CompositionPrincipes actifs
Donanemab (produit par génie génétique à partir de cellules ovariennes de hamster chinois (CHO)).
Excipients
Acide citrique
Polysorbate 80
Citrate de sodium dihydraté
Saccharose
Eau pour préparations injectables
Chaque flacon de 20 ml de Kisunla contient au total 11.5 mg de sodium.
Forme pharmaceutique et quantité de principe actif par unitéSolution à diluer pour perfusion (perfusion intraveineuse).
Un flacon de 20 ml de Kisunla contient 350 mg de donanemab (17.5 mg/ml).
La solution est limpide à opalescente, incolore à légèrement jaune ou légèrement brunâtre. La solution ne doit pas être utilisée si elle est trouble ou en présence de particules visibles.
Indications/Possibilités d’emploiKisunla est indiqué pour ralentir la progression d'une maladie d'Alzheimer symptomatique (Alzheimer's disease, AD) chez les patients adultes présentant une pathologie bêta-amyloïde typique d'Alzheimer confirmée et un diagnostic clinique de trouble cognitif léger (mild cognitive impairment, MCI) ou de démence légère, qui sont porteurs hétérozygotes ou non-porteurs de l'allèle ε4 du gène de l'apolipoprotéine E (ApoE ε4) (voir les rubriques "Contre-indications" et "Efficacité clinique" ).
Posologie/Mode d’emploiL'utilisation doit être initiée sous les directives et la supervision d'un médecin expérimenté dans le diagnostic et le traitement de la maladie d'Alzheimer. La perfusion de donanemab doit être instaurée et surveillée par du personnel médical. Un accès rapide à un IRM doit être assuré. Le traitement par donanemab doit avoir lieu sous la supervision d'une équipe pluridisciplinaire expérimentée et formée à la détection, à la surveillance et au traitement des ARIA et expérimentée dans la détection et le traitement des réactions liées à la perfusion.
Avant le début du traitement, il faut disposer:
d'une génotypisation de l'ApoE ε4, avec un conseil génétique dispensé au préalable conformément aux réglementations nationales ou locales en vigueur.
d'une imagerie par IRM initiale (ne remontant pas à plus de trois mois, selon un protocole standardisé)
d'un PET à l'amyloïde, avec quantification centiloïde
d'un Tau-PET (si disponible)
Un manuel de neuroimagerie et un manuel IRM pour le donanemab sont disponibles ci-dessous pour consultation.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Détection de l'amyloïde-bêta
La détection de l'amyloïde-bêta compatible avec une maladie d'Alzheimer doit être confirmée par une méthode validée (par ex. PET à l'amyloïde).
Posologie
Le donanemab est administré toutes les 4 semaines. La dose recommandée est de 350 mg pour la première utilisation, 700 mg pour la deuxième utilisation et 1050 mg pour la troisième utilisation (350/700/1050 mg), puis 1400 mg toutes les 4 semaines. Ce schéma posologique se base sur des données pharmacocinétiques et pharmacodynamiques de bridging provenant de l'étude de phase 3b TRAILBLAZER-ALZ-6 et de l'étude pivot de phase 3 TRAILBLAZER-ALZ-2. Cela ne correspond pas au schéma posologique des 3 premières utilisations décrit dans l'étude pivot de phase 3 (voir les rubriques "Pharmacocinétique" et "Efficacité clinique" ).
Durée du traitement
Le traitement doit être maintenu jusqu'à l'élimination des plaques amyloïdes, confirmée avec une méthode validée, sur une durée maximale de 18 mois. Il devrait être interrompu prématurément dès qu'une progression vers un stade clinique de la maladie supérieur est atteint. Aussi faut-il déterminer l'état cognitif avant le début du traitement, puis tous les trois mois durant le traitement au moyen de tests cognitifs appropriés et validés permettant de déterminer de manière fiable les stades de la maladie d'Alzheimer, et d'évaluer les symptômes cliniques. Les résultats sont à documenter par écrit.
La vérification de la cognition et de la progression des symptômes doit être effectuée afin d'évaluer si une progression de la maladie sous-jacente a eu lieu chez le patient et/ou si l'évolution clinique suggère plutôt que Kisunla n'a pas été efficace chez le patient.
Un PET à l'amyloïde est effectué entre le mois 6 et le mois 12, afin de vérifier la charge amyloïde et de mettre fin au traitement si le PET est négatif. Une durée de traitement de maximum 18 mois est recommandée. Si le PET est négatif avant 18 mois, que la réponse clinique disparaît ou que, dans le cas individuel, le stade de la maladie supérieur suivant est atteint, le traitement doit être arrêté.
Les avantages et les risques peuvent dépendre de la concentration initiale de Tau. Chez les patients avec une charge Tau faible à modérée, une efficacité numériquement supérieure a été observée par rapport à ceux avec une charge Tau élevée (voir la rubrique "Pharmacodynamique" ). Lors de la décision d'instaurer ou non le donanemab, les résultats de l'imagerie du Tau-PET devraient être pris en compte, si cette dernière a été réalisée.
Surveillance et interruption du traitement en cas d'anomalies d'imagerie liées à l'amyloïde.
Pendant tout le traitement par Kisunla, un accès à l'imagerie par résonance magnétique (IRM) doit être garanti.
Avant le début du traitement, il faut qu'une IRM cérébrale (ne remontant pas à plus de 3 mois) soit disponible en tant que valeur initiale. Une IRM est à effectuer respectivement avant la 2e utilisation, avant la 3e utilisation, avant la 4e utilisation et avant la 7e utilisation (mois 6). Chez les patients présentant des facteurs de risque d'ARIA, tels que les porteurs hétérozygotes de l'ApoE ε4 et/ou les patients ayant présenté précédemment des épisodes d'ARIA pendant le traitement, une IRM supplémentaire devrait être effectuée après une année de traitement (avant la douzième perfusion). Dès que les patients rapportent des symptômes évoquant un événement ARIA, une évaluation clinique incluant une IRM est à effectuer, quelle que soit la phase du traitement.
Le protocole de l'étude TRAILBLAZER-ALZ-2 (AACI) recommandait une IRM avec séquence FLAIR pour mettre en évidence les ARIA-E et un T2* Gradient-Recalled Echo pour mettre en évidence les ARIA-H. Une imagerie pondérée en susceptibilité est également acceptable pour détecter les ARIA-H.
Les recommandations pour interrompre le traitement chez les patients présentant des anomalies d'imagerie liées à l'amyloïde avec œdème (edema/effusion, ARIA-E) et avec hémorragies/dépôts d'hémosidérine (ARIA-H) sont mentionnées dans la Table 1.
Table 1: Recommandations posologiques pour les patients avec ARIA-E et ARIA-H
Symptômes cliniques Sévéritéb des ARIA-E et ARIA-H
à l'IRM
Léger Modéré Sévère
Asymptomatique Envisager la suspension du Suspendre le traitemen Arrêter le traitemen
traitement ta t
Symptomatique Suspendre le traitementa Suspendre le traitemen Arrêter le traitemen
ta t
a Suspension du traitement jusqu'à ce qu'un examen par IRM montre une résolution radiologique (ARIA-E) ou une stabilisation (ARIA-H) et que les symptômes, si présents, aient régressé. Un nouvel examen par IRM devrait être envisagé deux à quatre mois après la première présentation afin d'évaluer la résolution (ARIA-E) ou la stabilisation (ARIA-H). La reprise ou l'arrêt du traitement se fera en fonction de l'appréciation clinique. Avant la reprise du traitement, une nouvelle évaluation des facteurs de risque est à effectuer. Pour les ARIA-E, un traitement de soutien, y compris par corticostéroïdes, peut être appliqué. L'efficacité de ce traitement n'a toutefois pas été démontrée.
b Voir la Table 2 pour les critères de classification du degré de sévérité des ARIA à l'IRM.
En cas d'ARIA-E ou d'ARIA-H sévères sur le plan radiographique ou symptomatiques sévères au niveau clinique, le traitement par donanemab devrait être arrêté définitivement.
Le donanemab devrait être définitivement arrêté en cas d'ARIA-E cliniquement grave, d'ARIA-H grave ou d'hémorragie intracérébrale de plus de 1 cm. Après des épisodes récurrents d'ARIA symptomatiques au niveau clinique ou modérés ou sévères sur le plan radiologique, le traitement avec le donanemab devrait être arrêté définitivement.
Mode d'administration
Kisunla 350 mg doit être administré par perfusion intraveineuse uniquement. Le flacon est destiné à un usage unique. La perfusion est à administrer sur une durée d'au moins 30 minutes. Après la perfusion, les patients sont à observer pendant au moins 30 minutes. Pour les instructions sur la dilution du médicament avant son utilisation, voir rubrique "Remarques particulières, remarques concernant la manipulation" .
Les médicaments utilisés par voie parentérale doivent être contrôlés visuellement avant l'emploi pour s'assurer qu'ils ne présentent pas de particules ou de changement de couleur, pour autant que la solution et le récipient le permettent. Kisunla ne doit pas être utilisé s'il est trouble ou s'il contient des particules visibles.
Omission de dose
Si une perfusion a été manquée, rattraper l'administration dès que possible. On reprendra ensuite l'utilisation recommandée toutes les 4 semaines.
Patients âgés
Aucune adaptation posologique n'est nécessaire pour les personnes âgées. (voir la rubrique "Pharmacocinétique" )
Patients présentant une insuffisance rénale
Aucune adaptation posologique n'est nécessaire en cas d'insuffisance rénale légère à modérée. On ne dispose que de données limitées pour les patients présentant une insuffisance rénale sévère (voir rubrique "Pharmacocinétique" ).
Patients présentant une insuffisance hépatique
Aucune adaptation posologique n'est nécessaire en cas de légère insuffisance hépatique. On ne dispose que de données limitées pour les patients présentant une insuffisance hépatique modérée et il n'existe pas de données pour les patients présentant une insuffisance hépatique sévère (voir rubrique "Pharmacocinétique" ).
Patients sous plasmaphérèse concomitante
On fixera l'intervalle de temps entre l'utilisation du donanemab et la plasmaphérèse de façon à minimiser l'élimination du donanemab (voir la rubrique "Interactions" ).
Enfants et adolescents
Kisunla n'est pas enregistré pour l'utilisation chez les enfants et les adolescents. Aucune recommandation posologique n'est disponible.
Contre-indications-Hypersensibilité au donanemab ou à l'un des excipients (voir "Composition" ).
Résultats d'imagerie indiquant un risque augmenté d'ARIA ou d'hémorragie intracérébrale, tels que:
-Résultats initiaux d'IRM montrant
une hémorragie intracérébrale antérieure de plus de 1 cm
plus de 4 microhémorragies (définies comme ayant un diamètre ≤1 cm sur la séquence T2*),
sidérose superficielle, œdèmes vasogéniques (ARIA-E) ou autres résultats évoquant une angiopathie amyloïde cérébrale associées à une inflammation (CAA-ri).
plus de 2 infarctus lacunaires ou accidents vasculaires cérébraux touchant un large territoire vasculaire
hyperintensité sous-corticale sévère correspondant à un score Fazekas de 3
-Maladie grave de la substance blanche
-Autres pathologies graves pouvant entraîner un trouble cognitif
-Evidence d'une démence non liée à la maladie d'Alzheimer à l'IRM
-Accident vasculaire cérébral ou accident ischémique transitoire dans l'année précédant le début du traitement
-Hypertension insuffisamment contrôlée
-Trouble de la coagulation sanguine insuffisamment contrôlé (y compris numération plaquettaire <50 000 ou International Normalized Ratio [INR] >1.5 chez les patients ne prenant pas d'anticoagulants)
-Traitement par anticoagulants
-Lyse systémique par un thrombolytique
-Résultats médicaux instables interférant ou pouvant interférer avec le traitement par donanemab
-Tout résultat empêchant une évaluation IRM satisfaisante lors du monitoring de sécurité
-Épilepsie active dans l'année précédant le début du traitement
-Grossesse ou femmes en âge de procréer sans contraception adéquate
Mises en garde et précautionsProgramme d'accès contrôlé (Controlled access program)
Afin de garantir la sécurité et l'efficacité de l'utilisation du donanemab, l'initiation du traitement chez tous les patients doit s'effectuer par le biais d'un système d'enregistrement central mis en place dans le cadre d'un programme d'accès contrôlé.
Matériels éducationnels
Les médecins prescripteurs devraient être familiers avec le matériel éducationnel destiné à la détection et à la prise en charge des ARIA et discuter des avantages et des risques du traitement par donanemab avec le patient/son entourage. Les IRM ainsi que les signes et symptômes des éventuels effets secondaires doivent être expliqués au patient/à son entourage. En outre, il faut aussi expliquer à ces derniers quand une consultation en urgence doit avoir lieu. L'information destinée au patient et la carte patient sont à remettre et à expliquer au patient. Le patient doit toujours conserver la carte patient avec lui.
Anomalies d'imagerie liées à l'amyloïde (ARIA)
Des anomalies d'imagerie liées à l'amyloïde (amyloid-related imaging abnormalities, ARIA) graves ont été observées dans les études cliniques portant sur le donanemab, dont certaines ont eu une issue fatale (voir rubrique "Effets indésirables" ). Les ARIA englobent des œdèmes/effusions (ARIA-E, amyloid-related imaging abnormalities-edema/effusions, aussi connus sous le nom d'œdèmes vasogéniques cérébraux), et des hémorragies/dépôts d'hémosidérine (ARIA-H, amyloid-related imaging abnormalities hemorrhage/hemosiderin, incluant les microhémorragies cérébrales ≤10 mm et la sidérose superficielle corticale). Des hémorragies cérébrales d'une taille supérieure à 1 cm ont été observées. Les ARIA-H surviennent généralement conjointement avec les ARIA-E.
Les ARIA peuvent être identifiées par imagerie par résonance magnétique (IRM).
Des événements d'ARIA ont été observés de manière très fréquente dans les études cliniques sur le donanemab. La plupart des événements d'ARIA ont été observés pour la première fois dans les 24 semaines suivant le début du traitement et étaient généralement asymptomatiques. Toutefois, de rares événements graves engageant le pronostic vital, y compris des convulsions et un état de mal épileptique peuvent également survenir. La plupart des événements graves d'ARIA sont survenus au cours des 12 semaines suivant le début du traitement. Voir la rubrique "Effets indésirables" pour des informations sur l'incidence des ARIA. D'autres examens par IRM devraient être réalisés pendant le traitement par donanemab. Un examen par IRM doit être réalisé au début (dans les 3 mois précédant le début du traitement), avant la deuxième utilisation, avant la troisième utilisation, avant la quatrième utilisation et avant la septième utilisation. Chez les patients présentant des facteurs de risque d'ARIA, tels que les porteurs hétérozygotes de l'ApoE ε4 et/ou les patients ayant souffert de précédents événements d'ARIA, une IRM supplémentaire devrait être effectuée après une année de traitement (avant la douzième utilisation) (voir la rubrique "Posologie/Mode d'emploi" ). En cas d'apparition de symptômes d'ARIA, une IRM supplémentaire est indiquée. Les symptômes peuvent inclure céphalées, confusion, nausées, vomissements, incertitude, vertiges, tremblements, troubles visuels, troubles du langage, détérioration de la fonction cognitive, modifications de l'état de conscience et convulsions. Un événement d'ARIA devrait toujours être envisagé comme étiologie possible en présence de ces symptômes neurologiques. Habituellement, les symptômes liés aux ARIA se résolvent avec le temps. Après un premier événement d'ARIA, le taux de rechute lors d'une reprise du traitement est très fréquent: 24.3% pour les ARIA-E et 35.9% pour les ARIA-H (voir la rubrique "Effets indésirables" ).
Les ARIA-E se résolvent typiquement par la suite à l'IRM. Les ARIA-H peuvent persister et se stabiliser.
Pendant toute la durée du traitement, un accès en urgence à l'IRM doit être assuré.
Avant le début du traitement par donanemab, les bénéfices et les risques devraient être soigneusement évalués.
Surveillance des ARIA par IRM
Pour les recommandations sur l'imagerie par IRM, voir la rubrique "Posologie/Mode d'emploi" .
En cas de symptômes évocateurs d'un évènement d'ARIA, un examen clinique et une IRM supplémentaire doivent être effectués.
Recommandations d'interruption du traitement chez les patients présentant un ARIA.
Pour les recommandations d'interruption du traitement chez les patients présentant un ARIA-E et un ARIA-H, voir la rubrique "Posologie/Mode d'emploi" .
En cas d'ARIA-E grave, d'ARIA-H grave, d'hémorragies intracérébrales ≥1 cm ou d'épisode récurrent d'ARIA symptomatiques ou radiologiquement modéré ou sévère, le traitement par donanemab doit être définitivement arrêté.
Sévérité radiologique
La sévérité radiologique des ARIA sous donanemab a été classifiée selon les critères décrits dans la Table 2.
Table 2. Critères pour la classification IRM des ARIA
Type d'ARIA
Léger Modéré Sévère
ARIA-E Hyperintensité FLAIR Hyperintensité FLAIR de Hyperintensité FLAIR >10
limitée à une seule zone 5-10 cm dans sa plus cm associée à un
<5 cm au niveau du grande dimension, ou épaississement gyral et
sulcus et/ou de la sur plus d'une zone, à un effacement sulcal.
substance blanche du mesurant chacune <10 cm Une zone distincte/indépe
cortex/sous cortex ndante ou plus peuvent
être détectées
ARIA-H Microhémorrag ≤4 nouvelles microhémorra 5-9 nouvelles microhémor ≥10 nouvelles microhémorr
ies gies ragies agies
ARIA-H Sidérose 1 nouvelle zone focale 2 zones focales de > 2 zones focales de
superficiellea de sidérose superficielle sidérose superficielle sidérose superficielle
ou extension d'une zone nouvelle ou s'agrandissa nouvelles ou s'agrandissa
existante nt nt
Abréviations: FLAIR = Fluid-attenuated inversion recovery; ARIA-E = amyloid-related imaging abnormalities oedema/effusions, anomalies d'imagerie liées à l'amyloïde avec œdèmes/effusion; ARIA-H = amyloid-related imaging abnormalities haemorrhage/hemosiderin deposition, anomalies d'imagerie liées à l'amyloïde avec hémorragie/dépôts d'hémosidérine
Statut de porteur APOE ε4 et risque d'ARIA
Environ 15% des patients souffrant de maladie d'Alzheimer sont porteurs homozygotes de l'ApoE ε4. Dans un ensemble de données contrôlées par placebo, 16.8% (143/853) des patients dans le bras donanemab étaient porteurs homozygotes de l'apolipoprotéine E ε4 (ApoE ε4), 53% (452/853) étaient porteurs hétérozygotes et 29.9% (255/853) étaient non-porteurs. L'incidence des ARIA chez les porteurs homozygotes de l'ApoE ε4 était plus élevée (55.9% sous donanemab par rapport à 21.9% sous placebo) que chez les porteurs hétérozygotes (37.6% sous donanemab par rapport à 14.1% sous placebo) et que chez les non-porteurs (24.7% sous donanemab par rapport à 12.0% sous placebo). Chez les patients traités par donanemab, des ARIA-E symptomatiques sont survenus chez 8.4% des porteurs homozygotes de l'ApoE ε4, par rapport à 6.6% chez les porteurs hétérozygotes et à 3.9% chez les non-porteurs. Des événements graves d'ARIA ont été observés chez 2.8% des porteurs homozygotes de l'ApoE ε4, chez 1.8% des porteurs hétérozygotes et chez 0.8% des non-porteurs.
Des hémorragies intracrâniennes ont été rapportées dans la population indiquée chez 1,4 % (10/710) des hétérozygotes et non-porteurs d'ApoE ε4 après traitement avec donanemab par rapport à 0,8 % (6/728) des patients atteints de la maladie d'Alzheimer sous placebo. Des hémorragies intracérébrales de >1 cm de diamètre ont été observées chez 0,4 % (3/710) des patients traités avec donanemab et chez 0,3 % (2/728) de ceux sous placebo.
Avant le début du traitement, il est nécessaire de déterminer le statut de porteur ApoE ε4, afin d'établir le risque de développement d'ARIA. Avant le test de génotypisation, les médecins prescripteurs devraient discuter avec les patients du risque d'ARIA pour tous les génotypes et des conséquences du résultat du test génétique.
Une incidence plus élevée d'ARIA a également été observée chez les patients présentant des microhémorragies et/ou une sidérose superficielle avant le traitement.
Hémorragies intracérébrales
Dans une étude contrôlée par placebo (TRAILBLAZER-ALZ 2), des hémorragies intracérébrales de plus de 1 cm ont été observées chez 0.4% (3/853) des patients sous donanemab, par rapport à 0.2% (2/874) des patients sous placebo.
Des événements d'hémorragies cérébrales avec issue fatale ont été observés chez des patients traités par donanemab.
Chez des patients qui, dans l'étude TRAILBLAZER-ALZ-6, ont reçu pendant 24 semaines du donanemab à un dosage de 350/700/1050 mg puis 1400 mg toutes les 4 semaines, des hémorragies intracérébrales de plus de 1 cm ont été rapportées chez 0.9% (2/212) des patients.
Autres facteurs de risque d'hémorragies intracérébrales
Les patients étaient exclus de l'étude TRAILBLAZER-ALZ 2 si des résultats de neuro-imagerie suggéraient un risque accru d'hémorragies intracérébrales. Au nombre de ces résultats figuraient des résultats suggérant une angiopathie amyloïde cérébrale (hémorragie intracérébrale antérieure avec un diamètre de plus de 1 cm, plus de 4 microhémorragies, plus d'une zone avec une sidérose superficielle, un œdème vasogénique et une maladie sévère de la substance blanche) (voir la rubrique "Contre-indications" ). Ces lésions et d'autres (anévrisme, malformation vasculaire) peuvent potentiellement augmenter le risque d'hémorragie intracérébrale.
La présence d'un allèle ApoE ε4 est également associée à une angiopathie amyloïde cérébrale, pour laquelle il existe un risque accru d'hémorragies intracérébrales.
La prudence est de mise lorsque l'on envisage d'utiliser le donanemab chez des patients présentant des facteurs suggérant un risque accru d'hémorragies intracérébrales, et en particulier chez les patients sous traitement antithrombotique ou chez les patients avec des résultats IRM évoquant une angiopathie cérébrale amyloïde.
Traitement antithrombotique concomitant
Les patients ayant reçu du donanemab et un antithrombotique (acide acétylsalicylique, autres antiplaquettaires ou anticoagulants) n'ont pas montré d'augmentation de l'incidence des ARIA. La majorité des expositions aux antithrombotiques concernaient l'acide acétylsalicylique (80%). Le nombre des événements et l'exposition limitée aux antithrombotiques autres que l'acide acétylsalicylique limitent la possibilité de tirer des conclusions définitives sur le risque d'ARIA ou d'hémorragies intracérébrales chez les patients sous traitement antithrombotique. Des ARIA-H et des hémorragies intracérébrales de plus de 1 cm ayant été observées chez des patients sous donanemab, une précaution supplémentaire est de mise lorsqu'on envisage une administration concomitante d'antithrombotiques chez les patients qui sont déjà sous donanemab. L'utilisation concomitante d'acide acétylsalicylique et d'autres antiplaquettaires est admissible.
L'ARIA pouvant causer des déficits neurologiques focaux ressemblant à ceux d'un accident vasculaire cérébral ischémique, les médecins traitants devraient vérifier si de tels symptômes pourraient être attribués à un événement d'ARIA. Une lyse systémique avec un thrombolytique ainsi qu'un traitement par anticoagulants ne doivent pas être effectués pendant un traitement par donanemab (voir la rubrique "Contre-indications" ).
Rapport bénéfice-risque individuel basé sur la pathologie Tau
Le rapport bénéfice-risque peut dépendre de la concentration initiale de Tau. Une efficacité numériquement supérieure a été observée chez les patients avec une valeur faible à modérée de Tau, par rapport à ceux qui avaient une valeur élevée de Tau (voir la rubrique "Efficacité clinique" ). L'efficacité clinique chez les patients sans Tau ou avec une concentration très faible de Tau n'a pas été étudiée. Le résultat d'un test de la pathologie Tau devrait, s'il a été obtenu, être pris en compte lors de la discussion individuelle avec le patient sur les avantages et les risques.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité, y compris une anaphylaxie et un angio-œdème, ont été observées lors de l'utilisation du donanemab (voir la rubrique "Effets indésirables" ). Les signes et symptômes de ces réactions sont érythème, frissons, nausées, vomissements, sueurs, céphalées, sensation d'oppression dans la poitrine, dyspnée et modifications de la tension artérielle. Ces réactions peuvent être sévères voire engager le pronostic vital et elles surviennent habituellement pendant la perfusion ou dans les 30 minutes qui la suivent. Dès l'observation de signes ou symptômes compatibles avec une réaction d'hypersensibilité, la perfusion doit être immédiatement arrêtée et un traitement approprié doit être instauré. Chez les patients avec des antécédents d'hypersensibilité au donanemab ou à l'un des excipients, le donanemab est contre-indiqué. En cas de réaction liée à la perfusion non grave, il convient de réduire la vitesse de perfusion ou d'arrêter la perfusion et de mettre en place un traitement approprié correspondant à l'indication clinique. Une prémédication par antihistaminiques, paracétamol ou corticostéroïdes avant l'utilisation suivante peut être envisagée.
Réactions liées à la perfusion
Lors de l'administration de donanemab, des réactions liées à la perfusion (IRR) et une anaphylaxie ont été observées (voir la rubrique "Effets indésirables" ). Ces réactions peuvent être sévères voire engager le pronostic vital et elles surviennent habituellement pendant la perfusion ou dans les 30 minutes qui la suivent. Les signes et symptômes de ces réactions sont érythème, frissons, nausées, vomissements, sueurs, céphalées, sensation d'oppression dans la poitrine, dyspnée et modifications de la tension artérielle. Pour des informations sur la fréquence des réactions liées à la perfusion, voir la rubrique "Effets indésirables" .
En cas de réactions graves liées à la perfusion ou d'anaphylaxie, il convient d'arrêter immédiatement l'administration de donanemab et d'instaurer un traitement approprié. Après une réaction liée à la perfusion de degré 3 ou supérieur sans amélioration ou résolution après traitement, le traitement par donanemab doit être arrêté définitivement.
En cas d'une réaction non grave liée à la perfusion, le débit de la perfusion peut être réduit ou la perfusion interrompue et, en cas d'indication clinique, un traitement approprié instauré.
En présence de signes de lésion tissulaire dans le cadre d'une réaction d'hypersensibilité (par ex. arthrite, glomérulonéphrite ou mononeuropathie multiple) le traitement par donanemab doit être arrêté définitivement.
Immunogénicité
Dans les études cliniques contrôlées par placebo, 88.1% des patients traités par donanemab ont développé des anticorps dirigés contre le donanemab (anti-drug antibodies, ADA). Chez tous ces patients, il s'agissait d'anticorps neutralisants. Tous les patients présentant des IRR avaient des ADA. Un titre d'ADA plus élevé était associé à une incidence accrue d'IRR et d'hypersensibilité liée à la perfusion.
Patients exclus du programme d'études cliniques
Les patients souffrant d'un syndrome de Down peuvent présenter un taux plus élevé de CAA (angiopathie amyloïde cérébrale) et d'ARIA. Dans les études cliniques menées avec le donanemab, ces patients n'ont pas été étudiés. La sécurité et l'efficacité du donanemab chez ces patients ne sont pas connues.
Sodium
Ce médicament contient 11.5 mg de sodium par flacon de 350 mg de donanemab (20 ml). Une dose de 1400 mg de donanemab (4 flacons) contient 46 mg de sodium, ce qui correspond à 2% de l'apport alimentaire quotidien maximal de sodium recommandé par l'OMS de 2 g de sodium par pour un adulte.
InteractionsAucune étude formelle pour identifier les interactions médicamenteuses n'a été réalisée pour le donanemab. Sur la base des caractéristiques du donanemab, on ne s'attend pas à des interactions pharmacocinétiques.
L'utilisation concomitante de donanemab et d'immunoglobulines intraveineuses, de plasmaphérèse ou d'immunoadsorption peut entraîner une diminution de l'efficacité du donanemab. On fixera l'intervalle entre ces utilisations de façon à minimiser l'élimination du donanemab. Après l'administration de donanemab, on attendra au moins neuf semaines avant d'appliquer l'une de ces thérapies. Après un traitement antérieur avec des immunoglobulines intraveineuses, on respectera un intervalle de temps de trois semaines avant d'administrer le donanemab. Chez les patients qui nécessitent régulièrement l'une de ces thérapies, cette interaction sera prise en compte à l'avance dans les décisions thérapeutiques et il sera décidé en fonction de la nécessité médicale (voir la rubrique "Posologie/Mode d'emploi" ).
Grossesse, allaitementFemmes en âge de procréer
Le statut de grossesse devrait être vérifié chez les femmes en âge de procréer avant l'instauration d'un traitement par donanemab. Les femmes en âge de procréer doivent utiliser une méthode contraceptive efficace pendant le traitement et jusqu'à deux mois après le traitement.
Grossesse
On ne dispose pas de données sur l'utilisation du donanemab chez la femme enceinte. Aucune étude de développement embryofœtal n'a été menée chez l'animal avec le donanemab. Il est établi que l'IgG humaine traverse le placenta après le premier trimestre de la grossesse. Par conséquent, le donanemab peut potentiellement passer de la mère au fœtus en développement. L'utilisation du donanemab pendant la grossesse n'est pas recommandée.
Allaitement
Aucune étude sur la lactation n'a été menée avec le donanemab chez l'animal. Il est établi que l'immunoglobuline G humaine (IgG) peut passer dans le lait maternel. Par conséquent, le donanemab peut être transmis de la mère à l'enfant qu'elle allaite. Les risques pour un enfant allaité ne sont pas connus. Les femmes qui allaitent ne doivent recevoir le donanemab que si le bénéfice possible l'emporte sur le risque pour la mère et l'enfant.
Fertilité
On ne dispose pas de données sur les effets du donanemab sur la fertilité humaine. Il n'a pas été mené d'études sur le donanemab chez l'animal afin d'examiner de possibles limitations de la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesIl n'a pas été mené d'études pour évaluer les effets du donanemab sur l'aptitude à la conduite et à l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Dans une étude contrôlée par placebo (TRAILBLAZER-ALZ 2, phase 3) menée chez des patients présentant un trouble cognitif léger ou une démence légère dus à la maladie d'Alzheimer( Alzheimer's Disease, AD)), 853 adultes ont reçu au moins une dose de donanemab. Parmi ces patients, 710 faisaient partie de la population indiquée (porteurs hétérozygotes ou non-porteurs de l'ApoE ε4): 29.9% (255/853) de non-porteurs, 53.0% (452/853) d'hétérozygotes et 0.4% (3/853) de génotype inconnu.
Les effets indésirables rapportés le plus fréquemment dans la population indiquée ont été ARIA-E (20.6%), ARIA-H (27.6%), céphalées (14.6%) et réactions liées à la perfusion (8.3%). Les effets indésirables graves les plus importants étaient les ARIA-E graves (1.3%), les ARIA-H graves (0.3%) et les réactions d'hypersensibilité graves, y compris des réactions liées à la perfusion (0.4%). Une anaphylaxie a été rapportée occasionnellement (0.4%) (voir "Mises en garde et précautions" ).
Liste des effets secondaires dans la population totale
Les effets secondaires relevés dans des études cliniques sont présentés par classes de systèmes d'organes selon la classification MedDRA. Au sein de chaque classe de systèmes d'organes, les effets secondaires sont présentés par ordre de fréquence, les plus fréquents en premier lieu. Pour chaque groupe de fréquence, les effets secondaires sont classés par ordre décroissant de gravité. Pour chaque effet secondaire, la catégorie de fréquence se base sur la définition suivante: très fréquent (≥1/10); fréquent (≥1/100, <1/10); occasionnel (≥1/1000, <1/100); rare (≥1/10 000, <1/1000); très rare (<1/10 000).
Affections du système nerveux
Très fréquents: ARIA-Ea,b (24.0%), ARIA-Ha,b,c (31.4%), céphalées (14.0%)
a Évalués par IRM.
b Symptômes associés possibles: céphalées, confusion, nausées, vomissements, incertitude
, vertiges, tremblements, troubles visuels, troubles du language, détérioration de la fonction cognitive, modification de l'état de conscience, convulsions
c Inclut les microhémorragies et les sidéroses superficielles
Occasionnels: hémorragie intracérébrale (> 1cm)d
d Hémorrhagie cérébrale et accident vasculaire cérébral hémorragique
Affections gastro-intestinales
Fréquents: nausées/vomissements
Affections du système immunitaire
Fréquents: hypersensibilité
Occasionnels: réaction anaphylactique
Troubles généraux et anomalies au site d'administration
Fréquents: réactions liées à la perfusione, hypersensibilité
Occasionnels: réaction anaphylactique
e Inclut érythème, frissons, nausées, vomissements, sueurs, céphalées, sensation d'oppression dans la poitrine, dyspnée et modifications de la tension artérielle.
Description d'effets indésirables sélectionnés
Anomalies d'imagerie liées à l'amyloïde (ARIA) et hémorragie intracérébrale dans la population de patients indiquée
TRAILBLAZER-ALZ-2
Des ARIA (ARIA-E ou ARIA-H) ont été observées chez 33 % (234/710) des patients (hétérozygotes ou non-porteurs) sous donanemab, par rapport à 13.5 % (98/728) des patients sous placebo dans l'étude pivot contrôlée par placebo (schéma posologique de 700 mg de donanemab toutes les 4 semaines pour les 3 premières administrations, puis 1400 mg de donanemab toutes les 4 semaines). Des ARIA symptomatiques sont survenus chez 6.1 % des patients sous donanemab. Des ARIA cliniquement graves ont été rapportées chez 1.4% (10/710) des patients sous donanemab. Trois participants (0.4%) ont eu des ARIA graves et sont décédés.
Un événement d'ARIA-E (amyloid-related imaging abnormalities-edema/effusions, aussi connu sous le nom d'œdèmes vasogéniques cérébraux) a été observé chez 20.6 % des patients sous donanemab, par rapport à 1.8 % des patients sous placebo. La sévérité radiologique maximale des ARIA-E était léger chez 6.2%, modéré chez 12.7% et sévère chez 1.4% des patients. La durée médiane jusqu'à la régression radiologique des ARIA-E était d'environ 8.3 semaines. Des ARIA-E symptomatiques ont été rapportés chez 5.6% des patients sous donanemab dans les études contrôlées avec placebo.
Les symptômes cliniques d'ARIA-E ont régressé chez environ 80% des patients. La durée médiane jusqu'à la régression des symptômes cliniques des ARIA-E était d'environ 3.9 semaines.
Chez les porteurs hétérozygotes, des ARIA-E ont été rapportés chez 23.2% (105/452) des patients sous donanemab, par rapport à 2.1% (10/474) des patients sous placebo. La sévérité radiologique maximale des ARIA-E sous donanemab chez les porteurs hétérozygotes était léger chez 6.6%, modéré chez 14.2% et sévère chez 2.0% des patients. Des ARIA-E symptomatiques ont été rapportés chez 6.6% des porteurs hétérozygotes sous donanemab. Chez les nonporteurs, des ARIA-E ont été rapportées chez 15.7% (40/255) des patients sous donanemab, contre 0,8% (2/250) des patients sous placebo. La sévérité radiologique maximale des ARIA-E sous donanemab chez les nonporteurs était léger chez 5.1%, modéré chez 10.2% et sévère chez 0.4% des patients. Des ARIA-E symptomatiques ont été rapportés chez 3.9% des nonporteurs sous donanemab.
Des ARIA-H (amyloid-related imaging abnormalities hemorrhage/hemosiderin, incluant les microhémorragies cérébrales et la sidérose superficielle corticale) peuvent survenir spontanément et indépendamment du traitement chez les patients souffrant de maladie d'Alzheimer. Des ARIA-H ont été rapportés chez 27.6% des patients sous donanemab, par rapport à 12.2% des patients sous placebo. La sévérité radiologique maximale des ARIA-H était légère chez 14.4%, modérée chez 5.5% et sévère chez 7.6% des patients. Des ARIA-H symptomatiques ont été rapportées chez 1.1% des patients sous donanemab, par rapport à 0.3% des patients sous placebo. Des ARIA-H isolés (c.-à-d. des ARIA-H sans ARIA-E concomitants) ont été rapportés chez 12.4% des patients sous donanemab, par rapport à 11.5 % des patients sous placebo.
Chez les porteurs hétérozygotes, des ARIA-H ont été rapportés chez 32.5% (147/452) des patients sous donanemab, par rapport à 12.9% (61/474) des patients sous placebo. La sévérité radiologique maximale des ARIA-H chez les porteurs hétérozygotes était légère chez 15.0%, modérée chez 7.7% et sévère chez 9.5 % des patients. Des ARIA-H symptomatiques ont été rapportés chez 1.5% des porteurs hétérozygotes sous donanemab, par rapport à 0.2% sous placebo. Des ARIA-H isolés ont été rapportés chez 14.2% des porteurs hétérozygotes sous donanemab, contre 11.2% sous placebo. Chez les nonporteurs, des ARIA-H ont été rapportés chez 18.8% (48/255) des patients sous donanemab, contre 11.2% (28/250) des patients sous placebo. La sévérité radiologique maximale des ARIA-H chez les nonporteurs était légère chez 12.9%, modérée chez 1.6% et sévère chez 4.3% des patients. Des ARIA-H symptomatiques ont été rapportés chez 0.4% des nonporteurs sous donanemab, par rapport à 0.4% sous placebo. Des ARIA-H isolés ont été rapportés chez 9.0% des nonporteurs sous donanemab, contre 11.2% sous placebo.
Dans les études contrôlées par placebo, la majorité des premiers événements radiologiques d'ARIA sont apparus tôt durant le traitement (dans les 24 semaines après le début du traitement), bien que des ARIA puissent survenir à tout moment pendant le traitement et que des patients puissent présenter plus d'un épisode.
Des hémorragies intracérébrales de plus de 1 cm ont été observées chez 0.4% (3/710) des patients sous donanemab et chez 0.3% (2/728) des patients sous placebo. Chez un participant sous donanemab dans l'étude pivot, qui avait une sidérose superficielle déjà présente au début de l'étude, un événement d'ARIA-H avec hémorragie intracérébrale associée a eu une issue fatale.
TRAILBLAZER-ALZ-6
A la semaine 76 de TRAILBLAZER-ALZ-6, des ARIA (ARIA-E ou ARIA-H) ont été rapportés chez 28.8% des patients (hétérozygotes ou non-porteurs) ayant reçu une posologie de 350/700/1050 mg puis de 1400 mg de donanemab toutes les 4 semaines (n=191). Des événements d'ARIA cliniquement graves ont été rapportés chez 0.5% des patients sous donanemab.
Des ARIA-E ont été rapportés chez 14.7% des patients sous donanemab. La sévérité radiologique maximale des ARIA-E a été léger chez 5.8%, modéré chez 8.9% et sévère chez 0% des patients. La durée médiane jusqu'à la résolution des ARIA-E a été d'environ 8.4 semaines. Des ARIA-E symptomatiques sont survenus chez 3.1 % des patients sous donanemab. Les symptômes cliniques dus aux ARIA-E se sont résolus chez 83.3% des patients, avec un temps médian de 1.6 semaine jusqu'à la résolution des symptômes.
Des ARIA-H ont été observés chez 25.1% des patients sous donanemab. La sévérité radiologique maximale des ARIA-H était léger chez 18.3%, modéré chez 3.1% et sévère chez 3.7% des patients. Des ARIA-H symptomatiques ont été rapportées chez 0.5% des patients sous donanemab.
Chez les patients porteurs hétérozygotes et non-porteurs de l' ApoE-ε4, une hémorragie intracérébrale après l'utilisation du donanemab a été rapportée chez 1% (2/191) des patients. Parmi ceux-ci, 0.5% des patients (1/191) sous donanemab a présenté une hémorragie intracérébrale plus grande que 1 cm. Ce participant avec un ARIA-E avait été traité par thrombolytique suite à des symptômes ressemblant à ceux d'un accident vasculaire cérébral et a développé une hémorragie intracérébrale mortelle.
Réactions liées à la perfusion
Dans TRAILBLAZER-ALZ-2, des réactions liées à la perfusion ont été observées chez 8.7% des patients sous donanemab, par rapport à 0.5% des patients sous placebo. Une anaphylaxie a été rapportée occasionnellement (0.4%). De réactions liées à la perfusion graves ou une hypersensibilité sont apparues chez 0.4% des patients sous donanemab, par rapport à 0.1% des patients sous placebo. La majorité des réactions liées à la perfusion et des réactions d'hypersensibilité sont survenues le plus souvent pendant les 4 premières utilisations, bien que de telles réactions soient possibles à tout moment.
Sous donanemab, il y a eu des arrêts de traitement en raison d'IRR (3.6%), d'hypersensibilité (0.5%) et d'anaphylaxie (0.4%), alors qu'il n'y en a pas eu sous placebo.
Une nouvelle exposition a provoqué une IRR/hypersensibilité subséquente chez 47.5% des patients, avec unesévérité et une nature des symptômes généralement comparables à ceux du premier événement.
Les médications prophylactiques administrées avant le traitement suivant n'ont pas eu d'effet sur les IRR.
Immunogénicité
Dans les études cliniques contrôlées par placebo, 88.1% des patients sous donanemab ont développé des anticorps dirigés contre le médicament (anti-drug antibodies, ADA) et tous les patients avec des ADA avaient des anticorps neutralisants. Bien que l'exposition au donanemab diminue avec l'augmentation du titre d'ADA, le développement d'ADA n'était pas associé à une perte d'efficacité clinique du donanemab. Tous les patients qui ont rapporté des réactions liées à la perfusion avaient des ADA.
Un titre plus élevé d'ADA était associé à une incidence plus élevée de réactions liées à la perfusion/de réactions immédiates d'hypersensibilité.
L'annonce de suspicion d'effets secondaires après l'enregistrement est d'une grande importance. Elle permet une surveillance continue du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes dosages uniques allant jusqu'à 40 mg/kg (environ 2800 mg chez une personne de 70 kg) ont été utilisés. Chez 2 patients sur 4, des ARIA-E sont survenus avec cette dose, qui se sont résolus par la suite. En cas de surdosage, un traitement de soutien doit être instauré.
Propriétés/EffetsCode ATC
N06DX05
Groupe pharmaco-thérapeutique: système nerveux, psychoanaleptiques, médicaments anti-démence, autres médicaments anti-démence.
Le Donanemab est un anticorps monoclonal humanisé recombinant produit dans des cellules ovariennes de hamster chinois (Chinese Hamster Ovary, CHO).
Mécanisme d'action
Le donanemab est un anticorps monoclonal d'immunoglobuline 1 (IgG1) dirigé contre la forme tronquée N terminale insoluble modifiée avec du pyroglutamate de la protéine bêta-amyloïde (N3pG Aβ), qui n'est présente que dans les plaques amyloïdes du cerveau. Le donanemab se lie à N3pG Aβ et aide à la clairance des plaques par phagocytose médiée par les microglies.
Pharmacodynamique
Chez les patients traités par donanemab, une réduction des plaques amyloïdes cérébrales a été observée par tomographie par émission de positrons (PET) à l'amyloïde. Le donanemab a réduit la physiopathologie Tau mesurée par la P-Tau217 dans le plasma.
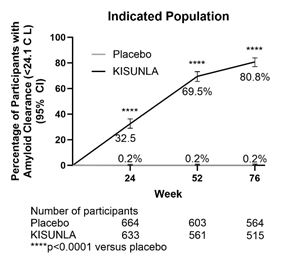
Le pourcentage de patients traités par donanemab ayant atteint une clairance amyloïde (amyloid clearance, c'est-à-dire inférieure à 24.1 centiloïdes) dans l'étude TRAILBLAZER-ALZ 2 était de 32.5% à la semaine 24, de 69.5% à la semaine 52 et de 80,8% à la semaine 76 dans la population indiquée.
Dans l'étude TRAILBLAZER-ALZ 2, la différence entre le donanemab et le placebo concernant la modification de la concentration d'amyloïde entre la valeur initiale et la semaine 76 était statistiquement significative dans la population indiquée (-89.24 centiloïdes).
Dans l'étude TRAILBLAZER-ALZ 6, une réduction similaire des plaques amyloïdes a été observée à la semaine 24 pour la posologie de 350/700/1050 mg, puis de 1400 mg toutes les 4 semaines, par rapport à celle observée avec la posologie de 700 mg pour les trois premières perfusions, puis de 1400 mg toutes les 4 semainesétudiée dans l'étude pivot.
L'exposition au donanemab a diminué avec l'augmentation du titre d'ADA. Une réduction de bêta-amyloïde a été constatée indépendamment du titre d'ADA. Une association entre la présence d'ADA et les résultats sur l'iADRS et le CDR-SB n'a pas été obeservée (voir la rubrique "Mises en garde et précautions" et la rubrique "Effets indésirables" ).
Efficacité clinique
La sécurité et l'efficacité du donanemab ont été évaluées dans le cadre d'une étude de phase 3 (TRAILBLAZER-ALZ 2). L'étude a été menée en double aveugle, contrôlée par placebo et en groupes parallèles. Elle a été réalisée auprès de patients âgés de 60 à 85 ans atteints de maladie d'Alzheimer symptomatique débutante (trouble cognitif léger, Mild Cognitive Impairment, MCI) ou de démence légère due à une maladie d'Alzheimer et ayant un score MMSE de 20 à 28 inclus. Les patients devaient présenter des signes de pathologie bêta-amyloïde confirmés par PET à l'amyloïde. Les patients présentaient en outre des signes de dépôt pathologique de Tau sur le PET au flortaucipir.
À l'appui des résultats de cette étude, il y a des données d'une étude de phase 2 (TRAILBLAZER-ALZ) de conception comparable. Les posologies utilisées dans cette étude se distinguent de la posologie enregistrée (voir la rubrique "Pharmacocinétique" ).
Pour évaluer la sécurité, les patients ont été observés pendant une période allant jusqu'à 76 semaines ou jusqu'à la fin du traitement plus 57 jours après la dernière dose.
TRAILBLAZER-ALZ 2 a été conçue à l'origine comme une étude de phase 2 et elle a été modifiée plus tard en étude de phase 3. Les principales modifications ont été: ajout d'une phase de titration avec 700 mg pour les trois premières utilisations; adaptation de l'étude de phase 2 en une étude de phase 3, notamment avec une augmentation de la taille de l'échantillon et une modification de l'analyse principale de "CDR-SB dans la population totale ou dans la population avec Tau intermédiaire" en "iADRS dans la population avec Tau intermédiaire" ; ajout d'une phase d'extension à long terme, pour évaluer l'efficacité et la sécurité du donanemab au cours du temps.
Étude de phase 3 TRAILBLAZER-ALZ 2
Dans cette étude, 1736 patients ont été randomisés selon un rapport de 1:1 pour recevoir une perfusion intraveineuse de 700 mg de donanemab toutes les 4 semaines pour les 3 premières utilisations, puis 1400 mg toutes les 4 semaines (N=860) ou de placebo (N=876), pour une durée totale allant jusqu'à 72 semaines. L'étude incluait une phase d'extension en double aveugle sur 78 semaines. L'utilisation a été poursuivie jusqu'à la fin de l'étude ou jusqu'à la dissolution (clairance) des plaques amyloïdes, définie comme une concentration en plaques inférieure à 25 centiloïdes lors de deux PET à l'amyloïde consécutifs ou lorsqu'un seul PET mesurait une concentration en plaques inférieure à 11 centiloïdes.
De plus, la suspension du traitement était autorisée en cas d'ARIA du au traitement. Si les patients recevaient déjà un traitement symptomatique (inhibiteurs de l'acétylcholinestérase et/ou inhibiteurs du N-méthyle-D-aspartate, mémantine) lors de l'inclusion dans l'étude, ces traitements pouvaient être poursuivis. Les traitements symptomatiques pouvaient être complétés ou modifiés durant l'étude, à la discrétion du médecin investigateur. L'étude a exclu les patients présentant un ARIA-E préexistant avec plus de 4 microhémorragies, plus d'une zone de sidérose superficielle, toute hémorragie intracérébrale >1 cm ou une maladie sévère de la substance blanche. À l'inclusion, l'âge moyen était de 73 ans, avec une fourchette de 59 à 86 ans, pour un poids moyen (SD) initial de 71.7 kg (15.7), un changement graduel et progressif des fonctions mnésiques pendant au moins 6 mois et un score MMSE (Mini-Mental State Examination) de 22.29 (3.88). 57.4% des participants étaient des femmes, 91.5% des Caucasiens, 5.7% étaient d'origine ethnique hispanique ou latino-américaine, 6.0% d'origine asiatique et 2.3% étaient Afro-américains. 80.0% des patients ont été inclus en Amérique du Nord, 13.9% en Europe, 5.1% au Japon et 1.0% en Australie. 29% étaient non-porteurs de l'ApoE ε4, 54% hétérozygotes et 17% homozygotes.
55.6% des patients étaient sous AChEI, 20.3% sous mémantine. 61% des patients recevaient soit un AChEI soit de la mémantine.
La moyenne (mean, SD) de l'amyloïde en à l'inclusion était de 102.5 (34.5) centiloïdes.
Deux populations d'analyse principale ont été définies lors du screening par Tau-PET au flortaucipir: 1) Une population avec des concentrations faibles à modérées de Tau (68.2% du total), et 2) la population combinée avec soit une concentration faible à modérée, soit une concentration élevée de Tau (31.8% du total).
Dans l'ensemble, 24.7% des patients ont arrêté prématurément le traitement (29.3% sous donanemab, 20.1% sous placebo).
Le critère d'efficacité principal était le changement de la cognition et de la fonction mesuré avec le score iADRS (integrated Alzheimer's Disease Rating Scale) entre l'inclusion et la semaine 76. L'iADRS est un examen combiné de la cognition et des fonctions du quotidien incluant des items du sous-score de l'Alzheimer's Disease Assessment Scale (ADAS-Cog13, nombre de points 0-85) et de l'Alzheimer's Disease Cooperative Study - instrumental Activities of Daily Living (ADCS-iADL, nombre de points 0-59), qui mesurent des domaines-clés de l'évolution de la maladie d'Alzheimer. Le score total varie de 0 à 144, les scores les plus bas indiquant des performances cognitives et fonctionnelles plus faibles. Les autres critères d'évaluation de l'efficacité comprenaient le CDR-SB (Clinical Dementia Rating Scale - Sum of Boxes), l'ADAS-Cog13 et l'ADCS-iADL.
La Table 3 ci-dessous présente les résultats importants de l'étude pour la population indiquée.
Dans la population indiquée, 717 participants ont été randomisés pour recevoir du donanemab, 414 étaient de sexe féminin et 303 de sexe masculin, 70 avaient <65 ans, 320 avaient de 65 à 74 ans et 327 avaient 75 ans ou plus.
Dans la population indiquée, 730 participants ont été randomisés pour recevoir du placebo, 426 étaient de sexe féminin et 304 de sexe masculin, 66 avaient <65 ans, 311 avaient de 65 à 74 ans et 353 avaient 75 ans ou plus.
Table 3: Résultats de l'analyse d'efficacité à la semaine 76 de l'étude TRAILBLAZER-ALZ 2 sur le donanemab dans la population indiquée (hétérozygotes et non-porteurs de l'ApoE ε4)
Critère clinique Porteurs hétérozygotes del'ApoE ε4 et
non-porteurs de l'ApoE ε4
Dona Placebo
iADRS (NCS)
Moyenne à l'inclusion (SD) 104.66 (14.12) 103.83 (14.03)
Changement (LS mean) par rapport à -10.21 (0.57) -13.59 (0.55)
l'inclusion
Différence par rapport au placebo 3,38 [1,83, 4,92]
[IC à 95 %]
Femmes
Moyenne à l'inclusion (SD) 104.92 (14.02) 103.75 (13.38)
Changement (LS mean) par rapport à -10.98 (0.76) -14.77 (0.72)
l'inclusion
Différence par rapport au placebo 3,80 [1,77, 5,83]
[IC à 95 %]
Hommes
Moyenne à l'inclusion (SD) 104.32 (14.26) 103.95 (14.87)
Changement (LS mean) par rapport à -9.18 (0.87) -11.92 (0.85)
l'inclusion
Différence par rapport au placebo 2,73 [0,36, 5,10]
[IC à 95 %]
<65 ans
Moyenne à l'inclusion (SD) 105.19 (15.46) 107.14 (14.19)
Changement (LS mean) par rapport à -12.74 (1.77) -14.21 (1.86)
l'inclusion
Différence par rapport au placebo 1,47 [-3,51, 6,45]
[IC à 95 %]
65 à 74 ans
Moyenne à l'inclusion (SD) 106.05 (12.88) 105.09 (13.59)
Changement (LS mean) par rapport à -9.33 (0.86) -13.69 (0.83)
l'inclusion
Différence par rapport au placebo 4,35 [2,03, 6,68]
[IC à 95 %]
>75 ans
Moyenne à l'inclusion (DS) 103.20 (14.83) 102.11 (14.19)
Changement (LS mean) par rapport à -10.48 (0.86) -13.37 (0.80)
l'inclusion
Différence par rapport au placebo 2,89 [0,61, 5,17]
[IC à 95 %]
CDR-SB (MMRM)
Moyenne à l'inclusion (SD) 3.96 (2.10) 3.94 (2.04)
Changement (LS mean) par rapport à 1.67 (0.11) 2.43 (0.10)
l'inclusion
Différence par rapport au placebo -0,77 [-1,04, -0,49]
[IC à 95 %]
Femmes
Moyenne à l'inclusion (DS) 4.03 (2.11) 3.97 (1.98)
Changement (LS mean) par rapport à 1.66 (0.12) 2.49 (0.15)
l'inclusion
Différence par rapport au placebo -0,83 [-1,20, -0,45]
[IC à 95 %]
Hommes
Moyenne à l'inclusion (SD) 3.87 (2.08) 3.89 (2.13)
Changement (LS mean) par rapport à 1.48 (0.15) 2.15 (0.14)
l'inclusion
Différence par rapport au placebo -0,66 [-1,07, -0,25]
[IC à 95 %]
<65 ans
Moyenne à l'inclusion (DS) 3.81 (2.05) 3.80 (1.54)
Changement (LS mean) par rapport à 2.53 (0.40) 2.81 (0.31)
l'inclusion
Différence par rapport au placebo -0,28 [-1,19, 0,63]
[IC à 95 %]
65 à 74 ans
Moyenne à l'inclusion (SD) 3.78 (1.88) 3.66 (1.92)
Changement (LS mean) par rapport à 1.46 (0.14) 2.55 (0.16)
l'inclusion
Différence par rapport au placebo -1,09 [-1,50, -0,68]
[IC à 95 %]
>75 ans
Moyenne à l'inclusion (DS) 4.17 (2.30) 4.21 (2.19)
Changement (LS mean) par rapport à 1.48 (0.15) 2.04 (0.16)
l'inclusion
Différence par rapport au placebo -0,56 [-0,96, -0,16]
[IC à 95 %]
ADAS-Cog13 (NCS)
Moyenne à l'inclusion (SD) 28.43 (8.91) 29.00 (8.93)
Changement (LS mean) par rapport à 5.37 (0.31) 7.06 (0.29)
l'inclusion
Différence par rapport au placebo -1,69 [-2,52, -0,86]
[IC à 95 %]
Femmes
Moyenne à l'inclusion (SD) 28.40 (9.15) 29.45 (8.61)
Changement (LS mean) par rapport à 5.59 (0.40) 7.20 (0.39)
l'inclusion
Différence par rapport au placebo -1,61 [-2,71, -0,52]
[IC à 95 %]
Hommes
Moyenne à l'inclusion (SD) 28.46 (8.60) 28.40 (9.32)
Changement (LS mean) par rapport à 5.09 (0.47) 6.88 (0.45)
l'inclusion
Différence par rapport au placebo -1,79 [-3,07, -0,52]
[IC à 95 %]
<65 ans
Moyenne à l'inclusion (SD) 27.82 (9.67) 27.11 (9.40)
Changement (LS mean) par rapport à 8.34 (0.94) 7.90 (0.96)
l'inclusion
Différence par rapport au placebo 0,44 [-2,19, 3,06]
[IC à 95 %]
65 à 74 ans
Moyenne à l'inclusion (SD) 27.82 (8.80) 28.74 (9.39)
Changement (LS mean) par rapport à 4.86 (0.45) 7.51 (0.44)
l'inclusion
Différence par rapport au placebo -2,66 [-3,89, -1,42]
[IC à 95 %]
> 75 ans
Moyenne à l'inclusion (SD) 29.16 (8.83) 29.59 (8.36)
Changement (LS mean) par rapport à 5.21 (0.46) 6.47 (0.43)
l'inclusion
Différence par rapport au placebo -1,26 [-2,49, -0,04]
[IC à 95 %]
Abréviations: ApoE-ε4 = allèle de sous-type 4 du gène qui code pour l'apolipoprotéine de classe E; CDR-SB = Clinical Dementia Rating Scale – Sum of boxes; IC = intervalle de confiance; Dona = donanemab; iADRS = integrated Alzheimers Disease Rating Scale; LSM = Least-Square mean, moyenne selon la méthode des moindres carrés; MMRM = Mixed Model for Repeated Measures, modèle mixte pour mesures répétées; SD = standard deviation.
Biomarqueurs
Le pourcentage de patients sous donanemab présentant une clairance amyloïde (c.-à-d. avec un PET à l'amyloïde au-dessous de 24.1 centiloïdes ou avec une lecture visuelle négative) dans l'étude TRAILBLAZER-ALZ 2 est présenté dans la Figure 2.
Sous donanemab, une réduction de la concentration plasmatique de P-tau217 (Log 10) a été observée par rapport au placebo. Dans la population avec une concentration faible à modérée de Tau (498 patients sous donanemab vs. 494 patients sous placebo), la variation moyenne (LS mean ± SE) était de -0.19 ± 0.012 et de -0.26 ± 0.015 aux semaines 24 et 76 par rapport au placebo (p<0.0001 pour les deux points temporels). En accord avec ces données, la population combinée présentait une variation moyenne (Log LS mean ± SE) de -0.17± 0.011 et -0.23± 0.013 aux semaines 24 et 76 par rapport au placebo (p<0.0001 pour les deux points temporels).
Figure 2. Pourcentage de patients dans la population indiquée sous donanemab ayant atteint une clairance des plaques amyloïdes mesurée par PET à l'amyloïde sur une période de 76 semaines dans l'étude TRAILBLAZER-ALZ 2.
Population avec une concentration élevée de Tau dans la population indiquée
Dans la population avec une concentration élevée de Tau (218 patients sous donanemab et 235 patients sous placebo), le donanemab a ralenti de 8% (1.55 ± 1.66 [p=0.351) le déclin clinique mesuré avec l'iADRS et de 18% (-0.60 ± 0.28 [p=0.032]) avec le CDR-SB à la semaine 76 par rapport au placebo.
Étude de phase 3 TRAILBLAZER-ALZ-6
La posologie de 350/700/1050 mg de donanemab puis 1400 mg toutes les 4 semaines a été étudiée dans une étude multicentrique randomisée, en double aveugle de phase IIIb, menée chez des adultes atteints de maladie d'Alzheimer symptomatique débutante (MCI dû à une maladie d'Alzheimer ou démence légère due à la maladie d'Alzheimer, score MMSE de 20 à 28 inclus) et présentant des signes de pathologie bêta-amyloïde confirmée par PET à l'amyloïde.
843 patients ont été randomisés dans un rapport de 1:1:1:1 et ont reçu quatre posologies différentes sur un total de 72 semaines, à savoir 700 mg lors des trois premières perfusions, puis 1400 mg toutes les 4 semaines (n=207), ou l'une des trois posologies alternatives (y compris la posologie de 350/700/1050 mg, puis de 1400 mg toutes les 4 semaines; n=212), la même quantité totale de médicament étant finalement utilisée dans tous les schémas.
Le critère principal de l'étude a été le pourcentage de participants avec toute apparition d'ARIA-E jusqu'à la semaine 24. Les résultats ont montré qu'un ARIA-E est apparu chez 14% des patients ayant reçu 350/700/1050 mg puis 1400 mg toutes les 4 semaines jusqu'à la semaine 24, par rapport à 24% des patients ayant reçu 700/700/700 mg puis 1400 mg toutes les 4 semaines, ce qui correspond à un risque relatif réduit de 41%. La diminution des plaques amyloïdes observée à la semaine 24 était similaire quelle que soit la posologie.
PharmacocinétiqueLa pharmacocinétique (PK) de KISUNLA a été caractérisée à partir de données acquises après une administration unique et d'administrations multiples. Lors d'une utilisation toutes les 4 semaines, il y a une accumulation de <1.3 fois; les expositions à l'équilibre sont atteintes après une administration unique. Lors d'administrations uniques allant de 350 mg à 2800 mg (environ le double de la dose recommandée de 1400 mg pour un poids corporel de 70 kg) et d'administrations multiples de 350 et 1400 mg, les expositions (Cmax et AUC) ont augmenté de façon proportionnelle. Une exposition similaire a été observée avec une posologie de 350/700/1050 mg puis de 1400 mg toutes les 4 semaines, en comparaison avec la posologie utilisé dans les études d'efficacité clinique (700 mg pour les trois premières perfusions, puis 1400 mg toutes les 4 semaines).
Le schéma de titration recommandé de l'étude TRAILBLAZER-ALZ-6 et le schéma de titration étudié dans l'étude pivot de phase 3 TRAILBLAZER-ALZ-2 se fondent sur des données de bridging PK/PD. Les dosages cumulés observés dans l'étude TRAILBLAZER-ALZ-6 (semaine 0 à 12), les AUC cumulées (semaine 0-12) et les concentrations moyennes à l'équilibre (Cav,ss) pour le schéma de titration recommandé et pour le schéma de titration examiné dans l'étude pivot de phase 3 étaient similaires et superposables. Pour l'exposition (Cav,ss), la non-infériorité a été montrée, définie comme une limite inférieure de l'intervalle de confiance à 90% du rapport géométrique moyen ≥0.8 pour le schéma de titration recommandé, par rapport au schéma de titration examiné dans l'étude pivot de phase 3. Des données de confirmation ont montré une PD observée similaire (réduction des plaques amyloïdes) aux semaines 24 et 52.
Absorption
Le donanemab n'est utilisé que par voie intraveineuse.
Distribution
Après administration intraveineuse, le donanemab est éliminé en deux phases. Le volume central de distribution est de 3.36 l avec 18.7% de variabilité interindividuelle. Le volume périphérique de distribution est de 4.83 l avec 93.9% de variabilité interindividuelle.
Métabolisme
Le donanemab est un anticorps monoclonal. Il est attendu qu'il soit dégradé en petits peptides et acides aminés par des voies cataboliques de la même façon que l'IgG endogène. Aussi n'y a-t-il pas d'inhibition métabolique ni d'induction de voies enzymatiques. Il n'est pas attendu que le donanemab soit métabolisé par des enzymes du cytochrome-P450 responsables du métabolisme et de l'élimination de petites molécules. On ne s'attend donc pas à ce qu'il y ait des métabolites actifs.
Élimination
La demi-vie du donanemab est d'environ 12.1 jours. La clairance du donanemab est de 0.0255 l/h (24.9% de variabilité interindividuelle).
Cinétique pour certains groupes de patients
Âge, sexe et masse corporelle
Des analyses pharmacocinétiques de population ont montré que l'âge, le sexe ou l'origine ethnique n'avaient pas d'influence sur la pharmacocinétique du donanemab. Bien que le poids corporel ait eu un effet sur la clairance et le volume de distribution, les variations qui en résultent ne permettent pas de conclure à la nécessité d'une adaptation posologique.
Insuffisance rénale et hépatique
Des analyses pharmacocinétiques de population ont montré qu'une insuffisance rénale et hépatique n'avait pas d'influence sur la pharmacocinétique du donanemab.
Données précliniquesPharmacologie de sécurité / Toxicité après administration répétée
Sur la base des études conventionnelles lors d'une administration répétée, les données précliniques ne permettent pas d'identifier des dangers particuliers pour l'être humain. Des études visant à tester une éventuelle carcinogénicité, génotoxicité ou altération de la fertilité du donanemab n'ont pas été réalisées.
Remarques particulièresIncompatibilités
Non pertinent
Influence sur les méthodes de diagnostic
Inconnue
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur le récipient.
Remarques particulières concernant le stockage
Flacon non ouvert
Conserver au réfrigérateur (2 ºC à 8 ºC) jusqu'à l'utilisation.
Le médicament peut être conservé non refroidi jusqu'à 3 jours à température ambiante (20 °C à 25 °C).
Conserver le flacon dans son carton, pour le protéger de la lumière.
Ne pas congeler. Ne pas secouer.
Conserver hors de portée des enfants.
Après dilution
La stabilité chimique et physique de la solution diluée a été montrée pour une durée de 72 heures à une température de 2 à 8°C ou 12 heures à température ambiante (20°C à 25°C).
La durée de la perfusion est incluse dans la durée de conservation.
D'un point de vue microbiologique, la solution diluée doit être utilisée immédiatement. Si la solution n'est pas utilisée immédiatement, les conditions de conservation et la durée de conservation sont de la responsabilité de l'utilisateur et elles ne doivent normalement pas dépasser 24 heures à 2 à 8°C, sauf si la dilution a eu lieu dans des conditions d'asepsie contrôlées et validées. Pour autant que la dilution ait eu lieu dans des conditions d'asepsie contrôlées et validées, la solution peut être conservée refroidie jusqu'à 72 heures à 2°C à 8°C ou jusqu'à 12 heures à température ambiante (20°C à 25°C).
Ne pas congeler la solution une fois qu'elle a été préparée.
Remarques concernant la manipulation
La solution pour perfusion Kisunla doit être préparée et administrée par du personnel qualifié, à l'aide d'une technique aseptique.
Laissez le donanemab s'équilibrer pendant environ 30 minutes à température ambiante avant la préparation.
Vérifiez le contenu du flacon afin de détecter d'éventuelles particules et changements de couleur. Jetez le flacon si vous constatez la présence de particules ou de changements de couleur.
Après dilution et préparation dans une solution injectable de chlorure de sodium à 0.9% (9 mg/ml), le donanemab est administré par perfusion intraveineuse.
-Calculez le volume nécessaire pour la préparation de la solution pour perfusion:Pour 350 mg de donanemab: 20 mlPour 700 mg de donanemab: 40 mlPour 1050 mg de donanemab: 60 mlPour 1400 mg de donanemab: 80 ml
-Prélevez le volume requis de donanemab et diluez-le dans une poche à perfusion contenant une solution injectable de chlorure de sodium à 0.9% (9 mg/ml), de sorte que la concentration finale soit de 4 mg/ml à 10 mg/ml. Utilisez uniquement une solution injectable de chlorure de sodium à 0.9% (9 mg/ml) pour la préparation.
-Retournez délicatement la poche à perfusion pour mélanger la solution.
-Perfusez la solution diluée pendant 30 minutes au moins.
-Administrez la totalité de la solution pour perfusion.
À la fin de la perfusion, rincez la tubulure de la perfusion avec une solution injectable de chlorure de sodium à 0.9% (9 mg/ml).
Après la perfusion, les patients doivent rester sous observation pendant au moins 30 minutes.
Numéro d’autorisation69523 (Swissmedic)
PrésentationKisunla, solution à diluer pour perfusion (perfusion intraveineuse)
avec 350 mg de donanemab dans 20 ml: 1 flacon (A)
Titulaire de l’autorisationEli Lilly (Suisse) SA, 1214 Vernier/GE.
Mise à jour de l’informationJanvier 2026
Manuel de neuro-imagerie pour le donanemab - guide de l'imagerie neuronale
Abréviations et définitions
Terme Définition
2D bidimensionnelle
3DT1 tridimensionnelle pondérée en T1
Aβ bêta-amyloïde
AD Alzheimer's Disease, maladie d'Alzheimer
ApoE apolipoprotéine E
ARIA Amyloid-Related Imaging Abnormalities, anomalies de l'imagerie liées à
l'amyloïde
CL Centiloïde
DWI Diffusion Weighted Imaging, imagerie pondérée en diffusion
FLAIR Fluid-Attenuated Inversion Recovery, atténuation fluidique par
inversion-récupération
FORE Fourier rebinning
GRE écho de gradient
IRM imagerie par résonance magnétique
IV intraveineux
MNI Montreal Neurologic Institute
MUBADA analyse discriminante barycentrique de multiblocs
NFT Neurofibrillary Tangles, enchevêtrement neurofibrillaire
OSEM Ordered Subset Expectation Maximisation, algorithme d'attente maximisation
PET Positronen-Emissions-Tomographie (tomographie par Emission de Positrons)
RAMLA Row-Action Maximum Likelihood Algorithm, algorithme d'action par ligne pour
estimation de la probabilité maximale
ROI Region Of Interest, zone d'intérêt
SUVr Standardised Uptake Value ratio, rapport de valeurs de fixation normalisées
Introduction
Les plaques amyloïdes (agrégats extracellulaires insolubles de peptides Aβ) et les NFT (agrégats de tau hyperphosphorylés, anormalement repliés) sont des indicateurs importants de la maladie d'Alzheimer (AD) et ils sont cruciaux pour le diagnostic neuropathologique de la maladie d'Alzheimer (Hyman et al. 2012; Jack et al. 2024). L'accumulation de peptide Aβ dans le parenchyme cérébral est le signe le plus précoce de l'AD et elle peut entraîner des modifications pathologiques subséquentes, y compris une tauopathie (Jack et al. 2024). Dans certains cas, une pathologie tau peut survenir indépendamment de l'accumulation d'Aβ, ce qui suggère l'existence à la fois de tauopathies indépendantes de l'amyloïde et de tauopathies facilitées par l'amyloïde (van der Kant et al. 2020).
Le donanemab se lie à la forme tronquée N-terminale de la protéine bêta-amyloïde et il contribue à la clairance des plaques par phagocytose médiée par les microglies (DeMattos et al. 2012). Par conséquent, la mesure des pathologies amyloïdes et tau avant et après traitement par donanemab a été incluse dans des études cliniques sur le donanemab (Mintun et al. 2021; Sims et al. 2023). Le PET constitue un outil de référence permettant de visualiser et de quantifier à la fois les plaques d'Aβ et les dépôts de NFT in vivo.
L'IRM vise deux objectifs principaux relatifs aux patients qui subissent une évaluation et un traitement avec des thérapies ciblant l'amyloïde (By et al. 2025):
évaluation de résultats d'exclusion, et
surveillance de la sécurité visant à déterminer la présence d'anomalies de neuro-imagerie apparaissant durant le traitement, en particulier les ARIA induites par des traitements ciblant l'amyloïde.
Se basant sur ces considérations, le programme de neuro-imagerie TRAILBLAZER-ALZ 2 a inclus des mesures de PET amyloïde et tau, ainsi que plusieurs mesures d'IRM (Tableau 1). Il faut relever que les évaluations spécifiques des scans PET et IRM, y compris les évaluations visuelles et quantitatives, ont été réalisées de façon centralisée par les fournisseurs de PET ou d'IRM.
Un premier scan a été effectué avant le traitement lors du screening ou au début des études, afin de déterminer l'éligibilité à l'étude ou pour la surveillance du traitement. Un génotypage d'APOE a également été effectué sur un échantillon de sang lors du screening, à moins que des lois et réglementations nationales spécifiques n'interdisent ce type de test.
Ce manuel de neuro-imagerie met l'accent sur les principaux éléments des procédures d'imagerie par PET et IRM appliquées dans l'étude TRAILBLAZER-ALZ 2. Des détails supplémentaires ont été spécifiés dans les procédures standard d'utilisation des fournisseurs d'imagerie et dans les manuels d'imagerie du site.
Tableau 1. Rôle des modalités d'imagerie dans l'étude TRAILBLAZER-ALZ 2
Modalité Screening/Valeur à Pendant la phase d'observation
l'inclusion
PET amyloïde Présence d'une Concentration en plaques
pathologie amyloïde amyloïdes et modification de la
≥37 CL (Q) concentration en plaques
amyloïdes cérébrales par rapport
à la valeur à l'inclusion (Q)
PET à tau Présence d'une Modification de la concentration
pathologie tau (VQ) en plaques amyloïdes par rapport
à la valeur à l'inclusion (Q)
IRM 3DT1 Image de référence anatomique Modification des
pour la quantification par PET à mesures volumétrique
tau s d'IRM par rapport
aux mesures à
l'inclusion (Q)
2D Axial FLAIR Détection d'œdème Détection d'œdème cérébral et de
(Atténuation fluidiq cérébral et de lésions de la substance blanche
ue par inversion-réc lésions de la (V)
upération (FLAIR) substance blanche
en 2D (V)
2D Axial T2* Écho Détection de dépôts Détection de dépôts d'hémosidérine
de gradient (GRE) d'hémosidérine et et d'autres types de lésions
d'autres types de hémorragiques (V)
lésions hémorragique
s (V)
2D Axial T2* Écho Détection de lésions Détection de lésions de la
de spin rapide/turbo de la substance substance blanche et autres
blanche et autres résultats d'IRM (V)
résultats d'IRM (V)
Imagerie 2D axiale Détection d'infarctu Détection d'infarctus aigus et
pondérée en diffusio s aigus et d'acciden d'accidents vasculaires cérébraux
n (DWI) ts vasculaires (V)
cérébraux (V)
Abréviations: 2D = bidimensionnelle; 3DT1 = tridimensionnelle pondérée en T1; CL = Centiloïdes; IRM = imagerie par résonance magnétique; PET = tomographie par émission de positrons; Q = évaluation quantitative à l'aide d'un logiciel spécial de traitement et d'analyse de l'image; V = évaluation visuelle; VQ = évaluation visuelle et quantitative.
Manuel de neuro-imagerie
PET amyloïde
Des scans de PET amyloïde avec soit le 18F-florbetapir soit le 18F-florbetaben comme radiotraceurs ont été utilisés pour la détermination de l'éligibilité des participants à l'étude, par évaluation de leur niveau de pathologie amyloïde. Pour les participants éligibles à l'étude, des scans de PET amyloïde de suivi ont été effectués à certains intervalles de temps entre les visites de l'étude, afin d'évaluer
a.le taux d'amyloïde post-traitement et de passer du traitement par le donanemab au placebo, selon une procédure en aveugle, si des critères prédéterminés de PET amyloïde étaient remplis et
b.la variation des dépôts de plaques amyloïdes entre l'inclusion et la semaine 76.
Pour le scan avec le 18F-florbetapir, les participants ont reçu un bolus par injection unique d'une dose cible de 370 MBq (10 mCi) de 18F-florbetapir, suivie d'un lavage avec une solution physiologique, puis ont été soumis à un scan de 20 minutes (4 acquisitions x 5 minutes) initié environ 50 minute après l'injection. Pour le scan avec 18F-florbetaben, les participants ont reçu un bolus par injection unique d'une dose cible de 300 MBq (8.1 mCi) de 18F-florbetaben, suivie d'un lavage avec une solution physiologique, puis ont été soumis à un scan de 20 minutes (4 acquisitions x 5 minutes) initié environ 90 minutes après l'injection.
Les images de PET ont été reconstituées au moyen de protocoles spécifiques des scanners, avec les sélections suivantes de paramètres: algorithme de reconstruction itérative (FORE, OSEM ou RAMLA), 3 à 6 itérations, 16 à 33 sous-ensembles. Des filtres gaussiens post-reconstruction de 3 à 5 mm ont été appliqués (à l'exception des scanners Philips où le réglage "Normal" ou "Fin" des paramètres de relaxation a été utilisé).
Dans l'étude TRAILBLAZER-ALZ 2, seules des évaluations quantitatives des images de PET amyloïde ont été réalisée.
1.1.1 Scan PETamyloide au screening ou au début de l'étude
Les images de PET de 5 minutes ont été corrigées pour le mouvement puis moyennées en une seule image statique (Shcherbinin et al. 2016; Pontecorvo et al. 2019). L'image moyenne de PET amyloïde obtenue au screening ou au début de l'étude a été normalisée spatialement par rapport au modèle TEP Amyloïde de référence dans l'espace cérébral standard (espace MNI). Aucune image d'IRM n'a été utilisées pour effectuer une évaluation quantitative de l'éligibilité.
Dans l'étude TRAILBLAZER-ALZ 2, l'évaluation quantitative des images obtenues tant avec le 18F-florbetapir qu'avec le 18F-florbetaben s'est faite selon la technique la plus courante d'estimation du SUVr. Le SUVr représente le rapport du signal moyen de PET dans une région néocorticale composite (région cible) sur le signal moyen de PET dans une région de référence qui est typiquement épargnée par les plaques amyloïdes, par exemple l'espace MNI. Pour le calcul du SUVr, toutes les ROI cibles et de référence ont été appliquées sur l'image de PET du participant normalisée dans l'espace MNI. Selon une méthodologie de PET amyloïde développée précédemment, le SUVr a été calculé comme une moyenne non pondérée de 6 régions corticales (mésiale orbitale frontale, cingulaire antérieure, précunéus, cingulaire postérieure, pariétale et temporale) et l'ensemble du cervelet a été utilisé comme région de référence (Joshi et al. 2015).
Pour harmoniser les mesures de concentrations d'amyloïde effectuées à l'aide des différents traceurs de PET, le 18Fflorbetapir ou le 18F-florbetaben, des procédures précédemment validées ont été utilisées pour convertir les mesures de SUVr spécifiques d'un traceur en un cadre commun de référence, l'échelle CL (Klunk et al. 2015). L'échelle CL permet une interprétation du SUVr de PET amyloïde en fonction de 2 points d'ancrage: 0 CL (signal moyen de PET amyloïde chez des individus jeunes [≤45 ans], sans handicap cognitif) et 100 CL (signal moyen de PET amyloïde chez des patients atteints de maladie d'Alzheimer, au stade de démence légère à modérée) (Klunk et al. 2015; Iaccarino et al. 2025). Différentes formules de transformation ont été utilisées pour convertir les mesures de SUVr au 18Fflorbetapir et au 18F-florbetaben en unités CL, ce qui a permis l'application de seuils de CL identiques pour les deux traceurs de PET amyloïde.
Dans l'étude TRAILBLAZER-ALZ 2, les participants aptes à l'étude avaient une pathologie amyloïde (≥37 CL) évaluée avec le 18F-florbetapir ou le 18F-florbetaben.
1.1.2 Scan PET amyloïde pendant la période d'observation
Pendant la période d'observation de l'étude TRAILBLAZER-ALZ 2, des scans PET amyloïde ont été effectués à 24, 52 et 76 semaines. De façon analogue aux analyses de screening ou au début de l'étude, les images de PET amyloïde de suivi ont été corrigées pour le mouvement et moyennées. Cependant, la normalisation spatiale par rapport au cadre de PET amyloïde de référence s'est effectuée différemment, pour l'évaluation
a.des concentrations post-traitement d'amyloïde et du passage du traitement de donanemab au placebo selon une procédure en aveugle, si les critères prédéterminés de PET àl'amyloïde étaient remplis,
b.des modifications des dépôts de plaques amyloïdes cérébrales entre l'inclusion et la semaine 76.
Pour l'évaluation des concentrations d'amyloïde post-traitement, l'image de suivi moyennée a été directement normalisée en fonction du cadre de PET amyloïde de référence, de façon analogue à la procédure appliquée au scan de screening ou au début de l'étude. Si la concentration de plaque amyloïde à 24 ou 52 semaines était inférieur à 11 CL sur l'un des scans de PET amyloïde ou inférieur à 25, mais supérieur à 11 CL lors de 2 scans de PET amyloïde consécutifs, le donanemab était remplacé par le placebo dans une procédure en aveugle (Sims et al. 2023).
Pour évaluer la modification des dépôts de plaques amyloïdes entre l'inclusion et la semaine 76 pour chaque participant, l'image moyennée a tout d'abord été alignée sur l'image de PET amyloïde correspondante à l'inclusion, puis sur le cadre cérébral standard, au moyen des mêmes paramètres de co-enregistrement que ceux du scan PET à l'inclusion. Outre la modification de la concentration de plaques amyloïdes par rapport à l'inclusion, le pourcentage de participants qui obtenaient une clairance de l'amyloïde (<24.1 CL) a été calculé. Un seuil de 24.1 CL a été utilisé, sur la base d'une discrimination précédemment établie entre des plaques absentes ou rares et des plaques modérées à fréquentes dans des données confirmées à l'autopsie entre cas positifs pour l'amyloïde et cas négatifs pour l'amyloïde (Joshi et al. 2015; Navitsky et al. 2018).
Aucune image d'IRM n'a été utilisée pour effectuer une évaluation quantitative des images de suivi de PET à l'amyloïde.
PET tau
Le PET tau avec le 18F-flortaucipir a été utilisée pour déterminer l'éligibilité à l'étude par l'évaluation du taux de pathologie tau du participant. Pour les patients aptes à l'étude, des scans de PET à tau de suivi ont été réalisés à la semaine 76, afin d'évaluer l'effet du donanemab sur les dépôts cérébraux de tau, par rapport au placebo.
Dans l'étude TRAILBLAZER-ALZ 2, les participants ont reçu un bolus par injection IV unique d'une dose cible de 370 MBq (10 mCi) de 18F-flortaucipir, suivie d'un lavage avec une solution physiologique, puis un scan dynamique du cerveau de 30-minute (6 acquisitions x 5 minutes) a été obtenu, environ 75 minutes après l'injection.
Les images de PET ont été reconstituées au moyen de protocoles spécifiques du scanner, avec la sélection suivante de paramètres: algorithme de reconstruction itérative (FORE, OSEM ou RAMLA), 3 à 6 itérations, 16 à 33 sous-ensembles. Des filtres gaussiens post-reconstruction de 3 à 5 mm ont été appliqués (à l'exception des scanners Philips où le réglage "Normal" ou "Fin" des paramètres de relaxation a été utilisé).
Pour l'analyse quantitative des images du flortaucipir, les images de PET de 5 minutes ont été corrigées pour le mouvement ainsi que corrigées pour le temps d'acquisition, puis moyennées en une seule image statique. Une correction pour le temps d'acquisition a été appliquée car les valeurs de SUVr avec le flortaucipir peuvent ne pas avoir atteint un plateau stable 75 minutes après l'injection, mais plutôt continuer à augmenter tout au long de la fenêtre potentielle d'imagerie (Shcherbinin et al. 2016). Par conséquent, des différences dans les intervalles de temps d'acquisition écoulé entre l'injection et le scan dans tous les scans réalisés à l'inclusion et à la semaine 76 pourraient affecter les analyses de SUVr. Les détails de la correction pour le temps d'acquisition ont été décrits précédemment (Pontecorvo et al. 2019).
1.1.3 Scan PET tau au screening ou au début de l'étude
Les scans au screening resp. au début de l'étude réalisés avec le flortaucipir ont été évalués à la fois visuellement et quantitativement afin de confirmer la présence d'une pathologie tau chez les participants à l'étude TRAILBLAZER-ALZ 2.
Les scans ont été interprétés par examen visuel comme présentant des schémas régionaux de captation du traceur qui étaient des schémas tau modérés (τAD+) ou avancés (τAD++) ou qui n'étaient pas des schémas tau (schéma tau négatif [τAD-) compatibles avec une maladie d'Alzheimer (Fleisher et al. 2020; Lu et al. 2021). Le schéma tau τAD- a été défini comme une absence d'augmentation de l'activité néocorticale ou comme une augmentation de l'activité néocorticale limitée aux régions mésiale temporale, antérolatérale temporale et/ou frontale. Le schéma tau τAD+ modéré a été défini comme une augmentation de l'activité néocorticale dans la région temporale postérolatérale ou occipitale. Le schéma tau τAD++ avancé a été défini comme soit une augmentation de l'activité néocorticale dans la région pariétale ou précunéus soit comme une augmentation de l'activité dans la région frontale, avec en plus des augmentations dans les régions temporale postéro-latérale, pariétale ou occipitale. Les lecteurs étaient des médecins qui possédaient une expertise dans la/les indication(s) de la maladie spécifique(s) de l'étude et qui avaient de l'expérience dans l'analyse d'essais cliniques. Avant d'évaluer des images de l'étude, les lecteurs avaient suivi un entraînement spécifique, avec un nouvel entraînement pendant l'étude en fonction des besoins.
L'évaluation quantitative d'images de 18F-flortaucipir prises tant à l'inclusion que pendant le suivi dans l'étude TRAILBLAZER-ALZ 2 s'est faite selon la technique la plus couramment utilisée, une estimation du SUVr. Le SUVr représente le rapport du signal moyen de PET dans une région néocorticale composite (région cible) sur le signal moyen de PET dans une région de référence qui est typiquement épargnée par les NFT, par exemple le cervelet ou la substance blanche. Pour le calcul du SUVr, toutes les ROI cibles et de référence ont été appliquées sur l'image de PET du participant normalisée dans l'espace MNI. Diverses techniques de normalisation spatiale et diverses mesures du SUVr ont été appliquées pour la prise de décision de l'éligibilité à l'étude au moyen des scans à la sélection/inclusion, et pour l'évaluation des variations des dépôts de tau pour chaque participant au moyen des scans de suivi.
Pour l'évaluation quantitative de l'éligibilité à l'étude, l'image moyenne de flortaucipir à la sélection/inclusion a été normalisée spatialement par rapport au modèle de PET tau de référence dans l'espace MNI. Aucune image de RM n'a été utilisée pour effectuer une évaluation quantitative de l'éligibilité à l'étude. Le taux néocortical global de tau a été mesuré quantitativement en utilisant un SUVr tau néocortical pondéré signature de la maladie d'Alzheimer publié précédemment (MUBADA, Devous et al. 2018) par rapport à une intensité du signal de référence dans la substance blanche spécifique du sujet (PERSI, Southekal et al. 2018).
La décision relative à l'éligibilité à l'étude (évaluation de la positivité tau) s'est basée sur des évaluations tant visuelles que quantitatives (voir Figure 1). Selon la méthode d'évaluation combinée de l'aptitude tant visuelle que quantitative (tauVQ), les participants à TRAILBLAZER-ALZ 2 qui avaient soit (1) des schémas τAD+ et un SUVr tau pondéré signature d'AD ≥1.10 soit (2) des schémas τAD++, ont été considérés comme ayant un niveau de tau positif. Les scans avec le schéma τAD- ont été considérés comme ayant un niveau de tau inapproprié et ont été exclus de l'étude sans quantification de l'image. Si un scan τAD+ ne pouvait pas être soumis à une analyse quantitative, il était analysé par évaluation visuelle afin de déterminer l'éligibilité à l'étude, ce qui était effectué par un évaluateur indépendant.

Abréviations: SUVr= standardised uptake value ratio (rapport de valeur de fixation normalisée).
Figure 1. Évaluation par PET à tau de l'éligibilité à l'étude TRAILBLAZER-ALZ 2.
1.1.4 Scan PET tau pendant la période d'observation
Dans l'étude TRAILBLAZER-ALZ 2, des scans PET à tau de suivi ont été réalisés à la semaine 76.
Pour l'étude de PET à tau longitudinale, une normalisation spatiale plus rigoureuse impliquant une image d'IRM 3D T1 individuelle à l'inclusion a été appliquée. Tout d'abord, l'image de PET tau moyennée à l'inclusion a été coenregistrée avec l'image d'IRM 3D T1 du participant. L'image de PET à tau à 18 mois a été corrigée pour le mouvement et le temps et moyennée, puis alignée tout d'abord sur l'image de PET à l'inclusion puis sur l'image d'IRM à l'inclusion correspondante, avec les mêmes paramètres de co-enregistrement que ceux du scan PET à tau à l'inclusion. Finalement, l'image d'IRM 3D T1 à l'inclusion a été normalisée en fonction de l'espace MNI et la même matrice de transformation IRM-vers-atlas a été utilisée pour la normalisation spatiale des images de PET avec flortaucipir précédemment coenregistrées tant à l'inclusion qu'à 18 mois.
Pour les analyses longitudinales de PET tau, des valeurs régionales et globales de SUVr relatives à la région de référence précédemment développée (Pontecorvo et al. 2017) pour la substance grise cérébelleuse ont été utilisées. Dans la région de référence, une région de substance grise cérébelleuse dérivée de la ROI du cortex cérébelleux a été modifiée par un transfert de 6 mm en direction inférieure. Cette modification a été effectuée afin d'éviter un possible chevauchement avec des zones corticales inférieures et le liquide cérébrospinal supratentoriel. Dans les analyses longitudinales, les ROI cibles ont compris toutes les régions de marquage anatomique automatisé (Tzourio-Mazoyer et al. 2002) ainsi que des régions sélectionnées additionnelles. En particulier, le MUBADA SUVr (Devous et al. 2018) pour ce qui est de la région de référence grise cérébelleuse a été utilisé pour mesurer les variations des dépôts tau néocorticaux globaux.
IRM
L'IRM a été effectuée lors des visites de screening et de suivi. Des évaluations de l'éligibilité à l'étude ont été réalisées afin d'évaluer des critères potentiels d'exclusion basés sur l'IRM. Lors des visites pendant la période d'observation, des mesures volumétriques par IRM ont été réalisées afin d'évaluer l'effet du donanemab par rapport au placebo sur les volumes des régions cérébrales. En outre, des évaluations de la sécurité ont été effectuées lors de chaque visite afin d'apprécier la survenue de résultats d'IRM.
Les scanners d'IRM pouvant être utilisés dans l'étude avaient uniquement une intensité du champ magnétique de 1.5 tesla ou de 3.0 tesla.
Chaque procédure d'IRM a porté sur les séquences d'IRM suivantes:
-3D T1: Magnetisation-Prepared Rapid Gradient-Echo (Siemens)/Inversion Recovery-Prepped Fast Spoiled Gradient-Recalled Echo (General Electric)/3D Turbo Field Echo (Philips)
-2D FLAIR
-2D Axial T2* Écho de gradient (GRE)
-2D Axial T2 Écho de spin rapide/turbo, et
2D axial DWI3DT1Les buts de ces séquences sont présentés dans le Tableau 1. Les paramètres exacts d'acquisition dépendaient du fabricant, du modèle de scanner, du matériel informatique et du logiciel installés, ainsi que de l'intensité du champ magnétique. Tous les scanners ont été qualifiés par le fournisseur d'IRM et les paramètres d'acquisition devaient satisfaire aux exigences fixées par le vendeur d'IRM.
1.1.5 IRM au screening ou au début de l'étude
Les principaux critères d'exclusion basés sur l'IRM comprenaient (Sims et al. 2023)
présence d'ARIA d'œdème/effusion (2D axiale FLAIR)
plus de 4 microhémorragies cérébrales (2D axiale T2* GRE)
plus d'une zone de sidérose superficielle (2D axiale T2* GRE)
une hémorragie intracérébrale de plus de 1 cm (2D axiale T2* GRE), et
maladie grave de la substance blanche (modifications de la substance blanche en lien avec l'âge = 3 dans un hémisphère quelconque des régions évaluées à l'intérieur du paradigme d'évaluation, 2D axiale T2 écho de spin rapide/turbo; Wahlund et al. 2001).
1.1.6 Scan IRM pendant la période d'observation
Des scans IRM pendant la période d'observation ont été réalisés à la semaine 4, à la semaine 12, à la semaine 24, à la semaine 52 et à la semaine 76 et lors d'un arrêt prématuré de l'étude. Des visites non planifiées ont été également effectuées en cas de besoin.
Lors des visites pendant la période d'observation, les évaluations ont compris des contrôles de la sécurité visant à évaluer la sécurité et latolérabilité du donanemab, vérifiées par IRM (ARIA et résultats radiologiques émergents tels qu'œdème cérébral, dépôts d'hémosidérine, maladie de la substance blanche ou toute autre anomalie à l'IRM et autres résultats secondaires).
La sécurité et la tolérabilité du donanemab ont été évaluées au moyen de scans IRM de suivi avec les paramètres suivants:
présence, degré de gravité radiographique et localisation des ARIA d'œdème/effusion (2D axiale FLAIR, 2D axiale T2)
présence, nombre et localisation de microhémorragies (2D axiale T2* GRE)
présence et localisation d'hémorragies intracérébrales plus grandes que 1 cm (2D axiale T2* GRE)
présence et localisation de sidérose superficielle (2D axiale T2* GRE)
présence et gravité radiographique d'ARIA-H hémorragique (GRE 2D axiale T2*)
présence et gravité radiographique d'une maladie de la substance blanche (FLAIR 2D axiale, 2D axiale T2), et
présence et localisation de toute autre anomalie à l'IRM (2D DWI, 2D axiale T2).
Tout participant chez qui une ARIA était détectée subissait une imagerie toutes les 4 à 6 semaines, jusqu'à résolution ou stabilisation (Sims et al. 2023). Les directives sur la gestion des ARIA et l'interruption du traitement dépendaient de la gravité radiographique et des symptômes. Si les perfusions étaient interrompues, il était conseillé aux investigateurs d'attendre la résolution des ARIA d'œdème/effusion sur l'imagerie radiographique et la stabilisation des ARIA de microhémorragies et de dépôts d'hémosidérine avant de reprendre les perfusions. Un arrêt définitif était conseillé en cas d'hémorragies intracérébrales plus grandes que 1 cm ou d'ARIA grave. Ce sont les investigateurs qui prenaient les décisions finales relatives à la gestion des ARIA. Les groupements de termes préférés d'ARIA, l'évaluation du degré de gravité et les définitions des événements ont été publiés (Zimmer et al. 2025).
Références
By S, Kahl A, Cogswell PM. Alzheimer's Disease clinical trials: what have we learned from magnetic resonance imaging. J Magn Reson Imaging. 2025;61(2):579-594. https://doi.org/10.1002/jmri.29462
DeMattos RB, Lu J, Tang Y, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76(5):908-920. https://doi.org/10.1016/j.neuron.2012.10.029
Devous MD Sr, Joshi AD, Navitsky M, et al. Test-retest reproducibility for the tau PET imaging agent flortaucipir F 18. J Nucl Med. 2018;59(6):937-943. https://doi.org/10.2967/jnumed.117.200691
Fleisher AS, Pontecorvo MJ, Devous MD, et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77(7):829-839. https://doi.org/10.1001/jamaneurol.2020.0528
Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1-13. https://doi.org/10.1016/j.jalz.2011.10.007
Iaccarino L, Burnham CS, Tunali I, et al. A practical overview of the use of amyloid-PET Centiloid values in clinical trials and research. NeuroImage: Clin. 2025;46:103765. https://doi.org/10.1016/j.nicl.2025.103765
Jack CR Jr, Andrews JS, Beach TG, et al. Revised criteria for diagnosis and staging of Alzheimer's disease: Alzheimer's Association Workgroup. Alzheimers Dement. 2024;20(8):5143-5169. https://doi.org/10.1002/alz.13859
Joshi AD, Pontecorvo MJ, Lu M, et al. A semiautomated method for quantification of F 18 florbetapir PET images. J Nucl Med. 2015;56(11):1736-1741. https://doi.org/10.2967/jnumed.114.153494
Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1):1-15.e154. https://doi.org/10.1016/j.jalz.2014.07.003
Lu M, Pontecorvo MJ, Devous MD, et al.; AVID Collaborators. Aggregated tau measured by visual interpretation of flortaucipir positron emission tomography and the associated risk of clinical progression of mild cognitive impairment and Alzheimer disease: results from 2 phase III clinical trials. JAMA Neurol. 2021;78(4):445–453. https://doi.org/10.1001/jamaneurol.2020.5505
Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691-1704. https://doi.org/10.1056/NEJMoa2100708
Navitsky M, Joshi AD, Kennedy I, et al. Standardization of amyloid quantitation with florbetapir standardized uptake value ratios to the Centiloid scale. Alzheimers Dement. 2018;14(12):1565-1571. https://doi.org/10.1016/j.jalz.2018.06.1353
Pontecorvo MJ, Devous MD Sr, Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140(3):748-763. https://doi.org/10.1093/brain/aww334
Pontecorvo MJ, Devous MD, Kennedy I, et al. A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer's disease dementia. Brain. 2019;142(6):1723-1735. https://doi.org/10.1093/brain/awz090
Shcherbinin S, Schwarz AJ, Joshi A, et al. Kinetics of the tau PET tracer 18F-AV-1451 (T807) in subjects with normal cognitive function, mild cognitive impairment, and Alzheimer disease. J Nucl Med. 2016;57(10):1535-1542. https://doi.org/10.2967/jnumed.115.170027
Sims JR, Zimmer JA, Evans CD, et al.; TRAILBLAZER-ALZ 2 Investigator. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512-527. https://doi.org/10.1001/jama.2023.13239
Southekal S, Devous MD Sr, Kennedy I, et al. Flortaucipir F 18 quantitation using parametric estimation of reference signal intensity. J Nucl Med. 2018;59(6):944-951. https://doi.org/10.2967/jnumed.117.200006
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage. 2002;15:273-289. https://doi.org/10.1006/nimg.2001.0978
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21-35. https://doi.org/10.1038/s41583-019-0240-3
Wahlund LO, Barkhof F, Bazekas F, et al. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke. 2001;32(6):1318-1322. https://doi.org/10.1161/01.STR.32.6.1318
Zimmer JA, Ardayfio P, Wang H, et al. Amyloid-related imaging abnormalities with donanemab in early symptomatic Alzheimer disease: secondary analysis of the TRAILBLAZER-ALZ and ALZ 2 randomized clinical trials. [published online ahead-of-print, 2025 March 10]. JAMA Neurol. 2025;e250065. https://doi.org/10.1001/jamaneurol.2025.0065
Mise à jour de l'information
Juillet 2025
Résumé du protocole d'IRM pour donanemab
Abréviations et définitions
Terme Définition
1.5T intensité du champ magnétique de 1.5 tesla
2D bidimensionnelle
3T intensité du champ magnétique de 3 tesla
3DT1 tridimensionnelle pondérée en T1
DWI Diffusion Weighted Imaging, imagerie pondérée en diffusion
FLAIR Fluid-Attenuated Inversion Recovery, atténuation fluidique par inversion-récupération
GRE Gradient Echo (écho de gradient)
IRM imagerie par résonance magnétique
PET positron emission tomography, tomographie par émission de positrons
Introduction/contexte
L'IRM chez les patients au screening ou en suivi pendant le traitement par donanemab visait à deux objectifs principaux:
recherche de signes d'exclusion, et
surveillance de sécurité visant à déterminer la présence d'anomalies de neuro-imagerie apparaissant durant le traitement, en particulier les anomalies d'imagerie liées à l'amyloïde et les hémorragies intracérébrales plus grandes que 1 cm.
Le présent résumé du protocole d'IRM met l'accent sur les principaux éléments des procédures d'imagerie par résonance magnétique.
Les modalités d'IRM appliquées dans l'étude TRAILBLAZER-ALZ 2 sont présentées dans le Tableau 1.
Tableau 1. Rôle des modalités d'IRM dans l'étude TRAILBLAZER-ALZ 2
Modalité Screening/Valeur à l'inclusion Pendant la phase d'observation
3DT1 Image de référence anatomique Modification des mesures
pour la quantification par TEP volumétriques d'IRM par rapport
tau aux mesures à l'inclusion (Q)
2D Axial FLAIR (Atténuatio Détection d'œdème cérébral et de Détection d'œdème cérébral et de
n fluidique par inversion- lésions de la substance blanche lésions de la substance blanche (V)
récupération) (V)
2D Axial T2* Écho de Détection de dépôts d'hémosidérin Détection de dépôts d'hémosidérine
gradient (GRE) e et d'autres types de lésions et d'autres types de lésions
hémorragiques (V) hémorragiques (V)
2D Axial T2 Écho de spin Détection de lésions de la Détection de lésions de la
rapide/turbo substance blanche et autres substance blanche et autres
résultats d'IRM (V) résultats d'IRM (V)
Imagerie 2D axiale Détection d'infarctus aigus et Détection d'infarctus aigus et
pondérée en diffusion d'accidents vasculaires d'accidents vasculaires cérébraux
(DWI) cérébraux (V) (V)
Abréviations: 2D = bidimensionnelle; 3DT1 = tridimensionnelle pondérée en T1; IRM = imagerie par résonance magnétique; TEP = tomographie par émission de positrons; Q = évaluation quantitative à l'aide d'un logiciel spécial de traitement et d'analyse de l'image; V = évaluation visuelle.
Résumé du protocole d'IRM pour le donanemab
Les scanners d'IRM pouvant être utilisés dans l'étude TRAILBLAZER-ALZ 2 avaient uniquement une intensité du champ magnétique de 1.5 tesla ou de 3.0 tesla. Chaque procédure d'IRM a porté sur les séquences d'IRM suivantes, en ordre chronologique d'acquisition:
-3DT1 (3 à 4 min): Magnetization-Prepared Rapid Gradient-Echo (MP-RAGE, Siemens)/Inversion Recovery-Prepped Fast Spoiled Gradient-Recalled Echo (General Electric)/3D Turbo Field Echo (Philips)
-2D FLAIR (2.5 à 4.5 min)
-2D Axial T2* Écho de gradient (GRE) (3 à 4 min)
-2D Axial T2 Écho de spin rapide/turbo (1 à 2 min), et
-2D axiale DWI (moins de 1 min).
Les buts de ces séquences sont présentés dans le Tableau 1. Les paramètres exacts d'acquisition dépendaient
du fabricant
du modèle de scanner
du matériel informatique et du logiciel installés et
de l'intensité du champ magnétique
Les paramètres spécifiques des scanners d'IRM utilisés dans l'étude TRAILBLAZER-ALZ 2 sont la propriété du fabricant du scanner d'IRM et ils ont été standardisés pour tous les fabricants d'IRM et pour toutes les intensités du champ magnétique, afin de permettre de comparer les images acquises sur des modèles différents de scanners et avec des intensités différentes de champ magnétique. Les séquences/modalités utilisées pour les évaluations de la sécurité dans les essais cliniques avec le donanemab sont employées en routine dans les protocoles neurodégénératifs d'IRM (Di Muzio et al. 2015; Benzinger and Jindal 2023; Harper et al. 2014).
Les recommandations consensuelles publiées pour les protocoles cliniques se trouvent ci-dessous (Benzinger; Cogswell et al 2022; Cogswell et al 2025; Lawson et al 2023; Philips). Il y a compatibilité avec les images acquises pendant les études. Les détails des paramètres fixés pour chaque séquence sont joints en annexe. Le Tableau 2 fournit des liens vers des tables de référence contenant les paramètres.
Tableau 2. Table des matières des tableaux avec les paramètres d'IRM
Intensité du champ magnétique (T) Tableau
GE 1.5 T Annexe 1
GE 3 T Annexe 2
Siemens 1.5 T Annexe 3
Siemens 3 T Annexe 4
Philips 1.5T Annexe 5
Philips 3T Annexe 6
Abréviations: GE = General Electric; T = Tesla.
Références
Benzinger TLS, Jindal S. MR imaging of neurodegeneration. In: Cross DJ, Mosci K, Minoshima S, eds. Molecular Imaging of Neurodegenerative Disorders. Cham: Springer; 2023:169-181. https://doi.org/10.1007/978-3-031-35098-6_11
Benzinger TL. ASNR-Recommended AD Therapeutic Imaging Protocols. Accessed November 21, 2025. https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Cogswell PM, Andrews TJ, Barakos JA, et al. Alzheimer disease anti-amyloid immunotherapies: imaging recommendations and practice considerations for monitoring of amyloid-related imaging abnormalities. Am J Neuroradiol. 2025;46(1):24-32. https://doi.org/10.3174/ajnr.A8469
Cogswell PM, Barakos JA, Barkhof F, et al. Amyloid-related imaging abnormalities with emerging Alzheimer disease therapeutics: detection and reporting recommendations for clinical practice. Am J Neuroradiol. 2022;43(9):E19-E35. https://doi.org/10.3174/ajnr.A7586
Di Muzio B, Campos A, Murphy A, et al. Neurodegenerative protocol (MRI). Radiopaedia.org Published August 11, 2015. Updated July 26, 2024. Accessed November 21, 2025. https://radiopaedia.org/articles/neurodegenerative-protocol-mri?lang=us
Harper L, Barkhof F, Scheltens P, et al. An algorithmic approach to structural imaging in dementia. J Neurol Neurosurg Psychiatry. 2014;85(6):692–698. https://doi.org/10.1136/jnnp-2013-306285.
Lawson S, Fofana A, Banerjee S. Optimizing the ARIA imaging protocol on GE HealthCare MR systems. 2023. Accessed November 21, 2025. https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Philips. Alzheimer's Disease Anti-Amyloid Immunotherapies (ARIA) 1.5T. Accessed November 21, 2025. https://www.mriclinicalcasemap.philips.com/global/case/291/i
Annexes: Manuel abrégé pour les paramètres d'IRM
Annexe 1 General Electric 1.5T
Paramètre T1 FLAIR T2* T2 DWI
Mode d'imagerie 3D 2D 2D 2D 2D
Séquence d'impulsions MP-RAGE FSE-XL Écho de gradient FSE Écho de spin
Orientation - Axiale Axiale Axiale Axiale
FOV (champ de vision, cm) 25.6 22.0 22.0 22.0 24.0
Épaisseur de la coupe (mm) 0.5 4.0 4.0 4.0 4.0
Distance entre les coupes (mm) - 0.4 0.4 0.4 0.4
Nombre de coupes 1 (slab) 36 36 36 36
Matrice de fréquences 256 300 224 300 120
Matrice de phases 256 200 200 200 160
Phase FOV 1.0 1.0 0.9 1.0 1.0
NEX 1.0 1.0 1.0 1.0 2.0
Angle de bascule (°) 12.0 160 (auto) 20.0 160 (auto) -
TE (ms) 3.1 130.0 25.0 130.0 Minimum
TR (ms) 7.7 10000.0 600.0 10000.0 9163.0
TI (ms) 799 2687 - 2687 -
Longueur du train d'écho - 20 - 20 -
Saturation de graisse/eau - Graisse - Graisse Graisse
Le protocole complet est disponible sous: https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Annexe 2 General Electric 3.0T
Paramètre T1 FLAIR T2* T2 DWI
Mode d'imagerie 3D 2D 2D 2D 2D
Séquence d'impulsions MP-RAGE FSE-XL Écho de gradient FSE Écho de spin EPI
Orientation - Axiale Axiale Axiale Axiale
FOV (champ de vision, cm) 25.6 22.0 22.0 22.0 24.0
Épaisseur de la coupe (mm) 0.5 4.0 4.0 4.0 4.0
Distance entre les coupes (mm) - 0.4 0.4 0.4 0.4
Nombre de coupes 1 (slab) 36 36 36 36
Matrice de fréquences 256 300 300 300 160
Matrice de phases 256 200 220 200 160
Phase FOV 1.0 0.9 0.9 0.9 1.0
NEX 1.0 1.0 1.0 1.0 1.0
Angle de bascule (°) 8.0 160 (auto) 20.0 160 (auto) -
TE (ms) 3.0 130.0 15.0 130.0 Minimum
TR (ms) 7.3 9000.0 800.0 9000.0 7500.0
TI (ms) 1000 2473 - 2473 -
Longueur du train d'écho - 23 - 23 -
Saturation de graisse/eau - Graisse - Graisse Graisse
Le protocole complet est disponible sous: https://signapulse.gehealthcare.com/autumn-2023/optimizing-the-aria-imaging-protocol-on-ge-healthcare-mr-systems
Annexe 3 Siemens 1.5 T
Paramètre T1 FLAIR T2* T2 DWI
Mode 3D 2D 2D 2D 2D
Séquence MPRAGE 3D FLAIR TSE 2D Echo de gradient TSE 2D Écho de spin EPI 2D
T2* 2D
Orientation Sagittale Transversale (axiale Transversale (axiale Transversale (axiale Transversale (axiale
) ) ) )
Taille du voxel (mm) 1.3 × 1.3 × 1.3 0.5 × 0.5 × 4.0 0.5 × 0.5 × 4.0 0.9 × 0.9 × 5.0 1.9 × 1.9 × 4.0
Lecture du FOV (mm) 240 240 240 240 240
Épaisseur de la 1.3 4.0 4.0 5.0 4.0
coupe (mm)
Coupes 192 30 30 28 30
TR (ms) 2400 10000 608 4700 4700
TE (ms) 2.24 114 35 111 79
TI (ms) 1000 2600 - - -
Angle de bascule (°) 8 150 20 180 -
Largeur de bande 190 130 130 190 1220
(Hz/Px)
Accélération GRAPPA 2 GRAPPA 2 GRAPPA 2 GRAPPA 2 GRAPPA 2
Moyennes 1 1 1 1 1
Concaténations 1 2 2 2 1
Facteur turbo/EPI 224 (turbo) 16 (turbo) - 26 (turbo) 128 (EPI)
Saturation de Standard Forte saturation de Standard Standard Forte saturation de
graisse graisse graisse
Le protocole complet est disponible sous: https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Annexe 4 Siemens 3 T
Paramètre T1 FLAIR T2* T2 DWI
Mode 3D 2D 2D 2D 2D
Séquence MPRAGE 3D FLAIR TSE 2D Echo de gradient TSE 2D Écho de spin EPI 2D
T2* 2D
Orientation Sagittale Transversale Transversale Transversale Transversale
Taille du voxel (mm) 1.0 × 1.0 × 1.0 0.5 × 0.5 × 4.0 0.5 × 0.5 × 4.0 0.9 × 0.9 × 5.0 1.9 × 1.9 × 4.0
Lecture du FOV (mm) 256 240 240 230 240
Épaisseur de la 1.0 4.0 4.0 5.0 4.0
coupe (mm)
Coupes 176 30 30 25 30
TR (ms) 2300 9000 597 5140 6700
TE (ms) 2.98 96.0 35.0 78.0 72.0
TI (ms) 900 2500 - - -
Angle de bascule (°) 9 150 20 120 -
Largeur de bande 240 199 200 260 1698
(Hz/Px)
Accélération GRAPPA 2 GRAPPA 3 GRAPPA 2 GRAPPA 3 GRAPPA 2
Moyennes 1 1 1 1 1
Concaténations 1 2 2 2 1
Facteur turbo/EPI 176 (turbo) 16 (turbo) - 26 (turbo) 128 (EPI)
Saturation de Nulle Forte saturation de Nulle Nulle Forte saturation de
graisse graisse graisse
Le protocole complet est disponible sous: https://www.magnetomworld.siemens-healthineers.com/clinical-corner/protocols/neurology-neurography/asnr
Annexe 5 Philips 1.5T
Paramètre T1 FLAIR T2* DWI
Mode 3D 2D 2D 2D
Type de séquence FFE, TFE, T1W 3D IR, TSE, FLAIR FFE, T2* SE, DWI, EPI
Orientation Sagittale Transversale (axiale Transversale (axiale Transversale (axiale
) ) )
Taille du voxel (mm) 1.05 × 1.05 × 1.10 0.90 × 1.12 × 4.00 0.90 × 1.12 × 4.00 1.51 × 2.17 × 4.00
Recon taille du 0.64 × 0.64 × 1.10 0.65 × 0.65 × 4.00 0.90 × 0.90 × 4.00 1.20 × 1.20 × 4.00
voxel (mm)
FOV (mm) FH 256, AP 238, RL AP 230, RL 183, FH AP 230, RL 183, FH RL 230, AP 230, FH
159 129 129 129
Épaisseur de la 1.10 4.00 4.00 4.00
coupe (mm)
Coupes 145 26 26 26
TR (ms) 7.4 11000 910 3498
TE (ms) 3.4 150 28 85
TI (ms) 950 2800 - -
Angle de bascule (°) 8 60/120 18 90
Largeur de bande 217.1 217.0 108.7 18.9
(Hz)
Accélération SENSE (2.2) SENSE (1) SENSE (1.5) SENSE (2)
Echo train/facteur TFE: 216 TSE: 41 - EPI: 53
EPI
Saturation de Nulle SPIR (forte) Nulle SPIR (forte)
graisse
Déplacement eau 1.0 Maximum 2.0 11.482
graisse (pixels)
Moyennes 1 2 2 1
Le protocole complet est disponible sous: https://www.mriclinicalcasemap.philips.com/global/case/291/i
Annexe 6 Philips 3T
Paramètre T1 FLAIR T2* DWI
Mode 3D 2D 2D 2D
Séquence FFE, TFE, T1W 3D IR, TSE, FLAIR FFE, T2* SE, DWI, EPI
Orientation Sagittale Transversale (axiale Transversale (axiale Transversale (axiale
) ) )
Taille du voxel (mm) 1.00 × 1.00 × 1.00 0.65 × 1.00 × 4.00 0.90 × 1.12 × 4.00 1.31 × 1.32 × 4.00
Recon taille du 0.47 × 0.47 × 1.00 0.45 × 0.45 × 4.00 0.53 × 0.53 × 4.00 0.96 × 0.96 × 4.00
voxel (mm)
FOV (mm) FH 240, AP 240, RL AP 230, RL 186.9, AP 230, RL 183.1, RL 230, AP 230, FH
170 FH 139 FH 139 119
Épaisseur de la 1.00 4.00 4.00 4.00
coupe (mm)
Coupes 170 28 28 24
TR (ms) 6.6 8000 835 2965
TE (ms) 3.0 135 16 55
TI (ms) 1060 2500 - -
Angle de bascule (°) 8 100 18 90
Largeur de bande 271.3 227.3 217.0 13.2
(Hz)
Accélération SENSE (AP 1, RL 2.2) SENSE (RL 2.4) Nulle SENSE (AP 2.2)
Écho train/facteur TFE: 204 TSE: 35 - EPI: 79
EPI
Saturation de Nulle SPIR (faible) Nulle SPIR (forte)
graisse
Déplacement eau 1.6 1.911 2.002 32.925
graisse (pixels)
Moyennes 1 2 1 1
Le protocole complet est disponible sous: https://www.mriclinicalcasemap.philips.com/global/case/291/i
Mise à jour de l'information
Janvier 2025
|