CompositionPrincipes actifs
Lenalidomidum.
Excipients
Dosage Gélules
2,5 mg 5 mg 7,5 mg 10 mg 15 mg 20 mg 25 mg
Lactose (sous forme 73,5 mg 147,0 mg 144,5 mg 294,0 mg 289,0 mg 244,5 mg 200,0 mg
de lactose anhydre)
Cellulose microcristalline, stéarate de magnésium, croscarmellose sodique.
Enveloppe des gélules; gélatine, dioxyde de titane, oxyde de fer (uniquement pour les gélules de 2,5 mg, de 7,5 mg, de 10 mg et de 20 mg), carmin indigo E132 (uniquement pour les gélules de 2,5 mg, de 10 mg, de 15 mg et de 20 mg).
Encre d'impression: gomme laque, propylène glycol, oxyde de fer noir, hydroxyde de potassium. Une capsule dure contient au maximum 0,2 mg (capsules dures 2,5 mg) ou au maximum 0,4 mg (capsules dures 5 mg et 7,5 mg) ou au maximum 0,8 mg (capsules dures 10 mg, 15 mg, 20 mg et 25 mg) de sodium.
Forme pharmaceutique et quantité de principe actif par unitéGélules de 2,5 mg, de 5 mg, de 7,5 mg, de 10 mg, de 15 mg, de 20 mg et de 25 mg.
Indications/Possibilités d’emploiRevlimid en association avec le bortézomib et la dexaméthasone est indiqué chez les patients adultes pour le traitement du myélome multiple non préalablement traité.
Revlimid est indiqué pour le traitement d'entretien du myélome multiple chez les patients adultes après une autogreffe de cellules souches.
Revlimid en association avec la dexaméthasone ou Revlimid en association avec le melphalan et la prednisone, respectivement suivi d'un traitement d'entretien par Revlimid, est indiqué pour le traitement du myélome multiple non préalablement traité chez les patients adultes non éligibles à une greffe.
Revlimid, en association avec la dexaméthasone, est indiqué pour le traitement des patients souffrant d'un myélome multiple qui ont déjà reçu au moins un traitement médicamenteux antérieurement.
Revlimid est indiqué pour le traitement des patients qui présentent une anémie dépendante de transfusion à la suite d'un syndrome myélodysplasique avec un risque faible ou intermédiaire 1, en relation avec une anomalie cytogénétique comportant une délétion 5q, accompagnée ou non d'autres anomalies cytogénétiques.
Revlimid est indiqué pour le traitement des patients qui présentent un lymphome des cellules du manteau (LCM) récidivant ou réfractaire après un traitement précédent par le bortézomib et une association chimiothérapie/rituximab.
Revlimid est indiqué, en association avec le rituximab (anticorps anti-CD20), pour le traitement des patients adultes présentant un lymphome folliculaire (de grade 1 à 3a) en rechute ou réfractaire (voir "Propriétés/Effets" ).
Posologie/Mode d’emploiLe traitement doit être initié et surveillé par un hématologue ou oncologue expérimenté.
Myélome multiple
Revlimid en association avec le bortézomib et la dexaméthasone chez les patients atteints d'un myélome multiple non préalablement traité
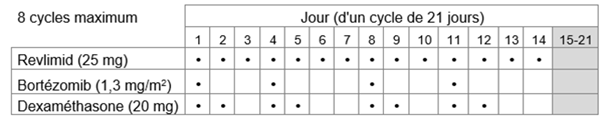
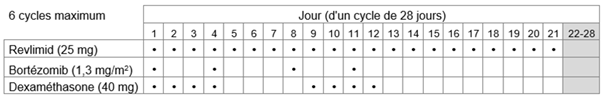
-Traitement initial: Revlimid en association avec le bortézomib et la dexaméthasone
Le traitement par Revlimid en association avec le bortézomib et la dexaméthasone ne doit pas être instauré si la numération des polynucléaires neutrophiles (PNN) est < 1,0 x 109/l et/ou si la numération plaquettaire est < 50 x 109/l.
La dose initiale recommandée est de 25 mg de Revlimid par voie orale en une prise par jour soit
a.les jours 1 à 14 de chaque cycle de traitement de 21 jours, soit
b.les jours 1 à 21 de chaque cycle de traitement de 28 jours.
Le bortézomib doit être administré par injection sous-cutanée (1,3 mg/m2 de surface corporelle) deux fois par semaine les jours 1, 4, 8 et 11 d'un cycle de 21 jours ou d'un cycle de 28 jours.
La dose recommandée de dexaméthasone est de
a.20 mg par voie orale une fois par jour les jours 1, 2, 4, 5, 8, 9, 11 et 12 ou
b.40 mg par voie orale une fois par jour les jours 1 à 4 et 9 à 12 de chaque cycle.
Huit cycles de 21 jours ou six cycles de 28 jours maximum (traitement initial de 24 semaines) sont recommandés.
Tableau 1: Schéma posologique recommandé pour Revlimid en association avec le bortézomib et la dexaméthasone
Ou
-Poursuite du traitement chez les patients qui n'ont pas reçu une autogreffe de cellules souches: Revlimid en association avec la dexaméthasone jusqu'à la progression de la maladie
Continuation du traitement avec 25 mg de Revlimid par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours en association avec la dexaméthasone. La dose recommandée de dexaméthasone est de 40 mg par voie orale en une prise par jour les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Le traitement peut être poursuivi jusqu'à la progression de la maladie ou la survenue d'une intolérance.
-Poursuite du traitement: autogreffe de cellules souches
Chez les patients dont le traitement par une autogreffe de cellules souches est poursuivi, une mobilisation des cellules souches hématopoïétiques devrait survenir au cours des premiers 4 cycles du traitement initial.
Revlimid chez les patients ayant reçu une autogreffe de cellules souches
Le traitement d'entretien par Revlimid doit être instauré après une bonne récupération hématologique après une autogreffe de cellules souches. Le traitement par Revlimid ne doit pas être initié si la PNN est < 1,0 x 109/l et/ou si la numération plaquettaire est < 75 x 109/l.
Posologie recommandée
La dose initiale recommandée est de 10 mg de Revlimid par voie orale en une prise par jour de façon continue (les jours 1 à 28 de chaque cycle de 28 jours) jusqu'à la progression de la maladie ou survenue d'une intolérance. Après 3 cycles de traitement d'entretien de 28 jours par Revlimid de façon continue, la dose peut être augmentée à 15 mg par voie orale en une prise par jour si elle est tolérée.
Revlimid administré en association avec la dexaméthasone jusqu'à la progression de la maladie chez les patients non traités non éligibles à une greffe
Le traitement par Revlimid ne doit pas être initié si la PNN est < 1,0 x 109/l et/ou si la numération plaquettaire est < 50 x 109/l.
Posologie recommandée
La dose initiale recommandée est de 25 mg de Revlimid par voie orale en une prise par jour, les jours 1 à 21 de chaque cycle de 28 jours. La dose recommandée de dexaméthasone est de 40 mg en une prise par jour par voie orale les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Le traitement par Revlimid et la dexaméthasone peut être poursuivi jusqu'à la progression de la maladie ou une toxicité.
Revlimid en association avec le melphalan et la prednisone suivis d'une monothérapie d'entretien chez les patients non traités non éligibles à une greffe
Le traitement par Revlimid ne doit pas être initié si la numération des polynucléaires neutrophiles (PNN) est < 1,5 x 109/l et/ou si la numération plaquettaire est < 75 x 109/l.
Posologie recommandée
Les doses initiales recommandées sont de 10 mg par jour de Revlimid par voie orale les jours 1 à 21 de chaque cycle de 28 jours pendant 9 cycles au maximum, de 0,18 mg/kg de melphalan par voie orale les jours 1 à 4 de chaque cycle de 28 jours et de 2 mg/kg de prednisone par voie orale les jours 1 à 4 de chaque cycle de 28 jours.
Les patients ayant terminé 9 cycles ou qui ne peuvent pas terminer le traitement en association en raison d'une toxicité sont traités par Revlimid seul, administré à la dose de 10 mg par jour par voie orale les jours 1 à 21 de chaque cycle de 28 jours jusqu'à la progression de la maladie.
Revlimid administré en association avec la dexaméthasone chez des patients souffrant d'un myélome multiple ayant reçu au moins un traitement antérieur
La dose initiale recommandée est de 25 mg de Revlimid par voie orale une fois par jour les jours 1 à 21 de cycles consécutifs de 28 jours. Pendant les 4 premiers cycles de traitement, la dose recommandée de dexaméthasone est de 40 mg par voie orale, une fois par jour, aux jours 1 à 4, 9 à 12 et 17 à 20 de chaque cycle de 28 jours, puis de 40 mg lors des jours 1 à 4. Le traitement doit être poursuivi jusqu'à la progression de la maladie ou survenue d'une toxicité inacceptable.
Syndrome myélodysplasique
La dose initiale recommandée est de 10 mg de Revlimid par voie orale une fois par jour pendant les jours 1 à 21 de cycles consécutifs de 28 jours. L'arrêt du traitement pour efficacité insuffisante est recommandé lorsque 16 semaines après le début du traitement par Revlimid, aucune réponse, même minime, correspondant à une amélioration de 50% au moins n'est constatée.
Lymphome des cellules du manteau récidivant ou réfractaire
La dose initiale recommandée est de 25 mg de Revlimid par voie orale une fois par jour les jours 1 à 21 de cycles consécutifs de 28 jours. Le traitement doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Lymphome folliculaire (LF) en association avec le rituximab (schéma R2)
Le traitement par Revlimid ne doit pas être initié si le nombre absolu de neutrophiles est < 1 x 109/l et/ou si la numération plaquettaire est < 50 x 109/l, à moins que ces diminutions ne soient secondaires à un envahissement médullaire induit par le lymphome.
La dose initiale recommandée de Revlimid est de 20 mg par voie orale en une prise par jour les jours 1 à 21 de chaque cycle de 28 jours, pendant 12 cycles de traitement au maximum. La dose initiale recommandée de rituximab est de 375 mg/m2 par voie intraveineuse (IV) une fois par semaine au cours du cycle 1 (jours 1, 8, 15 et 22) et le jour 1 de chaque cycle de 28 jours pendant les cycles 2 à 5.
Adaptation de la dose
La posologie de Revlimid ou d'autres médicaments utilisés dans le cadre d'un traitement d'association (dexaméthasone, melphalan, prednisone, bortézomib, rituximab) doit être adaptée en fonction des résultats cliniques et des valeurs de laboratoire.
En ce qui concerne les adaptations posologiques liées à la toxicité d'autres médicaments que Revlimid utilisés dans le cadre d'un traitement en association, il convient de consulter l'information pour le professionnel de chaque médicament.
Hématotoxicité
Ajustements posologiques recommandés pendant le traitement et lors de la reprise du traitement
Il est recommandé d'ajuster les doses comme indiqué ci-dessous en fonction de l'indication pour prendre en charge les thrombocytopénies ou neutropénies de grade 3 ou 4, ainsi que les autres toxicités de grade 3 ou 4, jugées comme étant liées au lénalidomide.
Revlimid en association avec le bortézomib et la dexaméthasone chez les patients atteints d'un myélome multiple non préalablement traité
Paliers de réduction de la posologie
Lénalidomide
Dose initiale 25 mg
Palier de dose -1 20 mg
Palier de dose-2 15 mg
Palier de dose -3 10 mg
Palier de dose -4 5 mg
Palier de dose -5 2,5 mg par jour ou 5 mg toutes les 48 h
Thrombocytopénie
Modification de la Action recommandée
numération plaquettaire
Chute < 30 x 109/l Arrêt du traitement par le lénalidomide et surveillance hebdomadaire de
l'hémogramme complet
Retour ≥50 x 109/l Reprendre le lénalidomide au palier de dose -1
Pour toute nouvelle Interrompre le traitement par le lénalidomide
rechute < 30 x 109/l
Retour ≥50 x 109l/l Reprendre le lénalidomide à la dose immédiatement inférieure. Ne pas
prendre une dose inférieure à 2,5 mg une fois par jour.
Neutropénie
Modification de la numération des Action recommandée a
neutrophiles
Première chute < 0,5 x 109/l ou Interrompre le traitement par le lénalidomide et surveillance
neutropénie fébrile (fièvre ≥38 hebdomadaire de l'hémogramme complet Reprendre le
°C; < 1 x 109/l) Retour ≥1 x 109/l lénalidomide au palier de dose -1
Pour toute nouvelle chute < 0,5 x Interrompre le traitement par le lénalidomide Reprendre le
109/l ou neutropénie fébrile lénalidomide à la dose immédiatement inférieure. Ne pas
Retour à ≥1 x 109/l prendre une dose inférieure à 2,5 mg une fois par jour.
a À l'appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Revlimid chez les patients ayant reçu une autogreffe de cellules souches
Paliers de réduction
de la posologie
Dose initiale (10 mg) En cas d'augmentation de la dose
(15 mg)a
Palier de dose -1 5 mg une fois par jour de façon continue 10 mg en une prise une par jour
de façon continue
Palier de dose -2 5 mg une fois par jour aux jours 1 à 21 de 5 mg en une prise une fois par
chaque cycle de 28 jours jour de façon continue
Palier de dose -3 Sans objet 5 mg en une prise une par jour
aux jours 1 à 21 de chaque cycle
de 28 jours
Ne pas diminuer la dose en dessous de 5 mg
une fois par jour aux jours 1 à 21 de
chaque cycle de 28 jours
a Après trois cycles de 28 jours en traitement d'entretien continu par Revlimid, la dose peut être augmentée à 15 mg par voie orale en une prise par jour si le traitement est bien toléré.
Thrombocytopénie
Modification de la Action recommandée
numération plaquettaire
Chute < 30 x 109/l Interrompre le traitement par le lénalidomide et surveillance
hebdomadaire de l'hémogramme complet
Retour ≥30 x 109/l Reprendre le lénalidomide au palier de dose -1
Pour toute nouvelle rechute Interrompre le traitement par le lénalidomide
< 30 x 109/l
Retour ≥30 x 109l/l Reprendre le lénalidomide à la dose immédiatement inférieure
Neutropénie
Modification de la numération Action recommandéea
des neutrophiles
Chute < 0,5 x 109/l Interrompre le traitement par le lénalidomide et surveillance
hebdomadaire de l'hémogramme complet
Retour ≥0,5 x 109/l Reprendre le lénalidomide au palier de dose -1
Pour toute nouvelle rechute < Interrompre le traitement par le lénalidomide
0,5 x 109/l
Retour ≥0,5 x 109/l Reprendre le lénalidomide à la dose immédiatement inférieure
a À l'appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Revlimid administré en association avec la dexaméthasone chez des patients non préalablement traités et non éligibles à une greffe.
Paliers de réduction de la posologie
Lénalidomide Dexaméthasone
Dose initiale 25 mg 40 mg
Palier de dose 1 20 mg 20 mg
Palier de dose 2 15 mg 12 mg
Palier de dose 3 10 mg 8 mg
Palier de dose 4 5 mg 4 mg
Palier de dose 5 2,5 mg par jour ou 5 mg toutes les 48 heures NA
Thrombocytopénie
Modification de la Action recommandée
numération plaquetta
ire
Chute < 25 x 109/l Arrêter l'administration de lénalidomide pendant le reste du cyclea.
Retour ≥50 x 109/l Reprendre le lénalidomide à une dose diminuée de 5 mg par rapport à la dose
précédente. Après une dose de 5 mg, reprendre le lénalidomide à une dose de
2,5 mg par jour ou de 5 mg toutes les 48 heures. Ne pas descendre en dessous
de 2,5 mg par jour ou de 5 mg toutes les 48 heures.
ª En cas de toxicité dose-limitante (TDL) survenant à partir du 15e jour d'un cycle, le traitement par le lénalidomide doit être interrompu pendant au moins le reste du cycle de 28 jours en cours.
Neutropénie
Modification de la numération des neutrophiles Action recommandéea
Première chute < 0,5 x 109/l ou neutropénie fébrile Interrompre le traitement par le
(fièvre ≥38 °C; < 1 x 109/l) Retour ≥1 x 109/l, la lénalidomide. Reprendre le lénalidomide à la
neutropénie étant la seule toxicité observée dose initiale en une prise par jour.
Retour ≥0,5 x 109/l, en cas d'autres toxicités Reprendre le lénalidomide au palier de dose
hématologiques dépendantes de la dose autres que la 1 en une prise par jour.
neutropénie
Toute nouvelle chute < 0,5 x 109/l Retour ≥0,5 x Interrompre le traitement par le
109/l lénalidomide. Reprendre le lénalidomide à la
dose immédiatement inférieure en une prise
par jour.
a À l'appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Si la dose de lénalidomide a été diminuée en raison d'une TDL hématologique, le traitement pourra être repris au palier de dose immédiatement supérieur (jusqu'à la dose initiale) à l'appréciation du médecin si la poursuite du traitement par lénalidomide/dexaméthasone a induit une amélioration de la fonction médullaire (absence de TDL pendant au moins deux cycles consécutifs et PNN ≥1,5 x 109/l avec plaquettes ≥100 x 109/l au début d'un nouveau cycle au palier de dose actuel).
Revlimid en association avec le melphalan et la prednisone suivis d'une monothérapie d'entretien chez les patients non éligibles à une greffe
Paliers de réduction de la
posologie
Lénalidomide Melphalan Prednisone
Dose initiale 10 mg 0,18 mg/kg 2 mg/kg
Palier de dose 1 7,5 mg par jour ou 15 mg 0,14 mg/kg 1 mg/kg
toutes les 48 h
Palier de dose 2 5 mg 0,10 mg/kg 0,5 mg/kg
Palier de dose 3 2,5 mg par jour ou 5 mg NA 0,25 mg/kg
toutes les 48 h
Thrombocytopénie
Modification de la Action recommandée
numération plaquettaire
Première chute < 25 x Interrompre le traitement par lénalidomide. Reprendre le lénalidomide et
109/l Retour ≥25 x 109/l le melphalan au palier de dose 1.
Toute nouvelle chute < Interrompre le traitement par le lénalidomide. Reprendre le lénalidomide
30 x 109/l Retour ≥30 x à la dose immédiatement inférieure (palier de dose 2 ou 3) en une prise
109/l par jour.
Neutropénie
Modification de la numération des Action recommandéea
neutrophiles
Première chute < 0,5 x 109/lª Retour ≥0,5 x Interrompre le traitement par le lénalidomide.
109/l, la neutropénie étant la seule Reprendre le lénalidomide à la dose initiale en une
toxicité observée prise par jour.
Retour ≥0,5 x 109/l, en cas d'autres Reprendre le lénalidomide au palier de dose 1 en
toxicités hématologiques dépendantes de la une prise par jour.
dose autres que la neutropénie
Toute nouvelle chute < 0,5 x 109/l Retour Interrompre le traitement par lénalidomide.
≥0,5 x 109/l Reprendre le lénalidomide à la dose immédiatement
inférieure en une prise par jour.
ª À l'appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
Myélome multiple avec au moins un traitement antérieur, syndrome myélodysplasique et lymphome à cellules du manteau
Pour les indications de MM chez les patients ayant reçu au moins un traitement antérieur et de SMD en présence d'une thrombocytopénie avec réduction du nombre de plaquettes à < 25 x 109/l, ou en présence d'une neutropénie avec une chute de la valeur à < 0,5 x 109/l, le traitement par le lénalidomide doit être suspendu.
Pour l'indication de LCM, en présence d'une thrombocytopénie avec réduction du nombre des plaquettes à < 50 x 109/l, ou en présence d'une neutropénie avec une chute de la valeur à < 0,5 x 109/l, ou avec une chute de la valeur à < 1 x 109/l pendant au moins 7 jours ou avec une chute de la valeur à < 1 x 109/l liée à une température ≥38,5 °C, le traitement par le lénalidomide doit être suspendu.
Après normalisation du nombre de thrombocytes/neutrophiles, le traitement devrait être poursuivi à la dose immédiatement inférieure suivante. En cas de survenue à nouveau de l'effet toxique, la dose doit être réduite. En cas de toxicité à la dose la plus faible, le traitement par le lénalidomide doit être arrêté.
Chez les patients atteints de myélome multiple ayant reçu au moins un traitement antérieur, la première réduction de dose est de 15 mg par jour, en cas d'un nouvel effet toxique, 10 mg et puis 5 mg par jour.
Dans le syndrome myélodysplasique, la première réduction de dose est de 5 mg par jour; en cas de nouvelle survenue, une deuxième réduction de dose de 2,5 mg par jour ou de 5 mg tous les deux jours est recommandée. Une troisième réduction de dose à 5 mg deux fois par semaine est recommandée en cas de nouvel effet toxique.
Le passage à un autre traitement doit être envisagé chez les LCM-patients qui n'ont pas présenté de réponse après la prise pendant plus de 3 mois d'une faible dose de Revlimid.
Lymphome folliculaire (LF)
Paliers de réduction de la
posologie
Lénalidomide
Dose initialea 20 mg en une prise par jour les jours 1 à 21, tous les 28 jours
Palier de dose -1 15 mg en une prise par jour les jours 1 à 21, tous les 28 jours
Palier de dose -2 10 mg en une prise par jour les jours 1 à 21, tous les 28 jours
Palier de dose -3 5 mg en une prise par jour les jours 1 à 21, tous les 28 jours
Palier de dose -4 b 2,5 mg en une prise par jour les jours 1 à 21, tous les 28 jours, ou
5 mg toutes les 48 heures
a Pour la dose initiale chez les patients insuffisants rénaux, voir la rubrique ci-dessous.
b Uniquement pour une dose initiale ajustée chez les patients souffrant d'insuffisance rénale modérée.
Thrombocytopénie
Modification de la Action recommandée
numération plaquetta
ire
Première chute < 50 Interrompre le traitement par le lénalidomide et réaliser un hémogramme
x 109/l Retour ≥50 complet chaque semaine Reprendre le lénalidomide au palier de dose -1 aux
x 109/l jours 1-21 du cycle de 28 jours
Toute nouvelle Interrompre le traitement par le lénalidomide et surveillance hebdomadaire de
chute < 50 x 109/l l'hémogramme complet. Reprendre le lénalidomide à la dose immédiatement
Retour ≥50 x 109/l inférieure (palier de dose -2 ou -3 en une prise par jour). Ne pas descendre
en dessous du palier de dose -3. Si la dose initiale était de 10 mga, ne pas
descendre en dessous du palier de dose -4.
a voir la rubrique "Patients insuffisants rénaux"
Neutropénie
Modification de la numération Action recommandéea
des neutrophiles
Première chute < 1,0 x 109/l Interrompre le traitement par le lénalidomide et contrôler
pendant au moins 7 jours OU l'hémogramme complet au moins tous les 7 jours
Chute < 1,0 x 109/l accompagnée
de fièvre ≥38,5 °C) OU Chute <
0,5 x 109/l
Retour ≥1,0 x 109/l Reprendre le lénalidomide à la dose immédiatement inférieure
(palier de dose -1)
Toute nouvelle chute 1,0 x 109/l Interrompre le traitement par le lénalidomide et réaliser un
pendant au moins 7 jours ou hémogramme complet au moins tous les 7 jours Reprendre le
chute < 1,0 x 109/l accompagnée lénalidomide à la dose immédiatement inférieure (palier de dose
de fièvre (température ≥38,5 °C) -2 ou -3). Ne pas descendre en dessous du palier de dose -3. Si
ou chute < 0,5 x 109/l Retour la dose initiale était de 10 mgb, ne pas descendre en dessous
≥0,5 x 109/l du palier de dose -4.
ª À l'appréciation du médecin, si la neutropénie est la seule toxicité quel que soit le palier de dose, ajouter un facteur de croissance granulocytaire (G CSF) et maintenir la dose de lénalidomide.
b Voir la rubrique "Patients insuffisants rénaux"
Autres motifs
Le traitement doit être interrompu en cas de survenue d'une éruption cutanée sans desquamation (avec formation de vésicules) de grade 3, d'une neuropathie de grade 3 ou d'une réaction allergique de grade 2. Le traitement peut être réinstauré à la dose immédiatement inférieure après régression jusqu'à ≤ grade 1.
Le traitement par Revlimid doit être arrêté en cas de survenue d'une éruption cutanée avec desquamation (accompagnée d'une formation de vésicules), d'une éruption cutanée sans desquamation (accompagnée d'une formation de vésicules) de grade 4, d'une neuropathie de grade 4 ou d'une réaction allergique ≥ grade 3.
Le traitement doit être interrompu en cas de survenue d'une constipation (≥ grade 3) et il convient d'instaurer un traitement de la constipation. Le traitement par Revlimid peut être réinstauré à la dose immédiatement inférieure après régression de la constipation jusqu'à ≤ grade 2.
Le traitement par Revlimid doit être interrompu en cas de survenue d'une thrombose veineuse/embolie (≥ grade 3) et il convient d'instaurer un traitement anticoagulant. La reprise du traitement doit être décidée par le médecin (maintien du palier de dose).
Revlimid doit être arrêté en cas d'angioedème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, de suspicion de syndrome de Stevens-Johnson (SJS), de nécrolyse épidermique toxique (NET) ou d'une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS) et ne doit pas être repris après l'arrêt de ces réactions.
Autres effets toxiques de grade 3 ou 4
En cas de survenue d'autres effets toxiques de grade 3 ou 4, imputés à Revlimid, le traitement doit être interrompu et après diminution de l'effet toxique à un grade ≤2, il doit être repris à la dose immédiatement inférieure selon l'appréciation du médecin.
Posologies spéciales
Patients insuffisants hépatiques
Revlimid n'a pas été étudié chez des patients souffrant d'insuffisance hépatique; il n'existe pas de recommandations posologiques spécifiques pour ce groupe de patients.
Patients insuffisants rénaux
En cas d'insuffisance rénale légère (cl. créat. 80-60 ml/min), aucune adaptation de la dose n'est nécessaire.
Pour les patients présentant un myélome multiple avec une dose initiale de 25 mg, pour les patients atteints d'un lymphome folliculaire avec une dose initiale de 20 mg et pour les patients présentant un myélome multiple ou un syndrome myélodysplasique avec une dose initiale de 10 mg, on recommande en début et en cours de traitement pour les patients en insuffisance rénale modérée (30 ≤ ClCr < 60 ml/min) ou sévère (ClCr < 30 ml/min) ou encore en insuffisance rénale au stade terminal les adaptations de la dose suivantes:
Fonction rénale Adaptation de la dose
(ClCr)
Dose initiale 25 mg Dose initiale 20 mg Dose initiale 10 mg
Fonction rénale 25 mg par jour 20 mg par jour 10 mg par jour
normale/insuffisance
rénale légère
(ClCr ≥60 ml/min)
Insuffisance rénale 10 mga par jour 10 mgc par jour 5 mg par jour
modérée (30 ≤ ClCr
< 60 ml/min)
Insuffisance rénale 7,5 mg par jour ou 5 mg par jour 2,5 mg par jour ou 5 mg
sévère (ClCr < 30 15 mgb toutes les 48 toutes les 48 heures
ml/min, pas de heures
dialyse nécessaire)
Insuffisance rénale 5 mg par jour. Les 5 mg par jour Les 2,5 mg par jour ou 5 mg trois
au stade terminal jours de dialyse, la jours de dialyse, la fois par semaine. Les jours
(ClCr < 30 ml/min, dose doit être dose doit être de dialyse, la dose doit être
dialyse nécessaire) administrée après la administrée après la administrée après la dialyse.
dialyse. dialyse.
a Après deux cycles, la dose peut être augmentée à 15 mg par jour dans le cas où le patient ne répond pas au traitement et s'il tolère le médicament.
b La dose peut être augmentée à 10 mg par jour dans le cas où le patient tolère le médicament.
c Après deux cycles, la dose peut être augmentée à 15 mg par jour dans le cas où le patient tolère le médicament.
Dans le traitement des patients atteints d'un LCM, on peut s'attendre à un effet de la fonction rénale sur les concentrations plasmatiques de Revlimid, la substance active, similaire à l'effet observé chez les patients atteints d'un MM, d'un SMD et d'un LF. Il convient d'envisager une réduction de la dose correspondante chez les patients atteints d'un LCM et présentant une insuffisance rénale. On note qu'il ne faut pas dépasser la dose initiale de 10 mg chez les patients dont la clairance de la créatinine est entre 30 et 60 ml/min.
Patients âgés
Aucune adaptation posologique n'est nécessaire. Toutefois, sachant que les patients âgés peuvent présenter une fonction rénale réduite, la fonction rénale devrait être surveillée à intervalles réguliers. Revlimid a été utilisé dans des études cliniques chez des patients jusqu'à un âge de 95 ans.
Myélome multiple non préalablement traité chez les patients non éligibles à une greffe
Chez les patients âgés de plus de 75 ans traités par le lénalidomide en association avec la dexaméthasone, la dose initiale de dexaméthasone est de 20 mg par jour, les jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Aucun ajustement de la posologie n'est recommandé chez les patients âgés de plus de 75 ans traités par le lénalidomide en association avec le melphalan et la prednisone.
Enfants et adolescents
Revlimid n'a pas fait l'objet d'études chez les patients pédiatriques. Pour cette raison, Revlimid ne devrait pas être utilisé dans ce groupe d'âge.
Mode d'administration
Les gélules de Revlimid doivent être prises avec un peu d'eau, au cours ou en dehors des repas, toujours environ à la même heure. Elles ne doivent être ni ouvertes, ni croquées. Les mains doivent être lavées immédiatement après tout contact avec les gélules. Il faut veiller à ne pas aspirer la poudre contenue dans les gélules (par exemple à partir d'une gélule endommagée) et à éviter tout contact de la poudre avec la peau et les muqueuses. Si un contact avec la peau s'est néanmoins produit, la zone en question doit être lavée à l'eau et au savon. Dans le cas d'un contact avec les yeux, rincer à l'eau claire.
Si la prise d'une dose de Revlimid a été oubliée et que moins de 12 heures se sont écoulées depuis cet oubli, la dose peut encore être prise. La dose ne doit pas être prise si l'heure de prise habituelle remonte à plus de 12 heures. Le patient doit attendre jusqu'au lendemain et prendre la dose suivante à l'heure habituelle. Il ne faut pas prendre 2 doses en une fois.
Contre-indicationsGrossesse.
Femmes en mesure de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies (voir "Mises en garde et précautions" ).
Hypersensibilité au lénalidomide ou à l'un des excipients.
Mises en garde et précautionsProgramme de prévention de la grossesse
Programme pour les patientes
Les conditions du programme de prévention de la grossesse doivent être remplies chez toutes les patientes, sauf chez celles dont l'incapacité de procréer est démontrée.
Critères pour l'évaluation du potentiel de procréer
Toute patiente ou conjointe de patient est considérée comme capable de procréer, à moins qu'elle ne remplisse au moins l'une des conditions suivantes:
-Âge ≥50 ans et aménorrhée naturelle depuis ≥1 an*
-Déficience ovarienne prématurée avérée
-Antécédent de salpingo-oophorectomie bilatérale, de ligature des trompes bilatérale ou d'hystérectomie
-Génotype XY, syndrome de Turner, aplasie de l'utérus
* Une aménorrhée après un traitement anticancéreux n'exclut pas la capacité de procréer.
Consultation
Le lénalidomide est contre-indiqué chez les femmes capables de procréer qui ne remplissent pas toutes les conditions ci-dessous:
-La patiente comprend le risque tératogène attendu auquel serait exposé un enfant à naître.
-Elle comprend la nécessité d'une contraception fiable sans interruption commencée 4 semaines avant le début du traitement, poursuivie pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement.
-Même en cas d'aménorrhée, toute patiente en âge de procréer doit respecter strictement toutes les recommandations fournies pour une contraception efficace.
-Elle doit être capable de respecter les exigences d'une méthode de contraception fiable.
-Elle est informée et a compris les conséquences d'une grossesse ainsi que la nécessité de consulter rapidement le médecin si une grossesse est suspectée.
-Elle comprend la nécessité de faire des tests de grossesse toutes les 4 semaines et accepte de s'y soumettre.
-Elle a confirmé avoir compris les risques et mesures de sécurité nécessaires dans le cadre d'un traitement par le lénalidomide.
Chez les femmes en mesure de procréer, le médecin prescripteur doit s'assurer que
-La patiente remplit les conditions indiquées ci-dessus.
-La patiente remplit les conditions spécifiées par le programme de prévention de la grossesse, y compris confirmation d'une compréhension suffisante.
-La patiente a appliqué des mesures contraceptives suffisantes au moins 4 semaines avant le début du traitement, continue à appliquer des mesures contraceptives suffisantes pendant toute la durée du traitement, pauses de traitement comprises, et continuera à les appliquer encore au moins 4 semaines au-delà de la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le lénalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement.
-Un test de grossesse fait avant le traitement soit négatif.
Contraception
Les femmes en mesure de procréer doivent appliquer une méthode de contraception fiable pendant les 4 semaines précédant le début du traitement, pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le lénalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement. Si une méthode de contraception fiable n'a pas été utilisée auparavant, la patiente doit être adressée à un service médical de consultation capable de conseiller et d'informer la patiente de façon approfondie pour le choix d'une méthode contraceptive fiable.
Les méthodes de contraception suivantes peuvent être considérées comme fiables:
- Méthodes indépendantes de la patiente:
-Implant
-Acétate de médroxyprogestérone retard
-Stérilisation
- Méthodes dépendantes de la patiente:
-Renoncement total aux rapports hétérosexuels.
-Rapports hétérosexuels uniquement avec un partenaire stérilisé par une vasectomie attestée par deux examens confirmant l'absence de spermatozoïdes.
-Contraceptifs oraux à la progestérone seule.
En raison du risque accru de thromboembolies veineuses sous lénalidomide, les contraceptifs oraux œstro-progestatifs ne sont pas recommandés. Si une patiente utilise déjà des contraceptifs oraux œstro-progestatifs, il convient de considérer le passage à une autre méthode de contraception. Le risque de thromboembolies veineuses persiste pendant 4 à 6 semaines après l'arrêt du contraceptif oral œstro-progestatif. Si aucune autre méthode ne peut être utilisée, on considérera une prévention antithrombotique pendant la durée d'utilisation du contraceptif oral œstro-progestatif. La patiente doit être dûment informée du risque de thromboembolie veineuse.
Les dispositifs intra-utérins (stérilets) sont associés à un risque accru d'infections lors de la mise en place et peuvent provoquer des saignements vaginaux ou règles irrégulières. Ces méthodes ne sont donc pas recommandées.
Test de grossesse
Des tests de grossesse avec une sensibilité d'au moins 25 UI/ml hCG doivent être effectués chez les femmes capables de procréer.
Chaque cas d'une patiente dont le test de grossesse est positif doit, sans attendre, être reporté au Swiss Teratogen Information Service (STIS) à Lausanne, au moyen du formulaire Swissmedic "Annonce d'effets indésirables suspectés d'un médicament (EI)" .
- Avant le début du traitement
Un test de grossesse sous contrôle médical doit être réalisé lors de la consultation où le lénalidomide est prescrit ou au cours des trois jours suivant la visite chez le médecin prescripteur. À la date du test, la patiente doit avoir appliqué une méthode contraceptive fiable depuis au moins 4 semaines. Le test doit assurer que la patiente n'est pas enceinte au début du traitement par le lénalidomide.
- Avant le début du traitement chez les patientes nécessitant un traitement immédiat
Une détermination quantitative du taux sérique de hCG doit être faite immédiatement. Après 7 jours d'application d'une méthode de contraception efficace, associée à l'utilisation de préservatifs, le test doit être répété. Si les deux tests confirment que la patiente n'est pas enceinte, le traitement peut être commencé.
- Pendant et après le traitement
Un test de grossesse doit être fait toutes les 4 semaines pendant le traitement, et 4 semaines après la fin du traitement. Ces tests de grossesse doivent être faits pendant les visites chez le médecin pour la prescription du lénalidomide ou dans les trois jours précédant une telle visite.
Les tests de grossesse et la prescription et remise du lénalidomide doivent de préférence avoir lieu le même jour. Le lénalidomide doit être remis au plus tard sous 7 jours à compter de la date de prescription.
Programme chez les patients de sexe masculin
Chez les patients de sexe masculin, les données cliniques attestent du passage de la substance dans le sperme pendant la prise de Revlimid. Les patients dont la partenaire est en mesure de procréer doivent de ce fait utiliser des préservatifs lors des rapports sexuels pendant le traitement par Revlimid et au moins pendant les 7 jours après l'arrêt du traitement. Les hommes traités par Revlimid doivent remplir les conditions suivantes:
-Ils doivent avoir compris le risque tératogène attendu s'ils ont des rapports sexuels avec une femme en mesure de procréer.
-Ils doivent avoir compris et accepté qu'ils doivent utiliser des préservatifs pour tous les rapports sexuels avec une femme en mesure de procréer pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 7 jours suivant la fin du traitement.
Le médecin prescripteur doit assurer que les patients de sexe masculin ont compris et accepté d'utiliser des préservatifs pendant toute la durée du traitement, pauses du traitement compris, et pendant les 7 jours suivant la fin du traitement, s'ils ont des rapports sexuels avec une femme en mesure de procréer.
Les patients ne doivent en aucun cas faire un don de sperme pendant leur traitement par Revlimid ou pendant les 7 jours suivant la fin de ce traitement.
Précautions supplémentaires
Les patients et patientes doivent être instruits de ne jamais donner ce médicament à d'autres personnes et de rapporter les gélules non utilisées à leur médecin ou à leur pharmacien à la fin du traitement.
Les patients ne doivent pas donner leur sang pendant le traitement ni pendant au moins 1 semaine après l'arrêt du lénalidomide.
Autres mises en garde et précautions
Neutropénies et thrombocytopénies
Les neutropénies et les thrombocytopénies font partie des principales toxicités susceptibles de limiter la dose de lénalidomide. C'est pourquoi un hémogramme complet – avec hémogramme différentiel, numération plaquettaire, hémoglobinémie et hématocrite – doit être fait.
Il est possible qu'une interruption du traitement et/ou une diminution de la dose soit souhaitable (voir "Posologie/Mode d'emploi" ). Il convient de surveiller les signes d'une infection chez les patients atteints de neutropénie. Il est conseillé aux patients et aux médecins d'être attentifs aux signes et symptômes évocateurs d'une hémorragie, y compris les pétéchies et l'épistaxis, notamment chez les patients recevant un traitement concomitant susceptible d'augmenter le risque de saignements. Il convient de prendre les mesures nécessaires en cas d'observation d'une telle toxicité.
Chez les patients atteints d'un myélome multiple non préalablement traité éligibles à une greffe et qui reçoivent Revlimid en association avec le bortézomib et la dexaméthasone, l'hémogramme complet doit être contrôlé tous les 7 jours (une fois par semaine) au cours du premier cycle de traitement et ensuite avant le début de chaque cycle consécutif. Des contrôles mensuels réguliers sont nécessaires (chaque 4 semaines) en cas de poursuite du traitement par Revlimid en association avec la dexaméthasone.
Chez les patients atteints d'un myélome multiple qui prennent Revlimid après une autogreffe de cellules souches, l'hémogramme complet doit être surveillé tous les 7 jours (une fois par semaine) pendant les deux premiers cycles de 28 jours, toutes les 2 semaines (jour 1 et jour 15) pendant le troisième cycle de 28 jours et ensuite tous les 28 jours (4 semaines).
Chez les patients atteints d'un myélome multiple non préalablement traité non éligibles à une greffe et qui reçoivent Revlimid en association avec le melphalan et la prednisone, l'hémogramme complet doit être surveillé tous les 7 jours (1 semaine) au cours du premier cycle (28 jours), tous les 14 jours (2 semaines) jusqu'à l'achèvement de 9 cycles et ensuite tous les 28 jours (4 semaines).
Chez les patients atteints d'un myélome multiple non préalablement traité non éligibles à une greffe et qui reçoivent Revlimid en association avec la dexaméthasone, l'hémogramme complet doit être contrôlé tous les 7 jours (chaque semaine) au cours des 2 premiers cycles, les jours 1 et 15 du cycle 3, et ensuite tous les 28 jours (4 semaines).
Chez les patients atteints d'un myélome multiple ayant reçu au moins un traitement antérieur et qui reçoivent Revlimid en association avec la dexaméthasone, l'hémogramme complet doit être surveillé tous les 14 jours (2 semaines) au cours des premières 12 semaines du traitement et ensuite une fois par mois.
Chez les patients qui reçoivent Revlimid en raison d'un syndrome myélodysplasique et qui sont porteurs d'une anomalie cytogénétique comportant une délétion 5q, l'hémogramme complet doit être surveillé chaque semaine au cours des premières 8 semaines du traitement et ensuite une fois par mois.
Chez les patients sous Revlimid en raison d'un LCM, l'hémogramme complet doit être évalué chaque semaine au cours du premier cycle (28 jours), toutes les deux semaines au cours des cycles 2-4, et ensuite une fois par mois.
Les patients atteints d'un lymphome folliculaire préalablement traité et qui reçoivent l'association Revlimid et rituximab doivent être surveillés une fois par semaine pendant les trois premières semaines du cycle 1 (28 jours), puis tous les 14 jours au cours des cycles 2 à 4, puis au début de chaque cycle suivant.
Infection avec ou sans neutropénie
Les patients atteints de myélome multiple ont tendance à développer des infections, dont des pneumonies. Il a été observé une fréquence plus élevée d'infections chez les patients traités par le lénalidomide en association avec la dexaméthasone que chez les patients recevant l'association MPT. Des infections de grade ≥3 sont survenues dans le contexte d'une neutropénie chez moins d'un tiers des patients. Les patients présentant des facteurs de risque connus d'infection doivent être surveillés étroitement. Tous les patients doivent être informés qu'ils doivent consulter un médecin sans attendre au premier signe d'infection (par exemple toux, fièvre, etc.), ce qui permettra une prise en charge précoce pour en diminuer la sévérité.
De rares cas de réactivation de l'hépatite B ont été rapportés à la suite du traitement par le lénalidomide chez des patients présentant des antécédents d'infection par le virus de l'hépatite B (VHB). Certains de ces cas ont évolué vers une insuffisance hépatique aiguë et ont conduit à l'arrêt du traitement par le lénalidomide et à l'instauration d'un traitement antiviral adéquat. La sérologie VHB doit être déterminée avant l'initiation du traitement par le lénalidomide. Chez les patients ayant un résultat positif au dépistage du virus de l'hépatite B, une consultation chez un médecin spécialisé dans le traitement de l'hépatite B est recommandée. La prudence s'impose en cas d'administration de lénalidomide chez des patients préalablement infectés par le VHB. Ces patients doivent être étroitement surveillés tout au long du traitement afin de détecter les signes et symptômes d'infection active par le VHB.
La proportion d'intolérance au traitement (événements indésirables de grade 3 ou 4, événements indésirables graves, arrêts du traitement) a été plus élevée chez les patients âgés de plus de 75 ans présentant une maladie de stade ISS (International staging system) III et ayant un indice de performance ECOG ≥2 ou une ClCr < 60 ml/min lorsque le lénalidomide était administré en association. La capacité des patients à tolérer le lénalidomide en association doit être évaluée attentivement en tenant compte de l'âge et de la présence d'autres comorbidités.
Événements thrombo-emboliques artériels et veineux
Chez les patients atteints de myélome multiple, l'administration du lénalidomide avec de la dexaméthasone ou d'autres chimiothérapies (p.ex. melphalan et prednisone) est associée à un risque accru d'événements thrombo-emboliques veineux (principalement des thromboses veineuses profondes et des embolies pulmonaires). Le risque thrombo-embolique veineux est plus faible chez les patients atteints d'un myélome multiple en traitement d'entretien après une autogreffe de cellules souches ainsi que chez les patients atteints de syndrome myélodysplasique ou porteurs d'un lymphome à cellules du manteau traités par le lénalidomide en monothérapie et les patients atteints de lymphome folliculaire traités par une association R2.
Il existe un risque accru d'événements thrombo-emboliques artériels (essentiellement des infarctus du myocarde et des accidents vasculaires cérébraux) chez les patients atteints d'un myélome multiple traités par le lénalidomide en association avec la dexaméthasone et dans une moindre mesure par le lénalidomide en association avec le melphalan et la prednisone.
Le risque thrombo-embolique artériel est plus faible chez les patients atteints d'un myélome multiple traités par le lénalidomide en traitement d'entretien après une autogreffe de cellules souches que chez les patients atteints d'un myélome multiple traités par le lénalidomide en association (soit avec la dexaméthasone, soit avec le melphalan et la prednisone).
Les patients présentant des facteurs de risque connus de survenue de thromboembolie, y compris un antécédent de thrombose, doivent donc être étroitement surveillés. Les patients doivent être informés qu'ils doivent consulter un médecin en cas de symptômes comme par exemple essoufflement, toux, douleurs thoraciques, ou douleurs et/ou gonflement des bras et des jambes. Des mesures doivent être prises pour essayer de réduire au minimum tous les facteurs de risque modifiables (par exemple le tabagisme, l'hypertension et l'hyperlipidémie).
L'administration concomitante de substances stimulant l'érythropoïèse ou des antécédents d'événements thrombo-emboliques peuvent également augmenter les risques de thrombose chez ces patients. Par conséquent, les substances stimulant l'érythropoïèse et les autres substances pouvant accroître les risques de thrombose, comme les traitements hormonaux substitutifs, doivent être utilisées avec précaution chez les patients atteints de myélome multiple traités par le lénalidomide et la dexaméthasone. Un taux d'hémoglobine supérieur à 11 g/dl doit conduire à l'arrêt des substances stimulant l'érythropoïèse.
La prescription d'antithrombotiques en prophylaxie est recommandée, en particulier chez les patients présentant des facteurs de risque thrombo-embolique supplémentaires.
La décision concernant les mesures thérapeutiques de prophylaxie anti-thrombotique doit être prise après une évaluation soigneuse individuelle de chaque patient.
En cas d'événement thrombo-embolique, le traitement par lénalidomide doit être interrompu et une thérapie anticoagulante standard doit être mise en œuvre. Une fois le patient stabilisé, le traitement par le lénalidomide peut si nécessaire être repris tout en poursuivant le traitement anticoagulant.
Hypertension artérielle pulmonaire
Des cas d'hypertension artérielle pulmonaire, dont certains d'issue fatale, ont été rapportés chez des patients traités par lénalidomide. Il convient donc de rechercher tout signe et symptôme de maladie cardiopulmonaire sous-jacente chez le patient avant et pendant un traitement par lénalidomide.
Infarctus du myocarde
Des cas d'infarctus du myocarde ont été rapportés chez les patients recevant du lénalidomide, notamment chez ceux qui présentent des facteurs de risque connus. Une surveillance étroite s'impose chez les patients présentant des facteurs de risque connus (parmi lesquels un antécédent de thrombose). Des mesures doivent être prises pour essayer de réduire au minimum tous les facteurs de risque modifiables (par exemple le tabagisme, l'hypertension artérielle et l'hyperlipidémie).
Cancers secondaires au traitement
En raison d'un petit nombre de cas, dans les études cliniques menées chez des patients recevant l'association lénalidomide/dexaméthasone et ayant déjà reçu un traitement pour leur myélome, on a observé un déséquilibre numérique par rapport aux témoins. Il s'agit essentiellement d'épithélioma basocellulaire ou spinocellulaire de la peau.
Dans des études cliniques réalisées chez des patients présentant un myélome multiple nouvellement diagnostiqué, on a observé une augmentation des formes invasives de cancer primitif secondaire, y compris de leucémie aiguë myéloblastique (LAM) et de syndromes myélodysplasiques (SMD), les cas étant diagnostiqués chez des patients qui recevaient du lénalidomide en association avec du melphalan (fréquence de 5,3%) ou directement après un traitement par melphalan à forte dose et une autogreffe de cellules souches (fréquence de 7,5%). La fréquence des cas de LAM et de SMD observée dans le bras lénalidomide/dexaméthasone était de 0,4%.
Des cas de lymphomes à cellules B (y compris de maladie de Hodgkin) ont été observés dans les études cliniques dans lesquelles les patients avaient reçu Revlimid après une autogreffe de cellules souches.
Une augmentation de cancers secondaires au traitement a été observée chez les patients qui avaient reçu le lénalidomide immédiatement après une forte dose intraveineuse de melphalan et une autogreffe de cellules souches (fréquence de 7,7%).
Chez les patients atteints d'un myélome multiple nouvellement diagnostiqué qui avaient reçu le lénalidomide en association avec le bortézomib et la dexaméthasone, la fréquence des cancers hématologiques secondaires allait de 0,0% jusqu'à 0,8% et la fréquence des cancers secondaires solides de 0,4% jusqu'à 4,5%.
Chez les patients atteints d'un LF qui avaient été traités par une association de lénalidomide et rituximab, la fréquence des cancers hématologiques secondaires était de 0,7% et la fréquence des cancers secondaires solides de 1,4%.
Tant les bénéfices obtenus avec le lénalidomide que le risque de survenue d'un cancer secondaire doivent être pris en compte avant d'instaurer le traitement par lénalidomide. Le médecin doit évaluer soigneusement les patients avant et pendant le traitement en utilisant les méthodes habituelles de dépistage des cancers pour surveiller le développement de cancers secondaires et, le cas échéant, instaurer un traitement.
Maladies hépatiques
Chez les patients qui reçoivent un traitement par le lénalidomide en association avec la dexaméthasone, on a rapporté la survenue d'une insuffisance hépatique, dont des cas d'issue fatale: insuffisance hépatique aiguë, hépatite toxique, hépatite cytolytique, hépatite cholestatique et hépatite mixte cytolytique/cholestatique ont été signalées. Les mécanismes de l'hépatotoxicité sévère d'origine médicamenteuse ne sont toujours pas connus, bien que dans certains cas, une maladie hépatique virale préexistante, des taux d'enzymes hépatiques élevés dès le départ et parfois un traitement par des antibiotiques peuvent constituer des facteurs de risque.
Des valeurs anormales de la fonction hépatique ont souvent été rapportées, qui étaient généralement asymptomatiques et réversibles après l'arrêt du traitement. Dès que les paramètres de la fonction hépatique reviennent aux valeurs de départ, la reprise du traitement à une dose inférieure peut alors être envisagée.
Le lénalidomide est excrété par les reins. Chez les patients insuffisants rénaux, il est important d'adapter la posologie afin d'éviter d'atteindre des taux plasmatiques susceptibles de majorer le risque d'effets indésirables hématologiques plus fréquents ou d'hépatotoxicité. Il est dès lors recommandé de surveiller la fonction hépatique, en particulier en cas d'antécédents ou d'infections hépatiques virales concomitantes ou lorsque le lénalidomide est associé à des médicaments connus pour induire des troubles de la fonction hépatique.
Réactions allergiques et réactions cutanées graves
Des œdèmes de Quincke, des anaphylaxies et de graves réactions dermatologiques, notamment le syndrome de Stevens-Johnson (SJS), de nécrolyse épidermique toxique (NET) et une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS) ont été rapportés. Le DRESS peut se manifester sous forme d'éruption cutanée ou de dermatite exfoliative associée à une éosinophilie, une fièvre et/ou une lymphadénopathie avec complications systémiques comme une hépatite, une néphrite, une pneumopathie inflammatoire, une myocardite et/ou une péricardite. Ces événements peuvent être fatals. Il convient d'envisager l'interruption ou l'arrêt définitif de Revlimid en cas d'éruption cutanée de grade 2 ou 3. En cas d'angioedème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, de suspicion d'un SJS, d'une TEN ou d'un DRESS, le traitement par Revlimid doit être arrêté. Après l'interruption en raison de ces réactions, le traitement ne doit pas être repris. Les patients ayant présenté une éruption cutanée sévère de grade 4 liée à un traitement par le thalidomide ne doivent pas être traités par Revlimid.
Syndrome de lyse tumorale
Un syndrome de lyse tumorale (SLT) peut survenir, y compris chez les patients atteints d'un lymphome. Des cas de SLT mortels ont été rapportés pendant le traitement par Revlimid.
Les patients présentant un tel risque sont ceux ayant une forte masse tumorale avant le traitement. Ces patients doivent faire l'objet d'une surveillance étroite, surtout au cours du premier cycle ou lors d'une augmentation de la posologie et des précautions appropriées doivent être prises.
Réaction de flambée tumorale
Il est conseillé de procéder à une surveillance étroite et à un examen approfondi en vue de déceler la survenue d'une réaction de flambée tumorale (TFR). Une flambée tumorale peut simuler une progression de la maladie (progression, PD) et entraîner le décès.. Dans l'étude MCL-001 en vue de l'obtention de l'AMM, près de 10% des patients ont présenté une TFR. Tous les cas ont été classés selon un degré de sévérité 1 ou 2 et ont été considérés comme liés au traitement.
Le taux de TFR rapporté dans l'étude NHL-007 était de 13,0%, dont un événement correspondait au grade 3. Dans l'étude NHL-008, le taux était de 4,0%, avec un événement considéré comme grave parmi les autres événements rapportés de degré 1 et 2. La plupart des événements sont survenus au cours du cycle 1. Chez les patients atteints de TFR de grade 1 et 2, le traitement par le lénalidomide peut être poursuivi sans interruption ou modification, à la discrétion du médecin. Pour traiter les symptômes de la TFR, les patients présentant une TFR de grade 1 et 2 ont été traités dans les études cliniques MCL-001, NHL-007 et NHL-008 par des corticostéroïdes, des anti-inflammatoires non stéroïdiens (AINS) et/ou des analgésiques narcotiques. Chez les patients présentant une TFR de grade 3 ou 4, le traitement par le lénalidomide doit être interrompu jusqu'à ce que la TFR retombe à ≤ grade 1. Pour le traitement des symptômes, les patients peuvent être traités conformément aux indications données pour une TFR de grade 1 et 2.
Décès prématurés chez les patients atteints d'un LCM
Dans l'étude MCL-002, il a été observé globalement une augmentation apparente des décès prématurés (au cours des 20 premières semaines). Les patients ayant une charge tumorale élevée avant l'initiation du traitement présentent un risque accru de décès prématuré: des décès prématurés ont été rapportés chez 16/81 patients (20 %) du bras lénalidomide et 2/28 patients (7 %) du bras contrôle. Les chiffres correspondants sur 52 semaines étaient de 32/81 patients (40 %) et 6/28 patients (21 %).
Réactions de rejet après transplantation d'organe
Dans le cadre de l'expérience post-autorisation de mise sur le marché, des cas de rejet de transplantation d'organe ont été rapportés lors de l'utilisation de Revlimid, dont certains d'évolution fatale. Dans la majorité des cas, la réaction de rejet est survenue dans les deux mois ayant suivi le début du traitement par Revlimid. Les facteurs ayant pu contribuer au rejet de la transplantation d'organe dans les cas rapportés sont: la maladie sous-jacente (p.ex. amyloïdose), des infections intercurrentes et une interruption ou réduction récente du traitement immunosuppresseur. Le caractère limité des données de sécurité collectées après la mise sur le marché ne permet pas de faire une évaluation fiable du taux d'incidence des réactions de rejet de transplantation d'organe. La prise de Revlimid a généralement été arrêtée définitivement après la survenue de la réaction de rejet. Avant d'instaurer un traitement par Revlimid, son bénéfice doit être évalué au regard du risque de rejet possible de transplantation d'organe chez des patients transplantés.
Troubles de la fonction thyroïdienne
Une hypothyroïdie ainsi qu'une hyperthyroïdie ont été observées pendant le traitement par le lénalidomide (voir "Effets indésirables" ). C'est pourquoi, avant le début du traitement par Revlimid, il est conseillé de stabiliser au mieux les maladies concomitantes qui pourraient avoir un effet sur la fonction thyroïdienne. Il est conseillé de surveiller la fonction thyroïdienne avant le début du traitement et pendant le traitement.
Électrophysiologie cardiaque
Un allongement de l'intervalle QTc a été observé sous lénalidomide. Un traitement concomitant avec des médicaments susceptibles de prolonger l'intervalle QT ou un traitement de patients souffrant d'un syndrome du QT long exige un maximum de prudence et des contrôles réguliers par ECG (cf. "Propriétés/Effets" ).
Effets immunosuppresseurs
Le lénalidomide a des effets immunosuppresseurs puissants. Par conséquent, la prudence est de mise lors d'une administration concomitante avec d'autres agents immunomodulateurs. L'efficacité de certains vaccins peut être affectée. En raison du risque d'infection, l'administration de vaccins vivants doit être évitée pendant le traitement par le lénalidomide.
Traitement en association
Pour des précisions relatives à d'autres médicaments utilisés en association avec le lénalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Intolérance au lactose
Les gélules de Revlimid contiennent du lactose. Les patients souffrant de troubles héréditaires rares d'intolérance au galactose, de déficit de lactase de Lapp ou de malabsorption de glucose et de galactose ne doivent pas prendre ce médicament.
Ce médicament renferme moins de 1 mmol de sodium (23 mg) par gélule, ce qui signifie qu'il s'agit du médicament "sans sodium" .
InteractionsLe lénalidomide n'est pas dégradé par les enzymes de phase I et ne se lie qu'en faible mesure aux protéines plasmatiques. Les interactions dues au cytochrome P450 ou à la liaison aux protéines sont donc improbables.
Le lénalidomide étant soumis à une sécrétion tubulaire active, des interactions avec d'autres médicaments soumis à une sécrétion tubulaire active sont possibles. On ne dispose que de peu de données de valeurs accrues d'acide urique.
Le lénalidomide (10 mg) n'a pas influencé la pharmacocinétique d'une dose unique de warfarine R/S administrée en même temps. Une dose unique de 25 mg de warfarine R/S n'a pas influencé la pharmacocinétique du lénalidomide administré en même temps. Toutefois, on ignore si des interactions surviennent lors de l'utilisation clinique. Une surveillance étroite du taux sanguin de warfarine est donc conseillée pendant le traitement.
Un traitement aux coumarines n'est pas recommandé en raison du risque élevé de thrombocytopénie.
La dexaméthasone (40 mg/jour) n'avait aucun effet sur la pharmacocinétique de Revlimid.
L'administration concomitante du lénalidomide à 10 mg/jour a accru l'exposition plasmatique à la digoxine (0,5 mg, dose unique) de 14% avec un IC (intervalle de confiance) à 90% [0,52–28,2%]. On ignore si les effets seraient différents dans un contexte thérapeutique (doses du lénalidomide plus élevées et administration concomitante de dexaméthasone). Il est donc conseillé de surveiller la concentration de la digoxine pendant le traitement par le lénalidomide.
L'administration concomitante de doses répétées de quinidine, un inhibiteur de la P-gp (600 mg deux fois par jour) n'a pas eu d'effet cliniquement pertinent sur la pharmacocinétique du lénalidomide (25 mg). L'administration concomitante de lénalidomide (25 mg) et de temsirolimus, un inhibiteur/substrat de la P-gp (25 mg), ne modifie pas la pharmacocinétique des deux médicaments.
Les substances stimulant l'érythropoïèse ou d'autres substances qui peuvent augmenter le risque de thrombose, comme par exemple un traitement hormonal de substitution, doivent être utilisées avec prudence chez les patients et les patientes atteints/atteintes de myélome multiple qui reçoivent du lénalidomide associé à la dexaméthasone.
Grossesse, allaitementGrossesse
On ne dispose pas de données cliniques sur l'utilisation du lénalidomide chez la femme enceinte. Le lénalidomide est structurellement proche du thalidomide. Le thalidomide est un tératogène connu, provoquant des anomalies congénitales graves, potentiellement létales chez l'enfant à naître après exposition pendant la grossesse. Dans une étude sur le développement embryonnaire/fœtal chez le singe femelle gravide, le lénalidomide a donné lieu à des malformations chez la progéniture (voir également "Données précliniques" ). Il faut s'attendre à ce que le lénalidomide ait un effet tératogène chez l'homme. Détails du programme de contraception: voir "Mises en garde et précautions" .
Pour les patients de sexe masculin, voir la section "Mises en garde et précautions" .
Allaitement
On ne sait pas si le lénalidomide passe dans le lait maternel. Par conséquent, Revlimid ne doit pas être utilisé chez les femmes qui allaitent. Une femme qui allaite ne pourra être traitée par Revlimid qu'après le sevrage du bébé.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été réalisée sur les effets éventuels sur l'aptitude à la conduite ou à l'utilisation de machines. Les effets indésirables de Revlimid peuvent englober de la fatigue, un engourdissement, une somnolence et une vue trouble. La conduite de véhicules ou l'utilisation de machines devront donc se faire avec précaution.
Effets indésirablesMyélome multiple
Patients atteints d'un myélome multiple non traité éligibles à une greffe et qui ont reçu le lénalidomide en association avec le bortézomib et la dexaméthasone
Dans les études PETHEMA GEM2012 (données regroupées des bras A et B (RVd), n=458) et IFM 2009 (bras A (RVd), n=356) l'effet indésirable sévère suivant a été le plus fréquemment observé (≥5%) avec le lénalidomide en association avec le bortézomib et la dexaméthasone:
pneumonie (5,9%) dans l'étude PETHEMA GEM2012.
Dans l'étude PETHEMA GEM2012, les effets indésirables les plus fréquemment observés avec le lénalidomide en association avec le bortézomib et la dexaméthasone administrés par voie sous-cutanée étaient la neuropathie périphérique (35,2%), la neutropénie (31,9%) et la thrombocytopénie (25,3%).
Dans l'étude IFM 2009, les effets indésirables les plus fréquemment observés avec le lénalidomide en association avec le bortézomib et la dexaméthasone administrés par voie intraveineuse étaient la neuropathie périphérique (54,8%) et la lymphopénie (52,2%).
Patients atteints d'un myélome multiple traités par le lénalidomide après une autogreffe de cellules souches
Dans deux études de Phase III, à deux bras, en double aveugle, contrôlées par placebo (IFM 2005-02 et CALGB 100104), 517 patients ont reçu le lénalidomide et 501 patients le placebo. Les effets indésirables de l'étude CALGB 100104 incluaient non seulement les événements survenus au cours de la période de traitement d'entretien mais aussi les événements rapportés après un traitement par MFD/AGCS. Dans l'étude IFM 2005-02, les données concernant les effets indésirables ne concernaient que la période de traitement d'entretien.
Les effets indésirables graves observés plus fréquemment (≥5%) avec le lénalidomide en traitement d'entretien qu'avec le placebo ont été:
pneumonies (10,6%; terme combiné)
infections pulmonaires (9,4%)
Dans les deux études, les effets indésirables observés plus fréquemment avec le lénalidomide en traitement d'entretien qu'avec le placebo ont été: neutropénie (79,0%), thrombocytopénie (72,3%), diarrhée (54,5%), bronchite (47,4%), rhinopharyngite (34,8%), spasmes musculaires (33,4%), éruption cutanée (31,7%), leucopénie (31,7%), asthénie (29,7%), toux (27,3%), infections des voies respiratoires supérieures (26,8%), fatigue (22,8%), gastroentérite (22,5%), anémie (21,0%) et fièvre (20,5%).
Patients atteints d'un myélome multiple non préalablement traité non éligibles à une greffe et traités par le lénalidomide en association avec le bortézomib et la dexaméthasone
Dans l'étude SWOG S0777 (bras B (RVd), n=262), les effets indésirables sévères observés avec le lénalidomide en association avec le bortézomib et la dexaméthasone par voie intraveineuse étaient plus fréquents (≥5%) que ceux observés avec le lénalidomide en association avec la dexaméthasone:
hypotension (6,5%), infection pulmonaire (5,7%), déshydratation (5,0%).
Les effets indésirables plus fréquemment observés avec le lénalidomide en association avec le bortézomib et la dexaméthasone qu'avec le lénalidomide en association avec la dexaméthasone étaient la fatigue (73,7%), la neuropathie périphérique (71,8%), la thrombocytopénie (57,6%), la constipation (56,1%) et l'hypocalcémie (50,0%).
Myélome multiple non préalablement traité chez les patients traités par le lénalidomide en association avec la dexaméthasone à faible dose
Dans une étude de phase III à trois bras, en ouvert, 535 patients ont reçu le lénalidomide en association avec de la dexaméthasone à faible dose jusqu'à la progression de la maladie (Rd), 541 patients ont été traités par le lénalidomide en association avec de la dexaméthasone à faible dose jusqu'à l'achèvement de 18 cycles de 28 jours (Rd18) et 547 patients ont reçu l'association de melphalan, prednisone et thalidomide (MPT).
Les effets indésirables graves observés plus fréquemment (≥5%) avec l'association lénalidomide + dexaméthasone à faible dose (Rd et Rd18) qu'avec l'association melphalan, prednisone et thalidomide (MPT) ont été:
pneumonie (9,8%);
insuffisance rénale (y compris aiguë) (6,3%).
Les effets indésirables observés plus fréquemment dans les bras Rd ou Rd18 que dans le bras MPT ont été: diarrhée (45,5%), fatigue (32,8%), dorsalgies (32,0%), asthénie (28,2%), insomnies (27,6%), éruption cutanée (24,3%), diminution de l'appétit (23,1%), toux (22,7%), pyrexie (21,4%) et spasmes musculaires (20,5%).
Myélome multiple non préalablement traité chez les patients traités par le lénalidomide en association avec le melphalan et la prednisone
Une étude de phase III en double aveugle, contrôlée contre placebo, à triple bras, a évalué la sécurité et l'efficacité de l'association de melphalan, prednisone et lénalidomide (MPR) suivie d'une monothérapie d'entretien par lénalidomide. 152 patients ont reçu l'association MPR par voie orale en traitement d'induction, suivie du lénalidomide en traitement d'entretien (MPR+R), 153 patients ont reçu l'association MPR par voie orale en traitement d'induction, suivie d'un placebo en traitement d'entretien (MPR+p), et 154 patients ont reçu l'association MPp par voie orale (MP + placebo) en traitement d'induction, suivie d'un placebo en traitement d'entretien (MPp+p).
Les effets indésirables graves observés plus fréquemment (≥5%) avec l'association melphalan, prednisone et lénalidomide suivie du lénalidomide en traitement d'entretien (MPR+R) ou melphalan, prednisone et lénalidomide suivie du placebo (MPR+p) qu'avec l'association melphalan, prednisone et placebo suivie du placebo (MPp+p) ont été:
neutropénie fébrile (6,0%);
anémie (5,3%).
Les effets indésirables observés plus fréquemment dans les bras MPR+R ou MPR+p que dans le bras MPp+p ont été: neutropénie (83,3%), anémie (70,7%), thrombocytopénie (70,0%), leucopénie (38,8%), constipation (34,0%), diarrhée (33,3%), éruptions cutanées (28,9%), pyrexie (27,0%), œdème périphérique (25,0%), toux (24,0%), diminution de l'appétit (23,7%) et asthénie (22,0%).
Myélome multiple chez les patients ayant reçu au moins un traitement antérieur
Dans les études de phase III, contrôlées contre placebo, 353 patients ont reçu l'association lénalidomide-dexaméthasone et 350 patients l'association placebo-dexaméthasone. Au moins un effet indésirable a été observé chez 325 patients (92%) sous lénalidomide-dexaméthasone et chez 288 patients (82%) sous placebo-dexaméthasone.
Les effets indésirables les plus graves ont été des thromboembolies veineuses (thromboses veineuses profondes, embolies pulmonaires) et des neutropénies de grade 4.
Les effets indésirables les plus souvent observés sous lénalidomide-dexaméthasone ont été: neutropénie (39,4%; grade 4: 5,1%), thrombocytopénie (18,4%, grades 3/4: 9,9%), épuisement (27,2%), constipation (23,5%), crampes musculaires (20,1%), asthénie (17,6%), anémie (17,0%), diarrhée (14,2%) et éruptions cutanées (10,2%), ainsi qu'insomnies (26,7%) et faiblesse musculaire (10,1%). La neutropénie et la thrombocytopénie se sont manifestées de façon dose-dépendante et ont pu être traitées efficacement par réduction de la dose.
Syndrome myélodysplasique
Dans une étude de phase III, contrôlée contre placebo, 69 patients ont reçu une fois par jour 10 mg de lénalidomide et 67 ont reçu le placebo.
Les effets indésirables les plus graves observés ont été les thrombo-embolies veineuses (thrombose veineuse profonde, embolie pulmonaire), neutropénie de grade 3-4, neutropénie fébrile et thrombocytopénie de grade 3-4.
Les effets indésirables observés les plus fréquents dans le groupe lénalidomide étaient les suivants: neutropénie (76,8%; grade 3-4: 75,4%), thrombocytopénie (49,3%; grade 3-4: 40,6%), diarrhée (37,7%), prurit (27,5%), nausées (20,3%), fatigue (18,8%), constipation (17,4%), spasmes musculaires (17,4%), fièvre (15,9%), rhinopharyngite (14,5%), bronchite (14,5%) et céphalées (14,5%). La neutropénie et la thrombocytopénie qui sont survenues étaient principalement doses dépendantes et ont été traitées avec succès par une réduction de la dose.
Lymphome des cellules du manteau
Dans l'étude sur le LCM en vue de l'obtention de l'AMM, un total de 134 patients a reçu au moins une dose de Revlimid.
Les infections représentaient le type le plus fréquent d'événements indésirables graves. Parmi les infections graves, la pneumonie a été la plus fréquemment rapportée.
Les effets indésirables observés les plus fréquents étaient: pneumonie (14,2%; grade 3-4: 9%), infections des voies respiratoires hautes (12,7%), neutropénie (48,5%; grade 3-4: 43,3%), thrombocytopénie (35,8%, grade 3-4: 27,6%), anémie (30,6%; grade 3-4: 11,2%), leucopénie (14,9%; grade 3-4: 6,7%), perte d'appétit (14,2%), hypokaliémie (12,7%; grade 3-4: 2,2%), perte de poids (12,7%), toux (28,4%), dyspnée (17,9%; grade 3-4: 6%), diarrhée (31,3%; grade 3-4: 6%), nausée (29,9%), constipation (15,7%), vomissement (11,9%), éruption cutanée (22,4%; grade 3-4: 1,5%), prurit (17,2%), douleurs dorsales (13,4%; grade 3-4: 1,5%), spasmes musculaires (12,7%), fatigue (33,6%; grade 3-4: 6,7%), fièvre (23,1%; grade 3-4: 2,2%), œdème périphérique (15,7%), et asthénie (14,2%; grade 3-4: 3%).
Lymphome folliculaire (LF)
Le profil de sécurité global du lénalidomide en association avec le rituximab chez les patients présentant un lymphome folliculaire préalablement traité est issu des données de 146 patients ayant participé à l'étude NHL-007 et de 177 patients de l'étude NHL-008.
Les effets indésirables les plus graves observés étaient les suivants: neutropénie fébrile (2,7%), embolie pulmonaire (2,7%) et pneumonie (2,7%).
Les effets indésirables observés les plus fréquents dans le groupe lénalidomide-rituximab étaient: neutropénie (58,2%), diarrhée (30,8%), leucopénie (28,8%), constipation (21,9%), toux (21,9%) et fatigue (21,9%).
Les effets indésirables qui ont été observés chez les patients atteints de myélome multiple, de syndrome myélodysplasique, de lymphome des cellules du manteau et de lymphome folliculaire ont été listés ci-dessous par système d'organes et fréquence de survenue. Dans chaque groupe de fréquence, les effets indésirables sont indiqués par sévérité décroissante.
Incidences: très fréquents: > 1/10; fréquents: > 1/100, < 1/10; occasionnels: > 1/1000, < 1/100; rares: > 1/10000, < 1/1000; très rares: < 1/10000.
Infections et infestations
Très fréquents: Bronchite (47,4%), rhinopharyngite (34,8%), infections des voies respiratoires supérieures (26,8%), gastroentérite (22,5%), infections neutropéniques (17,9%), pneumonie (17,1%), rhinite (15,0%), sinusite (14,0%), grippe (13,3%), infections urinaires (11,6%).
Fréquents: Bactériémie, septicémie, infections locales et systémiques (bactériennes, virales, fongiques), cellulite, candidose orale, infections des voies respiratoires, infection des poumons, infection des voies respiratoires inférieures, entérocolite infectieuse.
Occasionnels: Pneumonie atypique, pneumonie à Pneumocystis carinii, endocardite subaiguë, herpès ophtalmique, zona, infections des oreilles, candidose œsophagienne, réactivation du virus* (virus de l'hépatite B ou herpès zoster).
Très rares: Leucoencéphalopathie multifocale progressive*.
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
Très fréquent: Réaction de flambée tumorale (13,0%).
Fréquents: Leucémie aiguë myéloblastique, syndrome myélodysplasique, carcinome à cellules squameuses de la peau, carcinome basocellulaire, syndrome de lyse tumorale.
Occasionnels: Leucémie aiguë à cellules T.
Affections hématologiques et du système lymphatique
Très fréquents: Neutropénie (79,0%), thrombocytopénie (72,3%), lymphopénie (52,2%), anémie (43,8%), leucopénie (36,0%), neutropénie fébrile (17,4%).
Fréquents: Pancytopénie.
Occasionnels: Granulocytopénie, anémie hémolytique, prolongation du temps de coagulation, monocytopénie, leucocytose, lymphadénopathie.
Affections du système immunitaire
Occasionnel: Hypogammaglobulinémie acquise, œdème de Quincke*, maladie aiguë du greffon contre l'hôte*.
Rare: Anaphylaxie*.
Indéterminés: Rejet de greffe d'organe*.
Affections endocriniennes
Fréquent: Syndrome cushingoïde.
Occasionnels: Insuffisance surrénalienne, hypothyroïdie, hyperthyroïdie, taux de TSH réduit ou accru, hirsutisme.
Troubles du métabolisme et de la nutrition
Très fréquents: Hypocalcémie (50,0%), perte d'appétit (34,4%), hyponatrémie (30,5%), hypokaliémie (29,0%), déshydratation (16,4%), perte de poids (13,5%), hyperglycémie (11,7%), hypoglycémie (10,7%).
Fréquents: Anorexie, hypomagnésiémie, rétention liquidienne prise de poids, surcharge en fer, hypophosphatémie, hypercalcémie, hyperuricémie.
Occasionnels: Acidose métabolique, diabète sucré, hypoalbuminémie, cachexie, goutte, hyperphosphatémie, appétit accru.
Affections psychiatriques
Très fréquents: Insomnies (32,8%), dépressions (10,9%).
Fréquents: États de confusion, hallucinations, fluctuations d'humeur, anxiété, irritabilité, somnolence.
Occasionnels: Troubles psychotiques, hypomanie, idées fixes, baisse de la libido, changements de la personnalité, nervosité, agression, cauchemars.
Affections du système nerveux
Très fréquents: Neuropathie périphérique (71,8%), perturbation du sens gustatif (30,2%), vertiges (29,4%), paresthésies (22,5%), céphalées (15,4%).
Fréquents: Troubles circulatoires du cerveau, syncope, engourdissement, tremblements, troubles de la mémoire, névralgies, dysesthésie, neuropathie périphérique sensitive.
Occasionnels: Accident vasculaire cérébral, leuco-encéphalopathie, troubles de la parole, troubles de l'attention, troubles de l'équilibre, troubles moteurs, paresthésie orale, hyperactivité psychomotrice, anosmie, ataxie, dyskinésie, dysfonction motrice, syndrome myasthénique.
Affections oculaires
Très fréquent: Vision trouble (16,0%), cataracte (13,7%).
Fréquents: Troubles de la vue, larmoiement, conjonctivite.
Occasionnels: Cécité, artériosclérose rétinienne, thrombose veineuse rétinienne, kératite, irritation des yeux, sécheresse oculaire.
Affections de l'oreille et du labyrinthe
Fréquent: Vertige.
Occasionnels: Surdité, diminution de l'ouïe, acouphènes, douleurs dans les oreilles.
Affections cardiaques
Fréquents: Fibrillation auriculaire, infarctus du myocarde*, insuffisance cardiaque.
Occasionnels: Insuffisance cardiaque congestive, insuffisance valvulaire, flutter auriculaire, trigéminisme ventriculaire, bradycardie, tachycardie, allongement de l'intervalle QT, œdème pulmonaire, arythmie.
Affections vasculaires
Très fréquents: Hypotension (16,4%), thromboses veineuses profondes (10,2%).
Fréquents: Hypertension, bouffées de chaleur, hématome.
Occasionnels: Collapsus circulatoire, ischémie, phlébite.
Affections respiratoires, thoraciques et médiastinales
Très fréquents: Dyspnée (30,5%), toux (29,4%).
Fréquents: Embolie pulmonaire, crise d'étouffements, douleurs pleurétiques, hypoxie pharyngite, épistaxis, rhinorrhée, dysphonie, enrouement, hoquet.
Occasionnels: Asthme, douleurs dans la poitrine, hypertension artérielle pulmonaire.
Rare: Pneumonie interstitielle.
Affections hépatobiliaires
Très fréquents: Résultats anormaux des tests de la fonction hépatique, par exemple taux accrus d'ALAT (alaninaminotransférase) (25,6%), taux accrus d'ASAT (aspartataminotransférase) (21,4%) ou hyperbilirubinémie (15,2%); taux accru de phosphatase alcaline dans le sang (25,2%).
Fréquents: Lésion hépatocellulaire, hépatotoxicité, taux élevé de bilirubine dans le sang
Occasionnel: Insuffisance hépatique.
Indéterminés: Insuffisance hépatique aiguë, hépatite toxique, hépatite cytolytique, hépatite cholestatique, hépatite mixte cytolytique/cholestatique.
Affections gastrointestinales
Très fréquents: Constipation (56,1%), diarrhées (54,5%), nausées (37,4%), dyspepsie (19,1%), vomissements (17,6%), douleurs abdominales (14,7%), stomatite (12,2%), sécheresse buccale (11,5%).
Fréquents: Occlusion de l'intestin grêle, gastrite, distension de l'abdomen, douleurs épigastriques, ballonnements.
Occasionnels: Hémorragies gastro-intestinales, colite, proctite, dysphagie, hémorroïdes, douleurs buccales, saignements des gencives.
Rare: Pancréatite*.
Affections de la peau et du tissu sous-cutané
Très fréquents: Éruption cutanée (31,7%), prurit (27,5%), sécheresse cutanée (10,6%).
Fréquents: Œdèmes du visage, érythème, folliculite, hyperpigmentation, exanthème, sueurs profuses, chute des cheveux, sueurs nocturnes.
Occasionnels: Érythème noueux, urticaire, eczéma, hyperkératose, peau fissurée, acné, lichen scléreux, réaction de photosensibilité, sensation de brûlure cutanée, desquamation.
Rares: Syndrome de Stevens-Johnson*, nécrolyse épidermique toxique*.
Très rares: Réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS)*.
Affections musculosquelettiques et systémiques
Très fréquents: Spasmes musculaires (33,4%), douleurs dorsales (33,2%), faiblesse musculaire (24,4%), crampes musculaires (20,1%), arthralgie (19,0%), douleurs des membres (17,9%), myalgies (14,9%), douleurs dans la musculature squelettique (14,8%), douleurs osseuses (11,8%), douleurs thoraciques musculo-squelettiques (11,3%).
Fréquents: Myopathie, gonflements périphériques, douleurs de la nuque.
Occasionnels: Ostéonécrose, atrophie musculaire, spondylite, articulations enflées, raideur de la musculature squelettique, gonflements localisés.
Affections du rein et des voies urinaires
Fréquents: Insuffisance rénale, défaillance rénale (y compris aiguë), lésion rénale aiguë.
Occasionnels: Mictions fréquentes, nécrose tubulaire rénale, rétention urinaire, syndrome de Fanconi acquis, incontinence urinaire.
Affections des organes de reproduction et du sein
Fréquents: Dysfonction érectile, gynécomastie, métrorragie, mamelons douloureux.
Troubles généraux et anomalies au site d'administration
Très fréquents: Fatigue (73,7%), œdème périphérique (46,6%), asthénie (29,7%), fièvre (23,1%).
Fréquents: Chute, frissons, douleurs thoraciques non cardiaques, contusion, malaise.
Occasionnels: Sensation de soif, sensation de froid.
* = Données de pharmacovigilance
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les études, la toxicité dose-limitante était principalement de type hématologique. En cas de surdosage, des contrôles (cliniques, laboratoire) sont indiqués ainsi que des mesures de soutien symptomatiques.
Le lénalidomide est peu dialysable.
Propriétés/EffetsCode ATC
L04AX04
Mécanisme d'action
Le lénalidomide est un dérivé du thalidomide. Il constitue un mélange racémique. Il possède aussi bien des propriétés immunomodulatrices que des propriétés anti-angiogénétiques.
Le lénalidomide se lie à la protéine intracellulaire, cereblon (CRBN). Elle fait partie du complexe ubiquitine ligase E3, qui regroupe les protéines DDB1 (DNA Damage-Binding Protein 1), CUL4 (Cullin 4) et Roc1. Les ubiquitines ligases E3 sont responsables de la polyubiquitination d'une série de protéines substrats variées et pourraient expliquer les effets pléiotropes cellulaires observés sous traitement par le lénalidomide.
Le lénalidomide inhibe la libération de cytokines pro-inflammatoires, dont le facteur de nécrose tumorale α (TNF-α) et les interleukines 1β (IL-1β), 6 (IL-6) et 12 (IL-12) des cellules mononucléaires du sang périphérique stimulées par des lipopolysaccharides (LPS), et provoque une formation accrue de la cytokine antiinflammatoire IL-10 dans les cellules stimulées par LPS.
Il induit la production d'IL-2 et d'interféron 1γ (IFN-1γ) et stimule la prolifération des cellules T ainsi que l'activité cytotoxique des cellules tueuses naturelles.
Le lénalidomide inhibe la prolifération de différentes lignées de cellules cancéreuses hématopoïétiques.
Dans le lymphome folliculaire, l'association lénalidomide-rituximab augmente la cytotoxicité à médiation cellulaire dépendante des anticorps (ADCC) qui est médiée par les cellules tueuses naturelles, la formation de synapses immunes et l'apoptose directe, ce qui lui confère une activité antitumorale supérieure à celle des monothérapies.
Dans les modèles d'angiogenèse in vitro, le lénalidomide inhibe l'angiogenèse par une inhibition du développement de micro-vaisseaux et de canaux cellulaires endothéliaux ainsi que par une inhibition de la migration de cellules endothéliales. Le lénalidomide inhibe également la formation du facteur pro-angiogénétique VEGF dans les cellules PC-3 des cancers de la prostate.
Pharmacodynamique
Analyse de l'intervalle QT par électrophysiologie cardiaque
Aucun allongement de l'intervalle QTc n'a été observé lors de l'administration d'une dose unique de 10 mg ou 50 mg de lénalidomide à des sujets masculins en bonne santé.
Efficacité clinique
Expérience clinique du lénalidomide en association avec le bortézomib et la dexaméthasone chez les patients atteints d'un myélome multiple non préalablement traité, éligibles à une greffe
L'efficacité (conformément aux critères de réponse de l'International Myeloma Working Group, IMWG) et la sécurité du lénalidomide en association avec le bortézomib et la dexaméthasone (RVd) ont été évaluées dans deux études cliniques multicentriques de phase III: PETHEMA GEM2012 et IFM 2009.
PETHEMA GEM2012
PETHEMA GEM2012 était une étude multicentrique, randomisée, ouverte et contrôlée, de phase III, comparant 2 régimes de conditionnement (busulfan-melphalan et MEL200) administrés avant la greffe à des patients qui avaient reçu RVd en tant que traitement initial. RVd avait été administré pendant 6 cycles de 4 semaines (24 semaines). Les patients ont reçu 25 mg/jour de lénalidomide par voie orale les jours 1 à 21, 1,3 mg/ m2 de bortézomib par voie sous-cutanée les jours 1, 4, 8 et 11 ainsi que 40 mg/jour de dexaméthasone par voie orale les jours 1 à 4, 9 à 12 de cycles consécutifs de 28 jours. À la suite du traitement initial, les patients ont reçu soit un régime de conditionnement par busulfan-melphalan soit par MEL200 (randomisation selon le rapport 1:1) et une AGSC. Par ailleurs, les patients ont reçu deux cycles de traitement supplémentaires (8 semaines) par RVd à la suite de l'AGSC. Au total, 458 patients ont été inclus dans l'étude.
Dans l'étude PETHEMA GEM2012, à la fin du traitement initial par RVd, les taux de VGPR ou mieux étaient à 67%, les taux de CR à 33% et 47% (217/458) des participants à l'étude étaient MRM-négatifs. 64% (196/305) des participants avec des taux de VGPR ou mieux étaient MRM-négatifs (sensibilité de 10-4). Les taux de VGPR ou mieux après la greffe étaient à 75%, les taux de CR à 44% et 59% (287/458) des participants à l'étude étaient MRM-négatifs. 79% (271/344) des participants avec des taux de VGPR ou mieux étaient MRM-négatifs (sensibilité de 10-4).
IFM 2009
IFM 2009 était une étude multicentrique, randomisée, ouverte, contrôlée, de phase III visant à comparer RVd avec et sans AGSC en traitement initial chez des patients éligibles à une greffe atteints de myélome multiple non préalablement traité. Les patients ont reçu 25 mg/jour de lénalidomide par voie orale les jours 1 à 14, 1,3 mg/m2 de bortézomib par voie intraveineuse les jours 1, 4, 8 et 11 ainsi que 20 mg/jour de dexaméthasone par voie orale les jours 1, 2, 4, 5, 8, 9, 11 et 12 de cycles consécutifs de 21 jours. RVd a été administré en 8 cycles de 3 semaines (24 semaines) sans AGSC immédiate (bras A) ou en trois cycles de 3 semaines (9 semaines) avant l'AGSC (bras B). Les patients dans le bras B ont reçu en outre deux cycles de 3 semaines supplémentaires de RVd après l'AGSC. Au total, 700 patients ont été inclus dans l'étude.
Dans l'étude IFM 2009, les taux de VGPR ou mieux à la fin du traitement initial étaient à 68% et les taux de CR à 31%. 57% (136/237) des participants avec des taux de VGPR ou mieux étaient MRM-négatifs (sensibilité de 10-4).
Expérience clinique du lénalidomide chez les patients atteints d'un myélome multiple après une autogreffe de cellules souches
L'efficacité et la sécurité d'emploi du lénalidomide ont été étudiées dans deux études de phase III multicentriques, randomisées en double aveugle, contrôlées par placebo, en deux bras, en groupes parallèles: CALGB 100104 et IFM 2005-02. Le critère d'évaluation principal des deux études était la survie sans progression (SSP).
CALGB 100104
Des patients âgés de 18 à 70 ans présentant un myélome multiple progressif nécessitant un traitement et sans progression antérieure après un traitement initial ont été inclus dans l'étude.
Dans les 90 à 100 jours suivant l'AGCS, les patients ont été randomisés selon un rapport 1:1 pour recevoir le traitement d'entretien par le lénalidomide ou être dans le bras placebo. La dose d'entretien de lénalidomide était de 10 mg une fois par jour les jours 1 à 28 de chaque cycle de 28 jours (augmentée jusqu'à 15 mg une fois par jour après 3 mois si elle est tolérée) et le traitement était poursuivi jusqu'à la progression de la maladie.
Au total, 460 patients ont été randomisés: 231 patients dans le bras lénalidomide et 229 dans le bras placebo. Les caractéristiques démographiques et de la pathologie étaient comparables entre les deux bras.
L'aveugle de l'étude a été levé sur les recommandations du comité de surveillance des données, lorsque le seuil d'une analyse intermédiaire préprogrammée de la SSP a été dépassé. Après la levée de l'aveugle, les patients du bras placebo ont été autorisés à passer dans le bras lénalidomide avant la progression de leur maladie.
Au gel des données le 1er février 2016, la durée médiane de la période de suivi était de 81,9 mois. Le risque de progression de la maladie ou de décès était réduit de 39% en faveur du lénalidomide (RR = 0,61, IC à 95% = 0,48 - 0,76; p < 0,001). La survie sans progression médiane était de 56,9 mois dans le bras lénalidomide contre 29,4 mois dans le bras placebo.
Dans l'analyse de la SG, le RR observé était de 0,61 (IC à 95% = 0,46 - 0,81) pour le lénalidomide contre le placebo et suggérait une réduction du risque de décès de 39%. La survie globale médiane était de 111,0 mois dans le bras lénalidomide, contre 84,2 mois dans le bras placebo.
IFM 2005-02
Des patients âgés de moins de 65 ans lors du diagnostic qui avaient subi une chimiothérapie à forte dose suivie d'une AGCS et qui présentaient au moins une maladie stable au moment de la récupération hématologique, ont été inclus dans l'étude.
Dans les 6 mois après l'AGCS, les patients ont été randomisés pour recevoir le traitement d'entretien par le lénalidomide ou dans le bras placebo. Après deux cycles de consolidation par le lénalidomide (25 mg/jour les jours 1 à 21 d'un cycle de 28 jours), la dose d'entretien du lénalidomide était de 10 mg une fois par jour (les jours 1 à 28 d'un cycle de 28 jours, augmentée jusqu'à 15 mg une fois par jour après 3 mois si elle était tolérée). Le traitement avait été poursuivi jusqu'à la progression de la maladie.
Au total, 614 patients ont été randomisés: 307 patients dans le bras lénalidomide et 307 dans le bras placebo.
Le traitement a été interrompu après avoir observé un déséquilibre dans l'apparition de cancers secondaires chez les 119 participants à l'étude restant ayant reçu le traitement d'entretien par le lénalidomide (durée minimum de traitement de 27 mois).
Au gel des données le 1er février 2016, la durée médiane de la période de suivi était de 96,7 mois. Le risque de progression de la maladie ou de décès était réduit de 43% en faveur du lénalidomide (RR = 0,57, IC à 95% = 0,42 - 0,76; p < 0,001). La survie sans progression médiane était de 44,4 mois dans le bras lénalidomide contre 23,8 mois dans le bras placebo.
Dans l'analyse de la SG, le RR observé était de 0,90 (IC à 95% = 0,72 - 1,13) pour le lénalidomide contre le placebo. La survie globale médiane était de 105,9 mois dans le bras lénalidomide, contre 88,1 mois dans le bras placebo.
Expérience clinique avec le lénalidomide en association avec le bortézomib et la dexaméthasone chez des patients atteints d'un myélome multiple non préalablement traité non éligibles à une greffe
L'étude SWOG S0777 a évalué l'administration supplémentaire de bortézomib à un traitement de base par le lénalidomide et la dexaméthasone en tant que traitement initial, suivi d'un traitement supplémentaire par Rd jusqu'à la progression de la maladie chez des patients atteints d'un myélome multiple non préalablement traité, pour lesquels une greffe de cellules souches n'était pas imminente.
Les patients dans le bras de traitement sous lénalidomide, bortézomib et dexaméthasone (RVd) ont reçu 25 mg/jour de lénalidomide par voie orale les jours 1 à 14, 1,3 mg/m2 de bortézomib par voie intraveineuse les jours 1, 4, 8 et 11 ainsi que 20 mg/jour de dexaméthasone par voie orale les jours 1, 2, 4, 5, 8, 9, 11 et 12 de cycles consécutifs de 21 jours pendant un maximum de huit cycles de 21 jours (24 semaines). Les patients dans le bras de traitement avec le lénalidomide et la dexaméthasone (Rd) ont reçu 25 mg/jour de lénalidomide par voie orale les jours 1 à 21 et 40 mg/jour de dexaméthasone par voie orale les jours 1, 8, 15 et 22 de cycles consécutifs de 28 jours pendant un maximum de six cycles de 21 jours (24 semaines). Les patients dans les deux bras de traitement ont pris continuellement le schéma Rd: 25 mg/jour de lénalidomide par voie orale les jours 1 à 21 et 40 mg/jour de dexaméthasone par voie orale les jours 1, 8, 15 et 22 de cycles consécutifs de 28 jours. Il avait été prévu de poursuivre le traitement jusqu'à la progression de la maladie.
Le critère d'efficacité principal dans l'étude était la survie sans progression (SSP). Au total, 523 patients ont été inclus dans l'étude, dont 263 patients randomisés dans le bras RVd et 260 dans le bras Rd. Les caractéristiques démographiques et pathologiques initiales des patients étaient bien équilibrées dans les deux bras.
Les résultats de la SSP (analyse du Comité d'adjudication indépendant (IRAC), règles de censure de l'EMA), avec une date d'échéance du 1er décembre 2016 et une période de suivi de l'évolution médiane des participants survivants de 60,6 mois, ont indiqué une réduction de 24% du risque de progression de la maladie ou de décès en faveur de RVd (HR = 0,76; IC à 95%: 0,62; 0,94). La SSP médiane était de 41,7 mois (IC à 95%: 33,1; 51,5) dans le bras RVd contre 29,7 mois (IC à 95%: 24,2; 37,8) dans le bras Rd.
Chez les participants dans le bras RVd, une réduction du risque de décès de 28% a été observée par rapport au bras Rd (HR = 0,72; IC à 95% = 0,56 à 0,94). La SG médiane était globalement de 89,1 mois (IC à 95%: 76,1; non évaluable) dans le bras RVd, comparé à 67,2 mois (IC à 95%: 58,4; 90,8) dans le bras Rd. Les taux de VGPR ou mieux dans le bras RVd (58%) étaient également plus élevés que dans le bras Rd (32%).
Expérience clinique du lénalidomide en association avec la dexaméthasone chez des patients non préalablement traités et non éligibles à une greffe
La sécurité et l'efficacité du lénalidomide ont été évaluées dans une étude de phase III multicentrique, randomisée en ouvert, en trois bras (MM-020), menée chez des patients âgés de 65 ans ou plus ou chez les patients âgés de moins de 65 ans, qui n'étaient pas éligibles à une greffe de cellules souches parce qu'ils l'avaient refusée ou ne pouvaient pas en bénéficier en raison du coût ou pour d'autres motifs. L'étude (MM-020) visait à comparer l'association de lénalidomide et dexaméthasone (Rd) administrée pendant deux durées différentes (jusqu'à la progression de la maladie [bras Rd] ou pendant un maximum de 18 cycles de 28 jours [72 semaines, bras Rd18]) à l'association de melphalan, prednisone et thalidomide (MPT) administrée pendant un maximum de 12 cycles de 42 jours (72 semaines). Les patients ont été randomisés (1:1:1) dans l'un des trois bras de traitement. Les patients ont été stratifiés au moment de la randomisation en fonction de l'âge (≤75 ans versus > 75 ans), du stade (stades ISS I et II versus stade III) et du pays. Les patients des bras Rd et Rd18 ont pris 25 mg de lénalidomide une fois par jour, les jours 1 à 21 des cycles de 28 jours. La dexaméthasone 40 mg était prise une fois par jour, les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Dans les bras Rd et Rd18, la dose initiale et le schéma posologique étaient ajustés en fonction de l'âge et de la fonction rénale. Les patients âgés de plus de 75 ans ont reçu une dose de 20 mg de dexaméthasone une fois par jour, les jours 1, 8, 15 et 22 de chaque cycle de 28 jours. Tous les patients ont reçu une thromboprophylaxie (héparine de bas poids moléculaire, warfarine, héparine, aspirine à faible dose) pendant l'étude.
Le critère d'efficacité principal dans l'étude était la survie sans progression (progression free survival, PFS). Au total, 1 623 patients ont été inclus dans l'étude, dont 535 patients randomisés dans le bras Rd, 541 patients dans le bras Rd18 et 547 patients dans le bras MPT. Les caractéristiques démographiques et pathologiques initiales des patients étaient bien équilibrées dans les trois bras. En général, les patients de l'étude présentaient une maladie de stade avancé: sur la population totale de l'étude, 41% présentaient une maladie de stade ISS III et 9% une insuffisance rénale sévère (clairance de la créatinine [ClCr] < 30 ml/min). L'âge médian était de 73 ans dans les trois bras.
La survie sans progression était significativement supérieure dans le bras Rd (26,0 mois) par rapport au bras MPT (21,9 mois), avec un HR de 0,69 (IC 95%: 0,59-0,80; p = 0,001) indiquant une réduction de 31% du risque de progression ou de décès. Le taux de survenue de l'événement décès entrant dans le critère de survie sans progression était identique (10%) dans les deux bras de traitement sur la durée de l'étude. On a observé dans le bras Rd une amélioration de la médiane de survie sans progression de 4,3 mois par rapport au bras MPT. Le taux de réponse du myélome constaté dans le bras Rd était significativement plus élevé que dans le bras MPT (75,1% versus 62,3%; p < 0,00001), sachant que 15,1% des patients ont obtenu une réponse complète dans le bras Rd contre 9,3% dans le bras MPT. La durée médiane jusqu'à la première réponse était de 1,8 mois dans le bras Rd et 2,8 mois dans le bras MPT.
Au moment de l'analyse de la survie globale (SG), la durée médiane d'observation de l'ensemble des patients survivants était de 37,0 mois; le nombre de décès était de 574, soit un taux de survenue du dernier événement d'intérêt de 64% (574/896). Le HR observé était de 0,78 en faveur du bras Rd par rapport au bras MPT (IC 95% = 0,64, 0,96; valeur nominale de p = 0,01685), indiquant une réduction du risque de mortalité de 22%.
Expérience clinique du lénalidomide en association avec le melphalan et la prednisone chez des patients non préalablement traités et non éligibles à une greffe
La sécurité et l'efficacité du lénalidomide ont été évaluées dans une étude de phase III multicentrique randomisée en double aveugle, en trois bras (MM-015), menée chez des patients qui étaient âgés de 65 ans et plus et qui avaient une créatininémie < 2,5 mg/dl. L'étude visait à comparer l'association de lénalidomide plus melphalan et prednisone (MPR) avec ou sans traitement d'entretien par le lénalidomide en monothérapie jusqu'à la progression de la maladie à l'association de melphalan et prednisone administrée pendant 9 cycles au maximum. Les patients ont été randomisés selon un rapport 1:1:1 dans l'un des trois bras de traitement: bras MPR+R – association MPR par voie orale en traitement d'induction suivie du lénalidomide en traitement d'entretien (R); bras MPR+p – association MPR par voie orale en traitement d'induction suivie d'un placebo en traitement d'entretien (p); bras MPp+p – association MPp (MP + placebo) par voie orale en traitement d'induction, suivie d'un placebo en traitement d'entretien (p).
La survie sans progression évaluée par une revue indépendante en aveugle était significativement supérieure pour le schéma MPR+R par rapport au bras MPp+p, avec un HR de 0,388 (IC 95% = 0,274, 0,550) indiquant une réduction de 61% du risque de progression de la maladie pour le schéma MPR+R par rapport au bras MPp+p.
Expériences cliniques chez des patients atteints d'un myélome multiple ayant déjà reçu au moins un traitement antérieur
Deux études multicentriques randomisées, par groupes parallèles, en double aveugle, contrôlées contre placebo, ont été effectuées selon le même plan (MM-009 aux États-Unis et au Canada, MM-010 en Europe, en Israël et en Australie): 353 et 351 patients respectivement, souffrant d'un myélome multiple déjà traité par le passé par un ou plusieurs régimes de chimiothérapie, ont été traités soit par le lénalidomide + dexaméthasone, soit par la dexaméthasone seule.
Une analyse effectuée sur les données regroupées des deux études a révélé un délai médian de progression (TTP) de 48 semaines (IC à 95%: 41,1 – 60,1) chez les patients traités par l'association lénalidomide/dexaméthasone et de 20,1 semaines (IC à 95%: 19,9 – 20,7) chez les patients traités par l'association placebo/dexaméthasone. La médiane de survie sans progression (PFS) était de 47,3 semaines (IC à 95%: 36,9 – 58,4) contre 20,1 semaines (IC à 95%: 18,1 – 20,3). La survie totale (OS) chez les patients traités par l'association lénalidomide/dexaméthasone était significativement plus élevée avec 90,3 semaines contre 80,2 semaines, p = 0,015 (les patients du bras placebo pouvaient, après progression, c'est-à-dire après levée de l'insu, passer dans le bras du produit actif; 50% ont été traités par l'association lénalidomide/dexaméthasone).
La durée médiane de traitement a été de 28,1 semaines (min: 0,1 - max: 110,7).
Expériences cliniques dans le syndrome myélodysplasique
Dans une étude de phase II, multicentrique, en ouvert, à un seul bras (MDS-003 en Allemagne et aux États-Unis), 120 patients qui présentaient une dépendance transfusionnelle érythrocytaire sur la base d'un syndrome myélodysplasique de risque faible ou intermédiaire 1 et qui étaient porteurs d'une anomalie cytogénétique comportant une délétion 5q avec ou sans autre anomalie cytogénétique ont été traités par 10 mg de lénalidomide. La médiane de la durée de traitement était de 52,5 semaines. Le taux d'indépendance transfusionnelle (> 56 jours) atteignait 62,8%. L'augmentation du taux médian d'hémoglobine était de 5,9 g/dl. La durée médiane de la réponse atteignait 97 semaines. Une réponse cytogénétique clairement prononcée a été observée chez 34,6% des patients et une réponse cytogénétique moins prononcée a été observée chez 38,5% des patients.
Dans une étude de phase III, multicentrique, en double aveugle, contrôlée contre placebo, à triple bras (MDS-004 en Europe et en Israël), 138 patients présentant une dépendance avérée transfusionnelle érythrocytaire sur la base d'un syndrome myélodysplasique de risque faible ou intermédiaire 1 et qui étaient porteurs d'une anomalie cytogénétique comportant une délétion 5q avec ou sans autre anomalie cytogénétique ont été randomisés vers 10 mg, 5 mg de lénalidomide ou vers le placebo. La durée de la phase en double aveugle était de 16 à 52 semaines. Le taux d'indépendance transfusionnelle (> 182 jours) dans le groupe 10 mg était de 56,1%. Les taux correspondants d'indépendance transfusionnelle dans le groupe de traitement 5 mg et dans le groupe placebo atteignaient respectivement 41,3% et 5,9%. La durée médiane de réponse dans le groupe 10 mg était de 106 semaines; dans les groupes 5 mg et placebo celle-ci n'a en revanche pas pu être évaluée. Une réponse cytogénétique clairement prononcée ou moins prononcée a été observée chez respectivement 24,0% et 17,1% (10 mg); 10,9%, et 6,5% (5 mg) et 0% et 0% (placebo) des patients.
Le taux d'indépendance transfusionnelle (> 56 jours) était de 61,0% dans le groupe 10 mg, avec une augmentation médiane de l'hémoglobine de 6,3 g/dl. Les taux correspondants d'indépendance transfusionnelle et l'augmentation de l'hémoglobine dans les groupes 5 mg de lénalidomide et dans le groupe placebo étaient respectivement de 50,0% et 7,8% et 5,1 g/dl et 2,3 g/dl.
Expérience clinique du lymphome des cellules du manteau
L'étude MCL-001 était une étude de phase II, multicentrique, non contrôlée portant sur une monothérapie par le lénalidomide visant à évaluer la sécurité d'emploi et l'efficacité du lénalidomide chez des patients atteints d'un lymphome des cellules du manteau, lesquels avaient présenté une récidive ou qui étaient réfractaires après un traitement par le bortézomib ou un schéma posologique contenant du bortézomib. Seuls les patients présentant une translocation confirmée ou une surexpression de la cycline ainsi que les patients non candidats à une greffe de cellules souches ont été admis dans l'étude. Le lénalidomide a été administré aux jours 1–21 de cycles de traitement consécutifs de 28 jours jusqu'à la progression de la maladie, la survenue d'une toxicité inacceptable ou le retrait du consentement.
Une des conditions de participation à l'étude était un traitement préalable par une anthracycline ou par la mitoxantrone, le cyclophosphamide, le rituximab et le bortézomib seuls ou en association.
Ont été inclus les patients dont le nombre absolu de neutrophiles était ≥1 500 cellules/mm3, dont le nombre de thrombocytes était ≥60 000 cellules/mm3, dont la SGOT/AST ou SGPT/ALT sérique était < 3,0 x ULN (limite supérieure de la normale), sauf en cas de preuves documentées d'atteinte hépatique par le lymphome, dont la bilirubine sérique totale était < 1,5 x ULN, sauf dans le syndrome de Gilbert ou autre atteinte hépatique documentée par le lymphome, et dont la clairance de la créatinine calculée (selon la formule de Cockcroft-Gault) était > 30 ml/min.
Les principaux critères d'efficacité de l'étude MCL-001 étaient le taux de réponse globale (ORR) et la durée de la réponse (DOR). Le tableau suivant donne un aperçu des résultats de l'efficacité pour le groupe de patients en ITT (Intent to Treat en français "en intention de traiter" ) conformément à l'évaluation du IRC (Comité de contrôle indépendant). La durée médiane jusqu'à la réponse était de 2,2 mois (1,7 à 13,1 mois). La survie globale médiane était de 19,0 mois (IC à 95% 12,5; 22,9 mois). Pour l'ensemble de la population de l'étude, la survie sans progression était de 3,95 mois.
Évaluation du taux de réponse (N = 134) N (%) IC à 95%
Taux de réponse globale (IWRC) (CR+CRu+PR) 37 (28) (20,2; 36,0)
Rémission complète (CR+CRu) 10 (7) (3,6; 13,3)
CR 2 (1)
CRu 8 (6)
Rémission partielle (PR) 27 (20)
Pathologie stable (SD) 39 (29)
Durée de la rémission (mois) Médiane IC à 95%
Durée de la réponse globale (CR + CRu + PR)N = 37 16,6 (7,7; 26,7
Dans la population ITT de l'étude MCL-002, il a été observé une augmentation apparente globale des décès au cours des 20 premières semaines dans le bras lénalidomide (22/170 patients [13 %]) par rapport au bras contrôle (6/84 patients [7 %]). Chez les patients ayant une charge tumorale élevée, les chiffres correspondants étaient de 16/81 patients (20 %) et 2/28 patients (7 %).
Expériences cliniques dans le lymphome folliculaire
NHL-007
L'étude CC-5013-NHL-007 (AUGMENT) est une étude de phase III multicentrique, randomisée, en double aveugle. L'efficacité et la sécurité du lénalidomide en association au rituximab (R2) ont été évaluées en comparaison avec le rituximab plus placebo chez des patients atteints d'un lymphome indolent en rechute/réfractaire.
Au total, 358 patients âgés de 18 ans et plus atteints de LF de grade 1, 2 ou 3a (n = 295) ou de lymphome de la zone marginale (LZM) confirmé histologiquement ont été randomisés selon un rapport 1:1. Les patients avaient reçu au moins un traitement antérieur par chimiothérapie systémique, immunothérapie ou immunochimiothérapie. Les patients devaient avoir reçu antérieurement au moins 2 doses de rituximab et ne devaient pas être réfractaires au rituximab.
Le lénalidomide était administré par voie orale à la dose de 20 mg en une prise par jour pendant les 21 premiers jours de chaque cycle de traitement de 28 jours, et ce pendant 12 cycles ou jusqu'à la survenue d'une toxicité inacceptable. La dose de rituximab était de 375 mg/m2 une fois par semaine au cours du cycle 1 (jours 1, 8, 15 et 22) et le jour 1 de chaque cycle de 28 jours pendant les cycles 2 à 5.
Le critère d'efficacité principal dans l'étude était la survie sans progression (SSP). La survie sans progression (SSP) médiane était significativement plus longue chez les patients atteints de FL dans le bras R2 (39,4 mois; IC 95%: 25,1; NE) que dans le bras contrôle (13,8 mois; IC 95%: 11,2; 16,0); le risque de récidive a été réduit de 60% (HR 0,40; IC 95%: 0,29; 0,55). Les résultats du critère principal d'évaluation étaient cliniquement et statistiquement significatifs.
Les patients du bras R2 atteints de LF affichaient en outre un taux de réponse globale (ORR) supérieur (ORR 80,3 %; IC 95%: 72,9; 86,4) par rapport à la monothérapie par rituximab (ORR 55,4; IC 95%: 47,0; 63,6). La durée de la réponse médiane était de 36,6 mois dans le bras R2 et de 15,5 mois dans le bras contrôle. La mortalité, exprimée par le taux de survie globale (SG) à 2 ans, a été réduite de 55% (HR 0,45; IC 95%:0,22; 0,92) dans le bras R2, c'est-à-dire que les pourcentages des patients en vie après 2 ans étaient de 94,8% dans le bras R2 contre 85,8% dans le bras rituximab seul.
NHL-008
L'étude NHL-008 est une étude de phase III randomisée en ouvert menée chez des patients (n = 232) atteints de lymphome folliculaire en rechute ou réfractaire (grade 1-3b), de lymphome de la zone marginale (LZM) ou de lymphome des cellules du manteau. Contrairement à l'étude NHL-007, des patients réfractaires au rituximab, c'est-à-dire n'ayant pas répondu au traitement ou ayant rechuté dans les 6 mois suivant le traitement par le rituximab ou réfractaires à la fois au rituximab et à la chimiothérapie, ont été inclus dans l'étude NHL-008.
Après une période de traitement initiale commune comportant 12 cycles de traitement par le rituximab plus le lénalidomide (R2), les patients étaient randomisés pour entrer dans le traitement d'entretien et recevoir soit l'association R2 (ou après le cycle 18 en option le lénalidomide en monothérapie) soit le rituximab en monothérapie.
Pendant la période de traitement d'induction, le lénalidomide était administré à la dose de 20 mg les jours 1 à 21 de chaque cycle de 28 jours, et ce pendant 12 cycles au maximum ou jusqu'à la survenue d'une toxicité inacceptable ou jusqu'au retrait du consentement à la participation à l'étude. La dose de rituximab était de 375 mg/m2 chaque semaine au cours du cycle 1 (jours 1, 8, 15 et 22) puis le jour 1 d'un cycle sur deux de 28 jours (cycles 3, 5, 7, 9 et 11) pendant 12 cycles de traitement au maximum.
Le critère principal d'évaluation de l'efficacité de la phase d'induction de l'étude reposait sur le taux de réponse globale, déterminé à l'aide des critères de réponse modifiés de l'International Working Group (IWG-RC) 1999. Les résultats présentés résultent de l'analyse intermédiaire portant sur la phase de traitement initiale R2.
Après la phase d'induction de 12 cycles, le taux de réponse globale de tous les participants à l'étude atteints de LF (n = 148) était de 70,3%; les patients réfractaires au rituximab (n = 60) avaient un taux de réponse globale de 58,3%, tandis que les patients non réfractaires au rituximab (n = 88) avaient un taux de réponse globale de 79,3%.
PharmacocinétiqueAbsorption
Le lénalidomide est rapidement absorbé, avec un Tmax de 1 heure. La biodisponibilité de la substance administrée par voie orale est d'environ 70%. La pharmacocinétique du lénalidomide est proportionnelle à la dose.
Chez les volontaires sains, l'administration au cours d'un repas hyperlipidique diminue la quantité absorbée, ce qui entraîne une diminution d'environ 20% de l'aire sous la courbe de concentration en fonction du temps (ASC) et une réduction de 50% de la Cmax plasmatique.
Les analyses pharmacocinétiques de population indiquent que le taux d'absorption orale du lénalidomide chez les patients atteints d'un LCM est comparable à celui observé chez les patients atteints de MM ou de SMD.
Distribution
La liaison du lénalidomide aux protéines plasmatiques est faible (< 30%). Il n'a pas été étudié si le lénalidomide franchit la barrière hémato-encéphalique.
Après administration d'une dose quotidienne de 25 mg, le lénalidomide passe dans le sperme (< 0,01% de la dose). Trois jours après l'arrêt du médicament, le lénalidomide n'est plus retrouvé dans le liquide séminal de sujets sains.
Métabolisme
Le lénalidomide est peu métabolisé et le processus de métabolisation ne fait pas intervenir d'enzyme de phase 1. Le principal composant retrouvé in vivo dans le sang humain est le lénalidomide sous forme inchangée. Les métabolites identifiés sont le 5-hydroxyl-lénalidomide et la N-acétyl-lénalidomide, chacun d'entre eux atteignant moins de 5% du taux sanguin de la substance-mère.
Élimination
Environ deux tiers d'une dose de lénalidomide sont éliminés sous une forme inchangée par voie rénale.
La clairance rénale du lénalidomide est supérieure au taux de filtration glomérulaire, le lénalidomide est donc au moins secrété activement dans une certaine mesure.
Aux doses thérapeutiques (jusqu'à 25 mg/jour), la demi-vie plasmatique est d'environ 3 heures chez les volontaires sains et de 3 à 5 heures chez les patients.
Les concentrations à l'état d'équilibre sont atteintes le 4e jour. Les doses multiples ne conduisent pas à une accumulation.
Cinétique pour certains groupes de patients
On ne dispose pas de données concernant la pharmacocinétique les patients pédiatriques.
Le lénalidomide est surtout éliminé sous forme de substance inchangée par filtration glomérulaire et sécrétion tubulaire active. Après une dose unique de 25 mg, en cas d'insuffisance rénale légère (cl. créat. 80-50 ml/min), l'AUC est augmentée de 25%, lors d'insuffisance rénale modérée (cl. créat. 50-30 ml/min) l'AUC est augmentée du triple, et lors d'insuffisance rénale sévère (cl. créat. < 30 ml/min), et/ou d'insuffisance rénale nécessitant une dialyse (période interdialysaire), d'un facteur 4 à 5. La demi-vie d'élimination est prolongée, lors d'insuffisance rénale modérée, d'une durée triple, passant à 9 à 10 heures.
Troubles de la fonction hépatique
Les analyses pharmacocinétiques de population incluaient des patients en insuffisance hépatique légère (N = 16, bilirubine totale > 1,0 à ≤1,5 x limite supérieure de la normale ou ASAT > limite supérieure de la normale) et ont montré l'absence d'influence sur la cinétique du lénalidomide. On ne dispose d'aucune donnée chez les patients en insuffisance hépatique moyenne ou forte.
Données précliniquesToxicité à court et à moyen terme
Le lénalidomide présente un faible potentiel de toxicités aiguës; chez des rongeurs, les plus faibles doses mortelles après une administration orale ont dépassé 2000 mg/kg. L'administration de lénalidomide à long terme a provoqué chez les rats une minéralisation des bassinets, plus marquée chez les femelles que chez les mâles. La dose sous laquelle aucun effet indésirable n'a été observé (no observed adverse effect level, NOAEL) est estimée à moins de 75 mg/kg chez le rat, ce qui correspond – sur la base de l'AUC – à 25 fois environ l'exposition par jour atteinte avec 25 mg par jour chez les patients humains. Chez des singes, l'administration de doses orales répétées a conduit à une réduction dose-dépendante du nombre de neutrophiles; cet effet est dû aux effets pharmacodynamiques du médicament. L'administration répétée de doses orales de 4 et de 6 mg/kg chez des singes sur une durée de jusqu'à 20 semaines a conduit à une mortalité et à une toxicité accrues (nette perte de poids, réduction des nombres de globules rouges, de globules blancs et de thrombocytes, saignements multiples d'organes, inflammations des voies digestives, atrophie du tissu lymphatique et de la moelle osseuse). L'administration de 1 et de 2 mg/kg/jour pendant 52 semaines a conduit chez des singes à une modification de la proportion de cellules dans la moelle osseuse et à une légère réduction du rapport entre cellules myéloïdes et érythroïdes, ainsi qu'à une atrophie du thymus. Sous 1 mg/kg/jour, une légère suppression du nombre de leucocytes a été observée. La dose NOAEL a été de 1 mg/kg/jour. À cette dose, l'exposition décrite par l'AUC correspond à l'exposition d'un patient humain lors d'un traitement par 25 mg par jour.
Mutagénicité/carcinogénicité
Les études sur le potentiel mutagène effectuées in vitro (mutations bactériennes, lymphocytes humains, lymphome de la souris, transformation cellulaire embryonnaire chez le hamster syrien) et in vivo (test du micronoyau chez des rats) n'ont montré aucun effet dû à la substance, ni sur le plan génétique, ni sur le plan chromosomique. Aucune étude n'a été effectuée sur le potentiel cancérigène du lénalidomide.
Toxicité sur la reproduction
Des études ont été effectuées sur la toxicité pour l'animal en phase de développement (effets tératogènes et toxicité embryonnaire/fœtale) chez le rat, le lapin et le singe. Au cours d'une étude chez le singe, le lénalidomide a été administré en doses pouvant aller jusqu'à 4 mg/kg/jour. Les résultats de l'étude indiquent que l'administration de lénalidomide à des singes femelles gravides a donné lieu à des malformations chez la progéniture comparables à celles causées par le thalidomide.
Chez la lapine ayant reçu des doses orales de 3, 10 et 20 mg/kg/jour, la toxicité développementale aux doses de 10 et 20 mg/kg/jour se caractérisait par une légère réduction du poids corporel du fœtus, une plus grande incidence de pertes post-implantation (résorptions précoces et tardives et morts intrautérines) ainsi que des particularités macroscopiques externes chez le fœtus liées à une morbidité et à des effets pharmacotoxiques causés par le lénalidomide (coloration violette de la peau sur tout le corps). Aux doses de 10 mg et 20 mg/kg/jour, on a observé des altérations des parties molles et du squelette chez le fœtus, lesquelles sont toutefois typiques de la race des lapines utilisées. Chez la lapine, les NOAELs maternels et pour le développement ont été établis à 3 mg/kg/jour pour le lénalidomide.
Comme l'ont démontré des études préalables sur le thalidomide chez le rat, une étude sur le développement embryonnaire/fœtal chez le rat auquel on avait administré jusqu'à 500 mg/kg/jour de lénalidomide n'a également pas démontré un effet tératogène. Aux doses de 100, 300 ou 500 mg/kg/jour, une toxicité maternelle minime a été observée, notamment une légère réduction passagère de la prise de poids moyenne et de l'ingestion d'aliments.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine, ne pas conserver au-dessus de 25 °C et tenir hors de la portée des enfants.
Remarques concernant la manipulation
Comme pour les cytostatiques, une prudence particulière est indiquée lors de la manipulation et de l'évacuation de Revlimid (cf. "Posologie/Mode d'emploi" ).
Numéro d’autorisation57712 (Swissmedic)
PrésentationRevlimid 2,5 mg: 21 gélules (A)
Revlimid 5 mg: 21 gélules (A)
Revlimid 7,5 mg: 21 gélules (A)
Revlimid 10 mg: 21 gélules (A)
Revlimid 15 mg: 21 gélules (A)
Revlimid 20 mg: 21 gélules (A)
Revlimid 25 mg: 21 gélules (A)
Titulaire de l’autorisationBristol-Myers Squibb SA, Steinhausen
Mise à jour de l’informationAoût 2022
|