ZusammensetzungWirkstoffe
Perampanel.
Hilfsstoffe
Filmtabletten:
2 mg: Lactose-Monohydrat 78.5 mg, Hydroxypropylcellulose niedrig subst., Povidon, Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, rotes und gelbes Eisenoxid (E172).
4 mg: Lactose-Monohydrat 157 mg, Hydroxypropylcellulose niedrig subst., Povidon, Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, rotes Eisenoxid (E172).
6 mg: Lactose-Monohydrat 151 mg, Hydroxypropylcellulose niedrig subst., Povidon, Cellulose mikrokrist., Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, rotes Eisenoxid (E172).
8 mg: Lactose-Monohydrat 149 mg, Hydroxypropylcellulose niedrig subst., Povidon, Cellulose mikrokrist., Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, rotes und schwarzes Eisenoxid (E172).
10 mg: Lactose-Monohydrat 147 mg, Hydroxypropylcellulose niedrig subst., Povidon, Cellulose mikrokrist., Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, gelbes Eisenoxid (E172), Indigocarmin (E132).
12 mg: Lactose-Monohydrat 145 mg, Hydroxypropylcellulose niedrig subst., Povidon, Cellulose mikrokrist. Magnesiumstearat, Hypromellose 2910, Talcum, Macrogol 8000, Titandioxid, Indigocarmin (E132).
Suspension zum Einnehmen:
Sorbitol-Lösung kristallisierend (enthält Sorbitol (E420) 175 mg), mikrokristalline Cellulose, Carmellose-Natrium, Poloxamer 188, Dimeticon, Polysorbat 65, Methylcellulose, Siliciumdioxid, Macrogolstearat, Benzoesäure (E210) <0.005 mg, Sorbinsäure (E200), Schwefelsäure, Citronensäure, Natriumbenzoat (E211) 1.10 mg, gereinigtes Wasser pro 1 ml. Natriumgehalt: 0.474 mg pro 1 ml Suspension.
Indikationen/AnwendungsmöglichkeitenFycompa ist angezeigt als:
Zusatztherapie fokaler Anfälle mit oder ohne sekundäre Generalisierung bei Epilepsiepatienten ab 4 Jahren.
Zusatztherapie bei primär generalisierten tonisch-klonischen Anfällen bei Epilepsiepatienten ab 7 Jahren.
Dosierung/AnwendungFycompa muss entsprechend dem individuellen Ansprechen des Patienten titriert werden, um das Verhältnis von Wirksamkeit und Verträglichkeit zu optimieren. Der Arzt oder die Ärztin muss auf Grundlage von Gewicht und Dosis die am besten geeignete Formulierung und Stärke verschreiben. Es sind verschiedene Formulierungen von Perampanel, einschliesslich einer Suspension zum Einnehmen, erhältlich. Zwischen den Dosiserhöhungen sollte immer jeweils ein Zeitintervall von mindestens einer oder mindestens zwei Wochen liegen in Abhängigkeit von der Basis- bzw. Begleitmedikation (siehe untenstehende Empfehlungen sowie auch Abschnitt «Interaktionen») sowie dem individuellen Nutzen-Risiko Verhältnis.
Fokale Anfälle:
Erwachsene, Jugendliche und Kinder ab 4 Jahren
In der folgenden Tabelle 1 werden die empfohlenen Dosierungen für Erwachsene, Jugendliche und Kinder ab 4 Jahren in der Behandlung fokaler Anfälle zusammengefasst. Die Behandlung mit Fycompa sollte mit der kleinsten in Tabelle 1 beschriebenen Dosis für das Patientenalter und -gewicht begonnen werden. Die Dosis kann dann je nach klinischem Ansprechen und der Verträglichkeit schrittweise erhöht werden (Titrationsschritte gemäss Tabelle 1 für das jeweilige Alter und Gewicht).
Tabelle 1: Fycompa Dosierung für fokale Anfälle
|
|
Erwachsene/Jugendliche
(ab 12 Jahren)
[Tagesdosis]
|
Kinder (4 – 11 Jahre); Gewicht:
[Tagesdosis]
| |
≥30 kg
|
20 bis < 30 kg
|
< 20 kg
| |
Anfangsdosis/Tag
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
1 mg (2 ml)
| |
Titrationsschema
(schrittweise Erhöhung)a
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
1 mg (2 ml)
| |
Empfohlene Erhaltungsdosis/Tag
|
4–8 mg (8 - 16 ml)
|
4–8 mg (8-16 ml)
|
4–6 mg (8-12 ml)
|
2–4 mg (4-8 ml)
| |
Titrationsschema
(schrittweise Erhöhung)a
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
0,5 mg (1 ml)
| |
Empfohlene Höchstdosis/Tag
|
12 mg (24 ml)
|
12 mg (24 ml)
|
8 mg (16 ml)
|
6 mg (12 ml)
| |
a
nicht häufiger als in wöchentlichen Intervallen, Details siehe Abschnitt «Intervalle der Aufdosierung» unten
|
Primär generalisierte tonisch-klonische Anfälle:
Erwachsene, Jugendliche und Kinder ab 7 Jahren
In der folgenden Tabelle 2 werden die empfohlenen Dosierungen für Erwachsene, Jugendliche und Kinder ab 7 Jahren in der Behandlung primär generalisierter tonisch-klonischer Anfälle zusammengefasst. Die Behandlung mit Fycompa sollte mit der kleinesten in Tabelle 2 beschriebenen Dosis für das Patientenalter und -gewicht begonnen werden. Die Dosis kann dann je nach klinischem Ansprechen und der Verträglichkeit schrittweise (Titrationsschritte gemäss Tabelle 2 für das jeweilige Alter und Gewicht) erhöht werden
Tabelle 2: Dosierung für Primär Generalisierte Tonisch-Klonische Anfälle
|
|
Erwachsene/Jugendliche
(ab 12 Jahren)
[Tagesdosis]
|
Kinder (7 – 11 Jahre); Gewicht:
[Tagesdosis]
| |
≥30 kg
|
20 bis < 30 kg
|
< 20 kg
| |
Anfangsdosis/Tag
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
1 mg (2 ml)
| |
Titrationsschema (schrittweise Erhöhung)a
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
1 mg (2 ml)
| |
Empfohlene Erhaltungsdosis/Tag
|
Bis zu 8 mg (16 ml)
|
4–8 mg (8-16 ml)
|
4–6 mg (8-12 ml)
|
2–4 mg (4-8 ml)
| |
Titrationsschema (schrittweise Erhöhung)a
|
2 mg (4 ml)
|
2 mg (4 ml)
|
1 mg (2 ml)
|
0,5 mg (1 ml)
| |
Empfohlene Höchstdosis/Tag
|
12 mg (24 ml)
|
12 mg (24 ml)
|
8 mg (16 ml)
|
6 mg (12 ml)
| |
a
nicht häufiger als in wöchentlichen Intervallen, Details siehe Abschnitt «Intervalle der Aufdosierung» unten
|
Intervalle der Aufdosierung
Bei Patienten, die gleichzeitig Arzneimittel einnehmen, welche die Halbwertszeit von Perampanel nicht verkürzen (siehe Abschnitt «Interaktionen»), sollte nicht früher als in 2-wöchigen Abständen titriert werden.
Bei Patienten, die gleichzeitig Arzneimittel einnehmen, welche die Halbwertszeit von Perampanel verkürzen (siehe Abschnitt «Interaktionen»), sollte nicht früher als in 1-wöchigen Abständen titriert werden.
Dosierungsempfehlung bei CYP3A-induzierender Begleitmedikation
Die Ansprechraten nach zusätzlicher Gabe von Perampanel in fixen Dosen waren geringer, wenn die Patienten gleichzeitig CYP 3A-induzierende Antiepileptika (Carbamazepin, Phenytoin, Oxcarbazepin) erhielten, als bei Patienten, die gleichzeitig mit nicht-enzyminduzierenden Antiepileptika behandelt wurden. Das Ansprechen der Patienten ist zu überwachen, wenn diese von gleichzeitig angewendeten nicht-enzyminduzierenden Antiepileptika auf enzyminduzierende Substanzen oder umgekehrt umgestellt werden. Je nach individuellem klinischem Ansprechen und der Verträglichkeit kann die Dosis um jeweils 2 mg erhöht oder reduziert werden.
Art der Anwendung
Fycompa sollte einmal täglich beim Schlafengehen eingenommen werden. Fycompa kann unabhängig von den Mahlzeiten eingenommen werden (siehe «Pharmakokinetik»). Ein Wechsel von der Behandlung mit Tabletten zur Suspension bzw. umgekehrt sollte mit Vorsicht erfolgen (siehe «Pharmakokinetik»).
Filmtabletten:
Die Filmtablette ist als Ganzes mit einem Glas Wasser einzunehmen; sie sollte weder zerkaut, noch zerstossen oder geteilt werden. Die Filmtabletten haben keine Bruchrille und können nicht halbiert werden.
Suspension:
Zum Abmessen Ihrer Dosis verwenden Sie bitte die mitgelieferte Dosierspritze. Nachfolgend finden Sie die Anleitung zur Verwendung von Adapter und Spritze:
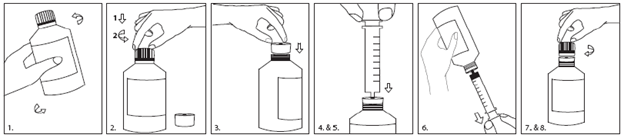
1.Vor Gebrauch mindestens 5 Sekunden schütteln.
2.Zum Öffnen der Flasche die Verschlusskappe herunterdrücken (1) und aufdrehen (2).
3.Flaschenadapter in den Flaschenhals einsetzen, sodass er dicht abschliesst.
4.Spritzenkolben ganz hinunterdrücken.
5.Die Spritze so weit wie möglich in die Adapteröffnung einführen.
6.Flasche umdrehen und die vorgeschriebene Menge Fycompa aus der Flasche in die Spritze aufziehen.
7.Flasche wieder umdrehen und die Spritze abnehmen.
8.Flaschenadapter in der Flasche lassen und die Verschlusskappe wieder aufsetzen.
Nach der Verabreichung der Suspension ziehen Sie den Kolben aus der Spritze und legen beide Bestandteile in heisses Seifenwasser. Danach Kolben und Spritzenkörper in sauberes Wasser legen, abspülen und an der Luft trockenen lassen, nicht mit einem Tuch trockenreiben.
Absetzen von Fycompa
Zur Minimierung einer möglichen Gefahr von Rebound-Anfällen wird ein ausschleichendes Absetzen empfohlen. Aufgrund seiner langen Halbwertszeit und des nachfolgend langsamen Rückgangs der Plasmakonzentrationen kann Perampanel jedoch auch abrupt abgesetzt werden, falls dies absolut notwendig ist.
Absetzen der Begleitmedikation
Beim Absetzen von gleichzeitig eingenommenen Enzyminduktoren kann die Perampanel-Plasmakonzentration erhöhen und eine Dosisanpassung erforderlich sein.
Vergessene Einnahme
Bei einmalig vergessener Einnahme sollte der Patient warten und seine nächste Dosis wie vorgesehen einnehmen, da Perampanel eine lange Halbwertszeit besitzt.
Wenn mehr als eine Dosis über einen zusammenhängenden Zeitraum von weniger als 5 Halbwertszeiten vergessen wurde (3 Wochen bei Patienten, die keine den Perampanel-Metabolismus induzierende Antiepileptika einnehmen, 1 Woche bei Patienten, die den Perampanel-Metabolismus induzierende Antiepileptika einnehmen (siehe «Interaktionen»), ist zu erwägen, die Behandlung von der letzten Dosisstufe ausgehend neu zu beginnen.
Wenn ein Patient Perampanel über einen zusammenhängenden Zeitraum von mehr als 5 Halbwertszeiten nicht mehr eingenommen hat, wird empfohlen, die weiter oben für die Behandlungseinleitung gegebenen Empfehlungen zu befolgen.
Jugendliche
Bei Jugendlichen ist auf Anzeichen von Veränderungen im Verhalten zu achten (siehe «Warnhinweise und Vorsichtsmassnahmen» zu Suizidalität und Aggression).
Pädiatrische Population
Die Sicherheit und Wirksamkeit von Fycompa bei Kindern unter 4 Jahren mit fokalen Anfällen sowie bei Kindern unter 7 Jahren mit primär generalisierten tonisch-klonischen Anfällen ist nicht gezeigt, es liegen keine Daten vor. Die Anwendung dieses Arzneimittels in dieser Altersgruppe wird nicht empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen» zu Suizidalität und Aggression).
Ältere Patienten (ab 65 Jahren)
In klinischen Studien mit Fycompa bei Epilepsie wurde keine ausreichende Zahl von Patienten ab 65 Jahren eingeschlossen, um feststellen zu können, ob diese anders als jüngere Patienten ansprechen. Das Nutzen-Risiko Verhältnis bzw. die Notwendigkeit der Anwendung von Perampanel bei älteren Patienten muss sehr sorgfältig abgewogen werden, da insbesondere bei polymedizierten, älteren Patienten zusätzlich das Potential für Arzneimittelinteraktionen in besonderer Weise zu berücksichtigen ist. Darüber hinaus siehe bitte auch «Warnhinweise und Vorsichtsmassnahmen», insbesondere zu Gleichgewichts- und Koordinationsstörungen sowie Sturzrisiko.
Patienten mit eingeschränkter Nierenfunktion
Bei Patienten mit leicht eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich. Die Anwendung von Fycompa bei Patienten mit mittelschwerer bis schwerer Nierenfunktionsstörung oder bei Hämodialysepatienten ist kontraindiziert (siehe auch Abschnitt «Kontraindikationen» sowie «Präklinische Daten»).
Patienten mit eingeschränkter Leberfunktion
Bei Patienten mit leicht und mässig eingeschränkter Leberfunktion sollten Dosiserhöhungen anhand des klinischen Ansprechens und der Verträglichkeit vorgenommen werden in einem Zeitintervall von mindestens zwei Wochen. Die Behandlung kann mit 2 mg (4 ml) begonnen und sollte in Dosisstufen von 2 mg (4 ml) jeweils im Abstand von mindestens 2 Wochen je nach Verträglichkeit und Wirksamkeit auftitriert werden bis auf höchstens 8 mg (16 ml). Die Perampanel-Dosis sollte bei Patienten mit leichter und mittelschwerer Leberfunktionsstörung 8 mg (16 ml) nicht überschreiten. Die Anwendung bei Patienten mit schwerer Leberfunktionsstörung ist kontraindiziert (siehe Abschnitt «Kontraindikationen» sowie «Präklinische Daten»).
KontraindikationenBekannte Überempfindlichkeit gegenüber Perampanel oder einem der Hilfsstoffe.
Mittelschwere oder schwere Nierenfunktionsstörung und bei Hämodialysepatienten.
Schwere Leberfunktionsstörung.
Warnhinweise und VorsichtsmassnahmenSuizidgedanken
Es gibt Hinweise, dass bei Epileptikern ein erhöhtes Risiko für Suizidalität besteht. Eine im Januar 2008 veröffentlichte Analyse der FDA (USA) bzgl. der Daten aus 199 placebokontrollierten klinischen Studien mit insgesamt 11 Antiepileptika fand für Patienten mit Epilepsie unter diesen Präparaten ein 3,6-fach höheres Risiko für Suizidalität als unter Placebo. Die einzelnen untersuchten Substanzen unterschieden sich dabei nicht in relevanter Weise bezüglich ihres Risikos für Suizidalität. In dieser Analyse war die Risikoerhöhung bei Patienten mit Epilepsie sogar stärker ausgeprägt als bei Patienten mit psychiatrischen Erkrankungen (wie z.B. bipolaren Störungen), wo eine Risikoerhöhung auf das 1,6-fache gefunden wurde. Insgesamt wurden unter den Antiepileptika in allen Indikationen Suizidgedanken oder suizidales Verhalten bei 0,43 % der Behandelten beobachtet, unter Placebo hingegen nur in 0,22 %.
Deshalb sollten Patienten (Kinder, Jugendliche und Erwachsene) auf Anzeichen von Veränderungen im Verhalten, z.B. erhöhte Irritierbarkeit, Gereiztheit, Veränderungen in der Stimmung, Feindseligkeit, neues Auftreten oder Verstärkung von aggressivem Verhalten bis hin zu Drohungen und fremdaggressiven Handlungen, sowie auf Anzeichen für das Auftreten von suizidalen Gedanken sehr gut und engmaschig überwacht werden.
Die bisher verfügbaren Daten können die Möglichkeit eines erhöhten Risikos für Fycompa nicht ausschliessen.
Patienten, die mit Fycompa behandelt werden, sollten daher insbesondere zu Beginn eines neuen Behandlungszyklus oder bei einer Dosis- bzw. Plasmaspiegeländerungen (Eintitrierung, Dosiserhöhungen, aber auch Ausschleichen sowie bei möglichen Veränderungen der Plasmaspiegel durch Veränderungen der Begleitmedikationen) hinsichtlich einer klinischen Verschlechterung (einschliesslich der Entwicklung von neuen Symptomen) und Suizidalität engmaschig überwacht werden.
Bestimmte Patientengruppen, wie Patienten mit suizidalem Verhalten oder Suizidgedanken in der Anamnese und junge Erwachsene, scheinen ein höheres Risiko für Suizidgedanken oder Suizidversuche aufzuweisen und sollten daher während der Behandlung streng überwacht werden. Dies gilt insbesondere auch für Patienten, bei welchen solche Symptome unmittelbar vor Einleitung der Therapie beobachtet wurden.
Bei Patienten mit einer klinischen Verschlechterung des Zustands (einschliesslich der Entwicklung von neuen Symptomen sowie Veränderungen im Verhalten) und/oder dem Auftreten von Suizidalität ist ein Wechsel der Therapie in Erwägung zu ziehen, insbesondere wenn diese Symptome ausgeprägt sind, abrupt auftreten oder nicht zur ursprünglichen Symptomatik des Patienten gehörten. In solchen Fällen kann auch ein Absetzen der Medikation erforderlich werden.
Es muss in jedem Fall umgehend eine angemessene Therapie in Betracht gezogen werden. Patienten und deren Betreuer sollten angewiesen werden, sofort fachärztlichen Rat einzuholen, wenn sich Anzeichen von suizidalen Gedanken oder suizidalem Verhalten oder auch fremdaggressiven Verhaltensweisen ergeben.
Dies gilt in besonderer Weise für die Gruppe der adoleszenten Patienten. Für Jugendliche (12 bis 18 Jahre) zeigte sich eine höhere Inzidenz neuropsychiatrischer Ereignisse, insbesondere ein vermehrtes Auftreten von Aggressionen, von psychotischen Störungen, von verstärkter Irritierbarkeit und suizidalem Verhalten.
Patienten (und deren Betreuer) sollten auf die Notwendigkeit einer Überwachung im Hinblick auf das Auftreten von Suizidgedanken, suizidalem Verhalten oder selbst – bzw. fremdschädigenden Absichten hingewiesen werden. Im Falle des Auftretens derartiger Symptome sollte sofort der Arzt konsultiert werden.
Schwere Hautreaktionen (SCARs)
Schwere Hautreaktionen (SCARs) einschliesslich Arzneimittelwirkung mit Eosinophilie und systemischen Symptomen (DRESS) und Stevens-Johnson-Syndrom (SJS), die lebensbedrohlich oder tödlich sein können, wurden in Zusammenhang mit der Anwendung von Perampanel berichtet (Häufigkeit unbekannt).
Bei Verschreibung sollten Patienten über Anzeichen und Symptome informiert und engmaschig bezüglich des Auftretens von Hautreaktionen überwacht werden.
DRESS-Symptome sind in der Regel, jedoch nicht ausschliesslich, Fieber, Ausschlag mit Beteiligung anderer Organsysteme, Lymphadenopathie, Anomalien bei Leberfunktionstests und Eosinophilie. Es muss beachtet werden, dass frühe Manifestationen einer Überempfindlichkeit, wie Fieber oder Lymphadenopathie, auftreten können, auch wenn kein Ausschlag festzustellen ist. DRESS kann durch eine lange Latenzzeit (von 2 bis 8 Wochen) zwischen Arzneimittelexposition und dem Auftreten der Krankheit charakterisiert sein. SJS-Symptome sind in der Regel, jedoch nicht ausschliesslich, Hautablösung (epidermale Nekrolyse/Blasenbildung) < 10 %, erythematöse Haut (konfluierend), schnelles Fortschreiten, schmerzhafte, atypische, zielscheibenartige Läsionen und/oder grossflächige dunkelrote Maculae oder grosse Erytheme (konfluierend), bullöse/erosive Beteiligung von mehr als 2 Schleimhäuten.
Wenn auf diese Reaktionen hinweisende Anzeichen und Symptome auftreten, sollte Perampanel sofort abgesetzt und eine alternative Behandlung erwogen werden (wie jeweils anwendbar). Wenn der Patient bei der Anwendung von Perampanel eine schwerwiegende Reaktion wie SJS oder DRESS entwickelt, darf bei diesem Patienten zukünftig keine Behandlung mit Perampanel mehr begonnen werden.
Absencen und myoklonische Anfälle
Absencen und myoklonische Anfälle sind zwei häufige Arten generalisierter Anfälle, die oft bei Patienten mit idiopathischer generalisierter Epilepsie auftreten. Es ist bekannt, dass andere Antiepileptika Anfälle dieser Arten induzieren oder verschlimmern. Patienten mit myoklonischen Anfällen und Absencen-Anfällen sollten während der Fycompa-Therapie überwacht werden.
Erkrankungen des Nervensystems
Perampanel kann Schwindel und Somnolenz hervorrufen und deshalb die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen stark beeinträchtigen (siehe Abschnitt «Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen»).
Aggression, Psychose
Über Fälle von Aggression, feindseligem und anormalem Verhalten bei Patienten mit Perampanel Therapie wurde berichtet. Bei Patienten, die im Rahmen von klinischen Studien mit Perampanel behandelt wurden, gibt es Berichte über das Auftreten von Aggression, Wut, Reizbarkeit und Psychosen. Es liegt eine Dosisabhängigkeit vor, da unter höheren Dosen häufiger darüber berichtet wurde. Diese wurden häufiger bei Jugendlichen als bei Erwachsenen beobachtet. Daher sollte die Dosis titriert werden und bei anhaltender Aggressionssymptomatik eine Dosisreduktion und ggf. Therapiebeendigung erwogen werden (siehe Abschnitt «Dosierung/Anwendung»). Diese Fälle und die Massnahmen sind vorab mit einem Facharzt zu besprechen und zu planen.
Die meisten dieser Ereignisse waren in den klinischen Studien leicht oder mässig ausgeprägt und bildeten sich entweder spontan oder nach Dosisanpassung wieder zurück. In einigen Fällen wurde aber über eine stark ausgeprägte Aggression berichtet, die zum Therapieabbruch führte. Dies umfasste auch Gedanken, wie anderen Menschen Schaden zuzufügen, körperliche Angriffe oder Drohverhalten. Patienten berichteten über Tötungsgedanken. Es wird empfohlen, Patienten (und Betreuer) zu Behandlungsbeginn über das mögliche Auftreten erhöhter Reizbarkeit, Aggression, Psychosen und depressiver Verstimmungen zu informieren. Bei auffälligen Veränderungen der Stimmungslage oder Verhaltensmuster sollten Patienten (und Betreuer) sofort einen Arzt oder eine medizinische Fachkraft verständigen. Wenn solche Symptome auftreten, sollte die Dosierung von Perampanel reduziert werden, und bei schweren Symptomen sollte das Arzneimittel sofort abgesetzt werden.
Sturzrisiko
Es besteht ein erhöhtes Sturzrisiko, insbesondere bei älteren Patienten. Die Ursache ist unklar, Gleichgewichtsstörungen, Schwindel und Benommenheit als mögliche Nebenwirkungen könnten beteiligt sein. Patienten und Betreuer müssen diesbezüglich aufmerksam sein, insbesondere zu Beginn der Behandlung und bei Änderungen der Dosis und/oder der Begleitmedikation (inklusive Änderungen der Dosis der Begleitmedikation).
Gleichzeitig angewendete Cytochrom P450-Induktoren oder -Inhibitoren
Wenn Cytochrom P450-Induktoren oder -Inhibitoren zusätzlich angewendet oder abgesetzt werden, sollten die Patienten hinsichtlich Verträglichkeit und klinischem Ansprechen engmaschig überwacht werden, da die Perampanel-Plasmaspiegel abfallen bzw. ansteigen können; die Perampanel-Dosis ist gegebenenfalls entsprechend anzupassen (siehe «Dosierung/Anwendung» und «Interaktionen»).
Hormonelle Kontrazeptiva
Aufgrund der nachgewiesenen Interaktion mit Levonorgestrel kann die Wirksamkeit gestagenhaltiger hormoneller Kontrazeptiva grundsätzlich herabgesetzt sein (siehe Abschnitt «Interaktionen»). Dies ist zu berücksichtigen und es wird empfohlen eine zusätzliche, nicht-hormonelle Verhütungsmethode (Intrauterinpessar (IUP), Kondom) anzuwenden.
Hepatotoxizität
Bei der Anwendung von Perampanel in Kombination mit anderen Antiepileptika wurden Fälle von Hepatotoxizität (hauptsächlich Anstieg der Leberwerte) berichtet. Wenn ein Anstieg der Leberwerte beobachtet wird, sollte eine Überwachung der Leberfunktion in Betracht gezogen werden.
Missbrauchspotential
Bei Patienten mit Suchtmittelmissbrauch in der Vorgeschichte ist Vorsicht geboten. Der Patient sollte auf Symptome eines Missbrauchs von Perampanel überwacht werden.
Inhaltsstoffe von speziellem Interesse
Fycompa Filmtabletten:
Lactose: Patienten mit der seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten die Filmtabletten nicht anwenden.
Fycompa Suspension:
Sorbitol: Die Fycompa Suspension enthält 175 mg Sorbitol pro ml, das eine Quelle für Fructose ist. Patienten mit hereditärer Fructoseintoleranz (HFI) dürfen dieses Arzneimittel nicht einnehmen. Sorbitol kann Magen-Darm-Beschwerden hervorrufen und kann eine leicht abführende Wirkung haben.
Benzoesäure/Benzoate: Die Fycompa Suspension enthält weniger als 1.105 mg Benzoesäure/Natriumbenzoat pro ml Suspension.
Natrium: Die Fycompa Suspension enthält weniger als 1 mmol Natrium (23 mg) pro ml, d.h. sie ist nahezu «natriumfrei».
InteraktionenPharmakokinetischen Interaktionen
In-vitro Daten
Arzneimittelmetabolismus Enzyminhibition
In humanen Lebermikrosomen hatte Perampanel (30 µmol/l) bei den wichtigsten hepatischen CYP- und UGT-Enzymen eine schwach hemmende Wirkung auf CYP2C8 und UGT1A9.
Arzneimittelmetabolismus Enzyminduktion
Verglichen mit Positivkontrollen (einschliesslich Phenobarbital, Rifampicin), wurde für Perampanel bei den wichtigsten hepatischen CYP und UGT-Enzymen eine schwach induzierende Wirkung auf CYP2B6 (30 µmol/l) und CYP3A4/5 (≥3 µmol/l) in gezüchteten humanen Hepatozyten gefunden.
In-vivo Daten
CYP3A-Substrate
Bei gesunden Probanden bewirkte Fycompa (6 mg/Tag über 20 Tage) eine Abnahme der AUC von Midazolam um 13 %. Eine grössere Abnahme der Exposition gegenüber Midazolam (oder anderen sensitiven CYP3A-Substraten) kann bei höheren Fycompa-Dosen nicht ausgeschlossen werden.
Cytochrom P450-Induktoren
Bei starken Induktoren von Cytochrom P450 wie Rifampicin und Hypericum ist mit einer Abnahme der Perampanel-Konzentrationen zu rechnen und die Möglichkeit von erhöhten Plasmakonzentrationen reaktiver Metaboliten ist in ihrer Gegenwart nicht auszuschliessen. Felbamat vermindert nachweislich die Konzentrationen bestimmter Arzneistoffe und könnte auch die Perampanel-Konzentrationen vermindern.
Cytochrom P450-Inhibitoren
Bei gesunden Probanden erhöhte der CYP3A4-Inhibitor Ketoconazol (400 mg/Tag über 10 Tage) die AUC von Perampanel um 20 % und verlängerte die Halbwertszeit von Perampanel um 15 % (67,8 h gegenüber 58,4 h). Stärkere Wirkungen können nicht ausgeschlossen werden, wenn Perampanel mit einem CYP3A-Inhibitor mit längerer Halbwertszeit als Ketoconazol kombiniert wird oder wenn der Inhibitor über eine längere Behandlungsdauer angewendet wird. Starke Inhibitoren anderer Cytochrom P450 Isoformen könnten die Perampanel Konzentrationen möglicherweise ebenfalls erhöhen.
Levodopa
Bei gesunden Probanden hatte Fycompa (4 mg/Tag über 19 Tage) keinen Einfluss auf die Cmax oder die AUC von Levodopa.
Antiepileptika
Mögliche Wechselwirkungen zwischen Fycompa (bis zu 12 mg/Tag) und anderen Antiepileptika (AED) wurden in klinischen Studien untersucht. Eine populations-pharmakokinetische Analyse von drei gepoolten Phase-3-Studien mit jugendlichen und erwachsenen Patienten mit fokalen Anfällen beurteilte den Effekt von Fycompa (bis zu 12 mg einmal täglich) auf die Pharmakokinetik von anderen Antiepileptika. In einer weiteren populations-pharmakokinetischen Analyse von gepoolten Daten aus 20 Phase 1 Studien mit gesunden Probanden, die Fycompa in Dosierungen bis zu 36 mg erhielten, einer Phase 2 und sechs Phase 3 Studien in pädiatrischen, jugendlichen und erwachsenen Patienten mit fokalen und sowie primär generalisierten tonisch-klonischen Anfällen unter Fycompa bis zu 16 mg täglich, wurde der Effekt von Antiepileptika auf die Perampanel Clearance untersucht. Die Auswirkungen dieser Wechselwirkungen auf die durchschnittliche Steady-State-Konzentration werden in der folgenden Tabelle zusammengefasst (die in der Tabelle angegebenen Werte basieren auf populationskinetischen Modellen).
|
Gleichzeitig angewendetes AED
|
Einfluss des AED auf die Perampanel-Konzentration
|
Einfluss von Perampanel auf die AED-Konzentration
| |
Carbamazepin
|
Abnahme auf ein Drittel
|
<10 % Abnahme
| |
Clobazam
|
kein Einfluss
|
<10 % Abnahme
| |
Clonazepam
|
kein Einfluss
|
kein Einfluss
| |
Lamotrigin
|
kein Einfluss
|
<10 % Abnahme
| |
Levetiracetam
|
kein Einfluss
|
kein Einfluss
| |
Oxcarbazepin
|
Abnahme auf die Hälfte
|
35 % Zunahme1)
| |
Phenobarbital
|
20% Abnahme
|
kein Einfluss
| |
Phenytoin
|
Abnahme auf die Hälfte
|
kein Einfluss
| |
Topiramat
|
20 % Abnahme
|
kein Einfluss
| |
Valproinsäure
|
kein Einfluss
|
<10 % Abnahme
| |
Zonisamid
|
kein Einfluss
|
kein Einfluss
| |
1) Der aktive Metabolit Monohydroxycarbazepin wurde nicht untersucht.
|
Basierend auf den Ergebnissen einer populations-pharmakokinetischen Analyse von Patienten mit fokalen Anfällen und Patienten mit primär generalisierten tonisch-klonischen Anfällen, war die Gesamtclearance von Fycompa erhöht, wenn es zusammen mit Carbamazepin (3-fach), Phenytoin oder Oxcarbazepin (2-fach), bekannte Induktoren von Metabolisierungsenzymen, angewendet wurde (siehe Abschnitt «Pharmakokinetik»). Dieser Effekt ist bei der zusätzlichen Anwendung bzw. beim Absetzen dieser Antiepileptika im Rahmen des Therapieschemas eines Patienten zu berücksichtigen und bei der Therapieführung zu beachten Clonazepam, Levetiracetam, Phenobarbital, Topiramat, Zonisamid, Clobazam, Lamotrigin und Valproinsäure hatten keine klinisch relevante Wirkung auf die Fycompa-Clearance.
In einer populationspharmakokinetischen Analyse von Patienten mit fokalen Anfällen, hatte Fycompa in der höchsten untersuchten Perampaneldosis (8 resp. 12 mg/Tag) keinen klinisch relevanten Einfluss auf die Clearance von Clonazepam, Levetiracetam, Phenobarbital, Phenytoin, Topiramat, Zonisamid, Carbamazepin, Clobazam, Lamotrigin und Valproinsäure.
Perampanel vermindert die Clearance von Oxcarbazepin um 26 %. Oxcarbazepin wird von der zytosolischen Reduktase rasch in den aktiven Metaboliten Monohydroxycarbazepin umgewandelt. Der Einfluss von Perampanel auf die Monohydroxycarbazepin-Konzentrationen ist nicht bekannt.
Perampanel wird unabhängig von anderen AED entsprechend der klinischen Wirkung dosiert.
Hormonelle Kontrazeptiva
Aufgrund der nachgewiesenen Interaktion mit Levonorgestrel kann die Wirksamkeit gestagenhaltiger hormonellerKontrazeptiva grundsätzlich herabgesetzt sein. Es wird empfohlen eine zusätzliche, nicht-hormonelle Verhütungsmethode (Intrauterinpessar (IUP), Kondom) anzuwenden.
Bei gesunden Frauen bewirkte Fycompa in einer kontrollierten klinischen Studie bei Gabe von 12 mg (jedoch nicht bei 4 oder 8 mg/Tag) über 21 Tage zusammen mit einem kombinierten hormonellen Kontrazeptivum nachweislich eine Abnahme der Levonorgestrel-Exposition (die mittleren Cmax- und AUC-Werte nahmen um jeweils 40 % ab). Die AUC-Werte von Ethinylestradiol wurden von Fycompa 12 mg nicht beeinflusst, während die Cmax um 18 % abnahm. Die Möglichkeit einer verminderten Wirksamkeit gestagenhaltiger Kontrazeptiva ist bei Mädchen oder Frauen im gebärfähigen Alter, die Fycompa benötigen, generell in angemessener Weise zu berücksichtigen (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Pädiatrische Population
Studien zur Erfassung von Wechselwirkungen wurden nur mit Erwachsenen durchgeführt. Die diesbezügliche Datenlage in der pädiatrischen Population ist begrenzt: In einer populations-pharmakokinetischen Analyse jugendlicher Patienten ≥12 Jahre und Kindern zwischen 4 und 11 Jahren wurden keine nennenswerten Unterschiede zwischen dieser Population im Vergleich zu der erwachsenen Population gefunden.
Pharmakodynamische Interaktionen
Alkohol
Die Wirkungen von Perampanel auf Tätigkeiten, die Aufmerksamkeit und Vigilanz erfordern, waren zu den Eigenwirkungen von Alkohol additiv oder sogar supraadditiv. Die Mehrfachdosierung von Perampanel 12 mg/Tag verstärkte die Wirkungen von Alkohol auch bezüglich Wachsamkeit und Aufmerksamkeit und erhöhte das Niveau von Wut, Verwirrtheit und Depression (siehe Abschnitt «Eigenschaften/Wirkungen»). Diese Wirkungen können auch beobachtet werden, wenn Fycompa in Kombination mit anderen, das Zentralnervensystem (ZNS) dämpfenden Substanzen, verwendet wird.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter und Kontrazeption
Die Anwendung von Fycompa bei Frauen im gebärfähigen Alter, die keine Kontrazeption anwenden, wird nicht empfohlen. Aufgrund der nachgewiesenen Interaktion mit Levonorgestrel kann die Wirksamkeit gestagenhaltiger hormoneller Kontrazeptiva herabgesetzt sein. Es wird empfohlen eine zusätzliche, nicht-hormonelle Verhütungsmethode (Intrauterinpessar (IUP), Kondom) anzuwenden.
Schwangerschaft
Bisher liegen nur begrenzte Erfahrungen (weniger als 300 Schwangerschafts-Ausgänge) über die Anwendung von Perampanel bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf teratogene Wirkungen bei Ratten oder Kaninchen, jedoch wurde bei Ratten bei Gabe maternal-toxischer Dosen Embryotoxizität beobachtet (siehe «Präklinische Daten»).
Stillzeit
Studien mit säugenden Ratten zeigten, dass Perampanel und/oder seine Metaboliten in die Milch übergehen (für Details siehe «Präklinische Daten»). Es ist nicht bekannt, ob Perampanel bei Menschen in die Muttermilch übergeht. Ein Risiko für das Neugeborene/Kleinkind kann nicht ausgeschlossen werden. Unter Abwägung des Nutzens des Stillens für das Kind und des Therapievorteils für die Frau muss entschieden werden, ob abgestillt oder die Therapie mit Fycompa abgebrochen wird.
Fertilität
Die Wirkung von Perampanel auf die Fertilität wurde beim Menschen nicht untersucht. In Tierstudien beobachtete Effekte hatten keine Auswirkungen auf die Fertilität (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenFycompa hat einen starken Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Perampanel kann unter anderem Benommenheit, Schwindel, Schläfrigkeit, Übelkeit, verstärkte Reizbarkeit und Irritierbarkeit hervorrufen und kann daher die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, stark beeinträchtigen. Die Patienten sind anzuweisen, so lange kein Fahrzeug zu führen, keine Maschinen zu bedienen und keine sonstigen potenziell gefährlichen Aktivitäten zu betreiben bzw. Tätigkeiten zu verrichten, bis untersucht bzw. bekannt ist, ob Perampanel ihre Fähigkeit zur Verrichtung dieser beeinflusst.
Unerwünschte WirkungenInsgesamt erhielten in allen kontrollierten und nicht-kontrollierten Studien mit Patienten mit fokalen Anfällen 1639 Patienten Perampanel, wovon 1'147 während 6 Monaten und 703 länger als 12 Monate behandelt wurden. In den kontrollierten und nicht-kontrollierten Studien mit Patienten mit primär generalisierten tonisch-klonischen Anfällen erhielten 114 Patienten Perampanel, davon 68 während 6 Monaten und 36 länger als 12 Monate.
Fokale Anfälle:
Unerwünschte Wirkungen, die zum Abbruch der Behandlung führten: In kontrollierten klinischen Studien der Phase 3 lag die Abbruchrate aufgrund von unerwünschten Wirkungen bei Patienten, die randomisiert wurden, um Perampanel in den empfohlenen Dosen von 4 mg, 8 mg und 12 mg/Tag zu erhalten, bei 1.7% (3/172), 4.2% (18/431) und 13.7% (35/255) und bei Patienten der Placebogruppe bei 1,4% (6/442). Schwindel und Somnolenz waren die häufigsten unerwünschten Wirkungen (>10 % in der gesamten Perampanel Gruppe und häufiger unter Placebo), die zum Therapieabbruch führten.
Primär generalisierte Anfälle:
In der kontrollierten Phase 3 Studie der primär generalisierten tonisch-klonischen Anfälle betrug die Abbruchrate in der für die Perampanel-Gruppe (bis 8 mg/d) aufgrund von unerwünschten Wirkungen 4,9 % (4/81), in der Placebo-Gruppe 1,2 % (1/82). Schwindel war die häufigste unerwünschte Wirkung, die zum Therapieabbruch führte (>2 % in der gesamten Perampanel Gruppe und häufiger unter Placebo).
In der nachfolgenden Liste sind die unerwünschten Wirkungen nach Organklasse und Häufigkeit geordnet. Die folgenden Häufigkeitsangaben wurden für die Klassifizierung der unerwünschten Wirkungen verwendet: sehr häufig: ≥1/10, häufig: ≥1/100, <1/10, gelegentlich: ≥1/1'000, <1/100, selten: ≥1/10'000, <1/1'000, nicht bekannt: Häufigkeit aufgrund der verfügbaren Daten nicht abschätzbar.
Stoffwechsel und Ernährungsstörungen
Häufig: verminderter Appetit, erhöhter Appetit.
Psychiatrische Erkrankungen
Häufig: Angst, Aggressivität, Wutgefühle, Verwirrtheit, Entwicklung depressiver Symptomatik.
Gelegentlich: Suizidalität, Risiko für Eigen- und Fremdgefährdung, Halluzinationen, Psychosen.
Erkrankungen des Nervensystems
Sehr häufig: Schwindel (32 %), Somnolenz (15 %).
Häufig: Ataxie, Gleichgewichtsstörungen, Dysarthrie, Reizbarkeit.
Augenerkrankungen
Häufig: Diplopie, verschwommenes Sehen.
Erkrankungen des Ohrs und des Labyrinths
Häufig: Vertigo.
Erkrankungen des Gastrointestinaltrakts
Häufig: Übelkeit.
Erkrankungen der Haut und des Unterhautgewebes
Nicht bekannt: Arzneimittelreaktion mit Eosinophilie und systemischen Symptomen (Drug Reaction with Eosinophilia and Systemic Symptoms [DRESS]), Stevens Johnson Syndrome (SJS).
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Rückenschmerzen.
Allgemeine Erkrankungen
Häufig: Fatigue, Gangstörungen.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: Stürze.
Untersuchungen
Häufig: Gewichtszunahme.
Pädiatrische Population
Basierend auf den klinischen Studiendaten von 196 Jugendlichen (12 bis 18 Jahre) unter Perampanel in Doppelblindstudien bei fokalen Anfällen und primär generalisierten tonisch-klonischen Anfällen ist das Sicherheitsprofil bei Jugendlichen vergleichbar mit dem von Erwachsenen, ausser betreffend Aggressivität, die bei Jugendlichen häufiger auftrat als bei Erwachsenen (siehe auch «Warnhinweise und Vorsichtsmassnahmen»).
Nach dem Stand der Daten aus klinischen Studien zu 180 pädiatrischen Patienten, die in einer multizentrischen, offenen Studie mit Perampanel behandelt wurden, war das Gesamtsicherheitsprofil von Kindern mit demjenigen von Jugendlichen und Erwachsenen vergleichbar, mit Ausnahme von Somnolenz, Reizbarkeit, Aggressionen und Agitiertheit, die in der Studie mit pädiatrischen Patienten häufiger beobachtet wurden als in den Studien mit Jugendlichen und Erwachsenen. Die vorliegenden Daten bei Kindern geben keinen Hinweis auf klinisch signifikante Auswirkungen auf Wachstums- und Entwicklungsparameter inklusive Körpergewicht, Körpergrösse, Schilddrüsenfunktion, Insulin-like Growth Factor-1 (IGF-1) Spiegel, Kognition (beurteilt mit Aldenkamp-Baker neuropsychological assessment schedule [ABNAS]), Verhalten (beurteilt mit Child Behavior Checklist [CBCL]) und Geschicklichkeit (beurteilt anhand Lafayette Grooved Pegboard Test [LGPT]). Langzeitwirkungen (mehr als 1 Jahr) in Bezug auf Lernfähigkeit, Intelligenz, Wachstum, endokrine Funktion und Pubertät von Kindern sind jedoch noch unbekannt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch
ÜberdosierungEs besteht nur eine limitierte klinische Erfahrung bezüglich Überdosierung von Perampanel bei Menschen. In einem Bericht über eine absichtliche Überdosierung, bei der eventuell bis zu 264 mg Fycompa eingenommen wurden, wies der Patient Bewusstseinstrübung, gesteigerte Erregbarkeit/Agitiertheit und aggressives Verhalten auf. Er konnte ohne Folgeerscheinungen wiederhergestellt werden.
Es gibt kein spezifisches Antidot für Perampanel. Allgemeine unterstützende Massnahmen inklusive Überwachung der Vitalzeichen und Beobachtung des klinischen Status des Patienten sind indiziert. Angesichts der langen Halbwertszeit könnte, die durch Perampanel hervorgerufenen Wirkungen verlängert sein. Aufgrund der niedrigen renalen Clearance sind spezielle Massnahmen wie forcierte Diurese, Dialyse oder Hämoperfusion wahrscheinlich nicht von Nutzen.
Eigenschaften/WirkungenATC-Code
N03AX22
Wirkungsmechanismus
Perampanel ist ein first-in-class, selektiver, nicht-kompetitiver Antagonist des ionotropen α-Amino-3-hydroxy-5-methyl-4-isoxazol-propionsäure (AMPA) Glutamat-Rezeptors auf post-synaptischen Neuronen. Glutamat ist der primäre exzitatorische Neurotransmitter im zentralen Nervensystem. Es wird angenommen, dass die Aktivierung von AMPA-Rezeptoren durch Glutamat für die meisten schnellen exzitatorischen synaptischen Übertragungen im Gehirn verantwortlich ist. In in-vitro Studien hemmte Perampanel die AMPA-induzierte Erhöhung von intrazellulärem Calcium.
Der exakte Mechanismus, durch welchen Perampanel in Menschen seine antiepileptische Wirkung entfaltet, ist noch nicht vollständig aufgeklärt.
Pharmakodynamische Wirkungen
Basierend auf den zusammengefassten Daten der 3 Wirksamkeitsstudien über fokale Anfälle wurde eine pharmakokinetisch-pharmakodynamische (Wirksamkeits-) Analyse durchgeführt. Eine solche wurde ebenfalls mit der Studie zu den primär generalisierten tonisch-klonischen Anfällen durchgeführt. In beiden Analysen korreliert die Perampanel-Exposition mit der Abnahme der Anfallshäufigkeit.
Psychomotorische Leistungsfähigkeit
In einer Studie mit gesunden Probanden wurden Standardbewertungsmethoden inklusive simuliertes Fahren verwendet, um die Wirkungen von Perampanel auf die psychomotorische Leistungsfähigkeit zu beurteilen. Einfache psychomotorische Aufgaben, die Fahrfähigkeit oder die sensorisch-motorische Koordination wurden durch tägliche Einzel- und Mehrfachdosen von 4 mg Perampanel nicht beeinträchtigt. Einzel- und Mehrfachdosen von 8 mg und 12 mg beeinträchtigten dosisabhängig die psychomotorische Leistungsfähigkeit. Die Fähigkeit ein Auto zu lenken, war nach Dosierung von 12 mg Perampanel vermindert, aber die Haltungsstabilität wurde nicht signifikant beeinträchtigt. Innerhalb von 2 Wochen nach Absetzen der Behandlung mit Perampanel erreichte die Leistungsfähigkeit wieder den Ausgangswert. Nachdem gesunden Probanden in der gleichen Studie Alkohol verabreicht wurde, um eine Blutkonzentration von 80 - 100 mg/100 ml zu erreichen, beeinträchtigte Perampanel die einfache psychomotorische Leistungsfähigkeit durchwegs nach Einzeldosen von 4 bis 12 mg (n=35) sowie nach 21 Tagen mit Mehrfachdosen von 12 mg/Tag (n=24). Die Wirkungen von Perampanel auf komplexe Tätigkeiten wie auf die Fahrtüchtigkeit waren additiv oder supraadditiv zu den beeinträchtigenden Wirkungen des Alkohols.
Kognitive Funktion
In einer Studie mit gesunden Probanden wurden Standardbewertungsmethoden verwendet, um die Wirkungen von Perampanel auf die Aufmerksamkeit und das Gedächtnis zu beurteilen. Nach einzelnen und mehrfachen Dosen von bis zu 12 mg Perampanel pro Tag wurde keine Beeinträchtigung gefunden.
In einer placebokontrollierten Studie bei jugendlichen Patienten zeigten Messungen anhand des Global Cognition Scores des Cognitive Drug Research (CDR) System keine signifikanten Veränderungen der Kognition für Perampanel gegenüber Placebo. In der offenen Verlängerungsphase dieser Studie wurden nach 52 Wochen Perampanel-Behandlung keine signifikanten Veränderungen des globalen CDR-System-Scores beobachtet (siehe Abschnitt «Kinder und Jugendliche» weiter unten).
In einer unverblindeten, unkontrollierten Studie mit pädiatrischen Patienten wurden nach der Zusatztherapie mit Perampanel keine klinisch relevanten Veränderungen der Kognition im Vergleich zur Ausgangslage, beurteilt über ABNAS, festgestellt (siehe auch «Eigenschaften/Wirkungen», «Pädiatrische Population»).
Aufmerksamkeit und Stimmung
Bei gesunden Probanden nahm der Grad der Aufmerksamkeit (Reaktionsbereitschaft) unter Perampanel in Dosierungen von 4 bis 12 mg/Tag dosisabhängig ab. Die Stimmung verschlechterte sich bei gesunden Probanden nur nach Dosen von 12 mg/Tag; die Stimmungsveränderungen waren gering und widerspiegelten eine allgemeine generelle Dämpfung. Die wiederholte Verabreichung von Perampanel 12 mg/Tag verstärkte auch die Wirkungen von Alkohol bezüglich Wachsamkeit und Aufmerksamkeit sowie die Intensität von Wutgefühlen, Verwirrtheit und Depression.
Kardiale Elektrophysiologie
Elektrokardiographische Wirkungen von Perampanel wurden in einer doppelblinden, randomisierten, Placebo- und mit Moxifloxazin kontrollierten klinischen Pharmakologiestudie bei gesunden Probanden bestimmt. Perampanel wurde in täglichen Dosen von bis zu 12 mg/Tag während 7 Tagen appliziert. Perampanel verlängerte das QTc Intervall nicht und es hatte keinen dosisabhängigen oder klinisch bedeutsamen Effekt auf die QRS-Dauer.
Klinische Wirksamkeit - fokale Anfälle
Der Nachweis der Wirksamkeit von Perampanel als Zusatztherapie bei fokalen Anfällen wurde in drei 19-wöchigen, randomisierten, doppelblinden, placebokontrollierten, multizentrischen Studien mit erwachsenen und jugendlichen Patienten bestimmt. Die Patienten wiesen fokale Anfälle mit oder ohne sekundäre Generalisierung auf und konnten mit einem bis drei gleichzeitig verabreichten Antiepileptika nicht angemessen kontrolliert werden.
Während einer 6-wöchigen Baseline-Periode mussten die Patienten mehr als fünf Anfälle und keine anfallsfreie Periode über 25 Tagen aufweisen. In diesen drei Studien betrug die mittlere Epilepsiedauer der Patienten ungefähr 21,06 Jahre. Zwischen 85,3 % und 89,1 % der Patienten nahmen zwei bis drei gleichzeitig verabreichte Antiepileptika in Kombination mit oder ohne gleichzeitiger Vagus-Stimulation ein.
Zwei Studien verglichen Perampanel in Dosen von 8 und 12 mg/Tag mit Placebo und die dritte Studie verglich Dosen von 2, 4 und 8 mg/Tag mit Placebo. Nach einer 6-wöchigen Baseline-Phase zur Ermittlung der Anfallshäufigkeit vor der Randomisierung wurden die Patienten in allen drei Studien randomisiert und bis zur randomisierten Dosis titriert. Während der Titrationsphase wurde in allen drei Studien die Behandlung mit 2 mg/Tag begonnen und in wöchentlichen Schritten von 2 mg/Tag bis zur Zieldosis erhöht. Patienten, welche nicht tolerierbare unerwünschte Wirkungen hatten, konnten bei der gleichen Dosis bleiben oder ihre Dosis auf die vorher verträgliche Dosis reduzieren. In allen drei Studien folgte nach der Titrationsphase eine 13-wöchige Erhaltungsphase, während derer Patienten auf einer stabilen Dosis von Perampanel bleiben sollten.
Die gepoolten 50%-Responderraten lagen unter Placebo bei 19%, unter 4 mg bei 29 %, unter 8 mg bei 35 % und unter 12 mg bei 35 %. Eine statistisch signifikante Wirkung hinsichtlich der Reduktion der Anfallshäufigkeit pro 28 Tage (von der Baseline- zur Behandlungsphase) im Vergleich zur Placebogruppe wurde unter der Behandlung mit Fycompa für die Dosierungen 4 mg/Tag (Studie 306), 8 mg/Tag (Studien 304, 305 und 306) und 12 mg/Tag (Studien 304 und 305) beobachtet. Diese Studien zeigen, dass die einmal tägliche Gabe von Perampanel in Dosen von 4 mg bis 12 mg in dieser Population als Zusatztherapie signifikant wirksamer war als Placebo.
Daten aus placebokontrollierten Studien belegen, dass bei einmal täglicher Gabe von Fycompa 4 mg eine Verbesserung der Anfallskontrolle beobachtet wird und dass dieser Nutzen bei Steigerung der Dosis auf 8 mg/Tag noch verstärkt wird. Im Gesamtkollektiv wurde für die 12 mg-Dosis im Vergleich zur 8 mg-Dosis kein Wirksamkeitsnutzen beobachtet. Ein Nutzen wurde unter der 12 mg-Dosis bei manchen Patienten beobachtet, welche die 8 mg-Dosis vertrugen und bei dieser Dosis ein unzureichendes klinisches Ansprechen aufwiesen.
Kinder und Jugendliche:
In den drei pivotalen, doppelblinden, placebokontrollierten Phase 3 Studien gab es insgesamt n = 143 Jugendliche im Alter zwischen 12 und 18 Jahren, die in diese Studien zusätzlich zu den erwachsenen Patienten aufgenommen worden waren. Die bei diesen Jugendlichen erhaltenen Ergebnisse waren mit denjenigen der erwachsenen Population vergleichbar.
Eine 19-wöchige, randomisierte, doppelblinde, placebokontrollierte Studie mit offener Verlängerungsphase (Studie 235) beurteilte die kurzfristigen Wirkungen von Fycompa auf die Kognition (Zieldosis: 8 bis 12 mg einmal täglich) während der Anwendung als Zusatztherapie bei 133 (Fycompa n=85, Placebo n=48) jugendlichen Patienten im Alter von 12 bis unter 18 Jahren mit nicht ausreichend kontrollierten fokalen Anfällen. Die kognitive Funktion wurde mithilfe des Global Cognition t-Scores des Cognitive Drug Research (CDR) System bewertet. Es handelt sich dabei um einen aus 5 Domänen zusammengesetzten Score, der Folgendes testet: Power of Attention (Aufmerksamkeitsleistung), Continuity of Attention (Aufmerksamkeitsdauer), Quality of Episodic Secondary Memory (Qualität des episodischen Sekundärspeichers), Quality of Working Memory (Qualität des Arbeitsgedächtnisses) und Speed of Memory (Gedächtnisgeschwindigkeit). Die mittlere Veränderung (SD) des CDR System Global Cognition t-Scores gegenüber dem Ausgangswert bis zum Ende der doppelblinden Behandlungsphase (19 Wochen) betrug 1,1 (7,14) in der Placebo-Gruppe und (minus) –1,0 (8,86) in der Perampanel-Gruppe, wobei der Unterschied zwischen den Behandlungsgruppen in Bezug auf den LS-Mittelwert (95 % KI) (minus) -2,2 (-5,2; 0,8) betrug. Es bestand kein statistisch signifikanter Unterschied zwischen den Behandlungsgruppen (p = 0,145). Die Global Cognition t-Scores des CDR System für Placebo und Perampanel betrugen 41,2 (10,7) bzw. 40,8 (13,0) bei der Ausgangswerterhebung. Bei Patienten, die Perampanel in der offenen Verlängerungsphase erhielten (n = 112), betrug die mittlere Veränderung (SD) des Global Cognition t-Scores des CDR System gegenüber dem Ausgangswert bis zum Ende der offenen Behandlung (52 Wochen) (minus) -1,0 (9,91). Dieses Ergebnis war nicht statistisch signifikant (p = 0,96). Nach bis zu 52 Wochen Behandlung mit Perampanel (n = 114) war keine Wirkung auf das Knochenwachstum zu beobachten. Nach bis zu 104 Wochen Behandlung zeigten sich keine Wirkungen auf Körpergewicht, Körpergrösse und sexuelle Entwicklung (n = 114).
Klinische Wirksamkeit – primär generalisierte tonisch-klonische Anfälle
In einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie wurde die Wirksamkeit von Perampanel bei Patienten ab 12 Jahren, die an idiopathisch generalisierter Epilepsie mit primär tonisch-klonischen Anfällen litten, untersucht.
Eingeschlossen wurden Patienten, die mit einem bis drei gleichzeitig verabreichten Antiepileptika während der 8-wöchigen Baseline-Periode mindestens 3 primär generalisierte tonisch-klonische Anfälle hatten. Die Studie umfasste 164 Patienten (Perampanel n = 82, Placebo n = 82), die über vier Wochen auf die Zieldosis von 8 mg/Tag, oder die höchst tolerierte Dosis, auftitriert wurden und über weitere 13 Wochen auf dem Dosislevel behandelt wurden. Die gesamte Behandlungsdauer betrug 17 Wochen. Die Studienmedikation wurde einmal täglich eingenommen.
Die 50%-Responderrate für primär generalisierte tonisch-klonische Anfälle während des Erhaltungstherapiezeitraums war in der Perampanel-Gruppe statistisch signifikant höher (58,0%) als in der Placebo-Gruppe (35,8%), p=0,0059. Die mediane prozentuale Veränderung der Häufigkeit von primär generalisierten tonisch-klonischen Anfällen pro 28-Tage-Zeitraum war während der Titrations- und Erhaltungstherapiephase (kombiniert) im Verhältnis zur Vorrandomisierung unter Perampanel grösser (-76,5%) als unter Placebo (-38,4%), p<0,0001. Während der 3-monatigen Erhaltungsphase wurden 30,9% (25/81) der Patienten, die in den klinischen Studien mit Perampanel behandelt wurden, frei von primär generalisierten tonisch-klonischen Anfällen, verglichen mit 12,3% (10/81) unter Placebo. Die Studie zeigt, dass die einmal täglich Gabe von 8 mg Perampanel als Zusatztherapie signifikant wirksamer als Placebo ist.
Offene Verlängerungsphase: Von den 140 Patienten, welche die kontrollierte Studie abschlossen, traten 114 (81,4%) in die offene Verlängerungsphase. Die Patienten auf Placebo wurden innerhalb von sechs Wochen auf Perampanel umgestellt. Gefolgt von einer Erhaltungstherapie (≥1 Jahr). In der Verlängerungsphase ≥1 Jahr erhielten 73,7% der Patienten mehr als 4 bis 8 mg/Tag und 16,7% 8 bis 12 mg/Tag.
Die Studie schloss 22 Jugendliche im Alter zwischen 12 und 18 Jahren ein. Die Ergebnisse dieser Population sind mit denen der Erwachsenen vergleichbar.
Andere Anfallstypen generalisierter Anfälle:
Die Wirksamkeit und Sicherheit von Perampanel bei Patienten mit myoklonischen Anfällen ist nicht belegt; mit den verfügbaren Daten kann keine abschliessende Beurteilung vorgenommen werden. Die Wirksamkeit von Perampanel in der Behandlung von Absencen wurde nicht gezeigt.
In der multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie 332 waren von den Patienten mit PGTC- und zusätzlichen myoklonischen Anfällen 16,7% (4/24) unter Perampanel verglichen mit 13,0% (3/23) unter Placebo anfallsfrei. Bei Patienten, die zusätzlich zu den PGTC-Anfällen auch Absencen hatten, waren 22,2% (6/27) auf Perampanel verglichen mit 12,1 % (4/33) auf Placebo anfallsfrei. Anfallsfrei (PGTC-, myoklonische Anfälle und Absencen) waren 23,5 % (19/81) der Patienten auf Perampanel verglichen mit 4.9% (4/81) auf Placebo.
Absetzen der AED-Begleitmedikation
In einer retrospektiven Studie zur klinischen Praxis wurden 51 Patienten mit Epilepsie, die Perampanel als Zusatztherapie erhielten, auf eine Perampanel-Monotherapie umgestellt. Die Mehrheit dieser Patienten hatte eine Vorgeschichte mit fokalen Anfällen. Von diesen Patienten kehrten 14 (27 %) in den Folgemonaten wieder zur Zusatztherapie zurück. 34 Patienten wurden für mindestens 6 Monate nachbeobachtet und 24 dieser Patienten (71 %) behielten die Perampanel-Monotherapie für mindestens 6 Monate bei. 10 Patienten wurden für mindestens 18 Monate nachbeobachtet und 3 dieser Patienten (30 %) behielten die Perampanel-Monotherapie für mindestens 18 Monate bei.
Pädiatrische Patienten
Eine unverblindete, unkontrollierte Studie (Studie 311) wurde durchgeführt, um das Expositions-Wirksamkeitsverhältnis von Perampanel in der Zusatztherapie bei 180 pädiatrischen Patienten zwischen 4 und 11 Jahren mit ungenügend kontrollierten fokalen bzw. primär generalisierten tonisch-klonischen Anfällen zu untersuchen. Die Patienten wurden über 11 Wochen auftitriert, bis zur Zieldosis von 8 mg/Tag oder der maximalen verträglichen Dosis (nicht über 12 mg/Tag) bei Patienten die keine CYP3A induzierenden Antiepileptika einnahmen (Carbamazepin, Oxcarbazepin, Eslicarbazepin und Phenytoin), bzw. bis 12 mg/Tag oder der maximal verträglichen Dosis (nicht über 16 mg/Tag) für Patienten, die zusätzlich CYP3A induzierende Antiepileptika einnahmen. Die am Ende der Titrationsphase erreichte Dosis wurde für 12 Wochen beibehalten (insgesamt 23 Wochen Exposition am Ende der Kernstudie). Patienten, die in die Verlängerungsphase gingen, wurden für weitere 29 Wochen behandelt (insgesamt 52 Wochen Exposition),
Bei Patienten mit fokalen Anfällen (n=148 Patienten) waren die mediane Veränderung der Anfallshäufigkeit pro 28 Tage, die ≥50% Ansprechrate und die anfallsfreie Rate nach 23 Wochen Perampanel Behandlung bei -40.1%, 46.6% (n=69/148) und 11.5% (n=17/148) für alle fokalen Anfälle. Die Behandlungswirkungen in Bezug auf die mediane Verringerung der Anfallshäufigkeit (Wochen 40–52: n = 108 Patienten, -69,4 %), die 50%-Responderrate (Wochen 40–52: 62,0 %, n = 67/108) und die Anfallsfreiheitsrate (Wochen 40–52: 13,0 %, n = 14/108) hielten nach der 52-wöchigen Perampanel-Behandlung an.
In einer Untergruppe von Patienten mit fokalen Anfällen und sekundär generalisierten Anfällen (N=54 Patienten), waren die betreffenden Werte -58.7%, 64.8% (n=35/54) und 18.5% (n=10/54) für sekundär generalisierte tonisch-klonische Anfälle. Die Behandlungswirkungen in Bezug auf die mediane Verringerung der Anfallshäufigkeit (Wochen 40–52: n = 41 Patienten, -73,8 %), die 50%-Responderrate (Wochen 40–52: 80,5 %, n = 33/41) und die Anfallsfreiheitsrate (Wochen 40–52: 24,4 %, n = 10/41) hielten nach der 52-wöchigen Perampanel-Behandlung an.
Bei Patienten mit primär generalisierten tonisch-klonischen Anfällen (n=22 Patienten, von den 19 Patienten 7 bis < 12 Jahre und 3 Patienten 4 bis < 7 Jahre alt waren) waren die mediane Veränderung der Anfallshäufigkeit pro 28 Tage, die ≥50% Ansprechrate und die anfallsfreie Rate bei -69.2%, 63.6% (n=14/22) und 54.5%(n=12/22). Die Behandlungswirkungen in Bezug auf die mediane Verringerung der Anfallshäufigkeit (Wochen 40–52: n = 13 Patienten, -100,0 %), die 50%-Responderrate (Wochen 40–52: 61,5 %, n = 8/13) und die Anfallsfreiheitsrate (Wochen 40–52: 38,5 %, n = 5/13) hielten nach der 52-wöchigen Perampanel-Behandlung an. Diese Ergebnisse sollten zurückhaltend interpretiert werden, da sie auf einer sehr geringen Anzahl von Patienten beruhen.
In einer Untergruppe von Patienten mit primär generalisierten tonisch-klonischen Anfällen mit idiopathischer generalisierter Epilepsie (IGE) (n=19 Patienten, von den 17 Patienten 7 bis < 12 Jahre und 2 Patienten 4 bis < 7 Jahre alt waren) wurden ähnliche Ergebnisse erzielt; die betreffenden Werte waren 56.5%, 63.2% (n=12/19) und 52.6% (n=10/19). Die Behandlungswirkungen in Bezug auf die mediane Verringerung der Anfallshäufigkeit (Wochen 40–52: n = 11 Patienten, -100,0 %), die 50%-Responderrate (Wochen 40–52: 54,5 %, n = 6/11) und die Anfallsfreiheitsrate (Wochen 40–52: 36,4 %, n = 4/11) hielten nach der 52-wöchigen Perampanel-Behandlung an. Diese Ergebnisse sollten zurückhaltend interpretiert werden, da sie auf einer sehr geringen Anzahl von Patienten beruhen.
Nach 23 bzw. 52 Wochen Perampanel Behandlung hat sich der Zustand von 42.6% (n=52/122) bzw. 53,8 % (n = 56/104) der Patienten mit fokalen Anfällen, von 43,8% (n=21/48) bzw. 61,5 % (n = 24/39) in der Untergruppe von fokalen Anfällen mit sekundärer Generalisierung, von 34.8% (n=8/23) bzw. 47,1 % (n = 8/17) mit primär generalisierten tonisch-klonischen Anfällen und von 35.3% (n=6/17) bzw. 58,3 % (n = 7/12) in der Untergruppe von primär generalisierten tonisch-klonischen Anfällen bei idiopathischer generalisierter Epilepsie (IGE) im Vergleich zur Ausgangslage sehr stark verbessert oder stark verbessert, beurteilt nach der Clinical Global Impression of Change (CGIC).
PharmakokinetikDie Pharmakokinetik von Perampanel wurde bei gesunden Erwachsenen (18 bis 79 Jahre), Erwachsenen und Jugendlichen und bei pädiatrischen Patienten mit fokalen und primär generalisierten tonisch-klonischen Anfällen sowie bei Erwachsenen mit Parkinson, diabetischer Neuropathie, Multipler Sklerose und Leberfunktionsstörungen untersucht.
Absorption
Perampanel wird nach oraler Applikation schnell und vollständig absorbiert und weist einen vernachlässigbaren First-Pass-Effekt auf (die absolute Bioverfügbarkeit beträgt ungefähr 100 %).
Nahrungsmittel beeinflussen das Ausmass der Absorption nicht, verlangsamen aber die Absorptionsrate. Bei gleichzeitiger Nahrungsaufnahme ist die maximale Plasmakonzentration verringert und im Vergleich zur Einnahme in nüchternem Zustand um 2 Stunden verzögert.
In einer populations-pharmakokinetischen Analyse von gepoolten Daten aus 20 Phase 1 Studien mit gesunden Probanden, die Perampanel in Dosierungen zwischen 0.2 und 36 mg entweder als Einzeldosis oder in mehrfachen Gaben erhielten, einer Phase 2 und fünf Phase 3 Studien mit Patienten mit fokalen Anfällen, die Perampanel in täglichen Dosierungen zwischen 2 und 16 mg erhielten und zwei Phase 3 Studien mit Patienten mit primär generalisierten tonisch-klonischen Anfällen, die Perampanel in täglichen Dosierungen zwischen 2 und 14 mg erhielten, wurde ein linearer Zusammenhang zwischen der Dosis und der Plasmakonzentration von Perampanel gefunden.
Perampanel Suspension zum Einnehmen und Perampanel Filmtabletten haben sich unter Nahrungskarenz auf mg/mg-Basis als bioäquivalent erwiesen. Wenn eine 12-mg-Einzeldosis beider Formulierungen mit einer fettreichen Mahlzeit eingenommen wurde, erreichte Perampanel Suspension zum Einnehmen die gleiche AUC0-inf und eine etwa 23 % geringere Cmax. Die maximalen Plasmaspiegel (Tmax) verzögerten sich um etwa 2 Stunden im Vergleich zu den Filmtabletten. Eine populationspharmakokinetische Analyse zeigte jedoch, dass unter simulierten Steady-State- Expositionsbedingungen die Cmax und AUC(0-24h) der Perampanel Suspension zum Einnehmen bioäquivalent zur Tablettenformulierung waren, sowohl unter Nahrungskarenz als auch nach Nahrungsaufnahme. Bei Gabe zusammen mit einer fettreichen Mahlzeit waren die Cmax und AUC0-inf einer 12-mg-Einzeldosis der Perampanel Suspension zum Einnehmen etwa 22 % bzw. 13 % niedriger im Vergleich zur Nahrungskarenz.
Distribution
Daten von in-vitro Studien weisen darauf hin, dass Perampanel zu ungefähr 95% an Plasmaproteine gebunden wird.
In-vitro Studien zeigen, dass Perampanel kein Substrat oder signifikanter Inhibitor der organische Anionen transportierenden Polypeptide (OATP) 1B1 und 1B3, der organischen Anionen-Transporter (OAT) 1, 2, 3 und 4, der organischen Kationen-Transporter (OCT) 1, 2 und 3 sowie der Efflux-Transporter P-Glykoprotein und Brustkrebs-Resistenz-Protein (BCRP) ist.
Metabolismus
Perampanel wird durch primäre Oxidation und fortlaufende Glukuronidierung umfassend metabolisiert. Der Metabolismus von Perampanel wird primär durch CYP3A4 und/oder CYP3A5 und zu kleinerem Anteil CYP1A2 und CYP2B6 vermittelt, basierend auf Resultaten von in-vitro Studien, in welchen rekombinantes humanes CYP und humane Lebermikrosomen verwendet wurden. Nach Anwendung von radioaktiv markiertem Perampanel wurden nur Spuren von Perampanel-Metaboliten im Plasma beobachtet.
Elimination
Nach Einnahme einer radioaktiv markierten Perampanel Dosis wurde bei 8 gesunden älteren Probanden 30 % der wiedergefundenen Radioaktivität im Urin und 70 % in den Fäzes gemessen. Die wiedergefundene Radioaktivität im Urin und in den Fäzes bestand hauptsächlich aus einer Mischung von oxidativen und konjugierten Metaboliten. In einer populations-pharmakokinetischen Analyse von zusammengefassten Daten aus 19 Phase I Studien betrug die mittlere t½ von Perampanel 105 Stunden. Bei einer Dosierung in Kombination mit dem potenten CYP3A Induktor Carbamazepin betrug die t½ 25 Stunden.
Kinetik spezieller Patientengruppen
Patienten mit eingeschränkter Leberfunktion
Die Pharmakokinetik von Perampanel nach einer einzigen 1 mg Dosis wurde in 12 Patienten mit schwacher und mässiger Leberfunktionsstörung (Child-Pugh A respektive B) evaluiert und mit 12 gesunden, demographisch vergleichbaren Probanden verglichen. Die mittlere scheinbare Clearance von ungebundenem Perampanel bei geringfügig beeinträchtigten Patienten lag bei 188 ml/min gegenüber 338 ml/min bei der Kontrollgruppe; bei den mässig beeinträchtigten Patienten lag sie bei 120 ml/min gegenüber 392 ml/min bei der Kontrollgruppe. Die t½ verlängerte sich bei geringfügig beeinträchtigten (306 h gegenüber 125 h) und mässig beeinträchtigten Patienten (295 h gegenüber 139 h) verglichen mit vergleichbaren gesunden Probanden der Kontrollgruppe.
Patienten mit eingeschränkter Nierenfunktion
Die Pharmakokinetik von Perampanel wurde bei Patienten mit Niereninsuffizienz nicht systematisch untersucht. Perampanel wird fast ausschliesslich durch Metabolisierung eliminiert, gefolgt von einer schnellen Ausscheidung der Metaboliten; nur Spuren von Perampanel-Metaboliten werden im Plasma beobachtet. In einer populations-pharmakokinetischen Analyse von Patienten mit fokalen Anfällen mit einer Kreatinin-Clearance im Bereich von 39 bis 160 ml/min, die Perampanel in Dosen bis zu 12 mg/Tag in placebokontrollierten klinischen Studien erhielten, wurde die Perampanel-Clearance durch die Kreatinin-Clearance nicht beeinflusst.
In einer populations-pharmakokinetischen Analyse von Patienten mit primär-generalisierten tonisch-klonischen Anfällen, die in einer placebokontrollierten Studie bis zu 8 mg/Tag Perampanel erhielten, wurde die Perampanel-Clearance durch die Kreatinin-Clearance nicht beeinflusst.
Geschlecht
In einer populations-pharmakokinetischen Analyse von Patienten mit fokalen Anfällen, die Perampanel in Dosen bis zu 12 mg/Tag und von Patienten mit primär generalisierten tonisch-klonischen Anfällen die Perampanel in Dosen bis zu 8 mg/Tag in placebokontrollierten klinischen Studien erhielten, war die Perampanel-Clearance bei Frauen (0.526 l/h) um 18.5% tiefer als bei Männern (0.646 l/h).
Ältere Patienten (ab 65 Jahren)
In einer populations-pharmakokinetischen Analyse von Patienten mit fokalen Anfällen (Alter 12 bis 74 Jahre) und mit primär generalisierten tonisch-klonischen Anfällen (Alter 12 bis 58 Jahre), die Perampanel in Dosen bis zu 8 mg/Tag respektive 12 mg/Tag in placebokontrollierten klinischen Studien erhielten, wurde kein signifikanter Effekt des Alters auf die Perampanel-Clearance gefunden. Eine Dosisanpassung für ältere Patienten ist nicht als notwendig erachtet.
Pädiatrische Patienten
In einer populationspharmakokinetischen Analyse gepoolter Daten von Kindern im Alter von 4 bis 11 Jahren, von Jugendlichen ab 12 Jahren und von Erwachsenen stieg die Perampanel-Clearance mit zunehmendem Körpergewicht an. Daher ist eine Dosisanpassung bei Kindern im Alter von 4 bis 11 Jahren mit einem Körpergewicht von < 30 kg erforderlich (siehe auch «Dosierung/Anwendung»).
Präklinische DatenSicherheitspharmakologie
In einer pharmakologischen Sicherheitsstudie an Ratten wurde ein dosisabhängiger Anstieg der Körpertemperatur (bis zu +0,6 °C bei 5 mg/kg) beobachtet. In Toxizitätsstudien wurde nach wiederholter Gabe manchmal eine Hypothermie bei höheren Dosierungen beobachtet. In klinischen Studien wurde keine eindeutige Erhöhung oder Verringerung der Körpertemperatur festgestellt.
Toxizität bei wiederholter Verabreichung
Die Leber ist als Target nach der wiederholten Gabe hoher Dosierungen in den Toxizitätsstudien ermittelt worden (erhöhtes Gewicht, Zellatrophie und/oder -nekrose). Basierend auf kovalenter Bindung an Makromoleküle akkumuliert Perampanel und erreicht hohe Gewebekonzentrationen in diesem Organ und interagiert mit Cytochrom P450- und UGT-Enzymen sowie dem OAT2-Transportprotein. Bezogen auf die systemische Exposition (AUC) bei an Tieren ermitteltem no-observed-adverse-effect level (NOAEL) und Patienten mit gestörter Leberfunktion beträgt der Sicherheitsabstand ≤1.
Mutagenität
Perampanel zeigte keine Gentoxizität in den durchgeführten in vitro- oder in vivo-Studien.
Karzinogenität
In einer 2-jährigen Kanzerogenitätsstudie an Ratten wurden Keratoakanthome (in männlichen Tieren) und Phäochromozytome (in weiblichen Tieren) in der Kontroll- und, mit höherer Inzidenz, den Perampanel-behandelten Gruppen beobachtet, bei fehlender statistischer Signifikanz. Die Inzidenz lag innerhalb des Bereiches für spontanes Auftreten in älteren Ratten.
Reproduktionstoxizität
In den Fruchtbarkeitsstudien an Ratten wurden bei Weibchen nach hohen Dosen (30 mg/kg; Sicherheitsabstand von <1 bezogen auf die systemische Exposition am no-observed-effect level, NOEL, und einer klinischen Dosis von 12 mg Perampanel/Tag) verlängerte und unregelmässige Zyklen beobachtet; jedoch beeinflussten diese Veränderungen die Fruchtbarkeit und die frühe embryonale Entwicklung nicht. Es gab keine Auswirkungen auf die männliche Fruchtbarkeit.
Der Übergang in die Muttermilch wurde in Ratten 10 Tage nach der Geburt gemessen. Die Maximalwerte wurden nach einer Stunde erreicht und waren 3,65-mal höher als die Plasmawerte. Perampanel überwindet die Plazentaschranke. Jedoch war der Transfer durch die Plazenta relativ gering; 0,09% oder weniger der applizierten Dosis wurde im Fötus nachgewiesen.
Tierexperimentelle Studien ergaben keine Hinweise auf teratogene Wirkungen bei Ratten oder Kaninchen, jedoch wurde bei Ratten bei Gabe maternal-toxischen Dosen Embryotoxizität beobachtet (Sicherheitsabstand <1 bezogen auf die systemische Exposition, AUC und einer klinischen Dosis von 12 mg Perampanel/Tag). In einer peri- und postnatalen Entwicklungstoxizitätsstudie in Ratten wurden bei toxischen Dosen abnormale Geburten und Pflegebedingungen beobachtet und die Anzahl von Totgeburten beim Nachwuchs war erhöht (Sicherheitsabstand von <1 bezogen auf die systemische Exposition, AUC, am NOEL und einer klinischen Dosis von 12 mg Perampanel/Tag). Zusätzlich neigte der Geburtenindex und der Lebensfähigkeitsindex 4 Tage nach der Geburt zu tiefen Werten, und es wurde eine Unterdrückung der Gewichtszunahme und eine verzögerte morphologische Differenzierung (verzögerte Öffnung der Vaginalöffnung oder Teilung der balanopreputial Hautfalte) beim Nachwuchs beobachtet. Jedoch wurden im Nachwuchs keine Auswirkungen auf Verhaltens- oder Reproduktionsfunktionen beobachtet.
Studien an Jungtieren
In Toxizitätstudien an juvenile Ratten und Hunden wurde eine erhöhte Empfindlichkeit gegenüber Perampanel im Vergleich zu erwachsenen Tieren bei ähnlicher systemischer Exposition beobachtet.
Weitere Daten (Phototoxizität)
In tierexperimentellen Studien wurde eine erhöhte Inzidenz von Hautreaktionen nach UV-Licht Exposition (Faltenbildung, Auftreten weisser oder erythemischer/geröteter Regionen), mit einem Sicherheitsabstand von ≤3 (bezogen auf die systemische Exposition, AUC, und einer klinischen Dosis von 12 mg Perampanel/Tag) beobachtet. Gewebe-Verteilungs-Studien mit 14C-Perampanel zeigten lange Retentionszeiten für die Radioaktivität (bis zu 110 Wochen) im Bulbus oculi bei Nagern und Nichtnagern. Zwar zeigten allgemeine Toxizitätsstudien bei wiederholter Verabreichung keine behandlungsbedingten ophthalmoskopische oder histopathologische Befunde am Auge.In der 2-jährigen Kanzerogenitätsstudie wurden allerdings eine dosisabhängige Keratitis (≥3 mg/kg/Tag, Sicherheitsabstand <1 bezogen auf die systemische Exposition, AUC und einer klinischen Dosis von 12 mg Perampanel/Tag) sowie Infiltrationen inflammatorischer Zellen/Ulzera der Haut (≥3 mg/kg/Tag, Sicherheitsabstand <1 bezogen auf die systemische Exposition, AUC und einer klinischen Dosis von 12 mg Perampanel/Tag) bei Mäusen beobachtet. Die in dieser Studie aufgetretenen nicht-neoplastischen Effekte am Urogenitaltrakt (Dilatationen und mesenchymale Läsionen) lassen sich mit Harnretention/Obstruktionen in Niere und Blase erklären.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Fycompa Suspension zum Einnehmen: ist nach dem Öffnen 90 Tage haltbar.
Besondere Lagerungshinweise
Nicht über 30 °C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer62440, 67665 (Swissmedic).
PackungenFycompa 2 mg: Packungen mit 7 Filmtabletten (B)
Fycompa 4 mg: Packungen mit 28 Filmtabletten (B)
Fycompa 6 mg: Packungen mit 28 Filmtabletten (B)
Fycompa 8 mg: Packungen mit 28 Filmtabletten (B)
Fycompa 10 mg: Packungen mit 28 Filmtabletten (B)
Fycompa 12 mg: Packungen mit 28 Filmtabletten (B)
Fycompa 0,5 mg/ml: 340 ml Suspension zum Einnehmen, in einer Flasche mit kindersicherem Verschluss; jede Packung enthält einen Adapter zum Eindrücken in die Flasche und zwei 20 ml Spritzen mit 0,5 ml-Skalierung.
ZulassungsinhaberinEisai Pharma AG, Zürich.
Stand der InformationDezember 2024
|