ZusammensetzungWirkstoffe
Blutgerinnungsfaktor VIII und von-Willebrand-Faktor vom Menschen
Hilfsstoffe
Albumin vom Menschen, Glycin, Lysinhydrochlorid, Natriumchlorid, Natriumcitrat-Dihydrat, Calciumchlorid-Dihydrat, Gesamtnatriumgehalt ca. 1.96 mg/ml
Lösungsmittel: Wasser für Injektionszwecke
Indikationen/AnwendungsmöglichkeitenTherapie und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor VIII-Mangel, Hämophilie A mit Faktor VIII-Inhibitor, erworbener Faktor VIII-Mangel aufgrund einer spontanen Entwicklung von Faktor VIII-Inhibitoren).
Von Willebrand-Jürgens-Syndrom mit Faktor VIII-Mangel.
Hinweis: Die Wirksamkeit und Sicherheit von IMMUNATE S/D bei von Willebrand-Jürgens Syndrom ist nur bei einer kleinen Anzahl von Patienten klinisch untersucht worden, dies gilt insbesondere für Typ 3.
Dosierung/AnwendungDie Behandlung muss unter Überwachung eines mit der Therapie von Hämophilie A vertrauten Arztes erfolgen.
Übliche Dosierung
Dosierung und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor VIII-Mangels, nach dem Ort und dem Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die verabreichten Faktor VIII-Einheiten werden in Internationalen Einheiten (I.E.) angegeben, entsprechend dem WHO-Standard für Faktor VIII-Produkte. Die Faktor VIII-Aktivität im Plasma wird entweder als Prozentsatz (relativ zum normalen menschlichen Plasma) oder in I.E. (relativ zum Internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine I.E. der Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem Milliliter normalem menschlichen Plasma.
Dosierung bei Hämophilie A
Die Berechnung der erforderlichen Dosis von Faktor VIII basiert auf dem empirischen Befund, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma um 1.5% bis 2% erhöht. Die erforderliche Dosis wird mit folgender Formel berechnet:
Erforderliche Einheiten (I.E.) = Körpergewicht (kg) x gewünschter Faktor VIII-Anstieg (%) x 0.5.
Dosis und Häufigkeit der Verabreichung sollen entsprechend der klinischen Wirksamkeit des Produktes im Einzelfall angepasst werden.
Blutungen und Operationen
Bei folgenden hämorrhagischen Ereignissen soll die Faktor VIII-Aktivität im entsprechenden Zeitraum nicht unter die angegebenen Plasmaspiegel (in % der Norm oder in I.E./dl) sinken. Die folgende Tabelle enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
|
Grad der Blutung / Art des chirurgi-schen Eingriffes
|
Erforderlicher Faktor VIII Plasmaspiegel (% oder I.E./dl)
|
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage)
| |
Blutung
Gelenkblutung im Frühstadium, Muskelblutungen oder Blutungen im Mund
|
20 – 40
|
Injektion alle 12 – 24 Stunden für mind. 1 Tag, bis die Blutung – angezeigt durch das Verschwinden der Schmerzen – steht oder Heilung erreicht ist.
| |
Ausgeprägtere Gelenkblutung, Muskelblutung oder Hämatom
|
30 – 60
|
Injektion alle 12 – 24 Stunden für 3 – 4 Tage oder länger wiederholen, bis die Schmerzen und die akute Beeinträchtigung beseitigt sind.
| |
Lebensbedrohliche Blutungen
|
60 – 100
|
Injektion alle 8 – 24 Stunden wiederholen bis die Gefahr für den Patienten vorüber ist.
| |
Chirurgische Eingriffe
Kleinere Eingriffe
Einschliesslich Zahnextraktion
|
30 – 60
|
Alle 24 Stunden für mind. 1 Tag bis die Wundheilung erreicht ist.
| |
Grössere Eingriffe
|
80 – 100
(prä- und postoperativ)
|
Injektion alle 8 – 24 Stunden bis zu angemessener Wundheilung wiederholen, dann Therapie für noch mind. 7 Tage fortsetzen, um eine Faktor VIII-Aktivität von 30 – 60% (I.E./dl) aufrecht zu erhalten.
|
Dosis und Häufigkeit der Verabreichung sollen entsprechend der klinischen Wirksamkeit des Produktes im Einzelfall angepasst werden. Unter bestimmten Umständen (z.B. bei Anwesenheit von niedrigen Titern an Faktor VII Inhibitoren), insbesondere zu Behandlungsbeginn, können höhere Dosierungen als berechnet notwendig sein.
Falls sich die Blutung mit der verordneten Dosis Faktor VIII nicht kontrollieren lässt, sollte man den Plasmaspiegel von Faktor VIII bestimmen und eine ausreichende Dosis IMMUNATE S/D verabreichen, um eine zufriedenstellende klinische Reaktion zu erreichen.
Während des Behandlungsverlaufes wird zur Steuerung der zu verabreichenden Dosis und der Häufigkeit der Injektionen eine angemessene Bestimmung der Faktor VIII-Plasmaspiegel empfohlen. Besonders bei grösseren chirurgischen Eingriffen ist eine genaue Überwachung der Substitutionstherapie durch Bestimmung der Faktor VIII-Aktivität im Plasma unerlässlich. Einzelne Patienten können sich in ihrer Reaktion auf Faktor VIII unterscheiden, verschiedene in vivo Recovery erreichen und unterschiedliche Halbwertszeiten aufweisen.
Langzeitprophylaxe
Zur Dauersubstitution von Blutungen bei Patienten mit schwerer Hämophilie A sollen Dosen zwischen 20 – 40 I.E. von Faktor VIII pro kg Körpergewicht im Abstand von 2 – 3 Tagen gegeben werden. In manchen Fällen, besonders bei jüngeren Patienten, können kürzere Dosierungsabstände oder höhere Dosen erforderlich sein.
Patienten mit Faktor VIII-Inhibitoren
Patienten sollten regelmässig auf die Bildung von Inhibitoren gegen Faktor VIII überwacht werden. Falls die erwarteten Faktor VIII-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird, muss ein Inhibitortest durchgeführt werden. Bei Patienten mit hohen Inhibitorwerten kann die Faktor VIII-Therapie nicht ansprechen und andere therapeutische Massnahmen müssen erwogen werden. Diese Therapien sollten nur von Ärzten durchgeführt werden, die über Erfahrung in der Behandlung von Patienten mit Hämophilie verfügen. Siehe auch «Warnhinweise und Vorsichtsmassnahmen».
Von Willebrand-Jürgens-Syndrom mit Faktor VIII-Mangel
IMMUNATE S/D ist zur Therapie und Prophylaxe von Faktor VIII bei Patienten mit von Willebrand-Jürgens-Syndrom angezeigt, bei denen die Faktor VIII-Aktivität vermindert ist und bei denen eine Behandlung mit Desmopressin unwirksam oder kontraindiziert ist. Für die Substitutionsbehandlung mit IMMUNATE S/D zur Kontrolle von Blutungen sowie zur perioperativen Blutungsprophylaxe gelten dieselben Richtlinien wie für Hämophilie A.
Die Erfahrungen bei Kindern sind limitiert.
Die Patienten müssen auf die Bildung von Inhibitoren gegen den von-Willebrand-Faktor überwacht werden, falls die erwarteten von-Willebrand-Faktor-Plasmaaktivitäten nicht erreicht werden oder die Blutung mit einer angemessenen Dosis nicht beherrscht wird.
In Patienten mit hohen von-Willebrand-Inhibitorwerten könnte die Therapie mit IMMUNATE S/D nicht wirksam sein und andere therapeutische Optionen sollten in Betracht gezogen werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Wird ein von-Willebrand-Faktor-Produkt verwendet, das Faktor VIII enthält, so sollte dem behandelnden Arzt bewusst sein, dass bei einer längerfristigen Behandlung der Faktor-VIII:C-Spiegel stark ansteigen kann. Um einen zu hohen Faktor-VIII:C-Spiegel zu vermeiden, sollte in Betracht gezogen werden, nach einer Behandlungsdauer von 24 bis 48 Stunden die Dosis zu reduzieren und/oder die Dosisintervalle zu verlängern.
Kinder und Jugendliche
Das Produkt sollte mit Vorsicht bei Kindern unter 6 Jahren angewendet werden, die noch wenig mit Faktor VIII-Produkten in Berührung gekommen sind. Es wurden keine formalen klinischen Studien in dieser Altersgruppe durchgeführt, jedoch lassen klinische Fallberichte auf Wirksamkeit und Verträglichkeit bei Kindern schliessen.
Art der Anwendung
Das Auflösen erfolgt wie im Abschnitt «Hinweise für die Handhabung» beschrieben. IMMUNATE S/D soll langsam intravenös verabreicht werden. Die Verabreichungsgeschwindigkeit sollte sich nach dem Wohlbefinden des Patienten richten und nicht mehr als 2 ml/min betragen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenWie bei allen intravenösen Substanzen können allergische Überempfindlichkeitsreaktionen auftreten. Die Patienten sollten über die möglichen Frühzeichen von Unverträglichkeitsreaktionen aufgeklärt werden, wie zum Beispiel Hypotonie, Tachykardie, Brustkorbschmerz, Dyspnoe, Ödeme (inklusive Gesichts- und Augenlidödeme), Urtikaria, Ausschlag, Hitzegefühl und Pruritus sowie Anaphylaxie bis hin zum anaphylaktischen Schock.
Die Patienten sollten angewiesen werden, beim Auftreten dieser Symptome die Behandlung abzubrechen und sofort ihren Arzt zu konsultieren. Die Behandlung des Schocks erfolgt nach den Regeln der modernen Schocktherapie.
Auch über andere infusionsbedingte Reaktionen wie Schüttelfrost, Fieber und Übelkeit wurde im Zusammenhang mit IMMUNATE S/D berichtet.
Da bei Verabreichung maximaler Tagesdosen die Natriummenge von 200 mg überschritten werden kann, können Personen, die unter einer niedrigeren Natriumdiät stehen, ungünstig beeinflusst werden.
250 I.E. und 500 I.E. Durchstechflasche:
IMMUNATE S/D enthält ca. 9.8 mg Natrium pro Durchstechflasche. Dies entspricht 0.5% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2g.
1000 I.E. Durchstechflasche:
IMMUNATE S/D enthält ca. 19.6 mg Natrium pro Durchstechflasche. Dies entspricht 1% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2g.
Die Bildung von neutralisierenden Antikörpern (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Patienten mit Hämophilie A. Diese Inhibitoren sind stets gegen die prokoagulatorische Aktivität von Faktor VIII gerichtete IgG Immunglobuline, die in Bethesda Einheiten (B.E.) pro ml Plasma mittels modifiziertem Bethesda Assay quantifiziert werden. Das Risiko, Inhibitoren zu entwickeln, korreliert mit dem Schweregrad der Erkrankung wie auch dem Ausmass der Exposition gegenüber dem Faktor VIII, wobei das Risiko innerhalb der ersten 50 Expositionstage am grössten ist. Das Risiko bleibt lebenslang bestehen, ist jedoch als seltenes Risik einzustufen. Patienten, die mit Koagulations-Faktor VIII vom Menschen behandelt werden, sollten sorgfältig klinisch und mit geeigneten Labortests hinsichtlich der Entwicklung von Inhibitoren überwacht werden. Siehe auch «Unerwünschte Wirkungen».
Das Risiko einer Inhibitor-Entwicklung hängt von einer Reihe von Faktoren ab, die mit speziellen Eigenschaften des jeweiligen Patienten zusammenhängen. Als wichtigste Risikofaktoren gelten unter anderem Art der Faktor VIII-Genmutation, Familienanamnese und ethnische Zugehörigkeit.
Inhibitoren wurden überwiegend bei nicht vorbehandelten Patienten berichtet.
IMMUNATE S/D wird aus humanem Plasma hergestellt. Standardmassnahmen zur Verhinderung von Infektionen (z.B durch Viren), die sich durch den Einsatz von Arzneimitteln ergeben, die aus Blut oder Blutplasma hergestellt werden, schliessen die Auswahl der Spender und das Screening der einzelnen Spenden und Plasmapools auf spezifische Infektionsmarker sowie den Einsatz effektiver Schritte zur Inaktivierung/Entfernung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln aus menschlichem Blut oder Blutplasma die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die ergriffenen Sicherheitsmassnahmen werden als wirksam gegen umhüllte Viren wie z.B. HIV, HBV und HCV sowie gegen das unbehüllte Hepatitis A Virus (HAV) betrachtet. Für Parvovirus B19 können die getroffenen Massnahmen von eingeschränktem Wert sein.
Parvovirus B19-Infektionen können schwerwiegende Folgen für schwangere Frauen (fötale Infektion) und für Personen mit Immunmangelkrankheiten oder gesteigerter Erythropoese (z.B. hämolytische Anämie) haben.
Bei einer längerfristigen Behandlung mit einem von-Willebrand-Faktor Produkt, das Faktor VIII enthält, kann der Faktor-VIII:C-Spiegel stark ansteigen. Bei der Behandlung von Patienten mit von-Willebrand-Syndrom besteht ein Risiko hinsichtlich des Auftretens thrombotischer Ereignisse, besonders bei Vorliegen von bekannten klinischen oder labortechnisch belegten Risikofaktoren. Deshalb müssen Risikopatienten auf frühe Zeichen von Thrombosen hin überwacht werden. Bei Patienten, bei denen in der Vergangenheit bereits venöse Thromboembolien aufgetreten sind, wurde ein endogener hoher Faktor-VIII-Spiegel mit einem erhöhten Risiko für spätere thrombotische Ereignisse in Verbindung gebracht.
Bei Patienten, die mit einem von-Willebrand-Faktor Produkt behandelt werden, das Faktor VIII enthält, sollte unbedingt der Faktor-VIII:C-Plasmaspiegel überwacht werden, um eine dauerhafte Erhöhung zu vermeiden, da dies mit einem erhöhten Risiko für thrombotische Ereignisse einhergeht.
Eine Prophylaxe gegenüber Thromboembolien sollte gemäss den aktuellen Empfehlungen durchgeführt werden.
Patienten mit von-Willebrand-Syndrom, besonders solche mit Typ 3, können neutralisierende Antikörper (Inhibitoren) gegen den von-Willebrand-Faktor entwickeln.
Solche Antikörper können auch eine Anaphylaxie auslösen. Patienten, bei denen es zu einer anaphylaktischen Reaktion kommt, sollten daher auf Inhibitoren gegen den von-Willebrand-Faktor untersucht werden.
Wenn der erwartete Anstieg der von-Willebrand-Restocetin-Cofaktor-Aktivität im Plasma nicht erreicht wird, oder wenn Blutungen nicht mit einer entsprechenden Dosis beherrscht werden können, sollen geeignete Tests auf die Anwesenheit von von-Willebrand-Inhibitoren durchgeführt werden. Es besteht die Möglichkeit, dass bei Patienten mit hohen Inhibitortitern die von-Willebrand-Therapie nicht effektiv ist und andere therapeutische Massnahmen in Betracht gezogen werden sollten. Die Behandlung solcher Patienten sollte unter der Leitung von Ärzten durchgeführt werden, die mit der Behandlung von Patienten mit Gerinnungsstörungen vertraut sind.
Für Patienten, die regelmässig Präparate aus menschlichem Blut oder Plasma erhalten, wird grundsätzlich eine Impfung gegen Hepatitis A und Hepatitis B empfohlen.
Es wird empfohlen, bei jeder Verabreichung von IMMUNATE S/D an einen Patienten den Namen und die Chargennummer des Präparates zu dokumentieren, um einen Zusammenhang zwischen Patient und Produktcharge herzustellen.
IMMUNATE S/D enthält Blutgruppen-Isoagglutinine (anti-A und anti-B). Bei Patienten mit Blutgruppe A, B oder AB kann es nach wiederholter Anwendung in kurzen Abständen oder nach der Anwendung sehr hoher Dosen zu einer Hämolyse kommen. Sehr hohe Dosen innerhalb kurzer Zeit werden möglicherweise im Rahmen einer Immuntoleranztherapie zur Behandlung einer Hämophilie A mit Faktor-VIII-Inhibitor angewendet.
Vor der Anwendung von IMMUNATE S/D muss unbedingt sichergestellt werden, dass es sich bei der Gerinnungsstörung tatsächlich um einen Faktor-VIII-Mangel (Hämophilie A) oder einen Mangel an von-Willebrand-Faktor (von-Willebrand-Syndrom) handelt.
InteraktionenEs wurden keine Wechselwirkungsstudien durchgeführt.
Schwangerschaft, StillzeitReproduktionsstudien an Tieren wurden mit Faktor VIII nicht durchgeführt. Aufgrund des seltenen Auftretens der Hämophilie A bei Frauen liegen über die Anwendung von Faktor VIII während der Schwangerschaft und Stillzeit keine Erfahrungen vor. Faktor VIII sollte daher in der Schwangerschaft und Stillzeit nur bei eindeutiger Indikationsstellung angewendet werden.
Informationen zu Parvovirus B19-Infektionen siehe «Warnhinweise und Vorsichtsmassnahmen».
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs ist kein Einfluss auf die Verkehrstüchtigkeit und das Bedienen von Maschinen bekannt.
Unerwünschte WirkungenDie nachstehend aufgelisteten unerwünschten Wirkungen basieren auf klinischen Studien und Spontanmeldungen nach der Zulassung von IMMUNATE S/D. Die Häufigkeit wurden ermittelt unter Verwendung der folgenden Kriterien: sehr häufig (≥10%), häufig (≥1% bis <10%), gelegentlich (≥0.1% bis<1%), selten (≥0.01% bis <0.1%), sehr selten (<0.01%).
Klinische Studien
Die Inzidenz aller nachstehenden aus klinischen Studien gemeldeten unerwünschten Wirkungen war «gelegentlich» (≥0.1% bis <1%).
Erkrankungen des Immunsystems
Gelegentlich: Allergische Reaktionen
Unerwünschte Wirkungen nach Markteinführung
Spontanmeldungen nach der Zulassung
Die Inzidenzrate war «sehr selten» (<0.01%) für alle nachstehend gemeldeten unerwünschten Wirkungen.
Erkrankungen des Blutes und des Lymphsystems
Gerinnungsstörungen, Faktor VIII Inhibition
Psychiatrische Erkrankungen
Unruhe
Herzerkrankungen
Herzklopfen, Tachykardie
Erkrankungen des Gastrointestinaltrakts
Erbrechen, Übelkeit
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Brustkorbschmerz, Brustkorbbeschwerden, Ödeme (Gesicht und periphere Ödeme), Irritationen an der Injektionsstelle (inklusiv Brennen), Schüttelfrost, Schmerzen, Fieber
Erkrankungen des Immunsystems
Überempfindlichkeit
Erkrankungen des Nervensystems
Kopfschmerzen, Schwindel, Paraesthesie
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Husten, Dyspnoe
Gefässerkrankungen
Hypotonie, Hitzegefühl, Blässe
Augenerkrankungen
Konjunktivitis, Augenlidödem
Erkrankungen der Haut und des Unterhautgewebes
Erythem, Exanthem, Neurodermatitis, Pruritus, Ausschlag, erythematöser Ausschlag, papulöser Ausschlag, Urtikaria, Hyperhidrosis
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Myalgie
Die folgenden unerwünschten Wirkungen wurden bisher nicht rapportiert, könnten jedoch mit IMMUNATE S/D auftreten:
Erkrankungen des Blutes und des Lymphsystems
Hämolyse in Patienten mit Blutgruppe A, B oder AB
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Vermindertes therapeutisches Ansprechen
Beschreibung ausgewählter Nebenwirkungen
In seltenen Fällen sind Überempfindlichkeits- oder allergische Reaktionen aufgetreten (einschliesslich Angioödem, Brennen und Stechen an der Injektionsstelle, Frösteln, Erythema, generalisierte Urticaria, Kopfschmerzen, Nesselausschlag, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Erbrechen, Keuchatmung und Inhibition des von-Willebrand-Faktors). In einigen Fällen kann dies in eine schwere Anaphylaxie münden (inklusive Schock). Den Patienten soll angeraten werden bei Auftreten dieser Symptome ihren Arzt zu kontaktieren
Fieber wurde in seltenen Fällen beobachtet.
Bei Patienten mit Hämophilie A, die neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII bilden, manifestiert sich dieser Zustand als eine unzureichende klinische Antwort. In diesen Fällen wird empfohlen, ein Hämophilie-Zentrum zu besuchen.
Nach der Applikation von sehr hohen Dosen (z.B. wenn Faktor VIII Plasma Spiegel über 100% erreicht werden sollen) kann bei Patienten mit Blutgruppe A, B oder AB aufgrund des Gehaltes an Isoagglutininen eine Hämolyse auftreten.
Für Informationen zur viralen Sicherheit siehe «Warnhinweise und Vorsichtsmassnahmen».
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden bisher keine Symptome durch Überdosierung mit Koagulations-Faktor VIII vom Menschen beschrieben.
Grundsätzlich besteht aber ein Risiko für thromboembolische Ereignisse. Bei Patienten mit Blutgruppe A, B oder AB besteht ein Hämolyse-Risiko.
Siehe «Warnhinweise und Vorsichtsmassnahmen».
Eigenschaften/WirkungenATC-Code
B02BD06
Pharmakotherapeutische Gruppe: Anti-Hämorrhagikum: Blut Gerinnungsfaktor VIII.
Wirkungsmechanismus
Der Faktor VIII / von Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von Willebrand-Faktor) mit unterschiedlichen physiologischen Funktionen.
Der aktivierte Faktor VIII wirkt als Cofaktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X aus Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Dieses setzt dann Fibrin aus Fibrinogen frei und die Gerinnselbildung kann erfolgen. Hämophilie A ist eine geschlechtsgebundene erbliche Störung der Blutgerinnung aufgrund erniedrigter Faktor VIII:C Spiegel. Dies führt, entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Durch die Substitutionstherapie wird der Faktor VIII-Plasmaspiegel erhöht, wodurch eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung erfolgt.
Der von Willebrand Faktor (vWF) vermittelt, zusätzlich zu seiner Rolle als Faktor VIII stabilisierendes Protein, die Adhäsion von Thrombozyten an den Orten einer Gefässverletzung, spielt eine Rolle bei der Thrombozytenaggregation und ist unentbehrlich zur Substitutionstherapie bei von Willebrand Patienten.
PharmakokinetikEine pharmakokinetische Studie mit 18 Patienten zeigte nach vollständiger Analyse die unten beschriebenen Ergebnisse.
Folgende Tabelle zeigt die pharmakokinetischen Eigenschaften von Blutgerinnungsfaktor VIII:
|
Parameter
|
Anzahl
|
Mittelwert
|
SD
|
Median
|
90% CI
| |
AUC0-48h ([I.E.xh]/ml)
|
18
|
11.4
|
2.8
|
11.6
|
10.9 – 12.7
| |
AUC0-∞ ([I.E.xh]/ml)
|
18
|
12.2
|
3.1
|
12.4
|
11.1 – 13.2
| |
Cmax (I.E./ml)
|
18
|
1.0
|
0.3
|
0.9
|
0.8 – 1.0
| |
Tmax (h)
|
18
|
0.3
|
0.1
|
0.3
|
0.3 – 0.3
| |
Terminale Halbwertszeit (h)
|
18
|
12.7
|
3.2
|
12.2
|
10.8 – 15.3
| |
Clearance (ml/h)
|
18
|
283
|
146
|
232
|
199 – 254
| |
Mittlere Verweilzeit (h)
|
18
|
15.3
|
3.6
|
15.3
|
12.1 – 17.2
| |
Vss (ml)
|
18
|
4166
|
2021
|
3613
|
2815 – 4034
| |
Incremental Recovery ([I.E./ml]/
[I.E./kg])
|
18
|
0.020
|
0.006
|
0.019
|
0.016 – 0.020
|
Folgende Tabelle beschreibt die pharmakokinetischen Eigenschaften des vWF:Ag:
|
Parameter
|
Anzahl
|
Median
|
90% CI
| |
AUC0-∝ ([I.E.xh]/ml)
|
15
|
24.6
|
12.8 – 48.3
| |
Cmax (I.E./ml)
|
17
|
1.40
|
1.15 – 1.51
| |
Tmax (h)
|
17
|
0.28
|
0.25 – 1.00
| |
Terminale Halbwertszeit (h)
|
16
|
13.6
|
10.5 – 47.2
| |
Clearance (ml/h)
|
15
|
136
|
68 – 178
| |
Mittlere Verweilzeit (h)
|
15
|
23.1
|
12.4 – 57.1
| |
Vss (ml)
|
15
|
3156
|
2391 – 4672
| |
Incremental Recovery ([I.E./ml]/
[I.E./kg])
|
17
|
0.028
|
0.024 – 0.030
|
Präklinische DatenDer in IMMUNATE S/D enthaltene humane Blutgerinnungsfaktor VIII ist ein physiologischer Bestandteil des menschlichen Plasmas und verhält sich wie der körpereigene Blutgerinnungsfaktor VIII.
Präklinische Daten, basierend auf Studien zur akuten Toxizität, lokalen Verträglichkeit und Immunogenität, weisen auf keine besondere Gefahr für den Menschen hin.
Sonstige HinweiseInkompatibilitäten
IMMUNATE S/D darf nicht mit anderen Arzneimitteln oder Lösungsmitteln gemischt werden, da dadurch die Sicherheit oder Wirksamkeit beeinträchtigt werden könnte. Es darf ausschliesslich der mitgelieferte Gerätesatz benutzt werden, da die Therapie als Folge einer Adsorption des Koagulations-Faktor VIII an der Innenoberfläche einiger Infusionssets versagen kann.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden (siehe «Lagerungshinweise»). Nach Auflösen bei Raumtemperatur (15-25 °C) lagern und innerhalb von 3 Stunden verabreichen. Aus mikrobiologischer Sicht sollte das Produkt unmittelbar nach der Rekonstitution appliziert werden.
Besondere Lagerungshinweise
Zwischen 2° und 8° C lagern. Nicht einfrieren. In der Originalpackung lagern zwecks Lichtschutz.
Innerhalb der Laufzeit kann das Produkt einmalig bei Raumtemperatur (höchstens 25°C) bis zu 6 Monate gelagert werden. Der Beginn der Lagerung bei Raumtemperatur ist unterhalb des aufgedruckten Verfalldatums einzutragen. Nach der Lagerung bei Raumtemperatur darf das Produkt nicht wieder gekühlt werden, es ist entweder zu verbrauchen oder zu entsorgen.
Ausserhalb der Reichweite von Kindern aufzubewahren.
Hinweise für die Handhabung
IMMUNATE S/D sollte erst unmittelbar vor der Anwendung rekonstituiert werden. Die Lösung sollte sofort verbraucht werden, da kein Konservierungsmittel enthalten ist. Die Lösung vor der Anwendung auf sichtbare Partikel und Verfärbung prüfen. Trübe Lösungen oder solche mit Niederschlag sind zu verwerfen. Es wird empfohlen, einen gemeinsamen venösen Zugang vor und nach der Verabreichung von IMMUNATE S/D mit einer salinen Lösung zu spülen.
Auflösen der Trockensubstanz: Auf aseptische Arbeitsweise achten wie nachfolgend beschrieben:
1.Lösungsmittel (sterilisiertes Wasser für Injektionszwecke) in der ungeöffneten Lösungsmittelflasche auf Raumtemperatur (höchstens 37°C) erwärmen.
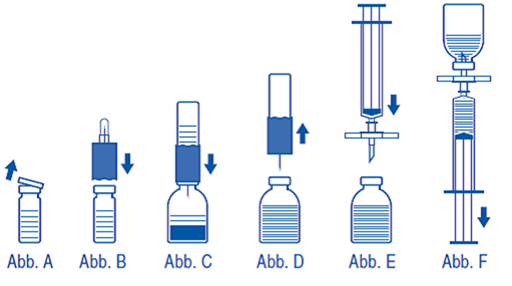
2.Schutzkappen von Präparat- und Lösungsmittelflasche entfernen (Abb. A) und die Gummistopfen beider Flaschen desinfizieren.
3.Transferset mit der gewellten Seite auf die Lösungsmittelflasche setzen und eindrücken (Abb. B).
4.Schutzhülle von der anderen Seite des Transfersets abziehen. Freies Kanülenende nicht berühren!
5.Transferset mit aufgesetzter Wasserflasche von oben in die Präparatflasche einstechen (Abb. C). Durch das in der Präparatflasche bestehende Vakuum wird das Lösungsmittel angesaugt.
6.Nach etwa 1 Minute das Transferset samt Lösungsmittelflasche von der Präparatflasche abziehen (Abb. D). Da das Präparat sich schnell löst, ist – wenn überhaupt – nur ein leichtes Schwenken der Präparatflasche erforderlich. DEN INHALT DER PRÄPARATFLASCHE NICHT SCHÜTTELN. DIE PRÄPARATFLASCHE ERST UNMITTELBAR VOR DER ENTNAHME DES INHALTES UMDREHEN.
7.Parenterale Produkte wie IMMUNATE S/D sollen nach der Auflösung und vor der Anwendung visuell auf Partikel und Verfärbungen überprüft werden. Auch wenn die Auflösungsvorschrift strikt befolgt wird, können fallweise wenige kleine Partikel sichtbar sein. Das beigepackte Filterset entfernt diese Partikel. Die auf der Packung angegebene Konzentration des arzneilich wirksamen Bestandteils wird dadurch nicht reduziert.
Auf aseptische Arbeitsweise achten wie nachfolgend beschrieben:
1.Um zu verhindern, dass vom Stopfen ausgestochene Gummipartikel verabreicht werden (Gefahr von Mikroembolien) ist zur Entnahme des gelösten Präparats das beigepackte Filterset zu benutzen. Filterset auf die beigepackte Einmalspritze setzen und in den Gummistopfen einstechen (Abb. E).
2.Durch zwischenzeitliches Lockern der Spritze vom Filterset wird die Präparatflasche belüftet, wodurch eventuell entstandener Schaum zusammenfällt. Daraufhin die Injektionslösung durch das Filterset in die Spritze aufziehen (Abb. F).
3.Das Filterset von der Spritze abziehen und die Lösung langsam mit dem beigepackten Infusionsset (bzw. Einmalnadel) intravenös verabreichen (maximale Injektionsrate: 2 ml pro Minute).
Unverbrauchte Lösung, leere Fläschchen, benützte Nadeln und Spritzen sind fachgerecht zu entsorgen.
Zulassungsnummer52715 (Swissmedic)
PackungenIMMUNATE S/D 250 I.E.
Durchstechflasche mit 250 I.E. Faktor VIII lyophilisiert
Durchstechflasche mit 5 ml Wasser für Injektionszwecke
Gerätesatz zur Auflösung und Injektion
IMMUNATE S/D 500 I.E.
Durchstechflasche mit 500 I.E. Faktor VIII lyophilisiert
Durchstechflasche mit 5 ml Wasser für Injektionszwecke
Gerätesatz zur Auflösung und Injektion
IMMUNATE S/D 1000 I.E.
Durchstechflasche mit 1000 I.E. Faktor VIII lyophilisiert
Durchstechflasche mit 10 ml Wasser für Injektionszwecke
Gerätesatz zur Auflösung und Injektion
Abgabekategorie: B
ZulassungsinhaberinTakeda Pharma AG, 8152 Opfikon
Stand der InformationMärz 2024
|