Composition
Principes actifs
Sébétralstat
Excipients
Noyau du comprimé : Cellulose microcristalline, croscarmellose sodique (équivalent à 2,3 mg de sodium), povidone K30, stéarate de magnésium.
Pelliculage : Copolymère greffé d’alcool polyvinylique et de macrogol, talc, dioxyde de titane (E171), monocaprylocaprate de glycérol (type 1), alcool polyvinylique, oxyde de fer jaune (E172), oxyde de fer noir (E172) , maltodextrine, gomme de guar, hypromellose, triglycérides à chaîne moyenne.
Forme pharmaceutique et quantité de principe actif par unité
Comprimé pelliculé.
Chaque comprimé pelliculé contient 300 mg de sébétralstat.
Ekterly est un comprimé biconvexe ovale de couleur jaune portant le logo "K" de KalVista gravé sur une face et la mention "300" sur l’autre face.
Indications/Possibilités d’emploi
Ekterly est indiqué dans le traitement des crises aiguës d’angiœdème héréditaire (AOH) chez les adultes et les adolescents âgés de 12 ans et plus.
Posologie/Mode d’emploi
Posologie
La dose recommandée d’Ekterly est de 300 mg, à administrer dès l’apparition des premiers signes d’une crise. Une dose supplémentaire d’un comprimé peut être prise au besoin.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie d’Ekterly n’est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (classe A ou B de Child-Pugh). L’utilisation d’Ekterly chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) n’est pas recommandée (voir la rubrique "Pharmacocinétique" ).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la posologie n’est nécessaire chez les patients présentant une insuffisance rénale (voir la rubrique "Pharmacocinétique" ).
Patients recevant des inhibiteurs puissants du CYP3A4
Chez les patients recevant des inhibiteurs puissants du CYP3A4, la posologie recommandée pour le traitement d’une crise d’AOH est une dose unique de 300 mg.
Patients recevant des inducteurs modérés ou puissants du CYP3A4
Chez les patients recevant un inducteur modéré ou puissant du CYP3A4, la posologie recommandée pour le traitement d’une crise d’AOH est une dose unique de 900 mg (3 comprimés de 300 mg).
Patients âgés
Seules des données limitées portant sur un total de 10 participants âgés de 65 ans et plus sont disponibles (2 traités par sébétralstat 300 mg et 8 traités par sébétralstat 600 mg). Ces données suggèrent qu’aucun ajustement de la posologie n’est nécessaire dans cette population (voir rubrique "Pharmacocinétique" ).
Population pédiatrique
La sécurité et l’efficacité du sébétralstat chez les enfants âgés de moins de 12 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale. Les comprimés pelliculés peuvent être pris à jeun ou au cours des repas.
Contre-indications
Hypersensibilité au principe actif ou à l’un des excipients mentionnés selon la composition.
Mises en garde et précautions
Crises laryngée : Conseiller aux patients de consulter immédiatement un médecin après le traitement d’une crise laryngée par Ekterly.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’estàdire qu’il est essentiellement "sans sodium" .
In vitro, le sébétralstat est un inhibiteur de MATE1, MATE2-K et OCT2 et l’administration concomitante peut augmenter l’exposition à ces transporteurs tels que la metformine. Les médecins doivent envisager de surveiller le lactate sanguin et la fonction rénale des patients qui présentent un risque plus élevé d’acidose lactique (voir la rubrique "Interactions" ).
Interactions
Effets d’autres médicaments sur la pharmacocinétique du sébétralstat
Le sébétralstat est principalement métabolisé par le CYP3A4 (voir la rubrique "Pharmacocinétique" ).
Des études cliniques d’interaction ont été menées pour évaluer l’effet des inhibiteurs et inducteurs puissants, modérés et faibles du CYP3A4 sur la pharmacocinétique du sébétralstat. Les résultats de ces études sont décrits ci-dessous et présentés dans le Tableau 1, qui montre les rapports de moyenne géométrique (RMG) pour les paramètres pharmacocinétiques pendant l’administration avec ou sans traitement concomitant avec des intervalles de confiance à 90 %.
Inhibiteur du CYP3A4
L’administration d’itraconazole, un inhibiteur puissant du CYP3A4, a entraîné une augmentation de 135 % de la Cmax d’une dose de 600 mg de sébétralstat et de 420 % de l’ASC. L’administration de vérapamil, un inhibiteur modéré du CYP3A, a entraîné une augmentation de 76 % de la Cmax d’une dose de 600 mg de sébétralstat et de 102 % de l’ASC. L’utilisation concomitante de cimétidine (inhibiteur faible du CYP3A4) n’a pas entraîné d’augmentation de la Cmax ou de l’ASC d’une dose de 600 mg de sébétralstat.
Chez les patients recevant un inhibiteur puissant du CYP3A4 (par exemple clarithromycine, itraconazole, kétoconazole, ritonavir), la posologie recommandée pour le traitement d’une crise d’AOH est une dose unique de 300 mg. Aucun ajustement de la posologie n’est nécessaire chez les patients recevant des inhibiteurs faibles ou modérés du CYP3A4.
Inducteurs du CYP3A4
L’administration de phénytoïne, un inducteur puissant du CYP3A4, a entraîné une diminution de 66 % de la Cmax d’une dose de 600 mg de sébétralstat et de 83 % de l’ASC. L’administration d’éfavirenz, un inducteur modéré du CYP3A4, a entraîné une diminution de 63 % de la Cmax d’une dose de 600 mg de sébétralstat et de 79 % de l’ASC. L’utilisation concomitante de modafinil, un inducteur faible du CYP3A4, a entraîné une diminution de 11 % de la Cmax d’une dose de 600 mg de sébétralstat et de 21 % de l’ASC.
Chez les patients utilisant des inducteurs puissants ou modérés du CYP3A4 (par exemple rifampicine, carbamazépine, phénytoïne, phénobarbital, éfavirenz), la posologie recommandée pour le traitement d’une crise d’AOH est une dose unique de 900 mg (3 comprimés de 300 mg). Aucun ajustement de la posologie n’est nécessaire lors de l’utilisation concomitante d’inducteurs faibles du CYP3A4.
Tableau 1. Interactions entre le sébétralstat et d’autres médicaments (médiés par le CYP3A4)
Principe actif (schéma Effet sur la concentration de Recommandation pour
posologique) sébétralstat RMG (IC à 90 %) l’utilisation concomitante
(Mécanisme d’interaction possible)
Itraconazole (200 mg une ASC0-t : 521,07 (455,27-596,37) Une dose unique de 300 mg de
fois par jour pendant 6 ASC0-inf : 520,13 (455,82-593,53) sébétralstat chez des
jours) Sébétralstat (600 mg Cmax : 235,37 (193,18-286,78) patients prenant un puissant
en une seule prise) (Inhibition du CYP3A4) inhibiteur du CYP3A4
Vérapamil (240 mg une fois ASC0-t : 203,62 (183,20-226,32) Pas d’ajustement de la
par jour pendant 6 jours) ASC0-inf : 202,15 (182,53-223,88) posologie chez les patients
Sébétralstat (600 mg en une Cmax : 176,37 (147,00-211,60) prenant un inhibiteur modéré
seule prise) (Inhibition du CYP3A4) du CYP3A4
Cimétidine (800 mg en une ASC0-t : 88,68 (74,28-105,86) Pas d’ajustement de la
seule prise) Sébétralstat ASC0-inf : 87,78 (73,41-104,96) posologie chez les patients
(600 mg en une seule prise) Cmax : 77,93 (63,47-95,67) (Aucun prenant un inhibiteur faible
effet observé) du CYP3A4
Phénytoïne (100 mg 3 fois ASC0-t : 20,89 (16,86-25,87) Une dose unique de 900 mg de
par jour pendant 15 jours) ASC0-inf : 20,82 (16,81-25,78) Cmax sébétralstat chez des
Sébétralstat (600 mg en une : 36,66 (28,15-47,75) (Induction patients prenant un puissant
seule prise) du CYP3A4) inducteur du CYP3A4
Éfavirenz (600 mg une fois ASC0-t : 16,44 (13,65-19,81) Une dose unique de 900 mg de
par jour pendant 14 jours) ASC0-inf : 17,28 (14,43-20,68) Cmax sébétralstat chez des
Sébétralstat (600 mg en une : 33,64 (24,48-46,24) (Induction patients prenant un inducteur
seule prise) du CYP3A4) modéré du CYP3A4
Modafinil (200 mg une fois ASC0-t : 78,79 (64,90-95,67) Pas d’ajustement de la
par jour pendant 15 jours) ASC0-inf : 78,58 (64,76-95,35) Cmax posologie chez les patients
Sébétralstat (600 mg en une : 88,96 (63,87-123,90) (Induction prenant un inducteur faible
seule prise) du CYP3A4) du CYP3A4
Le sébétralstat est un substrat de la glycoprotéine P (P-gp) et de la Breast Cancer Resistance Protein (BCRP) in vitro. Le sébétralstat n’est pas un substrat de OATP1B1, OATP1B3, OAT1, OAT3, OCT2 et MATE1 in vitro. Il s’agit d’un substrat limite de MATE2-K, mais cela n’est pas considéré comme cliniquement pertinent.
Une étude clinique d’interaction a été menée pour évaluer l’effet des inhibiteurs de la P-gp et de la BCRP sur la pharmacocinétiqu du sébétralstat. Les résultats de cette étude sont décrits ci-dessous et présentés dans le Tableau 2, qui montre les rapports de moyenne géométrique (RMG) pour les paramètres pharmacocinétiques pendant l’administration avec ou sans traitement concomitant avec des intervalles de confiance à 90 %.
Inhibiteurs de la P-gp
L’administration de quinidine, un inhibiteur de la P-gp, a entraîné une diminution de 18 % de la Cmax d’une dose de 600 mg de sébétralstat et de 14 % de l’ASC. L’exposition au sébétralstat peut être augmentée par l’utilisation concomitante des inhibiteurs de la P-gp, mais aucun ajustement de la posologie n’est nécessaire.
Inhibiteurs de la BCRP
L’administration d’eltrombopag, un inhibiteur de la BCRP, a entraîné une diminution de 12 % de la Cmax d’une dose de 600 mg de sébétralstat, par contre l’ASC est restée identique. La concentration maximale du sébétralstat peut être augmentée par l’utilisation concomitante des inhibiteurs de la BCRP, mais aucun ajustement de la posologie n’est nécessaire.
Tableau 2. Interactions entre le sébétralstat et d’autres médicaments (médiés par les transporteurs)
Principe actif (schéma Effet sur la concentration de Recommandation pour
posologique) sébétralstat RMG (IC à 90 %) l’utilisation concomitan
(Mécanisme d’interaction possible) te
Quinidine (300 mg 1 heure avant ASC0-t : 114,52 (97,09-135,06) Pas d’ajustement de la
et 3 heures après la dose de ASC0-inf : 114,18 (97,07-134,31) posologie chez les
sébétralstat) Sébétralstat (600 Cmax : 235,37 117,75 (88,23-157,14) patients prenant un
mg en une seule prise) (Inhibition de la P-gp) inhibiteur de la P-gp
Eltrombopag (75 mg une fois par ASC0-t : 103,16 (88,32-120,50) Pas d’ajustement de la
jour pendant 8 jours) ASC0-inf : 102,61 (88,15-119,45) posologie chez les
Sébétralstat (600 mg en une seule Cmax : 111,56 (85,98-144,77) patients prenant un
prise) (Inhibition de la BCRP) inhibiteur de la BCRP
Effets du sébétralstat sur la pharmacocinétique d’autres médicaments
Aucune étude d’interaction n’a été réalisée pour évaluer l’effet du sébétralstat sur d’autres médicaments. Du fait de l’utilisation intermittente du sébétralstat et de son absorption et élimination rapides, le potentiel d’induction d’interactions avec des substrats des enzymes du cytochrome et des transporteurs d’Ekterly est faible.
Les études in vitro indiquent que le sébétralstat inhibe les cytochromes 2C9 et 3A4 (CI50 de 30,1 et 120 µM respectivement), ainsi que les transporteurs BCRP, OATP1B1, OATP1B3, OAT3, OCT2, MATE1 et MATE2-K (CI50 de 82,3, 86,3, 51,3, 19,2, 5,28, 8,05 et 7,76 µM respectivement). Aucune donnée d’interaction clinique n’est disponible. Le potentiel d’interaction doit être pris en compte lorsque le sébétralstat est administré à des patients prenant des substrats de ces enzymes et des transporteurs, en particulier les substrats à marge thérapeutique étroite. Si possible, les substrats de ces médicaments et transporteurs ne doivent pas être pris au même moment de la journée que celui de la prise du sébétralstat pour traiter une crise d’AOH afin de réduire au maximum le potentiel d’interaction.
Les études in vitro indiquent que le sébétralstat inhibe les UGT 1A4 et 1A9 (CI50 de 57,5 et 31,1 µM respectivement). L’inhibition de l’UGT1A6, UGT2B7, UGT1A1, et UGT1A3 (CI50 de > 100 µM, > 100 µM, 87,9 µM et 90,1 µM) par le sébétralstat était absente ou faible. Aucune donnée d’interaction clinique n’est disponible. In vitro, le sébétralstat n’est pas un inhibiteur de la CYP1A2, 2B6, 2C8, 2C19 et 2D6 ou des transporteurs P-gp ou OAT1. Aucune inhibition dépendant du temps des enzymes du cytochrome n’a été observée. In vitro, l’induction du CYP3A4 a été observée à des concentrations de ³10 µM ; in vitro, l’induction de CYP1A2, 2B6, 2C8, 2C9 et 2C19 à 100 µM était absente ou minimale. Étant donnée l’utilisation intermittente du sébétralstat, l’induction in vitro du CYP3A4 n’est pas considérée comme cliniquement pertinente.
Grossesse/allaitement
Grossesse
Il n’existe pas de données concernant l’emploi d’Ekterly chez la femme enceinte. Des études chez les rates enceintes indiquent que l’administration quotidienne de sébétralstat à des expositions supérieures aux expositions cliniques était associée à une toxicité embryo-fœtale. Une étude chez les lapines a donné des résultats équivoques (voir "Données précliniques" ).
Par mesure de précaution, il est préférable d’éviter l’utilisation d’Ekterly pendant la grossesse et chez les femmes en âge de procréer qui n’utilisent pas une méthode de contraception efficace et médicalement appropriée.
Allaitement
On ne sait pas si le sébétralstat ou ses métabolites sont excrétés dans le lait maternel. Les données disponibles pour les animaux ont mis en évidence l’excrétion de sébétralstat et/ou de ses métabolites dans le lait (voir "Données précliniques" ).
Un risque pour le nouveau-né/nourrisson ne peut être exclu.
Il faut décider soit d’interrompre l’allaitement, soit d’interrompre/de renoncer au traitement par Ekterly, en tenant compte du bénéfice de l’allaitement pour l’enfant par rapport au bénéfice du traitement pour la femme.
Fertilité
Il n’existe pas de données concernant les effets d’Ekterly sur la fertilité humaine. Aucun effet sur la fertilité n’a été observé dans les expérimentations animales (voir "Données précliniques" ).
Effet sur l’aptitude à la conduite et l’utilisation de machines
Ekterly n’a aucune influence ou une influence négligeable sur l’aptitude à la conduite et l’utilisation de machines.
Effets indésirables
Résumé du profil de sécurité
Au total, Ekterly a été administré chez 411 volontaires sains et 239 patients atteints d’angiœdème héréditaire. Dans les études cliniques utilisées pour l’enregistrement, 1945 crises d’AOH ont été traitées par Ekterly.
Les céphalées sont l’effet indésirable le plus fréquent chez les patients atteints d’AOH recevant Ekterly (rapportées par 9,2 % des patients). En général, les événements de céphalées signalés étaient de sévérité légère à modérée, non graves, et se sont résolus sans intervention.
Liste tabulée des effets indésirables
La fréquence des effets indésirables présentés dans le tableau est définie selon la convention suivante :
très fréquent (≥1/10)
fréquent (≥1/100 à <1/10)
occasionnel (≥1/1000 à <1/100)
rare (≥1/10 000 à <1/1000)
très rare (<1/10 000).
Tableau 3. Synthèse des effets indésirables par classe de systèmes d’organes et fréquence
Classe de systèmes d’organes Effet indésirable Fréquence Affections du système nerveux Céphalées Fréquent
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Aucun cas de surdosage n’a été rapporté dans les études cliniques.
Propriétés/Effets
Code ATC
B06AC08
Classe pharmacothérapeutique : Autres agents hématologiques, médicaments utilisés dans l’angiœdème héréditaire.
Mécanisme d’action
Le sébétralstat est un inhibiteur compétitif réversible de la kallikréine plasmatique. La kallikréine plasmatique est une protéase à sérine qui clive le kininogène de haut poids moléculaire (KHPM) libérant de la bradykinine (BK) ce qui augmente la perméabilité vasculaire par l’activation des récepteurs de BK causant les œdèmes. Le sébétralstat inhibe le clivage du KHPM et la libération de BK, empêchant l’activation des récepteurs de BK et arrêtant la progression des crises d’AOH. Le sébétralstat inhibe également le mécanisme de rétrocontrôle positif du système kininekallikréine par la kallikréine plasmatique, ce qui diminue la synthèse de facteur XIIa et la libération supplémentaire de kallikréine plasmatique.
Pharmacodynamique
L’inhibition dépendante de la concentration de la kallikréine plasmatique, mesurée comme la réduction de l’activité enzymatique spécifique, était rapide, avec une inhibition quasi complète de la kallikréine plasmatique dès 15 minutes suivant l’administration chez les patients atteints d’AOH.
Électrophysiologie cardiaque
Après administration de 3000 mg d’Ekterly chez des volontaires sains (dose correspondant à 5 fois la dose maximale recommandée) dans le cadre d’une étude de l’intervalle QT, l’augmentation maximale moyenne de l’intervalle QTc était de 10,4 ms (limite supérieure de l’intervalle de confiance à 90 % = 15,3 ms). L’augmentation de l’intervalle QTc dépendait de la concentration.
Efficacité clinique
L’efficacité d’Ekterly dans le traitement des crises aiguës chez les adultes et les adolescents âgés de 12 ans et plus atteints d’angiœdème héréditaire (AOH) a été évaluée dans une étude croisée randomisée en double aveugle, contrôlée contre placebo, à trois périodes (KONFIDENT).
Sur les 136 patients randomisés dans l’étude, 110 patients ont traité au moins une crise aiguë, soit un total cumulé de 264 crises (87 ont été traitées avec une dose de 300 mg d’Ekterly, 93 avec une dose de 600 mg d’Ekterly et 84 avec le placebo). Les crises étaient d’intensité légère à très sévère et touchaient toutes les régions anatomiques.
Si nécessaire, une dose supplémentaire pouvait être prise après le traitement de chaque crise. Le critère d’évaluation principal était le délai jusqu’au début du soulagement des symptômes, évalué en utilisant l’échelle d’appréciation globale du changement par le patient (Patient Reported Global Impression of Change – PGI-C). Pour l’échelle PGI-C, les patients devaient évaluer leurs symptômes de la crise sur une échelle à 7 points (de "Très forte aggravation" à "Très forte amélioration" ). Pour atteindre le critère d’évaluation principal, le patient devait indiquer une réponse positive et maintenue sur l’échelle PGI-C dans les 12 heures.
Par rapport au placebo, un délai significativement plus court jusqu’au début du soulagement des symptômes a été observé avec la dose de 300 mg d’Ekterly (valeur de p ajustée par correction de Bonferroni < 0,0001) (Tableau 4, figure 1).
Figure 1. Étude KONFIDENT – Courbe de Kaplan-Meier du délai jusqu’au début du soulagement des symptômes dans les 12 heures suivant l’administration
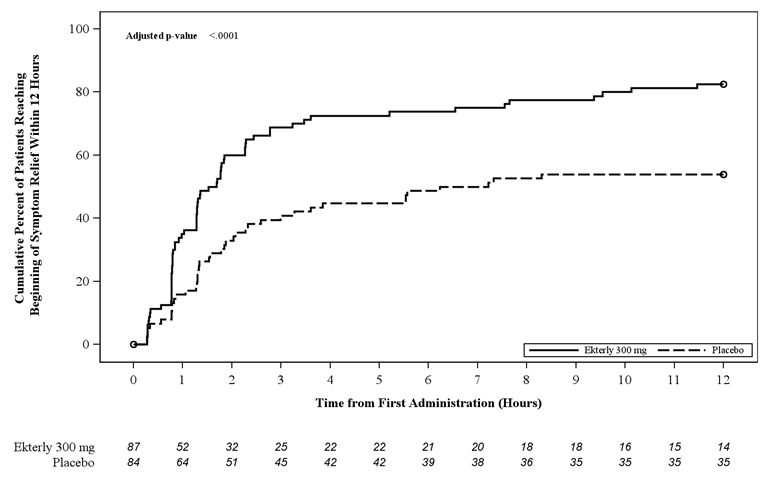
Tableau 4. Étude KONFIDENT – Délai jusqu’au début du soulagement des symptômes dans les 12 heures suivant l’administration
300 mg d’Ekterly Placebo N 87 84 Médiane (IC à 95 %) 1,61 (1,28 ; 2,27) 6,72 (2,33 ; NE)
NE = non évaluable à 12 heures
Le premier principal critère d’évaluation secondaire était le délai jusqu’à la réduction de la sévérité de la crise sur l’échelle d’appréciation globale de la sévérité par le patient (Patient Global Impression of Severity – PGI-S). Par rapport au placebo, un délai significativement plus court jusqu’à la diminution de la sévérité a été observé avec la dose de 300 mg d’Ekterly (valeur de p ajustée = 0,0036) (figure 2).
Figure 2. Étude KONFIDENT – Courbe de Kaplan-Meier du délai jusqu’à la réduction de la sévérité de la crise dans les 12 heures suivant l’administration

Le deuxième principal critère d’évaluation secondaire était le délai jusqu’à la résolution complète de la crise, déterminée par l’indication "Aucun symptôme" sur l’échelle PGI-S. Par rapport au placebo, un délai significativement plus court jusqu’à la résolution complète de la crise a été observé avec la dose de 300 mg d’Ekterly (valeur de p ajustée = 0,0022) (figure 3).
Figure 3. Étude KONFIDENT – Courbe de Kaplan-Meier du délai jusqu’à la résolution complète de la crise dans les 24 heures suivant l’administration

Par rapport au placebo, le traitement par Ekterly a réduit l’anxiété cumulée sur 12 heures après l’administration.
Dans l’étude KONFIDENT, les résultats du critère d’efficacité principal et des principaux critères d’efficacité secondaires observés dans tous les sousgroupes, incluant les sousgroupes définis en fonction du sexe, du groupe ethnique, de l’âge, de la sévérité des crises à l’inclusion, de la localisation des crises à l’inclusion, du délai entre le début de la crise et le traitement, de l’utilisation d’un traitement prophylactique au long cours et de la région, concordaient avec les résultats observés dans la population totale.
Dans l’étude en ouvert KONFIDENT-S, des crises récurrentes ont été traitées par Ekterly pendant jusqu’à deux ans. Au total, 1706 crises ont été traitées chez 134 patients (dont 23 adolescents). Le nombre médian de crises traitées était de 8 (intervalle : 1 à 61 crises). Le délai médian entre le début de la crise et le traitement était de 10 minutes. Chez les adolescents, le délai médian entre le début de la crise et le traitement était de 4 minutes. Les résultats d’efficacité concordaient avec ceux de l’étude KONFIDENT (Tableau 4). L’efficacité a été maintenue lors de traitements répétés.
Quatre crises d’AOH avec atteinte laryngée ont été traitées dans l’étude KONFIDENT (deux avec la dose de 300 mg, deux avec la dose de 600 mg). Dans l’étude en ouvert KONFIDENT-S, 32 crises avec atteinte laryngée ont été traitées avec la dose de 600 mg. Les résultats en termes de délai jusqu’au début du soulagement des symptômes étaient comparables à ceux observés chez les patients présentant des crises sans atteinte laryngée. Il n’a pas été rapporté de cas de difficultés à avaler les comprimés d’Ekterly.
Population pédiatrique
L’étude KONFIDENT incluait 13 adolescents âgés de 12 à moins de 18 ans. La sécurité et l’efficacité chez les patients adolescents étaient cohérentes avec celles observées chez les adultes.
La sécurité et l’efficacité d’Ekterly chez les patients pédiatriques âgés de moins de 12 ans n’ont pas été établies.
Pharmacocinétique
Absorption
Après administration à jeun d’une dose de 300 mg, les concentrations plasmatiques maximales de sébétralstat étaient atteintes en une heure environ.
Effet des aliments
Il n’a pas été observé de différence au niveau de l’ASC du sébétralstat après administration d’une dose de 600 mg d’Ekterly avec un repas hyperlipidique, mais une diminution de 29 % de la Cmax et une augmentation du Tmax médian de 2 heures ont été observées.
Distribution
La liaison aux protéines plasmatiques humaines est d’environ 77 %. Après administration d’une dose de 600 mg de sébétralstat radiomarqué, le rapport des concentrations sang/plasma de la radioactivité était d’environ 0,65. Après administration d’une dose de 300 mg, la moyenne géométrique du volume apparent de distribution (Vz/F) était de 208 litres.
Métabolisme
Le sébétralstat est principalement métabolisé par le CYP3A4. Après administration d’une dose de 600 mg de sébétralstat radiomarqué, le sébétralstat représentait 64,1 % de l’ASC0-24 de la radioactivité plasmatique totale, avec 11 métabolites représentant chacun 0,39 % à 7,1 % de l’ASC0-24 de la radioactivité totale. Le métabolite prédominant dans le plasma n’a pas d’activité pharmacologique.
Élimination
Après administration d’une dose de 300 mg, la moyenne géométrique de la demivie d’élimination du sébétralstat était de 3,7 heures. La moyenne géométrique de la clairance apparente (CL/F) était de 38,5 litres/heure.
Excrétion
Après administration d’une dose de 600 mg de sébétralstat radiomarqué chez des hommes volontaires sains, environ 32 % de la radioactivité ont été excrétés dans les urines et 63 % dans les fèces. Environ 8,7 % et 12,5 % de la dose ont respectivement été récupérés dans les urines et les fèces sous forme de sébétralstat inchangé. Le sébétralstat est principalement éliminé dans les fèces par métabolisme hépatique.
Linéarité/nonlinéarité
La Cmax était proportionnelle à la dose dans la plage de doses de 5 mg à 600 mg ; l’augmentation de l’ASC était plus que proportionnelle à la dose, probablement en raison de l’apparition d’une phase d’élimination terminale plus longue à des doses élevées.
Cinétique pour certains groupes de patients
Dans l’analyse pharmacocinétique de population, aucune influence du sexe, de l’âge (12 à 68 ans), du poids (44 à 135 kg) ou de l’origine ethnique n’a été mise en évidence sur la clairance ou le volume de distribution du sébétralstat.
Troubles de la fonction hépatique
La pharmacocinétique du sébétralstat administré à la dose de 600 mg a été étudiée chez des patients présentant une insuffisance hépatique légère ou modérée (classe A ou B de Child-Pugh). Chez les patients présentant une insuffisance hépatique légère, la Cmax était augmentée de 7 % et l’ASC de 16 % par rapport aux sujets ayant une fonction hépatique normale. Chez les patients atteints d’insuffisance hépatique modérée, la Cmax était augmentée de 63 % et l’ASC de 100 %.
La pharmacocinétique du sébétralstat n’a pas été étudiée chez les patients présentant une insuffisance hépatique sévère.
Troubles de la fonction rénale
La pharmacocinétique du sébétralstat n’a pas été étudiée chez les patients présentant une insuffisance rénale. Le sébétralstat n’étant pas majoritairement éliminé par les reins et n’étant pas utilisé comme traitement à long terme, aucun effet pertinent d’une insuffisance rénale sur la pharmacocinétique du sébétralstat n’est attendu.
Patients âgés
Seules des données limitées portant sur un total de 10 participants âgés de 65 ans et plus sont disponibles (2 traités par sébétralstat 300 mg et 8 traités par sébétralstat 600 mg). Ces données suggèrent qu’un ajustement de la posologie n’est pas nécessaire dans cette population.
Données précliniques
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité et cancérogenèse n’ont pas révélé de risque particulier pour l’homme.
Le potentiel cancérogène du sébétralstat a été évalué dans une étude de 26 semaines chez la souris transgénique rasH2 et dans une étude de 104 semaines chez le rat. Aucune augmentation de l’incidence des tumeurs malignes n’a été observée et aucun effet cancérogène n’a été mis en évidence chez les deux espèces animales, quelle que soit la dose. L’exposition aux doses les plus élevées (en lien avec l’ASC du sébétralstat plasmatique libre) chez les souris mâles et femelles représentait respectivement 0,2 fois et 0,4 fois l’exposition humaine à la dose maximale recommandée chez l’homme (DMRH) et 5,7 fois chez les rats.
Une étude sur le développement embryofœtal menée chez des rates gravides ayant reçu quotidiennement du sébétralstat à des doses (sur la base de l’ASC plasmatique libre) équivalant à trois fois la DMRH n’a révélé aucun signe de nocivité pour le fœtus en développement. À une exposition plus élevée (sur la base de l’ASC plasmatique libre) de 12 fois la DMRH, on a observé des pertes embryofœtales et une faible incidence de malformations (fentes palatines et anomalies du septum ventriculaire). Aucun effet n’a été observé dans une étude sur le développement prénatal et postnatal chez le rat, dans laquelle l’exposition chez les rates gravides (sur la base de l’ASC plasmatique libre) était au moins de trois fois la DMRH.
Une étude sur le développement embryofœtal avec une administration quotidienne a été menée chez des lapines gravides ayant reçu des doses (sur la base de l’ASC plasmatique libre) équivalant à 6,8 fois la DMRH. Une faible incidence de malformations majeures a été observée dans tous les groupes de doses de sébétralstat. Cependant, aucune relation dose-réponse n’a été observée et toutes les malformations se trouvaient dans les limites des données historiques de contrôle. Par conséquent, l’association avec le sébétralstat est équivoque et la pertinence clinique est incertaine. Le lapin ne fait pas partie des espèces pharmacologiquement pertinentes.
Le sébétralstat n’a eu aucun effet sur l’accouplement et la fertilité chez les souris mâles et femelles à des expositions (sur la base de l’ASC plasmatique libre) de 7,7 fois l’exposition à la DMRH.
Après administration d’une dose unique de sébétralstat radiomarqué chez des rates allaitantes, les concentrations de la radioactivité totale étaient comparables dans le lait et le plasma, la concentration maximale étant observée une heure postdose. À l’heure 24 postdose, les valeurs de la radioactivité dans le lait et le plasma étaient proches de celle induite par le rayonnement ambiant.
Remarques particulières
Incompatibilités
Non pertinent.
Durée de conservation
Ce médicament ne doit pas être utilisé au-delà de la date indiquée après la mention "EXP" sur l’emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30 °C.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation
69540 (Swissmedic)
Présentation
Boîtes de 4 comprimés [B] (non commercialisées actuellement)
Boîtes de 6 comprimés [B]
Les comprimés sont emballés dans des plaquettes en OPA/alu/PVC avec pellicule en aluminium (1 comprimé par plaquette).
Titulaire de l’autorisation
KalVista Pharmaceuticals Switzerland GmbH
Baarerstrasse 135
6300, Zug
Suisse
Mise à jour de l’information
Juillet 2025