ZusammensetzungWirkstoffe
Belimumab.
Hilfsstoffe
Zitronensäure-Monohydrat, Natriumcitrat-Dihydrat, Saccharose, Polysorbat 80.
Indikationen/AnwendungsmöglichkeitenBenlysta ist indiziert:
·zur Verminderung der Krankheitsaktivität bei Patienten ab 5 Jahren mit aktivem, Autoantikörper-positivem systemischem Lupus erythematodes (SLE), die eine Basistherapie erhalten
·zur Behandlung von Lupusnephritis bei erwachsenen Patienten, die eine Standardtherapie erhalten.
Benlysta wurde bei Patienten mit schwerem, aktivem Lupus des Zentralnervensystems nicht untersucht.
Dosierung/AnwendungAllgemeine Hinweise
Zu den Wirkungen von Belimumab bei Patienten mit schwerem, aktivem zentralnervösem Lupus liegen keine oder keine hinreichenden Daten vor. Zur Behandlung dieser Erkrankungen kann Benlysta daher nicht empfohlen werden.
Eine Therapie mit Benlysta sollte unbedingt unter Aufsicht einer Ärztin bzw. eines Arztes mit Erfahrung in der Behandlung von SLE Patienten eingeleitet werden. Notfallmassnahmen zur Behandlung von Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, müssen zur Verfügung stehen.
Benlysta wird als intravenöse Infusion verabreicht und muss vor der Anwendung rekonstituiert und verdünnt werden (siehe «Hinweise für die Handhabung»). Vor der intravenösen Infusion von Benlysta kann eine Prämedikation mit einem oralen Antihistaminikum, mit oder ohne Antipyretikum, erfolgen. Benlysta muss über einen Zeitraum von 1 Stunde infundiert werden (siehe «Warnhinweise und Vorsichtsmassnahmen»). Benlysta darf nicht als schnelle intravenöse Infusion oder Bolusinfusion verabreicht werden.
Die Patienten sollten während der Verabreichung von Benlysta und auch über einen angemessenen Zeitraum danach überwacht werden.
Die Infusionsgeschwindigkeit kann vermindert bzw. die Infusion gestoppt werden, wenn sich beim Patienten eine Infusionsreaktion entwickelt. Die Infusion ist umgehend zu beenden, wenn es beim Patienten zu einer potentiell lebensbedrohlichen unerwünschten Reaktion kommt (siehe «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen»).
Übliche Dosierung
Erwachsene
SLE
Die empfohlene Dosierung beträgt 10 mg/kg an den Tagen 0, 14 und 28 und anschliessend eine Dosis von 10 mg/kg in 4-wöchigen Abständen.
Falls nach sechsmonatiger Behandlung mit Benlysta keine Verbesserung der Krankheitskontrolle feststellbar ist, sollte ein Abbruch der Behandlung in Erwägung gezogen werden.
Lupusnephritis
Die empfohlene Dosierung beträgt 10 mg/kg an den Tagen 0, 14 und 28 und anschliessend eine Dosis von 10 mg/kg in 4-wöchigen Abständen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Umstellung von intravenöser auf subkutane Verabreichung
SLE
Wenn ein Patient mit SLE von der intravenösen Verabreichung von Benlysta auf die subkutane Verabreichung umgestellt wird, wird die erste subkutane Injektion zu 200 mg ungefähr 2 Wochen nach der letzten intravenösen Dosis verabreicht (siehe. «Pharmakokinetik»).
Lupusnephritis
Wird ein Patient mit Lupusnephritis von der intravenösen Verabreichung von Benlysta auf die subkutane Verabreichung umgestellt, wird die erste subkutane Dosis zu 200 mg ein bis zwei Wochen nach der letzten intravenösen Dosis verabreicht. Diese Umstellung kann zu einem beliebigen Zeitpunkt nach Verabreichung der ersten beiden intravenösen Dosen erfolgen (siehe «Pharmakokinetik»).
Kinder und Jugendliche
SLE
Die empfohlene Dosierung für Kinder ab 5 Jahren und Jugendliche beträgt 10 mg/kg an den Tagen 0, 14 und 28 und anschliessend eine Dosis von 10 mg/kg in 4-wöchigen Abständen.
Falls nach sechsmonatiger Behandlung mit Benlysta keine Verbesserung der Krankheitskontrolle feststellbar ist, sollte ein Abbruch der Behandlung in Erwägung gezogen werden.
Es liegen keine Daten zur Wirksamkeit und Sicherheit für Kinder unter 6 Jahren mit SLE vor.
Lupusnephritis
Die Wirksamkeit und Sicherheit von Belimumab bei Kindern und Jugendlichen unter 18 Jahren wurde nicht untersucht. Daher wird Benlysta nicht zur Anwendung bei Kindern und Jugendlichen mit Lupusnephritis empfohlen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Es wurden keine formalen Studien mit Belimumab bei Patienten mit eingeschränkter Leberfunktion durchgeführt. Bei Patienten mit eingeschränkter Leberfunktion ist jedoch wahrscheinlich keine Dosisänderung erforderlich (siehe «Kinetik spezieller Patientengruppen»).
Patienten mit Nierenfunktionsstörungen
Es wurden keine formalen Studien mit Belimumab bei Patienten mit eingeschränkter Nierenfunktion durchgeführt.
Belimumab wurde bei einer begrenzten Anzahl von SLE-Patienten mit eingeschränkter Nierenfunktion untersucht. Bei Patienten mit eingeschränkter Nierenfunktion ist keine Anpassung der Dosis erforderlich (siehe «Kinetik spezieller Patientengruppen»).
Ältere Patienten
Obwohl nur eingeschränkt Daten vorliegen, wird keine Dosisanpassung empfohlen (siehe «Kinetik spezieller Patientengruppen»).
KontraindikationenÜberempfindlichkeit gegenüber Belimumab oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenDie verfügbaren Daten bestätigen die Sicherheit und Wirksamkeit von Rituximab bei gleichzeitiger Verabreichung mit Belimumab bei Patienten mit SLE nicht (vgl. «Eigenschaften/Wirkungen - Klinische Wirksamkeit»).
Vorsicht ist geboten, wenn Benlysta in Kombination mit anderen B-Zell-gerichteten Therapien verabreicht wird.
Infusionsreaktionen und Überempfindlichkeit
Bei der Verabreichung von Benlysta kann es zu systemischen Infusionsreaktionen und Überempfindlichkeitsreaktionen kommen, die schwer oder tödlich sein können und die auch verzögert an den darauffolgenden Tagen auftreten können.
Im Fall einer schweren Reaktion muss die Gabe von Benlysta abgebrochen und eine geeignete medizinische Therapie eingeleitet werden. In den drei vor der Zulassung durchgeführten klinischen SLE Studien zur intravenösen Verabreichung wurde ein endgültiger Behandlungsabbruch bei 0,4% der behandelten Patienten berichtet. Patienten mit multiplen Arzneimittelallergien oder signifikanter Überempfindlichkeit in der Vorgeschichte können ein erhöhtes Risiko haben.
Diese Reaktionen entwickelten sich häufiger bei den ersten beiden Gaben und traten bei nachfolgenden Verabreichungen tendenziell seltener auf.
Ein verspätetes Auftreten von Überempfindlichkeitsreaktionen ist beobachtet worden, ebenso ein Wiederauftreten von klinisch bedeutsamen Reaktionen nach initialem Abklingen der Symptome nach Behandlung.
Patienten unter Benlysta sind darauf hinzuweisen, dass am Tag der Verabreichung oder noch mehrere Tage danach Überempfindlichkeitsreaktionen auftreten können, und müssen über die möglichen Zeichen und Symptome und die Möglichkeit eines erneuten Auftretens aufgeklärt werden. Die Patienten sollen auf die potentiellen Risiken solcher Reaktionen aufmerksam gemacht werden sowie auf die Wichtigkeit hingewiesen werden, unverzüglich ärztlichen Rat einzuholen. Entsprechende Symptome sind z.B. anaphylaktische Reaktion, Bradykardie, Hypotonie, Angioödem und Atemnot. Verspätete bzw. verzögerte Überempfindlichkeitsreaktionen können auftreten, die sich mit der Symptomatik Ausschlag bzw. Urtikaria, Übelkeit, Fieber, Fatigue, Myalgie, Kopf- und Gliederschmerzen sowie Gesichtsödem ankündigen können. Patienten sollen angewiesen werden, alle Symptome solcher Reaktionen umgehend zu melden.
In klinischen Studien wurde über Infusionsreaktionen bei 17% der Patienten unter Belimumab und bei 15% der Patienten unter Placebo berichtet (einschliesslich schwerwiegender Fälle von Anaphylaxie, Hypotonie, Angioödem, Urtikaria oder Ausschlag, Pruritus und Dyspnoe, Bradykardie, Myalgie, Kopfschmerzen, Fieber, Hypertonie, Schwindelgefühl und Arthralgie). Es kam bei weniger als 1% der Patienten zu schwerwiegenden Infusions- und Überempfindlichkeitsreaktionen. Deshalb sollten die Patienten während der Verabreichung von Benlysta und über einen angemessenen Zeitraum danach überwacht werden.
Vor der Infusion von Benlysta kann eine Prämedikation mit einem oralen Antihistaminikum, mit oder ohne Antipyretikum, erfolgen. Ob eine Prämedikation die Häufigkeit und Schwere von Infusionsreaktionen verringert, ist nicht hinreichend geklärt.
Infektionen
Wie bei anderen Immunmodulatoren kann auch der Wirkmechanismus von Belimumab das Risiko einer Infektionsentwicklung erhöhen, einschliesslich opportunistischer Infektionen.
In einer Placebo-kontrollierten Phase-IV Sicherheitsstudie mit intravenös verabreichtem Belimumab traten im Verlauf eines Jahres tödliche Infektionen bei Patienten unter der Behandlung mit Belimumab häufiger auf im Vergleich zur Placebo-Gruppe (0.45% unter Belimumab, 0.15% unter Placebo). Patienten mit einer aktiven akuten oder chronischen Infektion bis 60 Tage vor Studienbeginn, die eine spezifische Therapie verlangte, waren von der Studie ausgeschlossen. Die tödlichen Infektionen unter Belimumab traten überwiegend in den ersten 20 Behandlungswochen auf und betrafen gehäuft Patienten mit hoher Krankheitsaktivität (SELENA SLEDAI ≥10) und Patienten unter einer Begleittherapie mit Kortikosteroiden (>7.5 mg/Tag Prednisonäquivalenzdosis). Nach einem Jahr war die Gesamtmortalität unter der Behandlung mit Belimumab und Placebo vergleichbar. Insgesamt war die Inzidenz schwerwiegender Infektionen in den Belimumab- und den Placebogruppen ähnlich.
Patienten, die unter der Behandlung mit Benlysta eine Infektion entwickeln, sind engmaschig zu überwachen und ein Abbruch der immunsuppressiven Therapie ist zu erwägen. Ärzte sollen den Patienten raten, ärztliche Hilfe aufzusuchen, wenn sie Symptome einer Infektion entwickeln. Vorsicht ist geboten, wenn die Anwendung von Benlysta bei Patienten mit früheren schwerwiegenden oder chronischen Infektionen in Erwägung gezogen wird. Bei Patienten mit einer aktiven, klinisch relevanten Infektion sollte keine Therapie mit Benlysta neu eingeleitet werden.
Progressive multifokale Leukenzephalopathie (PML)
Bei SLE-Patienten unter immunsuppressiver Pharmakotherapie, unter anderem mit Benlysta, wurde über das Auftreten von progressiver multifokaler Leukenzephalopathie (PML) mit der Folge neurologischer Ausfälle und bisweilen tödlichem Ausgang berichtet. Grundsätzlich sollte bei Patienten, bei denen erstmals neurologische Anzeichen und Symptome auftreten oder bei denen sich bereits bestehende derartige Störungen verschlechtern, die Möglichkeit einer PML in Betracht gezogen werden. Betroffene Patienten sollten, wenn klinisch angezeigt, zur Beurteilung an einen Neurologen oder einen anderen qualifizierten Facharzt überwiesen werden. Im Falle eines Verdachts auf PML muss die immunsuppressive Therapie, einschliesslich Benlysta, unterbrochen werden, bis eine PML ausgeschlossen wurde. Wenn sich eine PML bestätigt, muss die immunsuppressive Therapie, einschliesslich Benlysta, abgesetzt werden.
Risiko maligner Erkrankungen
Wie bei anderen Immunmodulatoren kann auch der Wirkmechanismus von Belimumab potenziell das Risiko einer Entwicklung maligner Erkrankungen erhöhen. In klinischen Studien wurde in Bezug auf die Häufigkeit maligner Erkrankungen kein Unterschied zwischen den Behandlungsgruppen mit Belimumab und den Placebogruppen festgestellt.
Impfungen
30 Tage vor oder während der Behandlung mit Benlysta dürfen keine Lebendimpfstoffe verabreicht werden, da die klinische Sicherheit nicht nachgewiesen wurde. Zur Sekundärübertragung einer Infektion von Personen, die Lebendimpfstoffe erhalten haben, auf Patienten unter Benlysta liegen keine Daten vor. In Anbetracht seines Wirkungsmechanismus könnte Belimumab Auswirkungen auf die Immunisierungsreaktion haben. Daher sollte erwogen werden, zwischen der Auffrischimpfung und dem Behandlungsbeginn mit Benlysta einen angemessenen Zeitraum verstreichen zu lassen.
In einer Studie, in der die Impfantwort auf einen 23-valenten Pneumokokken-Impfstoff untersucht wurde, fielen jedoch die Immunantworten nach 4 Wochen auf die unterschiedlichen Serotypen bei SLE-Patienten unter Belimumab und SLE-Patienten, die zum Impfzeitpunkt keine Behandlung erhielten, insgesamt ähnlich aus. Die Aufrechterhaltung der Impfantwort über längere Zeit wurde in dieser Studie nicht untersucht. Eingeschränktes Datenmaterial weist darauf hin, dass Belimumab keinen relevanten Einfluss auf den Erhalt einer protektiven Immunreaktion auf Immunisierungen besitzt, die vor der Gabe von Belimumab erfolgt sind.
Zur Impfantwort und der Erhaltung einer protektiven Immunreaktion bei Kindern und Jugendlichen unter einer Behandlung mit Belimumab liegen keine schlüssigen Daten vor.
Psychiatrische Erkrankungen, Depression und Suizidalität
In kontrollierten klinischen Studien mit intravenöser und subkutaner Verabreichung wurde bei Patienten, die mit Belimumab behandelt wurden, häufiger über psychiatrische Erkrankungen (Depression, Suizidgedanken und suizidales Verhalten) berichtet, darunter ein Suizid bei einem Patienten, der 10 mg/kg erhalten hatte, und ein Suizid bei einem Patienten unter 1 mg/kg (siehe «unerwünschte Wirkungen»).
Die Ärztin bzw. der Arzt muss vor einer Behandlung mit Belimumab anhand der Krankengeschichte des Patienten und seines aktuellen psychiatrischen Status das Risiko einer Depression bzw. eines Suizids sorgfältig abwägen und den Patienten während der Behandlung entsprechend überwachen. Die Ärztin bzw. der Arzt muss den Patienten (und gegebenenfalls Betreuer) anweisen, bei neuen oder sich verschlechternden psychiatrischen Symptomen Kontakt mit ihr bzw. ihm aufzunehmen. Bei Patienten, bei denen solche Symptome auftreten, ist eine sorgfältige Nutzen-Risiko-Abwägung einer Weiterbehandlung mit Belimumab vorzunehmen.
Systemischer Lupus erythematodes bei Kindern und Jugendlichen
Kinder zeigen häufiger einen atypischen Verlauf der Krankheit, insbesondere sind schwerere Krankheitsschübe zu erwarten. Im Gegensatz zum polygenetisch bedingten systemischen Lupus erythematodes bei Erwachsenen können dem Krankheitsbild in der pädiatrischen Population, insbesondere bei jüngeren Kindern unter 12 Jahren, monogenetische Defekte des Immunsystems zugrunde liegen, die eine spezifische zielgerichtete Therapie erfordern. Bei Kindern mit systemischem Lupus erythematodes sollten solche genetische Defekte abgeklärt werden, bevor eine Therapie mit Benlysta erwogen wird.
InteraktionenEs wurden keine Interaktionsstudien mit Belimumab und anderen Arzneimitteln durchgeführt.
In klinischen Studien mit SLE-Patienten hat die gleichzeitige Anwendung von Mycophenolat-Mofetil, Cyclophosphamid, Azathioprin und Hydroxychloroquin keinen wesentlichen Einfluss auf die Pharmakokinetik von Belimumab (bei intravenöser oder subkutaner Verabreichung). Auch ein breites Spektrum von anderen Begleitmedikationen (nichtsteroidale Antirheumatika, Aspirin und HMG-CoA-Reduktasehemmer) wirkte sich nicht signifikant auf die Pharmakokinetik von Belimumab aus. In der populationspharmakokinetischen Analyse zeigten sich Hinweise, dass die Koadministration von Steroiden und ACE-Hemmern mit intravenös verabreichtem Belimumab, nicht aber mit subkutan verabreichtem Belimumab, zu einem signifikanten Anstieg der systemischen Clearance von Belimumab von 6,0% respektive 8,5% führt.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Empfängnisverhütung für Frauen
Wenn aufgrund einer Nutzen/Risiko-Abwägung (siehe unten), die Verhütung einer Schwangerschaft angezeigt ist, sollten Frauen im gebärfähigen Alter unter der Behandlung mit Benlysta sowie bis zum Ablauf von mindestens vier Monaten nach der letztmaligen Verabreichung von Benlysta angemessene Vorkehrungen zur Empfängnisverhütung treffen.
Schwangerschaft
Es liegen nur eingeschränkte Daten zur Anwendung von Belimumab bei Schwangeren vor. Post-Marketing-Daten aus einem prospektiven Schwangerschaftsregister haben Informationen zu Schwangerschaften von Frauen gesammelt, die Belimumab erhalten haben. Aufgrund der erreichten kleinen Stichprobengrösse können aus diesem Register keine endgültigen Schlussfolgerungen hinsichtlich eines möglichen Risikos von Geburtsfehlern nach einer Exposition gegenüber Belimumab gezogen werden.
Immunglobulin-G-(IgG-)Antikörper einschliesslich Belimumab können die Plazenta passieren. Benlysta darf während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen für die Mutter das potenzielle Risiko für den Foetus rechtfertigt.
Bei Frauen unter Belimumab sind kongenitale Missbildungen beobachtet worden. Aufgrund der limitierten Datenlage ist jedoch ein kausaler Bezug nicht schlüssig zu beurteilen.
Tierstudien ergaben keine direkten oder indirekten schädlichen Wirkungen im Sinne einer maternalen Toxizität oder im Hinblick auf den Schwangerschaftsverlauf oder die embryofetale Entwicklung. Behandlungsbedingte Befunde waren auf reversible Verminderungen der B-Zellen bei jungen Affen beschränkt (siehe «Präklinische Daten»). Ein Rückgang der B-Zellen bei Säuglingen kann die Impfantwort beeinträchtigen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Stillzeit
Die Sicherheit von Benlysta bei Anwendung während der Stillzeit ist nicht belegt. Zur Ausscheidung von Belimumab in die Muttermilch oder zur systemischen Resorption von Belimumab nach oraler Einnahme liegen keine Daten vor. Bei Javaneraffen wurde Belimumab jedoch in die Muttermilch ausgeschieden.
Bei stillenden Müttern wird empfohlen, unter der Behandlung mit Benlysta das Abstillen in Erwägung zu ziehen und dabei sowohl den Nutzen des Stillens für das Kind als auch den Nutzen der Therapie für die Frau sowie allfällige mögliche, durch Benlysta oder die zugrundeliegende mütterliche Erkrankung bedingte unerwünschte Wirkungen auf das gestillte Kind zu berücksichtigen. Bezüglich Risikobeurteilung für das gestillte Kind ist allenfalls ein Pädiater einzubeziehen.
Fertilität
Zu den Wirkungen von Belimumab auf die Fertilität beim Menschen liegen keine Daten vor. Zu den Wirkungen auf die männliche und weibliche Fertilität wurden keine Tierstudien durchgeführt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zur Untersuchung der Wirkung von Belimumab auf die Fahrtüchtigkeit oder die Fähigkeit zum Bedienen von Maschinen durchgeführt. Die Pharmakologie von Belimumab lässt nicht auf eine Beeinträchtigung solcher Tätigkeiten schliessen.
Bei der Einschätzung der Fähigkeit des Patienten zur Ausführung von Tätigkeiten, die Urteilsvermögen sowie motorische und kognitive Fertigkeiten erfordern, sind der klinische Zustand des Patienten sowie das Sicherheitsprofil von Belimumab zu berücksichtigen.
Unerwünschte WirkungenErwachsene
Die Sicherheit von Belimumab bei SLE-Patienten wurde in 3 vor der Zulassung durchgeführten placebokontrollierten Studien mit intravenöser Verabreichung, einer nachfolgenden regionalen placebokontrollierten Studie mit intravenöser Verabreichung und einer placebokontrollierten Studie mit subkutaner Verabreichung sowie in zwei nach Markteinführung durchgeführten placebokontrollierten Studien mit intravenöser Verabreichung untersucht. Die Sicherheit bei Patienten mit Lupusnephritis wurde in einer placebokontrollierten Studie mit intravenöser Verabreichung evaluiert.
Die unten beschriebenen Daten beruhen auf der intravenösen Behandlung von 674 SLE-Patienten aus den drei vor der Zulassung durchgeführten klinischen Studien und 470 Patienten aus der nachfolgenden placebokontrollierten Studie (10 mg/kg verabreicht über einen Zeitraum von 1 Stunde an den Tagen 0, 14, 28 sowie anschliessend alle 28 Tage über einen Zeitraum von 52 Wochen) und der subkutanen Behandlung von 556 Patienten mit SLE (200 mg einmal wöchentlich bis zu 52 Wochen). Die für die intravenöse Behandlung aufgeführten Sicherheitsdaten umfassen auch Daten, die bei manchen Patienten mit SLE über den Zeitraum von 52 Wochen hinausgehen. Darüber hinaus sind Daten von 224 Patienten mit Lupusnephritis enthalten, die Benlysta intravenös (10 mg/kg für bis zu 104 Wochen) erhielten. Daten aus Meldungen nach dem Inverkehrbringen wurden ebenfalls berücksichtigt. Die meisten Patienten erhielten darüber hinaus eine oder mehrere der folgenden Begleitmedikationen zur Behandlung von SLE: Kortikosteroide, Immunmodulatoren, Anti-Malaria-Mittel, nichtsteroidale Antirheumatika.
Unerwünschte Wirkungen werden im Folgenden nach MedDRA-Systemorganklasse und Häufigkeit angegeben. Es wurden die folgenden Häufigkeitskategorien verwendet:
Sehr häufig: ≥1/10
Häufig: ≥1/100 und <1/10
Gelegentlich: ≥1/1'000 und <1/100
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektionen (70%).
Häufig: Nasopharyngitis, Bronchitis, Pharyngitis, Zystitis, Gastroenteritis viral.
Erkrankungen des Blut- und Lymphsystems
Häufig: Leukopenie.
Herzerkrankungen
Gelegentlich: Bradykardien.
Erkrankungen des Immunsystems
Häufig: Überempfindlichkeitsreaktion.
Gelegentlich: Anaphylaktische Reaktion, Angioödem, Hypogammaglobulinämie.
Verspätete bzw. verzögerte Überempfindlichkeitsreaktionen können auftreten, die sich mit der Symptomatik Ausschlag bzw. Urtikaria, Übelkeit, Fieber, Fatigue, Myalgie, Kopf- und Gliederschmerzen sowie Gesichtsödem ankündigen können.
Psychiatrische Erkrankungen
Häufig: Depression.
Gelegentlich: Suizidgedanken, suizidales Verhalten.
Erkrankungen des Nervensystems
Häufig: Migräne.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Nausea (15%), Diarrhö (12%).
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag, Urtikaria.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Gliederschmerzen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Pyrexie, Infusions- oder Injektionsreaktionen, Schlaflosigkeit.
Kinder und Jugendliche
Das Nebenwirkungsprofil bei pädiatrischen Patienten basiert auf Sicherheitsdaten aus einer 52-wöchigen placebokontrollierten Studie, in der 53 Patienten mit SLE neben Begleitmedikationen Benlysta 10 mg/kg intravenös an den Tagen 0, 14 und 28 sowie anschliessend alle 28 Tage erhielten. Das Sicherheitsprofil bei pädiatrischen Patienten stimmte mit dem in klinischen Studien an Erwachsenen beobachteten überein.
Beschreibung ausgewählter Nebenwirkungen
Immunogenität
In den beiden Phase-III-Studien mit intravenöser Verabreichung bei SLE entwickelten 4 von 563 (0,7%) Patienten der Gruppe mit 10 mg/kg und 27 von 559 (4,8%) Patienten der Gruppe mit 1 mg/kg persistierende Anti-Belimumab-Antikörper. Die für die Gruppe mit 10 mg/kg berichtete Häufigkeit liegt möglicherweise unter der tatsächlichen Häufigkeit, da die Sensitivität des Assays in Gegenwart hoher Arzneimittelkonzentrationen vermindert ist.
Neutralisierende Antikörper wurden bei 3 SLE Patienten unter Belimumab 1 mg/kg (intravenös) festgestellt.
In der Phase-III-Studie mit subkutaner Verabreichung wurde in den 556 Patienten mit SLE, welche während der 52-wöchigen placebokontrollierten Periode Belimumab (200 mg einmal wöchentlich) erhielten, keine Bildung von Anti-Belimumab-Antikörpern beobachtet.
Angesichts der geringen Anzahl von Patienten, die positiv auf Antikörper gegen Belimumab getestet wurden, lassen sich hinsichtlich des Effekts der Immunogenität auf die Pharmakokinetik von Belimumab keine definitiven Schlüsse ziehen.
In der Lupusnephritis-Studie, in der 224 Patienten Benlysta 10 mg/kg intravenös erhielten, wurden keine Anti-Belimumab-Antikörper nachgewiesen.
In einer Studie an 53 pädiatrischen Patienten mit SLE kam es in keinem Fall zur Entwicklung von Antikörpern gegen Belimumab.
Todesfälle
In den kontrollierten klinischen Studien mit intravenöser Verabreichung wurden bei Patienten mit SLE unter bis zu 52-wöchiger Behandlung 14** Todesfälle verzeichnet: 6/673 Patienten unter 1 mg/kg Belimumab, 0/111 Patienten unter 4 mg/kg Belimumab, 6/674 Patienten unter 10 mg/kg Belimumab und 3/675 Patienten unter Placebo. Die häufigsten Todesursachen entsprachen den bei einer SLE-Population zu erwartenden, u.a. Infektion, Lupus-/Krankheitsschub, kardiovaskuläre Ursachen und Suizid. Bei den Patienten mit bis zu 76-wöchiger Behandlung traten keine weiteren Todesfälle ein.
[** nach Studienende verstarb ausserdem ein weiterer Patient der Gruppe mit 1 mg/kg]
In der Phase-III-Studie mit subkutaner Verabreichung bei SLE traten in der doppelblinden Phase insgesamt 5 Todesfälle auf, davon 2 (0,7%) in der Placebogruppe und 3 (0,5%) in der Gruppe mit Belimumab 200 mg subkutan. Die drei Todesfälle in der Gruppe mit Belimumab gingen jeweils auf eine Infektion zurück, die 2 Todesfälle in der Placebogruppe waren durch Herzstillstand und Thrombozytopenie bedingt.
Infektionen
In den drei vor der Marktzulassung durchgeführten klinischen Studien mit intravenöser Verabreichung betrug die Inzidenz von Infektionen insgesamt 70% in der Gruppe unter Belimumab und 67% in der Gruppe unter Placebo. Infektionen, die bei mindestens 3% der Patienten unter Belimumab und mindestens 1% häufiger als bei Patienten unter Placebo auftraten, waren Nasopharyngitiden, Bronchitiden, Pharyngitiden, Zystitiden und virale Gastroenteritiden. Schwerwiegende Infektionen traten bei 5% der Patienten sowohl unter Belimumab als auch unter Placebo auf. Die häufigsten schwerwiegenden Infektionen waren Pneumonien und Harnwegsinfektionen (Häufigkeit 0,5%). Ein Teil der Infektionen verlief schwer oder tödlich.
In klinischen Studien über 18 Monate (gepoolte Daten für intravenöse und subkutane Verabreichung) wurden schwerwiegende opportunistische Infektionen unter Belimumab numerisch häufiger beobachtet (10/2'014, 0,5% gegenüber 0/955 unter Placebo).
In einer randomisierten (1:1), doppelblinden, placebokontrollierten, 52-wöchigen Post-Marketing-Sicherheitsstudie (BEL115467) mit 4003 SLE-Patienten, in der die Mortalität und spezifische unerwünschte Ereignisse bei Erwachsenen untersucht wurden, traten bei 3,7% der Patienten, die Belimumab 10 mg/kg intravenös erhielten, und bei 4,1% der Patienten, die Placebo erhielten, schwere Infektionen auf. Tödliche Infektionen traten bei 0,45% (9/2002) der Belimumab-Patienten und bei 0,15% (3/2001) der Placebo-Patienten auf, während die Inzidenz der Gesamtmortalität in der Belimumab-Gruppe bei 0,50% (10/2002) und in der Placebo-Gruppe bei 0,40% (8/2001) lag.
In der Lupusnephritis-Studie erhielten die Patienten eine Standard-Basistherapie (siehe «Klinische Studien»). Bei 13,8% der Patienten, die Belimumab erhielten, und bei 17,0% der Patienten, die Placebo erhielten, traten schwerwiegende Infektionen auf. Tödliche Infektionen wurden bei 0,9% (2/224) der Patienten unter Belimumab und bei 0,9% (2/224) der Patienten unter Placebo beobachtet.
Psychiatrische Erkrankungen
In den drei vor der Zulassung durchgeführten klinischen Studien bei SLE mit intravenöser Verabreichung (n=2133) wurden psychiatrische Ereignisse häufiger unter Benlysta (16%) berichtet als unter Placebo (12%), wobei es sich hauptsächlich um depressionsbezogene Ereignisse (6,3% unter Benlysta und 4,7% unter Placebo), Schlaflosigkeit (6,0% unter Benlysta und 5,3% unter Placebo) und Angst (3,9% unter Benlysta und 2,8% unter Placebo) handelte. Schwerwiegende psychiatrische Ereignisse wurden bei 0,8% der Patienten unter Benlysta (0,6% bei 1 mg/kg, 1,2% bei 10 mg/kg) und bei 0,4% der Patienten unter Placebo berichtet. Eine schwerwiegende Depression trat bei 0,4% (6/1458) der Patienten unter Benlysta und bei 0,1% (1/675) der Patienten unter Placebo auf. Bei Patienten unter Benlysta wurden zwei Suizide (0,1%) verzeichnet.
In einer nach Markteinführung durchgeführten randomisierten, doppelblinden, placebokontrollierten SLE-Studie mit intravenöser Verabreichung von Belimumab 10 mg/kg wurde bei 1,0% (20/2002) der Patienten unter Belimumab und bei 0,3% (6/2001) der Patienten unter Placebo über schwerwiegende psychiatrische Ereignisse berichtet. Eine schwerwiegende Depression wurde bei 0,3% (7/2002) der Patienten unter Belimumab und bei <0,1% (1/2001) der Patienten unter Placebo beobachtet. Die allgemeine Inzidenz von schwerwiegenden Suizidgedanken, schwerwiegendem suizidalem Verhalten oder schwerwiegender Selbstverletzung ohne Suizidabsicht betrug 0,7% (15/2002) in der Belimumab-Gruppe und 0,2% (5/2001) in der Placebogruppe.
Auf der Columbia-Suicide-Severity-Rating-Scale (C-SSRS) gaben 2,4% (48/1974) der Patienten unter Belimumab Suizidgedanken oder suizidales Verhalten an, verglichen mit 2,0% (39/1988) der Patienten unter Placebo. Über Suizid wurde in keiner Gruppe berichtet.
In der oben aufgeführten SLE-Studie mit intravenöser Verabreichung wurden Patienten mit psychiatrischen Erkrankungen in der Anamnese nicht ausgeschlossen.
In der klinischen SLE-Studie mit subkutaner Verabreichung dagegen wurden Patienten mit anamnestisch bekannten psychiatrischen Erkrankungen ausgeschlossen, und über schwerwiegende psychiatrische Ereignisse wurde bei 0,2% (1/556) der Patienten unter Belimumab und bei keinem der Patienten unter Placebo berichtet. Schwerwiegende depressionsbedingte Ereignisse oder Suizide kamen in keiner Gruppe vor. Auf der C-SSRS gaben 1,3% (7/554) der Patienten unter Belimumab und 0,7% (2/277) der Patienten unter Placebo Suizidgedanken oder suizidales Verhalten an.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungZur Überdosierung von Belimumab liegen eingeschränkte Erfahrungen vor. Die im Zusammenhang mit Überdosierungsfällen berichteten unerwünschten Wirkungen reflektierten das bekannte Sicherheitsprofil von Belimumab.
Eigenschaften/WirkungenATC-Code
L04AG04
Wirkungsmechanismus
Der B-Lymphozyten-Stimulator (BLyS, auch als BAFF und TNFSF13 bezeichnet) gehört zur Familie der Tumornekrosefaktor-(TNF-)Liganden und ist das Zielmolekül von Belimumab. BLyS hemmt die B-Zell-Apoptose und stimuliert die B-Zell-Proliferation und die Differenzierung von B-Zellen zu Immunglobulin produzierenden Plasmazellen.
BLyS wird bei Patienten mit SLE überexprimiert. Es besteht ein starker Zusammenhang zwischen der SLE-Krankheitsaktivität (gemessen anhand des «Safety of Estrogen in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index» [SELENA-SLEDAI]) und den BLyS-Plasmaspiegeln.
Belimumab ist ein humaner monoklonaler IgG1λ-Antikörper von ca. 147 kDa, der spezifisch an löslichen humanen BLyS bindet und dessen biologische Aktivität hemmt. Belimumab bindet nicht direkt an B-Zellen, sondern bewirkt über Bindung und Neutralisation von BLyS eine Hemmung des Überlebens von B-Zellen – einschliesslich der autoreaktiven B-Zellen – und vermindert die Differenzierung von B-Zellen zu Immunglobulin produzierenden Plasmazellen.
Pharmakodynamik
In Studien mit intravenöser oder subkutaner Verabreichung gingen die medianen IgG-Konzentrationen bei Patienten mit SLE unter Belimumab bis Woche 52 um 11-15% zurück, bei Patienten unter Placebo um 2,5-0,7%.
Bei SLE-Patienten mit Anti-dsDNA-Antikörpern zu Studienbeginn war unter Belimumab bereits in Woche 4 eine Reduktion feststellbar; bis Woche 52 waren 16-18% der Patienten unter Belimumab gegenüber 7-15% der Patienten unter Placebo Anti-dsDNA-negativ.
Bei SLE-Patienten mit niedrigen Komplementspiegeln zu Studienbeginn führte die Behandlung mit Belimumab zu einem Anstieg der Komplementspiegel (C3 und C4), der bereits in Woche 4 zu beobachten war und im weiteren Verlauf anhielt. Bis Woche 52 hatten sich die C3- und C4-Spiegel bei 38-42% bzw. 44-53% der Patienten unter Belimumab, jedoch nur bei 17-21% bzw. 19-20% der Patienten unter Placebo normalisiert.
Unter Belimumab waren in Woche 52 die zirkulierenden B-Zellen, transitionale, naive und aktivierte B-Zellen, Plasmazellen und das SLE-B-Zell-Subset vermindert. Reduktionen von naiven, Plasma- und kurzlebigen Plasmazellen sowie des SLE-B-Zell-Subsets waren bereits in Woche 8 feststellbar. Die Gedächtniszellen stiegen anfänglich an und kehrten bis Woche 52 langsam wieder auf die Ausgangswerte zurück.
In einer unkontrollierten Langzeit-Verlängerungsstudie bei SLE mit intravenöser Verabreichung wurden B-Zellen (unter anderem naive, aktivierte, Plasmazellen und die SLE-B-Zell-Untergruppe) sowie die IgG-Konzentrationen unter laufender Behandlung mehr als 7 Jahre lang beobachtet. Es wurde ein deutlicher, anhaltender und progressiver Rückgang unterschiedlicher B-Zell-Untergruppen festgestellt, der zu einer medianen Abnahme der naiven B-Zellen um 87%, der B-Gedächtniszellen um 67%, der aktivierten B-Zellen um 99% und der Plasmazellen um 92% nach mehr als 7-jähriger Behandlung führte. Nach ca. 7 Jahren wurde eine mediane Abnahme der IgG-Konzentrationen um 28% beobachtet, wobei 1,6% der Patienten einen Rückgang der IgG-Konzentrationen auf unter 400 mg/dL zeigten. Die Inzidenz der gemeldeten unerwünschten Wirkungen blieb im Studienverlauf insgesamt stabil oder ging leicht zurück.
Bei Patienten mit Lupusnephritis kam es nach der Behandlung mit Benlysta (10 mg/kg intravenös) oder Placebo zu einem Anstieg der Serum-IgG-Spiegel, der mit einer verminderten Proteinurie assoziiert war. Im Vergleich zu Placebo wurde in der Benlysta-Gruppe ein geringerer Anstieg des Serum-IgG-Spiegels beobachtet, wie bei dem bekannten Mechanismus von Belimumab zu erwarten war. In Woche 104 betrug der mediane prozentuale Anstieg der IgG-Spiegel gegenüber dem Ausgangswert 17% für Benlysta und 37% für Placebo. Die beobachteten Verringerungen der Autoantikörper, Erhöhungen des Komplements und Verringerungen der zirkulierenden gesamten B-Zellen und B-Zell-Untergruppen stimmten mit den SLE-Studien überein.
Das in einer Studie bei pädiatrischen Patienten mit SLE beobachtete pharmakodynamische Ansprechen stand im Einklang mit den Daten von Erwachsenen.
Klinische Wirksamkeit
Intravenöse Infusion bei Erwachsenen mit SLE
In einer Phase-II-Studie in 449 Patienten, behandelt mit intravenös verabreichtem Belimumab 1, 4 vs. 10 mg/kg, wurde ein Effekt nur in ANA positiven Patienten beobachtet. In zwei randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien (1 mg vs. 10 mg/kg) wurden 1'684 Patienten mit einer aktiven SLE-Erkrankung mit SELENA-SLEDAI-Score ≥6 und ANA-Titer ≥1:80 und/oder positiv für Anti-dsDNA [≥30 Einheiten/mL]) aufgenommen. Patienten mit schwerer, aktiver Lupusnephritis und schwerem, zentralnervösem Lupus waren von der Teilnahme ausgeschlossen. Ausgeschlossen waren ausserdem Patienten, die positiv auf HIV-Antikörper, Hepatitis-B-Oberflächenantigene oder Hepatitis-C-Antikörper getestet wurden, sowie Transplantationspatienten und Patienten mit Hypogammaglobulinämie oder IgA-Mangel.
Die Patienten erhielten eine SLE-Basistherapie mit Kortikosteroiden, Anti-Malaria-Mittel, NSAR oder anderen Immunsuppressiva. Ausschlusskriterien waren eine vorausgegangene B-Zell-gerichtete Therapie, ein anderes experimentelles biologisches Arzneimittel im vorausgegangenen Jahr, HIV-Infektion, und aktive Hepatitis-B oder Hepatitis-C. Sowohl Studie 1 (über 76 Wochen) als auch Studie 2 (über 52 Wochen) hatten die primären Endpunkte zu Woche 52, bestehend aus einem zusammengesetzten Endpunkt (SLE-Responder-Index) und definiert als das Erfüllen aller nachfolgenden Kriterien in Woche 52 gegenüber Studienbeginn:
·Verminderung des SELENA-SLEDAI-Score um ≥4 Punkte und
·kein neuer Organbefall im BILAG-A-Score bzw. mehr als 1 neuer Organbefall im BILAG-B-Score (BILAG=British Isles Lupus Assessment Group)
·keine Verschlechterung (Anstieg um ≤0,30 Punkte) des PGA-Scores (PGA=Physician's Global Assessment)
Der SLE-Responder-Index stützt sich auf den SELENA-SLEDAI-Score als objektives Mass für die Verminderung der globalen Krankheitsaktivität, den BILAG-Index zum Ausschluss einer relevanten Verschlechterung in einem spezifischen Organsystem und den PGA-Score, um sicherzustellen, dass Verbesserungen der Krankheitsaktivität nicht auf Kosten des Allgemeinzustands des Patienten erreicht werden.
Studie 1 (HGS1006-C1056) wurde vorwiegend in Nordamerika und Westeuropa durchgeführt. Nach ihrer ethnischen Abstammung waren die Teilnehmer wie folgt verteilt: 70% Weisse/Kaukasier, 14% Schwarze/Afroamerikaner, 13% Ureinwohner Alaskas oder Amerikas und 3% Asiaten. Als Basisbehandlung wurden unter anderem Kortikosteroide (76%), Immunsuppressiva (56%) und Anti-Malaria-Mittel (63%) eingesetzt.
Studie 2 (HGS1006-C1057) wurde in Südamerika, Osteuropa, Asien und Australien durchgeführt. Nach ihrer ethnischen Abstammung waren die Teilnehmer wie folgt verteilt: 38% Asiaten, 26% Weisse/Kaukasier, 32% Ureinwohner Alaskas oder Amerikas und 4% Schwarze/Afroamerikaner. Als Basisbehandlung wurden unter anderem Kortikosteroide (96%), Immunsuppressiva (42%) und Anti-Malaria-Mittel (67%) eingesetzt.
Das mediane Alter der Patienten betrug in beiden Studien 37 Jahre (Bereich: 18 bis 73 Jahre), die Patienten waren mehrheitlich (94%) weiblich. Bei der Voruntersuchung wurden die Patienten nach dem Schweregrad der Krankheit – auf Grundlage ihres SELENA-SLEDAI-Scores (≤9 vs. ≥10) –, der Proteinurie (<2 g pro 24 Stunden vs. ≥2 g pro 24 Stunden) und ihrer ethnischen Abstammung stratifiziert und anschliessend per Randomisierung einer Behandlung mit Belimumab 1 mg/kg, Belimumab 10 mg/kg oder Placebo zusätzlich zur Basistherapie zugeteilt. Die Patienten erhielten die Studienmedikation intravenös über 1 Stunde an den Tagen 0, 14 und 28 und anschliessend alle 28 Tage über 48 oder 72 Wochen.
Belimumab induzierte in beiden Studien zu Woche 52 signifikante Verbesserungen des SLE-Responder-Index sowie dessen einzelnen Komponenten, siehe Tabelle 1. In Studie 1 zu Woche 76 unterschied sich der SLE-Responder-Index (SRI) mit Belimumab nicht signifikant von dem mit Placebo (39% bzw. 32%).
Tabelle 1: Ansprechrate zu Woche 52
|
Ansprechen
|
Studie 1
|
Studie 2
| |
Placebo
(n=275)
|
Belimumab
1 mg/kg*
(n=271)
|
Belimumab
10 mg/kg
(n=273)
|
Placebo
(n=287)
|
Belimumab
1 mg/kg*
(n=288)
|
Belimumab
10 mg/kg
(n=290)
| |
SLE-Responder-Index
|
33,8%
|
40,6%
(p=0,104)
|
43,2%
(p=0,021)
|
43,6%
|
51,4%
(p=0,013)
|
57,6%
(p=0,0006)
| |
Komponenten des SLE-Responder-Index
| |
Prozent Patienten mit Verminderung des SELENA-SLEDAI ≥4
|
35,6%
|
42,8%
|
46,9%
(p=0,006)
|
46,0%
|
53,1%
|
58,3%
(p=0,0024)
| |
Prozent Patienten ohne Verschlechterung nach BILAG-Index
|
65,1%
|
74,9%
|
69,2%
(p=0,32)
|
73,2%
|
78,5%
|
81,4%
(p=0,018)
| |
Prozent Patienten ohne Verschlechterung nach PGA
|
62,9%
|
72,7%
|
69,2%
(p=0,13)
|
69,3%
|
78,8%
|
79,7%
(p=0,0048)
|
* Die 1 mg/kg Dosis ist nicht zugelassen.
In einer gepoolten Analyse der beiden Studien belief sich der prozentuale Anteil der Patienten, die bei Studienbeginn >7,5 mg/Tag Prednison (oder Äquivalent) erhalten hatten und deren durchschnittliche Kortikosteroid-Dosis während der Wochen 40 bis 52 gegenüber Studienbeginn um mindestens 25% auf ≤7,5 mg/Tag Prednisonäquivalent reduziert wurde, auf 17,9% in der Gruppe mit Belimumab bzw. 12,3% in der Placebogruppe (p=0,0451).
Schwere SLE-Schübe mit einem Anstieg des SELENA-SLEDAI-Scores auf >12 waren ausgeschlossen. Das Risiko schwerer Schübe <12 war während des 52-wöchigen Beobachtungszeitraums in der Belimumab-Gruppe gegenüber der Placebogruppe um 36% vermindert (Hazard-Ratio=0,64; p=0,0011).
Die Wirksamkeit und Sicherheit von Benlysta in Kombination mit einem einzigen Zyklus Rituximab wurde in einer 104-wöchigen randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie mit 292 Patienten (BLISS-BELIEVE) untersucht. Der primäre Endpunkt war der Anteil an Patienten mit dem Status einer kontrollierten Erkrankung, definiert als SLEDAI-2K-Score ≤2, der ohne Immunsuppressiva und mit Kortikosteroiden in einer Dosis äquivalent zu ≤5 mg Prednison pro Tag in Woche 52 erreicht wurde. Dies wurde bei 19,4% (n = 28/144) der mit Benlysta in Kombination mit Rituximab behandelten Patienten und bei 16,7% (n = 12/72) der mit Benlysta in Kombination mit Placebo behandelten Patienten erreicht (Odds-Ratio 1,27; 95%-KI: 0,60; 2,71; p = 0,5342). Bei Patienten, die mit Benlysta in Kombination mit Rituximab behandelt wurden, wurden Nebenwirkungen (91,7% vs. 87,5%), schwere Nebenwirkungen (22,2% vs. 13,9%) und schwere Infektionen (9,0% vs. 2,8%) häufiger beobachtet als bei Patienten, die mit Benlysta in Kombination mit Placebo behandelt wurden.
Intravenöse Infusion bei Lupusnephritis
Die Wirksamkeit und Sicherheit von Belimumab 10 mg/kg intravenös verabreicht über einen Zeitraum von 1 Stunde an den Tagen 0, 14 und 28 und in der Folge alle 28 Tage wurde in einer 104-wöchigen randomisierten (1:1) doppelblinden placebokontrollierten Phase-III-Studie (BEL114054) mit 448 Patienten mit aktiver Lupusnephritis bewertet.
Bei den Patienten lag zum Zeitpunkt des Screenings eine klinische SLE-Diagnose entsprechend den Klassifizierungskriterien des ACR, eine durch Biopsie bestätigte Lupusnephritis Klasse III, IV und/oder V sowie eine aktive Nierenerkrankung vor, die eine Standardtherapie erforderlich machte (Kortikosteroide mit Mycophenolat-Mofetil für Induktion und Erhaltung oder Cyclophosphamid für die Induktions-, gefolgt von Azathioprin für die Erhaltungstherapie). Die Studie wurde in Asien, Nordamerika, Südamerika und Europa durchgeführt. Das Patientenalter betrug im Median 31 Jahre (Spanne: 18 bis 77 Jahre). Die Teilnehmenden waren mehrheitlich weiblich (88%).
Primärer Wirksamkeitsendpunkt war das primäre renale Ansprechen (Primary Efficacy Renal Response, PERR) in Woche 104, definiert als ein Ansprechen in Woche 100, bestätigt durch eine Wiederholungsmessung der folgenden Parameter in Woche 104: Urinprotein/Kreatinin-Verhältnis (uPCR) ≤0,7 und geschätzte glomeruläre Filtrationsrate (eGFR) ≥60 ml/min/1,73 m2 oder kein Abfall der eGFR von >20% im Vergleich zum Wert vor dem Schub.
Zu den wichtigsten sekundären Endpunkten gehörten:
·vollständiges renales Ansprechen (Complete Renal Response, CRR), definiert als ein Ansprechen in Woche 100, bestätigt durch eine Wiederholungsmessung der folgenden Parameter in Woche 104: uPCR <0,5 und eGFR ≥90 ml/min/1,73 m2 oder kein Abfall der eGFR von >10% im Vergleich zum Wert vor dem Schub
·PERR in Woche 52
·Zeit bis zum nierenbezogenen Ereignis oder Tod (nierenbezogenes Ereignis definiert als erstes Ereignis einer terminalen Niereninsuffizienz, Verdopplung des Serum-Kreatininwerts, Verschlechterung der Nierenfunktion [definiert als zunehmende Proteinurie und/oder eingeschränkte Nierenfunktion] oder Erhalt einer unzulässigen Therapie zur Behandlung von Nierenerkrankungen)
Hinsichtlich der PERR- und CRR-Endpunkte wurden Patienten, die die Studie frühzeitig abbrachen oder unzulässige Medikamente erhielten, als Non-Responder betrachtet. Für eine Einstufung als Responder hinsichtlich der betreffenden Endpunkte musste die Steroidbehandlung ab Woche 24 auf ≤10 mg/Tag reduziert werden.
Der Anteil der Patienten, die in Woche 104 PERR erreichten, war in der Belimumab-Gruppe signifikant höher als in der Placebo-Gruppe. Auch hinsichtlich der wichtigsten sekundären Endpunkte zeigten sich mit Belimumab signifikant bessere Ergebnisse als mit Placebo (Tabelle 2).
Tabelle 2: Wirksamkeitsergebnisse bei erwachsenen Lupusnephritis-Patienten
|
Wirksamkeitsendpunkt
|
Placebo
n=223
|
Belimumab
10 mg/kg
n=223
|
beobachteter Unterschied vs. Placebo
|
Odds/Hazard Ratio vs. Placebo
(95%-KI)
|
P-Wert
| |
PERR in Woche 1041
Responder
|
32,3%
|
43,0%
|
10,8%
|
OR 1,55
(1,04, 2,32)
|
0,0311
| |
PERR-Komponenten
| |
Urinprotein/Kreatinin-Verhältnis ≤0,7
|
33,6%
|
44,4%
|
10,8%
|
OR 1,54
(1,04, 2,29)
|
0,0320
| |
eGFR ≥60 ml/min/1,73 m2
oder kein eGFR-Abfall von >20% im Vergleich zum Wert vor dem Schub
|
50,2%
|
57,4%
|
7,2%
|
OR 1,32
(0,90, 1,94)
|
0,1599
| |
kein Therapieversagen
|
74,4%
|
83,0%
|
8,5%
|
OR 1,65
(1,03, 2,63)
|
0,0364
| |
CRR in Woche 1041
Responder
|
19,7%
|
30,0%
|
10,3%
|
OR 1,74
(1,11, 2,74)
|
0,0167
| |
CRR-Komponenten
| |
Urinprotein/Kreatinin-Verhältnis <0,5
|
28,7%
|
39,5%
|
10,8%
|
OR 1,58
(1,05, 2,38)
|
0,0268
| |
eGFR ≥90 ml/min/1,73 m2
oder kein eGFR-Abfall von >10% im Vergleich zum Wert vor dem Schub
|
39,9%
|
46,6%
|
6,7%
|
OR 1,33
(0,90, 1,96)
|
0,1539
| |
kein Therapieversagen
|
74,4%
|
83,0%
|
8,5%
|
OR 1,65
(1,03, 2,63)
|
0,0364
| |
PERR in Woche 521
Responder
|
35,4%
|
46,6%
|
11,2%
|
OR 1,59
(1,06, 2,38)
|
0,0245
| |
Zeit bis zum nierenbezogenen Ereignis oder Tod1
Anteil der Patienten mit Ereignis2
|
28,3%
|
15,7%
|
-
|
|
| |
Zeit bis zum Ereignis [Hazard Ratio (95%-KI)]
|
|
|
-
|
0,51
(0,34, 0,77)
|
0,0014
| |
1
PERR in Woche 104 war die primäre Wirksamkeitsanalyse; CRR in Woche 104, PERR in Woche 52 und Zeit bis zum nierenbezogenen Ereignis oder Tod waren Teil der vorab festgelegten Testhierarchie.
2 Bei Ausschluss der Todesfälle aus der Analyse (einer bei Belimumab, zwei bei Placebo) lag der Anteil der Patienten mit nierenbezogenem Ereignis mit Belimumab bei 15,2% und mit Placebo bei 27,4% (HR=0,51; 95%-KI: 0,34, 0,78).
|
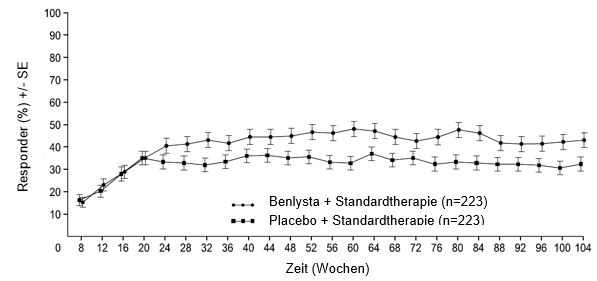
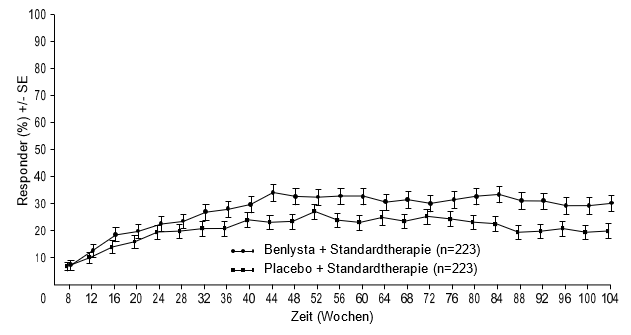
Ab Woche 24 erreichte verglichen mit Patienten unter Placebo ein numerisch grösserer Anteil der Patienten unter Belimumab PERR. Der betreffende Behandlungsunterschied blieb bis Woche 104 hindurch erhalten. Ab Woche 12 erreichte verglichen mit Patienten unter Placebo ein numerisch grösserer Anteil der Patienten unter Belimumab CRR. Der betreffende numerische Unterschied blieb bis Woche 104 hindurch erhalten (Abbildung 1).
Abbildung 1: Ansprechraten von Erwachsenen mit Lupusnephritis nach Visite
Primäres renales Ansprechen (Primary Efficacy Renal Response, PERR)
Komplettes renales Ansprechen (Complete Renal Response, CRR)
In deskriptiven Subgruppenanalysen wurden die PERR- und CRR-Raten nach Induktionstherapie (Mycophenolat oder Cyclophosphamid), Biopsieklasse (Klasse III oder IV, Klasse III + V oder Klasse IV + V oder Klasse V) und uPCR-Werten zu Studienbeginn (<3 g/g oder ≥3 g/g; Post-hoc-Analyse) untersucht (Abbildung 2).
Abbildung 2: Odds Ratio für PERR und CRR in Woche 104 in den verschiedenen Subgruppen
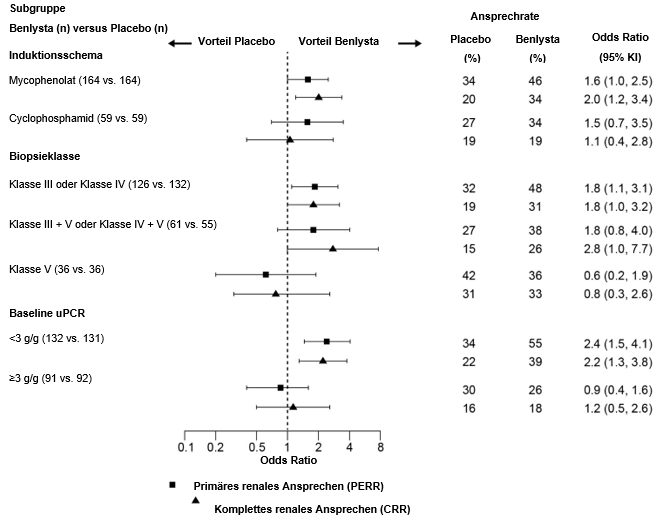
¹ Klasse III=Fokal proliferative Lupusnephritis; Klasse IV=Diffus proliferative Lupusnephritis; Klasse V=Membranöse Lupusnephritis; Klasse III + V=Gemischt membranös-fokal proliferative Lupusnephritis; Klasse IV + V=Gemischt membranös-diffus proliferative Lupusnephritis.
² Baseline-Urinprotein/Kreatinin-Verhältnis (uPCR) war eine Post-hoc-Analyse.
Andere besondere Patientengruppen
An den kontrollierten klinischen Studien nahmen zu wenige Männer oder Patienten im Alter von über 65 Jahren teil, um stichhaltige Schlüsse über die Auswirkungen von Geschlecht oder Alter auf die klinischen Outcomes ziehen zu können.
Intravenöse Infusion bei Kindern und Jugendlichen mit SLE (Benlysta wurde bei Kindern und Jugendlichen mit schwerem, aktivem Lupus des Zentralnervensystems oder schwerer, aktiver Lupusnephritis nicht untersucht).
Die Sicherheit und Wirksamkeit von Benlysta wurden in einer 52-wöchigen randomisierten, doppelblinden, placebokontrollierten Studie (BEL114055) an 93 pädiatrischen Patienten mit klinisch gemäss den Klassifizierungskriterien des American College of Rheumatology diagnostiziertem SLE untersucht. Die Patienten wiesen eine aktive SLE-Erkrankung auf, die definiert war als SELENA-SLEDAI-Score von mindestens 6 und Autoantikörper-Positivität zum Screeningzeitpunkt, entsprechend der Definition in den Studien an Erwachsenen. Die Patienten standen unter einem stabilen SLE-Behandlungsregime (Standardbehandlung). Ein- und Ausschlusskriterien entsprachen den Studien in Erwachsenen. Patienten mit schwerer aktiver Lupusnephritis, schwerem aktivem ZNS-Lupus, primärem Immundefekt, IgA-Mangel oder akuten oder chronischen behandlungsbedürftigen Infektionen wurden von der Studie ausgeschlossen. Die Studie wurde nicht für statistische Vergleiche herangezogen und alle Daten sind deskriptiv.
Diese Studie wurde in den USA, in Südamerika, Europa und Asien durchgeführt. Das Patientenalter betrug im Median 15,0 Jahre (Spanne: 6 bis 17 Jahre). In der Gruppe der 5- bis 11-Jährigen erhielten 10 Patienten Belimumab und 3 Patienten Placebo; der SELENA-SLEDAI-Score lag zwischen 4 und 13. In der Gruppe der 5-11-Jährigen waren die drei jüngsten Patienten 6 und 7 Jahre alt beim Screening. Alle drei waren in 52 Woche keine SRI-Responder, zwei davon unter einer Therapie mit Belimumab. Die übrigen Kinder in dieser Gruppe waren 9 Jahre oder älter.
In der Gruppe der 12- bis 17-Jährigen (n=79) lag der SELENA-SLEDAI-Wert zwischen 4 und 20. Die Teilnehmenden waren mehrheitlich weiblich (94,6%).
Primärer Wirksamkeitsendpunkt war der SLE-Responder-Index (SRI) nach 52 Wochen, wie in den Studien mit intravenöser Verabreichung an Erwachsene beschrieben. Unter Benlysta erreichte ein höherer Anteil der pädiatrischen Patienten ein SRI-Ansprechen als unter Placebo,. Das Ansprechen bezüglich der Einzelkomponenten des Endpunkts stimmte mit dem SRI-Gesamtansprechen überein.
Tabelle 3: Pädiatrische Ansprechrate nach Woche 52
|
Ansprechen
|
Placebo
(n=391)
|
Benlysta
10 mg/kg
(n=53)
| |
SLE-Responder-Index
Odds Ratio (95%-KI) vs. Placebo
|
43,6%
|
52,8%
1,49 (0,64; 3,46)
| |
Komponenten des SLE-Responder-Index
| |
Prozentualer Anteil Patienten mit SELENA-SLEDAI-Rückgang ≥4
Odds Ratio (95%-KI) vs. Placebo
|
43,6%
|
54,7%
1,62 (0,69; 3,78)
| |
Anteil Patienten ohne Verschlechterung gemäss BILAG-Index (%)
Odds Ratio (95%-KI) vs. Placebo
|
61,5%
|
73,6%
1,96 (0,77; 4,97)
| |
Anteil Patienten ohne Verschlechterung gemäss PGA (%)
Odds Ratio (95%-KI) vs. Placebo
|
66,7%
|
75,5%
1,70 (0,66; 4,39)
|
¹ Bei den Analysen wurden alle Probanden ausgeschlossen, bei denen eine Ausgangsmessung für eine der Komponenten fehlte (1 für Placebo).
Pädiatrische Patienten unter Benlysta 10 mg/kg wiesen ein um 64% geringeres Risiko von schweren Schüben auf als die Placebogruppe (Hazard Ratio 0,36; 95%-KI: 0,15–0,86). Unter den Patienten, die einen schweren Schub erlitten, trat der erste schwere Schub in der Placebogruppe im Median an Tag 113 ein, in der Benlysta-Gruppe an Tag 150. Dies stand im Einklang mit den Ergebnissen der klinischen Studien mit intravenöser Verabreichung an Erwachsene.
Auf Grundlage der Ansprechkriterien für den juvenilen SLE gemäss Paediatric Rheumatology International Trials Organisation/American College of Rheumatology (PRINTO/ACR) zeigte unter Benlysta ein höherer Anteil von pädiatrischen Patienten eine Verbesserung als unter Placebo.
PharmakokinetikSLE Studien
Die unten beschriebenen intravenösen pharmakokinetischen Parameter basieren auf Schätzungen der Populationsparameter bei den 563 Patienten mit SLE, die in den beiden Phase-III-Studien Belimumab 10 mg/kg intravenös (an den Tagen 0, 14, 28 sowie anschliessend alle 28 Tage über einen Zeitraum von bis zu 52 Wochen) erhalten hatten.
Die nachfolgend behandelten pharmakokinetischen Parameter bei subkutaner Verabreichung basieren auf populationspharmakokinetischen Schätzungen anhand der Daten von 661 Teilnehmern, darunter 554 SLE-Patienten und 107 gesunde Personen, die Belimumab subkutan erhielten.
Absorption
Nach intravenöser Verabreichung werden die maximalen Serumkonzentrationen (Cmax) von Belimumab in der Regel bei Infusionsende oder kurz danach beobachtet. Die Cmax betrug 313 µg/mL im Kumulationsgleichgewicht. Dieser Wert basiert auf der Simulation des Konzentrations-Zeit-Profils mit den typischen Parameterwerten des populationspharmakokinetischen Modells.
Distribution
Belimumab wurde mit einem Gesamtverteilungsvolumen von ca. 5 L in die Gewebe verteilt.
Metabolismus
Belimumab ist ein Protein und wird über den Abbau in kleine Peptide und einzelne Aminosäuren metabolisiert.
Elimination
Nach intravenöser Verabreichung nahmen die Belimumab-Konzentrationen im Serum biexponentiell ab, mit einer Distributionshalbwertszeit von 1,75 Tagen und einer terminalen Halbwertszeit von 19,4 Tagen. Die systemische Clearance belief sich auf 215 mL/Tag.
Lupusnephritis-Studie
Eine populations-pharmakokinetische Analyse wurde bei 224 erwachsenen Patienten mit Lupusnephritis durchgeführt, die Benlysta 10 mg/kg intravenös erhielten (an den Tagen 0, 14, 28, und dann alle 28 Tage bis zu 104 Wochen). Bei Patienten mit Lupusnephritis war aufgrund der Aktivität der Nierenerkrankung die Belimumab-Clearance anfänglich höher als die in SLE-Studien beobachtete. Nach 24 Wochen Behandlung und während der gesamten restlichen Studiendauer waren die Belimumab-Clearance und -Exposition jedoch ähnlich hoch wie bei erwachsenen Patienten mit SLE, die Belimumab 10 mg/kg intravenös erhielten.
Auf der Grundlage der populations-pharmakokinetischen Modellrechnung und Simulation wird davon ausgegangen, dass die durchschnittlichen Steady State-Konzentrationen bei einmal wöchentlicher subkutaner Verabreichung von Belimumab 200 mg bei Erwachsenen mit Lupusnephritis ähnlich hoch sind wie bei Erwachsenen mit Lupusnephritis, die Belimumab 10 mg/kg alle 4 Wochen intravenös erhalten.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Es wurden keine formalen Studien zur Beurteilung der Auswirkungen einer Leberfunktionseinschränkung auf die Pharmakokinetik von Belimumab durchgeführt. Immunglobuline wie Belimumab werden durch das retikulo-endotheliale System abgebaut. Es ist daher unwahrscheinlich, dass sich Veränderungen der Leberfunktion auf die Elimination von Belimumab auswirken.
Nierenfunktionsstörungen
Es wurden keine formalen Studien zur Beurteilung der Auswirkungen einer Nierenfunktionseinschränkung auf die Pharmakokinetik von Belimumab durchgeführt. Während der klinischen Entwicklung wurde Belimumab (intravenös und subkutan) bei einer limitierten Zahl von SLE-Patienten mit Nierenfunktionseinschränkung untersucht (Kreatinin-Clearance <60 mL/min, darunter einige Patienten mit einer Kreatinin-Clearance <30 mL/min). Eine Proteinurie (≥2 g/Tag) und eine verminderte Kreatinin-Clearance führten zu keinen klinisch relevanten Veränderungen der Clearance von Belimumab. Daher wird bei Patienten mit Nierenfunktionseinschränkung keine Dosisanpassung empfohlen.
Ältere Patienten
Belimumab wurde bei einer eingeschränkten Zahl älterer Patienten untersucht. Das Alter hatte laut der populationspharmakokinetischen Analyse keinen Einfluss auf die Exposition gegenüber Belimumab. Allerdings lässt sich ein Einfluss des Alters angesichts der geringen Anzahl der untersuchten Personen ab 65 Jahren nicht definitiv ausschliessen.
Kinder und Jugendliche
Die pharmakokinetischen Parameter basieren auf individuellen Parameter-Schätzwerten aus einer populationspharmakokinetischen Analyse der Daten von 53 Teilnehmenden einer Studie an pädiatrischen Patienten mit SLE. Die Belimumab-Expositionen bei pädiatrischen und erwachsenen SLE-Patienten waren vergleichbar.
Sonstige Patientenmerkmale
Geschlecht, Rasse oder ethnische Abstammung hatten keinen signifikanten Einfluss auf die Pharmakokinetik von Belimumab. Dem Einfluss der Körpermasse auf die Belimumab-Exposition nach intravenöser Verabreichung wird durch eine gewichtsadaptierte Dosierung Rechnung getragen.
Umstellung von intravenöser auf subkutane Verabreichung
SLE
Patienten mit SLE, bei denen der Wechsel von 10 mg/kg intravenös alle 4 Wochen auf 200 mg subkutan wöchentlich unter Verwendung eines Übergangsintervalls von 1 bis 4 Wochen erfolgte, hatten vor der ersten subkutanen Dosis Belimumab-Serumkonzentrationen, die nahe an ihrer möglichen subkutanen Steady-State Tal-Konzentration lagen (siehe «Dosierung/Anwendung»). Auf der Grundlage von Simulationen mit populations-pharmakokinetischen Parametern waren die durchschnittlichen Steady-State Belimumab-Konzentrationen für 200 mg subkutan wöchentlich ähnlich wie 10 mg/kg intravenös alle 4 Wochen.
Lupusnephritis
Basierend auf populations-pharmakokinetischen Simulationen wird für Patienten mit Lupusnephritis, die von 10 mg/kg intravenös ein bis zwei Wochen nach Abschluss der ersten beiden intravenösen Dosierungen auf 200 mg subkutan wöchentlich übergehen, ähnliche durchschnittliche Belimumab-Serumkonzentration vorhergesagt wie für Patienten, denen alle 4 Wochen 10 mg/kg intravenös verabreicht wird (siehe «Dosierung/Anwendung»).
Präklinische DatenIn den Studien zur Toxizität bei wiederholter Verabreichung und zur Reproduktionstoxizität liessen die präklinischen Daten keine besonderen Risiken für den Menschen erkennen.
Toxizität bei wiederholter Gabe
Die intravenöse und subkutane Verabreichung führte bei Affen zu der erwarteten Abnahme der peripheren B-Zellen und der B-Zellen lymphatischer Gewebe, ohne damit einhergehende toxikologische Befunde.
Genotoxizität
Da es sich bei Belimumab um einen monoklonalen Antikörper handelt, wurden keine Studien zur Gentoxizität durchgeführt.
Kanzerogenität
Es wurden keine Studien zur Kanzerogenität durchgeführt.
Reproduktionstoxizität
Es wurden keine Studien zum Einfluss auf die (männliche oder weibliche) Fertilität vorgenommen.
In Reproduktionsstudien wurde trächtigen Javaneraffen bis zu 21 Wochen lang alle 2 Wochen Belimumab 150 mg/kg als intravenöse Infusion verabreicht (etwa das 9-Fache der erwarteten maximalen Exposition beim Menschen). Die Behandlung mit Belimumab war nicht mit direkten oder indirekten schädlichen Auswirkungen im Sinne einer maternalen Toxizität, Entwicklungstoxizität oder Teratogenität assoziiert. Behandlungsbedingte Befunde beschränkten sich auf die zu erwartende reversible Verminderung der B-Zellen sowohl bei den Mutter- als auch bei den Jungtieren sowie auf die reversible Verminderung von IgM bei den Jungtieren. Nach dem Absetzen von Belimumab erholte sich die Zahl der B-Zellen bei erwachsenen Affen bis etwa 1 Jahr post partum bzw. bei den Jungtieren bis zum Ende des 3. Lebensmonats wieder. Die IgM-Spiegel bei in utero exponierten Jungtieren normalisierten sich bis zum Ende des 6. Lebensmonats.
Sonstige HinweiseInkompatibilitäten
Belimumab ist nicht kompatibel mit Glucose 5%.
Benlysta darf nur entsprechend den Anweisungen zubereitet und angewendet werden (siehe «Hinweise für die Handhabung»).
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Rekonstituierte und verdünnte Lösung: Bis zu 8 Stunden nach der Rekonstitution bei 2-8°C lagern. Vor direktem Sonnenlicht schützen.
Besondere Lagerungshinweise
Ungeöffnete Durchstechflaschen
Zwischen 2°C und 8°C lagern. Nicht einfrieren.
Vor Licht schützen. Bis zum Gebrauch im Originalkarton aufbewahren. Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Rekonstitution und Verdünnung
Benlysta enthält kein Konservierungsmittel; daher müssen Rekonstitution und Verdünnung unter aseptischen Bedingungen erfolgen.
Die Durchstechflasche 10 – 15 Minuten lang Raumtemperatur annehmen lassen.
Bei der Rekonstitution und Verdünnung wird zum Durchstechen des Stopfens der Durchstechflasche eine Nadel der Grösse 21–25 G empfohlen.
Den Inhalt der 120 mg-Einmal-Durchstechflasche mit 1,5 mL sterilem Wasser für Injektionszwecke auf eine Endkonzentration von 80 mg/mL Belimumab rekonstituieren. Den Inhalt der 400-mg-Einmal-Durchstechflasche mit 4,8 mL sterilem Wasser für Injektionszwecke auf eine Endkonzentration von 80 mg/mL Belimumab rekonstituieren.
Der Strahl des sterilen Wassers muss dabei auf die seitliche Wand der Durchstechflasche gerichtet werden, um eine Schaumbildung weitestgehend zu vermeiden. Die Durchstechflasche 60 Sekunden lang vorsichtig schwenken. Während der Rekonstitution Durchstechflasche bei Raumtemperatur belassen und alle 5 Minuten 60 Sekunden lang vorsichtig schwenken, bis sich das Pulver aufgelöst hat. Nicht schütteln.
Falls für die Rekonstitution von Benlysta ein mechanisches Rekonstitutionsgerät verwendet wird, dürfen 500 U/min nicht überschritten werden; die Durchstechflasche darf nicht länger als 30 Minuten geschwenkt werden.
Die Rekonstitution ist in der Regel innerhalb von 10 bis 15 Minuten nach Zugabe des sterilen Wassers abgeschlossen, kann aber unter Umständen bis zu 30 Minuten in Anspruch nehmen. Die rekonstituierte Lösung vor direktem Sonnenlicht schützen.
Nach vollständiger Rekonstitution muss die Lösung opaleszent und farblos bis blassgelb erscheinen und darf keine Schwebeteilchen aufweisen. Kleine Luftbläschen sind jedoch zu erwarten und vertretbar.
Das rekonstituierte Produkt wird mit Natriumchlorid 0,9%, Natriumchlorid 0,45% oder Ringer-Lactat auf 250 mL verdünnt.
Bei Patienten mit einem Körpergewicht von 40 kg oder darunter kann der Einsatz von Infusionsbeuteln bzw. Infusionsflaschen mit 100 mL der genannten Lösungsmittel in Betracht gezogen werden, wobei die Endkonzentration von Belimumab im Infusionsbeutel bzw. in der Infusionsflasche 4 mg/mL nicht überschreiten darf.
Glucose 5% ist nicht kompatibel mit Belimumab und darf daher nicht verwendet werden.
Aus einem Infusionsbeutel oder einer Infusionsflasche mit 250 mL (oder 100 mL) Natriumchlorid 0,9%, Natriumchlorid 0,45% oder Ringer-Lactat das Volumen entnehmen und verwerfen, das dem Volumen der rekonstituierten Belimumab-Lösung für die erforderliche Dosis entspricht. Dann das erforderliche Volumen der rekonstituierten Belimumab-Lösung in den Infusionsbeutel bzw. die Infusionsflasche übertragen. Zum Mischen der Lösung den Beutel bzw. die Flasche vorsichtig umdrehen. Allfällige Lösungsreste in der Durchstechflasche sind zu verwerfen.
Die Belimumab-Lösung vor der Verabreichung visuell auf Schwebeteilchen und Verfärbungen prüfen. Wenn Schwebeteilchen oder Verfärbungen erkennbar sind, muss die Lösung verworfen werden.
Wenn die rekonstituierte Lösung nicht sofort verwendet wird, muss sie vor direktem Sonnenlicht geschützt und im Kühlschrank bei 2°C bis 8°C gelagert werden. Mit Natriumchlorid 0,9%, Natriumchlorid 0,45% oder Ringer-Lactat verdünnte Lösungen können bei 2°C bis 8°C aufbewahrt werden.
Zwischen der Rekonstitution und dem Ende der Infusion dürfen insgesamt nicht mehr als 8 Stunden verstreichen.
Verabreichung
Benlysta muss über einen Zeitraum von 1 Stunde infundiert werden.
Benlysta darf nicht gleichzeitig mit anderen Arzneimitteln über dieselbe intravenöse Infusionsleitung verabreicht werden. Es wurden keine Studien zur physikalischen oder biochemischen Kompatibilität bei gleichzeitiger Anwendung von Benlysta mit anderen Arzneimitteln durchgeführt.
Es wurden keine Inkompatibilitäten zwischen Belimumab und Polyvinylchlorid- oder Polyolefinbeuteln festgestellt.
Zulassungsnummer61532 (Swissmedic).
PackungenDurchstechflasche à 120 mg (A).
Durchstechflasche à 400 mg (A).
ZulassungsinhaberinGlaxoSmithKline AG, 6340 Baar.
Stand der InformationJuli 2025
|