|
| Home - Ipro sur JERAYGO comprimés pelliculés |
| Information professionnelle sur JERAYGO comprimés pelliculés: | Idorsia Pharmaceuticals Ltd |
Propriétés/EffetsCode ATC Traitement Partie 1 (4 semaines Partie 2 (32 semaine Partie 3 (12 semaine
) s) s)
Conception double aveugle, simple aveugle sevrage en double
contrôlé par placebo aveugle, contrôlé
, randomisé (1:1:1) par placebo, randomi
sé (1:1)
Durée Semaine 0 à Semaine Semaine 4 à Semaine Semaine 36 à Semaine
4 36 48
Traitement d’appoint Aprocitentan 25 mg N = 243 N = 243 N = N = 704 N = 307 N = 307
au traitement de Aprocitentan 12,5 244
fond* mg Placebo
* ARA, ICC et un diurétique. Différence par
rapport au placebo
Bras de traitement N Référence# Moyenne Moyenne des MC Moyenne des MC Valeur de p
PASass (critère Moyenne des MC (LC Moyenne des MC (LC
d’évaluation princip à 97,5 %) à 97,5 %)
al)
12,5 mg 243 153,2 -15,3 (-17,4 à -3,8 (-6,8 à -0,8) 0,0042*
-13,2)
25 mg 243 153,3 -15,2 (-17,3 à -3,7 (-6,7 à -0,8) 0,0046*
-13,1)
Placebo 244 153,3 -11,5 (-13,6 à -9,4) - -
PADass Moyenne des MC (LC Moyenne des MC (LC
à 95 %) à 95 %)
12,5 mg 243 87,9 -10,4 (-11,6 à -9,3) -3,9 (-5,6 à -2,3) < 0,0001
25 mg 243 87,7 -11,0 (-12,1 à -9,8) -4,5 (-6,1 à -2,9) < 0,0001
Placebo 244 87,1 -6,5 (-7,6 à -5,3) - -
# Valeur de référenc
e observée (baseline
). * Statistiquemen
t significatif au
niveau de 2,5 %
prédéfini dans la
stratégie d’analyse.
LC = limite de
confiance ; Moyenne
des MC = moyenne
des moindres carrés
; PADass = pression
artérielle diastoliq
ue en position
assise ; PASass =
pression artérielle
systolique en
position assise.
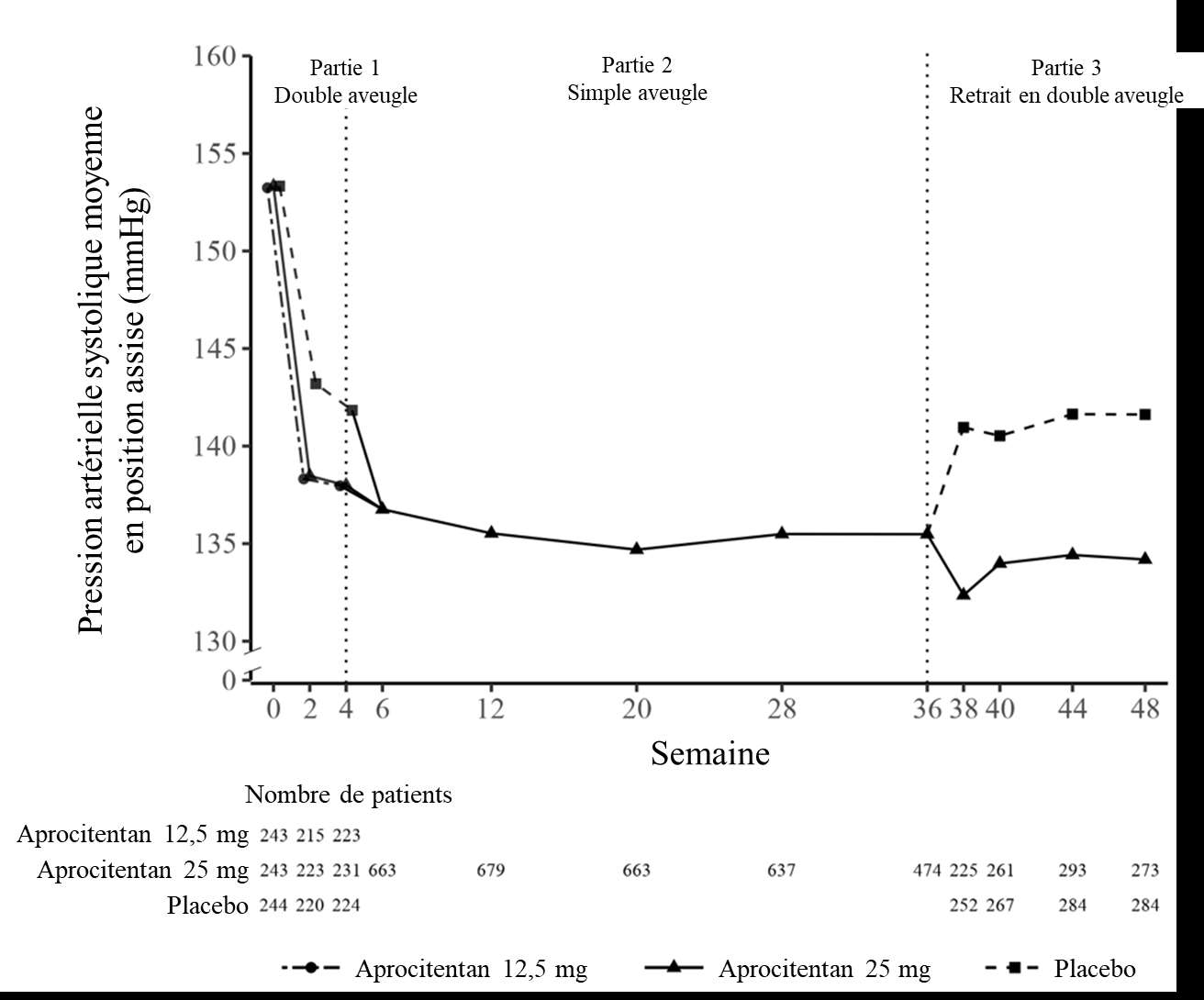
La persistance de l’effet antihypertenseur de l’aprocitentan a été montrée dans le traitement de sevrage en double aveugle (partie 3). Après une nouvelle randomisation, chez les patients recevant le placebo, la PASass moyenne a augmenté, tandis que chez les patients recevant l’aprocitentan 25 mg, l’effet moyen sur la PASass était stable, avec comme résultat une différence statistiquement significative. L’effet du traitement sur la PADass était constant (Tableau 4). Différence par
rapport au placebo
Bras de traitement N Référence du sevrage Moyenne des MC (LC Moyenne des MC (LC Valeur de p
en double aveugle# à 95 %) à 95 %)
Moyenne
PASass (critère
secondaire d’évaluat
ion clé)
25 mg 307 135,3 -1,5 (-3,0 à 0,0) -5,8 (-7,9 à -3,7) < 0,0001*
Placebo 307 136,4 4,4 (2,9 à 5,8) - -
PADass
25 mg 307 76,1 -0,5 (-1,5 à 0,5) -5,2 (-6,6 à -3,8) < 0,0001
Placebo 307 76,3 4,7 (3,7 à 5,7) - -
# Valeur de référenc
e observée (baseline
). Référence du
sevrage en double
aveugle : Semaine
36. * Statistiquemen
t significatif au
seuil de 5 % prédéfi
ni dans la stratégie
d’analyse. LC =
limite de confiance
; Moyenne des MC =
moyenne des moindres
carrés ; PADass =
pression artérielle
diastolique en
position assise ;
PASass = pression
artérielle systoliqu
e en position
assise.
L’effet était également constant entre la PAS et la PAD obtenues par une mesure ambulatoire de la pression artérielle (MAPA) et évaluées pendant la journée, pendant la nuit et sur des périodes de 24 h à la Semaine 4 (Figure 1) et à la Semaine 40.
MAPA = mesure ambulatoire de la pression artérielle ; PA = pression artérielle ; LC = limites de confiance ; PAD = pression artérielle diastolique ; Diff. moy. MC = différence moyenne des moindres carrés par rapport au placebo ; PAS = pression artérielle systolique.
| |

| ||||||||||||
|
|