Zusammensetzung
Wirkstoffe
Brolucizumab ist ein humanisiertes monoklonales einkettiges Fv(scFv)-Antikörperfragment mit einem Molekulargewicht von ca. 26 kDa, das in Zellen von Escherichia coli durch rekombinante DNA-Technologie hergestellt wird.
Hilfsstoffe
2.58 mg/ml Natriumcitrat, 58 mg/ml Saccharose, 0.2 mg/ml Polysorbat 80, Natriumhydroxid (zur Einstellung des pH-Werts auf ca. 7,2) und Wasser für Injektionszwecke.
Indikationen/Anwendungsmöglichkeiten
Beovu ist indiziert für die Behandlung der neovaskulären (feuchten) altersbedingten Makuladegeneration (AMD).
Dosierung/Anwendung
Durchstechflasche zum einmaligen Gebrauch oder Fertigspritze zum einmaligen Gebrauch. Nur zur intravitrealen Anwendung. Jede Durchstechflasche oder Fertigspritze darf nur für die Behandlung eines einzelnen Auges verwendet werden.
Beovu muss von einem qualifizierten Arzt verabreicht werden.
Übliche Dosierung
Die empfohlene Dosis für Beovu beträgt 6 mg (0,05 ml), verabreicht als intravitreale Injektion, wobei die ersten drei Injektionen im Abstand von 4 Wochen (monatlich) erfolgen. Danach wird Beovu alle 12 Wochen (3 Monate) verabreicht. Das Behandlungsintervall kann auf alle 8 Wochen (2 Monate) angepasst werden (siehe «Eigenschaften/Wirkungen»). Der Arzt kann die Behandlungsintervalle je nach Krankheitsaktivität individuell festlegen, gemessen an der Sehschärfe bzw. Anatomische Parameter.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Es wurden keine Studien bei Patienten mit eingeschränkter Leberfunktion durchgeführt.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit eingeschränkter Nierenfunktion wird keine Dosisanpassung empfohlen. Es liegen nur limitierte Daten zu Patienten mit moderat eingeschränkter Nierenfunktion und keine Daten zu Patienten mit stark eingeschränkter Nierenfunktion vor (siehe «Eigenschaften/Wirkungen»).
Ältere Patienten
Bei Patienten im Alter von 65 Jahren und älter ist keine Dosisanpassung erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Beovu bei Kindern und Jugendlichen ist nicht erwiesen.
Art der Anwendung
Wie alle Arzneimittel zur intravitrealen Anwendung sollte Beovu vor der Verabreichung einer Sichtkontrolle unterzogen werden (siehe «Hinweise für die Handhabung»).
Die intravitreale Injektion muss unter aseptischen Bedingungen durchgeführt werden. Dies beinhaltet eine chirurgische Handdesinfektion, sterile Operationshandschuhe, ein steriles Abdecktuch sowie ein steriles Lidspekulum (oder ein vergleichbares Instrument). Instrumente zur Durchführung einer sterilen Parazentese sollten als Vorsichtsmassnahme vorhanden sein. Vor der intravitrealen Injektion sollte eine gründliche Anamnese hinsichtlich möglicher Überempfindlichkeitsreaktionen erhoben werden (siehe «Kontraindikationen»). Vor der Injektion sind eine adäquate Anästhesie und die Desinfektion der periokularen Haut, des Augenlids und der Augenoberfläche mittels eines topischen Breitspektrum-Antiseptikums durchzuführen.
Für Informationen zur Vorbereitung von Beovu siehe Hinweise für Handhabung (siehe «Sonstige Hinweise»).
Die Injektionsnadel sollte 3,5 bis 4,0 mm posterior zum Limbus in den Glaskörper eingebracht werden. Dabei sollte der horizontale Meridian vermieden und in Richtung Bulbusmitte gezielt werden. Danach kann das Injektionsvolumen von 0,05 ml langsam injiziert werden. Die nachfolgenden Injektionen müssen an unterschiedlichen Stellen der Sklera erfolgen.
Die Sicherheit und Wirksamkeit der Behandlung mit Beovu an beiden Augen gleichzeitig wurde nicht untersucht.
Kontraindikationen
·Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
·Bestehende okulare oder periokulare Infektion bzw. Verdacht darauf.
·Bestehende intraokulare Entzündung.
Warnhinweise und Vorsichtsmassnahmen
Mit der intravitrealen Injektion in Zusammenhang stehende Reaktionen
Intravitreale Injektionen, einschliesslich jener von Beovu, werden mit Endophthalmitis, intraokulare Entzündung und Netzhautablösung in Verbindung gebracht (siehe «Unerwünschte Wirkungen»). Beovu muss immer unter aseptischen Injektionsbedingungen verabreicht werden. Die Patienten sollten angewiesen werden, mögliche Symptome eines der oben aufgeführten Ereignisse unverzüglich zu melden.
Eine vorübergehende Zunahme des intraokularen Drucks wurde innerhalb der ersten 30 Minuten nach der Injektion beobachtet, ähnlich wie bei der intravitrealen Verabreichung anderer VEGF-Inhibitoren (siehe «Unerwünschte Wirkungen»). Anhaltender erhöhter intraokularer Druck wurde ebenfalls berichtet. Sowohl der intraokulare Druck als auch die Perfusion der Durchtrittsstelle des Nervus opticus müssen kontrolliert und bei Bedarf behandelt werden.
Arterielle thromboembolische Ereignisse
Es besteht ein potentielles Risiko für arterielle thromboembolische Ereignisse bei der intravitrealen Applikation von VEGF-Inhibitoren. Bei Patienten mit einem bekannten Risiko für Schlaganfälle oder Myocardinfarkt ist das Risiko möglicherweise erhöht.
Interaktionen
Es wurden keine formalen Studien zur Erfassung von Interaktionen durchgeführt.
Schwangerschaft/Stillzeit
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während der Behandlung mit Beovu und bis mindestens einen Monat nach Beendigung der Behandlung mit Beovu eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Es liegen keine hinreichenden und gut kontrollierten Studien zur Verabreichung von Beovu an schwangere Frauen vor. Reproduktionsstudien an Tieren wurden nicht durchgeführt. Das potenzielle Risiko der Verwendung von Beovu während der Schwangerschaft ist unbekannt. Aufgrund des anti-VEGF-Wirkmechanismus muss Brolucizumab jedoch als potenziell teratogen und embryo-/fetotoxisch eingestuft werden. Somit darf Beovu während der Schwangerschaft nicht verabreicht werden, es sei denn es ist unbedingt notwendig.
Stillzeit
Es ist nicht bekannt, ob Brolucizumab nach Verabreichung von Beovu in die Muttermilch übergeht. Es liegen keine Daten über die Wirkungen von Beovu auf das gestillte Kind oder die Milchproduktion vor. Aufgrund des Potenzials für unerwünschte Arzneimittelwirkungen bei gestillten Kindern wird das Stillen während der Behandlung und bis mindestens einen Monat nach Beendigung der Behandlung mit Beovu nicht empfohlen.
Fertilität
Es liegen keine entsprechenden Daten vor.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Nach der intravitrealen Injektion von Beovu und der damit verbundenen Augenuntersuchung kann die Sehfähigkeit der Patienten vorübergehend beeinträchtigt sein. Die Patienten müssen daher angewiesen werden, erst dann wieder am Verkehr teilzunehmen oder Maschinen zu bedienen, wenn die Sehfunktion ausreichend wieder hergestellt ist.
Unerwünschte Wirkungen
Insgesamt 1'088 mit Beovu behandelte Patienten bildeten die Sicherheitspopulation in den beiden Phase-III-Studien HAWK und HARRIER. Die kumulative Exposition gegenüber Beovu betrug 96 Wochen und 730 Patienten wurden mit der empfohlenen Dosis von 6 mg behandelt.
Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (bei über 5% der mit Beovu 6 mg behandelten Patienten) waren verminderte Sehschärfe (7,3%), Katarakt (7,0%), Bindehautblutung (6,3%) und Mouches volantes (5,1%).
Seltener auftretende schwerwiegende unerwünschte Arzneimittelwirkungen, die bei weniger als 1% der mit Beovu 6 mg behandelten Patienten berichtet wurden, waren Endophthalmitis, Erblindung, Verschluss einer Netzhautarterie und Netzhautablösung.
Unerwünschte Arzneimittelwirkungen aus klinischen Studien sind nach Häufigkeit aufgelistet, wobei die häufigsten unerwünschten Arzneimittelwirkungen zuerst genannt werden. Darüber hinaus basiert die jeweilige Häufigkeitskategorie für jede unerwünschte Arzneimittelwirkung auf der folgenden Konvention (CIOMS III): sehr häufig (≥1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1'000 bis <1/<100), selten (≥1/10'000 bis <1/1'000), sehr selten (<1/10'000).
Erkrankungen des Immunsystems
Häufig: Überempfindlichkeita
Augenerkrankungen
Häufig: Sehschärfe vermindert, Katarakt, Bindehautblutung, Mouches volantes, Augenschmerzen, Netzhautblutung, Glaskörperablösung, Intraokulare Drucksteigerung, Konjunktivitis, Einriss des retinalen Epithelpigments, verschwommenes Sehen, Uveitis, Hornhautabschürfungen, Keratitis punctata, Iritis, Netzhauteinriss
Gelegentlich: Bindehauthyperämie, Tränensekretion verstärkt, Erblindung, Verschluss einer Netzhautarterie, anomale Sinnesempfindungen des Auges, Endophthalmitis, Netzhautablösung, Abhebung des retinalen Pigmentepithels, Vitritis, Entzündung der vorderen Augenkammer, Iridozyklitis, Vorderkammerflackern, Hornhautödem, Glaskörperblutung
a Einschliesslich Urtikaria, Ausschlag, Pruritus, Erythem.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht auch bei Patienten, die mit Beovu behandelt, werden, potenziell die Gefahr, dass eine Immunantwort ausgelöst wird. Die Immunogenität von Beovu wurde in Serumproben untersucht. Die Immunogenitätsdaten geben den Prozentsatz der Patienten wieder, deren Testergebnisse in Immunoassays als positiv für Antikörper gegen Beovu angesehen wurden. Der Nachweis einer Immunantwort ist stark von der Sensitivität und Spezifität der verwendeten Assays, dem Handling der Proben, dem Zeitpunkt der Probenentnahme, den Begleitmedikamenten und der Grunderkrankung abhängig. Aus diesen Gründen kann der Vergleich der Inzidenz der Antikörper gegen Beovu mit der Inzidenz der Antikörper gegen andere Produkte irreführend sein. Bei therapienaiven Patienten wurden bereits vor Behandlungsbeginn Antikörper, darunter auch Einzelkettenantikörper, gegen eine Vielzahl von biotechnologisch hergestellten therapeutischen Proteinen nachgewiesen.
Die Inzidenz von Antikörpern gegen Brolucizumab vor Behandlungsbeginn betrug 35 – 52%. Nach der Verabreichung von Beovu wurden bei 23 – 25% der Patienten über 88 Wochen behandlungsbedingte Antikörper gegen Brolucizumab nachgewiesen.
Das Auftreten von Antikörpern gegen Brolucizumab war nicht mit einer Beeinflussung der klinischen Wirksamkeit verbunden. Bei Patienten mit behandlungsbedingten Antikörpern wurde eine höhere Anzahl von intraokularen Entzündungsereignissen beobachtet. Die klinische Bedeutung von Antikörpern gegen Brolucizumab für die Sicherheit ist derzeit noch unklar.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Eine Überdosierung mit mehr als dem empfohlenen Injektionsvolumen kann zu einer Erhöhung des Augeninnendrucks führen. Daher sollte im Falle einer Überdosierung der Augeninnendruck überwacht und je nach Einschätzung durch den behandelnden Arzt gegebenenfalls behandelt werden.
Eigenschaften/Wirkungen
ATC-Code
S01LA06
Wirkungsmechanismus
Die erhöhte Signalgebung über den VEGF-A-Pfad (vaskulärer endothelialer Wachstumsfaktor A) ist mit einer pathologischen okularen Angiogenese und einem Netzhautödem verbunden. Brolucizumab bindet mit hoher Affinität an VEGF-A-Isoformen (z.B. VEGF110, VEGF121 und VEGF165) und verhindert so, dass VEGF-A an seine Rezeptoren VEGFR-1 und VEGFR-2 bindet. Durch Inhibierung der Bindung an VEGF-A unterdrückt Brolucizumab die Endothelzell-Proliferation, wodurch die pathologische Neovaskularisierung reduziert und die Gefässpermeabilität verringert werden.
Pharmakodynamik
Die neovaskuläre (feuchte) altersbedingte Makuladegeneration (AMD) ist durch eine pathologische choroidale Neovaskularisation (CNV) gekennzeichnet. Die Leckage von Blut und Flüssigkeit infolge von CNV kann zu einer Verdickung oder einem Ödem der Netzhaut bzw. sub-/intraretinalen Blutungen führen, was den Verlust der Sehschärfe verursacht.
In den Studien HAWK und HARRIER waren verwandte anatomische Parameter Teil der Beurteilungen der Krankheitsaktivität, die als Grundlage für Behandlungsentscheidungen dienten. Eine Reduktion der zentralen retinalen Netzhautdicke (central subfield thickness, CST) sowie des Vorhandenseins intraretinaler/subretinaler Flüssigkeit (IRF/SRF) oder subretinaler Pigmentepithel-(sub-RPE)-Flüssigkeit wurde bereits 4 Wochen nach Behandlungsbeginn und bis zu Woche 48 und Woche 96 bei Patienten beobachtet, die mit Beovu behandelt wurden.
In diesen Studien wurde bei mit Beovu behandelten Patienten bereits 12 Wochen nach Behandlungsbeginn sowie in Wochen 48 und 96 nach Behandlungsbeginn eine Reduktion der CNV-Läsionsgrösse beobachtet.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Beovu wurde in zwei randomisierten, multizentrischen, doppelblinden, aktivkontrollierten Phase-III-Studien (HAWK und HARRIER) bei Patienten mit neovaskulärer AMD untersucht. Total wurden 1'817 Patienten im Rahmen dieser Studien zwei Jahre lang behandelt (1'088 mit Beovu und 729 mit Aflibercept). Das Alter der Patienten lag zwischen 50 und 97 Jahren, mit einem Mittelwert von 76 Jahren.
In der HAWK-Studie wurden die Patienten im Verhältnis 1:1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
1.Beovu 3 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
2.Beovu 6 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
3.Aflibercept 2 mg, verabreicht alle 8 Wochen («q8w») nach den ersten 3 monatlichen Dosen.
In der HARRIER-Studie wurden die Patienten im Verhältnis 1:1 randomisiert und einem der folgenden Dosierungsschemata zugewiesen:
1.Beovu 6 mg, verabreicht alle 12 oder 8 Wochen («q12w/q8w») nach den ersten 3 monatlichen Dosen.
2.Aflibercept 2 mg, verabreicht alle 8 Wochen («q8w») nach den ersten 3 monatlichen Dosen.
In beiden Studien wurden Brolucizumab-Patienten im Anschluss an die ersten 3 monatlichen Dosen (Wochen 0, 4 und 8) alle 12 Wochen behandelt, mit der Option, basierend auf der Krankheitsaktivität auf ein 8-wöchiges Behandlungsintervall umzustellen. Die Krankheitsaktivität wurde von einem Arzt im ersten 12-Wochen-Intervall (in Wochen 16 und 20) und bei jeder nachfolgend geplanten 12-Wochen-Behandlungsvisite beurteilt. Patienten, die bei einem dieser Besuche Krankheitsaktivität zeigten (z.B. verminderte Sehschärfe, erhöhte zentrale retinale Netzhautdicke (CST) oder Vorhandensein retinaler Flüssigkeiten (IRF/SRF, Sub-RPE), wurden auf ein 8-wöchiges Behandlungsintervall umgestellt.
Ergebnisse
Der primäre Wirksamkeitsendpunkt der Studien war die Veränderung der bestmöglich korrigierten Sehschärfe (BCVA) gegenüber Baseline in Woche 48, gemessen mithilfe der ETDRS-Buchstabentafeln, mit dem primären Ziel, die Nichtunterlegenheit von Beovu gegenüber Aflibercept nachzuweisen. In beiden Studien wurde die Nichtunterlegenheit in Bezug auf die Wirksamkeit von Beovu (verabreicht nach einem 12-/8-wöchigen Behandlungsschema) gegenüber Aflibercept 2 mg, verabreicht alle 8 Wochen, nachgewiesen.
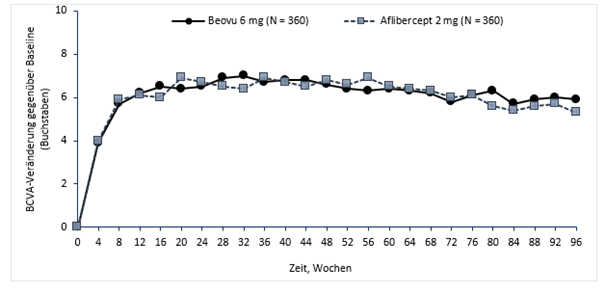
In der Studie HAWK erreichten die Patienten in Woche 48 in der Beovu-6 mg- und Aflibercept-Gruppe eine mittlere Veränderung gegenüber Baseline von +6,6 Buchstaben bzw. +6,8 Buchstaben (p <0,0001). Die mittlere Veränderung gegenüber der Baseline in der Beovu-3-mg-Gruppe lag bei +6,1 Buchstaben (p = 0,0003). Der Anteil der Patienten, die einen Zugewinn an Sehschärfe von mindestens 15 Buchstaben gegenüber Baseline erreichten, betrug 33,6% in der Brolucizumab-Gruppe gegenüber 25,4% in der Aflibercept-Gruppe. Der Anteil der Patienten, die 15 Buchstaben oder mehr Sehschärfe gegenüber dem Ausgangswert verloren, betrug 6,4% in der 6 mg Brolucizumab-Gruppe gegenüber 5,5% in der Aflibercept-Gruppe.
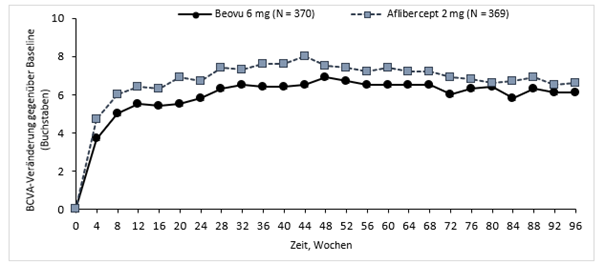
In der Studie HARRIER erreichten die Patienten in Woche 48 in der Beovu- und Aflibercept-Gruppe eine mittlere Veränderung gegenüber Baseline von +6,9 Buchstaben bzw. +7,6 Buchstaben (p <0,0001). Der Anteil der Patienten, die einen Zugewinn an Sehschärfe von mindestens 15 Buchstaben gegenüber Baseline erreichten, lag in der Brolucizumab-Gruppe bei 29,3% gegenüber 29,9% in der Aflibercept-Gruppe. Der Anteil der Patienten, die gegenüber dem Ausgangswert 15 Buchstaben oder mehr Sehschärfe verloren, betrug 3,8% in der 6 mg Brolucizumab-Gruppe gegenüber 4,8% in der Aflibercept-Gruppe
Der im ersten Jahr beobachtete Zugewinn an Sehschärfe wurde im zweiten Jahr beibehalten.
Abbildung 0-1: Mittlere Veränderung der Sehschärfe gegenüber Baseline bis Woche 96 in den Studien HAWK und HARRIER
HAWK
HARRIER
In den Studien HAWK und HARRIER erreichten 56% bzw. 51% der Patienten, die mit Beovu 6 mg in einem 12-wöchigen Behandlungsintervall behandelt wurden, in Woche 48 diesen Zugewinn an Sehschärfe (mittlere Veränderung im Vergleich zum Ausgangswert), und 45% bzw. 39% der Patienten in Woche 96.
Von den Patienten, die während des ersten 12-wöchigen Behandlungsintervalls als geeignet für dieses Behandlungsintervall identifiziert worden waren, wurde bei 85% bzw. 82% das 12-wöchige Behandlungsintervall bis Woche 48 beibehalten. Bei 82% bzw. 75% der Patienten, die in Woche 48 mit dem 12-wöchigen Behandlungsintervall behandelt worden waren, wurde das 12-wöchige Behandlungsintervall von Woche 48 bis Woche 96 beibehalten.
Die Behandlungseffekte stimmten in den auswertbaren Untergruppen (z.B. Alter, Geschlecht, ethnische Zugehörigkeit, Sehschärfe bei Baseline, Netzhautdicke bei Baseline, Läsionstyp, Läsionsgrösse, Flüssigkeitsstatus) in beiden Studien weitgehend mit den Ergebnissen in der Gesamtpopulation überein.
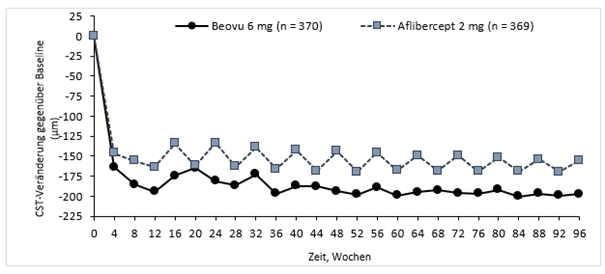
Die Krankheitsaktivität wurde anhand von Veränderungen der Sehschärfe bzw. morphologischen Kriterien beurteilt, einschliesslich der zentralen retinalen Netzhautdicke (CST) und des Vorhandenseins retinaler Flüssigkeiten (IRF/SRF, Sub-RPE). In Woche 16, als die Krankheitsaktivität erstmalig zur Bestimmung des Behandlungsintervalls bewertet wurde, zeigten statistisch weniger mit Beovu 6 mg behandelte Patienten eine Krankheitsaktivität als mit Aflibercept 2 mg behandelte Patienten (24% vs 35% in HAWK, p=0,0013; 23% vs 32% in HARRIER, p=0,0021). Die Krankheitsaktivität wurde jeweils über den Verlauf der gesamten Studien bewertet. Die morphologischen Kriterien der Krankheitsaktivität waren in Woche 48 und in Woche 96 in der Beovu-Gruppe im Vergleich zu Aflibercept geringer (Tabelle 0-2).
Tabelle 0-1: Bewertung der Krankheitsaktivität in den Studien HAWK und HARRIER bis Woche 96
|
|
|
HAWK |
|
HARRIER |
| ||
|
Wirksamkeitsergebnis (präspezifizierte sekundäre Endpunkte) |
In Woche |
Beovu 6 mg |
Aflibercept |
Unterschied (95%-KI) |
Beovu 6 mg |
Aflibercept |
Unterschied (95%-KI) |
|
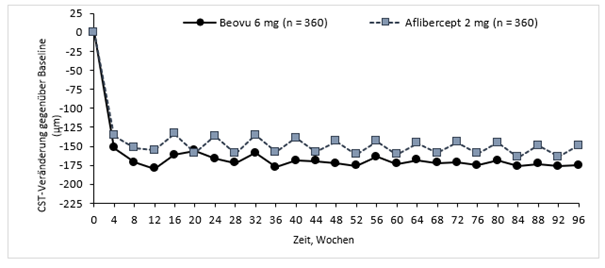
Mittlere CST-Veränderung gegenüber Baseline (µm) |
16 c) |
-161,4 |
-133,6 |
-27,8 |
-174,4 |
-134,2 |
-40,2 |
|
48 |
-172,8 |
-143,7 |
-29,0 |
-193,8 |
-143,9 |
-49,9 | |
|
96 |
-174,8 |
-148,7 |
-26,0 |
-197,7 |
-155,1 |
-42,6 | |
CST: zentrale retinale Netzhautdicke, IRF/SRF: intraretinale/subretinale Flüssigkeit, RPE: retinales Pigmentepithel
a) Sekundärer Endpunkt in HARRIER, bestätigende Analyse in HAWK. Einseitige p-Werte für die Überlegenheit von Brolucizumab
b) Sekundärer Entpunkt in HAWK und HARRIER; zweiseitige p-Werte
c) Bis Woche 16 war die Behandlungsexposition identisch, was einen abgestimmten Vergleich von Beovu mit Aflibercept ermöglichte.
Abbildung 0-2: Veränderung der zentralen retinalen Netzhautdicke ab Baseline bis Woche 96 in den Studien HAWK und HARRIER
HAWK
HARRIER
In beiden Studien führte die Behandlung mit Beovu zu klinisch relevanten Veränderungen gegenüber Baseline in Bezug auf den präspezifizierten sekundären Wirksamkeitsendpunkt «von Patienten berichtete Ergebnisse», die mithilfe des Fragebogens zur Augengesundheit des nationalen Augeninstituts der USA, NEI VFQ-25, aufgezeichnet wurden. Die Grössenordnung dieser Veränderungen war vergleichbar mit veröffentlichten Studien und entsprach einem Zugewinn von 15 Buchstaben bei der bestmöglich korrigierten Sehschärfe (BCVA). Der Nutzen der von den Patienten berichteten Ergebnisse wurde im zweiten Jahr aufrechterhalten.
Es wurden keine klinisch relevanten Unterschiede zwischen Beovu und Aflibercept hinsichtlich der Veränderungen des NEI VFQ-25-Gesamtscores und der Subskalen zwischen Baseline und Woche 48 festgestellt (generelle Sehkraft, Augenschmerzen, Nahsicht, Fernsicht, soziale Funktionsfähigkeit, psychisches Befinden, Schwierigkeiten bei der Ausübung sozialer Rollen, Abhängigkeit von anderen, Autofahren, Farbsehen und peripheres Sehen).
Pharmakokinetik
Absorption
Beovu wird direkt in den Glaskörper verabreicht, um lokale Effekte im Auge zu erzielen.
Distribution
Nach intravitrealer Verabreichung von 6 mg Brolucizumab pro Auge an nAMD-Patienten betrug die mittlere Cmax von freiem Brolucizumab im Serum 49,0 ng/ml (Spanne: 8,97 bis 548 ng/ml) und wurde innert eines Tages erreicht. Die mittlere AUC betrug 6000 h*ng/ml (Spanne: 1420 – 60400 h*ng/ml).
Metabolismus
Brolucizumab ist ein monoklonales Antikörperfragment und es wurden keine Studien zum Wirkstoffmetabolismus durchgeführt. Da Brolucizumab ein einkettiges Antikörperfragment ist, wird erwartet, dass freies Brolucizumab sowohl durch zielgerichtete Disposition über Bindung an das freie endogene VEGF, als auch durch passive renale Elimination und Verstoffwechselung durch Proteolyse eliminiert wird.
Elimination
Nach intravitrealer Injektion wurde Brolucizumab mit einer scheinbaren systemischen Halbwertszeit von 4,4 Tagen eliminiert. Es kam zu keiner Akkumulierung von Beovu im Serum, wenn dieses alle 4 Wochen intravitreal verabreicht wurde.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die Pharmakokinetik bei Patienten mit eingeschränkter Leberfunktion wurde nicht untersucht.
Nierenfunktionsstörungen
Die systemische Pharmakokinetik von Brolucizumab wurde bei nAMD-Patienten ausgewertet, von denen sowohl pharmakokinetische Daten zu Brolucizumab im Serum als auch Daten zur Kreatinin-Clearance von Brolucizumab verfügbar waren. Das Verhältnis des geometrischen Mittels (90% KI) bei Patienten mit milder (60 bis <90 ml/min (n=22)) bzw. moderater (30 to <60 ml/min (n=3)) Nierenfunktionsstörung im Vergleich zu Patienten mit normaler Nierenfunktion ist für Brolucizumab Cmax 1,4 (0,7; 2,9) bzw. 1,7 (1,0; 2,8) und das Verhältnis für AUCinf 1,4 (0,7; 2,9) bzw. 1.0 (0.5, 2.0). Es wurden keine Patienten mit schwerer (<30 ml/min) Nierenfunktionsstörung untersucht.
Ältere Patienten
Limitierte Daten zur Pharmakokinetik von Brolucizumab bei älteren Patienten erlauben keine Rückschlüsse bezüglich eines Alterseffekts auf die Pharmakokinetik von Brolucizumab.
Genetische Polymorphismen
Ethnische Gruppen
In einer Studie mit 24 kaukasischen und 26 japanischen Patienten wurden nach intravitrealer Injektion keine ethnischen Unterschiede hinsichtlich der systemischen pharmakokinetischen Eigenschaften beobachtet.
Präklinische Daten
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
Die intravitreale Injektion von Brolucizumab in Dosisstärken von bis zu 6 mg/Auge alle 4 Wochen über einen Zeitraum von 26 Wochen führte bei Cynomolgus-Affen zu keiner okulären oder systemischen Wirkung und war gut verträglich.
Mutagenität/Karzinogenität
Es wurden keine Studien zur Abklärung des mutagenen oder karzinogenen Potenzials von Beovu durchgeführt.
Reproduktionstoxizität
Es wurden keine Reproduktions- oder Fertilitätsstudien durchgeführt. Es wurde nachgewiesen, dass die VEGF-Inhibition die Follikelreifung, die Gelbkörperfunktion und die Fruchtbarkeit beeinflusst. Basierend auf dem Wirkmechanismus von VEGF-Inhibitoren besteht ein potenzielles Risiko für die weibliche Fortpflanzung und die embryo-fetale Entwicklung.
Sonstige Hinweise
Inkompatibilitäten
Da keine Verträglichkeitssstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Durchstechflasche resp. Fertigspritze mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Durchstechflasche: Im Originalkarton aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank (2–8 °C) lagern. Nicht einfrieren.
Die ungeöffnete Durchstechflasche kann vor der Anwendung bis zu 24 Stunden bei Raumtemperatur (25 °C) aufbewahrt werden.
Fertigspritze: In der versiegelten Blisterpackung in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank (2–8 °C) lagern. Nicht einfrieren.
Die ungeöffnete Blisterpackung kann vor der Anwendung bis zu 24 Stunden bei Raumtemperatur (25 °C) aufbewahrt werden.
Für weitere Informationen sehen Sie bitte «Hinweise für die Handhabung».
Zulassungsnummer
67245, 67244 (Swissmedic).
Packungen
1 Durchstechflasche zu 0.23 ml inklusive 1 Filternadel. [B]
1 Fertigspritze zu 0.165 ml. [B]
Zulassungsinhaberin
Novartis Pharma Schweiz AG, Risch; Domizil 6343 Rotkreuz.
Stand der Information
Januar 2020
Hinweise für die Handhabung
Zur Vorbereitung von Beovu für die intravitreale Anwendung folgen Sie bitte der Gebrauchsanweisung:
|
1 |
|
Ziehen Sie die Folie der Blisterpackung ab und nehmen Sie die Spritze unter aseptischen Bedingungen heraus. | |
|
2 |
|
|
Brechen Sie die Spritzenkappe ab (nicht drehen oder abschrauben). |
|
3 |
|
Befestigen Sie eine 30 G x ½″ Injektionsnadel unter aseptischen Bedingungen fest an der Spritze. | |
|
4 |
|
|
Um den Inhalt auf Luftblasen zu überprüfen, halten Sie die Spritze so, dass die Injektionsnadel nach oben zeigt. Sollten Luftblasen vorhanden sein, klopfen Sie vorsichtig mit dem Finger gegen die Spritze, bis die Luftblasen nach oben steigen. |
|
5 |
|
|
Halten Sie die Spritze auf Augenhöhe und drücken Sie den Kolben vorsichtig, bis sich der Rand unterhalb der Kuppel des Gummistopfens auf einer Linie mit der 0,05 ml-Dosierungsmarkierung befindet. Die Spritze ist nun bereit für die Injektion. |
|
6 |
|
Injizieren Sie die Lösung langsam, bis der Gummistopfen das Ende der Spritzenzylinders erreicht, damit das ganze Volumen von 0,05 ml verabreicht wird. Kontrollieren Sie, dass die ganze Dosis injiziert wurde, indem Sie überprüfen, ob der Gummistopfen das Ende des Spritzenzylinders erreicht hat. |