Zusammensetzung
Wirkstoffe
Secukinumab*.
*aus gentechnisch veränderten CHO (Chinese Hamster Ovary)-Zellen hergestellt.
Hilfsstoffe
Pulver zur Herstellung einer Injektionslösung
Saccharose, L-Histidin, L-Histidinhydrochlorid-Monohydrat, Polysorbat 80, pro vitro.
Injektionslösung in einer Fertigspritze bzw. im Fertigpen
Trehalose-Dihydrat, L-Histidin, L-Histidinhydrochlorid-Monohydrat, L-Methionin, Polysorbat 80, Wasser für Injektionszwecke q.s. ad solutionem pro 0.5 ml bzw. 1 ml bzw. 2 ml.
Darreichungsform und Wirkstoffmenge pro Einheit
Zur subkutanen Verabreichung.
Pulver zur Herstellung einer Injektionslösung
Jede Durchstechflasche enthält nach Rekonstitution mit 1 ml Wasser für Injektionszwecke 150 mg Secukinumab.
Injektionslösung in einer Fertigspritze bzw. im Fertigpen
Jede 2 ml Fertigspritze bzw. jeder 2 ml Fertigpen enthält 300 mg Secukinumab.
Jede 1 ml Fertigspritze bzw. jeder 1 ml Fertigpen enthält 150 mg Secukinumab.
Jede 0.5 ml Fertigspritze enthält 75 mg Secukinumab.
Indikationen/Anwendungsmöglichkeiten
Plaque-Psoriasis
Cosentyx/- SensoReady/- UnoReady ist zur Behandlung von erwachsenen und pädiatrischen Patienten ab 6 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis indiziert, die auf andere systemische Therapien einschliesslich Ciclosporin, Methotrexat oder PUVA nicht angesprochen haben, bei denen diese Therapien kontraindiziert sind oder die diese Therapien nicht tolerieren.
Psoriasis-Arthritis
Cosentyx/- SensoReady/- UnoReady, alleine oder in Kombination mit Methotrexat, ist zur Behandlung erwachsener Patienten mit aktiver Psoriasis-Arthritis indiziert, die unzureichend auf eine vorhergehende Therapie mit krankheitsmodifizierenden Antirheumatika (disease-modifying anti-rheumatic drugs, DMARD) angesprochen haben. Cosentyx/- SensoReady/- UnoReady verzögert die Progression struktureller Schäden und verbessert die körperliche Funktionsfähigkeit.
Axiale Spondyloarthritis (axSpA)
Ankylosierende Spondylitis (AS, Morbus Bechterew)
Cosentyx/- SensoReady ist für die Behandlung erwachsener Patienten mit schwerer, aktiver ankylosierender Spondylitis indiziert, die unzureichend auf eine konventionelle Therapie (beispielsweise NSAIDs) angesprochen haben.
Nicht-röntgenologische axiale Spondyloarthritis (nr-axSpA)
Cosentyx/- SensoReady ist für die Behandlung von schwerer aktiver nicht-röntgenologischer axialer Spondyloarthritis mit objektiven Anzeichen einer Entzündung mit erhöhtem C-reaktiven Protein (CRP) und Magnetresonanztomographie-(MRT)-Befund bei Erwachsenen indiziert, die unzureichend auf eine Therapie mit nichtsteroidalen entzündungshemmenden Medikamenten (NSAIDs) angesprochen haben.
Juvenile idiopathische Arthritis (JIA)
Enthesitis-assoziierte Arthritis (EAA)
Cosentyx/- SensoReady ist indiziert zur Behandlung von aktiver Enthesitis-assoziierter Arthritis bei Patienten ab 6 Jahren und älter, deren Erkrankung nur unzureichend auf nichtsteroidale entzündungshemmende Medikamente (NSAIDs) und krankheitsmodifizierende Antirheumatika (DMARDs) angesprochen hat. Siehe "Dosierung/Anwendung" und "Eigenschaften/Wirkungen" .
Juvenile Psoriasis-Arthritis (JPsA)
Cosentyx/- SensoReady ist indiziert zur Behandlung von aktiver juveniler Psoriasis-Arthritis bei Patienten ab 6 Jahren und älter, deren Erkrankung nur unzureichend auf nichtsteroidale entzündungshemmende Medikamente (NSAIDs) und krankheitsmodifizierende Antirheumatika (DMARDs) angesprochen hat. Siehe "Dosierung/Anwendung" und "Eigenschaften/Wirkungen" .
Hidradenitis suppurativa (HS)
Cosentyx ist indiziert zur Behandlung von erwachsenen Patienten mit aktiver mittelschwerer bis schwerer Hidradenitis suppurativa (Acne inversa), die unzureichend auf eine systemische Antibiotikatherapie angesprochen haben.
Dosierung/Anwendung
Die Anwendung von Cosentyx/- SensoReady/- UnoReady sollte unter Anleitung und Aufsicht eines Arztes angewendet werden, der Erfahrung in der Diagnose und Behandlung von Erkrankungen hat, bei denen Cosentyx/Cosentyx SensoReady/- UnoReady indiziert ist.
Cosentyx wird als subkutane Injektion verabreicht (zur Bioverfügbarkeit siehe Rubrik "Pharmakokinetik" ). Hautbezirke, die Anzeichen der Psoriasis aufweisen, sollten als Injektionsstellen vermieden werden.
Nach einem angemessenen Training zur Technik der subkutanen Selbstinjektion können sich Patienten Cosentyx/- SensoReady/- UnoReady selbst injizieren oder von einer Betreuungsperson injiziert bekommen, wenn ein Arzt bzw. eine Ärztin dies als angemessen erachtet. Der Arzt bzw. die Ärztin sollte jedoch eine angemessene Nachbeobachtung der Patienten sicherstellen. Die Patienten und/oder Betreuungspersonen sollten angewiesen werden, sich die vollständige Menge Cosentyx/- SensoReady/- UnoReady gemäss den Anweisungen in der Packungsbeilage zu injizieren.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Erwachsene Patienten
Plaque-Psoriasis
Die empfohlene Dosis beträgt 300 mg als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen. Jede 300-mg-Dosis wird in Form von einer subkutanen Injektion zu 300 mg oder zwei subkutanen Injektionen zu je 150 mg verabreicht.
Manche Patienten mit einem Körpergewicht von ≥90 kg können einen zusätzlichen Nutzen aus der Verabreichung von 300 mg alle 2 Wochen ziehen.
Psoriasis-Arthritis
Die empfohlene Dosis liegt bei 150 mg als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen. Die Dosis kann, abhängig vom klinischen Ansprechen, auf 300 mg erhöht werden.
Bei Patienten, die ungenügend auf Anti-TNF-alpha ansprechen (inadequate responders, IR) liegt die empfohlene Dosis bei 300 mg als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen. Jede 300-mg-Dosis wird als eine subkutane Injektion von 300 mg oder als zwei subkutane Injektionen von je 150 mg verabreicht.
Für Patienten mit begleitender mittelschwerer bis schwerer Plaque-Psoriasis siehe Dosierungs- und Anwendungsempfehlungen für Plaque-Psoriasis.
Axiale Spondyloarthritis (axSpA)
Ankylosierende Spondylitis (AS, Morbus Bechterew)
Die empfohlene Dosis liegt bei 150 mg als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen.
Nicht-röntgenologische axiale Spondyloarthritis (nr-axSpA)
Die empfohlene Dosis liegt bei 150 mg als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen.
Die vorliegenden Daten zu Psoriasis-Arthritis und axiale Spondyloarthritis deuten darauf hin, dass ein klinisches Ansprechen in der Regel innerhalb von 16 Behandlungswochen erzielt wird. Bei Patienten, die nach 16 Behandlungswochen nicht auf die Therapie angesprochen haben, sollte erwogen werden, die Behandlung abzusetzen. Bei manchen Patienten mit zunächst unvollständigem Ansprechen kann es im Verlauf bei Fortführung der Behandlung über 16 Wochen hinaus zu Verbesserungen kommen.
Hidradenitis suppurativa
Die empfohlene Dosis beträgt 300 mg Secukinumab, verabreicht als subkutane Injektion, zunächst als Initialdosis in den Wochen 0, 1, 2, 3 und 4, gefolgt von monatlichen Erhaltungsdosen. Bei fehlendem klinischen Ansprechen (HiSCR50) nach 32 Wochen kann die Erhaltungsdosis auf 300 mg alle 2 Wochen erhöht werden. Sollte darunter innert 16 Wochen kein klinisches Ansprechen erreicht werden, ist die Behandlung zu beenden. Jede 300-mg-Dosis wird entweder in Form einer subkutanen Injektion zu 300 mg oder von zwei subkutanen Injektionen zu je 150 mg verabreicht.
Pädiatrische Patienten
Plaque-Psoriasis
Die empfohlene Dosis ist abhängig vom Körpergewicht und wird als subkutane Injektion mit Startdosen in den Wochen 0, 1, 2, 3 und 4 verabreicht, gefolgt von monatlichen Erhaltungsdosen. Jede 75-mg-Dosis wird in Form einer subkutanen Injektion von 75 mg verabreicht. Jede 150-mg-Dosis wird in Form einer subkutanen Injektion von 150 mg verabreicht.
Tabelle 1: Empfohlene Dosierung für Plaque-Psoriasis bei Kindern und Jugendlichen
Körpergewicht zum Zeitpunkt der Dosierung Empfohlene Dosis < 50 kg 75 mg ≥50 kg 150 mg
Juvenile idiopathische Arthritis (JIA)
Enthesitis-assoziierte Arthritis (EAA) und juvenile Psoriasis-Arthritis (JPsA)
Die empfohlene Dosis richtet sich nach dem Körpergewicht. Für Patienten mit einem Körpergewicht von 15 kg bis < 50 kg beträgt die Dosis 75 mg. Für Patienten mit einem Körpergewicht von ≥50 kg beträgt die Dosis 150 mg. Cosentyx wird als subkutane Injektion in den Wochen 0, 1, 2, 3 und 4 verabreicht, gefolgt von monatlichen Erhaltungsdosen (alle 4 Wochen). Jede 75-mg-Dosis wird als eine subkutane Injektion von 75 mg verabreicht. Jede 150-mg-Dosis wird als eine subkutane Injektion von 150 mg verabreicht.
Dosisanpassung aufgrund unerwünschter Wirkungen/Interaktionen
Plaque-Psoriasis
Bei schwerwiegenden unerwünschten Wirkungen (siehe "Warnhinweise und Vorsichtsmassnahmen" ) soll eine temporäre Unterbrechung der Therapie bis zum Abklingen der unerwünschten Wirkung erwogen werden. Da in klinischen Studien, die insgesamt seltenen mukokutanen Candida-Infekte häufiger unter 300 mg auftraten, ist in schwerwiegenden Fällen eine Dosisreduktion auf 150 mg zu erwägen.
Patienten mit Leberfunktionsstörungen / Nierenfunktionsstörungen
Cosentyx/- SensoReady/- UnoReady wurde bei diesen Patientengruppen nicht spezifisch untersucht.
Ältere Patienten
Es liegen begrenzte Daten zur Anwendung von Cosentyx bei Psoriasis Arthritis Patienten über 75 Jahre und Patienten mit axialer Spondyloarthritis über 65 Jahre vor. Es liegen nur sehr begrenzte Daten zur Anwendung von Cosentyx bei Patienten mit Hidradenitis suppurativa über 65 Jahre vor. Es gibt jedoch keine Hinweise dafür, dass bei älteren Patienten eine andere Dosierung als bei jüngeren erwachsenen Patienten erforderlich ist.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit bei Kindern im Alter von unter 6 Jahren mit JIA der Kategorien EAA und JPsA ist nicht erwiesen.
Die Sicherheit und Wirksamkeit von Cosentyx wurde bei Kindern mit Plaque-Psoriasis im Alter von unter 6 Jahren nicht untersucht.
Die Sicherheit und Wirksamkeit bei Patienten unter 18 Jahren wurde in Hidradenitis suppurativa bisher nicht untersucht.
Kontraindikationen
Schwere Überempfindlichkeitsreaktionen gegenüber dem Wirkstoff oder einem der Hilfsstoffe (siehe "Zusammensetzung" , "Warnhinweise und Vorsichtsmassnahmen" sowie "Unerwünschte Wirkungen" ).
Schwere aktive Infektionen (z.B. aktive Tuberkulose, Sepsis, schwere opportunistische Infektionen)
Warnhinweise und Vorsichtsmassnahmen
Infektionen
Cosentyx/- SensoReady/- UnoReady kann das Infektionsrisiko erhöhen. In klinischen Studien wurden bei Patienten unter Cosentyx Infektionen beobachtet (siehe "Unerwünschte Wirkungen" ). Die meisten Infektionen waren von leichtem oder mittlerem Schweregrad. Erfahrungen bei Patienten mit bekannten aktiven HIV, HBV oder HCV Infektionen sind begrenzt. Daher soll vor der Anwendung von Cosentyx/- SensoReady/- UnoReady bei diesen Patienten der Nutzen der Therapie gegenüber den möglichen Risiken abgewogen werden.
In klinischen Studien wurden dosisabhängige Candida-Infektionen beobachtet, einschliesslich Oesophagus-Kandidiose. Es wurde jedoch keine systemische Ausweitung beobachtet, die Infekte waren mit Standard-Therapie beherrschbar und erforderten kein Absetzen der Behandlung. Daten aus klinischen Studien bis zu 1 Jahr zeigen kein erhöhtes Risiko für schwerwiegende opportunistische Infektionen. Die aktuelle Datenlage erlaubt keine Einschätzung des Langzeitrisikos für schwerwiegende opportunistische Infektionen.
Die Anwendung von Cosentyx/- SensoReady/- UnoReady bei Patienten mit einer chronischen Infektion oder rezidivierenden Infektionen in der Vorgeschichte sollte mit Vorsicht erwogen werden.
Die Patienten sollten angewiesen werden, bei Auftreten von Anzeichen oder Symptomen, die auf eine Infektion hindeuten, ärztlichen Rat einzuholen. Wenn ein Patient eine schwerwiegende Infektion entwickelt, ist er engmaschig zu überwachen; Cosentyx/- SensoReady/- UnoReady sollte bis zum Abklingen der Infektion nicht verabreicht werden.
Beurteilung Tuberkulose-Vorbehandlung
Tuberkulose (aktive und/oder latente Reaktivierung) kann bei mit Cosentyx/- SensoReady/- UnoReady behandelten Patienten auftreten. Patienten sollten vor Beginn der Behandlung mit Cosentyx/- SensoReady/- UnoReady auf eine Tuberkulose-Infektion untersucht werden. Patienten mit aktiver Tuberkulose sollte Cosentyx/- SensoReady/- UnoReady nicht verabreicht werden. Bei Patienten mit latenter Tuberkulose sollte vor Einleitung einer Therapie mit Cosentyx/- SensoReady/- UnoReady eine Anti-Tuberkulose-Behandlung erwogen werden. Bei Patienten mit latenter oder aktiver Tuberkulose in der Vorgeschichte, bei denen eine adäquate Behandlung nicht bestätigt werden kann, sollte vor Beginn der Behandlung mit Cosentyx/- SensoReady/- UnoReady eine Anti-Tuberkulose-Therapie erwogen werden. Die Patienten sollten während und nach der Behandlung engmaschig auf Anzeichen und Symptome einer aktiven Tuberkulose überwacht werden.
Reaktivierung von Hepatitis B
Eine Reaktivierung des Hepatitis B Virus kann bei mit Cosentyx/- SensoReady/- UnoReady behandelten Patienten auftreten. In Übereinstimmung mit klinischen Richtlinien für Immunsuppressiva sollten Patienten vor Beginn der Behandlung mit Cosentyx/- SensoReady/- UnoReady auf eine HBV-Infektion getestet werden. Bei Patienten mit aktiver Hepatitis B sollte Cosentyx/- SensoReady/- UnoReady nicht verabreicht werden. Patienten mit nachweislich positiver HBV-Serologie sollten während der Behandlung mit Cosentyx/- SensoReady/- UnoReady auf klinische Symptome und Laborbefunde einer HBV-Reaktivierung überwacht werden. Wenn während der Behandlung mit Cosentyx/- SensoReady/- UnoReady eine Reaktivierung von HBV auftritt, sollte ein Abbruch der Behandlung in Betracht gezogen und die Patienten gemäss klinischer Leitlinien behandelt werden.
Chronisch-entzündliche Darmerkrankungen
In klinischen Studien wurden Einzelfälle von chronisch-entzündlichen Darmerkrankungen beobachtet, die in einigen Fällen schwerwiegend verliefen. In den meisten Fällen handelte es sich um Exazerbationen eines vorbestehenden M. Crohn. Die Therapie ist in solchen Fällen sorgfältig zu reevaluieren und ein Therapieabbruch zu erwägen (s. "Dosierung/Anwendung" ). Secukinumab zeigte keine Wirksamkeit in einer Phase-2-Studie bei Patienten mit aktivem M. Crohn. Da Patienten mit Psoriasis inhärent ein höheres Risiko für chronisch-entzündliche Darmerkrankungen tragen, ist bei der gegenwärtigen Datenlage ein kausaler Bezug zu Cosentyx/- SensoReady/- UnoReady nicht schlüssig zu beurteilen.
Neu auftretende entzündliche Darmerkrankung
Fälle von neu auftretenden entzündlichen Darmerkrankungen wurden nach der Marktzulassung berichtet.
Maligne Erkrankungen
In klinischen Studien bis zu 1 Jahr zeigt die Behandlung mit Cosentyx/- SensoReady/- UnoReady kein erhöhtes Risiko für maligne Erkrankungen. Resultate der Untersuchungen zur Langzeitsicherheit sind noch nicht vorhanden.
Psoriasis-Patienten, die zuvor eine UV-Therapie erhalten haben, sollten vor und während der Behandlung mit Cosentyx/- SensoReady/- UnoReady auf das Vorliegen von Hauttumoren untersucht werden.
Überempfindlichkeitsreaktionen
In klinischen Studien wurden in seltenen Fällen anaphylaktische Reaktionen und Angioödeme bei Patienten beobachtet, die Cosentyx erhielten. Fälle von Angioödemen wurden auch nach der Markteinführung berichtet. Bei Auftreten einer anaphylaktischen oder einer anderen schwerwiegenden allergischen Reaktion sollte die Gabe von Cosentyx/- SensoReady/- UnoReady unverzüglich abgebrochen werden, und es sind geeignete Therapiemassnahmen einzuleiten.
Ekzematöser Ausschlag
In Berichten nach der Markteinführung wurde über das Auftreten schwerwiegender ekzematöser Ausschläge, einschließlich dermatitisähnlicher Ausschläge, dyshidrotischer Ekzeme und Erythrodermie (exfoliative Dermatitis) bei Patienten berichtet, die Cosentyx erhielten. Einige dieser Fälle erforderten eine Krankenhauseinweisung (siehe: "Unerwünschte Wirkungen" ). Der Beginn der ekzematösen Ausschläge variierte zwischen wenigen Tagen und mehreren Monaten nach der ersten Anwendung von Cosentyx. Es kann notwendig sein, die Behandlung mit Cosentyx zu unterbrechen, um den exzematösen Ausschlag zu behandeln. Bei einigen Patienten konnten die ekzematösen Ausschläge erfolgreich behandelt werden, während die Behandlung mit Cosentyx fortgeführt wurde.
Aufflammen der Psoriasis bei Absetzen der Therapie ( "Rebound" )
Bei Absetzen der Therapie bei Patienten, die primär angesprochen haben, ist das Risiko des Aufflammens der Psoriasis zu berücksichtigen. In einer Extensionsstudie mit Patienten, die primär angesprochen haben, wurde ein Aufflammen der Psoriasis sowohl nach Absetzen von 150 mg als auch 300 mg beobachtet: Bis 8 Wochen nach Absetzen der Therapie wurde bei 4% der mit 300 mg behandelten Patienten und 4.7% der mit 150 mg behandelten Patienten ein Aufflammen beobachtet. Aufflammen in Form einer Pustulären Psoriasis betraf 1.1% bzw. 0.7% der vorher mit 300 mg bzw. 150 mg behandelten Patienten.
Impfungen
Es liegen limitierte Daten zum Ansprechen auf Impfungen mit Lebendimpfstoffen oder inaktivierten Impfstoffen bei Patienten unter Behandlung mit Cosentyx/- SensoReady/UnoReady vor. Anhand der aktuellen Datenlage kann nicht beurteilt werden, inwieweit Cosentyx/- SensoReady/UnoReady die Immunantwort auf Neo- und/oder Booster-Antigene hemmt. Die Anwendung von attenuierten Lebendimpfstoffen wird während oder unmittelbar vor einer Cosentyx/- SensoReady/UnoReady Behandlung nicht empfohlen. Vor Einleitung der Therapie mit Cosentyx/- SensoReady/UnoReady wird empfohlen, den Impfstatus der Patienten entsprechend den aktuellen Impfleitlinien zu aktualisieren; dazu zählen je nach Situation auch Impfungen gegen Varizellen/Zosterinfektionen.
Kombination mit anderen Biologika
Die gleichzeitige Verabreichung von Cosentyx/- SensoReady/- UnoReady mit anderen Biologika wurde nicht untersucht und wird nicht empfohlen.
Latex-empfindliche Personen – betrifft nur Fertigspritze/Fertigpen
Die Nadelkappe der Fertigspritze kann Trockenkautschuk (Latex) enthalten. Die sichere Anwendung der Fertigspritze/Fertigpen bei Patienten mit einer Latex-Überempfindlichkeit ist nicht dokumentiert.
Interaktionen
Interaktionen mit Impfungen siehe "Warnhinweise und Vorsichtsmassnahmen" .
In einer Studie bei erwachsenen Patienten mit Plaque-Psoriasis wurden keine Interaktionen zwischen Secukinumab und Midazolam (CYP 3A4 Substrat) beobachtet. In Studien zu Psoriasis-Arthritis und axialer Spondyloarthritis wurde Cosentyx/-SensoReady/- UnoReady gleichzeitig mit Methotrexat und/oder Kortikosteroiden verabreicht; dabei wurden keine Interaktionen beobachtet.
Die Expression hepatischer CYP450-Enzyme wird durch Zytokine, die chronische Entzündungen stimulieren, unterdrückt. Daher könnte sich die Expression von CYP450 verändern, wenn eine Zytokinhemmung mit Secukinumab eingeleitet wird.
Wirkung von Cosentyx/- SensoReady/- UnoReady auf andere Arzneimittel
Patienten, die Arzneimittel einnehmen, deren Dosis individuell eingestellt wird und die durch CYP450 3A4, 1A2 oder 2C9 metabolisiert werden (z.B. Atorvastatin, Kalziumkanalblocker, Theophyllin, Acenocoumarol, Phenprocoumon, Phenytoin, Cyclosporin oder Benzodiazepine), sollten zu Beginn und Ende einer Therapie mit Secukinumab kontrolliert werden und die Dosis dieser Substanzen bei Bedarf angepasst werden. In Anbetracht seiner langen Eliminationshalbwertszeit kann die Wirkung von Secukinumab auf die Aktivität von CYP450-Enzymen nach Beendigung der Behandlung über mehrere Wochen bestehen bleiben.
Schwangerschaft, Stillzeit
Schwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung von Cosentyx/- SensoReady/- UnoReady während einer Schwangerschaft. Es ist bekannt, dass humanes IgG1 die Plazentaschranke überwindet, und Secukinumab ist ein IgG1 monoklonaler Antikörper. Daher besteht die Möglichkeit, dass Secukinumab von der Mutter auf den Fötus übergeht. Gebärfähige Frauen sollen während der Behandlung mit Cosentyx/- SensoReady/- UnoReady eine wirksame Empfängnisverhütung anwenden. Patientinnen sollen angewiesen werden für mindestens 20 Wochen nach der letzten Behandlung mit Cosentyx/- SensoReady/- UnoReady wirksame Verhütungsmethoden anzuwenden. Tierexperimentelle Studien zeigen keine direkten oder indirekten schädlichen Auswirkungen auf Schwangerschaft, Embryonalentwicklung, Entwicklung des Fötus, Geburt oder die postnatale Entwicklung (siehe "Präklinische Daten" ). Da sich anhand tierexperimenteller Reproduktionsstudien nicht immer die Reaktion beim Menschen vorhersagen lässt, sollte Cosentyx/- SensoReady/- UnoReady nur dann während einer Schwangerschaft angewendet werden, wenn der Nutzen gegenüber den möglichen Risiken eindeutig überwiegt.
Stillzeit
Es ist nicht bekannt, ob Secukinumab in die menschliche Muttermilch übergeht. Da viele Medikamente, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Wegen möglicher Schäden für den gestillten Säugling wird empfohlen während der Behandlung mit Cosentyx/- SensoReady/- UnoReady und während mindestens 20 Wochen nach der letzten Dosis nicht zu stillen. Daher soll die Entscheidung, entweder mit dem Stillen aufzuhören oder Cosentyx/-SensoReady/- UnoReady bei der Mutter abzusetzen unter Berücksichtigung der Vorteile des Stillens für das Kind und der Vorteile der Therapie für die Mutter, gefällt werden.
Fertilität
Die Wirkung von Cosentyx auf die Fertilität des Menschen ist nicht untersucht worden. Tierexperimentelle Studien zeigen keine direkten oder indirekten schädlichen Wirkungen in Bezug auf die Fertilität (siehe "Präklinische Daten" ).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Der Effekt von Cosentyx/- SensoReady/- UnoReady auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen wurde nicht untersucht.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
In verblindeten und offenen klinischen Studien wurden insgesamt über 20'000 Patienten mit verschiedenen Indikationen (Plaque-Psoriasis, Psoriasis-Arthritis, axiale Spondyloarthritis, Hidradenitis suppurativa und anderen Autoimmunerkrankungen) mit Cosentyx behandelt, was einer Exposition von 34'908 Patientenjahren entspricht. Von diesen Patienten waren 14'000 mindestens ein Jahr lang Cosentyx exponiert.
Zur Beurteilung der Sicherheit von Cosentyx in erwachsenen Patienten mit Plaque Psoriasis im Vergleich zu Placebo während bis zu 12 Wochen nach Einleitung der Behandlung wurden vier placebokontrollierte Phase-III-Studien gepoolt. Insgesamt 2'076 Patienten wurden beurteilt (692 Patienten unter 150 mg, 690 Patienten unter 300 mg und 694 Patienten unter Placebo).
Der Beurteilung der Sicherheit von Cosentyx in Patienten mit Psoriasis-Arthritis liegen fünf placebokontrollierte Studien mit 2'754 Patienten zugrunde (1'871 Patienten, die mit Cosentyx und 883 Patienten, die mit Placebo behandelt wurden), wobei die Placebo-kontrollierte Phase bis zu 24 Wochen dauerte, dies bei einer gesamten Studienexposition von 4'478 Patientenjahren. Das Sicherheitsprofil bei Patienten mit Psoriasis-Arthritis, die mit Cosentyx behandelt werden, entspricht dem bei Psoriasis.
Der Beurteilung der Sicherheit von Cosentyx in Patienten mit ankylosierender Spondylitis liegen zwei placebokontrollierte Studien mit 590 Patienten zugrunde (394 Patienten, die mit Cosentyx und 196 Patienten, die mit Placebo behandelt wurden), wobei die Placebo-kontrollierte Phase bis zu 24 Wochen, dies bei einer gesamten Studienexposition von 755 Patientenjahren (mittlere Expositionsdauer für Patienten, die mit Secukinumab behandelt wurden: 469 Tage in der AS1-Studie und 460 Tage in der AS2-Studie).
Die Sicherheitsbewertung von Cosentyx bei Patienten mit nicht-röntgenologischer axialer Spondyloarthritis (nr-axSpA) basiert auf einer placebokontrollierten Studie mit 555 Patienten (369 mit Cosentyx behandelte Patienten und 186 mit Placebo behandelte Patienten). Der placebokontrollierte Zeitraum betrug bis zu 52 Wochen betrug und die Gesamtstudienexposition von 588 Patientenjahren (mittlere Expositionsdauer bei mit Secukinumab behandelten Patienten: 395 Tage).
Das Sicherheitsprofil bei Patienten mit axialer Spondyloarthritis (ankylosierender Spondylitis und nicht-röntgenologischer axialer Spondyloarthritis), die mit Cosentyx behandelt werden, entspricht dem bei Psoriasis.
Unerwünschte Wirkungen bei Hidradenitis suppurativa
Cosentyx wurde in zwei placebokontrollierten Studien an 1'084 Patienten mit Hidradenitis suppurativa (721 Patienten unter Cosentyx und 363 unter Placebo) mit einer Studienexposition von insgesamt 825 Patientenjahren untersucht (mediane Dauer der Exposition bei den mit Secukinumab behandelten Patienten: 307 Tage). Der placebokontrollierte Zeitraum betrug bis zu 16 Wochen. Das Sicherheitsprofil, das bei den Patienten mit Hidradenitis suppurativa unter der Behandlung mit Cosentyx über die gesamte Behandlungsphase von bis zu 52 Wochen festgestellt wurde, entspricht dem bekannten Sicherheitsprofil bei Psoriasis.
Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen waren Infektionen der oberen Atemwege (am häufigsten Nasopharyngitis sowie Rhinitis). Die meisten dieser Ereignisse waren von leichtem oder mittlerem Schweregrad. In den placebokontrollierten Phasen der zulassungsrelevanten Phase-III-Studien betrug der Anteil der Patienten, die die Behandlung aufgrund unerwünschter Ereignisse abbrachen, rund 1.5 % im Cosentyx -Arm und 2.4 % im Placebo-Arm.
Unerwünschte Arzneimittelwirkungen aus klinischen Studien sind in der untenstehenden Liste nach MedDRA-Systemorganklassen geordnet aufgeführt. Innerhalb jeder Systemorganklasse sind die unerwünschten Arzneimittelwirkungen nach absteigender Häufigkeit angeordnet. In jeder Häufigkeitsgruppe sind die unerwünschten Arzneimittelwirkungen nach abnehmendem Schweregrad angegeben. Die Häufigkeitskategorien sind wie folgt definiert: "Sehr häufig" (≥1/10), "häufig" (<1/10, ≥1/100), "gelegentlich" (<1/100, ≥1/1'000, "selten" (<1/1'000, ≥1/10'000), "sehr selten" (<1/10'000).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektionen der oberen Atemwege (17.1%).
Häufig: Oraler Herpes.
Gelegentlich: Orale Candidose, Candidose des Oesophagus, Infektionen der unteren Atemwege, Tinea pedis.
Erkrankungen des Blutes- und Lymphsystems
Gelegentlich: Neutropenie.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Augenerkrankungen
Gelegentlich: Bindehautentzündung.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Rhinorrhö.
Erkrankungen des Gastrointestinaltrakts
Häufig: Diarrhö, Übelkeit.
Gelegentlich: Entzündliche Darmerkrankungen (einschliesslich Morbus Crohn und Colitis ulcerosa).
Leber und Gallenerkrankungen
Gelegentlich: Leberenzyme erhöht, Bilirubin erhöht.
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Urtikaria, Dermatitis (einschliesslich Ekzem)
Selten: Exfoliative Dermatitis (Fälle wurden bei Patienten mit diagnostizierter Psoriasis berichtet), Hypersensitivitätsvaskulitis, dyshidrotisches Ekzem.
Erkrankungen des Immunsystems
Selten: Anaphylaktische Reaktionen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Ermüdung.
Unerwünschte Wirkungen nach Markteinführung
Die folgenden unerwünschten Arzneimittelwirkungen traten nach der Markteinführung von Cosentyx auf und sind in spontanen Fallberichten und in der Fachliteratur beschrieben. Weil es sich dabei um freiwillige Angaben aus einer Population unbekannter Grösse handelt, ist es nicht möglich, eine verlässliche Schätzung ihrer Häufigkeit abzugeben, deshalb werden sie der Kategorie "nicht bekannt" zugeordnet. Unerwünschte Arzneimittelwirkungen sind nach MedDRA-Systemorganklassen geordnet aufgeführt. Innerhalb jeder Systemorganklasse sind die unerwünschten Arzneimittelwirkungen nach absteigender Schwere geordnet.
Infektionen und parasitäre Erkrankungen
Mukosale und kutane Candidose.
Erkrankungen der Haut und des Unterhautgewebes
Pyoderma gangraenosum, generalisierte exfoliative Dermatitis, Angioödem
Beschreibung ausgewählter Nebenwirkungen
Infektionen
In den placebokontrollierten Phasen der klinischen Studien zu Plaque Psoriasis (in welchen insgesamt 1'382 Patienten mit Cosentyx und 694 Patienten mit Placebo während bis zu 12 Wochen behandelt wurden) wurden bei 28.7% der mit Cosentyx behandelten Patienten im Vergleich zu 18.9% der mit Placebo behandelten Patienten Infektionen berichtet. Die meisten dieser Infektionen waren von leichtem oder mittlerem Schweregrad. Candida-Infektionen traten bei 1.2% der mit 300 mg Cosentyx, bei 0.4% der mit 150 mg Cosentyx, und bei 0.3% der mit Placebo behandelten Patienten auf. Schwerwiegende Infektionen traten bei 0.14% der mit Cosentyx und bei 0.3% der mit Placebo behandelten Patienten auf (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Über die gesamte Behandlungsphase (insgesamt 3'430 Patienten, die mehrheitlich während bis zu 52 Wochen mit Cosentyx behandelt wurden) wurden Infektionen bei 47.5% der mit Cosentyx behandelten Patienten (0.9 pro Patientenjahr der Nachbeobachtung) berichtet. Schwerwiegende Infektionen wurden bei 1.2% der mit Cosentyx behandelten Patienten berichtet (0.015 pro Patientenjahr der Nachbeobachtung).
Die Infektionsraten, die in klinischen Studien zu Psoriasis-Arthritis, axialer Spondyloarthritis (ankylosierender Spondylitis und nicht-röntgenologischer axialer Spondyloarthritis) beobachtet wurden, waren ähnlich wie jene, die bei Studien zu Psoriasis beobachtet wurden.
Aufgrund des Charakters der Hautläsionen sind Patienten mit Hidradenitis suppurativa anfälliger für Infektionen. In der placebokontrollierten Phase der klinischen Studien an Patienten mit Hidradenitis suppurativa (insgesamt wurden 723 Patienten mit Secukinumab und 363 Patienten mit Placebo behandelt, jeweils bis zu 16 Wochen lang) war die Anzahl Infektionen höher als in den Psoriasis-Studien (30,8 % der mit Secukinumab behandelten Patienten im Vergleich zu 31,7 % der Patienten, die mit Placebo behandelt wurden). Die meisten dieser Infektionen waren nicht schwerwiegend, von leichter oder mittelschwerer Ausprägung und erforderten weder einen Abbruch noch eine Unterbrechung der Behandlung. Über die gesamte Behandlungsphase (insgesamt 1062 Patienten, die während bis zu 52 Wochen mit Cosentyx behandelt wurden) wurden Infektionen bei 45.7% der mit Cosentyx behandelten Patienten (88.6 pro 100 Patientenjahre) berichtet. Schwerwiegende Infektionen wurden bei 3.2% der mit Cosentyx behandelten Patienten berichtet (4.2 pro 100 Patientenjahre).
Leberenzyme erhöht, Bilirubin erhöht
Während der Placebo-kontrollierten Phase in klinischen Studien zur Plaque-Psoriasis entwickelten 14.5% bzw. 5% der Patienten unter Cosentyx gegenüber 11.6% bzw. 2.5% unter Placebo isolierte und transiente Erhöhungen der Lebertransaminasen bzw. von Bilirubin bis zum 3-fachen des oberen Normwertes. Die Häufigkeiten unter Secukinumab und Etanercept waren vergleichbar.
Neutropenie
Neutropenie wurde häufiger mit Secukinumab als mit Placebo beobachtet. Die meisten Fälle waren mild, transient und reversibel. Neutropenie <1.0-0.5x109/l (CTCAE-Grad 3) wurde bei 18 von 3430 (0.5%) Patienten unter Secukinumab berichtet, mit keiner Dosisabhängigkeit und keinem zeitlichen Zusammenhang mit Infektionen in 15 von 18 Fällen. Es gab keine berichteten Fälle von schwerwiegenderer Neutropenie. In den übrigen 3 Fällen wurden nicht schwerwiegende Infektionen, die erfolgreich auf Standard-Therapie ansprachen und kein Absetzen von Cosentyx erforderten, berichtet.
Die Häufigkeit von Neutropenie bei Psoriasis-Arthritis, axialer Spondyloarthritis (ankylosierender Spondylitis und nicht-röntgenologischer axialer Spondyloarthritis) und Hidradenitis suppurativa war ähnlich wie bei Psoriasis.
Überempfindlichkeitsreaktionen
Über alle klinischen Studien wurden Urtikaria, seltene Fälle anaphylaktischer Reaktionen und Angioödemen in Patienten welche Cosentyx erhielten beobachtet. Fälle von Angioödemen wurden auch nach der Markteinführung berichtet.
Chronisch-entzündliche Darmerkrankungen
Über alle klinischen Studien wurden unter Secukinumab einzelne Fälle von chronisch-entzündlichen Darmerkrankungen in Risikopatienten beobachtet (siehe "Warnhinweise und Vorsichtsmassnahmen" ). Ein kausaler Zusammenhang mit Secukinumab kann bei der gegenwärtigen Datenlage nicht ausgeschlossen werden.
Immunogenität
In klinischen Studien zu Psoriasis, Psoriasis-Arthritis und axialer Spondyloarthritis (ankylosierender Spondylitis und nicht-röntgenologischer axialer Spondyloarthritis) sowie Hidradenitis suppurativa entwickelten während einer Behandlung von bis zu 52 Wochen weniger als 1% der mit Cosentyx behandelten Patienten Antikörper gegen Secukinumab. Bei etwa der Hälfte der während der Behandlung aufgetretenen gegen das Arzneimittel gerichteten Antikörper handelte es sich um neutralisierende Antikörper; dies war jedoch nicht mit einem Wirkungsverlust oder mit Abweichungen der pharmakokinetischen Parameter verbunden.
Pädiatrische Population
Plaque-Psoriasis
Die Sicherheit von Cosentyx wurde in zwei Phase-III-Studien bei pädiatrischen Patienten mit Plaque-Psoriasis untersucht. Bei der ersten Studie handelte es sich um eine doppelblinde, placebokontrollierte Studie mit 162 Patienten im Alter von 6 bis <18 Jahren mit schwerer Plaque-Psoriasis. Bei der zweiten Studie handelt es sich um eine offene Studie mit 84 Patienten im Alter von 6 bis <18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis. Das in diesen beiden Studien in insgesamt 287.4 Patientenjahren berichtete Sicherheitsprofil entsprach dem bei erwachsenen Plaque-Psoriasis-Patienten berichteten Sicherheitsprofil.
Juvenile idiopathische Arthritis (JIA)
Enthesitis-assoziierte Arthritis (EAA) und juvenile Psoriasis-Arthritis (JPsA)
Die Sicherheit von Cosentyx wurde auch in einer Phase-III-Studie an 86 pädiatrischen Patienten im Alter von 2 bis unter 18 Jahren mit JIA der Kategorien EAA und JPsA untersucht. Das Sicherheitsprofil dieser Studie stimmte mit dem Sicherheitsprofil bei erwachsenen Patienten überein.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
In den klinischen Studien wurden keine Fälle von Überdosierung berichtet.
In klinischen Studien wurden Dosen bis zu 30 mg/kg (d.h. ungefähr 2'000 bis 3'000 mg) intravenös ohne Auftreten einer dosislimitierenden Toxizität verabreicht.
Behandlung
Im Fall einer Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome unerwünschter Wirkungen zu überwachen und unverzüglich eine angemessene symptomatische Behandlung einzuleiten.
Eigenschaften/Wirkungen
ATC-Code
L04AC10
Wirkungsmechanismus
Secukinumab ist ein vollständig humaner IgG1-Antikörper der selektiv an das proinflammatorische Zytokin Interleukin-17A (IL-17A) bindet und es neutralisiert. Secukinumab wirkt gezielt auf IL-17A und hemmt dessen Interaktion mit dem IL-17-Rezeptor, der auf verschiedenen Zelltypen einschliesslich Keratinozyten exprimiert wird. Als Folge hemmt Secukinumab die Freisetzung von proinflammatorischen Zytokinen, Chemokinen und gewebeschädigenden Mediatoren und reduziert die IL-17A-vermittelte Beteiligung an autoimmunen und entzündlichen Erkrankungen. Klinisch relevante Secukinumab-Spiegel erreichen die Haut und reduzieren lokale Entzündungsmarker. Als eine direkte Folge reduziert die Behandlung mit Secukinumab Erythem, Verhärtung (Induration) und Schuppung (Desquamation) in den Läsionen der Plaque-Psoriasis. Bei Patienten mit Plaque-Psoriasis ist die IL-17A Produktion in den von Läsionen betroffenen Hautbereichen gegenüber Hautbereichen ohne Läsionen stark hochreguliert. Weiterhin wurden IL-17-bildende Zellen in der Gelenkflüssigkeit von Patienten mit Psoriasis-Arthritis häufiger festgestellt. Zudem waren die IL-17-bildenden Zellen bei Patienten mit axialer Spondyloarthritis auch im subchondralen Knochenmark der Facettengelenke deutlich erhöht. IL-17A ist in Läsionen bei Hidradenitis suppurativa im Vergleich zu Psoriasis Patienten und gesunden Kontrollpersonen deutlich hochreguliert und es wurden signifikant erhöhte IL-17A-Serumspiegel bei betroffenen Patienten festgestellt. Eine erhöhte Anzahl IL-17A produzierender Lymphozyten wurde auch bei Patienten mit nicht-röntgenologischer axialer Spondyloarthritis festgestellt.
IL-17A fördert zudem die Entzündungsreaktion im Gewebe, die Infiltration mit Neutrophilen, die Knochen- und Gewebszerstörung sowie das Gewebe-Remodeling einschliesslich Angiogenese und Fibrose.
Pharmakodynamik
In einer Studie mit Secukinumab waren die Spiegel der infiltrierenden Neutrophilen und verschiedene mit Neutrophilen assoziierte Marker, die in von Läsionen betroffenen Hautbereichen von Patienten mit Plaque-Psoriasis erhöht sind, nach ein- bis zweiwöchiger Behandlung signifikant reduziert.
Es wurde gezeigt, dass Secukinumab (innerhalb einer Behandlungszeit von ein bis zwei Wochen) den Spiegel des C-reaktiven Proteins senkt; das C-reaktive Protein ist ein Entzündungsmarker bei Psoriasis-Arthritis und axialer Spondylitis.
Klinische Wirksamkeit
Psoriasis
Erwachsene Patienten
Die Sicherheit und Wirksamkeit von Cosentyx wurde in vier randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien über 1 Jahr bei Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die auf eine Phototherapie oder systemische Therapie nicht angesprochen haben oder eine solche nicht tolerierten, beurteilt [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Wirksamkeit und Sicherheit von 150 mg und 300 mg Cosentyx wurden im Vergleich zu Placebo oder Etanercept beurteilt. Zudem wurde in einer Studie ein kontinuierliches Behandlungsregime im Vergleich zu einem Regime mit Therapieunterbruch in Woche 12 und einer Wiederaufnahme der Therapie "ondemand" bei klinischer Verschlechterung beurteilt [SCULPTURE]. In diesen Studien wurde jede 300 mg Dosis in Form von zwei subkutanen Injektionen zu 150 mg verabreicht.
Um eine unverzerrte Beurteilung der Wirksamkeit von Secukinumab in der Psoriasis Behandlung zu erhalten, war die gleichzeitige Anwendung einer systemischen oder topischen Psoriasismedikation oder Phototherapie während der Studien nicht erlaubt.
Von den 2'403 in die placebokontrollierten Studien eingeschlossenen Patienten waren 79% nicht biologisch vorbehandelt; 45% wiesen ein Therapieversagen unter einer nicht-biologischen Behandlung, 8% ein Therapieversagen unter einer biologischen Behandlung, 6% ein Therapieversagen unter einer anti-TNF-Behandlung und 2% ein Therapieversagen unter einer anti-p40 (anti-IL-12/IL-23)-Behandlung auf. Die Krankheitsmerkmale bei Baseline waren über alle Behandlungsgruppen hinweg im Allgemeinen vergleichbar: Der Medianwert des Psoriasis-Area-Severity-Index-Score (PASI) bei Baseline betrug 19 bis 20, der IGA-mod-2011-Score zum Baseline-Zeitpunkt lag zwischen "mittelschwer" (62%) und "schwer" (38%), der Medianwert der Body-Surface-Area (BSA) war bei Baseline ≥27 und der Medianwert des Dermatology-Life-Quality-Index-Score (DLQI) betrug 10 bis 12. Ungefähr 15 bis 25% der Patienten in den Phase-III-Studien wiesen bei Studienbeginn eine psoriatische Arthritis (PsA) auf.
In der Psoriasis-Studie 1 (ERASURE) wurden 738 Patienten beurteilt. Die Patienten, die einer Behandlung mit Cosentyx randomisiert zugeordnet wurden, erhielten Dosen zu 150 mg oder 300 mg in den Wochen 0, 1, 2, 3 und 4, gefolgt von derselben Dosis jeden Monat. Die Patienten, die einer Behandlung mit Placebo randomisiert zugeordnet wurden und in Woche 12 Non-Responder waren, wechselten anschliessend zu einer Behandlung mit Cosentyx (150 oder 300 mg) in den Wochen 12, 13, 14 und 15, gefolgt von derselben Dosis jeden Monat beginnend in Woche 16.
In der Psoriasis-Studie 2 (FIXTURE) wurden 1'306 Patienten beurteilt und neben der Placebo-Gruppe wurde ein Arm mit Etanercept als aktiver Komparator geführt. Die Behandlung mit Cosentyx und Placebo entsprach der Studie 1. Die Patienten, die einer Behandlung mit Etanercept randomisiert zugeordnet wurden, erhielten Dosen zu 50 mg zweimal wöchentlich während 12 Wochen, gefolgt von 50 mg jede Woche.
In der Psoriasis-Studie 3 (FEATURE) und Studie 4 (JUNCTURE) wurden 177 mit einer Fertigspritze bzw. 182 mittels Fertigpen behandelte Patienten nach 12-wöchiger Behandlung zur Beurteilung der Sicherheit, Verträglichkeit und Praktikabilität der Selbstverabreichung von Cosentyx mittels Fertigspritze mit einem Placebo verglichen. Die Behandlung mit Cosentyx und Placebo entsprach der Studie 1. In der Psoriasis-Studie 5 (SCULPTURE) wurden 966 Patienten beurteilt. Alle Patienten erhielten 150 mg oder 300 mg Cosentyx in den Wochen 0, 1, 2, 3, 4, 8 und 12 und wurden dann entweder einer Behandlung mit einem Erhaltungsregime mit kontinuierlicher Verabreichung derselben Dosis jeden Monat oder nach Therapieunterbruch einem Regime mit Wiederaufnahme der Therapie "on demand" bei klinischer Verschlechterung randomisiert zugeordnet. Die Patienten mit Therapieunterbruch und Wiederaufnahme "on demand" waren hinsichtlich der Aufrechterhaltung des Ansprechens den Patienten unter einem fixen monatlichen Erhaltungsregime unterlegen.
Die primären Endpunkte in den placebo- und aktiv kontrollierten Studien waren der Anteil der Patienten, die ein PASI-75-Ansprechen und ein "Investigator's Global Assessment (IGA)" -mod-2011-Ansprechen in den Kategorien "frei von" oder "nahezu frei von" im Vergleich zu Placebo in Woche 12 erreichten (siehe Tabelle 2 und Tabelle 3). Die maximale Ansprechrate wurde in Woche 16 erreicht, und die 300-mg-Dosis war über alle Studien hinweg überlegen.
Tabelle 2: Zusammenfassung des klinischen PASI-50/75/90/100-Ansprechens & IGA*-mod-2011-Ansprechens in den Kategorien "abgeheilt" oder "nahezu abgeheilt" in den Psoriasis-Studien 1, 3 und 4 (ERASURE, FEATURE und JUNCTURE)
Woche 12 Woche 16 Woche 52
Placebo n (%) (95% 150 mg n (%) (95% 300 mg n (%) (95% 150 mg n (%) (95% 300 mg n (%) (95% 150 mg n (%) (95% 300 mg n (%) (95%
CI) CI) CI) CI) CI) CI) CI)
Studie 1
Anzahl der Patienten 246 244 245 244 245 244 245
PASI-50-Ansprechen 22 (8.9%) (5.8, 203 (83.5%) (78.1, 222 (90.6%) (86.1, 212 (87.2%) (82.2, 224 (91.4%) (87.0, 187 (77%) (71.0, 207 (84.5%) (79.2,
13.4) 87.9) 93.8) 91.0) 94.5) 82.0) 88.7)
PASI-75-Ansprechen 11 (4.5%) (2.4, 8.1) 174 (71.6%)** 200 (81.6%)** 188 (77.4%) (71.5, 211 (86.1%) (81.0, 146 (60.1%) (53.6, 182 (74.3%) (68.3,
(65.4, 77.1) (76.1, 86.2) 82.4) 90.1) 66.2) 79.5)
PASI-90-Ansprechen 3 (1.2%) (0.3, 3.8) 95 (39.1%)** (33.0, 145 (59.2%)** 130 (53.5%) (47.0, 171 (69.8%) (63.6, 88 (36.2%) (30.2, 147 (60.0%) (53.6,
45.6) (52.7, 65.3) 59.9) 75.4) 42.6) 66.1)
PASI-100-Ansprechen 2 (0.8%) (0.1, 3.2) 31 (12.8%) (9.0, 70 (28.6%) (23.1, 51 (21.0%) (16.2, 102 (41.6%) (35.4, 49 (20.2%) (15.4, 96 (39.2%) (33.1,
17.8) 34.7) 26.8) 48.1) 25.9) 45.6)
IGA-mod-2011-Ansprec 6 (2.40%) (1.0, 5.5) 125 (51.2%)** 160 (65.3%)** 142 (58.2%) (51.7, 180 (73.5%) (67.4, 101 (41.4%) (35.2, 148 (60.4%) (54.0,
hen in den Kategorie (44.8, 57.6) (58.9, 71.2) 64.4) 78.8) 47.9) 66.5)
n "frei von" oder
"nahezu frei von"
Studie 3
Anzahl der Patienten 59 59 58 - - - -
PASI-50-Ansprechen 3 (5.1%) (1.3, 15.1) 51 (86.4%) (74.5, 51 (87.9%) (76.1, - - - -
93.6) 94.6)
PASI-75-Ansprechen 0 (0.0%) (0.0, 7.6) 41 (69.5%)** (56.0, 44 (75.9%)** (62.5, - - - -
80.5) 85.7)
PASI-90-Ansprechen 0 (0.0%) (0.0, 7.6) 27 (45.8%) (32.9, 35 (60.3%) (46.6, - - - -
59.2) 72.7)
PASI-100-Ansprechen 0 (0.0%) (0.0, 7.6) 5 (8.5%) (3.2, 19.4) 25 (43.1%) (30.4, - - - -
56.7)
IGA-mod-2011-Ansprec 0 (0.0%) (0.0, 7.6) 31 (52.5%)** (39.2, 40 (69.0%)** (55.3, - - - -
hen in den Kategorie 65.5) 80.1)
n "frei von" oder
"nahezu frei von"
Studie 4
Anzahl der Patienten 61 60 60 - - - -
PASI-50-Ansprechen 5 (8.2%) (3.1, 18.8) 48 (80.0%) (67.3, 58 (96.7%) (87.5, - - - -
88.8) 99.4)
PASI-75-Ansprechen 2 (3.3%) (0.6, 12.4) 43 (71.7%)** (58.4, 52 (86.7%)** (74.9, - - - -
82.2) 93.7)
PASI-90-Ansprechen 0 (0.0%) (0.0, 7.4) 24 (40.0%) (27.8, 33 (55.0%) (41.7, - - - -
53.5) 67.7)
PASI-100-Ansprechen 0 (0.0%) (0.0, 7.4) 10 (16.7%) (8.7, 16 (26.7%) (16.5, - - - -
29.0) 39.9)
IGA-mod-2011-Ansprec 0 (0.0%) (0.0, 7.4) 32 (53.3%)** (40.1, 44 (73.3%)** (60.1, - - - -
hen in den Kategorie 66.1) 83.5)
n "frei von" oder
"nahezu frei von"
* IGA mod 2011 ist
eine Skala mit 5
Kategorien mit "0 =
frei von" , "1 =
nahezu frei von" ,
"2 = leicht" , "3 =
mittelschwer" und
"4 = schwer" und
gibt die Gesamtbeurt
eilung des Psoriasis
-Schweregrads durch
den Arzt bzw. die
Ärztin im Hinblick
auf Verhärtung,
Erythem und Schuppun
g wieder. Der
Behandlungserfolg
"frei von" oder
"nahezu frei von"
bestand aus der
Abwesenheit von
Anzeichen einer
Psoriasis oder
normaler bis rosafar
bener Färbung der
Läsionen, fehlender
Verdickung der
Plaques und fehlende
r bis minimaler
herdförmiger Schuppu
ng. ** p-Werte
versus Placebo und
adjustiert hinsichtl
ich Multiplizität:
p<0,0001
Tabelle 3: Zusammenfassung des klinischen Ansprechens in Psoriasis-Studie 2 (FIXTURE)
Woche 12 Woche 16 Woche 52
Placebo n (%) (95% 150 mg n (%) (95% 300 mg n (%) (95% Etanercept n (%) 150 mg n (%) (95% 300 mg n (%) (95% Etanercept n (%) 150 mg n (%) (95% 300 mg n (%) (95% Etanercept n (%)
CI) CI) CI) (95% CI) CI) CI) (95% CI) CI) CI) (95% CI)
Anzahl der Patienten 324 327 323 323 327 323 323 327 323 323
PASI-50-Ansprechen 49 (15.1%) (11.5, 266 (81.3%) (76.6, 296 (91.6%) (87.9, 226 (70.0%) (64.6, 290 (88.7%) (84.6, 302 (93.5%) (90.1, 257 (79.6%) (74.7, 249 (76.1%) (71.1, 274 (84.8%) (80.3, 234 (72.4%) (67.2,
19.6) 85.3) 94.3) 74.9) 91.8) 95.8) 83.7) 80.6) 88.5) 77.2)
PASI-75-Ansprechen 16 (4.9%) (2.9, 8.1) 219 (67.0%)** 249 (77.1%)** 142 (44.0%) (38.5, 247 (75.5%) (70.4, 280 (86.7%) (82.4, 189 (58.5%) (52.9, 215 (65.7%)** 254 (78.6%)** 179 (55.4%) (49.8,
(61.5, 72.0) (72.0, 81.5) 49.6) 80.0) 90.1) 63.9) (60.3, 70.8) (73.7, 82.9) 60.9)
PASI-90-Ansprechen 5 (1.5%) (0.6, 3.8) 137 (41.9%) (36.5, 175 (54.2%) (48.6, 67 (20.7%) (16.5, 176 (53.8%) (48.3, 234 (72.4%) (67.2, 101 (31.3%) (26.3, 147 (45.0%) (39.5, 210 (65.0%) (59.5, 108 (33.4%) (28.4,
47.5) 59.7) 25.7) 59.3) 77.2) 36.7) 50.5) 70.2) 38.9)
PASI-100-Ansprechen 0 (0%) (0.0, 1.5) 47 (14.4%) (10.8, 78 (24.1%) (19.7, 14 (4.3%) (2.5, 7.3) 84 (25.7%) (21.1, 119 (36.8%) (31.6, 24 (7.4%) (4.9, 65 (19.9%) (15.8, 117 (36.2%) (31.0, 32 (9.9%) (7.0,
18.8) 29.3) 30.8) 42.4) 11.0) 24.7) 41.8) 13.8)
IGA-mod-2011-Ansprec 9 (2.8%) (1.4, 5.4) 167 (51.1%)** 202 (62.5%)** 88 (27.2%) (22.5, 200 (61.2%) (55.6, 244 (75.5%) (70.4, 127 (39.3%) (34.0, 168 (51.4%)** 219 (67.8%)** 120 (37.2%) (31.9,
hen in den Kategorie (45.5, 56.6) (57.0, 67.8) 32.5) 66.4) 80.1) 44.9) (45.8, 56.9) (62.4, 72.8) 42.7)
n "frei von" oder
"nahezu frei von"
** adjustierte
p-Werte versus
Etanercept: p=0,0250
Eine zusätzliche Psoriasis-Studie (CLEAR) beurteilte 676 Patienten. Secukinumab 300 mg erreichte die primären und sekundären Endpunkte dank der Überlegenheit gegenüber Ustekinumab in Bezug auf das Ausmass des PASI 90-Ansprechen in Woche 16 (primärer Endpunkt) und das Langzeit PASI 90 Ansprechen in Woche 52. Eine höhere Wirksamkeit von Secukinumab im Vergleich zu Ustekinumab für die Endpunkte PASI 75/90/100 und IGA mod 2011- 0 oder 1 Ansprechen ( "frei von" oder "nahezu frei von" ) war rasch sichtbar und setzte sich bis Woche 52 fort.
In dieser Studie wurde jede 300 mg Dosis in Form von zwei subkutanen Injektionen zu 150 mg verabreicht.
Tabelle 4: Zusammenfassung des klinischen Ansprechens in der CLEAR-Studie
Woche 16 Woche 52
Secukinumab 300 mg Ustekinumab* Secukinumab 300 mg Ustekinumab*
Anzahl der Patienten 334 335 334 335
PASI 75-Ansprechen 311 (93.1 %) 276 (82.4 %) 306 (91.6%) 262 (78.2%)
n (%)
PASI 90-Ansprechen 264 (79.0 %)** 192 (57.3 %) 250 (74.9%)*** 203 (60.6%)
n (%)
PASI 100-Ansprechen 148 (44.3 %) 95 (28.4 %) 150 (44.9%) 123 (36.7%)
n (%)
IGA-mod-2011-Ansprec 278 (83.2 %) 226 (67.5 %) 261 (78.1%) 213 (63.6%)
hen in den Kategorie
n "frei von" oder
"nahezu frei von" n
(%)
* Mit Secukinumab
behandelte Patienten
erhielten eine 300
mg Dosis in den
Wochen 0, 1, 2, 3
und 4, gefolgt von
derselben Dosis
jeden Monat bis
Woche 52. Mit
Ustekinumab behandel
te Patienten erhielt
en 45 mg oder 90 mg
in den Wochen 0 und
4 und dann alle 12
Wochen bis Woche 52
(dosiert nach
Gewicht und nach
zugelassener Dosieru
ng) ** p-Werte
versus Ustekinumab:
p<0.0001 für den
primären Endpunkt
PASI 90 in Woche 16
*** p-Werte versus
Ustekinumab: p=0.000
1 für den sekundären
Endpunkt PASI 90
in Woche 52
Cosentyx war bei nicht biologisch vorbehandelten Patienten, bei Biologika-vorbehandelten Patienten sowie bei Patienten mit einem Therapieversagen unter einem Biologikum wirksam. Die Ansprechraten bezüglich der primären Endpunkte, PASI 75 und IGA 0 oder 1, mit 300 mg Cosentyx lagen bei Patienten nach Versagen einer früheren TNF-Hemmer-Therapie bei 67.7% und 54.1% im Vergleich zu 78.5% und 56.9% bei Patienten ohne TNF-Hemmer-Vortherapie.
Cosentyx war in der Dosierung 300 mg mit einem raschen Einsetzen der Wirkung mit einer Reduktion des mittleren PASI in Woche 3 um 50% verbunden.
In allen Phase-III-Studien zu Plaque-Psoriasis wurden ungefähr 15 bis 25% Patienten mit gleichzeitig bestehender psoriatischer Arthritis bei Studienbeginn eingeschlossen. Die Verbesserungen des PASI 75 waren bei dieser Patientengruppe mit den Verbesserungen in der Gesamtgruppe der Patienten mit Plaque-Psoriasis vergleichbar.
Spezifische Lokalisation/Formen der Plaque-Psoriasis
In einer zusätzlichen, placebokontrollierten Studie wurde eine Verbesserung bei der Nagel-Psoriasis (TRANSFIGURE, 198 Patienten) festgestellt. In der TRANSFIGURE-Studie zeigte Secukinumab eine statistisch signifikant überlegene Wirkung gegenüber Placebo in Woche 16 (46,1 % für 300 mg, 38,4 % für 150 mg vs. 11,7 % für Placebo) in Bezug auf die Verbesserung des Nail Psoriasis Severity Index (NAPSI-Ansprechen, Index für den Schweregrad der Nagel-Psoriasis) bei Patienten mit moderater bis schwerer Plaque-Psoriasis mit einer Beteiligung der Nägel.
In weiteren klinischen Studien wurden auch Verbesserungen bei Nagel-Psoriasis, Befall der Kopfhaut und palmoplantaren Befall beobachtet.
In diesen Studien wurde jede 300 mg Dosis in Form von zwei subkutanen Injektionen zu 150 mg verabreicht.
Lebensqualität / Patientenberichtete Outcomes
Für den DLQI (Dermatology Life Quality Index) wurden statistisch signifikante Verbesserungen in Woche 12 (Studien 1-4) gegenüber dem Ausgangswert gezeigt; die Verbesserungen blieben über 52 Wochen (Studien 1 und 2) bestehen.
Statistisch signifikante Verbesserungen der patientenberichteten Anzeichen und Symptome Juckreiz, Schmerzen und Schuppung in Woche 12 gegenüber dem Ausgangswert im Vergleich zu Placebo (Studien 1 und 2) wurden anhand des validierten Psoriasis Symptom Diary© gezeigt.
Im DLQI wurden in Woche 4 bei den mit Secukinumab behandelten Patienten im Vergleich zu den mit Ustekinumab behandelten Patienten (CLEAR) statistisch signifikante Verbesserungen gegenüber dem Ausgangswert festgestellt und diese Verbesserungen blieben bis zu 52 Wochen erhalten.
Im Psoriasis Symptom Diary wurden bei den mit Secukinumab behandelten Patienten im Vergleich zu den mit Ustekinumab behandelten Patienten statistisch signifikante Verbesserungen der patientenberichteten Anzeichen und Symptome Juckreiz, Schmerzen und Schuppung in Woche 16 und in Woche 52 (CLEAR) festgestellt.
Pädiatrische Patienten
Schwere Plaque-Psoriasis
In einer 52-wöchigen, randomisierten, doppelblinden, Placebo- und Etanercept-kontrollierten Phase-III-Studie wurden 162 pädiatrische Patienten im Alter von 6 bis < 18 Jahren mit schwerer Plaque-Psoriasis (definiert anhand eines PASI-Score ≥20, eines IGA-mod-2011-Score von 4 und einer betroffenen Körperoberfläche von ≥10 %) untersucht, für die eine systemische Therapie in Frage kam. Ungefähr 43 % der Patienten wurden zuvor mit Phototherapie behandelt, 53 % mit konventionellen systemischen Therapien und 3 % mit Biologika. 9 % der Patienten hatten begleitend eine Psoriasis-Arthritis-Erkrankung.
Die Patienten wurden für eine der folgenden vier Behandlungen randomisiert:
-Niedrige Dosierung Secukinumab (75 mg bei einem Körpergewicht < 50 kg oder 150 mg bei einem Körpergewicht ≥50 kg) in den Wochen 0, 1, 2, 3 und 4, gefolgt von der gleichen Dosis alle 4 Wochen
-Hohe Dosierung Secukinumab (75 mg bei einem Körpergewicht < 25 kg, 150 mg bei einem Körpergewicht zwischen ≥25 kg und < 50 kg oder 300 mg bei einem Körpergewicht ≥50 kg) in den Wochen 0, 1, 2, 3 und 4, gefolgt von der gleichen Dosis alle 4 Wochen
-Placebo in den Wochen 0, 1, 2, 3 und 4, gefolgt von der gleichen Dosis alle 4 Wochen
-Etanercept (0.8 mg/kg) wöchentlich (bis zu einem Maximum von 50 mg)
Patienten, die auf Placebo randomisiert wurden und die in Woche 12 nicht angesprochen hatten, wurden entweder in die Secukinumab-Gruppe mit niedriger oder mit hoher Dosierung (Dosierung basierend auf der Körpergewichtsgruppe) eingeteilt und erhielten das Studienmedikament in den Wochen 12, 13, 14 und 15, gefolgt von der gleichen Dosis alle 4 Wochen, beginnend in Woche 16.
Die co-primären Endpunkte waren der Anteil an Patienten, die von der Baseline bis Woche 12 eine Verbesserung des PASI-Scores um mindestens 75 % (PASI-75-Ansprechen) und einen IGA-mod-2011-Score von "symptomfrei" oder "fast symptomfrei" (0 oder 1) mit einer Verbesserung um mindestens 2 Punkte erreichten.
Während des 12-wöchigen placebokontrollierten Zeitraums war die Wirksamkeit bei der niedrigen und bei der hohen Dosierung von Secukinumab im Hinblick auf die co-primären Endpunkte vergleichbar. Die Schätzungen der Odds Ratio zugunsten beider Secukinumab-Dosierungen waren sowohl für das PASI-75-Ansprechen als auch für das IGA-mod-2011-Ansprechen von "symptomfrei" oder "fast symptomfrei" (0 oder 1) klinisch relevant und statistisch signifikant.
Alle Patienten wurden nach der ersten Dosis 52 Wochen lang bezüglich der Wirksamkeit und Sicherheit nachbeobachtet. Der Anteil der Patienten, die ein PASI-75-Ansprechen und ein IGA-mod-2011-Ansprechen von "symptomfrei" oder "fast symptomfrei" (0 oder 1) erreichten, zeigte bereits einen Unterschied zwischen den Secukinumab-Behandlungsgruppen und Placebo in Woche 4, wobei der Unterschied in Woche 12 sich vergrösserte. Das Ansprechen blieb über den gesamten Zeitraum von 52 Wochen erhalten. Die PASI-50-, PASI-90- und PASI-100- Werte und der Anteil der Patienten mit Children's Dermatology Life Quality Index-(CDLQI-)Scores von 0 oder 1 verbesserte sich ebenfalls und wurde über den gesamten Zeitraum von 52 Wochen aufrechterhalten.
Nach Woche 12 war die Wirksamkeit sowohl der niedrigen als auch der hohen Dosierung von Secukinumab vergleichbar, obwohl die Wirksamkeit der hohen Dosierung bei Patienten mit einem Körpergewicht von ≥50 kg höher war. Die Sicherheitsprofile der niedrigen Dosierung und der hohen Dosierung waren vergleichbar.
Die Ergebnisse zur Wirksamkeit in den Wochen 12 sind in Tabelle 5 dargestellt.
Tabelle 5: Zusammenfassung des klinischen Ansprechens bei Kindern und Jugendlichen mit schwerer Psoriasis in den Wochen 12*
Ansprech-kriterium Behandlungsvergleich "Test" "Kontrolle" Odds-Ratio
"Test" vs. "Kontroll n/m** (%) n/m** (%) Schätzer (95%-KI) p-Wert
e"
in Woche 12***
PASI 75 Secukinumab niedrige 32/40 (80.0) 6/41 (14.6) 25.78 (7.08,114.66) <0.0001
Dosierung vs.
Placebo
Secukinumab hohe 31/40 (77.5) 6/41 (14.6) 22.65 (6.31,98.93) <0.0001
Dosierung vs.
Placebo
IGA 0/1 Secukinumab niedrige 28/40 (70.0) 2/41 (4.9) 51.77 (10.02,538.64) <0.0001
Dosierung vs.
Placebo
Secukinumab hohe 24/40 (60.0) 2/41 (4.9) 32.52 (6.48,329.52) <0.0001
Dosierung vs.
Placebo
PASI 90 Secukinumab niedrige 29/40 (72.5) 1/41 (2.4) 133.67 (16.83,6395.2 <0.0001
Dosierung vs. 2)
Placebo
Secukinumab hohe 27/40 (67.5) 1/41 (2.4) 102.86 (13.22,4850.1 <0.0001
Dosierung vs. 3)
Placebo
* Bei fehlenden
Daten erfolgte eine
Imputation als
Non-Responder ** n
= Anzahl der Respond
er, m = Anzahl der
auswertbaren Patient
en *** verlängertes
Zeitfenster für
Visiten in Woche 12
Odds Ratio, 95
%-Konfidenzintervall
und p-Wert stammen
aus einem exakten
Regressionsmodell
mit Behandlungsgrupp
e, Körpergewichtskat
egorie bei Baseline
und Alterskategorie
als Faktoren
Ein höherer Anteil der mit Secukinumab behandelten pädiatrischen Patienten berichtete über eine Verbesserung der gesundheitsbezogenen Lebensqualität, gemessen an einem CDLQI-Score von 0 oder 1 im Vergleich zu Placebo in Woche 12.
Mittelschwere bis schwere Plaque-Psoriasis
In einer offenen, zweiarmigen, parallelen, multizentrischen Phase-III-Studie wurden 84 pädiatrische Patienten im Alter von 6 bis <18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis (definiert durch einen PASI-Score ≥12, einen IGA-mod-2011-Score ≥3 sowie eine betroffene Körperoberfläche ≥10 %) untersucht, für die eine systemische Therapie in Frage kam.
Die Patienten wurden für eine Behandlung mit Secukinumab in den Wochen 0, 1, 2, 3 und 4, gefolgt von der gleichen Dosis alle 4 Wochen, in folgender Weise randomisiert:
-Niedrige Dosierung Secukinumab (75 mg bei einem Körpergewicht < 50 kg oder 150 mg bei einem Körpergewicht ≥50 kg),
-Hohe Dosierung Secukinumab (75 mg bei einem Körpergewicht < 25 kg, 150 mg bei einem Körpergewicht zwischen ≥25 kg und < 50 kg oder 300 mg bei einem Körpergewicht ≥50 kg).
Die co-primären Endpunkte waren der Anteil der Patienten, die von der Baseline bis Woche 12 eine Verbesserung des PASI-Scores um mindestens 75 % (PASI-75-Ansprechen) und einen IGA-mod-2011-Score von "symptomfrei" oder "fast symptomfrei" (0 oder 1) mit einer Verbesserung um mindestens 2 Punkte erreichten.
Die Wirksamkeit sowohl der niedrigen als auch der hohen Dosierung von Secukinumab war vergleichbar und zeigte für die co-primären Endpunkte eine klinisch relevante Verbesserung im historischen Vergleich gegenüber Placebo.
Alle Patienten wurden nach der ersten Verabreichung mindestens 24 Wochen lang bezüglich der Wirksamkeit untersucht. Die Wirksamkeit (definiert als PASI-75-Ansprechen und IGA-mod-2011-Ansprechen von "symptomfrei" oder "fast symptomfrei" [0 oder 1]) wurde bereits in Woche 2 beobachtet. Der Anteil der Patienten, die ein PASI-75-Ansprechen und ein IGA-mod-2011-Ansprechen von "symptomfrei" oder "fast symptomfrei" (0 oder 1) erreichten, stieg während des gesamten Zeitraums von 24 Wochen an. Verbesserungen des PASI 90 und PASI 100 wurden ebenfalls in Woche 12 beobachtet und erhöhte sich über den gesamten 24-Wochen-Zeitraum.
Nach Woche 12 war die Wirksamkeit sowohl der niedrigen als auch der hohen Dosierung von Secukinumab vergleichbar. Die Sicherheitsprofile der niedrigen Dosierung und der hohen Dosierung waren ebenfalls vergleichbar.
Die Ergebnisse zur Wirksamkeit in den Wochen 12 und 24 sind in Tabelle 6 dargestellt.
Tabelle 6: Zusammenfassung des klinischen Ansprechens bei mittelschwerer bis schwerer pediatrischen Psoriasis in den Wochen 12* und 24*
Woche 12 Woche 24
Secukinumab niedrige Secukinumab hohe Secukinumab niedrige Secukinumab hohe
Dosis Dosis Dosis Dosis
Anzahl an Patienten 42 42 42 42
PASI-75-Ansprechen 39 (92.9%) 39 (92.9%) 40 (95.2%) 40 (95.2%)
n (%)
IGA-mod-2011-Ansprec 33 (78.6%) 35 (83.3%) 37 (88.1%) 39 (92.9%)
hen "symptomfrei"
oder "fast symptomfr
ei" n (%)
PASI-90-Ansprechen 29 (69.0%) 32 (76.2%) 37 (88.1%) 37 (88.1%)
n (%)
PASI-100-Ansprechen 25 (59.5%) 23 (54.8%) 28 (66.7%) 28 (66.7%)
n (%)
* Bei fehlenden
Daten erfolgte eine
Imputation als
Non-Responder
Dosisflexibilität bei Plaque-Psoriasis
In einer randomisierten, doppelblinden, multizentrischen Studie mit 331 Patienten wurde die Wirksamkeit, Sicherheit und Verträglichkeit von Cosentyx 300 mg, angewendet alle 4 Wochen, im Vergleich zu Cosentyx 300 mg, angewendet alle 2 Wochen, bei erwachsenen Patienten mit einem Körpergewicht ≥90 kg und mittelschwerer bis schwerer Plaque-Psoriasis untersucht. Die Patienten wurden im Verhältnis 1:1 wie folgt randomisiert:
-Secukinumab 300 mg in den Wochen 0, 1, 2, 3 und 4, gefolgt von derselben Dosis alle 2 Wochen bis Woche 52 (n=165).
-Secukinumab 300 mg in den Wochen 0, 1, 2, 3 und 4, gefolgt von der derselben Dosis alle 4 Wochen bis Woche 16 (n=166).
-Die Patienten, die für Secukinumab 300 mg alle 4 Wochen randomisiert worden waren und in Woche 16 ein PASI-90-Ansprechen erreicht hatten, erhielten bis Woche 52 weiterhin dasselbe Dosierungsschema. Die Patienten, die für Cosentyx 300 mg alle 4 Wochen randomisiert worden waren und in Woche 16 kein PASI-90-Ansprechen erreicht hatten, erhielten entweder weiterhin dasselbe Dosierungsschema oder wurden für die Zeit bis Woche 52 auf Cosentyx 300 mg alle 2 Wochen umgestellt.
Die primären und die wichtigsten sekundären Endpunkte waren der Anteil der Patienten, die in Woche 16 ein PASI-90-Ansprechen sowie ein Ansprechen von "symptomfrei" oder "fast symptomfrei" (0 oder 1) in den IGA-mod-2011-Kategorien erreicht hatten.
In Woche 16 war der Anteil der Patienten, die ein PASI-90-Ansprechen erreicht hatten, in der Gruppe, die mit dem Schema alle 2 Wochen (Q2W) behandelt worden war, höher als in der Gruppe, die mit dem Schema alle 4 Wochen (Q4W) behandelt worden war (73.2% bzw. 55.5%). Der Behandlungsunterschied war statistisch signifikant (einseitiger p-Wert = 0,0003).
Die Gruppe der Patienten, die kein PASI-90-Ansprechen erreicht hatten und in Woche 16 auf das Schema alle 2 Wochen umgestellt worden waren, wies in Woche 32 ein höheres PASI-90-Ansprechen auf als die Gruppe, bei der das Schema alle 4 Wochen beibehalten worden war (38,7 % vs. 16,5 %). Der Behandlungsunterschied war klinisch relevant aber nur explorativ.
Die Sicherheitsprofile der beiden Dosierungsschemata, Cosentyx 300 mg alle 4 Wochen und Cosentyx 300 mg alle 2 Wochen, waren bei Patienten mit einem Körpergewicht ≥90 kg vergleichbar und stimmten mit dem bei Psoriasis-Patienten angegeben Sicherheitsprofil überein.
Psoriasis-Arthritis
Es wurde bei erwachsenen Patienten mit aktiver psoriatischer Arthritis gezeigt, dass Cosentyx die Anzeichen und Symptome, die körperliche Funktionsfähigkeit und die gesundheitsbezogene Lebensqualität verbessert und ausserdem die Progressionsrate der peripheren Gelenkschädigung reduziert.
Die Sicherheit und Wirksamkeit von Cosentyx wurde in drei randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien bei 1'999 Patienten gezeigt, die trotz einer Behandlung mit nichtsteroidalen Antirheumatika (non-steroidal anti-inflammatory drug, NSAID), Kortikosteroiden oder krankheitsmodifizierenden Antirheumatika (disease-modifying anti-rheumatic drugs, DMARD) eine aktive Psoriasis-Arthritis (≥3 geschwollene und ≥3 druckschmerzempfindliche Gelenke) hatten. Die PsA-Diagnose der Patienten in diesen Studien wurde vor mindestens fünf Jahren gestellt. Die Mehrheit der Patienten hatte zudem eine Hautläsion aufgrund einer aktiven Psoriasis oder eine dokumentierte Psoriasis in der Vorgeschichte. Um eine unverzerrte Beurteilung der Wirksamkeit von Cosentyx in der Psoriasis Behandlung zu erhalten, war die gleichzeitige Anwendung einer topischen Kortikosteroidtherapie oder UV basierten Therapie während der Studien nicht erlaubt. Bei mehr als 61% bzw. 42% der PsA-Patienten lag bei Baseline jeweils Enthesitis bzw. Daktylitis vor. Die Anzahl der PsA Patienten mit axialer Beteiligung war für eine aussagekräftige Beurteilung zu gering.
Die Wirksamkeit und Sicherheit von Cosentyx in Dosen von 75 mg, 150 mg und 300 mg wurde gegenüber Placebo mit einer entweder intravenösen oder subkutanen Anfangsdosis beurteilt. In der Studie Psoriasis-Arthritis 1 (PsA1-Studie) bzw. der Studie Psoriasis-Arthritis-2 (PsA2-Studie) und Studie Psoriasis-Arthritis 3 (PsA3-Studie) wurden jeweils 29% bzw. 35% und 30% der Patienten zuvor mit Anti-TNF-alpha-Medikamenten behandelt, wobei diese Behandlung entweder aufgrund eines fehlenden Ansprechens oder aufgrund einer Unverträglichkeit abgesetzt wurde (Anti-TNF-alpha-IR-Patienten).
In der PsA1-Studie (FUTURE 1) wurden 606 Patienten bewertet; davon erhielten 60.7% begleitend MTX. Es wurden Patienten mit allen PsA-Untergruppen eingeschlossen, einschliesslich polyartikulärer Arthritis ohne Nachweis von Rheumaknoten (76.7%), Spondylitis mit peripherer Arthritis (18.5%), asymmetrischer peripherer Arthritis (60.2%), distaler interphalangealer Beteiligung (59.6%) und Arthritis mutilans (7.9%). Patienten, die randomisiert Cosentyx zugeteilt wurden, erhielten 10 mg/kg i.v. in Woche 0, 2 und 4, gefolgt von entweder monatlich 75 mg oder 150 mg s.c., beginnend in Woche 8. Patienten, die randomisiert Placebo zugeteilt wurden und auf die Behandlung nicht ansprachen, wechselten dann in Woche 16 zur Behandlung mit 75 mg oder 150 mg Cosentyx s.c. einmal monatlich. In Woche 24 wurden die verbliebenen Placebo-Patienten zur Behandlung mit 75 mg oder 150 mg Cosentyx s.c. überführt. Der primäre Endpunkt war das Ansprechen gemäss American College of Rheumatology (ACR) 20 in Woche 24.
In der PsA2-Studie (FUTURE 2) wurden 397 Patienten beurteilt, von denen 46.6% begleitend mit MTX behandelt wurden. Es wurden Patienten mit allen PsA-Untergruppen eingeschlossen, einschliesslich polyartikulärer Arthritis ohne Nachweis von Rheumaknoten (85.9%), Spondylitis mit peripherer Arthritis (21.7%), asymmetrischer peripherer Arthritis (64.0%), distaler interphalangealer Beteiligung (57.9%) und Arthritis mutilans (6.3%). Patienten, die randomisiert Cosentyx zugeteilt wurden, erhielten 75 mg, 150 mg oder 300 mg s.c. in Woche 0, 1, 2, 3 und 4, gefolgt von der gleichen monatlichen Dosis. Patienten, die randomisiert Placebo zugeteilt wurden und bis Woche 16 nicht auf die Behandlung ansprachen, wechselten dann in Woche 16 zur Behandlung mit 150 mg oder 300 mg Cosentyx s.c. einmal monatlich. In Woche 24 wurden die verbliebenen Placebo-Patienten zur Behandlung mit 150 mg oder 300 mg Cosentyx s.c. überführt. Der primäre Endpunkt war das Ansprechen gemäss ACR 20 in Woche 24.
In der PsA3-Studie (FUTURE 5) wurden 996 Patienten beurteilt, von denen 50.1% begleitend mit MTX behandelt wurden. Es wurden Patienten mit allen PsA-Untergruppen eingeschlossen, einschliesslich polyartikulärer Arthritis ohne Nachweis von Rheumaknoten (78.7%), Spondylitis mit peripherer Arthritis (19.8%), asymmetrischer peripherer Arthritis (65.0%), distaler interphalangealer Beteiligung (56.7%) und Arthritis mutilans (6.8%). Die Patienten wurden in folgende Gruppen randomisiert: Cosentyx 150 mg, Cosentyx 300 mg oder Placebo, jeweils s.c. in Woche 0, 1, 2, 3 und 4, gefolgt von der gleichen monatlichen Dosis, oder Cosentyx 150 mg einmal monatlich ohne initiale Sättigungsdosis. Patienten, die initial Placebo zugeteilt wurden und bis Woche 16 nicht auf die Behandlung ansprachen, wechselten dann in Woche 16 zur Behandlung mit Cosentyx (entweder 150 mg oder 300 mg s.c.) einmal monatlich. In Woche 24 wurden die verbliebenen Placebo-Patienten zur Behandlung mit Cosentyx (entweder 150 mg oder 300 mg) einmal monatlich überführt. Der primäre Endpunkt war das Ansprechen gemäss ACR 20 in Woche 16, und der wichtigste sekundäre Endpunkt war der Unterschied beim modifizierten Total Sharp Score (mTSS) in Woche 24 gegenüber der Baseline.
In der PsA4-Studie (FUTURE 3) wurden 414 Patienten beurteilt, von denen 47.6% begleitend mit MTX behandelt wurden. Patienten, die randomisiert Cosentyx zugeteilt wurden, erhielten 150 mg oder 300 mg s.c. in Woche 0, 1, 2, 3 und 4, gefolgt von der gleichen monatlichen Dosis. Patienten, die randomisiert Placebo zugeteilt wurden und bis Woche 16 nicht auf die Behandlung ansprachen, wechselten dann in Woche 16 zur Behandlung mit 150 mg oder 300 mg Cosentyx s.c. einmal monatlich. In Woche 24 wurden die verbliebenen Placebo-Patienten zur Behandlung mit 150 mg oder 300 mg Cosentyx s.c. überführt. Der primäre Endpunkt war das Ansprechen gemäss ACR 20 in Woche 24.
Klinisches Ansprechen
Anzeichen und Symptome
Die Behandlung mit Cosentyx führte in den Wochen 16, 24 und 52 im Vergleich zu Placebo zu einer signifikanten Verbesserung des Ausmasses der Krankheitsaktivität. Diese Messungen umfassten das Ansprechen der Gelenkssymptomatik in Bezug auf ACR20, ACR50, ACR70, das Ansprechen der Hautsymptomatik (Psoriasis-Area-and-Severity-Index, PASI) 75, PASI 90, sowie weitere Scores zur Krankheitsaktivität und Gesundheitszustand (Disease Activity Score, DAS28-CRP, Short Form Health Survey – Physical Component Summary; SF-36 PCS, Health Assessment Questionnaire – Disability Index, HAQ-DI) (siehe Tabelle 7).
Tabelle 7: Klinisches Ansprechen in den Studien PsA2 und PsA3 in den Wochen 16, 24 und 52
PsA2 PsA3 Placebo 150 mg1 300 mg1 Placebo 150 mg1 300 mg1 Anzahl der randomisi 98 100 100 332 220 222 erten Patienten ACR-20-Ansprechen n (%) Woche 16 18 (18.4%) 60 (60.0%***) 57 (57.0%***) 91◊ (27.4%) 122◊ (55.5%***) 139◊ (62.6%***) Woche 24 15◊ (15.3%) 51◊ (51.0%***) 54◊ (54.0%***) 78 (23.5%) 117 (53.2%***) 141 (63.5%***) Woche 52 - 64 (64.0%) 64 (64.0%) NA NA NA ACR-50-Ansprechen n (%) Woche 16 6 (6.1%) 37 (37.0%***) 35 (35.0%***) 27 (8.1%) 79 (35.9%***) 88 (39.6%***) Woche 52 - 39 (39.0%) 44 (44.0%) NA NA NA ACR-70-Ansprechen n (%) Woche 16 2 (2.0%) 17 (17.0%**) 15 (15.0%**) 14 (4.2%) 40 (18.2%***) 45 (20.3%***) Woche 52 - 20 (20.0%) 24 (24.0%) NA NA NA DAS28-CRP Woche 16 -0.50 -1.45*** -1.51*** -0.63 -1.29*** -1.49*** Woche 52 - -1.69 -1.78 NA NA NA PASI-75-Ansprechen n (%) Woche 16 3 (7.0%) 33 (56.9%***) 27 (65.9%***) 20 (12.3%) 75 (60.0%***) 77 (70.0%***) Woche 52 - 33 (56.9%) 30 (73.2%) - - - PASI-90-Ansprechen n (%) Woche 16 3 (7.0%) 22 (37.9%***) 18 (43.9%***) 15 (9.3%) 46 (36.8%***) 59 (53.6%***) Woche 52 - 25 (43.1%) 23 (56.1%) - - - Verschwinden Dactyli tis n (%)† Woche 16 10 (37%) 21 (65.6%*) 26 (56.5%) 40 (32.3%) 46 (57.5%***) 54 (65.9%***) Woche 52 - 21 (65.6%) 32 (69.6%) NA NA NA Verschwinden Enthesi tis n (%)‡ Woche 16 17 (26.2%) 32 (50.0%**) 32 (57.1%***) 68 (35.4%) 77 (54.6%***) 78 (55.7%***) Woche 52 - 31 (48.4%) 30 (53.6%) NA NA NA * p<0.05, ** p<0.01, *** p<0.001; gegenüber Placebo Alle p-Werte werden ohne Korrektur für multiples Testen wiedergegeben. Patienten mit fehlenden binären Endpunkten wurden als Non-Responder erfasst ( "Non-respo nder Imputation" ). NA: nicht verfügbar (Not Available); ACR: American College of Rheumatol ogy; PASI: Psoriasis Area and Severity Index; DAS: Disease Activity Score; BSA: Körperoberfläch e (Body Surface Area, BSA) ◊ Primäre r Endpunkt 1 Cosenty x 150 mg oder 300 mg s.c. in Woche 0, 1, 2, 3 und 4, gefolgt von der gleichen monatlichen Dosis. † Bei Patienten mit Daktylitis bei Baseline (n=27, 32 bzw. 46 in PsA2 und n=124, 80 bzw. 82 in PSA3) Vollständig es Abklingen der Daktylitis wurde in der Untergruppe von Patienten mit Dayktylitis bei Baseline beurteilt und ist als Patiente nanteil mit einem Leeds Dactylitis Index (LDI) Wert von Null ausgedrückt . ‡ Bei Patienten mit Enthesitis bei Baseline (n=65, 64 bzw. 56 in PsA2 und n=192, 141 bzw. 140 in PsA3) Vollständig es Abklingen der Enthesitis wurde in der Untergruppe von Patienten mit Enthesitis bei Baseline beurteilt und ist als Patiente nanteil mit einem Leeds Enthesitis Index (LEI) Wert von Null ausgedrückt .
Die Wirkung von Cosentyx trat in Woche 2 ein. Ein statistisch signifikanter Unterschied beim ACR 20 im Vergleich zu Placebo wurde in Woche 3 erreicht. In der Studie PsA2 wurde die Wirksamkeit bis Woche 104 aufrechterhalten (64.4% und 69.4% für 150 mg bzw. 300 mg).
In Woche 16 wiesen mit Cosentyx behandelte Patienten signifikante Verbesserungen der Anzeichen und Symptome auf, darunter ein signifikant höheres Ansprechen hinsichtlich ACR 20 (60.0 % und 57.0 % für 150 mg bzw. 300 mg) im Vergleich zum Placebo (18.4 %).
Der Anteil der Patienten, die pro Besuch ein ACR-20-Ansprechen zeigten, ist in Abbildung 1 dargestellt.
Abbildung 1: ACR20-Ansprechen in der PsA2-Studie im Verlauf der Zeit bis Woche 24
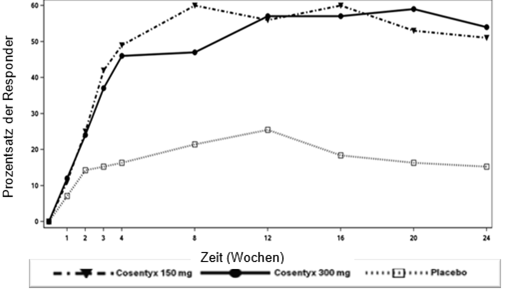
Bei den primären und wichtigen sekundären Endpunkten wurde bei den PsA-Patienten ein ähnliches Ansprechen beobachtet, unabhängig davon, ob sie begleitend MTX erhielten oder nicht.
Sowohl die bisher nicht mit Anti-TNF-alpha behandelten als auch die Anti-TNF-alpha–IR-Patienten, die mit Cosentyx behandelt wurden, zeigten in Woche 16 und 24 im Vergleich zu Placebo ein signifikant höheres ACR-20-Ansprechen, wobei das Ansprechen in der bisher nicht mit Anti-TNF-alpha behandelten Gruppe nummerisch höher war (Anti-TNF-alpha-unbehandelt in PsA2: 64% bzw. 58% bei 150 mg bzw. 300 mg verglichen mit Placebo 15.9%; Anti-TNF-alpha-IR: 30% bzw. 46% bei 150 mg bzw. 300 mg verglichen mit Placebo 14.3%.
Anti-TNF-alpha–IR-Patienten, die mit einer Dosis von 300 mg behandelt wurden, zeigten im Vergleich zu Placebo-Patienten eine höhere Ansprechrate gemäss ACR20 (p<0.05) und zeigte einen klinisch bedeutenden Nutzen gegenüber 150 mg bei ACR50, PASI75, PASI90, HAQ-DI, Daktylitis und Enthesitis.
Der Anteil der Patienten in PsA2, die ein modifiziertes Ansprechen gemäss PsA Response Criteria (PsARC) erreichten, war in Woche 24 in der Gruppe der mit Cosentyx behandelten Patienten höher (59.0% bzw. 61.0% bei 150 mg bzw. 300 mg) als in der mit Placebo behandelten (26.5%).
Die Ergebnisse der Komponenten der ACR-Kriterien des Ansprechens sind in Tabelle 8 dargestellt.
Tabelle 8: Differenz der Mittelwerte der ACR-Komponenten gegenüber Baseline in der PsA2-Studie in Woche 24
Placebo (N=98) 150 mg (N=100) 300 mg (N=100) Anzahl der geschwollenen Gelenke Baseline 12.1 11.9 11.2 Differenz -5.14 -6.32 -7.28* Anzahl der druckempfindlichen Gelenke Baseline 23.4 24.1 20.2 Differenz -4.28 -11.42*** -10.84** Beurteilung der Schmerzen durch den Patienten Baseline 55.4 58.9 57.7 Differenz -11.71 -23.39** -22.35** Gesamtbeurteilung durch den Patienten Baseline 57.6 62.0 60.7 Differenz -10.14 -25.78*** -26.70*** Gesamtbeurteilung durch den Arzt Baseline 55.0 56.7 55.0 Differenz -25.23 -32.97* -38.52*** Behinderungsindex (HAQ) Baseline 1.1684 1.2200 1.2828 Differenz -0.31 -0.48* -0.56** CRP (mg/dl) Baseline 7.71 14.15 10.69 hsCRP, (Verhältnis post-BSL/BSL) 0.75 0.55* 0.55* * p<0.05, ** p<0.01, *** p<0.001 basierend auf nominalem, jedoch nicht angepasstem p-Wert
In der PsA1-Studie zeigten mit Cosentyx behandelte Patienten in Woche 24 signifikant verbesserte PsA-Anzeichen und Symptome bei einem ähnlichen Ansprechen wie in der PsA2-Studie. Die Wirksamkeit wurde bis Woche 104 aufrechterhalten.
Radiographisches Ansprechen
In der PsA3-Studie wurde die strukturelle Schädigung radiographisch beurteilt und in Form des modifizierten Total Sharp Score (mTSS) und seiner Komponenten, des Erosion Score (ES) und des Joint Space Narrowing Score (JSN), ausgedrückt. Es wurden Röntgenaufnahmen der Hände, der Handgelenke und der Füsse bei Baseline, in Woche 16 und/oder in Woche 24 durchgeführt und von mindestens zwei Begutachtern, die im Hinblick auf die Behandlungsgruppe und die Nummer des Besuchs verblindet waren, unabhängig voneinander bewertet.
Durch die Behandlung mit Cosentyx 150 mg bzw. 300 mg wurde im Vergleich zur Behandlung mit Placebo die Progressionsrate der peripheren Gelenkschädigung, die anhand der Veränderung des mTSS in Woche 24 gegenüber der Baseline beurteilt wurde, signifikant reduziert (Tabelle 8).
Der Prozentsatz der Patienten ohne Krankheitsprogression (definiert als Veränderung beim mTSS von ≤0.5) von der Randomisierung bis Woche 24 lag bei 79.8%, 88.0% und 73.6% für Cosentyx 150 mg, 300 mg bzw. Placebo. Eine Hemmung der strukturellen Schädigung wurde unabhängig von einer eventuell vorhandenen begleitenden Anwendung von MTX bzw. dem TNF-Status festgestellt.
Durch die Behandlung mit Cosentyx 150 mg wurde bis Woche 24 im Vergleich zur Behandlung mit Placebo eine signifikant verminderte Progressionsrate der peripheren Gelenkschäden erreicht. Diese wurde anhand der Veränderung beim mTSS gegenüber der Baseline beurteilt (siehe Tabelle 9). Die Hemmung der strukturellen Schädigung wurde unter der Behandlung mit Cosentyx bis Woche 52 aufrechterhalten.
Tabelle 9: Änderung beim modifizierten Total Sharp Score in den Studien PsA3 und PsA1
PsA3
Placebo n=296 150 mg1 n=213 300 mg1 n=217
Total Score
Baseline 15.0 13.6 12.9
(SD) (38.2) (25.9) (23.7)
Durchschnittliche Änderung in 0.5 0.17* 0.08*
Woche 24
* p<0.05, basierend auf dem
nominalen p-Wert, der aber nicht
im Hinblick multiples Testen
korrigiert wurde. 1 Cosentyx 150
mg oder 300 mg s.c. in den Wochen
0, 1, 2, 3 und 4, gefolgt von der
gleichen monatlichen Dosis.
Axiale Manifestationen bei PsA
Eine randomisierte, doppelblinde, placebokontrollierte Studie (MAXIMISE) untersuchte die Wirksamkeit von Secukinumab bei 485 PsA-Patienten mit axialer Manifestation, die nicht mit Biologika vorbehandelt waren und unzureichend auf nichtsteroidale Antirheumatika (NSAID) ansprachen. Die primäre Variable einer mindestens 20%ige Verbesserung der ASAS-20-Kriterien (Assessment of Spondyloarthritis International Society, ASAS) in Woche 12 wurde erfüllt (siehe Tabelle 10).
Tabelle 10: Klinisches Ansprechen in der MAXIMISE-Studie in Woche 12
Placebo (n=164) 150 mg (n=157) 300 mg (n=164) ASAS-20-Ansprechen, % (95% CI) 31.2 (24.6; 38.7) 66.3 (58.4, 73.3)* 62.9 (55.2, 70.0)* * p<0.0001; gegenüber Placebo unter Verwendung der Mehrfach-Impu tation. ASAS: Assessment of Spondylo Arthritis International Society Criteria;
Eine Verbesserung des ASAS-20-Ansprechens für beide Secukinumab-Dosen wurde in Woche 4 beobachtet und blieb bis zu 52 Wochen erhalten.
Körperliche Funktionsfähigkeit und gesundheitsbezogene Lebensqualität
In der PsA2-Studie und der PsA3-Studie zeigten Patienten, die mit Cosentyx 150 mg und 300 mg behandelt wurden, in Woche 24 bzw. Woche 16 eine Verbesserung der körperlichen Funktionsfähigkeit im Vergleich zu Patienten, die mit Placebo behandelt wurden, gemessen anhand des Health Assessment Questionnaire – Disability Index (HAQ-DI). Die Verbesserungen in den HAQ-DI-Ergebnissen wurden unabhängig von einer vorherigen Exposition gegenüber Anti-TNF-alpha beobachtet. Vergleichbare Resultate wurden bei der PsA1-Studie beobachtet.
Die mit Cosentyx behandelten Patienten berichteten von deutlichen Verbesserungen der gesundheitsbezogenen Lebensqualität, gemessen anhand des Short Form (36) Health Survey – Physical Component Summary (SF-36 PCS) (p<0.001). Es gab auch statistisch signifikante Verbesserungen im FACIT-F (Functional Assessment of Chronic Illness Therapy – Fatigue) Score für 150 mg und 300 mg im Vergleich zu Placebo in Woche 24 (p<0.01), und diese Verbesserungen wurden in der Studie PsA2 bis Woche 104 aufrechterhalten.
Dosiseskalation auf 300 mg bei Patienten mit unbefriedigenden Ansprechen auf 150 mg.
Aus den Studien PsA1, PsA2 und PsA4 liegen auswertbare Daten zur Dosiseskalation von 150 mg zu 300 mg von 159 Patienten vor, die nach 92 bis 156 Wochen unbefriedigend angesprochen hatten. Dies entspricht 21.1% der in den Studien verbliebenen Patienten. Nach der Dosiseskalation der Secukinumab Dosis von 150 mg auf 300 mg wurde eine Verbesserung der ACR und PASI Ansprechrate und eine Reduktion des Anteils nicht ansprechender Patienten (< ACR20; < PASI75) beobachtet.
Axiale Spondyloarthritis (axSpA)
Ankylosierende Spondylitis (Morbus Bechterew)
Die Sicherheit und Wirksamkeit von Cosentyx wurde in zwei randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien bei 590 Patienten gezeigt, die trotz einer Behandlung mit nichtsteroidalen Antirheumatika (non-steroidal anti-inflammatory drug, NSAID), Kortikosteroiden oder krankheitsmodifizierenden Antirheumatika (disease-modifying anti-rheumatic drugs, DMARD) eine aktive ankylosierende Spondylitis (AS) mit einer Krankheitsaktivität definiert mittels Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) von ≥4 hatten. Die Patienten in diesen Studien wiesen im Mittel eine AS-Diagnose seit 2.7 bis 5.8 Jahren auf. Patienten mit kompletter Ankylosierung waren ausgeschlossen.
Für beide Studien war der primäre Endpunkt eine mindestens 20%ige Verbesserung gemäss den Kriterien der Assessment of Spondyloarthritis International Society (ASAS20-Ansprechen) in Woche 16.
In der Studie Ankylosierende Spondylitis 1 (AS1-Studie) bzw. der Studie Ankylosierende Spondylitis 2 (AS2-Studie) wurden 27.0% bzw. 38.8% der Patienten zuvor mit Anti-TNF-alpha-Medikamenten behandelt, und die Behandlung wurde entweder aufgrund eines fehlenden Ansprechens oder aufgrund einer Unverträglichkeit abgesetzt (Anti-TNF-alpha-IR-Patienten).
In der AS1-Studie (MEASURE 1) wurden 371 Patienten bewertet; davon erhielten 14.8%, 33.4% bzw. 13.5% begleitend MTX, Sulfasalazin bzw. Kortikosteroide. Patienten, die für Cosentyx randomisiert wurden, erhielten 10 mg/kg i.v. in Woche 0, 2 und 4, gefolgt von 75 mg oder 150 mg s.c. einmal monatlich. Patienten, die für Placebo randomisiert wurden und bis Woche 16 nicht ansprachen sowie bei Woche 24 alle übrigen Placebo Patienten, wechselten zur Behandlung mit 75 mg oder 150 mg Cosentyx s.c. einmal monatlich.
In der AS2-Studie (MEASURE 2) wurden 219 Patienten beurteilt, von denen 11.9%, 14.2% bzw. 8.2% begleitend jeweils mit MTX, Sulfasalazin bzw. Kortikosteroide behandelt wurden. Patienten, die für Cosentyx randomisiert wurden, erhielten 75 mg oder 150 mg s.c. in Woche 0, 1, 2, 3 und 4, gefolgt von der gleichen monatlichen Dosis. Patienten, die bei Baseline für Placebo randomisiert wurden, wurden in Woche 16 erneut randomisiert, um monatlich Cosentyx s.c. zu erhalten (entweder 75 mg oder 150 mg).
Klinisches Ansprechen
Anzeichen und Symptome
In der AS2-Studie führte die Behandlung mit Cosentyx 150 mg in Woche 16 zu der deutlichsten Verbesserung der Messgrössen der Krankheitsaktivität verglichen mit Placebo (siehe Tabelle 11).
Tabelle 11: Klinisches Ansprechen in der AS2-Studie in Woche 16
Ergebnis (p-Wert gegenüber Placebo (n = 74) 75 mg (n = 73) 150 mg (n = 72) Placebo) Wirksamkeit in Woche 16 ASAS20-Ansprechen, % 28.4 41.1 61.1*** ASAS40-Ansprechen, % 10.8 26.0 36.1*** hsCRP, (Verhältnis post-BSL/BSL)# 1.13 0.61 0.55*** ASAS5/6, % 8.1 34.2 43.1*** BASDAI, LS mittlere Veränderung -0.85 -1.92 -2.19*** ab Baseline-Wert† ASAS, partielle Remission, % 4.1 15.1 13.9 BASDAI50, % 10.8 24.7* 30.6** ASDAS-CRP "major improvement" ˆ 4.1 15.1* 25.0*** *p<0.05; **p<0.01; ***p< 0.001 gegenüber Placebo Alle p-Werte wurden für den Multiplikationstest basierend auf einer vordefinierte n Hierarchie angepasst, mit Ausnahme von BASDAI50 und ASDAS-CRP. Die nicht ansprechenden Patienten wurden für den fehlenden binären Endpunkt angerechnet. ASAS: Assessment of Spondylo Arthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: hochsensitives C-reaktives Protein; ASDA: Ankylosing Spondylitis Disease Activity Score; BSL: Baseline; LS: least square # hsCRP Baseline Mittelwerte sind 8.30, 5.30 bzw. 8.40 † BASDAI Baseline Mittelwerte sind 6.78, 6.57 bzw. 6.59 ˆASDAS-CRP "major improvement" definiert als Verbesserung ≥2 Einheiten
In Woche 16 führte die Behandlung mit Cosentyx 150 mg bei jeder Komponente der ASAS20 Kriterien des Ansprechens (Gesamtbeurteilung durch den Patienten, gesamte Wirbelsäulenschmerzen, BASFI und spinale Entzündung) zu klinisch relevanten Verbesserungen gegenüber der Baseline.
In der AS2-Studie führte die Behandlung mit Cosentyx 150 mg bereits 1 Woche nach Therapiebeginn im Vergleich zu Placebo zu einer signifikanten Verbesserung der ASAS20. Der Anteil der Patienten, die pro Besuch ein ASAS20-Ansprechen zeigten, ist in Abbildung 2 dargestellt.
Abbildung 2: ASAS20-Ansprechen in der AS2-Studie bis Woche 16
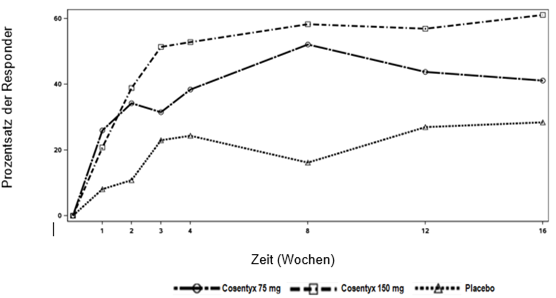
Das ASAS20-Ansprechen war in Woche 16 sowohl bei den bisher nicht mit Anti-TNF-alpha behandelten Patienten (68.2% gegenüber 31.1%; p<0.05) als auch bei den Anti-TNF-alpha-IR-Patienten (50.0% gegenüber 24.1%; p<0.05) bei Cosentyx 150 mg im Vergleich zu Placebo überlegen. Das ASAS20-Ansprechen hinsichtlich des HLA-B27-Status wurde in der AS2-Studie analysiert. 57/72 (79 %) zu 150 mg und 58/74 (78 %) zu Placebo randomisierte Patienten waren HLA B27-positiv. Für 3 Patienten in der 150-mg-Gruppe und 5 Patienten in der Placebo-Gruppe war HLA B27 nicht verfügbar.
Unter den Patienten mit verfügbaren ASAS20 Messwerten betrug das ASAS20 für die 150-mg-Kohorte in Woche 16 37/54 (68.5 %) für HLA-B27-positive und 5/9 (55.6 %) für HLA-B27-negative Patienten. Für Placebo betrug das ASAS20 in Woche 16 18/51 (35.3 %) für HLA-B27-positive und 3/8 (37.5 %) für HLA-B27-negative Patienten.
Die Antwort wurde in beiden Studien unter Cosentyx/- SensoReady bis Woche 52 in einer vergleichbaren Grössenordnung aufrechterhalten. Von den 72 Patienten die in der AS2- Studie für Cosentyx 150 mg randomisiert wurden, waren 61 (84.7%) in Woche 52 immer noch in Behandlung. 45 respektive 35 Patienten wiesen ein ASAS20/40 Ansprechen auf.
Wirbelsäulenbeweglichkeit:
Die Beweglichkeit der Wirbelsäule wurde anhand des BASMI (Bath Ankylosing Spondylitis Metrology Index) bis Woche 52 beurteilt. In der AS2-Studie (150 mg) und der AS1-Studie (75 mg und 150 mg) wurden in jeder der BASMI-Komponenten für die mit Cosentyx behandelten Patienten im Vergleich zu den mit Placebo behandelten Patienten in den Wochen 4, 8, 12 und 16 nummerisch Verbesserungen nachgewiesen (mit Ausnahme der lateralen lumbalen Flexion in Woche 4, 8 und 12 bei Patienten, die nach der intravenösen Anfangsdosis mit einer Dosis von 75 mg behandelt wurden).
Körperliche Funktionsfähigkeit und gesundheitsbezogene Lebensqualität
In der AS1 und AS2 Studie zeigten die mit Cosentyx 150 mg behandelten Patienten eine Verbesserung der gesundheitsbezogenen Lebensqualität, gemessen anhand des ASQoL (Ankylosing Spondylitis Quality of Life questionnaire) (p< 0.001) und der SF-36 Physical Component Summary (SF-36 PCS) (p< 0.001). Mit Cosentyx 150 mg behandelte Patienten zeigten im Vergleich zum Placebo auch signifikante Verbesserungen hinsichtlich der exploratorischen Endpunkte in Bezug auf die physische Funktionsfähigkeit gemessen anhand des BASFI-Index (Bath Ankylosing Spondylitis Functional Index), in Bezug auf Schmerzen gemessen anhand der Skala für globale und nächtliche Rückenschmerzen (Total and Nocturnal Back Pain) und in Bezug auf Erschöpfung gemessen anhand der FACIT-F-Skala (Functional Assessment of Chronic Illness Therapy – Fatigue). All diese Verbesserungen der Körperfunktionen wurden bis Woche 52 aufrechterhalten.
Nicht-röntgenologische Spondyloarthritis (nr-axSpA)
Die Sicherheit und Wirksamkeit von Secukinumab wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie bei 555 Patienten mit aktiver nicht-röntgenologischer axialer Spondyloarthritis (nr-axSpA) untersucht, die die ASAS-(Assessment of Spondylo Arthritis international Society)-Klassifikationskriterien für axiale Spondyloarthritis (axSpA) ohne Röntgennachweis von Veränderungen der Iliosakralgelenke erfüllten, die die modifizierten New-York-Kriterien für Spondylitis ankylosans (AS) erfüllen würden. Die in die Studie aufgenommenen Patienten hatten, eine mittels Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) definierte Krankheitsaktivität von ≥4, einen Schmerzgrad auf der visuellen Analogskala (VAS) für Rückenschmerzen gesamt von ≥40 (auf einer Skala von 0-100 mm), trotz aktueller oder früherer Therapie mit nichtsteroidalen Antirheumatika (NSAID), einen erhöhten C-reaktives Protein (CRP)-Wert und/oder Anzeichen einer Sakroiliitis in der Magnetresonanztomographie (MRT). Bei den Patienten in dieser Studie war die axSpA-Diagnose durchschnittlich vor 2,1 bis 3,0 Jahren gestellt worden und 54 % der Studienteilnehmer waren Frauen.
In der nr-axSpA-Studie 1 waren 9,7 % der Patienten zuvor bereits mit einem TNF-α-Inhibitor behandelt worden, der entweder aufgrund mangelnder Wirksamkeit oder aufgrund Unverträglichkeit abgesetzt wurde (Anti-TNFα-IR-Patienten).
In der nr-axSpA-Studie 1 (PREVENT) wurden 555 Patienten ausgewertet, von denen 9,9 % bzw. 14,8 % gleichzeitig MTX bzw. Sulfasalazin erhielten. In der doppelblinden Phase erhielten die Patienten 52 Wochen lang entweder Placebo oder Secukinumab. Zu Secukinumab randomisierte Patienten erhielten 150 mg subkutan in den Wochen 0, 1, 2, 3 und 4 und anschliessend die gleiche Dosis in monatlichen Abständen oder aber eine einmal monatliche Injektion von 150 mg Secukinumab. Der primäre Endpunkt bestand in einer mindestens 40%igen Verbesserung gemäss der Kriterien der Assessment of Spondyloarthritis International Society (ASAS40-Ansprechen) in Woche 16 bei TNFα-naiven Patienten.
Klinisches Ansprechen
Anzeichen und Symptome:
In der nr-axSpA-Studie 1 führte die Behandlung mit 150 mg Secukinumab in Woche 16 zu einer signifikanteren Verbesserung der Parameter der Krankheitsaktivität im Vergleich zu Placebo. Diese Parameter umfassen ASAS 40-Ansprechen, ASAS 5/6, BASDAI, BASDAI 50, hochsensitives C- CRP (hsCRP), ASAS 20-Ansprechen und ASAS partielle Remission im Vergleich zu Placebo in Woche 16 (Tabelle 12).
Tabelle 12: Klinisches Ansprechen in der nr-axSpA-Studie 1 in Woche 16
Ergebnis (p-Wert gegenüber Placebo) Placebo 150 mg1 Anzahl der randomisierten anti-TNFα-naiven Patienten 171 164 ASAS-40-Ansprechen, % 29,2 % 41,5 %* Gesamtzahl der randomisierten Patienten 186 185 ASAS-40-Ansprechen, % 28,0 % 40,0 %* ASAS 5/6, % 23,7 % 40,0 %** BASDAI, LS mittlere Änderung gegenüber Baseline -1,46 -2,35** BASDAI 50, % 21,0 % 37,3 %** hsCRP, (Post-BSL/BSL-Verhältnis) 0,91 0,64** ASAS-20-Ansprechen, % 45,7 % 56,8 %* ASAS partielle Remission, % 7,0 % 21,6 %** * p<0,05; **p<0.001 gegenüber Placebo Alle p-Werte wurden einer Adjustierung für multiples Testen auf der Grundlage einer vordefinierten Hierarchie unterzogen Bei fehlenden Daten für einen binären Endpunkt erfolgte eine Imputation als Non-Responder. 1 Secukinumab 150 mg s.c. in den Wochen 0, 1, 2, 3 und 4, und anschliessend die gleiche Dosis in monatlichen Abständen ASAS: Assessment of Spondylo Arthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: hochsensitives C-reaktives Protein; BSL: Baseline; LS: Kleinstes Quadrat
In einer explorativen Analyse profitierten Patienten, bei denen zu Studienbeginn sowohl ein abnormaler CRP-Wert als auch Entzündungsanzeichen gemäss MRT vorlagen, am meisten von Secukinumab.
Die Wirkung von Secukinumab 150 mg setzte in der nr-axSpA-Studie 1 bei Anti-TNFα-naiven Patienten hinsichtlich des ASAS-40-Ansprechens bereits in Woche 3 ein (überlegen gegenüber Placebo). Der prozentuale Anteil der Patienten, die unter den Anti-TNFα-naiven Patienten ein ASAS-40-Ansprechen erreichen, ist in Abbildung 3 im zeitlichen Verlauf der Besuchstermine dargestellt. Patienten, die mit Secukinumab behandelt wurden, zeigten im Vergleich zu Placebo bis Woche 52 weiterhin ein Ansprechen.
Abbildung 3: ASAS-40-Ansprechen von Anti-TNFα-naiven Patienten in der nr-axSpA-Studie 1 über den Zeitverlauf bis Woche 16
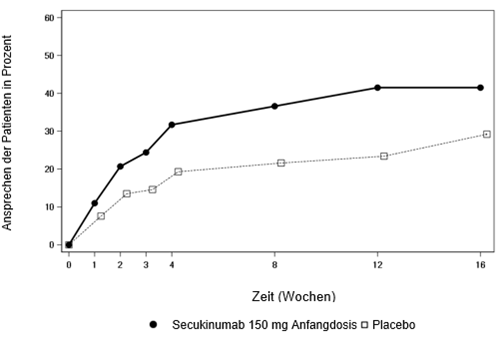
In einer explorativen Analyse der 54 Patienten mit vorhergehender anti-TNF-Behandlung wurde bei 6 von 21 Patienten unter Cosentyx und bei 2 von 15 Patienten unter Placebo bei Woche 16 ein Ansprechen beobachtet.
Körperliche Funktionsfähigkeit und gesundheitsbezogene Lebensqualität:
Mit Secukinumab 150 mg behandelte Patienten zeigten bis Woche 16 statistisch signifikante Verbesserungen im Vergleich zu mit Placebo behandelten Patienten in Bezug auf die körperliche Funktionsfähigkeit, wie mittels BASFI bewertet wurde (Woche 16: -1,75 gegenüber -1,01, p <0,01). Mit Secukinumab behandelte Patienten berichteten im Vergleich zu Placebo-behandelten Patienten in Woche 16 über signifikante Verbesserungen der gesundheitsbezogenen Lebensqualität, die mit den Fragebögen ASQoL (LS-Mittelwertänderung: Woche 16: -3,45 gegenüber -1,84, p <0,001) und SF-36 Summenscore der körperlichen Komponente (SF-36 PCS) (LS-Mittelwertänderung: Woche 16: 5,71 gegenüber 2,93, p <0,001) erfasst wurden. Diese Verbesserungen wurden bis in Woche 52 aufrechterhalten.
Entzündungshemmung in der Magnetresonanztomographie (MRT):
Die Anzeichen einer Entzündung wurden bei Baseline und in Woche 16 mittels MRT beurteilt und als Veränderung des Berlin-SI-Gelenk-Ödem-Scores für Iliosakralgelenke und des ASspiMRI-a-Scores und des Berlin-Wirbelsäulen-Scores für die Wirbelsäule ausgedrückt. Bei mit Secukinumab behandelten Patienten wurde eine Hemmung der Entzündungsanzeichen sowohl im Iliosakralgelenk als auch in der Wirbelsäule beobachtet. Die mittlere Veränderung gegenüber Baseline im Berlin-SI-Gelenk-Ödem-Scores betrug bei Patienten, die mit Secukinumab 150 mg (n = 180) behandelt wurden, -1,68, gegenüber -0,39 bei mit Placebo behandelten Patienten (n = 174) (p <0,0001).
Juvenile idiopathische Arthritis (JIA)
Enthesitis-assoziierte Arthritis (EAA) und juvenile Psoriasis-Arthritis (JPsA)
Die Wirksamkeit und Sicherheit von Secukinumab wurde bei 86 Patienten in einer dreiteiligen, doppelblinden, placebokontrollierten, ereignisgesteuerten, randomisierten Phase-III-Studie an Patienten im Alter von 2 bis < 18 Jahren mit aktiver EAA oder JPsA untersucht, die nach den modifizierten Kriterien für die Einteilung der JIA der International League of Associations for Rheumatology (ILAR) diagnostiziert wurde. Die Studie bestand aus einem offenen Teil (Teil 1), gefolgt von einem randomisierten Entzug (Teil 2) und einer anschliessenden offenen Behandlung (Teil 3). Die Untergruppen der JIA-Patienten bei Studienbeginn waren: 60,5% EAA und 39,5% JPsA.
Bei Baseline hatten 65,1% der Patienten MTX erhalten (63,5% (33 von 52) der EAA-Patienten und 67,6% (23 von 34) der JPsA-Patienten). Die Patienten erhielten eine Dosis von 75 mg, wenn sie < 50 kg wogen, oder 150 mg, wenn sie ≥50 kg wogen. Der primäre Endpunkt war die Zeit bis zum Krankheitsschub in Teil 2. Ein Krankheitsschub wurde definiert als eine ≥30%ige Verschlechterung in mindestens drei der sechs JIA-ACR-Ansprechkriterien und eine ≥30%ige Verbesserung in nicht mehr als einem der sechs JIA-ACR-Ansprechkriterien.
Im offenen Teil 1 der Studie erhielten alle Patienten Secukinumab bis Woche 12. Patienten, die in Woche 12 als Responder eingestuft wurden, traten in die doppelblinde Phase von Teil 2 ein und wurden im Verhältnis 1:1 randomisiert, um die Behandlung mit Secukinumab fortzusetzen oder eine Behandlung mit Placebo zu beginnen. Am Ende von Teil 1 zeigten 75 von 86 Patienten (90,4%) ein JIA-ACR-30-Ansprechen und nahmen an Teil 2 teil.
Die Studie erreichte ihren primären Endpunkt, indem sie eine statistisch signifikante Verlängerung der Zeit bis zum Krankheitsschub bei den mit Secukinumab behandelten Patienten im Vergleich zu Placebo nachwies. Das Risiko eines Krankheitsschubs wurde bei Patienten, die Secukinumab erhielten, im Vergleich zu Patienten, die Placebo erhielten, um 72% reduziert (Hazard Ratio der Krankheitsschübe = 0,28, 95-%-KI: 0,13 bis 0,63, p<0,001) (Abbildung 4). Im zweiten Teil der Studie traten bei insgesamt 21 Patienten in der Placebogruppe Schübe auf (11 JPsA und 10 EAA), verglichen mit 10 Patienten in der Secukinumab-Gruppe (4 JPsA und 6 EAA). Die Überlebensanalyse der Zeit bis zum Auftreten eines Krankheitsschubs zeigte, dass die Secukinumab-Behandlung im Vergleich zu Placebo in TP2 zu einer Verlängerung der Zeit bis zum Auftreten eines Krankheitsschubs führte, unabhängig von der Einnahme von MTX bei Baseline. Das Risiko eines Krankheitsschubs war bei den Probanden unter Secukinumab mit gleichzeitiger MTX-Behandlung um 35% (Hazard Ratio = 0,65, 95% CI: 0,23, 1,83) und bei den Probanden unter Sekukinumab ohne gleichzeitige MTX-Behandlung um 92% (Hazard Ratio = 0,08, 95% CI: 0,02, 0,32) geringer, verglichen mit den Probanden, die in TP2 randomisiert mit Placebo behandelt wurden.
Abbildung 4: Kaplan Meier Schätzungen der Zeit bis zum Krankheitsschub in Teil 2
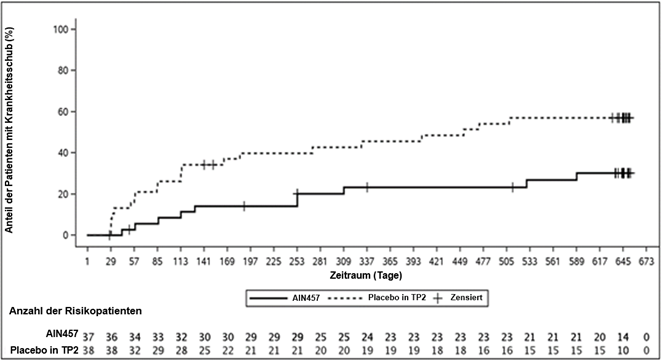
Hidradenitis suppurativa
Die Sicherheit und die Wirksamkeit von Secukinumab wurden bei 1'084 erwachsenen Patienten mit mittelschwerer bis schwerer Hidradenitis suppurativa (HS), die für eine systemische Therapie mit Biologika infrage kamen, in zwei randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien untersucht. Die Patienten, die in die HS-Studie 1 (SUNSHINE) bzw. die HS-Studie 2 (SUNRISE) aufgenommen wurden, wiesen zur Baseline das Hurley-Stadium I (4,6 % bzw. 2,8 %), II (61,4 % bzw. 56,7 %) oder III (34,0 % bzw. 40,5 %) auf und hatten mindestens fünf entzündliche Läsionen, die zwei anatomische Bereiche betrafen. Der Anteil der Patienten mit einem Körpergewicht von ≥90 kg betrug 54,7 % in der HS-Studie 1 und 50,8 % in der HS-Studie 2. Die Patienten in diesen Studien hatten die Diagnose einer mittelschweren bis schweren HS seit durchschnittlich 7,3 Jahren, und 56,3 % der Studienteilnehmer waren weiblich. In der HS-Studie 1 und der HS-Studie 2 wurden 82,3 % bzw. 83,6 % der Patienten zuvor mit systemischen Antibiotika behandelt und 23,8 % bzw. 23,2 % der Patienten wurden zuvor mit einem Biologikum behandelt und hatten dieses entweder wegen mangelnder Wirksamkeit oder wegen Unverträglichkeit abgesetzt (Biologika-exponierte Patienten).
In der HS-Studie 1 wurden 541 Patienten untersucht und in der HS-Studie 2 waren es 543 Patienten; von diesen erhielten 12,8 % bzw. 10,7 % gleichzeitig eine konstante Dosis von Antibiotika. In beiden Studien erhielten die Patienten, die für Secukinumab randomisiert worden waren, 300 mg Secukinumab subkutan in den Wochen 0, 1, 2, 3 und 4, gefolgt von 300 mg alle 2 Wochen (Q2W) oder alle 4 Wochen (Q4W). In Woche 16 wurden die Patienten, die für Placebo randomisiert worden waren, neu zugewiesen und erhielten in den Wochen 16, 17, 18, 19 und 20 jeweils 300 mg Secukinumab, gefolgt von entweder 300 mg Secukinumab Q2W oder 300 mg Secukinumab Q4W.
Der primäre Endpunkt in beiden Studien (HS-Studie 1 und HS-Studie 2) war der Anteil der Patienten, die in Woche 16 ein klinisches Ansprechen der Hidradenitis suppurativa erreichten. Dieses war definiert als eine mindestens 50%ige Abnahme der Gesamtzahl an Abszessen und entzündlichen Knoten und keine Zunahme der Anzahl der Abszesse und/oder der Anzahl der dränierenden Fisteln gegenüber der Baseline (HiSCR50). Der Rückgang der HS-bezogenen Hautschmerzen wurde als sekundärer Endpunkt anhand der gepoolten Daten aus der HS-Studie 1 und der HS-Studie 2 mithilfe einer numerischen Bewertungsskala (NRS) bei den Patienten beurteilt, die zur Baseline einen initialen Score von 3 oder höher aufwiesen.
In der HS-Studie 1 und der HS-Studie 2 erreichte ein signifikant höherer Anteil der Patienten, die mit Secukinumab 300 mg Q2W behandelt wurden, in Woche 16 ein HiSCR50-Ansprechen mit einer signifikanten Abnahme der Anzahl von Abszessen und entzündlichen Knoten (AN) im Vergleich zu Placebo, nicht aber mit Secukinumab 300mg Q4W im Vergleich zu Placebo in HS-Studie 1. In der HS Studie 2 wurde zudem ein statistisch signifikanter Unterschied im HiSCR50-Ansprechen und der Anzahl AN unter dem Secukinumab 300 mg Q4W-Regime im Vergleich zu Placebo beobachtet. In der Secukinumab 300 mg Q4W-Gruppe in der HS-Studie 2 und in der Secukinumab 300 mg Q2W-Gruppe in der HS-Studie 1 traten im Vergleich zu Placebo bis zu Woche 16 weniger Krankheitsschübe auf. Ein signifikant höherer Anteil der Patienten, die mit Secukinumab 300 mg Q2W behandelt wurden (gepoolte Daten), verzeichnete in Woche 16 im Vergleich zu Placebo einen klinisch bedeutsamen Rückgang der HS-bezogenen Hautschmerzen.
Tabelle 13: Klinisches Ansprechen in HS-Studie 1 und HS-Studie 2 in Woche 16¹
HS-Studie 1 HS-Studie 2
Placebo 300 mg Q4W 300 mg Q2W Placebo 300 mg Q4W 300 mg Q2W
Anzahl der randomisi 180 180 181 183 180 180
erten Patienten
HiSCR-50-Ansprechen, 33,7 41,8 45,0* 31,2 46,1* 42,3*
%
Anzahl der AN, % -24,3 -42,4 -46,8* -22,4 -45,5* -39,3*
LS-Mittelwert für
die Veränderung
gegenüber der
Baseline
Schübe, % 29,0 23,2 15,4* 27,0 15,6* 20,1
Gepoolte Daten
(HS-Studie 1 und
HS-Studie 2)
Placebo 300 mg Q4W 300 mg Q2W
Anzahl der Patienten 251 252 266
mit NRS ≥3 zur
Baseline
NRS-30-Ansprechen, % 23.0 33.5 36.6*
¹ Bei fehlenden
Daten wurde eine
Mehrfachimputation
durchgeführt *
Statistisch signifik
ant im Vergleich zu
Placebo auf der
Grundlage der
vordefinierten
Hierarchie mit
einem Gesamt-Alpha
= 0,05 AN: Abszesse
und entzündliche
Knötchen; HiSCR:
Klinisches Anspreche
n der Hidradenitis;
NRS: Numerische
Bewertungsskala
In beiden Studien setzte die Wirkung von Secukinumab bereits in Woche 2 ein, die Wirksamkeit nahm bis Woche 16 zu und hielt bis Woche 52 an.
Verbesserungen wurden bei den primären und wichtigen sekundären Endpunkten bei HS-Patienten unabhängig von einer vorangegangenen oder begleitenden Antibiotikabehandlung beobachtet. Das Ansprechen auf HiSCR50 wurde in Woche 16 sowohl bei Biologika naiven als auch bei Biologika exponierten Patienten verbessert. Grössere Verbesserungen in Woche 16 gegenüber dem Ausgangswert im Vergleich zu Placebo wurden nachgewiesen bei der gesundheitsbezogenen Lebensqualität, gemessen mit dem Dermatology Life Quality Index.
Pharmakokinetik
Absorption
Die Daten in diesem Abschnitt basieren auf einer populationspharmakokinetischen Analyse.
Nach Gabe einer subkutanen Einzeldosis von 150 mg bzw. 300 mg verabreicht als 2 Injektionen zu 150 mg an Patienten mit Plaque-Psoriasis erreichte Secukinumab Spitzenkonzentrationen im Serum von 13.7 ± 4.8 µg/ml bzw. 27.3 ± 9.5 µg/ml zwischen 5 und 6 Tagen nach Dosisgabe.
Nach Einleitung der wöchentlichen Dosierung während des ersten Monats betrug die Zeit bis zum Erreichen der maximalen Konzentration 31 bis 34 Tage.
Die Spitzenkonzentrationen im Steady-State (Cmax,ss) nach subkutaner Verabreichung von 150 mg bzw. 300 mg betrugen 27.6 µg/ml bzw. 55.2 µg/ml. Unter den monatlichen Dosierungsregimes wird der Steady-State nach 20 Wochen erreicht.
Nach wiederholten monatlichen Erhaltungsdosen wiesen Patienten einen im Vergleich zur Exposition nach einer Einzeldosis zweifachen Anstieg der Serum-Spitzenkonzentration auf.
Secukinumab wird nach subkutaner Injektion in den Bauch mit einer durchschnittlichen absoluten Bioverfügbarkeit von 73% absorbiert. Die Absorption von Secukinumab nach subkutaner Injektion in den Oberarm oder Oberschenkel wurde nicht untersucht.
Bei Patienten mit Plaque-Psoriasis war die systemische Exposition mit Secukinumab nach einer einmaligen subkutanen Injektion von 300 mg Injektionslösung in einer Fertigspritze ähnlich wie die zuvor beobachtete Exposition nach zwei Injektionen von 150 mg.
In der HS Studie 1 und in der HS Studie 2 betrug nach subkutaner Verabreichung von 300 mg in den Wochen 0, 1, 2, 3 und 4, die mittlere Talkonzentration im Steady-State (± SD) von Secukinumab in Woche 16 etwa 55,1 ± 26,7 µg/ml bzw. 58,1 ± 30,1 µg/ml für das Secukinumab 300 mg-Regime alle 2 Wochen bzw. 28,8 ± 18,3 µg/ml und 30,2 ± 18,0 µg/ml für das Secukinumab 300 mg-Regime alle 4 Wochen.
Distribution
Das mittlere Verteilungsvolumen während der terminalen Phase (Vz) nach Gabe einer intravenösen Einzeldosis betrug bei Patienten mit Plaque-Psoriasis zwischen 7.10 und 8.60 l. Dies deutet darauf hin, dass Secukinumab einer begrenzten Distribution in die peripheren Kompartimente unterliegt.
Ein und zwei Wochen nach Gabe einer subkutanen Einzeldosis von 300 mg Secukinumab (verabreicht als zwei Injektionen zu 150 mg) betrugen die Secukinumab-Konzentrationen in der interstitiellen Flüssigkeit in der Haut von Patienten mit Plaque-Psoriasis zwischen 28% und 39% der Konzentrationen im Serum.
Metabolismus
Keine Daten.
Elimination
Die mittlere systemische Clearance betrug bei Patienten mit Plaque-Psoriasis 0.19 l/T. Wie bei einem therapeutischen monoklonalen IgG1-Antikörper, der mit einem löslichen Target-Zytokin wie beispielsweise IL-17A interagiert, zu erwarten war, war die Clearance dosis- und zeitunabhängig.
Die geschätzte mittlere Eliminationshalbwertszeit bei Patienten mit Plaque-Psoriasis beträgt 27 Tage. Bei den einzelnen Patienten mit Plaque-Psoriasis lag die geschätzte Halbwertszeit zwischen 17 und 41 Tagen. In einer populationspharmakokinetischen Analyse lag die mittlere systemische Clearance nach subkutaner Verabreichung von 300 mg in den Wochen 0, 1, 2, 3 und 4, gefolgt von 300 mg alle 2 Wochen oder alle 4 Wochen, bei Patienten mit Hidradenitis suppurativa bei 0,26 l/Tag.
Die mittlere Eliminationshalbwertszeit, die anhand einer populationspharmakokinetischen Analyse abgeschätzt wurde, betrug bei Patienten mit Hidradenitis suppurativa 23 Tage.
Linearität/Nicht Linearität
Die Pharmakokinetik (PK) von Secukinumab nach Gabe einer Einzeldosis sowie nach Mehrfachgabe bei Patienten mit Plaque-Psoriasis wurde in mehreren Studien mit intravenösen Dosen zwischen 1 x 0,3 mg/kg und 3 x 10 mg/kg sowie mit subkutanen Dosen von 1 x 25 mg bis zu wiederholten Dosen von 300 mg bestimmt. Die Exposition verlief in allen Dosierungsregimes dosisproportional.
Die PK-Eigenschaften von Secukinumab, die bei Patienten mit Psoriasis-Arthritis und axialer Spondyloarthritis (ankylosierender Spondylitis und nicht-röntgenologischer axialer Spondyloarthritis) beobachtet wurden, waren ähnlich wie die bei Patienten mit Plaque-Psoriasis.
Leberfunktionsstörungen / Nierenfunktionsstörungen
Bei Patienten mit Leber- oder Niereninsuffizienz sind keine pharmakokinetischen Daten verfügbar.
Ältere Patienten
Von den 3'430 Patienten mit Plaque-Psoriasis, die in klinischen Studien gegenüber Secukinumab exponiert waren, waren insgesamt 230 Patienten 65 Jahre oder älter und 32 Patienten 75 Jahre oder älter. Von den 721 Patienten mit Hidradenitis suppurativa, die in klinischen Studien mit Cosentyx behandelt wurden, waren insgesamt 11 Patienten 65 Jahre alt oder älter und 0 Patienten 75 Jahre alt oder älter.
Von den 2'536 Patienten mit Psoriasis-Arthritis, die in klinischen Studien gegenüber Cosentyx exponiert waren, waren insgesamt 236 Patienten 65 Jahre oder älter und 25 Patienten 75 Jahre oder älter.
Von den 571 Patienten mit ankylosierender Spondylitis, die in klinischen Studien gegenüber Cosentyx exponiert waren, waren insgesamt 24 Patienten 65 Jahre oder älter und 3 Patienten 75 Jahre oder älter.
Von den 524 Patienten mit nicht-röntgenologischer axialer Spondyloarthritis, die in klinischen Studien Cosentyx erhalten haben, waren 9 Patienten 65 Jahre alt oder älter und 2 Patienten 75 Jahre alt oder älter.
Basierend auf einer populationspharmakokinetischen Analyse war die Clearance bei älteren Patienten mit der Clearance bei Patienten unter 65 Jahren vergleichbar.
Pädiatrische Patienten
Plaque psoriasis
In einem Pool der beiden pädiatrischen Studien wurde den Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis (im Alter von 6 bis <18 Jahren) Secukinumab im empfohlenen pädiatrischen Dosierungsschema verabreicht. In Woche 24 wiesen Patienten mit einem Körpergewicht von ≥25 und <50 kg eine mittlere ± SD Steady-State-Talkonzentration von 19.8 ± 6.96 μg/ml (n=24) nach 75 mg Secukinumab auf. Patienten mit einem Körpergewicht von ≥50 kg wiesen eine mittlere Steady-State-Talkonzentration von 27.3 ± 10.1 μg/ml (n=36) nach 150 mg Secukinumab auf. Die mittlere ± SD Steady-State-Talkonzentration bei Patienten mit einem Körpergewicht von <25 kg (n=8) betrug 32.6 ± 10.8 μg/ml in Woche 24 nach der 75-mg-Dosis. Die Bioverfügbarkeit von Secukinumab ist bei pädiatrischen Patienten etwa 26 % höher als bei Erwachsenen.
Juvenile idiopathische Arthritis (JIA): Enthesitis-assoziierte Arthritis (EAA) und juvenile Psoriasis-Arthritis (JPsA)
In einer pädiatrischen Studie wurde EAA- und JPsA-Patienten (im Alter von 2 bis unter 18 Jahren) Secukinumab in der empfohlenen pädiatrischen Dosierung verabreicht. In Woche 24 hatten Patienten mit einem Körpergewicht von < 50 kg und Patienten mit einem Körpergewicht ≥50 kg eine mittlere ± SA Steady-State-Talkonzentration von 25,2 ± 5,45 Mikrogramm/ml (n = 10) bzw. 27,9 ± 9,57 Mikrogramm/ml (n = 19).
Präklinische Daten
Präklinische Daten basierend auf Tests zur Gewebekreuzreaktivität, sicherheitspharmakologischen Untersuchungen, Toxizitätsstudien mit wiederholter Dosierung sowie Studien zur Reproduktionstoxizität, die mit Secukinumab oder einem murinen Anti-Maus-IL-17A-Antikörper durchgeführt wurden, erbrachten keine Hinweise auf eine spezielle Gefährdung des Menschen.
Da Secukinumab an IL-17A von Cynomolgus-Affen und Menschen bindet, wurde die Sicherheit von Secukinumab an Cynomolgus-Affen untersucht.
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
Nach subkutaner Gabe von Secukinumab an Cynomolgus-Affen während bis zu 13 Wochen und nach intravenöser Gabe während bis zu 26 Wochen [einschliesslich Beurteilungen der Pharmakokinetik, Pharmakodynamik, Immunogenität und Immuntoxizität (z.B. T-Zell-abhängige Antikörperantwort und NK-Zellaktivität)] wurden keine unerwünschten Wirkungen beobachtet. Die durchschnittlichen Serumkonzentrationen, die bei Affen nach 13 wöchentlichen subkutanen Dosen von 150 mg/kg beobachtet werden, sind 48-fach höher als die vorausgesagten durchschnittlichen Serumkonzentrationen, die bei Patienten mit Psoriasis unter der höchsten klinischen Dosis zu erwarten sind. Der Faktor, um den die Exposition erhöht wird, ist sogar noch grösser, wenn die durchschnittliche Serumkonzentration der 26-wöchigen intravenösen toxikologischen Studie bei Cynomolgus-Affen berücksichtigt wird. Antikörper gegen Secukinumab wurden nur bei einem von 101 Tieren nachgewiesen. Bei der Applikation von Secukinumab auf normale humane Gewebe wurde keine unspezifische Gewebekreuzreaktivität gezeigt.
Mutagenität / Karzinogenität
Es wurden keine präklinischen Studien zur Beurteilung des mutagenen oder karzinogenen Potentials von Secukinumab durchgeführt.
Reproduktionstoxizität
Bei Gabe während der Organogenese und der späten Trächtigkeit zeigte Secukinumab in einer Studie zur embryofötalen Entwicklung bei Cynomolgus-Affen keine maternale Toxizität, Embryotoxizität oder Teratogenität.
In Studien zur Fertilität und frühen embryonalen, prä- und postnatalen Entwicklung bei Mäusen wurden keine unerwünschten Wirkungen eines murinen Anti-Maus-IL-17A-Antikörpers beobachtet. Die in diesen Studien eingesetzte hohe Dosis war höher als die maximal wirksame Dosis in Bezug auf Unterdrückung und Aktivität von IL-17A.
Sonstige Hinweise
Inkompatibilitäten
Pulver zur Herstellung einer Injektionslösung:
Cosentyx darf mit keinen anderen Arzneimitteln oder Lösungsmitteln als sterilem Wasser für Injektionszwecke gemischt werden.
Injektionslösung (Fertigspritze und Fertigpen):
Diese Arzneimittel dürfen nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Arzneimittel sind für Kinder unerreichbar aufzubewahren.
Im Kühlschrank (2-8°C) lagern.
In der Originalpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Nur für die Fertigspritze und den Fertigpen:
Nicht einfrieren.
Falls nötig, kann Cosentyx/ -SensoReady/- UnoReady ungekühlt für einen einmaligen Zeitraum von bis zu 4 Tagen bei Raumtemperatur (nicht über 30°C) gelagert werden. Die Fertigspritze bzw. der Fertigpen müssen nach 4 Tagen entsorgt werden, falls sie ungekühlt aufbewahrt werden.
Für Pulver zur Herstellung einer Injektionslösung:
Die Zubereitung enthält kein Konservierungsmittel. Chemische und physikalische in-use Stabilität wurde für 24 Stunden bei 2-8°C gezeigt. Aus mikrobiologischen Gründen ist die gebrauchsfertige Zubereitung unmittelbar nach Anbruch zu verwenden.
Hinweise für die Handhabung
Das Pulver ist ein weisses festes Lyophilisat. Die Lösung in der Fertigspritze bzw. im Fertigpen ist farblos bis leicht gelblich.
Nach der Injektion darf die in der Durchstechflasche, Fertigspritze bzw. im Fertigpen verbliebene Lösung nicht verwendet werden und sollte entsprechend den lokalen Anforderungen entsorgt werden. Die Durchstechflasche, Fertigspritze bzw. der Fertigpen sind nur für den Einmalgebrauch bestimmt.
Detaillierte Gebrauchsanweisungen für das Pulver zur Herstellung einer Injektionslösung finden Sie untenstehend.
Zulassungsnummer
63295, 65225, 65226, 68324 (Swissmedic)
Packungen
Cosentyx Pulver zur Herstellung einer Injektionslösung in Durchstechflasche zu 150 mg: 1 [B].
Cosentyx Injektionslösung in einer Fertigspritze zu 75 mg: 1 [B].
Cosentyx Injektionslösung in einer Fertigspritze zu 150 mg: 1 und 2 [B].
Cosentyx Injektionslösung in einer Fertigspritze zu 300 mg: 1 [B].
Cosentyx SensoReady Injektionslösung im Fertigpen zu 150 mg: 1 und 2 [B].
Cosentyx UnoReady Injektionslösung im Fertigpen zu 300 mg: 1 [B].
Zulassungsinhaberin
Novartis Pharma Schweiz AG, Risch; Domizil: 6343 Rotkreuz
Stand der Information
Mai 2025
Gebrauchsanweisung für Cosentyx, Pulver zur Herstellung einer Injektionslösung
Die folgenden Angaben sind nur für Ärzte oder Fachpersonen aus dem Gesundheitswesen bestimmt.
Die Durchstechflasche für den einmaligen Gebrauch enthält 150 mg Cosentyx zur Rekonstitution mit 1 ml sterilem Wasser für Injektionszwecke (SWFI). Die Zubereitung der Lösung zur subkutanen Injektion soll ohne Unterbrechung und unter Einhaltung aseptischer Bedingungen erfolgen. Die Zubereitungszeit vom Durchstechen des Stopfens bis zum Ende der Rekonstitution beträgt durchschnittlich 20 Minuten und soll 90 Minuten nicht überschreiten.
Zur Zubereitung von Cosentyx, Pulver zur Herstellung einer Injektionslösung befolgen Sie bitte die folgenden Anweisungen:
Anweisungen zur Rekonstitution von Cosentyx, Pulver zur Herstellung einer Injektionslösung:
1.Bringen Sie die Durchstechflasche mit Cosentyx, Pulver zur Herstellung einer Injektionslösung auf Raumtemperatur und vergewissern Sie sich, dass das sterile Wasser für Injektionszwecke (SWFI) ebenfalls Raumtemperatur hat.
2.Ziehen Sie etwas mehr als 1.0 ml steriles Wasser für Injektionszwecke (SWFI) in eine graduierte 1-ml-Einwegspritze auf und adjustieren Sie das Volumen auf 1.0 ml.
3.Entfernen Sie die Kunststoffkappe von der Durchstechflasche.
4.Stechen Sie die Kanüle der Spritze durch die Mitte des Gummistopfens in die Durchstechflasche mit dem Cosentyx-Lyophilisatkuchen und rekonstituieren Sie den Lyophilisatkuchen, indem Sie 1.0 ml SWFI langsam in die Durchstechflasche injizieren. Der Strom des SWFI ist direkt auf den Lyophilisatkuchen zu richten.
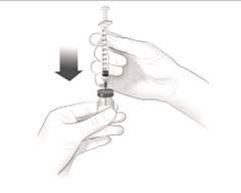
5.Neigen Sie die Durchstechflasche bis zu einem Winkel von etwa 45° und drehen Sie sie vorsichtig etwa 1 Minute lang zwischen den Fingerspitzen. Die Durchstechflasche nicht schütteln oder umdrehen.
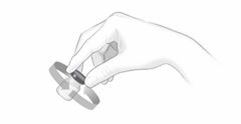
6.Lassen Sie die Durchstechflasche mindestens 10 Minuten lang bei Raumtemperatur stehen, um das Auflösen des Lyophilisats zu ermöglichen. Beachten Sie, dass Schaumbildung der Lösung auftreten kann.
7.Neigen Sie die Durchstechflasche bis zu einem Winkel von etwa 45° und drehen Sie sie vorsichtig etwa 1 Minute lang zwischen den Fingerspitzen. Die Durchstechflasche nicht schütteln oder umdrehen.
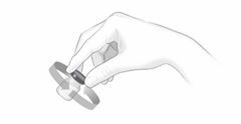
8.Lassen Sie die Durchstechflasche etwa 5 Minuten ungestört bei Raumtemperatur stehen. Die entstandene Lösung sollte klar sein. Die Farbe der Lösung kann zwischen farblos und leicht gelblich variieren. Wenn sich das lyophilisierte Pulver nicht vollständig gelöst hat oder die Flüssigkeit gut sichtbare Partikel enthält, trüb oder deutlich braun ist, darf die Lösung nicht verwendet werden.
9.Bereiten Sie die benötigte Anzahl an Durchstechflaschen (1 Durchstechflasche von 150 mg für die Dosis von 75 mg, 1 Durchstechflasche von 150 mg für die Dosis von 150 mg, 2 Durchstechflaschen von 150 mg für die Dosis von 300 mg) vor.
Nach der Zubereitung soll die Lösung zur subkutanen Injektion sofort verwendet werden, da sie kein Konservierungsmittel enthält. Nicht einfrieren.
Anweisungen zur Verabreichung der Cosentyx-Lösung
1.Neigen Sie die Durchstechflasche bis zu einem Winkel von etwa 45° und positionieren Sie die Nadelspitze an der tiefsten Stelle der Lösung in der Durchstechflasche, wenn Sie die Lösung in die Spritze aufziehen. Die Durchstechflasche NICHT umdrehen.
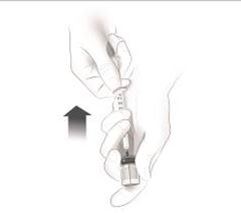
2.Für die Dosis von 150 mg und 300 mg, ziehen Sie mit einer geeigneten Kanüle (z.B. 21G x 2") vorsichtig etwas mehr als 1.0 ml der Lösung zur subkutanen Injektion aus der Durchstechflasche in eine graduierte 1-ml-Einwegspritze auf. Diese Kanüle wird nur zum Aufziehen von Cosentyx in die Einwegspritze verwendet. Bereiten Sie die benötigte Anzahl an Spritzen (1 Spritze für die Dosis von 150 mg, 2 Spritzen für die Dosis von 300 mg) vor. Bei einem Kind, das die 75-mg-Dosis erhält, ziehen Sie vorsichtig etwas mehr als 0.5 ml der Lösung für die subkutane Injektion auf und verwerfen den Rest sofort.
3.Klopfen Sie bei nach oben gerichteter Kanüle leicht gegen die Spritze, um etwaige Luftblasen nach oben zu befördern.
4.Tauschen Sie die aufgesetzte Kanüle gegen eine 27G x ½" Kanüle aus.

5.Drücken Sie die Luftblasen aus der Spritze heraus. Für die 150 mg Dosis schieben Sie den Kolben bis zur 1.0-ml-Markierung vor. Für die 75 mg Dosis schieben Sie den Kolben bis zur 0.5-ml-Markierung vor.
6.Reinigen Sie die Injektionsstelle mit einem Alkoholtupfer.
7.Die empfohlene Stelle für subkutane Injektion der Cosentyx-Lösung ist der Unterbauch (jedoch nicht der Bereich von 5 cm rund um den Nabel).
Alternativ können auch die Vorderseite der Oberschenkel oder die Aussenseiten der Oberarme verwendet werden, falls die empfohlene Injektionsstelle nicht geeignet ist (vgl. Rubrik "Pharmakokinetik" ).
Wählen Sie für jede Injektion eine andere Injektionsstelle. Injizieren Sie die Lösung nicht in Bereiche mit empfindlicher, verletzter, geröteter, schuppender oder verhärteter Haut. Vermeiden Sie Bereiche mit Narben oder Dehnungsstreifen.
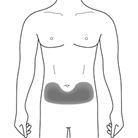
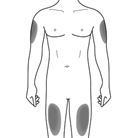
8.In der Durchstechflasche verbliebene Lösung darf nicht verwendet werden und sollte entsprechend den lokalen Anforderungen entsorgt werden. Die Durchstechflaschen sind nur für den Einmalgebrauch bestimmt. Werfen Sie die gebrauchte Spritze in einen Spritzenbehälter. Kanülen und gebrauchte Spritzen dürfen nicht wiederverwendet werden.