PharmakokinetikAllgemeine Einführung
Fentanyl ist stark lipophil und kann sehr rasch über die Mundschleimhaut und etwas langsamer über den konventionellen gastrointestinalen Weg resorbiert werden. Es wird einer First-Pass-Metabolisierung in Leber und Darm unterzogen. Die Metaboliten haben keinen Anteil an den therapeutischen Wirkungen von Fentanyl.
Bei Effentora kommt eine Freisetzungstechnik zum Einsatz, die sich einer Brause-Reaktion bedient, welche Rate und Menge des über die Wangenschleimhaut aufgenommenen Fentanyls steigert. Vorübergehende pH-Veränderungen, die mit der Brause-Reaktion einhergehen, können die Auflösung (bei einem niedrigeren pH-Wert) und die Membrandurchlässigkeit (bei einem höheren pH-Wert) optimieren.
Die Verweilzeit (definiert als die Zeitdauer, die die Tablette nach buccaler Anwendung benötigt, um zu zerfallen), beeinflusst nicht die frühe systemische Verfügbarkeit von Fentanyl. In einer Vergleichsstudie mit einer 400 µg Effentora Tablette, die entweder buccal (d.h. zwischen Wange und Zahnfleisch) oder sublingual appliziert wurde, wurden die Kriterien für Bioäquivalenz erfüllt.
Die Wirkung einer Nieren- oder Leberfunktionseinschränkung auf die Pharmakokinetik von Effentora wurde nicht untersucht.
Absorption
Nach Anwendung von Effentora in der Mundhöhle wird Fentanyl mit einer absoluten Bioverfügbarkeit von 65% leicht resorbiert. Das Resorptionsprofil von Effentora resultiert grösstenteils aus einer initial raschen Aufnahme über die Wangenschleimhaut, wobei die Plasmaspitzenkonzentrationen nach venöser Probenentnahme im Allgemeinen innerhalb einer Stunde nach Anwendung in der Mundhöhle erreicht werden. Etwa 50% der angewendeten Gesamtdosis wird rasch transmukosal absorbiert und wird systemisch verfügbar. Die verbleibende Hälfte der Gesamtdosis wird geschluckt und langsam aus dem Gastrointestinaltrakt resorbiert. Etwa 30% der geschluckten Menge (50% der Gesamtdosis) entgeht der First-Pass-Elimination in Leber und Darm und wird systemisch verfügbar.
Die wichtigsten pharmakokinetischen Parameter sind in der folgenden Tabelle aufgeführt.
Pharmakokinetische Parameter* bei erwachsenen Probanden, die Effentora erhalten
|
Pharmakokinetische Parameter
(Mittel)
|
Effentora 400 Mikrogramm
| |
Absolute Bioverfügbarkeit
|
65 % (± 20 %)
| |
Transmukosal aufgenommener Anteil
|
48 % (± 31,8 %)
| |
Tmax (Minuten) **
|
46,8 (20-240)
| |
Cmax (ng/ml)
|
1,02 (± 0,42)
| |
AUC0-tmax (ng x h/ml)
|
0,40 (± 0,18)
| |
AUC0-inf (ng x h/ml)
|
6,48 (± 2,98)
|
* basierend auf venösen Blutproben (Plasma). Die Fentanylcitrat-Konzentrationen waren im Serum höher als im Plasma: Serum AUC und Cmax waren ungefähr 20% bzw. 30% höher als Plasma AUC und Cmax. Der Grund dieses Unterschiedes ist unbekannt.
** Daten für Tmax sind als Median angegeben (Bereich).
In pharmakokinetischen Studien, die die absolute und relative Bioverfügbarkeit von Effentora und oral-transmukosal verabreichtem Fentanylcitrat (OTFC) verglichen, wies die Fentanylabsorptionsrate und -menge für Effentora eine 30% bis 50% höhere Exposition auf als die von oral-transmukosal verabreichtem Fentanylcitrat. Wenn von anderen oralen Fentanylcitrat-Arzneimitteln umgestellt wird, ist eine unabhängige Dosistitration mit Effentora erforderlich, da die Bioverfügbarkeit der Arzneimittel signifikant unterschiedlich ist. Jedoch kann bei diesen Patienten eine Anfangsdosis in Betracht gezogen werden, die höher als 100 Mikrogramm liegt.
Siehe Abbildung unten:
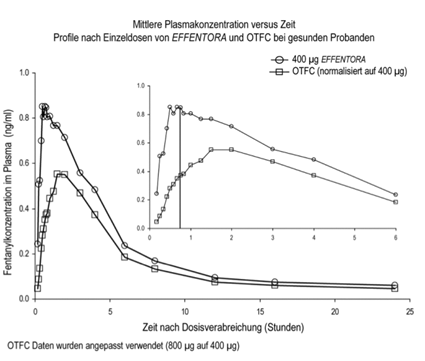
Unterschiede in der Verfügbarkeit von Effentora wurden in einer klinischen Studie bei Patienten mit einer Mukositis des Schweregrads 1 beobachtet. Bei Patienten mit Mukositis war die Cmax 1% und die AUC0-8 25% höher als bei denjenigen ohne Mukositis. Die beobachteten Unterschiede waren nicht klinisch signifikant.
Distribution
Fentanyl ist in hohem Masse lipophil und verteilt sich mit einem grossen scheinbaren Verteilungsvolumen gut über das Gefässsystem. Nach buccaler Anwendung von Effentora erfährt Fentanyl initial eine rasche Verteilung, die ein Gleichgewicht von Fentanyl zwischen Plasma und stark durchbluteten Geweben (Hirn, Herz und Lungen) darstellt. Anschliessend erfolgt eine Umverteilung von Fentanyl zwischen tiefem Kompartiment (Muskeln und Fett) und Plasma.
Die Plasmaproteinbindung von Fentanyl beträgt 80% bis 85%. Das Hauptbindungsprotein ist alpha-1-saures Glykoprotein, aber auch Albumin und Lipoproteine haben einen gewissen Anteil. Der freie Anteil von Fentanyl erhöht sich bei Vorliegen einer Azidose.
Metabolismus
Die Verstoffwechselungswege nach buccaler Anwendung von Effentora waren bislang nicht Gegenstand klinischer Studien. Fentanyl wird in der Leber und Darmschleimhaut durch CYP3A4 Isoform zu Norfentanyl metabolisiert. In Tierstudien ist Norfentanyl pharmakologisch nicht aktiv. Mehr als 90% der verabreichten Fentanyl-Dosis wird durch Biotransformation zu N-dealkylierten und hydroxylierten inaktiven Metaboliten eliminiert.
Elimination
Nach intravenöser Gabe von Fentanyl werden weniger als 7% der verabreichten Dosis unverändert im Urin ausgeschieden und nur etwa 1% finden sich unverändert in den Faeces wieder. Die Metaboliten werden vorwiegend über den Urin ausgeschieden, während die fäkale Exkretion weniger wichtig ist.
Nach der Anwendung von Effentora ist die terminale Eliminationsphase von Fentanyl das Ergebnis der Umverteilung zwischen Plasma und tiefem Kompartiment. Diese Phase der Elimination ist langsam und resultiert in einer medianen terminalen Eliminationshalbwertzeit t1/2 von etwa 22 Stunden nach buccaler Anwendung der efferveszierenden Formulierung und etwa 18 Stunden nach intravenöser Anwendung. Die Gesamtplasma-Clearance von Fentanyl nach intravenöser Anwendung beträgt etwa 42 l/h.
Linearität/Nicht-Linearität
Es konnte eine Dosisproportionalität von 100 Mikrogramm bis 1.000 Mikrogramm gezeigt werden.
|