ZusammensetzungWirkstoffe
Donanemab (gentechnologisch hergestellt unter Verwendung von Ovarialzellen des chinesischen Hamsters (CHO)).
Hilfsstoffe
Citronensäure
Polysorbat 80
Natriumcitrat-Dihydrat
Saccharose
Wasser für Injektionszwecke
Jede Kisunla 20 ml Durchstechflasche enthält insgesamt 11.5 mg Natrium.
Darreichungsform und Wirkstoffmenge pro EinheitKonzentrat zur Herstellung einer Infusionslösung (intravenöse Infusion).
Eine Kisunla 20 ml Durchstechflasche enthält 350 mg Donanemab (17.5 mg/ml).
Die Lösung ist klar bis opaleszierend, farblos bis leicht gelblich oder leicht bräunlich.Die Lösung darf nicht verwendet werden, wenn sie trüb ist oder sichtbare Partikel vorhanden sind.
Indikationen/AnwendungsmöglichkeitenKisunla ist indiziert zur Verlangsamung des Fortschreitens einer symptomatischen Alzheimer-Krankheit (Alzheimer's disease, AD) bei erwachsenen Patienten mit nachgewiesener Alzheimer-typischer Amyloid-beta Pathologie und einer klinischen Diagnose einer leichten kognitiven Störung (mild cognitive impairment, MCI) oder einer leichten Demenz, die heterozygote Träger oder Nicht-Träger des Apolipoprotein E ε4 (ApoE ε4) Allels sind (siehe Rubrik "Kontraindikationen" und "Klinische Wirksamkeit" ).
Dosierung/AnwendungDie Anwendung soll unter Anleitung und Aufsicht eines in Diagnose und Behandlung der Alzheimer-Krankheit erfahrenen Arztes begonnen werden. Die Infusion von Donanemab soll durch medizinisches Fachpersonal begonnen und überwacht werden. Ein zeitnaher Zugang zu einem MRT muss gewährleistet sein. Die Behandlung mit Donanemab soll unter Aufsicht eines erfahrenen, multidisziplinären Teams erfolgen, welches trainiert ist auf den Nachweis, Überwachung und Behandlung von ARIA und erfahren ist im Erkennen und Behandlung von infusionsbedingten Reaktionen.
Vor dem Behandlungsbeginn müssen vorliegen:
-ApoE ε4 Genotypisierung, mit zuvor angemessener Genberatung gemäss den geltenden nationalen oder lokalen Regelungen
-MRT-Bildgebung zu Beginn (nicht älter als 3 Monate, nach standardisiertem Protokoll)
-Amyloid-PET, ausgewertet mit der Centiloid-Methode
-Tau-PET (sofern verfügbar)
Ein Donanemab Neuroimaging Manual und ein MRT-Manual stehen weiter unten zur Konsultation zur Verfügung.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Nachweis von Amyloid-beta
Der Nachweis von Amyloid-beta vereinbar mit einer AD soll durch eine validierte Methode (z.B. Amyloid-PET) bestätigt werden.
Dosierung
Donanemab ist alle 4 Wochen zu verabreichen. Die empfohlene Dosis beträgt 350 mg für die erste Anwendung, 700 mg für die zweite Anwendung und 1050 mg für die dritte Anwendung (350/700/1050 mg), gefolgt von 1400 mg alle 4 Wochen. Dieses Dosierungsschema basiert auf pharmakokinetischen und pharmakodynamischen Bridging-Daten zwischen empfohlener Dosis aus der Phase 3b TRAILBLAZER-ALZ-6-Studie und der pivotalen Phase 3 TRAILBLAZER-ALZ-2-Studie. Es entspricht nicht dem Dosierungsschema der ersten 3 Anwendungen in der pivotalen Phase 3 Studie (siehe Rubriken "Pharmakokinetik" und "Klinische Wirksamkeit" ).
Therapiedauer
Die Behandlung soll bis zur Auflösung der Amyloid-Plaques, bestätigt durch eine validierte Methode, und bis zu maximal 18 Monate fortgesetzt werden.
Sie sollte vorzeitig abgebrochen werden, sobald eine Progression in ein höheres klinisches Krankheitsstadium stattgefunden hat. Deshalb ist der kognitive Status vor und nach Beginn der Therapie alle drei Monate mittels eines geeigneten, validierten kognitiven Testverfahrens, das die Stadien der Alzheimer-Krankheit zuverlässig erfasst, und eine Beurteilung der klinischen Symptome durchzuführen. Die Ergebnisse sind schriftlich zu dokumentieren.
Die Überprüfung der Kognition und der Progression der Symptome sollten durchgeführt werden, um zu beurteilen, ob beim Patienten eine Progression der Grunderkrankung stattgefunden hat und/oder ob der klinische Verlauf anderweitig nahelegt, dass Kisunla bei dem Patienten keine Wirksamkeit gezeigt hat.
Ein Amyloid-PET ist zwischen Monat 6 und Monat 12 durchzuführen, um die Amyloid-Last zu überprüfen und bei PET-Negativität die Behandlung zu beenden. Empfohlen wird eine Behandlungsdauer von maximal 18 Monaten. Wenn die PET-Negativität früher als 18 Monate erreicht wird, das klinische Ansprechen ausbleibt oder das im individuellen Fall nächsthöhere Erkrankungsstadium erreicht wird, muss die Behandlung abgebrochen werden.
Nutzen und Risiken können von der Tau-Anfangskonzentration abhängen. Bei Patienten mit niedriger bis mittlerer Tau-Last wurde im Vergleich zu hohem Tau eine numerisch höhere Wirksamkeit beobachtet (siehe Rubrik "Pharmakodynamik" ). Bei der Entscheidung, ob Donanemab angewendet werden soll oder nicht, sollten die Ergebnisse aus der Tau-PET-Bildgebung, sofern diese durchgeführt wurde, berücksichtigt werden.
Überwachung und Therapieunterbrechung bei Amyloid-bedingten Bildgebungsanomalien
Während der gesamten Behandlung mit Kisunla muss ein Zugang zu Magnetresonanztomographie (MRT) gewährleistet sein.
Vor Behandlungsbeginn soll eine MRT des Gehirns als Ausgangswert (nicht älter als 3 Monate) vorhanden sein. Ein MRT ist jeweils vor der 2. Anwendung, vor der 3. Anwendung, vor der 4. Anwendung und vor der 7. Anwendung (Monat 6) durchzuführen. Bei Patienten mit ARIA-Risikofaktoren, wie heterozygote Träger von ApoE ε4 und/oder Patienten mit früheren ARIA-Ereignissen während der Behandlung, sollte nach einjähriger Behandlung (vor der zwölften Infusion) ein zusätzliches MRT durchgeführt werden. Sofern Patienten über Symptome berichten, welche auf ARIA hindeuten, soll zu jedem Zeitpunkt der Behandlung eine klinische Untersuchung einschliesslich MRT-Bildgebung erfolgen.
Das Studienprotokoll der TRAILBLAZER-ALZ-2 Studie (AACI) empfahl ein MRT mit FLAIR-Sequenz zum Nachweis von ARIA-E, und T2* Gradient-Recalled Echo zum Nachweis von ARIA-H. Eine suszeptibilitätsgewichtete Bildgebung ist ebenfalls akzeptabel zum Nachweis von ARIA-H.
Die Empfehlungen für Therapieunterbrechungen bei Patienten mit Amyloid-bedingten Bildgebungsanomalien mit Ödemen (edema/effusion, ARIA-E) und mit Hämorrhagien/Hämosiderinablagerungen (ARIA-H) sind in Tabelle 1 aufgeführt.
Tabelle 1: Dosierungsempfehlungen für Patienten mit ARIA-E und ARIA-H
Klinische Symptome Schwereb der ARIA-E und ARIA-H im MRT
Leicht Mittelschwer Schwer
Asymptomatisch Therapieunterbrechung erwägen Therapie unterbrechena Therapie beenden
Symptomatisch Therapie unterbrechen a Therapie unterbrechen a Therapie beenden
a Unterbrechung der Behandlung, bis eine MRT-Untersuchung einen radiologischen Rückgang (ARIA-E) oder eine Stabilisierung (ARIA-H) zeigt, und die Symptome, falls vorhanden, zurückgegangen sind. Zwei bis vier Monate nach der Erstfeststellung sollte eine erneute MRT-Untersuchung in Betracht gezogen werden, zur Beurteilung des Rückgangs (ARIA-E) bzw. der Stabilisierung (ARIA-H). Die Wiederaufnahme oder das Absetzen der Therapie sollte nach klinischem Ermessen erfolgen. Vor der Wiederaufnahme der Behandlung ist eine erneute Bewertung der Risikofaktoren durchzuführen. Bei ARIA-E kann eine unterstützende Therapie, einschliesslich Kortikosteroide, erfolgen. Die Wirksamkeit dieser Behandlung wurde jedoch nicht nachgewiesen.
b Siehe Tabelle 2 für die Kriterien zur Klassifizierung von ARIA-Schweregraden im MRT
Bei radiographisch oder klinisch symptomatisch schweren ARIA-E oder ARIA-H ist die Therapie mit Donanemab dauerhaft abzubrechen.
Donanemab sollte nach klinisch schwerwiegenden ARIA-E, schwerwiegenden ARIA-H oder einer intrazerebralen Blutung grösser als 1 cm endgültig abgesetzt werden. Nach wiederkehrenden klinisch symptomatischen oder radiographisch mittelschweren oder schweren ARIA-Ereignissen sollte die Behandlung mit Donanemab dauerhaft abgesetzt werden.
Art der Anwendung
Kisunla 350 mg ist nur für die intravenöse Infusion. Die Durchstechflasche ist zum einmaligen Gebrauch bestimmt. Die Infusion soll über mindestens 30 Minuten erfolgen. Patienten sollen nach der Infusion über mindestens 30 Minuten beobachtet werden. Für Hinweise zur Verdünnung des Arzneimittels vor der Anwendung siehe Rubrik "Sonstige Hinweise, Hinweise für die Handhabung" .
Parenteral angewendete Arzneimittel sollen vor der Anwendung visuell auf Partikel und Verfärbungen geprüft werden, wenn immer die Lösung und das Behältnis dies erlauben. Kisunla darf nicht verwendet werden, wenn es trüb aussieht oder sichtbare Partikel enthält.
Versäumte Anwendung
Falls eine Infusion versäumt wurde, ist die Gabe so bald wie möglich nachzuholen. Anschliessend wird die empfohlene Anwendung alle 4 Wochen wieder aufgenommen.
Ältere Menschen
Es ist keine Dosisanpassung für ältere Menschen erforderlich. (siehe Rubrik "Pharmakokinetik" )
Patienten mit eingeschränkter Nierenfunktion
Bei leicht bis moderat eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich. Für Patienten mit schwer eingeschränkter Nierenfunktion liegen nur begrenzte Daten vor (siehe Rubrik "Pharmakokinetik" ).
Patienten mit eingeschränkter Leberfunktion
Bei leicht eingeschränkter Leberfunktion ist keine Dosisanpassung erforderlich. Es liegen nur begrenzte Daten für Patienten mit moderat eingeschränkter Leberfunktion und keine Daten für Patienten mit schwer eingeschränkter Leberfunktion vor (siehe Rubrik "Pharmakokinetik" ).
Patienten mit zeitgleicher Plasmapherese
Die Zeit zwischen der Anwendung von Donanemab und der Plasmapherese sollte abgestimmt werden, um die Eliminierung von Donanemab zu minimieren (siehe Rubrik "Interaktionen" ).
Kinder und Jugendliche
Kisunla ist nicht für die Anwendung bei Kindern und Jugendlichen zugelassen. Dosierungsempfehlungen sind nicht verfügbar.
Kontraindikationen-Überempfindlichkeit gegen Donanemab oder einen der Hilfsstoffe (siehe "Zusammensetzung" ).
Bildgebende Befunde, die auf ein erhöhtes Risiko für ARIA oder intrazerebrale Blutungen hinweisen, wie:
-MRT-Ausgangsbefunde mit
einer vorangegangenen intrazerebralen Blutung von mehr als 1 cm
mehr als 4 Mikroblutungen (definiert als <=1 cm im Durchmesser auf der T2*-Sequenz),
superfizielle Siderose, vasogene Ödeme (ARIA-E) oder andere Befunde, die auf eine zerebrale Amyloid-Angiopathie mit assoziierter Entzündung (CAA-ri) hindeuten.
-Mehr als 2 lakunäre Infarkte oder Schlaganfälle, die einen grossen Gefässbereich betreffen
-Schwere subkortikale Hyperintensität übereinstimmend mit einem Fazekas-Score von 3
-Schwere Erkrankung der weissen Substanz
-Andere schwerwiegende Pathologien, die zu einer kognitiven Beeinträchtigung führen können
-Evidenz einer nicht-AD Demenz im MRT
-Schlaganfall oder transiente ischämische Attacke innerhalb von 1 Jahr vor Behandlungsbeginn
-Schlecht kontrollierte Hypertonie
-Unzureichend kontrollierte Blutgerinnungsstörung (einschliesslich einer Thrombozytenzahl <50'000 oder einer International Normalized Ratio [INR] >1.5 bei Patienten ohne Anwendung von Antikoagulanzien)
-Behandlung mit Antikoagulanzien
-Systemische Lyse mit einem Thrombolytikum
-Instabile medizinische Befunde, die mit der Behandlung mit Donanemab interferieren, oder interferieren können
-Jeder Befund, der eine zufriedenstellende MRT-Bewertung für die Sicherheitsüberwachung verhindern könnte
-Aktive Epilepsie innerhalb von 1 Jahr vor Therapiebeginn
-Schwangerschaft oder Frauen im gebärfähigen Alter ohne angemessene Verhütung
Warnhinweise und VorsichtsmassnahmenKontrolliertes Zugangsprogramm (Controlled access programme)
Für die sichere und wirksame Anwendung von Donanemab soll der Behandlungsbeginn aller Patienten über ein zentrales Registrierungssystems erfolgen, das als Teil eines kontrollierten Zugangsprogramms eingerichtet wird.
Schulungsmaterialien
Verschreibende sollten mit dem Schulungsmaterial zur Erkennung und Behandlung von ARIA vertraut sein und die Vorteile und Risiken der Donanemab-Therapie mit dem Patienten/ Betreuer besprechen. MRT-Bildgebungen, Anzeichen oder Symptome möglicher Nebenwirkungen müssen dem Patienten/Betreuer erklärt werden. Ferner müssen diese aufgeklärt sein, wann eine notfallmässige Konsultation stattfinden muss. Die Patienteninformation sowie die Patientenkarte wird dem Patienten ausgehändigt und erklärt. Die Patientenkarte ist stets mitzuführen.
Amyloid-bedingte Bildgebungsanomalien (ARIA)
Schwerwiegende Amyloid-bedingte Bild-gebungssanomalien (amyloid-related imaging abnormalities, ARIA) wurden in klinischen Studien zu Donanemab beobachtet, einige davon mit fatalem Ausgang (siehe Rubrik "Unerwünschte Wirkungen" ). ARIA umfasst Ödeme/Ergüsse (ARIA-E, amyloid-related imaging abnormalities-edema/effusions, auch bekannt als zerebrale vasogene Ödeme) und Hämorrhagie/Hämosiderin-Ablagerung (ARIA-H, amyloid-related imaging abnormalities hemorrhage/hemosiderin, dies umfasst zerebrale Mikroblutungen ≤10 mm und kortikale superfizielle Siderose). Intrazerebrale Blutungen grösser 1 cm wurden beobachtet. ARIA-H treten im Allgemeinen in Verbindung mit ARIA-E auf.
ARIA können durch Magnetresonanztomographie (MRT) erkannt werden.
ARIA-Ereignisse wurden sehr häufig in den klinischen Studien zu Donanemab beobachtet. Die meisten ARIA-Ereignisse wurden erstmals innerhalb von 24 Wochen nach Behandlungsbeginn beobachtet und waren in der Regel asymptomatisch, allerdings können selten auch schwerwiegende und lebensbedrohliche Ereignisse, einschliesslich Krampfanfälle und Status epilepticus auftreten. Die meisten schwerwiegenden ARIA-Ereignisse traten innerhalb von 12 Wochen nach Behandlungsbeginn auf. Siehe Rubrik "Unerwünschte Wirkungen" für Informationen zur Inzidenz von ARIA. Während der Behandlung mit Donanemab sollten weitere MRT-Untersuchungen durchgeführt werden. Eine MRT-Untersuchung soll zu Beginn (innerhalb von 3 Monaten vor Behandlungsbeginn), vor der zweiten Anwendung, vor der dritten Anwendung, vor der vierten Anwendung und vor der siebten Anwendung durchgeführt werden. Bei Patienten mit ARIA-Risikofaktoren, wie heterozygote Träger von ApoE ε4 und/oder Patienten mit vorausgegangenen ARIA-Ereignissen während der früheren Behandlung sollte ein zusätzliches MRT nach einjähriger Behandlung (vor der zwölften Anwendung) durchgeführt werden (siehe Rubrik "Anwendung/Dosierung" ). Bei Auftreten von ARIA-Symptomen ist eine zusätzliche MRT-Untersuchung indiziert. Die Symptome können Kopfschmerzen, Verwirrtheit, Übelkeit, Erbrechen, Unsicherheit, Schwindel, Tremor, Sehstörungen, Sprachstörungen, verschlechterte kognitive Funktion, Bewusstseinsveränderungen und Krämpfe umfassen. ARIA sollte stets als mögliche Aetiologie bei diesen neurologischen Symptomen in Betracht gezogen werden. Üblicherweise gehen die mit ARIA zusammenhängenden Symptome mit der Zeit zurück. Nach einem ersten ARIA-Ereignis ist die Rückfallrate bei Wiederaufnahme der Behandlung sehr häufig: 24.3% mit ARIA-E und 35.9% mit ARIA-H (siehe Rubrik "Unerwünschte Wirkungen" ).
ARIA-E lösen sich im Verlauf typischerweise in der MRT-Bildgebung auf. ARIA-H können bestehen bleiben und sich stabilisieren.
Während der gesamten Behandlungsdauer muss ein notfallmässiger Zugang zu einer MRT-Bildgebung gewährleistet sein.
Vor Beginn der Behandlung mit Donanemab sollten Nutzen und Risiko sorgfältig bewertet werden.
MRT-Monitoring für ARIA
Für Empfehlungen zur MRT-Bildgebung siehe Rubrik "Anwendung/Dosierung" .
Bei ARIA verdächtigen Symptomen muss eine klinische Untersuchung sowie ein zusätzliches MRT erfolgen.
Empfehlungen zur Unterbrechung der Behandlung bei Patienten mit ARIA
Für Empfehlungen zur Unterbrechung der Behandlung bei Patienten mit ARIA-E und ARIA-H (siehe Rubrik "Anwendung/Dosierung" ). Bei schwerwiegenden ARIA-E, schwerwiegenden ARIA-H, intracerebralen Hämorrhagien ≥1cm, wiederholt auftretenden, symptomatischen oder radiographisch mässiggradigen oder schweren ARIA-Ereignissen ist die Behandlung mit Donanemab endgültig abzubrechen.
Radiologischer Schweregrad
Der radiologischer Schweregrad von ARIA unter Donanemab wurde nach den in Tabelle 2 beschriebenen Kriterien klassifiziert.
Tabelle 2: Kriterien für die Klassifizierung von ARIA im MRI
Art des ARIA
Leicht Mittelschwer Schwer
ARIA-E FLAIR Hyperintensität FLAIR Hyperintensität FLAIR Hyperintensität
begrenzt auf eine mit 5-10 cm in der >10 cm mit Schwellung
Lokalisation <5 cm in grössten Ausdehnung, von Gyri und Verstreichen
der weissen Substanz, oder an mehr als 1 der Sulci. Es lassen
von Sulci und/oder Stelle mit jeweils <10 sich eine oder mehrere
Kortex/Subkortex cm getrennte/unabhängige
Stellen feststellen
ARIA-H Mikroblutunge ≤4 neue Mikroblutungen 5-9 neue Mikroblutungen ≥10 neue Mikroblutungen
n
ARIA-H Superfizielle 1 neuer oder sich 2 neue oder sich >2 neue oder sich
Siderosea vergrössernder Herd mit vergrössernde Herde mit vergrössernde Herde mit
superfizieller Siderose superfizieller Siderose superfizieller Siderose
Abkürzungen: FLAIR = Fluid-attenuated inversion recovery; ARIA-E = amyloid-related imaging abnormalities oedema/effusions, Amyloid-bedingte Bildgebungsanomalie mit Ödemen/Ergüssen; ARIA-H = amyloid-related imaging abnormalities haemorrhage/hemosiderin deposition, Amyloid-bedingte Bildgebungsanomalien mit Hämorrhagie/Hämosiderin-Ablagerungen.
APOE ε4-Trägerstatus und Risiko für ARIA
Etwa 15% der Patienten mit Alzheimer-Krankheit sind homozygote Träger von ApoE ε4. Im placebokontrollierten Datensatz waren 16.8% (143/853) der Patienten im Donanemab-Arm homozygote Träger von Apolipoprotein E ε4 (ApoE ε4), 53% (452/853) waren heterozygote Träger und 29.9% (255/853) waren Nicht-Träger. Die Inzidenz von ARIA war bei homozygoten Trägern von ApoE ε4 höher (55.9% unter Donanemab gegenüber 21.9% unter Placebo) als bei heterozygoten Trägern (37.6% unter Donanemab gegenüber 14.1% unter Placebo) und bei Nicht-Trägern (24.7% unter Donanemab gegenüber 12.0% unter Placebo). Bei Patienten, die mit Donanemab behandelt wurden, traten bei 8.4% der homozygoten Träger von ApoE ε4 symptomatische ARIA-E auf, gegenüber 6.6% der heterozygoten Träger und 3.9% der Nicht-Träger. Schwerwiegende ARIA-Ereignisse wurden bei 2.8% der homozygoten Träger von ApoE ε4 beobachtet, bei 1.8% der heterozygoten Träger und 0.8% der Nicht-Träger.
Intrakranielle Hämorrhagien wurden in der indizierten Population bei 1.4% (10/710) der ApoE ε4 heterozygoten und nicht-Träger nach Behandlung mit Donanemab gegenüber 0.8% (6/728) der mit Placebo behandelten AD-Patienten berichtet. Intrazerebrale Blutungen >1cm Durchmesser wurden bei 0.4% (3/710) der mit Donanemab und bei 0.3% (2/728) mit Placebo behandelten Patienten beobachtet.
Vor dem Behandlungsbeginn ist die Bestimmung des ApoE ε4-Träger-Status erforderlich, um das Risiko für die Entwicklung einer ARIA zu ermitteln. Vor dem Genotyp-Test sollten verschreibende Ärzte mit den Patienten das Risiko von ARIA über alle Genotypen hinweg und die Auswirkungen des Gentestergebnisses besprechen.
Eine höhere Inzidenz von ARIA wurde auch bei Patienten beobachtet, die vor Behandlung Mikroblutungen und/oder superfizieller Siderose aufwiesen.
Intrazerebrale Blutungen
In einer placebokontrollierten Studie (TRAILBLAZER-ALZ 2) wurden intrazerebrale Blutungen grösser als 1 cm bei 0.4% (3/853) der Patienten nach Behandlung mit Donanemab im Vergleich zu 0.2% (2/874) mit Placebo beobachtet.
Fatale Ereignisse intrazerebraler Blutungen wurden bei Patienten mit Donanemab beobachtet.
Bei Patienten, die in der TRAILBLAZER-ALZ-6-Studie über 24 Wochen Donanemab in einer Dosierung von 350/700/1050 mg mit nachfolgend 1400 mg alle 4 Wochen erhalten haben, wurden bei 0.9% (2/212) intrazerebrale Blutungen grösser als 1 cm berichtet.
Andere Risikofaktoren für intrazerebrale Blutungen
Patienten waren von der Teilnahme an der Studie TRAILBLAZER-ALZ 2 ausgeschlossen, wenn Befunde aus dem Neuroimaging auf ein erhöhtes Risiko für intrazerebrale Blutungen hinwiesen. Dazu gehörten Befunde, die auf eine zerebrale Amyloidangiopathie hindeuten (frühere intrazerebrale Blutung mit einem Durchmesser von mehr als 1 cm, mehr als 4 Mikroblutungen, mehr als ein Bereich mit superfizieller Siderose, vasogenes Ödem und schwere Erkrankungen der weissen Substanz) (siehe Rubrik "Kontraindikationen" ). Diese und andere Läsionen (Aneurysma, vaskuläre Fehlbildung) können das Risiko einer intrazerebralen Blutung potenziell erhöhen.
Das Vorhandensein eines ApoE ε4-Allels ist ebenfalls mit einer zerebralen Amyloid-Angiopathie assoziiert, bei der ein erhöhtes Risiko für intrazerebrale Blutungen besteht.
Bei Patienten mit Faktoren, die auf ein erhöhtes Risiko für intrazerebrale Blutungen hindeuten, und insbesondere bei Patienten, die eine gerinnungshemmende Therapie benötigen, oder bei Patienten mit MRT-Befunden, die auf eine zerebrale Amyloid-Angiopathie hindeuten, ist Vorsicht geboten, wenn die Anwendung von Donanemab in Erwägung gezogen wird.
Begleitende antithrombotische Behandlung
Patienten, die Donanemab und ein Antithrombotikum (Acetylsalicylsäure, andere Thrombozytenaggregationshemmer oder Antikoagulantien) erhielten, zeigten keine erhöhte Inzidenz von ARIA. Die Mehrzahl der Antithrombotika-Expositionen betrafen Acetylsalicylsäure (80%). Die Anzahl der Ereignisse und die begrenzte Exposition gegenüber anderen Antithrombotika als Acetylsalicylsäure begrenzen abschliessende Schlussfolgerungen zum Risiko von ARIA oder intrazerebralen Blutungen bei Patienten unter antithrombotischer Medikation. Da bei Patienten unter Donanemab ARIA-H und intrazerebrale Blutungen grösser als 1 cm Durchmesser beobachtet wurden, ist zusätzliche Vorsicht geboten, wenn bei Patienten, die bereits mit Donanemab behandelt werden, eine begleitende Gabe von Antithrombotika in Erwägung gezogen wird. Die gleichzeitige Anwendung von Acetylsalicylsäure und anderen Thrombozytenaggregationshemmern ist zulässig.
Da ARIA fokale neurologische Defizite verursachen kann, die einem ischämischen Schlaganfall ähneln können, sollten die behandelnden Ärzte abwägen, ob solche Symptome auf ARIA zurückzuführen sein könnten. Eine systemische Lyse mit einem Thrombolytikum sowie die Behandlung mit Antikoagulanzien dürfen nicht während der Therapie mit Donanemab erfolgen (siehe Rubrik "Kontraindikationen" ).
Individuelles Nutzen-Risiko-Verhältnis basierend auf der Tau-Pathologie
Das Nutzen-Risiko-Verhältnis kann von der Tau-Ausgangskonzentration abhängen. Eine numerisch höhere Wirksamkeit wurde bei Patienten mit niedrigem bis mittlerem Tau-Wert im Vergleich zu hohem Tau-Wert beobachtet (siehe Rubrik "Klinische Wirksamkeit" ). Die klinische Wirksamkeit bei Patienten ohne oder mit sehr niedriger Tau-Konzentration wurde nicht untersucht. Der Befund eines Tau-Pathologie-Tests sollte, falls durchgeführt, im individuellen Nutzen-Risiko-Gespräch mit dem Patienten berücksichtigt werden.
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie und Angioödem wurden bei Anwendung von Donanemab beobachtet (siehe Rubrik "Unerwünschte Wirkungen" ). Zu den Anzeichen und Symptomen dieser Reaktionen gehören Erythem, Schüttelfrost, Übelkeit, Erbrechen, Schwitzen, Kopfschmerzen, Engegefühl in der Brust, Dyspnoe und Blutdruckveränderungen. Diese Reaktionen können schwer oder lebensbedrohlich sein und treten üblicherweise während oder innerhalb von 30 Minuten nach Infusion auf. Bei der ersten Beobachtung von Anzeichen oder Symptomen, die mit einer Überempfindlichkeitsreaktion vereinbar sind, ist die Infusion sofort abzubrechen und eine geeignete Therapie einzuleiten. Bei Patienten mit Überempfindlichkeit gegen Donanemab oder einen der Hilfstoffe in der Vorgeschichte ist Donanemab kontraindiziert. Bei einer nicht-schwerwiegenden infusionsbedingten Reaktion wird die Infusionsrate reduziert, oder die Infusion abgebrochen, und eine angemessene Behandlung entsprechend der klinischen Indikation begonnen. Eine Prämedikation mit Antihistaminika, Paracetamol oder Kortikosteroiden kann vor der nächsten Anwendung in Betracht gezogen werden.
Infusionsbedingte Reaktionen
Bei Gabe von Donanemab sind infusionsbedingte Reaktionen (IRR) und Anaphylaxie, beobachtet worden (siehe Rubrik "Unerwünschte Wirkungen" ). Diese Reaktionen können schwer oder lebensbedrohlich sein und treten üblicherweise während oder innerhalb von 30 Minuten nach Infusion auf. Zu den Anzeichen und Symptomen von diesen Reaktionen gehören Erythem, Schüttelfrost, Übelkeit, Erbrechen, Schwitzen, Kopfschmerzen, Engegefühl in der Brust, Dyspnoe, und Blutdruckveränderungen. Für Informationen zur Häufigkeit von infusionsbedingten Reaktionen siehe Rubrik "Unerwünschte Wirkungen" .
Bei schwerwiegenden infusionsbedingten Reaktionen oder Anaphylaxie ist die Gabe von Donanemab unverzüglich abzubrechen und eine angemessene Behandlung zu beginnen. Nach einer Infusionsbedingten Reaktion vom Grad 3 oder höher, die sich nach der Behandlung nicht bessert oder zurückbildet, muss die Therapie mit Donanemab dauerhaft beendet werden.
Im Falle einer nicht schwerwiegenden infusionsbedingten Reaktion kann die Infusionsrate reduziert oder die Infusion abgebrochen und bei klinischer Indikation eine geeignete Therapie eingeleitet werden.
Bei Anzeichen eines Gewebeschadens im Rahmen einer Hypersensitivitätsreaktion (z.B. Arthritis, Glomerulonephritis oder Mononeuritis multiplex) muss die Therapie mit Donanemab dauerhaft beendet werden.
Immunogenität
In den Placebo-kontrollierten klinischen Studien entwickelten 88,1% der mit Donanemab behandelten Patienten Antikörper auf Donanemab (anti-drug antibodies, ADA). In all diesen Patienten waren es neutralisierende Antikörper. Alle Patienten mit IRR hatten ADA. Ein höherer ADA-Titer war mit einer erhöhten Inzidenz an IRR und infusionsbedingter Hypersensitivität assoziiert.
Ausgeschlossene Patienten im klinischen Studienprogramm
Patienten mit Down-Syndrom können mit einer höheren Rate an CAA (zerebralen Amyloidangiopathien)- und ARIA-Ereignissen assoziiert sein. In den klinischen Studien mit Donanemab wurden diese nicht untersucht. Die Sicherheit und Wirksamkeit von Donanemab in diesen Patienten ist nicht bekannt.
Natrium
Dieses Arzneimittel enthält 11.5 mg Natrium pro Durchstechflasche zu 350 mg Donanemab (20 ml). Eine Dosis von 1400 mg Donanemab (4 Durchstechflaschen) enthält 46 mg Natrium, dies entspricht 2% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme von 2 g.
InteraktionenFür Donanemab wurden keine formalen Studien zur Erfassung von Arzneimittel-Wechselwirkungen durchgeführt. Auf Grundlage der Merkmale von Donanemab werden keine pharmakokinetischen Wechselwirkungen erwartet.
Die gleichzeitige Anwendung von Donanemab und intravenösen Immunglobulinen, Plasmapherese oder Immunadsorption kann zu einer verminderten Wirksamkeit von Donanemab führen. Die Zeit zwischen diesen Anwendungen sollte abgestimmt sein, um die Eliminierung von Donanemab zu minimieren. Nach der Gabe von Donanemab sollte mindestens neun Wochen abgewartet werden, bis eine solche Therapie durchgeführt wird. Nach einer vorausgegangenen Behandlung mit intravenösen Immunglobulinen sollte der zeitliche Abstand bis zur Donanemab-Gabe drei Wochen betragen. Bei Patienten, die regelmässig eine dieser Therapien benötigen, sollte diese Interaktion bei den therapeutischen Entscheidungen vorausschauend berücksichtigt und nach medizinischer Notwendigkeit entschieden werden (siehe Rubrik "Dosierung/Anwendung" ).
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Der Schwangerschaftsstatus von Frauen im gebärfähigen Alter sollte vor Einleitung der Behandlung mit Donanemab überprüft werden. Frauen im gebärfähigen Alter müssen während und bis zu zwei Monate nach der Behandlung eine wirksame Verhütung anwenden.
Schwangerschaft
Daten zur Anwendung von Donanemab bei Schwangeren liegen nicht vor. Mit Donanemab wurden keine embryofetalen Entwicklungsstudien bei Tieren durchgeführt. Humanes IgG ist nach dem ersten Schwangerschaftstrimenon bekanntermassen plazentagängig. Daher kann Donanemab potenziell von der Mutter auf den sich entwickelnden Fötus übergehen. Die Anwendung von Donanemab in der Schwangerschaft wird nicht empfohlen.
Stillzeit
Mit Donanemab wurden keine Laktationsstudien bei Tieren durchgeführt. Es ist bekannt, dass humanes Immunglobulin G (IgG) in die Muttermilch übergehen kann. Folglich kann Donanemab von der Mutter auf das gestillte Kind übertragen werden. Die Risiken für einen gestillten Säugling sind unbekannt. Stillende Frauen sollen Donanemab nur dann erhalten, wenn der mögliche Nutzen das potenzielle Risiko für Mutter und Kind überwiegt.
Fertilität
Daten zu den Wirkungen von Donanemab auf die menschliche Fertilität liegen nicht vor. Studien mit Donanemab bei Tieren zur Untersuchung möglicher Fertilitäts-Einschränkungen wurden nicht durchgeführt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenStudien zur Beurteilung der Wirkungen von Donanemab auf die Fahrtüchtigkeit und das Bedienen von Maschinen wurden nicht durchgeführt.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In einer placebokontrollierten Studie (TRAILBLAZER-ALZ 2, Phase 3) bei Patienten mit leichter kognitiver Störung oder leichter Demenz im Rahmen einer Alzheimer-Krankheit (Alzheimer Disease, AD), erhielten 853 Erwachsene mindestens eine Dosis Donanemab. Davon waren 710 Patienten in der indizierten Population (ApoE ε4 Heterozygote oder Nichtträger): 29.9% (255/853) Nichtträger, 53.0% (452/853) Heterozygote und 0.4% (3/853) Genotyp unbekannt.
Die am häufigsten berichteten unerwünschten Wirkungen in der indizierten Population waren ARIA-E (20.6%), ARIA-H (27.6%), Kopfschmerzen (14.6%) und infusionsbedingte Reaktionen (8.3%). Die wichtigsten schwerwiegenden unerwünschten Wirkungen waren schwerwiegende ARIA-E (1.3%), schwerwiegende ARIA-H (0.3%) und schwerwiegende Überempfindlichkeitsreaktion einschliesslich infusionsbedingte Reaktionen (0.4%). Anaphylaxie wurde gelegentlich berichtet (0.4%) (siehe "Warnhinweise und Vorsichtsmassnahmen" ).
Liste der Nebenwirkungen in der Gesamtpopulation
Die Nebenwirkungen aus klinischen Studien sind nach MedDRA-Systemorganklassen aufgeführt. Innerhalb einer Systemorganklasse werden die Nebenwirkungen nach Häufigkeit angegeben, die häufigsten zuerst. Innerhalb einer Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Die Häufigkeitskategorie basiert für jede Nebenwirkung auf der folgenden Häufigkeitsdefinition: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000)
Erkrankungen des Nervensystems
Sehr häufig: ARIA-Ea,b (24.0%), ARIA-Hab,c, (31.4%), Kopfschmerzen (14.0%)
a Wie durch MRT beurteilt.
b Mögliche assoziierte Symptome: Kopfschmerzen, Verwirrtheit, Übelkeit, Erbrechen, Unsicherheit, Schwindel, Tremor, Sehstörungen, Sprachstörungen, Verschlechterung der kognitiven Funktion, Bewusstseinsveränderungen, Krampfanfälle
c Beinhaltet Mikroblutungen und superfizielle Siderosen
Gelegentlich: intrazerebrale Blutung (> 1cm)d
d Zerebrale Hämorrhagie und hämorrhagischer Schlaganfall.
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit, Erbrechen
Erkrankungen des Immunsystems
Häufig: Überempfindlichkeit
Gelegentlich: Anaphylaktische Reaktion
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Infusionsbedingte Reaktionene, Überempfindlichkeit
Gelegentlich: Anaphylaktische Reaktion
e Umfasst Erythem, Schüttelfrost, Übelkeit, Erbrechen, Schwitzen, Kopfschmerzen, Engegefühl in der Brust, Atemnot und Blutdruckveränderungen.
Beschreibung ausgewählter Nebenwirkungen
Amyloid-bedingte Bildgebungsanomalien (ARIA) und intrazerebrale Hämorrhagie in der indizierten Patientenpopulation
TRAILBLAZER-ALZ-2
ARIA (ARIA-E oder ARIA-H) wurden bei 33 % (234/710) der Patienten (Heterozygote oder Nichtträger) unter Donanemab beobachtet, im Vergleich zu 13.5 % (98/728) der Patienten unter Placebo in der placebokontrollierten pivotalen Studie (Dosierungsschema 700mg Donanemab alle 4 Wochen für die ersten 3 Verabreichungen, danach 1400mg Donanemab alle 4 Wochen). Symptomatische ARIA traten bei 6.1 % der Patienten mit Donanemab auf. Klinisch schwerwiegende ARIA wurden bei 1.4% (10/710) der Patienten unter Donanemab berichtet. Drei Teilnehmer (0.4%) hatten schwerwiegende ARIA und starben.
ARIA-E (, amyloid-related imaging abnormalities-edema/effusions, auch bekannt als zerebrale vasogene Ödeme) wurde bei 20.6% der Patienten mit Donanemab beobachtet, im Vergleich zu 1.8% der Patienten mit Placebo. Der maximale radiologische Schweregrad der ARIA-E war leicht bei 6.2%, mittelschwer bei 12.7% und schwer bei 1.4% der Patienten. Die mediane Zeit bis zum radiologischen Rückgang der ARIA-E betrug etwa 8.3 Wochen. Symptomatische ARIA-E wurden bei 5.6% der Patienten unter Donanemab in den placebokontrollierten Studien berichtet.
Klinische Symptome von ARIA-E gingen bei etwa 80% der Patienten zurück. Die mediane Zeit bis zum Rückgang der klinischen Symptome der ARIA-E betrug etwa 3.9 Wochen.
Bei heterozygoten Trägern wurden ARIA-E bei 23.2% (105/452) der Patienten mit Donanemab berichtet, im Vergleich zu 2.1% (10/474) der Patienten mit Placebo. Der maximale radiologische Schweregrad der ARIA-E mit Donanemab bei heterozygoten Trägern war leicht bei 6.6% der Patienten, mittelschwer bei 14.2% und schwer bei 2.0%. Symptomatische ARIA-E berichteten 6.6% der heterozygoten Träger mit Donanemab. Bei Nicht-Trägern wurden ARIA-E bei 15.7% (40/255) der Patienten mit Donanemab berichtet, im Vergleich zu 0.8% (2/250) der Patienten mit Placebo. Der maximale radiologische Schweregrad der ARIA-E mit Donanemab bei Nicht-Trägern war leicht bei 5.1% der Patienten, mittelschwer bei 10.2% und schwer bei 0.4%. Symptomatische ARIA-E berichteten 3.9% der Nicht-Träger mit Donanemab.
ARIA-H (amyloid-related imaging abnormalities hemorrhage/hemosiderin, dies umfasst zerebrale Mikroblutungen und kortikale superfizielle Siderose) können bei Patienten mit AD spontan und unabhängig von der Behandlung auftreten. ARIA-H wurden bei 27.6 % der Patienten unter Donanemab im Vergleich zu 12.2% der Patienten unter Placebo berichtet. Der maximale radiologische Schweregrad der ARIA-H war leicht bei 14.4 %, mittelschwer bei 5.5 % und schwer bei 7.6% der Patienten. Symptomatische ARIA-H wurden bei 1.1% der Patienten unter Donanemab berichtet, im Vergleich zu 0.3% unter Placebo. Isolierte ARIA-H (d.h. ARIA-H ohne begleitende ARIA-E) wurden bei 12.4% der Patienten unter Donanemab berichtet im Vergleich zu 11.5% unter Placebo.
Bei heterozygoten Trägern wurden ARIA-H bei 32.5% (147/452) der Patienten mit Donanemab berichtet, im Vergleich zu 12.9% (61/474) der Patienten mit Placebo. Der maximale radiologische Schweregrad der ARIA-H bei heterozygoten Trägern war leicht bei 15.0% der Patienten, mittelschwer bei 7.7% und schwer bei 9.5%. Symptomatische ARIA-H berichteten 1.5% der heterozygoten Träger mit Donanemab im Vergleich zu 0.2% mit Placebo. Isolierte ARIA-H bei heterozygoten Trägern wurden bei 14.2% mit Donanemab im Vergleich zu 11.2% mit Placebo berichtet. Bei Nicht-Trägern wurden ARIA-H bei 18.8% (48/255) der Patienten mit Donanemab berichtet, im Vergleich zu 11.2% (28/250) der Patienten mit Placebo. Der maximale radiologische Schweregrad der ARIA-H bei Nicht-Trägern war leicht bei 12.9% der Patienten, mittelschwer bei 1.6% und schwer bei 4.3%. Symptomatische ARIA-H berichteten 0.4% der Nicht-Träger mit Donanemab im Vergleich zu 0.4% mit Placebo. Isolierte ARIA-H bei Nicht-Trägern wurden bei 9.0% mit Donanemab im Vergleich zu 11.2% mit Placebo berichtet.
Die Mehrzahl der ersten radiologischen ARIA-Ereignisse in den placebokontrollierten Studien traten früh unter Behandlung auf (innerhalb von 24 Wochen nach Behandlungsbeginn), wenngleich ARIA zu jedem Zeitpunkt während der Behandlung möglich sind und Patienten mehr als eine Episode haben können.
Intrazerebrale Blutungen grösser als 1 cm wurden bei 0.4% (3/710) der mit Donanemab behandelten Patienten und bei 0.3% (2/728) der mit Placebo behandelten Patienten beobachtet. Bei einem der mit Donanemab behandelten Teilnehmer der Zulassungsstudie mit einer zu Studienbeginn bestehenden superfiziellen Siderose verlief eine ARIA-H mit begleitender intrazerebraler Hämorrhagie tödlich.
TRAILBLAZER-ALZ-6
In Woche 76 der TRAILBLAZER-ALZ-6 waren ARIA (ARIA-E oder ARIA-H) bei 28.8% der Patienten (heterozygote oder Nicht-Träger) berichtet worden, die Donanemab im Dosierungsschema 350/700/1050 mg gefolgt von 1400 mg alle 4 Wochen erhalten hatten (n=191). Klinisch schwerwiegende ARIA-Ereignisse wurden bei 0.5% der Patienten mit Donanemab berichtet.
ARIA-E wurden bei 14.7% der Patienten mit Donanemab berichtet. Der höchste radiologische Schweregrad der ARIA-E war leicht bei 5.8% der Patienten, mittelschwer bei 8.9% und schwer bei 0%. Die mediane Zeit bis zum radiologischen Rückgang der ARIA-E betrug etwa 8.4 Wochen. Symptomatische ARIA-E wurden bei 3.1% der Patienten mit Donanemab berichtet. Klinische Symptome durch ARIA-E gingen bei 83.3% der Patienten zurück, mit einer medianen Zeit von 1.6 Wochen bis zum Symptomrückgang.
ARIA-H wurden bei 25.1% der Patienten mit Donanemab beobachtet. Der höchste radiologische Schweregrad der ARIA-H war leicht bei 18.3% der Patienten, mittelschwer bei 3.1% und schwer bei 3.7%. Symptomatische ARIA-H wurden bei 0.5% der Patienten mit Donanemab berichtet.
Bei heterozygoten und Nicht-Trägern von ApoE-ε4 wurde bei 1% (2/191) der Patienten nach Anwendung von Donanemab eine intrazerebrale Hämorrhagie berichtet. Davon hatten 0.5% (1/191) der Patienten mit Donanemab eine schwerwiegende intrazerebrale Hämorrhagie grösser als 1 cm. Dieser Teilnehmer mit ARIA-E hatte ein Thrombolytikum zur Behandlung von Schlaganfall-ähnlichen Symptomen erhalten und hatte eine tödliche intrazerebrale Hämorrhagie.
Infusionsbedingte Reaktionen
In der TRAILBLAZER-ALZ-2 wurden Infusionsreaktionen bei 8.7% der Patienten unter Donanemab im Vergleich zu 0.5% unter Placebo beobachtet. Anaphylaxie wurde gelegentlich berichtet (0.4%). Schwerwiegende Infusionsreaktionen oder Überempfindlichkeit traten bei 0.4% der Patienten unter Donanemab auf, im Vergleich zu 0.1% unter Placebo. Die Mehrzahl der Infusionsreaktionen und Überempfindlichkeitsreaktionen traten meistens während der ersten 4 Anwendungen von Donanemab auf, wenngleich solche Reaktionen jederzeit möglich sind.
Unter Donanemab-Therapie kam es zu Behandlungsabbrüchen aufgrund von IRR (3.6%), Überempfindlichkeit (0.5%) und Anaphylaxie (0.4%), nicht hingegen unter Placebo.
Eine erneute Exposition führte zu einer nachfolgenden IRR/Überempfindlichkeit in 47.5% der Patienten, wobei Schweregrad und Art der Symptome meist vergleichbar mit dem Erstereignis war.
Prophylaktische Medikationen vor nachfolgenden Therapien hatten keinen Einfluss auf IRR.
Immunogenität
In placebokontrollierten klinischen Studien entwickelten 88.1% der Patienten unter Donanemab gegen das Arzneimittel gerichtete Antikörper (anti-drug antibodies, ADA) und alle Patienten mit ADA hatten neutralisierende Antikörper. Wenngleich die Donanemab-Exposition mit zunehmenden ADA-Titern sinkt, war die Entwicklung von ADA nicht mit einem Verlust der klinischen Wirksamkeit von Donanemab verbunden. Alle Patienten, die infusionsbedingte Reaktionen berichteten, hatten ADA.
Ein höherer ADA-Titer war mit einer erhöhten Inzidenz von infusionsbedingten Reaktionen/sofortige Überempfindlichkeitsreaktionen verbunden.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEinmaldosierungen von bis zu 40 mg/kg (etwa 2800 mg bei einer Person mit 70 kg) wurden angewendet. Bei 2 von 4 Patienten traten mit dieser Dosis ARIA-E auf, welche wieder zurückgingen. Bei Überdosierung ist eine unterstützende Behandlung zu beginnen.
Eigenschaften/WirkungenATC-Code
N06DX05
Pharmakotherapeutische Gruppe: Nervensystem, Psychoanaleptika, Antidementiva, andere Antidementiva.
Donanemab ist ein rekombinanter, humanisierter, monoklonaler Antikörper, der in Ovarzellen des chinesischen Hamsters (Chinese Hamster Ovary, CHO) hergestellt wird.
Wirkungsmechanismus
Donanemab ist ein monoklonaler Immunoglobulin-gamma-1 (IgG1) Antikörper, der sich gegen die unlösliche, pyroglutamate-modifizierte, N-terminal verkürzte Form von Amyloid-beta (N3pG Aβ) richtet, die nur in Amyloid-Plaques des Gehirns vorkommt. Donanemab bindet an N3pG Aβ und unterstützt die Auflösung der Plaques durch Mikroglia-vermittelte Phagozytose.
Pharmakodynamik
Bei Patienten, die mit Donanemab behandelt wurden, wurde mit Hilfe von Amyloid-Positronenemissionstomographie (PET) eine Reduktion der zerebralen Amyloid-Plaques beobachtet. Donanemab reduzierte die Tau-Pathophysiologie, gemessen anhand von P-Tau217 im Plasma.
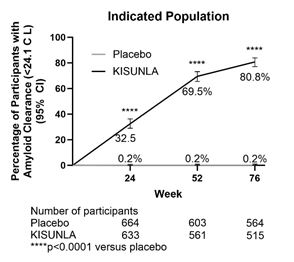
Der prozentuale Anteil der mit Donanemab behandelten Patienten, die in der Studie TRAILBLAZER-ALZ 2 Amyloid-Freiheit erreichten (Amyloid clearance, das heisst, weniger als 24.1 Centiloide), betrug 32.5% in Woche 24, 69.5% in Woche 52 und 80,8% in Woche 76 in der indizierten Population.
In der Studie TRAILBLAZER-ALZ 2 war der Unterschied zwischen Donanemab und Placebo für die Veränderung der Amyloid-Konzentration ab Ausgangswert in Woche 76 in der indizierten Population signifikant (-89.24 Centiloide).
In der Studie TRAILBLAZER-ALZ 6 wurde eine ähnliche Reduktion der Amyloid-Plaques in Woche 24 für das Dosierungsschema von 350/700/1'050 mg, gefolgt von 1'400 mg alle 4 Wochen beobachtet, verglichen mit dem Dosierungsschema von 700 mg für die ersten drei Infusionen, gefolgt von 1'400 mg alle 4 Wochen, welches in der pivotalen Studie untersucht wurde.
Die Donanemab-Exposition nahm mit zunehmendem ADA-Titer ab. Eine Amyloid-Beta-Reduktion wurde unabhängig vom ADA-Titer festgestellt. Es wurde kein Zusammenhang zwischen dem Vorhandensein von ADA und den Ergebnissen im iADRS und CDR-SB beobachtet (siehe auch Rubrik “Warnhinweise und Vorsichtmassnahmen”, sowie Rubrik "Unerwünschte Wirkungen" ).
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Donanemab wurden in einer Studie Phase 3 (TRAILBLAZER-ALZ 2) untersucht. Es war eine doppelblinde, placebokontrollierte Studie im Parallelgruppendesign. Durchgeführt wurde sie bei Patienten im Alter von 60 bis 85 Jahren mit früher symptomatischer AD (leichte kognitive Einschränkung, Mild Cognitive Impairment, MCI) oder leichter Demenz aufgrund von AD und einem MMSE-Wert von 20 bis einschliesslich 28. Die Patienten mussten Anzeichen einer Amyloid-beta-Pathologie aufweisen, bestätigt durch einen Amyloid-PET-Scan. Die Teilnehmer wiesen in einem Flortaucipir-PET-Scan zudem Anzeichen einer pathologischen Tau-Ablagerung auf.
Supportiv gibt es Daten aus einer Studie Phase 2 (TRAILBLAZER-ALZ) mit vergleichbarem Design, Die in diesen Studien verwendeten Dosierungsschemata unterscheiden sich vom zugelassenen Dosierungsschema (siehe Rubrik "Pharmakokinetik" ).
Für die Sicherheitsauswertung wurden die Patienten über bis zu 76 Wochen oder bis zum Ende der Behandlung plus 57 Tage nach der letzten Dosis beobachtet.
TRAILBLAZER-ALZ 2 war ursprünglich als Phase 2-Studie konzipiert und wurde später in eine Phase 3-Studie umgewandelt. Die wichtigsten Änderungen waren: Ergänzung einer Titrationsphase mit 700 mg für die ersten drei Anwendungen; Anpassungen einer Phase-2- zu einer Phase-3-Studie, einschliesslich der Erhöhung des Stichprobenumfangs und der Änderung der primären Analyse von "CDR-SB in der Gesamtpopulation oder der intermediären Tau-Population" zu "iADRS in der intermediären Tau-Population" ; Ergänzung einer Langzeit-Verlängerungsphase zur weiteren Beurteilung der Wirksamkeit und Sicherheit von Donanemab im zeitlichen Verlauf.
Studie Phase 3 TRAILBLAZER-ALZ 2
In dieser Studie wurden 1736 Patienten im Verhältnis 1:1 randomisiert und erhielten eine intravenöse Infusion mit 700 mg Donanemab alle 4 Wochen für die ersten 3 Anwendungen, danach 1400 mg alle 4 Wochen (N=860) oder Placebo (N=876), über bis zu 72 Wochen insgesamt. Die Studie beinhaltete eine doppelblinde Verlängerungsphase über 78 Wochen. Die Anwendung wurde fortgesetzt bis zum Ende der Studie oder bis zur Auflösung (Clearance) der Amyloid-Plaques, definiert als Nachweis einer Plaque-Konzentration von weniger als 25 Centiloiden aus zwei aufeinanderfolgenden Amyloid-PET-Scans oder ein einzelner PET-Scan mit einer Plaque-Konzentration von weniger als 11 Centiloiden.
Darüber hinaus war eine Dosisaussetzung für behandlungsbedingte ARIA zulässig. Patienten, die zu Studieneintritt bereits eine symptomatische Behandlung erhielten (Acetylcholinesterase-Inhibitoren [AChEI] und/oder N-Methyl-D-Aspartat-Inhibitoren, Memantin), konnten diese Behandlungen fortsetzen. Während der Studie konnten symptomatische Behandlungen im Ermessen des Studienarztes ergänzt oder verändert werden. Ein Studienausschluss erfolgte für Patienten mit vorbestehenden ARIA-E mit mehr als 4 Mikroblutungen, mehr als 1 Bereich mit superfizieller Siderose, jeglicher intrazerebraler Blutung >1 cm oder schwerer Erkrankung der weissen Substanz. Zu Studienbeginn betrug das mittlere Alter 73 Jahre mit einem Bereich von 59 bis 86 Jahre, bei einem mittleren (SD) Ausgangsgewicht von 71.7 kg (15.7), einer schrittweisen und progressiven Veränderung der Gedächtnis-Funktion über mindestens 6 Monate und einem MMSE (Mini-Mental State Examination) Score von 22.29 (3.88). 57.4% der Teilnehmer waren Frauen, 91.5% Weisse, 5.7% mit hispanischer oder lateinamerikanischer Ethnizität, 6.0% asiatischer Herkunft und 2.3% waren Schwarze. 80.0% der Patienten wurden in Nordamerika, 13.9% in Europa, 5.1% in Japan und 1.0% in Australien eingeschlossen. 29% waren ApoEε 4 Nichtträger, 54% Heterozygote und 17% Homozygote.
55.6% der Patienten erhielten AChEI, 20.3% Memantin. 61% der Patienten erhielten entweder AChEI oder Memantin.
Der Mittelwert (mean, SD) an Amyloid in Centiloiden betrug bei Baseline 102.5 (34.5).
Auf Grundlage des Tau-PET-Scans im Screening mit Flortaucipir gab es zwei primäre Analysenpopulationen: 1) Population mit niedrigen bis mittleren Tau-Konzentrationen (Anteil insgesamt 68.2%), und 2) die kombinierte Population mit niedrigen bis mittleren sowie hohen Tau-Konzentrationen (Anteil insgesamt 31.8%).
Insgesamt beendeten 24.7% der Patienten frühzeitig die Behandlung (29.3% unter Donanemab, 20.1% unter Placebo).
Der primäre Wirksamkeitsendpunkt war die Veränderung von Kognition und Funktion, gemessen anhand des iADRS (integrated Alzheimer's Disease Rating Scale) Scores von Beginn bis Woche 76. Der iADRS ist eine kombinierte Untersuchung von Kognition und täglicher Funktion und besteht aus Items aus der Kognitions-Subskala der Alzheimer's Disease Assessment Scale (ADAS-Cog13, Punktzahl 0-85) und der Alzheimer's Disease Cooperative Study - instrumental Activities of Daily Living (ADCS-iADL, Punktzahl 0-59) Skala, die Kerndomänen im Verlauf der klinischen AD messen. Der Gesamtscore reicht von 0 bis 144, wobei niedrigere Scores einer schlechteren kognitiven und funktionalen Leistung entsprechen. Zu den anderen Wirksamkeitsendpunkten gehörten CDR-SB (Clinical Dementia Rating Scale - Sum of Boxes), ADAS-Cog13 und ADCS-iADL.
Die nachfolgende Tabelle 3 zeigt wichtige Studienergebnisse in der indizierten Population.
In der indizierten Population wurden 717 Teilnehmer randomisiert und Donanemab zugeteilt, 414 waren weiblich und 303 männlich, 70 waren <65 Jahre alt, 320 waren 65-74 Jahre alt und 327 waren >= 75 Jahre alt.
In der indizierten Population wurden 730 Teilnehmer randomisiert und der Placebo-Gruppe zugeteilt, 426 waren weiblich und 304 männlich, 66 waren <65 Jahre alt, 311 waren 65-74 Jahre alt und 353 waren >= 75 Jahre alt.
Tabelle 3: Ergebnisse der Wirksamkeitsanalyse aus der Donanemab-Studie TRAILBLAZER-ALZ 2 in Woche 76 in der indizierten Population (heterozygote und Nichtträger von ApoE ε4)
Klinischer Endpunkt Heterozygote ApoE ε4-Träger und ApoE
ε4-Nichtträger
Dona Placebo
iADRS (NCS)
Mittlerer Ausgangswert (SD) 104.66 (14.12) 103.83 (14.03)
Veränderung (LS mean) ab -10.21 (0.57) -13.59 (0.55)
Ausgangswert
Unterschied gegenüber Placebo (95% 3.38 (1.83, 4.92)
CI)
Frauen
Mittlerer Ausgangswert (SD) 104.92 (14.02) 103.75 (13.38)
Veränderung (LS mean) ab -10.98 (0.76) -14.77 (0.72)
Ausgangswert
Unterschied gegenüber Placebo (95% 3.80 (1.77, 5.83)
CI)
Männer
Mittlerer Ausgangswert (SD) 104.32 (14.26) 103.95 (14.87)
Veränderung (LS mean) ab -9.18 (0.87) -11.92 (0.85)
Ausgangswert
Unterschied gegenüber Placebo (95 % 2.73 (0.36, 5.10)
CI)
<65 Jahre
Mittlerer Ausgangswert (SD) 105.19 (15.46) 107.14 (14.19)
Veränderung (LS mean) ab -12.74 (1.77) -14.21 (1.86)
Ausgangswert
Unterschied gegenüber Placebo (95% 1.47 (-3.51, 6.45)
CI)
65-74 Jahre
Mittlerer Ausgangswert (SD) 106.05 (12.88) 105.09 (13.59)
Veränderung (LS mean) ab -9.33 (0.86) -13.69 (0.83)
Ausgangswert
Unterschied gegenüber Placebo (95% 4.35 (2.03, 6.68)
CI)
>75 Jahre
Mittlerer Ausgangswert (SD) 103.20 (14.83) 102.11 (14.19)
Veränderung (LS mean) ab -10.48 (0.86) -13.37 (0.80)
Ausgangswert
Unterschied gegenüber Placebo (95% 2.89 (0.61, 5.17)
CI)
CDR-SB (MMRM)
Mittlerer Ausgangswert (SD) 3.96 (2.10) 3.94 (2.04)
Veränderung (LS mean) ab 1.67 (0.11) 2.43 (0.10)
Ausgangswert
Unterschied gegenüber Placebo (95% -0.77 (-1.04, -0.49)
CI)
Frauen
Mittlerer Ausgangswert (SD) 4.03 (2.11) 3.97 (1.98)
Veränderung (LS mean) ab 1.66 (0.12) 2.49 (0.15)
Ausgangswert
Unterschied gegenüber Placebo (95% -0.83 (-1.20, -0.45)
CI)
Männer
Mittlerer Ausgangswert (SD) 3.87 (2.08) 3.89 (2.13)
Veränderung (LS mean) ab 1.48 (0.15) 2.15 (0.14)
Ausgangswert
Unterschied gegenüber Placebo (95% -0.66 (-1.07, -0.25)
CI)
<65 Jahre
Mittlerer Ausgangswert (SD) 3.81 (2.05) 3.80 (1.54)
Veränderung (LS mean) ab 2.53 (0.40) 2.81 (0.31)
Ausgangswert
Unterschied gegenüber Placebo (95% -0.28 (-1.19, 0.63)
CI)
65-74 Jahre
Mittlerer Ausgangswert (SD) 3.78 (1.88) 3.66 (1.92)
Veränderung (LS mean) ab 1.46 (0.14) 2.55 (0.16)
Ausgangswert
Unterschied gegenüber Placebo (95% -1.09 (-1.50, -0.68)
CI)
>75 Jahre
Mittlerer Ausgangswert (SD) 4.17 (2.30) 4.21 (2.19)
Veränderung (LS mean) ab 1.48 (0.15) 2.04 (0.16)
Ausgangswert
Unterschied gegenüber Placebo (95% -0.56 (-0.96, -0.16)
CI)
ADAS-Cog13 (NCS)
Mittlerer Ausgangswert (SD) 28.43 (8.91) 29.00 (8.93)
Veränderung (LS mean) ab 5.37 (0.31) 7.06 (0.29)
Ausgangswert
Unterschied gegenüber Placebo (95% -1.69 (-2.52, -0.86)
CI)
Frauen
Mittlerer Ausgangswert (SD) 28.40 (9.15) 29.45 (8.61)
Veränderung (LS mean) ab 5.59 (0.40) 7.20 (0.39)
Ausgangswert
Unterschied gegenüber Placebo (95% -1.61 (-2.71, -0.52)
CI)
Männer
Mittlerer Ausgangswert (SD) 28.46 (8.60) 28.40 (9.32)
Veränderung (LS mean) ab 5.09 (0.47) 6.88 (0.45)
Ausgangswert
Unterschied gegenüber Placebo (95% -1.79 (-3.07, -0.52)
CI)
<65 Jahre
Mittlerer Ausgangswert (SD) 27.82 (9.67) 27.11 (9.40)
Veränderung (LS mean) ab 8.34 (0.94) 7.90 (0.96)
Ausgangswert
Unterschied gegenüber Placebo (95% 0.44 (-2.19, 3.06)
CI)
65-74 Jahre
Mittlerer Ausgangswert (SD) 27.82 (8.80) 28.74 (9.39)
Veränderung (LS mean) ab 4.86 (0.45) 7.51 (0.44)
Ausgangswert
Unterschied gegenüber Placebo (95% -2.66 (-3.89, -1.42)
CI)
> 75 Jahre
Mittlerer Ausgangswert (SD) 29.16 (8.83) 29.59 (8.36)
Veränderung (LS mean) ab 5.21 (0.46) 6.47 (0.43)
Ausgangswert
Unterschied gegenüber Placebo (95% -1.26 (-2.49, -0.04)
CI)
Abkürzungen: ApoE-ε4 = Allel-Subtyp 4 des Gens, das für das Apolipoprotein Klasse E kodiert; CDR-SB = Clinical Dementia Rating Scale – Sum of boxes; CI = confidence interval, Konfidenzintervall; Dona = Donanemab; iADRS = integrated Alzheimers Disease Rating Scale; LSM = Least-Square mean, Mittelwert nach der Methode der kleinsten Quadrate; MMRM = Mixed Model for Repeated Measures, gemischtes Modell für wiederholte Messungen; SD = standard deviation, Standardabweichung.
Biomarker
Der prozentuale Anteil der Patienten unter Donanemab mit Amyloid-Clearance (d.h. weniger als 24.1 Centiloide oder visuell negativ im Amyloid-PET-Scan) in der Studie TRAILBLAZER-ALZ 2 wird in Abbildung 2 dargestellt.
Unter Donanemab wurde eine Reduktion der P-tau217-Konzentration (Log 10) im Plasma beobachtet im Vergleich zu Placebo. In der Population mit niedriger bis mittlerer Tau-Konzentration (498 Patienten unter Donanemab vs. 494 Patienten unter Placebo) betrug die mittlere Veränderung (LS mean ± SE) -0.19 ± 0.012 und -0.26 ± 0.015 in den Wochen 24 und 76, im Vergleich zu Placebo (p<0.0001 zu beiden Zeitpunkten). Damit übereinstimmend zeigte die kombinierte Population eine mittlere Veränderung (Log LS mean ± SE) von -0.17± 0.011 und -0.23± 0.013 in den Wochen 24 und 76, im Vergleich zu Placebo (p<0.0001 zu beiden Zeitpunkten).
Abbildung 2: Prozentualer Anteil der Patienten in der indizierten Population unter Donanemab mit erreichter Amyloid-Plaque-Clearance gemessen anhand von Amyloid-PET über 76 Wochen in der Studie TRAILBLAZER-ALZ 2.
Population mit hoher Tau-Konzentration in der indizierten Population.
In der Population mit hoher Tau-Konzentration (218 unter Donanemab und 235 Patienten unter Placebo) zeigte Donanemab eine Verlangsamung der klinischen Verschlechterung um 8% (1.55 ± 1.66 [p=0.351) im iADRS und 18% (-0.60 ± 0.28 [p=0.032]) im CDR-SB in Woche 76 im Vergleich zu Placebo.
TRAILBLAZER-ALZ-6-Studie Phase-III
Das Dosierungsschema mit 350/700/1050 mg Donanemab und nachfolgend 1400 mg alle 4 Wochen wurde untersucht in einer multizentrischen, randomisierten, doppelblinden Studie der Phase IIIb bei Erwachsenen mit früher, symptomatischer AD (MCI aufgrund von AD oder leichte AD-Demenz, MMSE-Score 20 bis 28 einschliesslich) und Hinweis auf eine Amyloid-beta-Pathologie, bestätigt durch einen Amyloid-PET-Scan.
843 Patienten wurden im Verhältnis 1:1:1:1 randomisiert und erhielten Donanemab in vier unterschiedlichen Dosierungsschemata über insgesamt 72 Wochen, 700 mg in den ersten drei Infusionen, danach 1400 mg alle 4 Wochen (n=207), oder eines von drei alternativen Dosierungsschemata (einschliesslich das Dosierungsschema 350/700/1050 mg, gefolgt von 1400 mg alle 4 Wochen; n=212), wobei in allen Schemata insgesamt die gleiche Arzneimittelgesamtmenge angewendet wurde.
Der primäre Endpunkt der Studie war der Anteil der Teilnehmer mit jeglichem Auftreten von ARIA-E bis Woche 24. Die Ergebnisse zeigten, dass bei 14% der Patienten mit 350/700/1050 mg und nachfolgend 1400 mg alle 4 Wochen bis Woche 24 ein ARIA-E aufgetreten war, im Vergleich zu 24% der Patienten mit 700/700/700 mg und nachfolgend 1400 mg alle 4 Wochen, ein 41% geringeres relatives Risiko. Die in Woche 24 beobachtete Abnahme der Amyloid-Plaques war in allen Dosierungsschemata ähnlich
PharmakokinetikDie Pharmakokinetik (PK) von KISUNLA wurde anhand von Daten nach Einmal- und Mehrfachgabe charakterisiert. Bei Anwendung alle 4 Wochen erfolgt eine Akkumulation <1.3-fach; die Steady-state-Expositionen werden nach einer Einmalgabe erreicht. Bei Einmalgaben zwischen 350 mg bis 2800 mg (dem ~2-Fachen der empfohlenen Dosis von 1400 mg für ein Körpergewicht von 70 kg) und Mehrfachgaben von 350 und 1400 mg stiegen die Expositionen (Cmax und AUC) proportional. Eine ähnliche Exposition wurde bei einem Dosierungsschema von 350/700/1050 mg und dann 1400 mg alle 4 Wochen beobachtet, verglichen mit dem Dosierungsschema, das in den Studien zur klinischen Wirksamkeit verwendet wurde (700 mg für die ersten drei Infusionen, dann 1400 mg alle 4 Wochen).
Das empfohlene Titrationsschema aus der TRAILBLAZER-ALZ-6-Studie und das in der pivotalen Phase 3 TRAILBLAZER-ALZ-2-Studie untersuchte Titrationsschema stützen sich auf PK/PD Bridging-Daten. Die in der TRAILBLAZER-ALZ-6-Studie beobachteten kumulativen Dosierungen (Woche 0 bis 12), kumulativen AUC (Woche 0-12) und durchschnittlichen Konzentrationen im Steady-state (Cav,ss) für das empfohlene Titrationsschema und das in der Zulassungsstudie Phase 3 untersuchte Titrationsschemata waren ähnlich und überlappten. Für die Exposition (Cav,ss) wurde die Nicht-Unterlegenheit gezeigt, definiert als eine Untergrenze des 90% Konfidenzintervalls der geometrischen mittleren Ratio ≥0.8 für das empfohlene Titrationsschema im Vergleich zum in der Zulassungsstudie Phase 3 untersuchtem Titrationsschema. Unterstützende Daten zeigten eine ähnliche beobachtete PD (Amyloid-Plaque-Reduktion) in den Wochen 24 und 52.
Absorption
Donanemab wird nur intravenös angewendet.
Distribution
Nach intravenöser Anwendung wird Donanemab zweiphasig eliminiert. Das zentrale Verteilungsvolumen beträgt 3.36 l mit 18.7% inter-individueller Variabilität. Das periphere Verteilungsvolumen beträgt 4.83 l mit 93.9% inter-individueller Variabilität.
Metabolismus
Donanemab ist ein monoklonaler Antikörper. Es wird erwartet, dass es über katabole Wege in gleicher Weise wie endogenes IgG in kleine Peptide und Aminosäuren abgebaut wird. Daher gibt es keine metabolische Hemmung oder Induktion enzymatischer Wege. Es ist nicht davon auszugehen, dass Donanemab durch die Cytochrom-P450 Enzyme, die für den Metabolismus und die Eliminierung kleiner Moleküle verantwortlich sind, metabolisiert wird. Aktive Metabolite werden daher nicht erwartet.
Elimination
Die Halbwertszeit von Donanemab beträgt etwa 12.1 Tage. Die Clearance von Donanemab beträgt 0.0255 l/h (24.9% inter-individuelle Variabilität).
Kinetik spezieller Patientengruppen
Alter, Geschlecht und Körpergewicht
Populationspharmakokinetische Analysen zeigten, dass Alter, Geschlecht oder Herkunft keinen Einfluss auf die Pharmakokinetik von Donanemab hatten. Während das Körpergewicht Einfluss auf Clearance und Verteilungsvolumen hatte, lassen die daraus resultierenden Veränderungen nicht darauf schliessen, dass eine Dosisanpassung erforderlich ist.
Eingeschränkte Nieren- und Leberfunktion
Populationspharmakokinetische Analysen zeigten, dass eine eingeschränkte Nieren- und Leberfunktion keinen Einfluss auf die Pharmakokinetik von Donanemab hatte.
Präklinische DatenSicherheitspharmakologie / Toxizität nach wiederholter Gabe
Basierend auf den konventionellen Studien bei wiederholter Gabe lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Es wurden keine Studien durchgeführt, um Donanemab auf mögliche Karzinogenität, Genotoxizität oder Beeinträchtigung der Fruchtbarkeit zu testen.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend
Beeinflussung diagnostischer Methoden
Nicht bekannt
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit "EXP" bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ungeöffnete Durchstechflasche
Bis zur Verwendung im Kühlschrank lagern (2 ºC bis 8 ºC).
Das Arzneimittel kann ungekühlt bis zu 3 Tage bei Raumtemperatur (20 °C bis 25 °C) aufbewahrt werden.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Nicht einfrieren. Nicht schütteln.
Ausser Reichweite von Kindern aufbewahren.
Nach Verdünnung
Die chemische und physikalische Stabilität der verdünnten Lösung wurde für einen Zeitraum von 72 Stunden bei 2°C bis 8°C oder von 12 Stunden bei Raumtemperatur (20°C bis 25°C) gezeigt.
Die Infusionszeit ist Teil der Aufbewahrungszeit.
Aus mikrobiologischer Sicht ist die verdünnte Lösung sofort zu verwenden. Wird die Lösung nicht sofort verwendet, so liegen Lagerungsbedingungen und Dauer der Lagerung in der Verantwortung des Anwenders und dürfen im Normalfall nicht mehr als 24 Stunden bei 2 bis 8°C betragen, es sei denn die Verdünnung ist unter kontrollierten und validierten aseptischen Bedingungen erfolgt. Sofern die Verdünnung unter kontrollierten und validierten aseptischen Bedingungen erfolgt ist, kann die Lösung bis zu 72 Stunden gekühlt bei 2°C bis 8°C oder bis zu 12 Stunden bei Raumtemperatur (20°C bis 25°C) aufbewahrt werden.
Die zubereitete Lösung nicht einfrieren.
Hinweise für die Handhabung
Die Kisunla Infusionslösung soll durch qualifiziertes Fachpersonal unter Verwendung einer aseptischen Technik zubereitet und angewendet werden.
Lassen Sie Donanemab vor der Zubereitung über etwa 30 Minuten Raumtemperatur annehmen.
Überprüfen Sie den Inhalt der Durchstechflasche auf Partikel und Verfärbungen. Entsorgen Sie die Durchstechflasche, wenn Sie Partikel oder Verfärbungen feststellen.
Nach der Verdünnung und Zubereitung in Natriumchloridlösung 0.9% (9 mg/ml) zur Injektion wird Donanemab als intravenöse Infusion angewendet:
-Berechnen Sie das erforderliche Volumen von Donanemab zur Herstellung der Infusionslösung:Für 350 mg Donanemab: 20 mlFür 700 mg Donanemab: 40 mlFür 1050 mg Donanemab: 60 mlFür 1400 mg Donanemab: 80 ml
-Entnehmen Sie das benötigte Volumen von Donanemab und verdünnen Sie es weiter in einem Infusionsbeutel, der Natriumchloridlösung 0.9% (9 mg/ml) zur Injektion enthält, so dass die endgültige Konzentration 4 mg/ml bis 10 mg/ml beträgt. Verwenden Sie nur Natriumchloridlösung 0.9% (9 mg/ml) zur Injektion für die Zubereitung.
-Drehen Sie den Infusionsbeutel zum Mischen vorsichtig um.
-Infundieren Sie die verdünnte Lösung über einen Zeitraum von mindestens 30 Minuten
-Wenden Sie die gesamte Infusionslösung an.
Spülen Sie die Infusionsleitung am Ende der Infusion mit Natriumchloridlösung 0.9% (9 mg/ml) zur Injektion.
Patienten sollen nach der Infusion über mindestens 30 Minuten beobachtet werden.
Zulassungsnummer69523 (Swissmedic)
PackungenKisunla, Konzentrat zur Herstellung einer Infusionslösung (intravenöse Infusion)
mit 350 mg Donanemab in 20 ml: 1 Durchstechflasche (A)
ZulassungsinhaberinEli Lilly (Suisse) SA, 1214 Vernier/GE.
Stand der InformationJanuar 2026
Donanemab Neuroimaging Manual - Leitfaden für die neuronale Bildgebung
Abkürzungen und Definitionen
Begriff Definition
2D Zweidimensional
3DT1 Dreidimensional, T1-Wichtung
Aβ Beta-Amyloid
AD Alzheimer's disease, Alzheimer-Krankheit
ApoE Apolipoprotein E
ARIA Amyloid-related imaging abnormalities, Amyloid-bedingte Bildgebungsanomalien
CL Centiloid
DWI Diffusion Weighted Imaging, diffusionsgewichtete Bildgebung
FLAIR Fluid-Attenuated Inversion Recovery, FLAIR-Sequenz
FORE Fourier-Rebinning
GRE Gradientenecho
IV Intravenös
MNI Montreal Neurological Institute
MRT Magnetresonanztomographie
MUBADA Multiblock barycentric discriminant Analysis
NFT Neurofibrilläre Tangles
OSEM Ordered subset expectation Maximisation
PET Positronen-Emissions-Tomographie
RAMLA Row-action maximum likelihood Algorithm
ROI Region of Interest, Bereich von Interesse
SUVr Standardised uptake value Ratio
Einleitung
Amyloid-Plaques (extrazelluläre, unlösliche Aβ-Peptid-Agglomerate) und NFTs (hyperphosphorylierte, fehlgefaltete Tau-Agglomerate) sind wesentliche Indikatoren einer AD und entscheidend für die neuropathologische Diagnose der AD (Jack et al. 2024). Die Akkumulation von Aβ-Peptiden im Gehirnparenchym ist das früheste Anzeichen einer AD und kann nachfolgend zu pathologischen Veränderungen, einschliesslich einer Tauopathie führen (Jack et al. 2024). Manchmal kann eine Tau-Pathologie unabhängig von einer Aβ-Akkumulation auftreten, was darauf hindeutet, dass es Amyloid-unabhängige und Amyloid-vermittelte Tauopathien gibt (van der Kant et al. 2020).
Donanemab bindet an die N-terminal verkürzte Form des Beta-Amyloids und unterstützt die Plaque-Entfernung durch Mikroglia-vermittelte Phagozytose (DeMattos et al. 2012). Die klinischen Studien zu Donanemab umfassten daher Messungen der Amyloid- und ebenso der Tau-Pathologie vor und nach der Behandlung mit Donanemab (Mintun et al. 2021; Sims et al. 2023). PET dient als Referenz für die Visualisierung und Quantifizierung von Aβ-Plaques und NFT-Ablagerung in vivo.
MRT bei Patienten zur Vorbereitung auf eine oder zur Überwachung während einer gezielten Amyloid-Therapie hat zwei Hauptgründe (By et al. 2025):
-Untersuchung auf ausschliessende Befunde und
-Sicherheitsüberwachung zur Erkennung von während der Behandlung aufgetretenen neuronalen Bildgebungsanomalien, insbesondere von ARIA durch gezielte Amyloid-Therapien.
Aufgrund dieser Erwägungen umfasste das Programm für die neuronale Bildgebung in der TRAILBLAZER-ALZ 2 ein Amyloid- und ein Tau-PET, sowie mehrere MRT-Untersuchungen (Tabelle 1). Es sei angemerkt, dass die spezifischen Auswertungen aller PET- und MRT-Scans, einschliesslich die visuellen und quantitativen Bewertungen, zentral durch PET- bzw. MRT-Anbieter erfolgten. Ein Scan erfolgte vor der Behandlung im Screening oder zu Studienbeginn, um die Eignung festzustellen, oder zur Überwachung der Behandlung. Beim Screening erfolgte ausserdem eine APOE-Genotypisierung anhand von Blutproben, sofern solche Untersuchungen nicht durch länderspezifische Gesetze und Vorschriften untersagt waren.
Dieser Leitfaden für die neuronale Bildgebung beleuchtet wesentliche Elemente der PET- und MRT-Untersuchungen, die in der TRAILBLAZER-ALZ 2 durchgeführt wurden. Weitere Details wurden in den Standardarbeitsabläufen der Bildgebungsanbieter und in den Bildgebungshandbüchern vor Ort festgelegt.
Tabelle 1. Funktion der Bildgebungsmodalitäten in der Studie TRAILBLAZER-ALZ 2
Modalität Screening/Ausgangswert Während der Beobachtungspha
se
Amyloid-PET Vorhandensein einer Amyloid-Plaque-Konzentratio
Amyloid-Pathologie, ≥37 n und Veränderung der
CL (Q) Amyloid-Plaque-Konzentratio
n im Gehirn gegenüber dem
Ausgangswert (Q)
Tau-PET Vorhandensein einer Veränderung der Amyloid-Pla
Tau-Pathologie (VQ) que-Konzentration im
Gehirn gegenüber dem
Ausgangswert (Q)
MRT 3DT1 Anatomisches Referenzbild Veränderung der
für die Tau-PET-Quantifizie volumetrischen
rung MRT-Messung gegenüber
dem Ausgangswert (Q)
2D, axial, FLAIR Erkennung zerebraler Erkennung zerebraler Ödeme
Ödeme und Läsionen in und Läsionen in der
der weissen Substanz (V) weissen Substanz (V)
2D, axial, T2* GRE Erkennung von Hämosiderin Erkennung von Hämosiderinab
ablagerungen und anderen lagerungen und anderen
Arten hämorrhagischer Arten hämorrhagischer
Läsionen (V) Läsionen (V)
2D, axial, T2 Erkennung von Läsionen Erkennung von Läsionen in
Turbo/Fast Spin Echo in der weissen Substanz der weissen Substanz und
und anderen MRT-Befunden anderen MRT-Befunden (V)
(V)
2D, axial, DWI Erkennung von akuten Erkennung von akuten
Infarkten und Schlaganfäl Infarkten und Schlaganfälle
len (V) n(V)
Abkürzungen: 2D = Zweidimensional; 3DT1 = Dreidimensional, T1-Wichtung; CL = Centiloide; DWI = Diffusion Weighted Imaging, diffusionsgewichtete Bildgebung; GRE = Gradientenecho; MRT = Magnetresonanztomographie; PET = Positronen-Emissions-Tomographie; Q = Quantitative Beurteilung mit spezialisierter Bildverarbeitung und Analysesoftware; V = Visuelle Beurteilung; VQ = Visuelle und quantitative Beurteilung.
Leitfaden für die neuronale Bildgebung
Amyloid-PET
Amyloid-PET-Scans mit den radioaktiven Tracern 18F-Florbetapir oder 18F-Florbetaben wurden zur Feststellung der Eignung verwendet, indem der Grad der Amyloid-Pathologie eines Teilnehmers beurteilt wurde. Bei geeigneten Teilnehmern erfolgten weitere Amyloid-PET-Scans in bestimmten Studienvisitenintervallen, um
a.die Amyloid-Konzentrationen nach der Behandlung zu beurteilen und verblindet von Donanemab auf Placebo umzustellen, wenn zuvor festgelegte Amyloid-PET-Kriterien erreicht wurden und
b.die Veränderung der Amyloid-Plaque-Ablagerung im Gehirn in Woche 76 gegenüber dem Ausgangswert zu beurteilen.
Für den 18F-Florbetapir-Scan erhielten die Teilnehmer eine einzelne IV Bolusinjektion mit einer Zieldosis von 370 MBq (10 mCi) 18F-Florbetapir, gefolgt von einer Spülung mit Natriumchloridlösung; der Scan über 20 Minuten (4 Rahmen x 5 Minuten) begann etwa 50 Minuten nach der Injektion. Für den 18F-Florbetaben-Scan erhielten die Teilnehmer eine einzelne IV Bolusinjektion mit einer Zieldosis von 300 MBq (8.1 mCi) 18F-Florbetaben, gefolgt von einer Spülung mit Natriumchloridlösung; der Scan über 20 Minuten (4 Rahmen x 5 Minuten) begann etwa 90 Minuten nach der Injektion.
Die PET-Bilder wurden mit Scanner-spezifischen Protokollen und der folgenden Parameter-Auswahl rekonstituiert: iterativer Rekonstruktionsalgorithmus (FORE, OSEM oder RAMLA), 3 bis 6 Iterationen, 16 bis 33 Teildatensätze. Nach der Rekonstruktion wurden Gauss-Filter von 3 bis 5 mm eingesetzt (ausser bei Philips-Scannern, bei denen die Relaxationsparameter 'Normal' oder 'Scharf' verwendet wurden).
In der Studie TRAILBLAZER-ALZ 2 erfolgten nur quantitative Bestimmungen der Amyloid-PET-Bilder.
1.1.1 Amyloid-PET-Scan im Screening bzw. zu Studienbeginn
Die über 5 Minuten aufgenommenen PET-Bilder wurden für Bewegungen korrigiert und dann zu einem einzelnen statischen Bild gemittelt (Shcherbinin et al. 2016; Pontecorvo et al. 2019). Das gemittelte Amyloid-PET-Bild aus dem Screening bzw. vom Studienbeginn wurde räumlich auf die Referenz-Amyloid-PET-Vorlage im Standard-Gehirnraum (MNI-Space) normalisiert. MRT-Bilder wurden nicht für die quantitative Eignungsbeurteilungen verwendet.
Die quantitative Bewertung der 18F-Florbetapir- oder 18F-Florbetaben-Bilder in der TRAILBLAZER-ALZ 2 folgte der am häufigsten verwendeten Technik, einer SUVr-Schätzung. SUVr beschreibt das Verhältnis des durchschnittlichen PET-Signals in einer neokortikalen kombinierten Region (Zielregion), skaliert durch das durchschnittliche PET-Signal in einer Referenzregion, die typischerweise von Amyloid-Plaques verschont bleibt, zum Beispiel das Cerebellum. Zur Berechnung der SUVr wurden alle Ziel- und Referenz-ROIs auf das räumlich normalisierte PET-Bild im MNI-Bereich des Teilnehmers angewendet. Entsprechend der zuvor entwickelten Amyloid-PET-Methodik wurde SUVr als ungewichteter Durchschnitt aus 6 kortikalen Regionen berechnet (mesial-orbital-frontal, anterior-cingulär, Precuneus, posterior-cingulär, parietal und temporal), mit dem gesamten Cerebellum als Referenzregion (Joshi et al. 2015).
Zur Harmonisierung der Amyloid-Konzentrations-Messungen mit verschiedenen PET-Tracern, 18F-Florbetapir oder 18F-Florbetaben, wurden zuvor validierte Verfahren verwendet, um Tracer-spezifische SUVr-Messungen in einen gemeinsamen Bezugsrahmen, die CL-Skala, umzuwandeln (Klunk et al. 2015). Die CL-Skala ermöglicht die Interpretation der SUVr aus dem Amyloid-PET anhand von 2 Festpunkten: 0 CL (durchschnittliches Signal im Amyloid-PET bei jungen Menschen [≤45 Jahre], ohne kognitive Beeinträchtigung) und 100 CL (durchschnittliches Signal im Amyloid-PET von Patienten mit typischer leichter bis mittelschwerer AD) (Klunk et al. 2015; Iaccarino et al. 2025). Verschiedene Transformationsformeln wurden verwendet, um 18F-Florbetapir und 18F-Florbetaben SUVr-Messungen in CL-Einheiten umzuwandeln, was die Einführung identischer CL-Schwellenwerte für die beiden radioaktiven Amyloid-PET-Tracer ermöglichte.
In der TRAILBLAZER-ALZ 2 wurde bei den geeigneten Teilnehmern die Amyloid-Pathologie (≥37 CL) mit 18F-Florbetapir oder 18F-Florbetaben beurteilt.
1.1.2 Amyloid-PET-Scans während der Beobachtungszeit
Während der Beobachtungszeit erfolgten Amyloid-PET-Scans in den Wochen 24, 52 und 76 der TRAILBLAZER-ALZ 2. Wie bei den Auswertungen im Screening bzw. zu Studienbeginn wurden die während der Beobachtungszeit aufgenommenen Amyloid-PET-Bilder bewegungskorrigiert und dann gemittelt. Die räumliche Normalisierung auf die Referenz-Amyloid-PET-Vorlage wurde jedoch anders durchgeführt. Beurteilt wurde
a.die Amyloid-Konzentration nach der Behandlung, für die verblindete Umstellung von Donanemab auf Placebo, wenn zuvor festgelegte Amyloid-PET-Kriterien erreicht wurden und
b.die Veränderung der Amyloid-Plaque-Ablagerung im Gehirn bis Woche 76 gegenüber dem Ausgangswert.
Für die Bewertung der Amyloid-Konzentrationen nach der Behandlung wurde das gemittelte Bild aus der Beobachtungszeit direkt auf die Referenz-Amyloid-PET-Vorlage normalisiert, analog zum Verfahren, das beim Scan aus dem Screening bzw. zu Studienbeginn angewendet wurde.
Wenn die Amyloid-Plaque-Konzentration in den Wochen 24 oder 52 weniger als 11 CL in einem einzelnen Amyloid-PET-Scan oder weniger als 25, aber mehr als 11 CL in zwei aufeinanderfolgenden Amyloid-PET-Scans betrug, wurde Donanemab verblindet auf Placebo umgestellt (Sims et al. 2023).
Um beim einzelnen Teilnehmer die Veränderung der Amyloid-Plaque-Ablagerung im Gehirn von Beginn bis Woche 76 zu bewerten, wurde das gemittelte Bild zunächst auf das entsprechende Amyloid-PET-Anfangsbild und dann auf die Standard-Gehirnvorlage abgestimmt, wobei für die kombinierte Registrierung dieselben Parameter wie für das PET-Anfangsbild verwendet wurden. Zusätzlich zur Veränderung der Amyloid-Plaque-Konzentration gegenüber dem Ausgangswert wurde der prozentuale Anteil der Teilnehmer mit erreichter Amyloid-Clearance (<24.1 CL) berechnet. Ein Grenzwert von 24.1 CL wurde aufgrund der zuvor etablierten Unterscheidung von nicht vorhandenen oder wenigen versus mittleren bis häufigen Plaques in Autopsie-bestätigten Daten zwischen Amyloid-positiven und Amyloid-negativen Fällen verwendet (Joshi et al. 2015; Navitsky et al. 2018).
MRT-Bilder wurden nicht für quantitative Beurteilungen von während der Beobachtungszeit erstellten Amyloid-PET-Bildern verwendet.
Tau-PET
Tau-PET mit 18F-Flortaucipir wurden verwendet, um durch Beurteilung des Grads der Tau-Pathologie eines Teilnehmers die Eignung festzustellen. Bei geeigneten Teilnehmern erfolgten in Woche 76 Tau-PET-Scans während der Beobachtungszeit, um die Wirkung von Donanemab versus Placebo auf die Tau-Ablagerung im Gehirn zu beurteilen.
In der TRAILBLAZER-ALZ 2 erhielten die Teilnehmer eine einzelne IV Bolusinjektion mit einer Zieldosis von 370 MBq (10 mCi) 18F-Flortaucipir, gefolgt von einer Spülung mit Natriumchloridlösung; der dynamische Gehirn-Scan über 30 Minuten (6 Rahmen x 5 Minuten) erfolgte etwa 75 Minuten nach der Injektion.
Die PET-Bilder wurden mit Scanner-spezifischen Protokollen und der folgenden Parameter-Auswahl rekonstituiert: iterativer Rekonstruktionsalgorithmus (FORE, OSEM oder RAMLA), 3 bis 6 Iterationen, 16 bis 33 Teildatensätze. Nach der Rekonstruktion wurden Gauss-Filter von 3 bis 5 mm eingesetzt (ausser bei Philips-Scannern, bei denen die Relaxationsparameter 'Normal' oder 'Scharf' verwendet wurden).
Für die quantitativen Auswertungen der Flortaucipir-Bilder wurden die über 5 Minuten aufgenommenen PET-Bilder für Bewegungen und Aufnahmezeit korrigiert und dann zu einem einzelnen statischen Bild gemittelt. Die Korrektur für die Aufnahmezeit erfolgte, weil die SUVr-Werte mit Flortaucipir 75 Minuten nach der Injektion kein stabiles Plateau erreichen, sondern während des möglichen Bildgebungs-Fensters weiterhin ansteigen (Shcherbinin et al. 2016). Folglich könnten unterschiedliche Zeiten zwischen Injektion und Scan-Aufnahme zwischen den Scans zu Beginn und in Woche 76 die SUVr-Auswertungen beeinflussen. Details zur Korrektur der Aufnahmezeit wurden zuvor beschrieben (Pontecorvo et al. 2019).
1.1.3 Tau-PET-Scan im Screening bzw. zu Studienbeginn
Bei Teilnehmern der TRAILBLAZER-ALZ 2 wurden Flortaucipir-Scans aus dem Screening bzw. zu Studienbeginn visuell und quantitativ beurteilt, um die vorhandene Tau-Pathologie zu bestätigen.
Die Scans wurden mit visueller Untersuchung interpretiert, ob sie regionale Muster einer Tracer-Aufnahme aufweisen, die einem mässigen (τAD+) oder fortgeschrittenen (τAD++) Taumuster entsprechen, oder ohne einem mit einer AD übereinstimmenden Taumuster (negatives [τAD-] Taumuster) (Fleisher et al. 2020; Lu et al. 2021). Das τAD- Taumuster war definiert als kein Anstieg der neokortikalen Aktivität oder ein Anstieg der neokortikalen Aktivität, der auf mesial-temporale, anterolateral-temporale und/oder frontale Regionen begrenzt ist. Ein mässiges τAD+ Taumuster war definiert als eine gesteigerte neokortikale Aktivität posterolateral-temporal oder in der Occipitalregion. Ein fortgeschrittenes τAD++ Taumuster war definiert als entweder eine gesteigerte neokortikale Aktivität parietal oder in der Precuneus-Region oder eine gesteigerte Aktivität in der frontalen Region, zusammen mit Anstiegen in posterolateral-temporalen, parietalen oder occipitalen Regionen. Die Leser waren Ärzte mit Fachwissen in den studienspezifischen Krankheitsindikation(en) und mit Erfahrung in der Beurteilung klinischer Studien. Vor einer Auswertung der Studienbilder absolvierten die Leser eine spezielle Schulung, mit Nachschulung während der Studie bei Bedarf.
Die quantitative Bewertung von zu Studienbeginn oder während der Beobachtungszeit aufgenommenen 18F-Flortaucipir-Bildern in der TRAILBLAZER-ALZ 2 folgte der am häufigsten verwendeten Technik, einer SUVr-Schätzung. SUVr beschreibt das Verhältnis des durchschnittlichen PET-Signals in einer neokortikalen kombinierten Region (Zielregion) dar, skaliert durch das durchschnittliche PET-Signal in einer Referenzregion, die typischerweise von NFTs verschont bleibt, zum Beispiel das Cerebellum oder die weisse Substanz. Zur Berechnung der SUVr wurden alle Ziel- und Referenz-ROIs auf das räumlich normalisierte PET-Bild im MNI-Bereich des Teilnehmers angewendet. Verschiedene räumliche Normalisierungstechniken und SUVr-Messungen wurden angewandt, um anhand von Scans im Screening oder zu Beginn über die Eignung zu entscheiden und um anhand von Scans während der Beobachtungszeit beim einzelnen Teilnehmer Veränderungen der Tau-Ablagerung zu beurteilen.
Für die quantitative Beurteilung der Eignung wurde das im Screening bzw. zu Studienbeginn aufgenommene, gemittelte Flortaucipir-Bild räumlich auf die Referenz-Tau-PET-Vorlage im MNI-Raum normalisiert. MRT-Bilder wurden nicht für quantitative Beurteilungen der Eignung verwendet. Die globale neokortikale Tau-Konzentration wurde mit einem zuvor veröffentlichten, AD-Signatur-gewichteten neokortikalen Tau SUVr quantitativ gemessen (MUBADA, Devous et al. 2018), mit Bezug auf die Referenzsignalintensität in der subjektspezifischen weissen Substanz (PERSI, Southekal et al. 2018).
Die Entscheidung über die Eignung (Beurteilung als Tau-positiv) basierte auf visuellen und quantitativen Beurteilungen (siehe Abbildung 1). Entsprechend der kombinierten visuellen und quantitativen Eignungsmethode (tauVQ), wurde die Tau-Konzentration bei Teilnehmern der TRAILBLAZER-ALZ 2 bei entweder (1) τAD+ Muster mit einer AD-Signatur gewichteten neokortikalen Tau SUVr ≥1.10 oder (2) τAD++ Muster als positiv erachtet. Für Scans mit einem τAD- Muster wurde eine unzureichende Tau-Konzentration angenommen und diese ohne Bild-Quantifizierung von der Studie ausgeschlossen. Wenn ein τAD+ Scan nicht quantitativ ausgewertet werden konnte, wurde der Scan visuell ausgewertet, um die Eignung zu bestimmen, dies erfolgte durch einen unabhängigen Prüfer.

Abkürzungen: SUVr = Standardised uptake value Ratio.
Abbildung 1. Tau-PET zur Prüfung der Eignung in TRAILBLAZER-ALZ 2.
1.1.4 Tau PET-Scan während der Beobachtungszeit
Tau-PET-Scans während der Beobachtungszeit wurden in der TRAILBLAZER-ALZ 2 in Woche 76 durchgeführt.
Für die longitudinale Tau-PET-Analyse wurde eine strengere räumliche Normalisierung unter Einbeziehung eines individuellen, zu Beginn aufgenommenen 3DT1-MRT-Bildes implementiert. Zuerst wurde das gemittelte Tau-PET-Bild zu Studienbeginn zusammen mit dem 3DT1-MRT-Bild des Teilnehmers registriert. Das Tau-PET-Bild nach 18 Monaten wurde für Bewegung und Zeit korrigiert, gemittelt, zuerst am PET-Bild zu Studienbeginn ausgerichtet und dann am entsprechenden MRT-Bild zu Studienbeginn, mit Verwendung derselben Parameter für die kombinierte Registrierung wie im Tau-PET-Scan zu Studienbeginn. Schliesslich wurde das zu Beginn aufgenommene 3DT1-MRT-Bild auf den MNI-Raum normalisiert, dabei wurde für die räumliche Normalisierung der zuvor kombiniert registrierten Basislinie und 18-Monats-Flortaucipir-PET-Bilder dieselbe MRT-zu-Atlas-Transformationsmatrix verwendet.
Für longitudinale Tau-PET-Analysen wurden in Bezug auf die zuvor entwickelte (Pontecorvo et al. 2017) zerebellare graue Referenzregion regionale und globale SUVr-Werte verwendet. In der Referenzregion wurde eine aus der zerebellären crustaneous ROI stammende Region der zerebellären grauen Substanz modifiziert, wofür eine iSignalverlagerung inferior um 6 mm gewählt wurde. Diese Modifikation wurde durchgeführt, um eine mögliche Überscheidung mit inferioren kortikalen Bereichen und der supratentoriellen Zerebrospinalflüssigkeit zu vermeiden. In longitudinalen Auswertungen umfassten die Ziel-ROIs alle automatisierten anatomischen Markierungsbereiche (Tzourio-Mazoyer et al. 2002) sowie weitere, individuelle Bereiche. Insbesondere wurde MUBADA SUVr (Devous et al. 2018) für die zerebelläre graue Referenzregion verwendet, um die Veränderung der globalen neokortikalen Tau-Ablagerung zu messen.
MRT
Im Screening und bei Visiten während der Beobachtungszeit wurden MRT-Untersuchungen durchgeführt. Eignungsuntersuchungen erfolgten zur Bewertung möglicher MRT-basierter Ausschlusskriterien. Bei Visiten während der Beobachtungszeit erfolgten volumetrische MRT-Messungen, um die Wirkungen von Donanemab versus Placebo auf die Volumina von Gehirnregionen zu beurteilen. Zudem erfolgten bei allen Visiten Sicherheitsuntersuchungen zum Auftreten von MRT-Befunden.
Geeignete MRT-Scanner hatten ausschliesslich eine magnetische Feldstärke von 1.5 Tesla oder 3.0 Tesla.
Jede MRT-Untersuchung bestand aus den folgenden MRT-Sequenzen:
-3DT1: Magnetisation-Prepared Rapid Gradient-Echo (Siemens)/ Inversion Recovery-Prepped Fast Spoiled Gradient-Recalled Echo (General Electric)/3D Turbo Field Echo (Philips)
-2D FLAIR
-2D Axial T2* GRE
-2D Axial T2 Turbo/Fast Spin-Echo und
-2D Axial DWI
Die Ziele dieser Sequenzen werden in Tabelle 1 beschrieben. Die genauen Erfassungsparameter hingen von Hersteller, Scanner-Modell, installierter Hard- und Software sowie der magnetischen Feldstärke ab. Alle Scanner wurden vom MRT-Anbieter qualifiziert, und die Erfassungsparameter mussten die vom MRT-Anbieter festgelegten Anforderungen erfüllen.
1.1.5 MRT-Scan im Screening bzw. zu Studienbeginn
Zu den wesentlichen, MRT-basierten Ausschlusskriterien gehörten (Sims et al. 2023)
-Vorhandene ARIA mit Ödem/Erguss (2D Axial FLAIR)
-Mehr als 4 zerebrale Mikrohämorrhagien (2D Axial T2* GRE)
-Mehr als 1 Bereich mit superfizieller Siderose (2D Axial T2* GRE)
-Eine intrazerebrale Hämorrhagie grösser als 1 cm (2D Axial T2* GRE) und
-Schwere Erkrankung der weissen Substanz (altersabhängige Veränderungen der weissen Substanz = 3 in jeder Hemisphäre der im Bewertungsparadigma bewerteten Regionen, 2D Axial T2 Turbo/Fast Spin Echo; Wahlund et al. 2001).
1.1.6 MRT-Scan während der Beobachtungszeit
MRT-Scans während der Beobachtungszeit wurden in den Wochen 4, 12, 24, 52 und 76 bzw. bei frühzeitigem Abbruch durchgeführt. Bei Bedarf wurden ungeplante Visiten durchgeführt.
Die Untersuchungen bei Visiten während der Beobachtungszeit umfassten Sicherheitsuntersuchungen zur Beurteilung der Sicherheit und Verträglichkeit von Donanemab anhand von MRT (ARIA und aufgetretene radiologische Befunde wie zerebrale Ödeme, Hämosiderin-Ablagerungen, Erkrankung der weissen Substanz, andere MRT-Abweichungen oder andere Zufallsbefunde).
Die Sicherheit und Verträglichkeit von Donanemab wurden mit MRT-Scans beurteilt, die während der Beobachtungszeit mit den folgenden Parametern durchgeführt wurden:
-Vorhandensein, radiologischer Schweregrad und Lokalisation von ARIA mit Ödemen/Erguss (2D Axial FLAIR, 2D Axial T2)
-Vorhandensein, Anzahl und Lokalisation von Mikrohämorrhagien (2D Axial T2* GRE)
-Vorhandensein und Lokalisation von intrazerebralen Hämorrhagien grösser als 1 cm (2D Axial T2* GRE)
-Vorhandensein und Lokalisation einer superfiziellen Siderose (2D Axial T2* GRE)
-Vorhandensein und radiologischer Schweregrad von hämorrhagischen ARIA-H (2D Axial T2*GRE)
-Vorhandensein und radiologischer Schweregrad einer Erkrankung der weissen Substanz (2D Axial FLAIR, 2D Axial T2) und
-Vorhandensein und Lokalisation einer anderen MRT-Abweichung (2D DWI, 2D Axial T2).
Bei Teilnehmern, bei denen ein ARIA festgestellt wurde, erfolgte alle 4 bis 6 Wochen eine Bildgebung bis zum Rückgang oder zur Stabilisierung (Sims et al. 2023). Die Behandlung von ARIA und die Leitlinien für Therapieunterbrechungen waren abhängig vom radiologischen Schweregrad und den Symptomen. Bei Unterbrechung der Infusionen wurde den Prüfärzten empfohlen, bis zum Rückgang der ARIA mit Ödemen/Erguss im radiologischen Bild und zur Stabilisierung der ARIA mit Mikrohämorrhagie und Hämosiderinablagerung abzuwarten, bevor die Infusionen wieder aufgenommen wurden. Bei intrazerebralen Blutungen grösser als 1 cm oder schwerwiegenden ARIA wurde zum endgültigen Abbruch geraten. Die endgültige Entscheidung zur Behandlung von ARIA traf der Prüfarzt. Die Gruppierung der bevorzugten Begriffe (preferred Term) für ARIA, die Schweregradbewertung und die Ereignisdefinitionen wurden veröffentlicht (Zimmer et al. 2025).
Referenzen
By S, Kahl A, Cogswell PM. Alzheimer's Disease clinical trials: what have we learned from magnetic resonance imaging. J Magn Reson Imaging. 2025;61(2):579-594. https://doi.org/10.1002/jmri.29462
DeMattos RB, Lu J, Tang Y, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76(5):908-920. https://doi.org/10.1016/j.neuron.2012.10.029
Devous MD Sr, Joshi AD, Navitsky M, et al. Test-retest reproducibility for the tau PET imaging agent flortaucipir F 18. J Nucl Med. 2018;59(6):937-943. https://doi.org/10.2967/jnumed.117.200691
Fleisher AS, Pontecorvo MJ, Devous MD, et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77(7):829-839. https://doi.org/10.1001/jamaneurol.2020.0528
Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1-13. https://doi.org/10.1016/j.jalz.2011.10.007
Iaccarino L, Burnham CS, Tunali I, et al. A practical overview of the use of amyloid-PET Centiloid values in clinical trials and research. NeuroImage: Clin. 2025;46:103765. https://doi.org/10.1016/j.nicl.2025.103765
Jack CR Jr, Andrews JS, Beach TG, et al. Revised criteria for diagnosis and staging of Alzheimer's disease: Alzheimer's Association Workgroup. Alzheimers Dement. 2024;20(8):5143-5169. https://doi.org/10.1002/alz.13859
Joshi AD, Pontecorvo MJ, Lu M, et al. A semiautomated method for quantification of F 18 florbetapir PET images. J Nucl Med. 2015;56(11):1736-1741. https://doi.org/10.2967/jnumed.114.153494
Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1):1-15.e154. https://doi.org/10.1016/j.jalz.2014.07.003
Lu M, Pontecorvo MJ, Devous MD, et al.; AVID Collaborators. Aggregated tau measured by visual interpretation of flortaucipir positron emission tomography and the associated risk of clinical progression of mild cognitive impairment and Alzheimer disease: results from 2 phase III clinical trials. JAMA Neurol. 2021;78(4):445–453. https://doi.org/10.1001/jamaneurol.2020.5505
Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691-1704. https://doi.org/10.1056/NEJMoa2100708
Navitsky M, Joshi AD, Kennedy I, et al. Standardization of amyloid quantitation with florbetapir standardized uptake value ratios to the Centiloid scale. Alzheimers Dement. 2018;14(12):1565-1571. https://doi.org/10.1016/j.jalz.2018.06.1353
Pontecorvo MJ, Devous MD Sr, Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140(3):748-763. https://doi.org/10.1093/brain/aww334
Pontecorvo MJ, Devous MD, Kennedy I, et al. A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer's disease dementia. Brain. 2019;142(6):1723-1735. https://doi.org/10.1093/brain/awz090
Shcherbinin S, Schwarz AJ, Joshi A, et al. Kinetics of the tau PET tracer 18F-AV-1451 (T807) in subjects with normal cognitive function, mild cognitive impairment, and Alzheimer disease. J Nucl Med. 2016;57(10):1535-1542. https://doi.org/10.2967/jnumed.115.170027
Sims JR, Zimmer JA, Evans CD, et al.; TRAILBLAZER-ALZ 2 Investigator. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512-527. https://doi.org/10.1001/jama.2023.13239
Southekal S, Devous MD Sr, Kennedy I, et al. Flortaucipir F 18 quantitation using parametric estimation of reference signal intensity. J Nucl Med. 2018;59(6):944-951. https://doi.org/10.2967/jnumed.117.200006
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15:273-289. https://doi.org/10.1006/nimg.2001.0978
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21-35. https://doi.org/10.1038/s41583-019-0240-3
Wahlund LO, Barkhof F, Bazekas F, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. 2001;32(6):1318-1322. https://doi.org/10.1161/01.STR.32.6.1318
Zimmer JA, Ardayfio P, Wang H, et al. Amyloid-related imaging abnormalities with donanemab in early symptomatic Alzheimer disease: secondary analysis of the TRAILBLAZER-ALZ and ALZ 2 randomized clinical trials. [published online ahead-of-print, 2025 March 10]. JAMA Neurol. 2025;e250065. https://doi.org/10.1001/jamaneurol.2025.0065
Stand der Information
Juli 2025.
Zusammenfassung des MRT-Protokolls für Donanemab
Abkürzungen und Definitionen
Begriff Definition
1.5T Magnetische Feldstärke von 1.5 Tesla
2D Zweidimensional
3T Magnetische Feldstärke von 3 Tesla
3DT1 Dreidimensional, T1-Wichtung
DWI Diffusion Weighted Imaging, diffusionsgewichtete Bildgebung
FLAIR Fluid-Attenuated Inversion Recovery
GRE Gradientenecho
MRT Magnetresonanztomographie
PET Positronen-Emissions-Tomographie
Einleitung/Hintergrund
MRT bei Patienten zur Vorbereitung auf eine oder Überwachung während einer Behandlung mit Donanemab hatten zwei wesentliche Zwecke:
-Untersuchung auf ausschliessende Befunde und
-Sicherheitsüberwachung zur Feststellung von während der Behandlung aufgetretenen Abweichungen in der neuronalen Bildgebung, insbesondere Amyloid-bedingte Bildgebungsanomalien und intrazerebrale Hämorrhagien grösser als 1 cm.
Diese Zusammenfassung des MRT-Protokolls beleuchtet die wesentlichen Elemente der MRT-Untersuchung.
Tabelle 1. beschreibt die in der Studie TRAILBLAZER-ALZ 2 implementierten MRT-Modalitäten
Tabelle 1. Funktion der MRT-Modalitäten in der Studie TRAILBLAZER-ALZ 2
Modalität Screening/Ausgangswert Während der Beobachtungsphase
3DT1 Anatomisches Referenzbild für die Veränderung der volumetrischen
Tau-PET-Quantifizierung MRT-Messung gegenüber dem
Ausgangswert (Q)
2D FLAIR Erkennung zerebraler Ödeme und Erkennung zerebraler Ödeme und
Läsionen in der weissen Substanz (V) Läsionen in der weissen Substanz (V)
2D Axial T2* GRE Erkennung von Hämosiderinablagerungen Erkennung von Hämosiderinablagerungen
und anderen Arten hämorrhagischer und anderen Arten hämorrhagischer
Läsionen (V) Läsionen (V)
2D Axial T2 Turbo/Fa Erkennung von Läsionen in der weissen Erkennung von Läsionen in der weissen
st Spin Echo Substanz und anderen MRT-Befunden (V) Substanz und anderen MRT-Befunden (V)
2D Axial DWI Erkennung von akuten Infarkten und Erkennung von akuten Infarkten und
Schlaganfällen (V) Schlaganfällen(V)
Abkürzungen: 2D = zweidimensional; 3DT1 = dreidimensional, T1-Wichtung; DWI = Diffusion Weighted Imaging, diffusionsgewichtete Bildgebung; GRE = Gradientenecho; MRT = Magnetresonanztomographie; PET = Positronen-Emissions-Tomographie; Q = Quantitative Untersuchung mit spezialisierter Bildverarbeitung und Analysesoftware; V = Visuelle Beurteilung.
Zusammenfassung des MRT-Protokolls für Donanemab
Geeignete MRT-Scanner hatten ausschliesslich eine magnetische Feldstärke von 1.5 Tesla oder 3.0 Tesla. Jede MRT-Untersuchung bestand aus den folgenden MRT-Sequenzen in chronologischer Reihenfolge der Aufnahme:
-3DT1 (3 bis 4 Minuten): Magnetization-Prepared Rapid Gradient-Echo (Siemens)/ Inversion Recovery-Prepped Fast Spoiled Gradient-Recalled Echo (General Electric)/ 3D Turbo Field Echo (Philips)
-2D FLAIR (2.5 bis 4.5 Minuten)
-2D Axial T2* GRE (3 bis 4 Minuten)
-2D Axial T2 Turbo/Fast Spin-Echo (1 bis 2 Minuten) und
-2D Axial DWI (weniger als 1 Minute).
Die Ziele dieser Sequenzen werden in Tabelle 1 hervorgehoben. Die genauen Erfassungsparameter hingen ab von
-Hersteller
-Scanner-Modell
installierter Hard- und Software und
der magnetischen Feldstärke
Die für die MRT-Scanner speziellen Parameter, die in der TRAILBLAZER-ALZ 2 verwendet wurden, sind Eigentum des MRT-Herstellers und wurden zwischen MRT-Herstellern und Feldstärken standardisiert, um Bilder von verschiedenen Scanner-Modellen und Feldstärken vergleichen zu können. Sequenzen bzw. Modalitäten, die für Sicherheitsuntersuchungen in den klinischen Studien zu Donanemab verwendet wurden, werden in neurodegenerativen MRT-Protokollen routinemässig verwendet (Di Muzio et al. 2015; Benzinger and Jindal 2023; Harper et al. 2014).
Veröffentlichte Konsensempfehlungen für klinische Protokolle sind unten aufgeführt (Benzinger [WWW]; Cogswell et al 2022; Cogswell et al 2025; Lawson et al 2023; Philips [WWW]). Diese sind kompatibel mit den Bildern, die während der Studien aufgenommen wurden. Detaillierte Parametereinstellungen für jede Sequenz werden im Anhang bereitgestellt. Tabelle 2 enthält die Verknüpfung zu den Referenztabellen, die die Parameter enthalten.
Tabelle 2. Inhaltsverzeichnis der Tabellen mit den MRT-Parametern
Magnetische Feldstärke (T) Ort
GE 1.5 T Anhang 1
GE 3 T Anhang 2
Siemens 1.5 T Anhang 3
Siemens 3 T Anhang 4
Philips 1.5 T Anhang 5
Philips 3 T Anhang 6
Abkürzungen: GE = General Electric; T = Tesla.
Referenzen
Benzinger TLS, Jindal S. MR imaging of neurodegeneration. In: Cross DJ, Mosci K, Minoshima S, eds. Molecular Imaging of Neurodegenerative Disorders. Cham: Springer; 2023:169-181. https://doi.org/10.1007/978-3-031-35098-6_11
Benzinger TL. ASNR-Recommended AD Therapeutic Imaging Protocols. Accessed November 21, 2025. https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Cogswell PM, Andrews TJ, Barakos JA, et al. Alzheimer disease anti-amyloid immunotherapies: imaging recommendations and practice considerations for monitoring of amyloid-related imaging abnormalities. Am J Neuroradiol. 2025;46(1):24-32. https://doi.org/10.3174/ajnr.A8469
Cogswell PM, Barakos JA, Barkhof F, et al. Amyloid-related imaging abnormalities with emerging Alzheimer disease therapeutics: detection and reporting recommendations for clinical practice. Am J Neuroradiol. 2022;43(9):E19-E35. https://doi.org/10.3174/ajnr.A7586
Di Muzio B, Campos A, Murphy A, et al. Neurodegenerative protocol (MRI). Radiopaedia.org Published August 11, 2015. Updated July 26, 2024. Accessed November 21, 2025. https://radiopaedia.org/articles/neurodegenerative-protocol-mri?lang=us
Harper L, Barkhof F, Scheltens P, et al. An algorithmic approach to structural imaging in dementia. J Neurol Neurosurg Psychiatry. 2014;85(6):692–698. https://doi.org/10.1136/jnnp-2013-306285.
Lawson S, Fofana A, Banerjee S. Optimizing the ARIA imaging protocol on GE HealthCare MR systems. 2023. Accessed November 21, 2025. https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Philips. Alzheimer's Disease Anti-Amyloid Immunotherapies (ARIA) 1.5T. Accessed November 21, 2025. https://www.mriclinicalcasemap.philips.com/global/case/291/i
Anhänge: Kurze Leitfäden für die MRT-Parameter
Anhang 1. General Electric 1.5T
Parameter T1 FLAIR T2* T2 DWI
Bildmodus 3D 2D 2D 2D 2D
Pulssequenz MP-RAGE FSE-XL Gradientenecho FSE Spin-Echo
Orientierung - Axial Axial Axial Axial
FOV (cm) 25.6 22.0 22.0 22.0 24.0
Schnittdicke (mm) 0.5 4.0 4.0 4.0 4.0
Schichtabstände (mm) - 0.4 0.4 0.4 0.4
Anzahl der Schichten 1 (slab) 36 36 36 36
Frequenz-Matrix 256 300 224 300 120
Phasen-Matrix 256 200 200 200 160
Phase FOV 1.0 1.0 0.9 1.0 1.0
NEX 1.0 1.0 1.0 1.0 2.0
Kippwinkel (°) 12.0 160 (auto) 20.0 160 (auto) -
TE (ms) 3.1 130.0 25.0 130.0 Minimum
TR (ms) 7.7 10000.0 600.0 10000.0 9163.0
TI (ms) 799 2687 - 2687 -
Echo-Train Länge - 20 - 20 -
Fett/Wasser Sättigung - Fett - Fett Fett
Das vollständige Protokoll ist verfügbar unter: https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Anhang 2 General Electric 3.0T
Parameter T1 FLAIR T2* T2 DWI
Bildmodus 3D 2D 2D 2D 2D
Pulssequenz MP-RAGE FSE-XL Gradientenecho FSE Spin-Echo EPI
Orientierung - Axial Axial Axial Axial
FOV (cm) 25.6 22.0 22.0 22.0 24.0
Schnittdicke (mm) 0.5 4.0 4.0 4.0 4.0
Schichtabstand (mm) - 0.4 0.4 0.4 0.4
Anzahl der Schichten 1 (slab) 36 36 36 36
Frequenz-Matrix 256 300 300 300 160
Phasen-Matrix 256 200 220 200 160
Phase FOV 1.0 0.9 0.9 0.9 1.0
NEX 1.0 1.0 1.0 1.0 1.0
Kippwinkel (°) 8.0 160 (auto) 20.0 160 (auto) -
TE (ms) 3.0 130.0 15.0 130.0 Minimum
TR (ms) 7.3 9000.0 800.0 9000.0 7500.0
TI (ms) 1000 2473 - 2473 -
Echo-Train Länge - 23 - 23 -
Fett/Wasser Sättigung - Fat - Fat Fat
Das vollständige Protokoll ist verfügbar unter: https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Anhang 3. Siemens 1.5T
Parameter T1 FLAIR T2* T2 DWI
Modus 3D 2D 2D 2D 2D
Sequenz 3D MPRAGE 2D TSE FLAIR 2D T2* Gradientenech 2D TSE 2D EPI Spin-Echo
o
Orientierung Sagittal Transversal (axial) Transversal (axial) Transversal (axial) Transversal (axial)
Voxel-Grösse (mm) 1.3 × 1.3 × 1.3 0.5 × 0.5 × 4.0 0.5 × 0.5 × 4.0 0.9 × 0.9 × 5.0 1.9 × 1.9 × 4.0
Gelesenes FOV (mm) 240 240 240 240 240
Schnittdicke (mm) 1.3 4.0 4.0 5.0 4.0
Schichten 192 30 30 28 30
TR (ms) 2400 10000 608 4700 4700
TE (ms) 2.24 114 35 111 79
TI (ms) 1000 2600 - - -
Kippwinkel (°) 8 150 20 180 -
Bandbreite (Hz/Px) 190 130 130 190 1220
Beschleunigung GRAPPA 2 GRAPPA 2 GRAPPA 2 GRAPPA 2 GRAPPA 2
Durchschnitt 1 1 1 1 1
Verknüpfungen 1 2 2 2 1
Turbo/EPI Faktor 224 (Turbo) 16 (Turbo) - 26 (Turbo) 128 (EPI)
Fett-Sättigung Standard Fat Sat Strong Standard Standard Fat Sat Strong
Das vollständige Protokoll ist verfügbar unter: https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Anhang 4. Siemens 3T
Parameter T1 FLAIR T2* T2 DWI
Modus 3D 2D 2D 2D 2D
Sequenz 3D MPRAGE 2D TSE FLAIR 2D T2* Gradientenech 2D TSE 2D EPI Spin-Echo
o
Orientierung Sagittal Transversal Transversal Transversal Transversal
Voxel-Grösse (mm) 1.0 × 1.0 × 1.0 0.5 × 0.5 × 4.0 0.5 × 0.5 × 4.0 0.9 × 0.9 × 5.0 1.9 × 1.9 × 4.0
Gelesens FOV (mm) 256 240 240 230 240
Schnittdicke (mm) 1.0 4.0 4.0 5.0 4.0
Schichten 176 30 30 25 30
TR (ms) 2300 9000 597 5140 6700
TE (ms) 2.98 96.0 35.0 78.0 72.0
TI (ms) 900 2500 - - -
Kippwinkel (°) 9 150 20 120 -
Bandbreite (Hz/Px) 240 199 200 260 1698
Beschleunigung GRAPPA 2 GRAPPA 3 GRAPPA 2 GRAPPA 3 GRAPPA 2
Durchschnitt 1 1 1 1 1
Verknüpfungen 1 2 2 2 1
Turbo/EPI Faktor 176 (Turbo) 16 (Turbo) - 26 (Turbo) 128 (EPI)
Fett-Sättigung Keine Fat Sat Strong Keine Keine Fat Sat Strong
Das vollständige Protokoll ist verfügbar unter: https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Anhang 5. Philips 1.5T
Parameter T1 FLAIR T2* DWI
Modus 3D 2D 2D 2D
Sequenz-Typ 3D FFE, TFE, T1W IR, TSE, FLAIR FFE, T2* SE, DWI, EPI
Orientierung Sagittal Transversal (axial) Transversal (axial) Transversal (axial)
Voxel-Grösse (mm) 1.05 × 1.05 × 1.10 0.90 × 1.12 × 4.00 0.90 × 1.12 × 4.00 1.51 × 2.17 × 4.00
Recon Voxel-Grösse 0.64 × 0.64 × 1.10 0.65 × 0.65 × 4.00 0.90 × 0.90 × 4.00 1.20 × 1.20 × 4.00
(mm)
FOV (mm) FH 256, AP 238, RL AP 230, RL 183, FH AP 230, RL 183, FH RL 230, AP 230, FH
159 129 129 129
Schnittdicke (mm) 1.10 4.00 4.00 4.00
Schichten 145 26 26 26
TR (ms) 7.4 11000 910 3498
TE (ms) 3.4 150 28 85
TI (ms) 950 2800 - -
Kippwinkel (°) 8 60–120 18 90
Bandbreite (Hz) 217.1 217.0 108.7 18.9
Beschleunigung SENSE (2.2) SENSE (1) SENSE (1.5) SENSE (2)
Echo-Train/EPI TFE: 216 TSE: 41 - EPI: 53
Faktor
Fett-Sättigung Keine SPIR (stark) Keine SPIR (stark)
Wasser-Fett Verschie 1.0 Maximum 2.0 11.482
bung (Pixel)
Durchschnitt 1 2 2 1
Das vollständige Protokoll ist verfügbar unter: https://www.mriclinicalcasemap.philips.com/global/case/291/i
Anhang 6. Philips 3T
Parameter T1 FLAIR T2* DWI
Modus 3D 2D 2D 2D
Sequenz 3D FFE, TFE, T1W IR, TSE, FLAIR FFE, T2* SE, DWI, EPI
Orientierung Sagittal Transversal (axial) Transversal (axial) Transversal (axial)
Voxel-Grösse (mm) 1.00 × 1.00 × 1.00 0.65 × 1.00 × 4.00 0.90 × 1.12 × 4.00 1.31 × 1.32 × 4.00
Recon Voxel-Grösse 0.47 × 0.47 × 1.00 0.45 × 0.45 × 4.00 0.53 × 0.53 × 4.00 0.96 × 0.96 × 4.00
(mm)
FOV (mm) FH 240, AP 240, RL AP 230, RL 186.9, AP 230, RL 183.1, RL 230, AP 230, FH
170 FH 139 FH 139 119
Schnittdicke (mm) 1.00 4.00 4.00 4.00
Schichten 170 28 28 24
TR (ms) 6.6 8000 835 2965
TE (ms) 3.0 135 16 55
TI (ms) 1060 2500 - -
Kippwinkel (°) 8 100 18 90
Bandbreite (Hz) 271.3 227.3 217.0 13.2
Beschleunigung SENSE (AP 1, RL 2.2) SENSE (RL 2.4) Keine SENSE (AP 2.2)
Echo-Train/EPI TFE: 204 TSE: 35 - EPI: 79
Faktor
Fett-Sättigung Keine SPIR (schwach) Keine SPIR (stark)
Wasser-Fett Verschie 1.6 1.911 2.002 32.925
bung (Pixel)
Durchschnitt 1 2 1 1
Das vollständige Protokoll ist verfügbar unter: https://www.mriclinicalcasemap.philips.com/global/case/291/i
Stand der Information
Januar 2025.
|