CompositionPrincipes actifs
Cyclosilicate de zirconium sodique.
Excipients
Non pertinent.
Forme pharmaceutique et quantité de principe actif par unitéPoudre pour suspension buvable.
Le cyclosilicate de zirconium sodique est une poudre blanche cristalline insoluble.
LOKELMA 5 g: chaque sachet contient 5 g de cyclosilicate de zirconium sodique (contenant environ 400 mg de sodium).
Indications/Possibilités d’emploiLOKELMA est indiqué dans le traitement de l'hyperkaliémie chez l'adulte.
Posologie/Mode d’emploiLOKELMA ne doit pas être utilisé comme médicament d'urgence en cas d'hyperkaliémie menaçant le pronostic vital.
Instauration du traitement
Traitement de l'hyperkaliémie lors de la phase de correction initiale
La dose initiale recommandée de LOKELMA chez les patients ayant un taux sérique de potassium > 5,0 mmol/l est de 10 g, administrée trois fois par jour par voie orale sous forme d'une suspension dans l'eau. La normokaliémie (taux de potassium normal compris entre 3,5 et 5,0 mmol/l) souhaitée induite par ce schéma posologique est atteinte de manière caractéristique en 24 à 48 heures. Si le taux sérique de potassium est toujours supérieur à 5,0 mmol/l après 48 heures, 10 g trois fois par jour peuvent être administrés pour un jour supplémentaire (24 heures) avant d'instaurer le traitement d'entretien. Si la normokaliémie n'est pas atteinte après 3 jours, une autre approche thérapeutique doit être envisagée.
Traitement d'entretien
Traitement de l'hyperkaliémie lors de la phase d'entretien
Pour le traitement d'entretien, la dose minimale efficace pour prévenir la récidive de l'hyperkaliémie doit être établie. La dose journalière recommandée est de 5 g une fois par jour que l'on peut augmenter, si nécessaire, jusqu'à 10 g une fois par jour ou réduire jusqu'à 5 g un jour sur deux afin de maintenir un taux normal de potassium. La dose journalière à utiliser pour le traitement d'entretien ne doit pas dépasser 10 g une fois par jour.
Posologie usuelle
Adultes
Voir "Traitement d'entretien" .
Les taux sériques de potassium doivent être surveillés régulièrement pendant le traitement. La fréquence des contrôles dépend de plusieurs facteurs incluant l'administration d'un autre traitement médicamenteux, la progression d'une néphropathie chronique et l'apport alimentaire en potassium.
En cas d'apparition d'une hypokaliémie sévère, LOKELMA doit être arrêté et le patient doit être réévalué.
Instructions posologiques particulières
Patients sous dialyse chronique
Les patients sous dialyse doivent prendre LOKELMA uniquement les jours sans dialyse. La dose initiale recommandée est de 5 g une fois par jour. Cette dose peut être augmentée ou diminuée chaque semaine en fonction de la kaliémie en prédialyse, qui est déterminée après l'intervalle interdialytique long afin d'obtenir une normokaliémie (4,0 à 5,0 mmol/l). La dose peut être augmentée à intervalles d'une semaine de 5 g jusqu'à 15 g une fois par jour (les jours sans dialyse). Afin de maintenir la normokaliémie, le taux sérique de potassium doit être surveillé régulièrement (p.ex. mensuellement).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie n'est nécessaire chez les patients présentant une insuffisance hépatique.
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la posologie n'est nécessaire chez les patients présentant une insuffisance rénale et qui ne sont pas sous hémodialyse chronique.
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés.
Enfants et adolescents
La sécurité et l'efficacité de LOKELMA pour les enfants et les adolescents (âgés de moins de 18 ans) ne sont pas établies.
Prise retardée
Si la prise d'une dose a été oubliée, la dose usuelle suivante doit être prise à l'heure habituellement prévue.
Mode d'administration
Voie orale.
Le contenu entier du sachet doit être vidé dans un verre contenant environ 45 ml d'eau et être bien remué. La poudre ne se dissoudra pas. Le liquide insipide doit être bu pendant qu'il est encore trouble et que la poudre est en suspension. Si la poudre se dépose, il faut remuer à nouveau le liquide. Il faut s'assurer de la prise complète du contenu.
LOKELMA peut être pris indépendamment des repas.
Contre-indicationsHypersensibilité connue au principe actif.
Mises en garde et précautionsHypokaliémie
Une hypokaliémie peut survenir. Une titration de la dose peut être nécessaire pour prévenir une hypokaliémie modérée à sévère (voir "Posologie/Mode d'emploi" , "Traitement d'entretien" ). Chez les patients ayant un taux sérique de potassium < 3,0 mmol/l, LOKELMA doit être arrêté et le patient doit être réévalué.
Lorsque cela est indiqué cliniquement, le taux sérique de potassium doit être surveillé. Ceci peut être indispensable, par exemple, en cas de modifications de traitements médicamenteux impactant la kaliémie (p.ex. inhibiteurs du système rénine-angiotensine-aldostérone [SRAA] ou diurétiques). Si nécessaire, la dose de LOKELMA doit être titrée en conséquence.
Dégradation d'une insuffisance cardiaque préexistante
Les patients présentant une insuffisance cardiaque préexistante, en particulier, ceux chez lesquels un apport en sodium accru peut provoquer une surcharge hydrique et une décompensation, doivent être surveillés afin de détecter tout signe de dégradation de l'insuffisance cardiaque, notamment dyspnée croissante, œdèmes et prise de poids rapide et, le cas échéant, être traités selon la pratique clinique usuelle.
Sodium
Ce médicament contient respectivement environ 400 mg (LOKELMA 5 g) de sodium par sachet, ce qui équivaut à 20% de l'apport alimentaire journalier maximal de sodium recommandé par l'OMS. LOKELMA est considéré comme riche en sodium. Il convient d'en tenir compte, notamment chez les patients suivant un régime pauvre en sodium.
Allongement du QT
Pendant la phase de normalisation de l'hyperkaliémie, un allongement de l'intervalle QT peut être observé comme le résultat physiologique d'une diminution de la concentration du potassium dans le sérum.
Risque d'interactions avec les rayons X
Le cyclosilicate de zirconium sodique peut être opaque aux rayons X. Si le patient est exposé au niveau de l'abdomen aux rayons X, les radiologistes doivent le prendre en compte.
Perforation intestinale
Le risque de perforation intestinale lors de l'utilisation de LOKELMA est actuellement inconnu. Aucun cas de perforation intestinale n'a été signalé à ce jour avec LOKELMA. Étant donné que la perforation intestinale a été rapportée avec des polymères qui agissent sur le tube digestif, une attention particulière doit être accordée aux signes et symptômes liés à la perforation intestinale.
Limite des données cliniques
Hyperkaliémie sévère
L'expérience est limitée chez les patients présentant des concentrations sériques de potassium supérieures à 6,5 mmol/l.
Utilisation à long terme
Les études cliniques effectuées avec LOKELMA ne comprenaient pas d'expositions au médicament qui duraient plus d'un an.
InteractionsInteractions pharmacocinétiques
LOKELMA peut augmenter de manière transitoire le pH gastrique en résorbant les ions hydrogène, ce qui peut entraîner des changements au niveau de la solubilité et de la cinétique de résorption des médicaments co-administrés dont la biodisponibilité est dépendante du pH. LOKELMA doit être pris au moins 2 heures avant ou 2 heures après les médicaments oraux dont la biodisponibilité dépend de manière cliniquement pertinente du pH gastrique.
Lorsque LOKELMA est co-administré avec des médicaments oraux dont la biodisponibilité est indépendante du pH, un intervalle de temps entre les administrations n'est pas nécessaire.
Données in vivo
Dans le cadre d'une étude clinique d'interaction médicamenteuse, menée chez des sujets sains, la co-administration de LOKELMA avec l'amlodipine, le dabigatran, le clopidogrel, l'atorvastatine, le furosémide, le glipizide, la warfarine, le losartan ou la lévothyroxine n'a pas entraîné d'interactions médicamenteuses cliniquement pertinentes. Aucun ajustement posologique ou intervalle de temps entre les administrations n'est nécessaire pour ces médicaments (voir Tableau 1).
Tableau 1: Interactions entre LOKELMA et d'autres médicaments
Principe actif (schéma Répercussions sur la concentration du Recommandation pour une
posologique) médicamentGMR (IC à 90%) utilisation concomitante
Atorvastatine (dose Atorvastatine ASC: 103,99 Aucun ajustement posologique ou
unitaire de 10 mg) (95,04–113,78) Cmax: 168,54 intervalle de temps entre les
LOKELMA (dose unitaire (144,07–197,17) (Augmentation administrations n'est
de 10 g) transitoire du pH gastrique) recommandé.
Furosémide (dose Furosémide ASC: 106,13 (98,36–114,51) Aucun ajustement posologique ou
unitaire de 20 mg) Cmax: 166,15 (128,28–215,19) intervalle de temps entre les
LOKELMA (dose unitaire (Augmentation transitoire du pH administrations n'est
de 10 g) gastrique) recommandé.
Dabigatran (dose Dabigatran ASC: 59,08 (39,59–88,15) Aucun ajustement posologique ou
unitaire de 75 mg) Cmax: 57,41 (40,30–81,78) intervalle de temps entre les
LOKELMA (dose unitaire (Augmentation transitoire du pH administrations n'est
de 10 g) gastrique) recommandé.
Dans le cadre d'une autre étude d'interaction menée chez des volontaires sains, la co-administration de LOKELMA 15 g et de tacrolimus 5 mg a entraîné une diminution de l'ASC et de la Cmax du tacrolimus respectivement de 37% et 29%. Par conséquent, le tacrolimus doit être pris au moins 2 heures avant ou après LOKELMA. Lors de la même étude, la co-administration de LOKELMA et de ciclosporine n'a pas mis en évidence d'interaction cliniquement pertinente.
Interactions pharmacodynamiques
Non pertinent.
Effet de LOKELMA sur d'autres médicaments
Étant donné que LOKELMA n'est ni résorbé ni métabolisé par l'organisme, et qu'il ne se lie pas de manière significative à d'autres médicaments, ses effets sur les autres médicaments sont limités.
Le tableau ci-dessous donne des exemples de principes actifs qui doivent être pris 2 heures avant ou après LOKELMA afin d'éviter des interactions médicamenteuses éventuelles à type d'augmentation du pH gastrique.
Classe de principes Principe actif
actifs
Antifongiques azolés Kétoconazole, itraconazole, posaconazole
Agents anti-VIH Atazanavir, nelfinavir (non autorisé en Suisse), indinavir (non autorisé en
Suisse), ritonavir, saquinavir, raltégravir, lédipasvir, rilpivirine
Inhibiteurs de Erlotinib, dasatinib, nilotinib
tyrosine kinase
Effets d'autres médicaments sur LOKELMA
Étant donné que LOKELMA n'est ni résorbé ni métabolisé par l'organisme, aucun effet d'autres médicaments sur l'action pharmacologique de LOKELMA n'est attendu.
Grossesse, allaitementGrossesse
Il n'y a pas de données à ce jour sur l'utilisation du cyclosilicate de zirconium sodique chez la femme enceinte. Les études expérimentales animales n'ont pas révélé d'indices de toxicité pour la reproduction (voir "Données précliniques" ). Par mesure de précaution, l'utilisation de LOKELMA doit être évitée pendant la grossesse.
Allaitement
On ignore si le cyclosilicate de zirconium sodique est excrété dans le lait maternel humain ou animal. Compte tenu de ses propriétés physicochimiques, le cyclosilicate de zirconium sodique n'est pas résorbé dans la circulation systémique. Il ne devrait donc pas être excrété dans le lait maternel et l'exposition du nouveau-né/du nourrisson au cyclosilicate de zirconium sodique n'est pas attendue. La décision doit être prise soit d'arrêter l'allaitement, soit d'interrompre/d'arrêter le traitement par le cyclosilicate de zirconium sodique en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'existe pas de données cliniques sur l'effet de LOKELMA sur la fertilité humaine. Les études expérimentales animales effectuées avec le cyclosilicate de zirconium sodique n'ont pas mis en évidence d'effets sur la reproduction ou la fertilité (voir "Données précliniques" ).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesRésumé du profil de sécurité
Les effets secondaires les plus fréquemment observés étaient l'hypokaliémie (4,1%) et les événements liés à des œdèmes (5,7%).
Études cliniques
La sécurité de LOKELMA a été évaluée dans le cadre d'études cliniques portant sur la réduction de l'hyperkaliémie incluant 1760 patients, dont 507 patients exposés pendant un an.
Liste des effets indésirables
Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classe de systèmes d'organes (MedDRA). Les effets indésirables sont mentionnés selon les catégories suivantes: très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), occasionnel (≥1/1000 à < 1/100), rare (≥1/10'000 à < 1/1000), très rare (< 1/10'000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Troubles du métabolisme et de la nutrition
Fréquent: hypokaliémie.
Affections gastro-intestinales
Fréquent: constipation (au cours d'études cliniques conduites dans des pays à population majoritairement asiatique, la constipation est survenue à une fréquence estimée à 8,9% chez les patients sous LOKELMA et a disparu après ajustement de la dose ou arrêt du traitement).
Troubles généraux et anomalies au site d'administration
Fréquent: événements liés à des œdèmes, incluant surcharge liquidienne (rétention hydrique, œdème généralisé, hypervolémie, œdème localisé, œdème, œdème périphérique et gonflement périphérique; ces effets secondaires ont été observés uniquement durant la phase d'entretien).
Affections cardiaques
Très fréquent: dégradation d'une insuffisance cardiaque préexistante (13,6%).
Description de certains effets indésirables
Hypokaliémie
4,1% des patients traités par LOKELMA ont présenté une hypokaliémie avec une valeur de la kaliémie inférieure à 3,5 mmol/l qui a été résolue par un ajustement posologique ou l'arrêt de LOKELMA.
Événements liés à des œdèmes
Les effets indésirables les plus souvent décrits étaient les événements liés à des œdèmes rapportés chez 5,7% des patients traités par LOKELMA (respectivement 2,7%, 5,2% et 14,3% des patients randomisés pour recevoir LOKELMA 5 g, 10 g et 15 g, une fois par jour pendant une période maximale de 1 mois) et chez 1,7% des patients sous placebo. Chez 53% des patients, cet effet secondaire a été pris en charge en instaurant un traitement diurétique ou en ajustant la dose de diurétique; dans les autres cas, un traitement n'a pas été nécessaire.
Dégradation d'une insuffisance cardiaque préexistante
Selon une analyse groupée des études cliniques contrôlées contre placebo effectuées avec LOKELMA chez des patients non dialysés (études PRIORITIZE-HF, REALIZE-K, STABILIZE-CKD), une dégradation de l'insuffisance cardiaque s'est produite chez quelques patients atteints d'insuffisance cardiaque préexistante. Cet événement est survenu à une fréquence de 13,6% (30/220) avec le traitement par LOKELMA et à une fréquence de 5,7% (12/209) avec le placebo. Cet événement s'est résolu dans la plupart des cas après un traitement clinique adéquat sans nécessiter l'arrêt de LOKELMA (voir "Mises en garde et précautions" ).
Utilisation à long terme
Dans le cadre de 2 études cliniques avec une exposition à LOKELMA en phase ouverte jusqu'à 1 an chez 874 patients, les événements suivants ont été rapportés comme étant liés au médicament par les médecins investigateurs: événements gastro-intestinaux (constipation [2,9%], diarrhée [0,9%], douleur/distension abdominale [0,5%], nausées [1,6%], vomissements [0,5%]) et réactions d'hypersensibilité (éruption cutanée (0,3%) et prurit (0,1%)). Ces événements étaient d'intensité légère à modérée, aucun n'a été signalé comme étant grave et ils se sont généralement résolus alors que le patient poursuivait le traitement. Compte tenu de la conception de l'étude en phase ouverte, un lien de causalité entre ces événements et LOKELMA ne peut pas être clairement établi.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Le surdosage de LOKELMA peut provoquer une hypokaliémie.
Traitement
Le taux de potassium sérique doit être contrôlé et une supplémentation en potassium doit être administrée, si nécessaire.
Propriétés/EffetsCode ATC
V03AE10
Mécanisme d'action
Le cyclosilicate de zirconium sodique est une poudre inorganique, non polymère, non résorbée, avec une structure microporeuse uniforme qui capte préférentiellement le potassium en échange de cations hydrogène et sodium. In vitro, le cyclosilicate de zirconium sodique est hautement sélectif pour les ions potassium, y compris en présence d'autres cations, tels que le calcium et le magnésium. Le cyclosilicate de zirconium sodique capte le potassium dans l'ensemble du tube digestif et réduit la concentration de potassium libre dans la lumière gastro-intestinale, diminuant ainsi le taux de potassium sérique et augmentant l'excrétion fécale de potassium afin de traiter une hyperkaliémie.
Pharmacodynamique
Le cyclosilicate de zirconium sodique commence à réduire la concentration sérique de potassium 1 heure après l'ingestion et une normokaliémie peut être normalement obtenue en 24 à 48 heures. Le cyclosilicate de zirconium sodique ne modifie pas les concentrations sériques de calcium ou de magnésium ou l'excrétion urinaire de sodium. Il existe une corrélation étroite entre la concentration sérique initiale de potassium et l'importance de l'effet; les patients ayant une concentration initiale de potassium plus élevée obtiennent une réduction plus importante du potassium sérique. Une réduction de l'excrétion urinaire de potassium subsiste qui est une conséquence de la réduction de la concentration sérique de potassium. Dans le cadre d'une étude menée chez des sujets sains recevant LOKELMA 5 g ou 10 g une fois par jour pendant quatre jours, les réductions dose-dépendantes de la concentration sérique de potassium et de l'excrétion urinaire totale de potassium se sont accompagnées d'augmentations moyennes de l'excrétion fécale de potassium. Aucune variation statistiquement significative de l'excrétion urinaire de sodium n'a été observée.
Aucune étude n'a été réalisée pour évaluer la pharmacocinétique du cyclosilicate de zirconium sodique administré au cours ou en dehors des repas.
Il a été également démontré que le cyclosilicate de zirconium sodique fixe les ions ammonium in vitro et in vivo, éliminant ainsi les ions ammonium et augmentant les taux sériques de bicarbonate. Les patients traités par LOKELMA ont présenté une augmentation du taux de bicarbonate de 1,1 mmol/l à la dose de 5 g une fois par jour, de 2,3 mmol/l à la dose de 10 g une fois par jour et de 2,6 mmol/l à la dose de 15 g une fois par jour par comparaison avec une augmentation moyenne de 0,6 mmol/l pour les patients recevant le placebo. Dans un environnement où d'autres facteurs impactant la rénine et l'aldostérone n'étaient pas contrôlés, LOKELMA a démontré une variation dose-indépendante des taux sériques moyens d'aldostérone (intervalle: de -30% à -31%) en comparaison avec le groupe placebo (+14%). Aucun effet durable n'a été observé sur la pression artérielle systolique et diastolique.
De plus, une réduction moyenne de l'azote uréique sanguin (BUN) a été observée dans les groupes traités par 5 g (1,1 mg/dl) et 10 g (2,0 mg/dl) de LOKELMA trois fois par jour par comparaison avec une faible augmentation moyenne dans les groupes placebos (0,8 mg/dl) et les groupes traités par le cyclosilicate de zirconium sodique à faible dose (0,3 mg/dl).
Efficacité clinique
L'effet hypokaliémiant de LOKELMA a été démontré dans le cadre de trois études randomisées, en double aveugle, contrôlées contre placebo, chez des patients présentant une hyperkaliémie. Au cours des trois études, l'effet initial de LOKEMA pour corriger l'hyperkaliémie a été testé pendant une période de 48 heures. Deux études évaluant l'effet sur le maintien de la normokaliémie obtenue ont également été réalisées. Les études concernant le traitement d'entretien ont inclus des patients présentant une néphropathie chronique (58%), une insuffisance cardiaque (10%), un diabète sucré (62%) et des patients suivant un traitement par inhibiteurs du SRAA (68%). Ces études ont inclus 1760 patients ayant reçu des doses de LOKELMA; 507 d'entre eux pendant au moins 360 jours. De plus, l'efficacité et la sécurité de LOKELMA ont été évaluées dans le cadre d'une étude, en double aveugle, contrôlée contre placebo chez 196 patients hémodialysés chroniques présentant une hyperkaliémie qui ont reçu des doses de LOKELMA pendant 8 semaines.
Au cours des études, LOKELMA a réduit le taux sérique de potassium et maintenu des taux sériques de potassium normaux, indépendamment de la cause de l'hyperkaliémie, de l'âge, du sexe, de l'origine ethnique, des comorbidités ou de l'utilisation concomitante d'inhibiteurs du SRAA. Aucune restriction alimentaire n'a été imposée et il a été demandé aux patients de poursuivre leur régime alimentaire habituel sans modifications particulières.
Étude randomisée, en double aveugle, contrôlée contre placebo, en deux phases
Dans cette étude, 753 patients (âge moyen: 66 ans; fourchette: 22 à 93 ans) présentant une hyperkaliémie (5,0 à ≤6,5 mmol/l, potassium sérique moyen à l'inclusion: 5,3 mmol/l) ont été randomisés et répartis dans deux groupes pour recevoir soit un traitement par LOKELMA (1,25 g, 2,5 g, 5 g ou 10 g) soit un placebo, chacun étant administré trois fois par jour au cours des 48 premières heures. L'étude incluait des patients atteints de néphropathie chronique, d'insuffisance cardiaque, de diabète et des patients suivant un traitement par inhibiteurs du SRAA.
Le traitement par LOKELMA a induit une réduction dose-dépendante du taux sérique de potassium aux doses de 2,5 g, de 5 g et de 10 g qui a été observée en quelques heures après l'administration de la première dose (Tableau 3). Une réduction statistiquement significative de la kaliémie a été observée 1 heure après la prise de la première dose de 10 g de LOKELMA. À cette dose, la réduction moyenne du taux sérique de potassium était de 0,7 mmol/l, et 86% des patients ont obtenu une normokaliémie en 48 heures. Les patients qui avaient une kaliémie initiale plus élevée ont eu une réponse plus importante avec LOKELMA. Les patients qui avaient un taux de potassium supérieur à 5,5 mmol/l (valeur moyenne initiale: 5,8 mmol/l) avant le traitement ont obtenu une diminution moyenne de 1,1 mmol/l en 48 heures tandis que ceux dont la kaliémie initiale était inférieure ou égale à 5,3 mmol/l ont obtenu une diminution de 0,6 mmol/l à la dose la plus élevée.
Tableau 3: Phase de correction (étude 1): pourcentage de patients présentant une normokaliémie après 48 heures de traitement par LOKELMA
Dose de LOKELMA
(trois fois par
jour)
Placebo 1,25 g 2,5 g 5 g 10 g
N 158 154 141 157 143
Taux sérique initial 5,3 5,4 5,4 5,3 5,3
de potassium en
mmol/l
Normokaliémie après 48 51 68 78 86
48 heures en %
Valeur p par rapport NS < 0,001 < 0,001 < 0,001
au placebo
NS: non significatif
Les patients ayant obtenu une normokaliémie (taux de potassium compris entre 3,5 et 5,0 mmol/l) ont ensuite de nouveau été randomisés pour recevoir le traitement actif à la même dose ou le placebo une fois par jour pendant 12 jours. Durant cette phase de l'étude, les critères d'évaluation de l'efficacité prédéfinis ont été atteints dans les groupes recevant les doses de 2,5 g, de 5 g et de 10 g, par comparaison avec leurs groupes placebos respectifs. À la fin de la phase de traitement, après l'arrêt de LOKELMA, les taux de potassium sont revenus presque aux valeurs initiales.
Étude d'entretien multiphase, contrôlée contre placebo comportant une phase supplémentaire en ouvert
Lors de la phase de correction de l'étude, 258 patients présentant une hyperkaliémie (taux de potassium moyen à l'inclusion: 5,6 mmol/l, fourchette de 4,1 à 7,2 mmol/l) ont reçu 10 g de LOKELMA trois fois par jour pendant 48 heures.
Une réduction de la kaliémie a été observée 1 heure après l'administration de la première dose de 10 g de LOKELMA. La durée médiane jusqu'à l'obtention de la normokaliémie était de 2,2 heures, 66% des patients ayant obtenu une normokaliémie après 24 heures et 88% après 48 heures. Le taux sérique de potassium a diminué respectivement de 0,8, 1,2 et 1,5 mmol/l chez les patients dont la kaliémie était à l'inclusion respectivement < 5,5 mmol/l, de 5,5 à 5,9 mmol/l et ≥6 mmol/l.
Les patients ayant obtenu une normokaliémie (kaliémie comprise entre 3,5 et 5 mmol/l) ont été randomisés en double aveugle pour recevoir l'une des trois doses de LOKELMA (5 g (n = 45), 10 g (n = 51) ou 15 g (n = 56)) ou le placebo (n = 85), administrés une fois par jour pendant 28 jours (phase de sevrage randomisée en double aveugle).
La proportion de participants présentant une kaliémie moyenne < 5,1 mmol/l entre le jour 8 et le jour 29 de l'étude (période de trois semaines) a été plus importante aux doses de LOKELMA de 5 g, 10 g et 15 g une fois par jour (respectivement 80%, 90% et 94%) par rapport au placebo (46%). Dans les groupes de traitement 5 g, 10 g, 15 g une fois par jour par LOKELMA et dans le groupe placebo, la diminution moyenne du potassium sérique était respectivement de 0,77 mmol/l, 1,10 mmol/l, 1,19 mmol/l et 0,44 mmol/l et la proportion de participants toujours normokaliémiques était respectivement de 71%, 76%, 85% et 48%.
Résultats de la phase d'entretien (en ouvert) avec titration de la dose de LOKELMA: 123 patients ont été inclus dans la phase en ouvert de 11 mois. La proportion de participants ayant un taux sérique moyen de potassium < 5,1 mmol/l était de 88%. Le taux sérique moyen de potassium était de 4,66 mmol/l et la proportion des taux sériques de potassium en dessous de 3,5 mmol/l était inférieure à 1%. Chez 77% des participants, elle était comprise entre 3,5 et 5,1 mmol/l et chez 93% entre 3,5 et 5,5 mmol/l, indépendamment des autres facteurs qui pourraient influencer la kaliémie. Le traitement a été arrêté à la sortie de l'étude (jour 365).
Les estimations selon Kaplan-Meier du délai jusqu'à la rechute pendant la phase d'entretien ont démontré une dépendance dose-temps du délai jusqu'à la rechute, avec un délai médian pour la dose de 5 g de 4 à 21 jours en fonction des taux sériques de potassium à l'inclusion. La kaliémie doit être surveillée régulièrement et la dose de LOKELMA doit être titrée comme décrit dans la rubrique "Posologie/Mode d'emploi" .
La Figure 1: illustre le taux sérique moyen de potassium durant les phases de correction et d'entretien de l'étude

Étude chez des patients atteints de néphropathie chronique associée à une hyperkaliémie
Cette étude était une étude en double aveugle, contrôlée contre placebo, avec escalade de dose, menée chez 90 patients (60 patients traités par LOKELMA; 30 patients témoins) ayant un DFGe initial compris entre 30 et 60 ml/min/1,73 m2 et une hyperkaliémie (kaliémie à l'inclusion: 5,2 mmol/l, fourchette de 4,6 à 6 mmol/l). Les patients ont été randomisés pour recevoir des doses croissantes de LOKELMA (0,3 g, 3 g et 10 g) ou un placebo, administrés trois fois par jour au cours des repas pendant deux à quatre jours.
Le critère principal d'évaluation était le taux de variation de la kaliémie initiale et tout au long des 2 premiers jours de traitement. Le critère principal d'évaluation de l'étude a été atteint aux doses de 3 g et 10 g de LOKELMA par comparaison avec le placebo. LOKELMA administré à la posologie de 10 g et de 3 g a induit des réductions maximales moyennes de respectivement 0,92 mmol/l et 0,43 mmol/l. Le recueil des urines des 24 h a montré que LOKELMA diminuait l'excrétion urinaire du potassium par rapport à la valeur initiale de 15,8 mmol/24 h par comparaison avec une augmentation de 8,9 mmol/24 h avec le placebo (p < 0,001). L'excrétion du sodium est restée inchangée par comparaison avec le placebo (augmentation de 25,4 mmol/24 h avec 10 g par comparaison avec une augmentation de 36,9 mmol/24 h avec le placebo [NS]).
Étude randomisée, en double aveugle, contrôlée contre placebo chez des patients hémodialysés chroniques
Dans cette étude, 196 patients (âge moyen de 58 ans, fourchette de 20 à 86 ans) souffrant d'insuffisance rénale terminale sous dialyse stable depuis au moins 3 mois et présentant une hyperkaliémie prédialytique persistante ont été randomisés pour recevoir lors des jours sans dialyse LOKELMA 5 g ou un placebo une fois par jour. Lors de la randomisation, les taux moyens de potassium étaient de 5,8 mmol/l (fourchette de 4,2 à 7,3 mmol/l) dans le groupe LOKELMA et de 5,9 mmol/l (fourchette de 4,2 à 7,3 mmol/l) dans le groupe placebo. Pour atteindre un taux sérique de potassium prédialytique compris entre 4,0 et 5,0 mmol/l au cours de la période d'ajustement posologique (les 4 premières semaines), la dose pouvait être augmentée chaque semaine par paliers de 5 g jusqu'à 15 g une fois par jour en fonction de la kaliémie prédialytique après le LIDI. La dose atteinte à la fin de la période d'ajustement posologique a été maintenue tout au long de la phase d'évaluation de 4 semaines qui a suivi. À la fin de la phase d'ajustement posologique, respectivement 37%, 43% et 19% des patients étaient traités par LOKELMA 5 g, 10 g et 15 g. La proportion de répondeurs, définis comme étant les patients chez lesquels un taux sérique de potassium prédialytique compris entre 4,0 et 5,0 mmol/l après le LIDI a été maintenu pour au moins 3 des 4 périodes de dialyse et qui n'ont pas reçu de traitement de secours pendant la phase d'évaluation était de 41% dans le groupe LOKELMA et de 1% dans le groupe placebo (p < 0,001) (voir Figure 2).
Dans les analyses post hoc, la fréquence des taux sériques de potassium compris entre 4,0 et 5,0 mmol/l après le LIDI au cours de la phase d'évaluation était plus élevée dans le groupe LOKELMA. 24% des patients avaient atteint ces valeurs lors des 4 visites dans le groupe LOKELMA et aucun dans le groupe placebo. L'analyse post hoc a montré que la proportion de patients qui avaient un taux sérique de potassium compris entre 3,5 et 5,5 mmol/l pour au moins 3 périodes de dialyse sur 4 après le LIDI pendant la phase d'évaluation était de 70% dans le groupe LOKELMA et de 21% dans le groupe placebo.
À la fin du traitement, le taux sérique moyen de potassium postdialytique était de 3,6 mmol/l (fourchette de 2,6 à 5,7 mmol/l) dans le groupe LOKELMA et de 3,9 mmol/l (fourchette de 2,2 à 7,3 mmol/l) dans le groupe placebo. Il n'y avait aucune différence entre les groupes LOKELMA et placebo concernant la prise de poids interdialytique (IDWG = interdialytic weight gain). L'IDWG a été défini comme le poids prédialytique auquel était soustrait le poids postdialytique lors de la séance de dialyse précédente et a été mesuré après le LIDI.
Figure 2: Taux sérique moyen de potassium prédialytique au cours du temps chez des patients hémodialysés chroniques
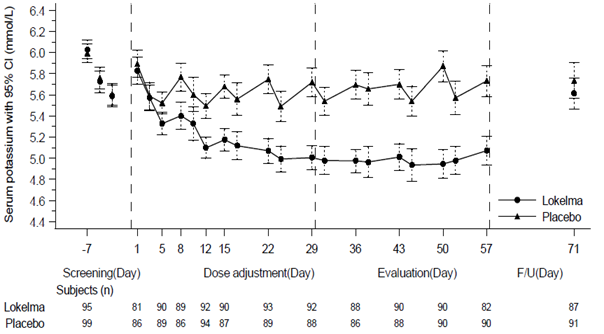
F/U – phase de suivi
Les barres d'erreur affichées correspondent à des intervalles de confiance à 95%.
n = nombre de patients pour lesquels il ne manquait pas de mesures du potassium lors d'une visite donnée.
Initiative de réduction de la kaliémie dans le cadre d'une étude multicentrique, randomisée, en double aveugle, contrôlée contre placebo, d'une durée de trois mois en groupes parallèles visant à optimiser le traitement par les inhibiteurs du SRAA en présence d'insuffisance cardiaque (étude PRIORITIZE HF)
Au cours de cette étude randomisée, en double aveugle, contrôlée contre placebo, il a été évalué si un schéma thérapeutique de LOKELMA permettrait une augmentation de la posologie des traitements par les inhibiteurs du système rénine-angiotensine-aldostérone (SRAA) pour atteindre les doses cibles après 3 mois vs placebo chez des patients souffrant d'insuffisance cardiaque et d'hyperkaliémie ou à haut risque d'hyperkaliémie. Le critère principal d'évaluation de l'étude était la proportion de patients dans les 4 catégories suivantes après 3 mois: pas d'inhibiteurs de l'enzyme de conversion de l'angiotensine (IECA)/d'antagonistes des récepteurs de l'angiotensine (ARA)/d'inhibiteurs des récepteurs de l'angiotensine et de la néprilysine (ARNI) ou leur administration à une dose inférieure à la dose cible et pas d'antagoniste des récepteurs des minéralocorticoïdes (ARM); IECA/ARA/ARNI à la dose cible et pas d'ARM; ARM à une dose inférieure à la dose cible; ARM à la dose cible.
Des patients présentant une insuffisance cardiaque de classe II à IV selon la classification fonctionnelle de la New York Heart Association (NYHA) et une FEVG ≤40%, un DFGe de 20 à 59 ml/min/1,73 m² et une kaliémie de 4,0 à 5,5 mmol/l ont été randomisés pour recevoir un traitement par LOKELMA ou un placebo (dans un rapport de 1:1) pendant 3 mois. Une augmentation de la posologie des inhibiteurs du SRAA aux doses recommandées par les directives était certes favorisée, mais n'était pas prescrite et des titrations des doses de LOKELMA ou du placebo ont été effectuées parallèlement afin d'éviter une hyperkaliémie.
L'étude a été arrêtée prématurément pendant la pandémie de Covid-19 en raison de problèmes de recrutement et de la difficulté à garantir une surveillance adéquate de la sécurité, étant donné que les patients ne pouvaient pas se présenter aux visites de l'étude et aux visites de contrôle des analyses de laboratoire. En conséquence, seulement 182 patients au lieu des 280 patients prévus ont été randomisés. L'arrêt prématuré de l'étude ne permet donc pas de conclusions sûres concernant les critères principaux d'efficacité et les autres critères.
Étude randomisée, en double aveugle, contrôlée contre placebo, effectuée en groupes parallèles portant sur la prise en charge de l'hyperkaliémie chez des patients souffrant d'insuffisance cardiaque symptomatique à fraction d'éjection réduite traités par la spironolactone (étude REALIZE-K)
Au cours de cette étude de sevrage de phase IV, randomisée, prospective, en double aveugle, l'efficacité et la sécurité de LOKELMA lors de l'optimisation du traitement par les antagonistes des récepteurs des minéralocorticoïdes (ARM) ont été évaluées chez des patients souffrant d'insuffisance cardiaque à fraction d'éjection réduite. Le critère d'évaluation principal était l'obtention d'une réponse optimale, définie comme un taux sérique de potassium normal (3,5 à 5,0 mEq/ml) à une dose de spironolactone ≥25 mg/jour sans la nécessité d'un traitement de secours en raison d'une hyperkaliémie.
Des adultes présentant un diagnostic confirmé d'insuffisance cardiaque (depuis ≥3 mois, fraction d'éjection ventriculaire gauche ≤40%) et des symptômes de classe II à IV selon la classification de la New York Heart Association (NYHA), traités par un inhibiteur de l'enzyme de conversion de l'angiotensine (IECA)/un antagoniste des récepteurs de l'angiotensine (ARA)/un inhibiteur des récepteurs de l'angiotensine et de la néprilysine (ARNI) et un bêtabloquant (s'il n'était pas contre-indiqué) à une dose stable depuis ≥4 semaines, ont été inclus dans cette étude. Les patients qui n'étaient pas traités par un antagoniste des récepteurs des minéralocorticoïdes (ARM) et les patients traités par la spironolactone ou l'éplérénone à une dose < 25 mg une fois par jour pouvaient participer à l'étude.
Les patients ont été sélectionnés et inclus dans une phase préliminaire en ouvert comportant deux cohortes. La cohorte 1 incluait des patients présentant une hyperkaliémie prévalente avérée (définie par un taux sérique de K+ de 5,1 à 5,9 mEq/l) et un DFGe ≥30 ml/min/1,73 m². Les patients de cette cohorte ont reçu LOKELMA pour corriger la concentration de potassium afin d'atteindre une normokaliémie; un traitement par la spironolactone a été ensuite instauré et la posologie a été augmentée conformément au protocole de l'étude. La cohorte 2 incluait des patients à haut risque d'hyperkaliémie (définie par un taux sérique de potassium > 5,0 mEq/l lors des 36 derniers mois et un DFGe ≥30 ml/min/1,73 m² OU un taux sérique de potassium de 4,5 à 5,0 mEq/l et un DFGe de 30 à 60 ml/min/1,73 m² OU un taux sérique de potassium de 4,5 à 5,0 mEq/l et un âge > 75 ans). Chez ces patients, le traitement par la spironolactone a été instauré ou sa posologie a été augmentée jusqu'à la dose cible; les patients qui présentaient une hyperkaliémie ont reçu LOKELMA pour corriger le taux de potassium afin d'obtenir une normokaliémie, tandis que les patients qui ne présentaient pas d'hyperkaliémie en l'espace de 4 semaines ont quitté l'étude.
Au cours de cette étude, l'administration de LOKELMA a entraîné plus fréquemment une réponse optimale concernant le critère d'évaluation principal que le placebo (OR 4,45 [IC à 95%: 2,89 à 6,86], p < 0,001, pourcentage évalué: 71% vs 36%). LOKELMA a induit également une amélioration des critères d'évaluation secondaires par comparaison avec le placebo, à savoir une normokaliémie à la dose randomisée de spironolactone et sans traitement de secours en raison d'une hyperkaliémie (OR 4,58 [IC à 95%: 2,78 à 7,55], p < 0,001; pourcentage évalué: 58% vs 23%); l'obtention d'une dose de spironolactone ≥25 mg/par jour (OR 4,33 [IC à 95%: 2,50 à 7,52], p < 0,001; pourcentage évalué: 81% vs 50%); le délai jusqu'au premier épisode d'hyperkaliémie (taux sérique de K+ > 5,0 mEq/l) (HR 0,51 [IC à 95%: 0,37 à 0,71], p < 0,001) et le délai jusqu'à la première diminution de la dose de spironolactone ou jusqu'à l'arrêt de la spironolactone en raison d'une hyperkaliémie (HR 0,37 [IC à 95%: 0,17 à 0,73], p = 0,006).
Étude randomisée, en double aveugle, contrôlée contre placebo évaluant l'effet de LOKELMA sur la progression de la néphropathie chronique chez des patients présentant une IRC et une hyperkaliémie ou un risque élevé d'hyperkaliémie (étude STABILIZE-CKD)
Il a été évalué dans le cadre de cette étude de sevrage de phase III, randomisée, en double aveugle, contrôlée contre placebo, effectuée en groupes parallèles, si LOKELMA en complément d'un traitement par un inhibiteur de l'enzyme de conversion de l'angiotensine (IEC)/un antagoniste des récepteurs de l'angiotensine (ARA) est supérieur au placebo pour ralentir la progression de l'IRC chez des patients présentant une hyperkaliémie ou à risque élevé d'hyperkaliémie. Les critères d'évaluation co-primaires étaient la baisse globale du DFGe (de la randomisation à la fin du traitement) et la baisse chronique du DFGe (à partir de 12 semaines après la randomisation jusqu'à la fin du traitement).
Les patients présentant un DFGe de 25 à 59 ml/min/1,73 m², un RACu de 200 à 5 000 mg/g et une hyperkaliémie (taux sérique de potassium [sK+] > 5,0 à ≤6,5 mmol/l) sous traitement adéquat/limité par un IECA/un ARA ou présentant une normokaliémie sous traitement limité par un IECA/un ARA, ont été inclus dans l'étude. Les patients souffrant d'insuffisance cardiaque congestive de classe III à IV selon la classification de la NYHA au moment de la sélection ou ayant des antécédents d'insuffisance cardiaque sévère ou symptomatique ont été exclus de l'étude.
L'étude a été arrêtée prématurément en raison de problèmes de recrutement, ayant pour conséquence une taille d'échantillon réduite de 760 patients randomisés au lieu des 1360 patients prévus et une durée de suivi écourtée après la randomisation (~8 à 9 mois [moyenne] au lieu des 24 mois prévus). L'arrêt prématuré de l'étude ne permet pas de tirer des conclusions fiables sur les critères d'efficacité primaires et autres.
Données à long terme
Étude multicentrique, multidoses, en ouvert, en deux phases évaluant la sécurité et l'efficacité
Les effets à long terme (jusqu'à 12 mois) de LOKELMA ont été évalués dans le cadre de cette étude menée chez 751 patients atteints d'hyperkaliémie (moyenne à l'inclusion 5,59 mmol/l; fourchette de 4,3 à 7,6 mmol/l). Les comorbidités étaient la néphropathie chronique (65%), le diabète sucré (64%), l'insuffisance cardiaque (15%) et l'hypertension artérielle (83%). L'utilisation de diurétiques et d'inhibiteurs du SRAA rapportée était de respectivement 51% et 70% des patients. Pendant la phase de correction, 10 g de LOKELMA ont été administrés trois fois par jour pendant au moins 24 heures et jusqu'à 72 heures.
Les patients ayant obtenu une normokaliémie (3,5 à 5,0 mmol/l inclus) en 72 heures ont été inclus dans la phase d'entretien de l'étude. Tous les patients de la phase d'entretien ont reçu LOKELMA à une dose initiale de 5 g une fois par jour, dose qui pouvait être augmentée par paliers de 5 g une fois par jour (jusqu'à une dose maximale de 15 g une fois par jour) ou diminuée (jusqu'à une dose minimale de 5 g un jour sur deux) selon le schéma de titration.
La normokaliémie a été obtenue chez 494/748 (66%), 563/748 (75%) et 583/748 (78%) des patients après respectivement 24, 48 et 72 heures d'administration lors de la phase de correction, avec une réduction moyenne du taux sérique de potassium de 0,81 mmol/l, 1,02 mmol/l et 1,10 mmol/l après respectivement 24 (n= 748), 48 (n= 104) et 72 (n= 28) heures. La normokaliémie était dépendante de la concentration de potassium à l'inclusion; les patients présentant les concentrations sériques de potassium à l'inclusion les plus élevées ayant obtenu la diminution la plus importante après avoir débuté le médicament à l'étude constituaient toutefois la proportion la plus faible de patients ayant obtenu une normokaliémie. Cent vingt-six patients avaient un taux sérique de potassium à l'inclusion ≥6,0 mmol/l (taux moyen de potassium à l'inclusion: 6,28 mmol/l). Ces patients avaient obtenu une réduction moyenne de 1,37 mmol/l à la fin de la phase de correction.
Tableau 4: Phase de correction (étude 4): proportion de patients ayant des concentrations sériques de potassium comprises entre 3,5 et 5,0 mmol/l inclus ou entre 3,5 et 5,5 mmol/l inclus, par jour d'étude lors de la phase de correction
Population ITT
LOKELMA 10 g trois
fois par jour (N =
749)
Phase de correction Potassium sérique: Potassium sérique:
(PC) 3,5 à 5,0 mmol/l 3,5 à 5,5 mmol/l
inclus inclus
n/N Proportion IC à 95% n/N Proportion IC à 95%
PC après 24 heures 494/748 0,660 0,625; 0,694 692/748 0,925 0,904; 0,943
PC après 48 heures 563/748 0,753 0,720; 0,783 732/748 0,979 0,965; 0,988
PC après 72 heures/
PC dernière dose 583/748 0,779 0,748; 0,809 738/748 0,987 0,976; 0,994
Remarque: Pour un patient, la valeur après l'administration de la dernière dose a été relevée plus de 1 jour après la dernière dose. Ce patient était donc éligible pour la population ITT de la phase de correction; cependant, le temps d'évaluation a été exclu de l'analyse.
La normokaliémie a pu être maintenue tant que les patients étaient sous traitement, mais la concentration sérique moyenne de potassium a augmenté après l'arrêt du traitement. Parmi les patients utilisant des inhibiteurs du SRAA à l'inclusion, 89% n'ont pas arrêté le traitement par les inhibiteurs du SRAA et 74% ont pu continuer à prendre la même dose pendant la phase d'entretien. Parmi les patients qui n'utilisaient pas d'inhibiteurs du SRAA à l'inclusion, 14% ont pu débuter ce traitement. La normokaliémie a pu être maintenue chez 75,6% des patients pendant la phase d'entretien malgré l'utilisation d'inhibiteurs du SRAA.
La Figure 3: illustre la concentration sérique moyenne de potassium durant les phases de correction et d'entretien de l'étude
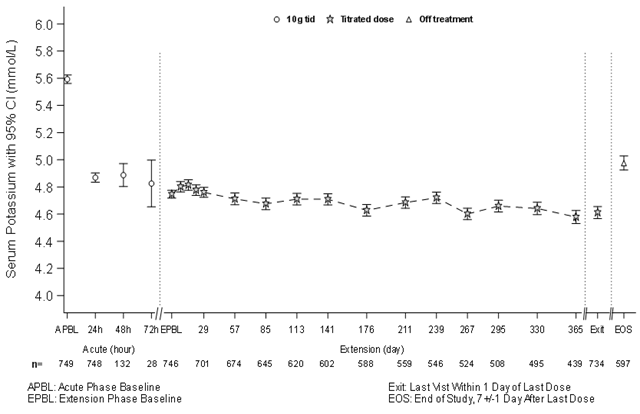
PharmacocinétiqueAbsorption
LOKELMA est une substance inorganique insoluble, qui ne subit pas de métabolisme enzymatique. De plus, les études cliniques ont montré que LOKELMA n'est pas résorbé dans la circulation systémique. Une étude d'équilibre de masse in vivo chez le rat a montré que le cyclosilicate de zirconium sodique était retrouvé dans les fèces, sans signe de résorption systémique. Compte tenu de ces facteurs et de l'insolubilité de la substance, aucune étude in vivo ou in vitro n'a été réalisée pour examiner son effet sur l'activité des enzymes du cytochrome P450 (CYP450) ou des transporteurs.
Distribution
Non pertinent.
Métabolisme
Non pertinent.
Élimination
LOKELMA est éliminé dans les fèces.
Données précliniquesLes données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en administration répétée, la toxicité pour la reproduction et le développement et la génotoxicité n'ont pas révélé de risque particulier pour l'homme.
Carcinogénicité
Des études à long terme évaluant le potentiel carcinogène du cyclosilicate de zirconium sodique n'ont pas été effectuées.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C.
Conserver hors de la portée des enfants.
Numéro d’autorisation67851 (Swissmedic)
PrésentationLOKELMA 5 g: emballage de 30 sachets (B).
Titulaire de l’autorisationAstraZeneca AG, 6340 Baar
Mise à jour de l’informationJanvier 2026
|