierlich überwachen, um zu überprüfen, ob die Behandlung die gewünschte Wirkung zeigt.
Wenn Sie eine grössere Menge von Mayzent eingenommen haben, als Sie sollten
Wenn Sie zu viele Mayzent-Tabletten eingenommen haben, oder versehentlich die erste Tablette aus der Packung mit der Behandlungsdosis anstatt der Starterpackung eingenommen haben, verständigen Sie unverzüglich Ihren Arzt oder Ihre Ärztin. Ihr Arzt bzw. Ihre Ärztin wird Sie möglicherweise überwachen.
Wenn Sie die Einnahme von Mayzent vergessen haben
Wenn Sie innerhalb der ersten 6 Behandlungstage die Einnahme einer Dosis an einem Tag vergessen haben, rufen Sie Ihren Arzt oder Ihre Ärztin an, bevor Sie die nächste Dosis einnehmen. Ihr Arzt bzw. Ihre Ärztin muss Ihnen dann ein neues Starter-Set verschreiben. Sie müssen erneut bei Tag 1 mit einer neuen Starterpackung beginnen.
Falls Sie eine Dosis ausgelassen haben, wenn Sie bereits die reguläre Behandlungsdosis erhalten (ab Tag 7), holen Sie die Eingabe nach, sobald es Ihnen aufgefallen ist. Sollte die Einnahme der nächsten Dosis kurz bevorstehen, lassen Sie die vergessene Dosis weg und setzen Sie Ihr Behandlungsschema wie gewohnt fort. Nehmen Sie nicht die doppelte Menge ein.
Wenn Sie die Einnahme von Mayzent an 4 oder mehr aufeinanderfolgenden Tagen aussetzen, müssen Sie die Behandlung erneut mit einer Starterpackung an Tag 1 beginnen.
Wenn Sie die Einnahme von Mayzent abbrechen
Sie dürfen die Einnahme von Mayzent nicht beenden oder Ihre Dosis ändern, ohne vorher mit Ihrem Arzt bzw. Ihrer Ärztin gesprochen zu haben.
Mayzent bleibt nach dem Absetzen der Behandlung noch mindestens 10 Tage lang im Blut nachweisbar. Die Anzahl Ihrer weissen Blutkörperchen (Lymphozytenzahl) kann bis zu 4 Wochen niedrig bleiben, und die in dieser Packungsbeilage beschriebenen Nebenwirkungen können weiterhin auftreten.
Wenn Sie 4 Tage oder mehr Tage nach dem Absetzen die Behandlung mit Mayzent wiederaufnehmen, müssen Sie die Behandlung erneut mit einer Starterpackung an Tag 1 beginnen.
Sagen Sie Ihrem Arzt bzw. Ihrer Ärztin sofort, wenn Sie glauben, dass sich Ihre MS verschlimmert, nachdem Sie die Behandlung mit Mayzent abgebrochen haben.
Kinder und Jugendliche (unter 18 Jahren)
Die Anwendung und Sicherheit von Mayzent bei Kindern unter 18 ist bisher nicht geprüft worden.
Ältere Patienten
Es gibt keine Erfahrungen mit Mayzent bei älteren Patienten und Patientinnen (über 61 Jahre).
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Mayzent haben?
Wie alle Arzneimittel kann Mayzent Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Folgende Nebenwirkungen können schwerwiegend sein oder werden:
Häufig (betrifft 1 bis 10 von 100 Behandelten)
·Ausschlag mit kleinen, mit Flüssigkeit gefüllten Bläschen, die auf geröteter Haut auftreten; als Anzeichen einer Virusinfektion, die schwerwiegend verlaufen kann (Herpes Zoster).
·Eine Hautkrebsart, die als Basalzellkarzinom bezeichnet wird und die oft als perlenartige Knötchen erscheint, aber auch andere Formen annehmen kann.
·Fieber, Halsschmerzen, Geschwüre der Mundschleimhaut aufgrund von Infektionen (Lymphopenie).
·Krämpfe, Krampfanfälle.
·Schattenbildung oder «blinder Fleck» im zentralen Sehbereich, verschwommenes Sehen, Probleme bei der Erkennung von Farben oder Details (Anzeichen einer Schwellung im zentralen Sehbereich der Netzhaut am Augenhintergrund: Makulaödem).
·Herzrhythmusstörungen (Atrioventrikulärer Block).
·Langsamer Herzschlag (Bradykardie).
Gelegentlich (betrifft 1 bis 10 von 1000 Behandelten)
·Eine Hautkrebsart, die als Plattenepithelzellkarzinom bezeichnet wird und die als festes rotes Knötchen, als Wunde mit einer Kruste oder als neue Wunde auf einer bestehenden Narbe auftreten kann.
·Eine Art von Hautkrebs, das sogenannte maligne Melanom, das sich in der Regel aus einem ungewöhnlichen Muttermal entwickelt. Mögliche Anzeichen für ein Melanom sind Muttermale, die im Laufe der Zeit ihre Grösse, Form, Höhe oder Farbe verändern können, oder neue Muttermale. Die Muttermale können jucken, bluten oder ulzerieren.
·Kryptokokkeninfektionen (eine Art Pilzinfektion) oder Virusinfektion (verursacht durch das Herpes- oder Varizella-Zoster-Virus), einschliesslich Meningitis und/oder Enzephalitis mit Symptomen wie Kopfschmerzen zusammen mit einem steifen Nacken, Lichtempfindlichkeit, Übelkeit und/oder Verwirrtheit.
Selten (betrifft 1 bis 10 von 10'000 Behandelten)
·Eine seltene Gehirninfektion, die als progressive multifokale Leukenzephalopatie (PML) bezeichnet wird. Die Symptome einer PML können denen einer MS ähneln, wie z.B. Schwäche oder Sehstörungen, Gedächtnisverlust, Probleme beim Denken oder Schwierigkeiten beim Gehen.
Wenn bei Ihnen eine der vorher genannten Nebenwirkungen auftritt, müssen Sie dies umgehend Ihrem Arzt bzw. Ihrer Ärztin mitteilen.
Andere mögliche Nebenwirkungen
Andere Nebenwirkungen sind nachfolgend aufgeführt. Wenn diese Nebenwirkungen schwerwiegend werden, teilen Sie dies bitte Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin mit.
Die meisten Nebenwirkungen sind leichter bis mittelschwerer Intensität und klingen in der Regel nach einigen Wochen unter der Behandlung wieder ab.
Sehr häufig (betrifft mehr als einen von 10 Behandelten)
·Kopfschmerzen
·Bluthochdruck (Hypertonie), der gelegentlich mit Symptomen wie Kopfschmerzen und Schwindel einhergeht.
·Erhöhte Werte im Leberfunktionstest, (erhöhte Leberwerte): erhöhte Werte der Enzyme Alanin-Aminotransferase (ALT), Gamma-Glutamyltransferase (GGT) und Aspartat-Aminotransferase (AST).
Häufig (betrifft 1 bis 10 von 100 Behandelten)
·Neu aufgetretene Leberflecke oder Muttermale (Nävi): kleine (Durchmesser von weniger als 1 cm) Flecken, Papeln oder Knötchen mit unscharfen Grenzen, die bläulich/schwarz bis braun, rosafarben oder hautfarben erscheinen können (melanozytäre Nävi).
·Schwindel
·Unwillkürliches Zittern des Körpers (Tremor)
·Durchfall
·Übelkeit
·Schmerzen in Händen oder Füssen
·Schwellungen an den Händen, Fussgelenken, Beinen oder Füssen (peripheres Ödem)
·Schwäche (Asthenie)
·Erniedrigte Werte im Lungenfunktionstest (erniedrigte Lungenfunktionswerte)
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, oder Apotheker bzw. Ihre Ärztin, Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Aufbrauchfrist nach Anbruch
Nach erstmaligem Öffnen 3 Monate haltbar bei Lagerung nicht über 25°C.
Entsorgungshinweis
Entsorgen Sie keine Arzneimittel in das Abwasser oder den Hausmüll. Abgelaufene oder nicht mehr benötigte Arzneimittel aus Haushalten können in Apotheken oder Sammelstellen abgegeben werden. Diese Massnahmen tragen zum Schutz der Umwelt bei.
Lagerungshinweis
Im Kühlschrank lagern (2°C bis 8°C).
In der Originalverpackung aufbewahren.
Arzneimittel für Kinder unzugänglich aufbewahren.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Mayzent enthalten?
Wirkstoffe
Siponimod (als Siponimod-Fumarsäure).
Hilfsstoffe
Tablettenkern:
Lactose-Monohydrat, mikrokristalline Cellulose, Crospovidon, Glyceroldibehenat und kolloidales wasserfreies Siliciumdioxid.
Jede Tablette zu 0.25 mg enthält 62.2 mg Lactose-Monohydrat.
Jede Tablette zu 1 mg enthält 61.4 mg Lactose-Monohydrat.
Jede Tablette zu 2 mg enthält 60.3 mg Lactose-Monohydrat.
Tablettenüberzug:
Polyvinylalkohol, Titandioxid (E171), rotes Eisenoxid (E172), Talkum, Lecithin aus Soja (E322), Xanthangummi, schwarzes Eisenoxid (E172, nur 0.25 mg- und 1 mg-Tabletten), gelbes Eisenoxid (E172, nur 2 mg-Tabletten).
Wo erhalten Sie Mayzent? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Starterpackung mit 12 Filmtabletten à 0.25 mg (B)
Packung mit 120 Filmtabletten à 0.25 mg (B)
Packung mit 28 Filmtabletten à 1 mg (B)
Packung mit 28 Filmtabletten à 2 mg (B)
Zulassungsnummer
67230 (Swissmedic)
Zulassungsinhaberin
Novartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz
Diese Packungsbeilage wurde im Juni 2025 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
TravoTim-Vision
Was sind TravoTim-Vision Augentropfen und wann werden sie angewendet?
TravoTim-Vision Augentropfen sind eine Kombination von Travoprost und Timolol. Travoprost ist ein Prostaglandin-Analog, welches den Abfluss des Kammerwassers erhöht und damit den Druck im Auge senkt. Timolol ist ein Betablocker, welcher die Produktion des Kammerwassers im Auge reduziert. Die beiden Substanzen wirken zusammen und reduzieren den Druck im Auge.
TravoTim-Vision Augentropfen sind zur Behandlung eines erhöhten Druckes im Auge bestimmt. Dieser erhöhte Druck kann eine Erkrankung namens Glaukom (grüner Star) hervorrufen.
TravoTim-Vision Augentropfen dürfen nur auf Verschreibung des Arztes oder der Ärztin angewendet werden.
Was sollte dazu beachtet werden?
Wenn Sie weiche Kontaktlinsen tragen, nehmen Sie die Linsen vor dem Eintropfen heraus. Lassen Sie nach dem Eintropfen 15 Minuten vergehen, bevor Sie die Kontaktlinsen wieder einsetzen.
Wann dürfen TravoTim-Vision Augentropfen nicht angewendet werden?
Wenn Sie allergisch gegenüber Prostaglandinen, Betablockern oder einem der sonstigen Bestandteile des Arzneimittels sind.
TravoTim-Vision Augentropfen sollen nicht angewendet werden bei:
·gegenwärtigen oder vergangenen Atemwegserkrankungen wie Asthma, Bronchitis, anderen schweren Atemwegserkrankungen, bronchialen Problemen, oder einer chronisch obstruktiven Lungenerkrankung;
·Herzproblemen wie verlangsamter Herzschlag, Herzinfarkt, oder anderen Problemen des Herzrhythmus;
·Frauen, die schwanger werden könnten, ohne dass ausreichende schwangerschaftsverhütende Massnahmen ergriffen wurden;
·getrübter Hornhaut;
·starkem allergischem Schnupfen.
Dürfen TravoTim-Vision Augentropfen während einer Schwangerschaft oder in der Stillzeit angewendet werden?
TravoTim-Vision Augentropfen dürfen während der Schwangerschaft oder Stillzeit nur mit ausdrücklicher Genehmigung Ihres Arztes oder Apothekers bzw. Ihrer Ärztin oder Apothekerin angewendet werden.
Wie verwenden Sie TravoTim-Vision Augentropfen?
Die übliche Dosierung
Erwachsene: Einen Tropfen in das (die) betroffene(n) Auge(n) tropfen, einmal täglich – am Morgen oder am Abend, allerdings immer zur selben Tageszeit.
Wenden Sie TravoTim-Vision Augentropfen nur dann an beiden Augen an, wenn Ihr Arzt bzw. Ihre Ärztin dies angewiesen hat. Verwenden Sie TravoTim-Vision Augentropfen so lange, wie von Ihrem Arzt bzw. Ihrer Ärztin verordnet.
Verwenden Sie TravoTim-Vision Augentropfen ausschliesslich zum Eintropfen in Ihre Augen.
Wie sind TravoTim-Vision Augentropfen anzuwenden?
(image)
·Entfernen Sie die umhüllende Folie unmittelbar vor dem Erstgebrauch von TravoTim-Vision Augentropfen (Abbildung 1). Nehmen Sie das Fläschchen heraus und schreiben Sie das Öffnungsdatum auf das Etikett.
·Nehmen Sie die TravoTim-Vision-Flasche und einen Spiegel zur Hand.
·Waschen Sie Ihre Hände.
·Schrauben Sie die Kappe ab.
·Halten Sie das Fläschchen mit der Spitze nach unten zwischen dem Daumen und den Fingern.
·Beugen Sie den Kopf zurück. Ziehen Sie das Augenlid mit sauberem Finger nach unten, bis ein Spalt zwischen Lid und Auge entsteht. Bringen Sie hierzu die Tropferspitze nahe an das Auge heran. Verwenden Sie einen Spiegel, wenn dies das Eintropfen erleichtert.
·Tropfen Sie nun, wie in Abbildung 2 dargestellt, ein.
·Berühren Sie jedoch weder das Auge, das Augenlid noch die Augenumgebung oder andere Oberflächen mit der Tropferspitze, da sonst Keime in die Augentropfen gelangen können.
·Üben Sie sanften Druck auf das Fläschchen aus, so dass sich ein Tropfen TravoTim-Vision Augentropfen löst (Abbildung 3).
·Nachdem Sie TravoTim-Vision Augentropfen angewendet haben, drücken Sie mit einem Finger etwa 2 Minuten lang auf den Augenwinkel neben der Nase (Abbildung 4). Das verhindert, dass zu viel TravoTim-Vision Augentropfen in den übrigen Körper gelangt.
·Wenn Sie die Tropfen für beide Augen verwenden, wiederholen Sie die Schritte am anderen Auge.
·Verschliessen Sie das Fläschchen sofort nach Gebrauch wieder fest.
Sollte ein Tropfen nicht ins Auge gelangt sein, tropfen Sie nach.
Wenn zu viel des Arzneimittels ins Auge gelangt ist, spülen Sie das Auge mit lauwarmem Wasser aus. Tropfen Sie nicht mehr nach, bis es Zeit für die nächste planmässige Anwendung ist.
Wenn Sie die Anwendung von TravoTim-Vision Augentropfen vergessen haben, setzen Sie die Behandlung planmässig fort. Tropfen Sie keine doppelte Dosis ein, wenn Sie die vorherige Anwendung vergessen haben.
Wenn Sie zusätzlich andere Augentropfen verwenden, lassen Sie zwischen der Anwendung von TravoTim-Vision Augentropfen und anderen Arzneimitteln 5 Minuten vergehen.
Die Anwendung und Sicherheit von Travoprost-Timolol Augentropfen bei Kindern und Jugendlichen ist bisher nicht geprüft worden. Bei diesen Altersgruppen wird das Arzneimittel daher nicht angewendet.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen können TravoTim-Vision Augentropfen haben?
Folgende Nebenwirkungen können bei der Anwendung von TravoTim-Vision auftreten:
Bei einigen Patienten bzw. Patientinnen, die TravoTim-Vision Augentropfen anwenden, kommt es zu lokalen Nebenwirkungen. Sie können unangenehm sein, die meisten gehen jedoch schnell vorüber.
Bleibende Hautveränderungen in der Augenregion, wie ein Dunklerwerden der Haut und der Lider oder Farbveränderungen der Iris können auftreten.
Wenn Sie besorgt sind, wenden Sie sich bitte an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin.
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
Auswirkungen auf das Auge: Augenrötung.
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Auswirkungen auf das Auge: Entzündungen am Auge, verschwommenes Sehen, Trockenheit des Auges, Augenschmerzen, Jucken am Auge, Fremdkörpergefühl am Auge, Augenirritation.
Auswirkungen auf den Körper: Nervosität, Schwindelgefühl, Kopfschmerzen, erhöhter oder verminderter Blutdruck, Schmerzen in Händen und Beinen.
Gelegentlich (betrifft 1 bis 10 von 1'000 Anwendern)
Auswirkungen auf das Auge: Entzündungen der Augenoberfläche, Entzündung der Iris, entzündete Bindehaut, Entzündung des Lids, Lichtempfindlichkeit, verminderte Sehkraft, Juckreiz am Augenlid, Entzündung der Augenliddrüsen, Müdigkeit der Augen, Blutungen im Auge, Augenschwellung, erhöhter Tränenfluss, Rötung des Lids, Verkrustung der Lidränder, vermehrtes Wimpernwachstum, Farbveränderung der Haut (Augenlid und um das Auge herum).
Auswirkungen auf den Körper: Allergie, Schwindel, Kopfschmerzen, Verlangsamung des Herzschlages, Kurzatmigkeit, Hautentzündung, starkes Haarwachstum, Atemnot, Husten.
Selten (betrifft 1 bis 10 von 10'000 Anwendern)
Auswirkungen auf das Auge: Hornhautveränderungen, Entzündung der Lidranddrüsen, Fehlstellung der Wimpern.
Auswirkungen auf den Körper: Veränderung der Stimme, Halsbeschwerden, Nesselsucht.
Weitere Nebenwirkungen bei Travoprost-Timolol Augentropfen, welche nach der Markteinführung gemeldet wurden und deren Häufigkeit jedoch nicht bekannt ist, beinhalten:
Auswirkungen auf das Auge: Schwellung der Netzhaut, Augenentzündung, Entzündungen der Augenoberfläche, Entzündung des Augenlids, erhöhter Tränenfluss, Schwellungen des Auges, Schwellungen des Lids, Augenrötung, Erschlaffung der Augenlider, eingefallene Augen, Farbveränderungen der Iris (der farbige Ring um die Pupille).
Allgemeine Nebenwirkungen: Dinge sehen, fühlen oder hören, die nicht da sind (Halluzination), Schwindel, Brustschmerzen, erhöhter Herzschlag, Atemnot, Husten, Asthma, Schwellung der Gliedmassen, niedriger Blutdruck, Geschmacksstörung, Hautausschlag, Haarausfall, Depression.
Bei der Anwendung von Augentropfen, die Travoprost oder Timolol enthalten, ist von folgenden weiteren Nebenwirkungen berichtet worden, von denen jedoch nicht bei Travoprost-Timolol Augentropfen berichtet wurde:
Auswirkungen auf das Auge: schmerzhaftes rotes Auge, erhöhter Tränenfluss, Rötungen, Schwellungen, Bläschen am Lid, Bindehautstörungen, Linsentrübung, Trauma am Auge nach einer Augenoperation, verminderte Hornhautempfindlichkeit, Doppeltsehen.
Auswirkungen auf den Körper: niedriger Blutzucker, Schlaflosigkeit, Albträume, Gedächtnisverlust, verminderter Blutfluss zum Gehirn, chronische neuromuskuläre Schwäche, Herzstolpern, Herzinfarkt, Herzstillstand, kalte Hände und Füsse, Geschmackstörung, Übelkeit, Bauchbeschwerden, Hautabschälung, Muskelschmerz, sexuelle Funktionsstörungen, verringertes sexuelles Verlangen, Verlust von Stärke und Energie, Halsschmerzen, Mundtrockenheit, Verstopfung, systemische allergische Reaktionen, Schwellung von Gesicht und Rachen, juckender Ausschlag, Ausschlag lokal und am ganzen Körper, Juckreiz, Atembeschwerden, Husten, Schwindel, Brustkorbschmerzen, Schwellungen, Schnupfen, Durchfall, Erbrechen.
Wenn Sie Nebenwirkungen bemerken, die hier nicht beschrieben sind, sollten Sie Ihren Arzt oder Apotheker, bzw. Ihre Ärztin oder Apothekerin informieren. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Aufbrauchfrist nach Anbruch
Vier Wochen nach dem ersten Öffnen sollten Sie die Augentropfen wegwerfen, da die Keimfreiheit dann nicht mehr garantiert ist. Notieren Sie sich das Anbruchdatum auf dem Etikett und der Faltschachtel.
Lagerungshinweis
Bewahren Sie das Arzneimittel bei Raumtemperatur (15–25 °C) und ausser Reichweite von Kindern auf. Bringen Sie das Arzneimittel nach Beendigung der Behandlung Ihrer Abgabestelle (Arzt, Apotheker bzw. Ärztin, Apothekerin) zum fachgerechten Entsorgen zurück.
Weitere Hinweise
Um eine Verunreinigung der Tropferspitze und der Augentropfen zu vermeiden, ist es wichtig, dass Sie die Lider und die umgebenden Augenpartien nicht mit der Tropferspitze der Flasche berühren. Die Tropferspitze sollte auch nicht mit den Händen in Kontakt kommen.
Flasche sofort nach Gebrauch gut verschliessen.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in TravoTim-Vision Augentropfen enthalten?
TravoTim-Vision sind sterile Augentropfen und enthalten als Wirkstoffe 40 µg Travoprost und 5 mg Timolol (als Timololmaleat) pro 1 ml.
TravoTim-Vision enthält als Konservierungsmittel Benzalkoniumchlorid sowie weitere Hilfsstoffe.
Wo erhalten Sie TravoTim-Vision Augentropfen? Welche Packungen sind erhältlich?
Sie erhalten TravoTim-Vision Augentropfen in Apotheken nur gegen ärztliche Verschreibung.
Es sind Packungen zu 2,5 ml und 3 x 2,5 ml erhältlich.
Zulassungsnummer
67232 (Swissmedic)
Zulassungsinhaberin
OmniVision AG, 8212 Neuhausen.
Diese Packungsbeilage wurde im März 2021 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Ezetimib Simvastatin Zentiva®
Qu'est-ce que l'Ezetimib Simvastatin Zentiva et quand doit-il être utilisé?
Ezetimib Simvastatin Zentiva contient deux principes actifs, la simvastatine et l'ézétimibe. Il provoque une baisse du cholestérol total trop élevé, du cholestérol LDL (mauvais cholestérol) et des triglycérides (substances apparentées aux lipides) et il élève le cholestérol HDL (bon cholestérol) dans le sang. Il est destiné aux patients dont les taux de cholestérol sont trop élevés et chez lesquels un régime seul ne provoque pas de réduction suffisante de ces valeurs.
Ezetimib Simvastatin Zentiva associe les mécanismes d'action complémentaires des deux principes actifs entraînant une baisse du cholestérol. Ezetimib Simvastatin Zentiva diminue l'absorption de cholestérol à partir de l'intestin grêle et inhibe la production endogène de cholestérol par le foie.
Votre médecin vous a prescrit Ezetimib Simvastatin Zentiva afin de diminuer vos taux sanguins trop élevés de cholestérol et de triglycérides. Le cholestérol est l'un des nombreux lipides que l'on trouve dans la circulation sanguine. Votre cholestérol total est constitué principalement de cholestérol LDL et de cholestérol HDL.
Ezetimib Simvastatin Zentiva ne doit être utilisé que sur prescription médicale.
De quoi faut-il tenir compte en dehors du traitement?
Avant d'instaurer le traitement par Ezetimib Simvastatin Zentiva, il faut suivre un régime hypocholestérolémiant et diminuer l'excès pondéral. Le régime pauvre en graisses et en cholestérol doit également être poursuivi durant le traitement par Ezetimib Simvastatin Zentiva.
Quand Ezetimib Simvastatin Zentiva ne doit-il pas être pris?
Ne prenez pas Ezetimib Simvastatin Zentiva si vous présentez une hypersensibilité (allergie) à l'ézétimibe, à la simvastatine ou à l'un des composants du médicament. Les patients souffrant d'une maladie du foie ou ceux qui, pour des raisons inconnues, présentent des taux sanguins trop élevés d'enzymes hépatiques, ne doivent pas utiliser Ezetimib Simvastatin Zentiva.
En outre, Ezetimib Simvastatin Zentiva ne doit pas être pris durant la grossesse ou en période d'allaitement. Il ne faut pas administrer Ezetimib Simvastatin Zentiva aux enfants et aux adolescents de moins de 18 ans, étant donné que l'on ne dispose pas encore d'expériences pour cette classe d'âge.
Ezetimib Simvastatin Zentiva ne doit pas être pris si vous utilisez un ou plusieurs des médicaments suivants: contre les mycoses, p.ex. itraconazole (Sporanox®), kétoconazole (Nizoral®), posaconazole (Noxafil®) ou voriconazole (Vfend®); inhibiteurs de la protéase du VIH, p.ex. indinavir (Crixivan®), nelfinavir (Viracept®), ritonavir (Norvir®), saquinavir (Invirase®, Fortovase®); médicaments pour le traitement des infections par le virus de l'hépatite C, p.ex. bocéprévir (Victrelis®) ou télaprévir (Incivo®); antibiotiques, p.ex. érythromycine (Erythrocin®), clarithromycine (Klacid®), télithromycine; acide fusidique systémique (Fucidin® comprimés pelliculés); certains antidépresseurs, p.ex. néfazodone (Nefadar®); médicaments contenant du cobicistat (Stribild®); gemfibrozil (Gevilon®), un fibrate hypocholestérolémiant; ciclosporine (Sandimmun®); danazol (Danatrol®).
Demandez l'avis de votre médecin si vous ne savez pas avec certitude si un médicament que vous prenez fait partie de la liste ci-dessus.
Quelles sont les précautions à observer lors de la prise d'Ezetimib Simvastatin Zentiva?
Veuillez informer votre médecin de tous les problèmes médicaux que vous présentez actuellement ou que vous avez présentés dans le passé, en particulier des problèmes et affections hépatiques ou des allergies.
Pendant la prise d'ézétimibe/simvastatine des valeurs élevées sont apparues dans de rares cas pour certains tests hépatiques (sans signes pathologiques). C'est pourquoi le médecin contrôlera périodiquement ces données de laboratoire au cours du traitement. Pour les patients ayant une consommation excessive d'alcool ou ceux dont les antécédents font état d'une maladie du foie, le médecin ne prescrira ce médicament qu'en observant des précautions particulières. Veuillez donc informer votre médecin si vous consommez de grandes quantités d'alcool ou si vous avez souffert d'une maladie du foie dans le passé.
Votre médecin vous surveillera étroitement au cours de votre traitement avec ce médicament afin de détecter le développement éventuel d'un diabète ou d'un risque de diabète. Si vous avez des taux sanguins accrus de glucose et de lipides et si vous présentez un excès de poids et une hypertension, vous êtes probablement à risque de développer un diabète.
Veuillez consulter immédiatement votre médecin si vous ressentez des douleurs, une sensibilité ou une faiblesse au niveau des muscles qui sont inexpliquables. Dans de rares cas, les problèmes musculaires peuvent être graves; y compris une dégradation musculaire qui peut entraîner des dommages rénaux et en outre la mort. Le risque d'une dégradation de muscles est plus élevé chez des patient(e)s qui prennent de doses d'Ezetimib Simvastatin Zentiva plus fortes, surtout avec un dosage de 10/80 mg, chez les patient(e)s âgé(e)s (65 ans ou plus), chez la femme et lors de lésions rénales préexistantes ou de problèmes de la glande thyroïde.
Etant donné que le risque de problèmes musculaires peut être plus marqué si vous prenez l'un des médicaments ou substances ci-après en même temps qu'Ezetimib Simvastatin Zentiva (voir sous «Quels effets secondaires Ezetimib Simvastatin Zentiva peut-il provoquer?»), il est particulièrement important que vous informiez votre médecin si vous prenez l'un des médicaments ou substances suivants: l'immunosuppresseur ciclosporine (Sandimmun Néoral®); le danazol (Danatrol®); des médicaments contre les mycoses, p.ex. l'itraconazole (Sporanox®), le kétoconazole (Nizoral®), le posaconazole (Noxafil®) ou le voriconazole (Vfend®); des fibrates, p.ex. le gemfibrozil (Gevilon®), le ciprofibrate (Hyperlipen®), le bézafibrate (Cedur®) ou le fénofibrate (Lipanthyl®); des antibiotiques comme l'érythromycine (Erythrocin®), la clarithromycine (Klacid®), la télithromycine et l'acide fusidique (Fucidin®); des inhibiteurs de la protéase du VIH, par ex. l'indinavir (Crixivan®), le nelfinavir (Viracept®), le ritonavir (Norvir®) et le saquinavir (Invirase®, Fortovase®); médicaments antivirales contre l'hépatite C, p.ex. le bocéprévir (Victrelis®), le télaprévir (Incivo®), elbasvir ou grazoprevir (les principes actifs de Zepatier®); l'antidépresseur néfazodone (Nefadar®); médicaments contenant du cobicistat (Stribild®); l'amiodarone (Cordarone®), un médicament destiné au traitement des arythmies cardiaques; des médicaments destinés au traitement de l'hypertension artérielle, de l'angine de poitrine ou d'autres affections cardiaques, par ex. le vérapamil (Isoptin®), le diltiazem (Dilzem®) ou l'amlodipine (Norvasc®); le lomitapide (un médicament utilisé pour le traitement d'une maladie génétique rare et sévère caractérisée par des taux de cholestérol anormaux); la daptomycine (un médicament utilisé pour le traitement des infections compliquées de la peau et de la structure de la peau et de la bactériémie); le ticagrelor (Brilique®, antiagrégant plaquettaire); le jus de pamplemousse (à éviter pendant la prise d'Ezetimib Simvastatin Zentiva).
Il est important que vous informiez votre médecin si vous prenez des anticoagulants (médicaments utilisés pour fluidifier le sang) comme la phenprocoumone (Marcoumar®) ou l'acénocoumarol (Sintrom®), de la colchicine (médicament pour traiter la goutte), ou si vous prenez de la niacine ou de la digoxine.
Certains de ces médicaments ont déjà été mentionnés ci-dessus dans le chapitre «Quand Ezetimib Simvastatin Zentiva ne doit-il pas être pris?».
Si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de prendre Ezetimib Simvastatin Zentiva.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu'il est essentiellement «sans sodium».
En raison des effets indésirables possibles, ce médicament peut affecter l'aptitude à la conduite et les capacités à utiliser des outils ou des machines.
Adressez-vous à votre médecin ou pharmacien avant de prendre Ezetimib Simvastatin Zentiva, si vous avez ou avez eu une myasthénie (maladie accompagnée d'une faiblesse musculaire générale, pouvant parfois toucher les muscles utilisés pour respirer) ou une myasthénie oculaire (maladie provoquant une faiblesse musculaire au niveau des yeux), car les statines peuvent parfois aggraver ces affections ou entraîner leur apparition (voir «Quels effets secondaires Ezetimib Simvastatin Zentiva peut-il provoquer?»).
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie, vous êtes allergique, vous prenez ou utilisez déjà d'autres médicaments (même en automédication!) en usage interne ou externe, ou vous êtes d'origine asiatique.
Veuillez aussi informer votre médecin qui vous prescrit un nouveau médicament que vous prenez Ezetimib Simvastatin Zentiva.
Ezetimib Simvastatin Zentiva peut-il être pris pendant la grossesse ou l'allaitement?
Ezetimib Simvastatin Zentiva ne doit pas être pris pendant la grossesse ou l'allaitement. Vous ne devrez pas non plus prendre Ezetimib Simvastatin Zentiva si vous prévoyez une grossesse ou si vous pensez que vous pourriez être enceinte. Si vous deviez tout de même tomber enceinte durant le traitement à Ezetimib Simvastatin Zentiva, arrêtez le traitement et prenez immédiatement contact avec votre médecin.
Comment utiliser Ezetimib Simvastatin Zentiva?
Avant d'instaurer le traitement à Ezetimib Simvastatin Zentiva, il faut suivre un régime hypocholestérolémiant approprié, qui devra être poursuivi durant le traitement.
·La dose usuelle au début du traitement se monte à 1 comprimé d'Ezetimib Simvastatin Zentiva 10/10 une fois par jour, le soir. En fonction de la situation et de la réponse au traitement, le médecin augmentera lentement la dose à 1 comprimé d'Ezetimib Simvastatin Zentiva 10/20, d'Ezetimib Simvastatin Zentiva 10/40 ou d'Ezetimib Simvastatin Zentiva 10/80.
·En raison d'un risque accru de problèmes musculaires, la dose de 10/80 mg d'Ezetimib Simvastatin Zentiva ne convient qu'aux patients ayant utilisé cette dose de façon chronique sans problèmes musculaires sérieux. Avec un dosage de 10/80 mg, les effets secondaires sont plus fréquents.
·Ezetimib Simvastatin Zentiva peut être pris indépendamment des repas.
·Si votre médecin vous a prescrit de prendre en même temps qu'Ezetimib Simvastatin Zentiva également un échangeur d'anions, par ex. la colestyramine (Quantalan®) ou le colestipol (Colestid®), prenez Ezetimib Simvastatin Zentiva au moins 2 heures avant ou au moins 4 heures après l'échangeur d'anions.
·Ezetimib Simvastatin Zentiva doit être pris conformément aux prescriptions de votre médecin. Poursuivez la prise des autres médicaments hypocholestérolémiants jusqu'à ce que votre médecin vous ordonne de les arrêter.
Il est important de prendre Ezetimib Simvastatin Zentiva tous les jours, exactement comme prescrit par votre médecin.
Même si vous prenez des médicaments destinés à abaisser vos taux de cholestérol, il est important de faire contrôler régulièrement vos taux de cholestérol. Vous devriez connaître vos taux de cholestérol ainsi que les valeurs que vous souhaitez atteindre et maintenir.
Que faut-il faire si vous prenez trop de médicaments?
Si vous avez pris plus de comprimés d'Ezetimib Simvastatin Zentiva que le nombre prescrit, veuillez vous adresser à votre médecin ou pharmacien.
Que faut-il faire si vous avez oublié une dose?
Essayez de prendre Ezetimib Simvastatin Zentiva en vous conformant aux prescriptions. Si toutefois vous deviez oublier une prise, ne prenez pas de dose supplémentaire, mais poursuivez la prise le lendemain, comme d'habitude.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Ezetimib Simvastatin Zentiva peut-il provoquer?
Fréquent (concerne 1 à 10 utilisateurs sur 100)
Douleurs musculaires (voir sous «Quelles sont les précautions à observer lors de la prise d'Ezetimib Simvastatin Zentiva?»).
Occasionnel (concerne 1 à 10 utilisateurs sur 1000)
Perte de poids, vertiges, maux de tête, picotements, douleurs abdominales, problèmes digestifs, ballonnements, nausées, vomissements, ventre gonflé, diarrhée, sécheresse buccale, brûlures d'estomac, éruption cutanée, démangeaisons, urticaire, douleurs articulaires, douleurs musculaires, sensibilité ou faiblesse musculaire, crispations musculaires, douleurs au niveau de la nuque, douleurs des extrémités supérieures et inférieures, douleurs dorsales, fatigue ou faiblesse inhabituelles, manque de forces, douleurs dans la poitrine, gonflements affectant en particulier les mains, les pieds et le visage, troubles du sommeil, insomnie.
Rare (concerne 1 à 10 utilisateurs sur 10'000)
Inflammations rhino-pharyngées, hypersensibilité, manque d'appétit, réduction de l'appétit, anxiété, dépression, libido réduite, manque d'énergie, accès de rougissement avec sensation de chaleur, tension artérielle accrue, problèmes respiratoires (y compris toux tenace et/ou détresse respiratoire ou fièvre), saignements du nez, gastrite, acné, chute des cheveux, transpiration excessive, psoriasis, douleur de flanc, articulations gonflées, douleurs musculaires, sensibilité et faiblesse de la musculature, besoin fréquent d'uriner, trouble érectile, sensation de soif.
En outre, les effets indésirables ci-après ont été constatés lors de l'emploi de comprimés d'ézétimibe (comprimés Ezetrol®) ou de simvastatine (comprimés pelliculés Zocor®) (des comprimés qui contiennent l'un des principes actifs d'Ezetimib Simvastatin Zentiva): problèmes de foie (parfois sérieux), constipation, renvois acides, réactions allergiques, y compris gonflement du visage, des lèvres, de la langue et/ou du pharynx, susceptibles de provoquer des difficultés respiratoires ou des problèmes de déglutition (nécessitant un traitement immédiat); éruptions cutanées, urticaire, éruptions de plaques rouges en relief (éventuellement de forme discoïde à lésion centrale); douleurs musculaires, sensibilité ou faiblesse musculaire (qui, dans de très rares cas, ne disparaissent pas après l'arrêt d'administration d'Ezetimib Simvastatin Zentiva); pancréatite, calculs biliaires, cholécystite, problèmes de mémoire, perte de mémoire, confusion. On a rapporté dans des cas isolés des problèmes concernant le tendon d'Achille, rarement associés à une rupture du tendon d'Achille.
Le développement d'un diabète est possible, surtout si vous avez des taux sanguins élevés de glucose et de lipides, si vous présentez un excès de poids et si votre tension artérielle est élevée. Votre médecin vous surveillera pour détecter les signes éventuels correspondants au cours de votre traitement avec ce médicament.
Si votre médecin vous a prescrit Ezetimib Simvastatin Zentiva, il souhaitera peut-être faire des tests sanguins pour vérifier la santé de votre foie avant que vous preniez Ezetimib Simvastatin Zentiva pour la première fois et dans le cas où des problèmes hépatiques surviennent pendant le traitement avec Ezetimib Simvastatin Zentiva. Contactez immédiatement votre médecin si vous développez les symptômes suivants de problèmes de foie: fatigue et faiblesse, manque d'appétit, douleurs dans l'abdomen supérieur, urines foncées, teinte jaune de la peau ou du blanc des yeux.
Fréquence inconnue (ne peut être estimée sur la base des données disponibles): Myasthénie (maladie provoquant une faiblesse musculaire générale, pouvant parfois toucher les muscles utilisés pour respirer). Myasthénie oculaire (maladie provoquant une faiblesse des muscles oculaires). Adressez-vous à votre médecin si vous présentez une faiblesse dans vos bras ou vos jambes qui s'aggrave après des périodes d'activité, une vision double ou des paupières tombantes, des difficultés à avaler ou un essoufflement.
Affection sévère avec forte desquamation et gonflement de la peau, formation de cloques sur la peau, dans la bouche, dans les yeux et au niveau des organes génitaux, et fièvre; éruption cutanée avec taches roses/rouges, notamment au niveau de la paume des mains ou de la plante des pieds, lesquelles peuvent former des cloques. Vous pouvez aussi présenter en même temps des symptômes de type grippal comme une fièvre, des frissons ou des douleurs musculaires; des symptômes de type grippal avec éruption cutanée, fièvre, gonflement des ganglions et résultats anormaux des analyses sanguines (y compris augmentation des globules blancs (éosinophilie) et des enzymes hépatiques).
Informez votre médecin ou votre pharmacien si vous remarquez l'un ou l'autre de ces symptômes ou si vous remarquez d'autres symptômes en rapport avec la prise d'Ezetimib Simvastatin Zentiva.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
A quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Conservez dans l'emballage d'origine, à une température pas au-dessus de 25 °C et hors de portée des enfants.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Ezetimib Simvastatin Zentiva?
Chaque comprimé d'Ezetimib Simvastatin Zentiva contient
Principes actifs
10 mg d'ézétimibe ainsi que respectivement 10 mg (Ezetimib Simvastatin Zentiva 10/10), 20 mg (Ezetimib Simvastatin Zentiva 10/20), 40 mg (Ezetimib Simvastatin Zentiva 10/40) ou 80 mg (Ezetimib Simvastatin Zentiva 10/80) de simvastatine
Excipients
Lactose monohydrate, hypromellose, croscarmellose sodique, cellulose microcrystalline, acide ascorbique, acide citrique anhydre, butylhydroxyanisole (E320), gallate de propyle (E310), stéarate de magnésium, oxyde de fer jaune (E172), oxyde de fer rouge (E172), oxyde de fer noir (E172)
Où obtenez-vous Ezetimib Simvastatin Zentiva? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Ezetimib Simvastatin Zentiva 10/10: plaquettes thermoformées de 28 et de 98 comprimés.
Ezetimib Simvastatin Zentiva 10/20: plaquettes thermoformées de 28 et de 98 comprimés.
Ezetimib Simvastatin Zentiva 10/40: plaquettes thermoformées de 28 et de 98 comprimés.
Ezetimib Simvastatin Zentiva 10/80: plaquettes thermoformées de 28 comprimés.
Numéro d'autorisation
67242 (Swissmedic).
Titulaire de l'autorisation
Helvepharm AG, Frauenfeld.
Cette notice d'emballage a été vérifiée pour la dernière fois en mars 2024 par l'autorité de contrôle des médicaments (Swissmedic).: | Janssen-Cilag AG | | Wie verwenden Sie Durogesic Matrix?Durogesic Matrix ist ein rechteckiges Matrixpflaster, das auf die Haut geklebt wird. Das transdermale Pflaster besteht aus zwei funktionalen Schichten. Die Oberseite besteht aus einer wasserundurchlässigen Trägerfolie. Darunter befindet sich eine Fentanyl-haltige, selbstklebende Matrixschicht. Diese Matrixschicht ist durch eine Abziehfolie bedeckt, die vor dem Gebrauch aufgrund der Schlitzung einfach zu entfernen ist.
Durch eine Polymer-Matrixschicht wird der Wirkstoff Fentanyl kontinuierlich freigesetzt. Der Wirkstoff dringt aus der Matrixschicht durch die Haut und gelangt von dort in die Blutbahn. Solange es auf der Haut haftet, gibt Durogesic Matrix fortwährend geringe Mengen von Fentanyl direkt an das Blut ab.
Durogesic Matrix ist in 5 verschiedenen Dosierungen bzw. Grössen erhältlich, 12 µg/h enthält die niedrigste, 100 µg/h die höchste Dosis. Die verschiedenen Dosierungsstärken sind gut ersichtlich und in unterschiedlichen Farben auf den Packungen aufgedruckt:
Durogesic Matrix 12 µg/h: orange;
Durogesic Matrix 25 µg/h: rosa;
Durogesic Matrix 50 µg/h: grün;
Durogesic Matrix 75 µg/h: blau;
Durogesic Matrix 100 µg/h: grau.
Die Dauer und Dosierung der Anwendung richtet sich nach der Art der zu behandelnden Schmerzen, nach dem Allgemeinzustand sowie der bisherigen Schmerzmedikation, und wird in jedem Fall von Ihrem Arzt/Ihrer Ärztin bestimmt und überwacht. Ihr Arzt/Ihre Ärztin wird ein für Sie resp. für Ihr Kind geeignetes Therapieschema empfehlen. Halten Sie sich genau an seine/ihre Vorschriften. Bitte fragen Sie bei Ihrem Arzt oder Apotheker resp. Ihrer Ärztin oder Apothekerin nach, wenn Sie sich nicht ganz sicher sind.
Wird nach längerer Anwendung Durogesic Matrix nicht mehr benötigt, so wird Ihr Arzt resp. Ihre Ärztin über die stufenweise Reduktion der Dosis sowie über das Absetzen der Behandlung entscheiden.
Anleitung zur Anwendung, Handhabung und Entsorgung
Anwendung und Wechseln der Pflaster
·Notieren Sie sich, wann das Pflaster appliziert worden ist (Tag, Datum und Uhrzeit), um nicht zu vergessen, wann es gewechselt werden muss.
·Jedes Pflaster enthält eine ausreichende Wirkstoffmenge für 3 Tage (72 Stunden).
·Wechseln Sie das Pflaster alle drei Tage.
·Entfernen Sie immer zuerst das alte Pflaster, bevor Sie ein neues Pflaster anbringen.
·Wechseln Sie das Pflaster alle 3 Tage (72 Stunden) immer zur selben Uhrzeit.
·Wenn mehrere Pflaster angewendet werden, müssen alle Pflaster zur selben Zeit gewechselt werden.
·Wenn das Pflaster direkter Wärmeeinwirkung ausgesetzt wird, kann sich die Wirkung von Durogesic Matrix verstärken. Verzichten Sie deshalb während der Behandlung mit Durogesic Matrix auf Wärmewickel, elektrische Heizdecken, heizbare Wasserbetten, Hitzelampen, Solarien, Wärmeflaschen, ausgedehnte heisse Bäder, Sauna, heisse Whirlpool-Bäder usw. sowie intensive Sonnenbestrahlung.
Applikationsstelle
·Bringen Sie das Pflaster nicht zweimal hintereinander an derselben Stelle an.
·Durogesic Matrix Pflaster sind auf einer flachen, gesunden, möglichst faltenfreien, nicht irritierten und nicht bestrahlten Hautstelle des Oberkörpers oder der Oberarme anzubringen.
Kinder
·Bei Kindern bringen Sie das Pflaster immer auf dem Rücken des Kindes an, wo es für das Kind schwer erreichbar ist und vom Kind nicht entfernt werden kann.
·Überprüfen Sie häufig, ob das Pflaster noch auf der Haut klebt.
·Es ist wichtig, dass das Kind das Pflaster nicht entfernt und es in den Mund nimmt, da dies lebensbedrohlich oder sogar tödlich sein könnte.
·Beobachten Sie das Kind sehr engmaschig 48 Stunden lang, nachdem:
·das erste Pflaster angebracht worden ist;
·ein Pflaster mit höherer Dosis angebracht worden ist.
Anbringen eines Pflasters
Schritt 1: Vorbereitung der Haut
·Achten Sie darauf, dass Sie das Pflaster auf ein unbehaartes oder von Haaren befreites (mit Schere, nicht rasieren!), gesundes Hautareal aufkleben.
·Vor dem Aufkleben sollte die Haut, falls notwendig, mit sauberem Wasser (keine Reinigungsmittel verwenden!) gereinigt und gut abgetrocknet werden.
·Die Hautstelle soll trocken und nicht mit Cremes, Öl, Lotionen, Puder oder Fett behandelt sein.
Schritt 2: Öffnen des Beutels
·Entnehmen Sie das Pflaster erst unmittelbar vor dem Aufkleben aus dem Schutzbeutel.
·Zur Entnahme des Pflasters wird der Schutzbeutel wie folgt geöffnet: Schneiden Sie den Schutzbeutel bei der Markierung am Rand (wird durch einen Pfeil auf dem Etikett des Pflasters angezeigt) ca. 2 mm ein. Reissen Sie anschliessend den Beutel vorsichtig von Hand entlang der Ränder auf und klappen Sie ihn wie Buchseiten auseinander.
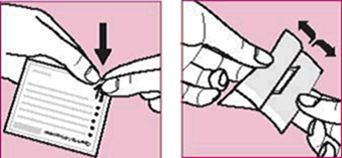
·Entnehmen Sie das Pflaster. Überprüfen Sie das Pflaster vor der Anwendung auf etwaige Beschädigungen. Pflaster, welche zerschnitten, geteilt oder in irgendeiner Weise beschädigt sind, sollen nicht verwendet werden.
·Das Pflaster klebt auf einer grösseren Schutzfolie, die zum leichteren Entfernen s-förmig gekerbt ist. Ziehen Sie die eine Hälfte der Schutzfolie von der Rückseite des Pflasters ab. Der freigelegte Teil des Pflasters kann nun auf die ausgewählte Hautstelle geklebt werden. Die zweite Hälfte der Schutzfolie kann entfernt werden. Das Berühren der klebenden Seite des Pflasters ist zu vermeiden.
Schritt 3: Abziehen und Andrücken
·Vermeiden Sie, die klebende Seite des Pflasters zu berühren.
·Drücken Sie das Pflaster für mindestens 30 Sekunden mit der flachen Hand fest auf die Haut.
·Achten Sie darauf, dass die Ränder des Pflasters gut angedrückt sind. Die Schutzfolie kann mit dem Hausmüll entsorgt werden.
·Waschen Sie Ihre Hände mit Wasser (keine Seife).
Schritt 4: Entsorgung des Pflasters
·Ziehen Sie das Pflaster ab und falten Sie es sofort mit der Klebeseite nach innen zusammen.
·Das Pflaster zurück in den Originalbeutel geben und nach Anweisung des Apothekers bzw. der Apothekerin entsorgen.
·Nicht benutzte Pflaster sollten in der Apotheke (im Krankenhaus) zurückgegeben werden.
·Die Pflaster für Kinder unzugänglich aufbewahren - auch benutzte Pflaster enthalten noch eine gewisse Menge an Wirkstoff, der für Kinder schädlich oder sogar tödlich sein kann.
·Waschen Sie Ihre Hände nach dem Entfernen des Pflasters mit Wasser (keine Seife).
Weitere Hinweise zur Anwendung
Wenn sich nach Abnahme des Matrixpflasters eventuell Rückstände auf der Haut befinden, können diese mit reichlich Wasser entfernt werden. Die Reinigung sollte keinesfalls mit Alkohol oder anderen Lösungsmitteln durchgeführt werden, da diese – bedingt durch die Wirkung des transdermalen Pflasters – durch die Haut gelangen könnten.
Bei Ersteinstellung von Durogesic Matrix und bei Umstellung von anderen Schmerzmitteln kann der maximale schmerzstillende Effekt erst nach ca. 24 Stunden beurteilt werden, da die Fentanyl-Spiegel im Blut nach der Erstanwendung langsam ansteigen. Demzufolge benötigen Sie eventuell am ersten Tag der Behandlung ein zusätzliches Schmerzmittel.
Nachdem Durogesic Matrix entfernt worden ist, nimmt die Wirkstoffkonzentration im Blut nur langsam innerhalb 1–2 Tagen ab, da der Wirkstoff noch aus der Haut resorbiert wird.
Informieren Sie Ihren Arzt/Ihre Ärztin, wenn Ihre Schmerzen während der Anwendung von Durogesic Matrix stärker werden. Er/Sie wird die Dosis von Durogesic Matrix anpassen. Ihr Arzt/Ihre Ärztin kann Ihnen auch die gleichzeitige Anwendung von mehreren Durogesic Matrix Pflastern verschreiben.
Ebenfalls kann Ihnen Ihr Arzt/Ihre Ärztin ein zusätzliches Schmerzmittel verschreiben, um gelegentliche Schmerzschübe zu mildern.
Wenn Sie eine grössere Menge von Durogesic Matrix angewendet haben, als Sie sollten
Eine Überdosierung mit Durogesic Matrix kann eine Gehirnerkrankung (bekannt als toxische Leukenzephalopathie) auslösen.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Durogesic Matrix haben?Wie andere ähnliche Schmerzmittel, kann Durogesic Matrix die Atemtätigkeit vermindern. Falls die Person, welche mit Durogesic Matrix behandelt wird, langsam oder schwach atmet, sollte unverzüglich der Arzt oder die Ärztin informiert werden. Halten Sie die Patientin/den Patienten wach, indem Sie mit ihr/ihm sprechen oder sie/ihn ab und zu schütteln.
Folgende Nebenwirkungen wurden im Zusammenhang mit der Anwendung von Durogesic Matrix berichtet:
Sehr häufig (betrifft mehr als einen von 10 Behandelten): Schläfrigkeit (15%), Kopfschmerzen (12%), Schwindel (13%), Übelkeit (36%), Erbrechen (23%), Verstopfung (23%).
Häufig (betrifft 1 bis 10 von 100 Behandelten: Überempfindlichkeit, Appetitlosigkeit, Schlaflosigkeit, Verwirrtheit, Depression, Angstzustände, Halluzinationen, Zittern, Taubheitsgefühl (Parästhesie), Drehschwindel (Vertigo), Herzklopfen (Palpitationen), zu schneller Herzschlag, Bluthochdruck, Atemnot (Dyspnoe), Durchfall, Bauchschmerzen, Mundtrockenheit, Verdauungsstörung (Dyspepsie), übermässiges Schwitzen, Juckreiz (Pruritus), Hautausschlag, Hautreaktionen (z.B. Hautrötung), unwillkürliche Muskelkontraktionen, Harnverhalten, Erschöpfung (Fatigue), Wasseransammlung im peripheren Gewebe, Kraftlosigkeit (Asthenie), Unwohlsein und Kältegefühl.
Gelegentlich (betrifft 1 bis 10 von 1000 Behandelten): Unruhe, Desorientierung, Euphorie, reduzierte Wahrnehmung von Sinnesreizen (Hypoästhesie), Krampfanfälle, Erinnerungslücken, verringerter Bewusstseinsgrad, Bewusstlosigkeit, verschwommenes Sehen, verlangsamter Herzschlag, blaurot verfärbte Lippen, erniedrigter Blutdruck, Atemprobleme (Atemdepression), Atemnot, geringer Sauerstoffgehalt im Blut (Hypoxie), Darmverschluss, Ekzem, Dermatitis, Hautfunktionsstörungen, Muskelzucken, Störungen der Sexualfunktion, Reaktion an der Applikationsstelle, grippeähnliche Symptome, Gefühl von Körpertemperaturschwankungen, Überempfindlichkeit an der Applikationsstelle und Entzugssymptome.
Selten (betrifft 1 bis 10 von 10'000 Behandelten): Engstellung der Pupille (Miosis), Atemstillstand, Hypoventilation, Vorstufe eines Darmverschlusses (Subileus), Probleme beim Transport von Speisen oder Getränken durch die Speiseröhre (Schluckstörungen) und Dermatitis oder Ekzem an der Applikationsstelle.
Häufigkeit nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden): allergische Reaktionen (anaphylaktischer Schock, anaphylaktische Reaktionen, anaphylaktoide Reaktion), Arzneimittelabhängigkeit, Atemaussetzer im Schlaf (Schlafapnoe), verlangsamte Atemfrequenz (Bradypnoe), Symptome im Zusammenhang mit einer Entzündung der Bauchspeicheldrüse (Pankreatitis) und des Gallengangsystems, wie starke Schmerzen im Oberbauch, die möglicherweise in den Rücken ausstrahlen, Übelkeit, Erbrechen oder Fieber, Fieber und Mangel an männlichen Geschlechtshormonen (Androgenmangel) sowie Hautverdünnung, -rötung oder -geschwüre an der Applikationsstelle.
Bei Umstellung von anderen stark wirksamen Opioiden auf Durogesic Matrix oder bei abruptem Abbruch der Therapie kann es zu schwerwiegenden Entzugserscheinungen, wie z.B. Übelkeit, Erbrechen, Durchfall, Angstzuständen und Kältezittern kommen.
Aus diesem Grund sollten Sie die Behandlung mit Durogesic Matrix nie von sich aus abbrechen. Falls Ihr Arzt/Ihre Ärztin entscheidet, dass die Therapie abgebrochen werden soll, so sollten Sie seine/ihre Anweisungen genau befolgen. Sollten Sie unter einer der oben aufgeführten unerwünschten Wirkungen leiden, informieren Sie Ihren Arzt resp. Ihre Ärztin.
Kinder und Jugendliche
Bei Kindern und Jugendlichen entsprach das Nebenwirkungsprofil dem der Erwachsenen. Neben den bei der Opioidbehandlung von Schmerzen bei schwer kranken Kindern üblicherweise zu erwartenden Nebenwirkungen wurden keine weiteren Risiken bekannt.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Falls irgendein anderes Krankheitszeichen auftritt, bei dem Sie einen Zusammenhang mit der Anwendung von Durogesic Matrix vermuten, sollten Sie Ihren Arzt oder Ihre Ärztin informieren.
Überdosierung: Wichtigstes Anzeichen einer Überdosierung ist abgeschwächte Atmung. Wenn die behandelte Person langsam oder schwach atmet, müssen sofort alle Durogesic Matrix Pflaster entfernt und unverzüglich der behandelnde Arzt oder die behandelnde Ärztin benachrichtigt werden. In der Zwischenzeit halten Sie die Patientin/den Patienten wach, indem Sie mit ihr/ihm sprechen oder sie/ihn ab und zu schütteln. Andere Anzeichen einer Überdosierung können auch Probleme mit dem «Nervensystem» einschliessen, die durch eine Schädigung der weissen Hirnsubstanz verursacht werden (bekannt als toxische Leukenzephalopathie).
Was ist ferner zu beachten?Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Bei Raumtemperatur (15 – 25 °C) lagern.
Im verschlossenen Beutel in der Originalverpackung aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Bewahren Sie das Arzneimittel an einem Ort, der für andere nicht zugänglich ist, sicher auf. Das Arzneimittel kann Menschen schaden, die es versehentlich anwenden oder die es mit Absicht anwenden, obwohl es ihnen nicht verschrieben wurde.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Durogesic Matrix enthalten?Wirkstoffe
1 Transdermales Pflaster Durogesic Matrix 12 µg/h (5,25 cm²) enthält als Wirkstoff 2,1 mg Fentanyl.
1 Transdermales Pflaster Durogesic Matrix 25 µg/h (10,5 cm²) enthält als Wirkstoff 4,2 mg Fentanyl.
1 Transdermales Pflaster Durogesic Matrix 50 µg/h (21 cm²) enthält als Wirkstoff 8,4 mg Fentanyl.
1 Transdermales Pflaster Durogesic Matrix 75 µg/h (31,5 cm²) enthält als Wirkstoff 12,6 mg Fentanyl.
1 Transdermales Pflaster Durogesic Matrix 100 µg/h (42 cm²) enthält als Wirkstoff 16,8 mg Fentanyl.
Hilfsstoffe
Wirkstoffhaltige Schicht: Adhäsives Polyacrylat.
Trägerschicht: Folie aus Polyester/Ethylenvinylacetat-Copolymer.
Schutzfolie: Folie aus silikonisiertem Polyester.
Drucktinten (auf der Trägerschicht):
Durogesic Matrix 12 µg/h: Orange Drucktinte.
Durogesic Matrix 25 µg/h: Rote Drucktinte.
Durogesic Matrix 50 µg/h: Grüne Drucktinte.
Durogesic Matrix 75 µg/h: Blaue Drucktinte.
Durogesic Matrix 100 µg/h: Graue Drucktinte.
Wo erhalten Sie Durogesic Matrix? Welche Packungen sind erhältlich?In Apotheken gegen ärztliche Verschreibung, die nur zum einmaligen Bezug berechtigt.
Durogesic Matrix 12 µg/h, Packung zu 5 Pflastern (zurzeit nicht im Handel).
Durogesic Matrix 25 µg/h, Packung zu 5 Pflastern (zurzeit nicht im Handel).
Durogesic Matrix 50 µg/h, Packung zu 5 Pflastern (zurzeit nicht im Handel).
Durogesic Matrix 75 µg/h, Packung zu 5 Pflastern (zurzeit nicht im Handel).
Durogesic Matrix 100 µg/h, Packung zu 5 Pflastern (zurzeit nicht im Handel).
Zulassungsnummer53904 (Swissmedic).
Diese Packungsbeilage wurde im März 2024 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft. |
|