ZusammensetzungWirkstoffe
Gadoteridolum
Hilfsstoffe
Calteridolum calcicum, Trometamolum, Acidum hydrochloridum / Natrii hydroxidum (ad pH), Aqua ad iniectabile q.s. ad solutionem pro 1 ml.
Natriumgehalt: Max. 0.0575 mg/ml unter Berücksichtigung der variablen Natriumhydroxid-Zusätze.
Indikationen/AnwendungsmöglichkeitenBeim Einsatz in der Magnetresonanz-Tomographie (MRT) ermöglicht ProHance eine Kontrastverstärkung des Gehirns, des Rückenmarks und des umgebenden Gewebes mit verbesserter Darstellung von Läsionen mit abnormer Vaskularität oder von Läsionen, die mutmasslich mit einer Zerstörung der Blut-Hirn-Schranke einhergehen.
ProHance wird auch bei Erwachsenen ab 18 Jahren für die Ganzkörper-MRT eingesetzt, wie z.B. Krankheitszustände an Kopf, Hals, Leber, Brust, dem muskuloskelettalen System sowie den Weichteilen.
ProHance sollte nur dann angewendet werden, wenn die diagnostische Information notwendig ist und mit einer Magnetresonanztomographie (MRT) ohne Kontrastmittelverstärkung nicht erhoben werden kann.
Dosierung/AnwendungÜbliche Dosierung
Es ist die geringstmögliche Dosis zu verwenden, mit der eine für diagnostische Zwecke ausreichende Kontrastverstärkung erzielt wird. Die Dosis wird abhängig vom Körpergewicht des Patienten berechnet und sollte die in diesem Abschnitt angegebene empfohlene Dosis pro Kilogramm Körpergewicht nicht überschreiten.
Die empfohlene Dosierung bei Kindern und Jugendlichen (Kontrastverstärkung des Gehirns, des Rückenmarks und des umgebenden Gewebes), sowie Erwachsenen beläuft sich auf 0,1 mmol/kg einer 0,5 M Lösung (0,2 mL/kg). In Ausnahmefällen wie z.B. Verdacht auf Gehirnmetastasen oder andere kontrastarme Läsionen kann die zusätzliche Gabe von 0.4 ml/kg (0.2 mmol/kg) gerechtfertigt sein.
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktionsstörungen
ProHance sollte bei Patienten mit schwerer eingeschränkter Nierenfunktion (GFR < 30 ml/min/1.73 m2) und bei Patienten in der perioperativen Phase einer Lebertransplantation nur nach sorgfältiger Nutzen-Risiko-Abwägung angewendet werden und nur, wenn die diagnostische Information notwendig ist und mit einer MRT ohne Kontrastmittelverstärkung nicht erhoben werden kann (siehe „Warnhinweise und Vorsichtsmassnahmen“). Falls die Anwendung von ProHance notwendig ist, sollte die Dosis 0.2 ml/kg (0.1 mmol/kg) Körpergewicht nicht übersteigen und während eines Scans sollte nicht mehr als eine Dosis angewendet werden. Da keine Informationen zur wiederholten Anwendung vorliegen, sollte die Injektion von ProHance nicht wiederholt werden, es sei denn, der Abstand zwischen den Injektionen beträgt mindestens 7 Tage.
Ältere Patienten
Es ist keine Dosisanpassung notwendig. Trotzdem ist bei älteren Patienten (65 Jahre und älter) Vorsicht geboten (siehe „Warnhinweise und Vorsichtsmassnahmen“).
Neugeborene bis zum Alter von 4 Wochen und Säuglinge bis zum Alter von 1 Jahr
Wegen der unreifen Nierenfunktion bei Neugeborenen bis zu einem Alter von 4 Wochen und bei Säuglingen bis zum Alter von 1 Jahr sollte ProHance bei diesen Patienten nur nach sorgfältiger Abwägung in einer Dosis von höchstens 0.2 ml/kg (0.1 mmol/kg) Körpergewicht angewendet werden. Während eines Scans sollte nicht mehr als eine Dosis angewendet werden. Da keine Informationen zur wiederholten Anwendung vorliegen, sollte die Injektion von ProHance nicht wiederholt werden, es sei denn, der Abstand zwischen den Injektionen beträgt mindestens 7 Tage.
1Von einem Ganzkörperscan bei Kindern und Jugendlichen unter 18 Jahren wird abgeraten.
Art der Anwendung
Zwei Stunden vor der Untersuchung soll der Patient nüchtern bleiben.
Um eine vollständige Injektion des Kontrastmittels sicherzustellen, sollte nach der Injektion mit 5 ml physiologischer Kochsalzlösung nachgespült werden. Die kontrastverstärkte MRT sollte binnen einer Stunde nach der Injektion von ProHance abgeschlossen sein.
Parenterale Präparate sollten vor der Verabreichung visuell auf Partikel und Verfärbungen überprüft werden, sofern Lösung und Behältnis dies zulassen. Unverbrauchte Restmengen an Kontrastmittel sind zu entsorgen.
KontraindikationenSchrittmacher, Clips.
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenDie für die Magnetresonanz-Bildgebung generell notwendigen Überlegungen und Vorkehrungen zur Sicherheit gelten auch beim Einsatz von ProHance als Kontrastmittel. Diagnostische Verfahren unter Verwendung von Kontrastmitteln sollten unter der Anleitung eines entsprechend ausgebildeten Arztes durchgeführt werden, der mit dem anzuwendenden Verfahren gründlich vertraut ist.
Patienten mit bekannter Arzneimittelallergie oder anderen überempfindlichkeits-ähnlichen Reaktionen müssen während der Behandlung sowie der Verabreichung des Kontrastmittels und, wenn dies vom Arzt auf Grund des Patientenzustandes als notwendig erachtet wird, darüber hinaus engmaschig beobachtet werden.
Wie bei anderen Gadolinium-haltigen Kontrastmitteln gibt es auch bei ProHance Berichte über anaphylaktische/anaphylaktoide Überempfindlichkeitsreaktionen. Diese Reaktionen zeigten sich mit unterschiedlichem Schweregrad, einschliesslich anaphylaktischem Schock und Tod. Sie betrafen ein oder mehrere Körpersysteme, vor allem das respiratorische, kardiovaskuläre und mukokutane System waren betroffen. Häufig beobachtete Symptome waren trockener Hals, Halsirritation, Dyspnoe, Engegefühl in der Brust, Hitzegefühl, Dysphagie, Brennen, Pharynx-, bzw. Larynxödem, Hypotension.
Über einen anaphylaktischen Schock wurde sehr selten beim Gebrauch von ProHance berichtet. Notwendige Medikamente und Geräte müssen vorhanden sein, falls schwerwiegende Reaktionen vorkommen.
Wie bei anderen Gadolinium-haltigen Kontrastmitteln sind bei Patienten mit einer niedrigen Reizschwelle für Krampfanfälle spezielle Vorsichtsmassnahmen notwendig. Diese beinhalten eine enge Überwachung dieser Patienten sowie die Verfügbarkeit von Ausrüstung und Medikamenten für eine unverzügliche Behandlung möglicher Krampfanfälle.
Risiken im Zusammenhang mit der intrathekalen Anwendung
Gadoteridol darf nicht intrathekal angewendet werden. Schwere lebensbedrohliche und tödliche Fälle, überwiegend mit neurologischen Reaktionen (z. B. Koma, Enzephalopathie, Krampfanfälle), wurden bei intrathekaler Anwendung berichtet.
Patienten mit Nierenfunktionsstörungen
Es wird empfohlen, vor der Anwendung von ProHance bei allen Patienten das Vorliegen einer Nierenfunktionsstörung durch Labortests abzuklären.
Da Gadoteridol durch glomeruläre Filtration ausgeschieden wird, ist bei Patienten mit stark eingeschränkter Nierenfunktion Vorsicht geboten (siehe auch „Pharmakokinetik” und “Spezielle Dosierungsanweisungen”).
In Zusammenhang mit der Anwendung einiger Gadolinium-haltiger Kontrastmittel wurde bei Patienten mit akuter oder chronischer schwerer Niereninsuffizienz (GFR < 30 ml/min/1.73 m2) über eine nephrogene systemische Fibrose (NSF) berichtet. Ein besonderes Risiko besteht auch bei Patienten, die sich einer Lebertransplantation unterziehen, da die Inzidenz eines akuten Nierenversagens in dieser Gruppe hoch ist. Da die Möglichkeit besteht, dass mit ProHance eine NSF auftritt, sollte es daher bei Patienten mit schwerer Einschränkung der Nierenfunktion und bei Patienten in der perioperativen Phase einer Lebertransplantation nur nach sorgfältiger Nutzen-Risiko-Abwägung angewendet werden und nur, wenn die diagnostische Information notwendig ist und mit einer MRT ohne Kontrastmittelverstärkung nicht erhoben werden kann.
Eine Hämodialyse kurz nach der Verabreichung von ProHance kann bei Patienten hilfreich sein, um ProHance aus dem Körper zu entfernen. Es gibt kein Anzeichen dafür, dass die Einleitung einer Hämodialyse zur Prävention oder Behandlung von NSF bei Patienten angezeigt ist, die sich nicht schon einer Hämodialyse unterziehen (vgl. „Pharmakokinetik: Kinetik spezieller Patientengruppen“).
Neugeborene und Säuglinge
Wegen der unreifen Nierenfunktion bei Neugeborenen bis zum Alter von 4 Wochen und bei Säuglingen bis zu einem Alter von 1 Jahr sollte ProHance bei diesen Patienten nur nach sorgfältiger Abwägung angewendet werden. Pharmakokinetische Daten zur Exposition von Gadoteridol liegen bei Kindern ab einem Alter von 5 Jahren vor.
Ältere Patienten
Da die renale Clearance von Gadoteridol bei älteren Menschen beeinträchtigt sein kann, ist es besonders wichtig, Patienten ab 65 Jahren bezüglich einer Nierenfunktionsstörung zu überprüfen.
Wird ProHance nicht mit Einmal-Spritzen injiziert, so ist peinlich darauf zu achten, dass die Spritzen nicht mit Spuren von Reinigungsmittelrückständen kontaminiert sind.
Falls nach klinischer Beurteilung des Arztes sequentielle oder wiederholte Untersuchungen erforderlich sind, sollten ausreichende Zeitabstände zwischen den Verabreichungen eingeplant werden, um eine normale Clearance der Substanz aus dem Körper zu ermöglichen.
Wie auch bei Anwendung von anderen Kontrastmitteln können bei Gadoteridol anaphylaktoide Reaktionen mit unter Umständen lebensbedrohlichen kardiovaskulären (Schock) oder respiratorischen (Larynx-Ödem, Bronchospasmus) sowie Abdominalsymptomen, Urtikaria, Angioödem oder neurologischen Komplikationen auftreten.
Bei jeder Untersuchung müssen deshalb die personellen Voraussetzungen für die Notfalltherapie gegeben sein und das notwendige Material verwendungsbereit sein (Sauerstoff, Adrenalin, Infusionsmaterial, Intubations- und Beatmungsmöglichkeiten, u.a.). Es ist unbedingt erforderlich, mit der Anwendung der Notfallmassnahmen vertraut zu sein. Nach Kontrastmittelgabe soll der Patient noch mindestens 30 bis 60 Minuten unter Aufsicht bleiben, da erfahrungsgemäss die Mehrzahl aller schweren Zwischenfälle innerhalb dieser Zeit auftritt.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosiervolumen, d.h. es ist nahezu „natriumfrei“.
InteraktionenArzneimittelinteraktionen mit Gadoteridol sind bisher nicht bekannt.
Weder durchgeführte klinische Studien noch Laborwerte ergaben signifikante Hinweise auf Interaktionen.
Schwangerschaft, StillzeitSchwangerschaft
Gadolinium-haltige Kontrastmittel können die Plazentaschranke passieren und zu einer Exposition des Fötus führen. Im Menschen liegen nur unzureichende klinische Daten vor, so dass keine abschliessende Beurteilung des Zusammenhangs von Gadolinium-haltigen Kontrastmitteln und schädlichen fetalen Folgen möglich ist.
Es liegen keine Erfahrungen mit der Anwendung von ProHance bei Schwangeren vor.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf die Reproduktionstoxizität (siehe „Präklinische Daten“). Gadoteridol darf während der Schwangerschaft nicht verwendet werden, es sei denn, dass eine Anwendung von Gadoteridol aufgrund des klinischen Zustands der Frau erforderlich ist.
Stillzeit
Gadolinium-haltige Kontrastmittel werden in sehr geringen Mengen in die Muttermilch ausgeschieden (siehe „Präklinische Daten“). In klinischen Dosen sind wegen der geringen in die Milch ausgeschiedenen Menge und der schwachen Resorption aus dem Darmtrakt keine Auswirkungen auf den Säugling zu erwarten. Ob das Stillen fortgesetzt oder nach der Verabreichung von ProHance für 24 Stunden unterbrochen wird, sollten der Arzt und die stillende Mutter entscheiden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenAnhand des pharmakokinetischen und pharmakodynamischen Profils wird keine oder eine vernachlässigbare Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen durch den Gebrauch von Gadoteridol erwartet.
Unerwünschte WirkungenDie nachfolgenden unerwünschten Wirkungen wurden nach der Anwendung von ProHance berichtet. Spontan gemeldete unerwünschte Wirkungen sind mit der Häufigkeit „Unbekannt“ aufgeführt.
Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1000, <1/100); selten (≥1/10‘000, <1/1000); sehr selten (<1/10'000), unbekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Erkrankungen des Immunsystems
Gelegentlich: anaphylaktische/anaphylaktoide Reaktionen
Unbekannt: Angioödema
Psychiatrische Erkrankungen
Selten: Ängstlichkeit
Erkrankungen des Nervensystems
Gelegentlich: Kopfschmerzen, Parästhesie, Schwindel, Geschmacksveränderung
Selten: mentaler Leistungsabfall, gestörte Koordination, Konvulsion
Unbekannt: Bewusstlosigkeit, Koma, vasovagale Reaktion
Augenerkrankungen
Gelegentlich: erhöhter Tränenfluss
Erkrankungen des Ohrs und des Labyrinths
Selten: Tinnitus
Herzerkrankungen
Selten: verlängertes P-R-Intervall, erhöhte Herzfrequenz, AV-Knotenrhythmus, Bradykardie
Unbekannt: Herzstillstand
Gefässerkrankungen
Gelegentlich: Hautrötung, Hypotonie, Hypertonie
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Selten: Laryngospasmus, Dyspnoe, Rhinitis, Husten, Apnoe, pfeifendes Atmen
Unbekannt: Atemstillstand, Lungenödem, akutes Atemnotsyndrom
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit
Gelegentlich: Mundtrockenheit, Erbrechen
Selten: Durchfall, Bauchschmerzen, Zungenödem, oraler Pruritus, Gingivitis, weicher Stuhl, Dysphagie, erhöhter Speichelfluss
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Pruritus, Ausschlag, Urtikaria
Selten: Gesichtsödem
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Selten: Steifheit
Erkrankungen der Nieren und Harnwege
Unbekannt: akutes Nierenversagen
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Gelegentlich: Schmerzen an der Injektionsstelle, Reaktionen an der Injektionsstelle (möglicherweise durch Paravasation), Asthenia
Selten: Brustschmerz, Pyrexie
Untersuchungen
Gelegentlich: erhöhte Herzfrequenz
Beschreibung ausgewählter Nebenwirkungen
Vasovagale Reaktionen
Vasovagale Reaktionen, die selten zu vasovagaler Synkope führten, wurden während oder sofort nach Gadoteridolanwendung berichtet. Dieser Zustand ist oft verbunden mit Angstgefühlen oder schmerzhaften bzw. unangenehmen Reizen (z.B. Nadelstich für IV-Platzierung). Häufige Symptome sind auch Übelkeit, Schwindel und Diaphorese.
In schweren Fällen können Synkopen auftreten. Die Patienten sind dann gewöhnlich blass und diaphoretisch bei verändertem Bewusstseinszustand und bradykard. Zusätzlich können Patienten häufig auch von unangenehmen Vorahnungen, Ruhelosigkeit, Schwäche und verstärktem Speichelfluss geplagt werden. Eine klare Erkennung dieser Reaktion und eine Differentialdiagnose von Überempfindlichkeits-, bzw. anaphylaktischen Reaktionen ist lebenswichtig, damit die richtigen Behandlungsmassnahmen angewandt werden können, um den vagalen Reiz umzukehren.
Anaphylaktische/anaphylaktoide Reaktionen
Wie mit anderen Gadolinium-haltigen Kontrastmitteln gab es Berichte über anaphylaktische/anaphylaktoide Überempfindlichkeitsreaktionen mit Gadoteridol. Diese Reaktionen zeigten sich mit unterschiedlichem Schweregrad, eingeschlossen anaphylaktischer Schock und Tod. Sie betreffen ein oder mehrere Körpersysteme, meistens das respiratorische, kardiovaskuläre und/oder mukokutane System. Häufig berichtete Symptome schliessen ein: Engegefühl im Hals, Halsirritation, Dyspnoe, Brustbeschwerden, Hitzegefühl, Dysphagie, Brennen, Rachen- oder Kehlkopfödem, Hypotonie.
Akutes Nierenversagen
Fälle von akutem Nierenversagen wurden bei Patienten mit vorbestehender schwerwiegender eingeschränkter Nierenfunktion berichtet.
Nephrogene systemische Fibrose
Einzelfälle von nephrogener systemischer Fibrose (NSF) wurden bei Gadoteridol-Anwendung berichtet, die meisten bei Patienten, denen gleichzeitig andere Gadolinium- haltige Kontrastmittel verabreicht wurden (vgl. „Warnhinweise und Vorsichtsmassnahmen“).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungBislang wurde kein Fall von Überdosierung bekannt, daher sind auch keine Symptome einer eventuellen Überdosierung bekannt. Gadoteridol kann durch Hämodialyse entfernt werden. Es gibt jedoch keine Hinweise dafür, dass eine Hämodialyse zur Prävention einer nephrogenen systemischen Fibrose (NSF) geeignet ist.
Eigenschaften/WirkungenATC-Code
V08CA04
Wirkungsmechanismus
ProHance ist eine klare, farblose bis leicht gelbliche, sterile, wässrige Lösung, die zur Injektion bestimmt ist. Unter Anwendungsbedingungen ist ProHance hypertonisch, wobei die Osmolarität etwa dem Zweifachen derjenigen des Plasmas entspricht. ProHance enthält keine Konservierungsstoffe.
Gadoteridol als Wirkstoff von ProHance ist eine paramagnetische Substanz und entwickelt als solche ein magnetisches Moment, sobald sie in ein magnetisches Feld eingebracht wird. Das von der paramagnetischen Substanz verursachte magnetische Moment führt zu einem relativ grossen lokalen magnetischen Feld, das die Relaxationsrate von Wasserprotonen in der Umgebung der paramagnetischen Substanz verlängern kann. Bei der Magnetresonanz-Bildgebung hängt die Darstellung von gesundem und pathologischem Hirngewebe zum Teil von Unterschieden der Radiofrequenz-Signalstärke ab, die auftreten bei Unterschieden 1) der Protonendichte; 2) der longitudinalen Spin-Gitter Relaxationszeiten (T1); und 3) der Spin-Spin oder transversalen Relaxationszeit. Sobald Gadoteridol in ein magnetisches Feld eingebracht wird, verkürzt es die T1-Relaxationszeiten in den Zielgeweben. In den empfohlenen Dosen wird der Effekt mit grösster Sensitivität in sogenannten T1-gewichteten Sequenzen beobachtet.
Ein Defekt der Blut-Hirn-Schranke oder der normalen Vaskularität erlaubt eine Kumulation von Gadoteridol bei Läsionen wie beispielsweise Neoplasmen, Abszessen und subakuten Infarkten.
Pharmakodynamik
siehe «Wirkungsmechanismus»
Klinische Wirksamkeit
keine Angaben
PharmakokinetikAbsorption
keine Angaben
Distribution
Nach intravenöser Verabreichung von Gadoteridol entspricht die Pharmakokinetik bei gesunden Probanden einem offenen Zwei-Kompartiment-Modell. Die mittlere Verteilungs-Halbwertszeit beträgt etwa 12 Minuten. Das Verteilungsvolumen von ca. 129 ml/kg entspricht demjenigen des extrazellulären Wassers. Untersuchungen an Ratten ergaben keine Hinweise auf eine Plasmaproteinbindung.
Metabolismus
Eine nachweisbare Biotransformation oder ein Abbau von Gadoteridol konnte nicht beobachtet werden.
Elimination
Die mittlere Eliminationshalbwertszeit von Gadoteridol beträgt etwa 1 Stunde und 34 Minuten. Die Substanz wird ausschliesslich mit dem Harn ausgeschieden (Ausscheidungsrate ca. 95% der innerhalb von 24 Stunden nach der Injektion ausgeschiedenen Dosis).
Bei Gadoteridol entspricht die renale Clearance im Wesentlichen der plasmatischen Clearance (ca. 1.41 ml/min/kg bzw. 1.50 ml/min/kg). Dieser Befund weist darauf hin, dass das Präparat hauptsächlich durch die Niere eliminiert wird. Die Clearance ähnelt derjenigen von Substanzen, die der glomerulären Filtration unterliegen.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Daten zur Kinetik bei Leberinsuffizienz liegen bisher nicht vor.
Nierenfunktionsstörungen
Bei Patienten mit mittelschwerer bis schwerer Niereninsuffizienz wird die Verteilungs-Halbwertszeit nur unwesentlich beeinträchtigt.
Die Eliminationshalbwertszeit erhöht sich im Vergleich zu gesunden Probanden mit 1 Stunde und 34 Minuten bei mittelschwerer Niereninsuffizienz auf 6 Stunden und 57 Minuten resp. auf 9 Stunden und 32 Minuten bei schwerer Beeinträchtigung der Nierenleistung.
Die Serumclearance beträgt 37.2 ml/min bei mittelschwerer und 16.0 ml/min bei schwerer Niereninsuffizienz.
Bei hämodialysepflichtigen Patienten wird ProHance effektiv hämodialysiert. Die Clearancerate von Gadoteridol beträgt 97% der Kreatininclearance und 71% der Blutharnstoff-Nitrogen-Clearance. Nach 3 Dialysesitzungen verbleiben noch ca. 2% der injizierten Dosis Gadoteridol im Blut.
Präklinische DatenMutagenität
Die mutagenen Eigenschaften von Gadoteridol wurden in einer Versuchsreihe von in-vivo Tests untersucht. Es ergaben sich keine Hinweise auf ein mutagenes Potential.
Karzinogenität
Es wurden keine Kanzerogenitätsuntersuchungen durchgeführt.
Reproduktionstoxizität
Untersuchungen zur Fertilität, Embryotoxizität und der peri-/postnatalen Phase mit i.v. verabreichtem Gadoteridol bei Ratten und Kaninchen ergaben im Dosisbereich von 6-10 mmol/kg (60-100-fache Humandosis) Hinweise auf paternal-, maternal- und embryotoxische Effekte.
Sonstige HinweiseInkompatibilitäten
ProHance darf nicht mit anderen Medikamenten gemischt werden.
Beeinflussung diagnostischer Methoden
Bis zum jetzigen Zeitpunkt keine bekannt.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit „EXP“ bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Bei Raumtemperatur (15-25°C) lagern.
Vor Licht geschützt aufbewahren.
Für Kinder unzugänglich aufbewahren.
Hinweise für die Handhabung
Nur zum Einmalgebrauch. Restmengen sowie anderes Material entsorgen.
ProHance darf nicht eingefroren werden. Eingefrorene Spritzen sollten verworfen werden.
Abziehetikette für das Patientendossier
Die Abziehetikette ist zur Rückverfolgung auf die Patientenakte zu kleben, um eine genaue Dokumentation des verwendeten Gadolinium-haltigen Kontrastmittels sicherzustellen. Die verwendete Dosis ist ebenfalls anzugeben.
ProHance Fertigspritze
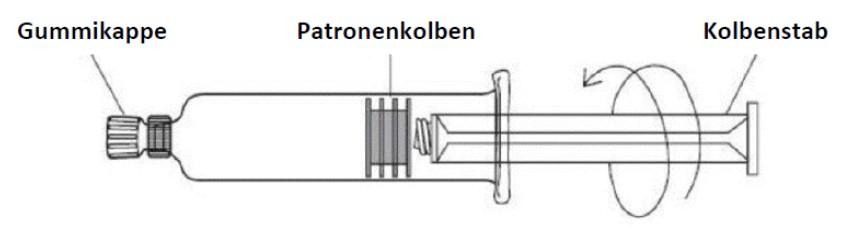
Befolgen Sie nachstehende Anweisungen sorgfältig:
1. Drehen Sie die Kolbenstange im Uhrzeigersinn, um das Gewindeende in den Kolben einzuschrauben, und schieben Sie sie einige Millimeter nach vorne, um jegliche Reibung zwischen dem Kolben und dem Glaszylinder der Spritze zu beseitigen.
2. Halten Sie die Spritze senkrecht, entfernen Sie die Gummikappe unter aseptischen Bedingungen von der Spritzenspitze, und setzen Sie die sterile Einweg-Kanüle oder den sterilen Einweg-Schlauch mit kompatiblem Luer-Anschluss mit einer Drehbewegung auf.
3. Halten Sie die Spritze weiterhin senkrecht, und drücken Sie den Kolben nach vorn, bis sämtliche Luft entwichen ist und Flüssigkeit an der Kanülenspitze austritt bzw. der Schlauch vollständig gefüllt ist. Führen Sie nach vorheriger Aspiration die Injektion durch. Zur Sicherstellung der vollständigen Kontrastmittelabgabe müssen Sie anschliessend physiologische Kochsalzlösung infundieren.
Zulassungsnummer52599, 52273 (Swissmedic)
PackungenFertigspritzen 10 ml zu 2.793 g (5.0 mmol): 1 und 5 [B].
Fertigspritzen 15 ml zu 4.1895 g (7.5 mmol): 1 und 5 [B].
Fertigspritzen 17 ml zu 4.7481 g (8.5 mmol): 1 und 5 [B].
Durchstechflaschen 5 ml zu 1.3965 g (2.5 mmol): 1, 5, 10, 20 [B].
Durchstechflaschen 10 ml zu 2.793 g (5.0 mmol): 1, 5, 10, 20 [B].
Durchstechflaschen 15 ml zu 4.1895 g (7.5 mmol): 1, 5, 10, 20 [B].
Durchstechflaschen 20 ml zu 5.586 g (10 mmol): 1, 5, 10, 20 [B].
Durchstechflaschen 50 ml zu 13.965 g (25 mmol): 1, 10 [B].
ZulassungsinhaberinBRACCO SUISSE SA, Cadempino
Stand der InformationJuni 2025
|