Eigenschaften/WirkungenATC-Code
A10BK02
Wirkungsmechanismus
Der Natrium-Glukose-Co-Transporter 2 (SGLT2), in den proximalen renalen Tubuli exprimiert, ist für einen Grossteil der Resorption der filtrierten Glukose aus dem Tubuluslumen verantwortlich. Canagliflozin ist ein oral aktiver Inhibitor von SGLT2. Durch die Inhibition von SGLT2 reduziert Canagliflozin die Reabsorption der filtrierten Glukose und senkt die renale Schwelle für Glukose (RTG), was zu einer erhöhten Glukoseausscheidung im Urin (UGE: urinary glucose excretion) führt. Hierdurch wird bei Patienten mit Diabetes mellitus Typ 2 der Glukosespiegel im Plasma gesenkt. Dieser Mechanismus ist nicht abhängig von Insulin. Die erhöhte Glukoseausscheidung im Urin (UGE) durch die Inhibition von SGLT2 führt darüber hinaus zu einer osmotischen Diurese, und über einen diuretischen Effekt zu einer Senkung des systolischen Blutdrucks. Die vermehrte Glukoseausscheidung im Urin (UGE) verursacht einen Kalorienverlust. Die UGE erhöht das Risiko für Harnwegsinfektionen und für genitale mykotische Infektionen.
In den Studien konnte unter Canagliflozin keine Glukose-Malabsorption beobachtet werden.
Canagliflozin erhöht die Zufuhr von Natrium in den distalen Tubulus durch Blockade der SGLT2-abhängigen Glukose- und Natrium-Reabsorption, wodurch das tubuloglomeruläre Feedback steigt; dies ist in präklinischen Diabetes-Modellen und klinischen Studien mit einer Verringerung des intraglomerulären Drucks sowie der Hyperfiltration und potenziell mit einer nierenprotektiven Wirkung assoziiert.
Pharmakodynamik
Nach ein- und mehrfachen oralen Dosen von Canagliflozin bei Patienten mit Diabetes mellitus Typ 2 wurden dosisabhängige Senkungen der RTG und Steigerungen der Glukoseausscheidung im Urin beobachtet. Bei einem Ausgangswert der RTG von ungefähr 13 mmol/l wurde unter der Dosis von 300 mg täglich bei Patienten mit Diabetes mellitus Typ 2 in Phase-1-Studien eine maximale Suppression der mittleren RTG über 24 Stunden auf ungefähr 4 bis 5 mmol/l beobachtet, was auf ein geringes Risiko für eine behandlungsinduzierte Hypoglykämie hindeutet. Die Reduktion der RTG führte bei Patienten mit Diabetes mellitus Typ 2, die entweder mit Canagliflozin 100 mg oder 300 mg behandelt wurden, zu einer erhöhten Glukoseausscheidung im Urin im Bereich zwischen 77 und 119 g/Tag über alle Phase-1-Studien hinweg. Die beobachtete Glukoseausscheidung im Urin entspricht einem Verlust zwischen 308 und 476 kcal/Tag. Die Senkungen der RTG und die Steigerungen der Glukoseausscheidung im Urin blieben bei Patienten mit Diabetes mellitus Typ 2 über eine 26-wöchige Behandlungsperiode hinweg erhalten. Nach einer leichten Zunahme (meist <400-500 ml) der täglichen Urinmenge nahm diese im Verlauf der ersten Tage nach Behandlungsbeginn wieder ab. Durch Canagliflozin erhöhte sich vorübergehend die Ausscheidung von Harnsäure im Urin (im Vergleich zum Ausgangswert am ersten Tag um 19% gesteigert, Abschwächung auf 6% an Tag 2 und 1% an Tag 13). In diesem Rahmen kam es zu einer nachhaltigen Senkung der Harnsäurespiegel im Serum von ungefähr 20%.
In einer Einzeldosisstudie an Patienten mit Diabetes mellitus Typ 2 führte die Verabreichung von 300 mg vor einer gemischten Mahlzeit zu einer Verzögerung der intestinalen Glukoseabsorption und zu einer Reduktion der postprandialen Blutzuckerwerte.
Kardiale Elektrophysiologie
In einer randomisierten, doppelblinden, Placebo-kontrollierten Studie mit positiver Kontrolle (Moxifloxacin) erhielten 60 gesunde Probanden eine orale Einzeldosis von Canagliflozin 300 mg, Canagliflozin 1'200 mg (das Vierfache der empfohlenen Maximaldosis), Moxifloxacin und Placebo. Es wurden weder unter der empfohlenen Dosis von 300 mg noch unter der Dosis von 1'200 mg bedeutsame Veränderungen des QTc-Intervalls beobachtet. Unter der Dosis von 1'200 mg betrugen die Spitzenkonzentrationen von Canagliflozin im Plasma ungefähr das 1,4-fache der Steady-State-Spitzenkonzentrationen unter einer einmal täglichen Dosis von 300 mg.
Klinische Wirksamkeit
Glykämische Wirksamkeit und Sicherheit
Insgesamt nahmen 10'285 Patienten mit Diabetes mellitus Typ 2 an neun doppelblinden, kontrollierten klinischen Wirksamkeits- und Sicherheitsstudien teil, in denen die Wirkung von Invokana auf die glykämische Kontrolle beurteilt wurde. Die Verteilung der ethnischen Zugehörigkeit entsprach 72% Weissen, 16% Asiaten, 4% Schwarzen und 8% Angehörigen anderer Gruppen. 16% der Patienten waren hispanischer Herkunft. Ungefähr 58% der Patienten waren männlich. Das Durchschnittsalter der Patienten betrug 59,6 Jahre (Bandbreite 21 bis 96 Jahre). 3'082 Patienten waren 65 Jahre und älter und 510 Patienten waren ≥75 Jahre. 58% der Patienten wiesen einen Body Mass Index (BMI) von 30 kg/m2 oder höher auf. Für die Untergruppe der Patienten mit mässiger Niereninsuffizienz mit einer Anfangs-eGFR von 30 bis <60 ml/min/1,73 m2 wurden Daten von 1'087 Patienten aus dem klinischen Entwicklungsprogramm gepoolt und analysiert.
Placebo-kontrollierte Studien
Invokana wurde als Monotherapie, als duale Therapie mit Metformin, als duale Therapie mit Sulfonylharnstoff, als dreifache Therapie mit Metformin und Sulfonylharnstoff, sowie als zusätzliche Therapie zu Insulin untersucht (Tabelle 5). Zusammenfassend führte die Behandlung mit Invokana im Vergleich zu Placebo zu klinisch und statistisch signifikanten (p<0,001) Ergebnissen bei der glykämischen Kontrolle, einschliesslich HbA1c, Anteil der Patienten mit einem HbA1c <7%, Veränderungen der Nüchternglukose (FPG) gegenüber dem Ausgangswert sowie postprandiale Glukosewerte (PPG) (nach 2 Stunden). Zusätzlich wurden relativ zu Placebo auch Abnahmen des Körpergewichts und des systolischen Blutdrucks beobachtet.
Tabelle 5: Wirksamkeitsresultate aus Placebo-kontrollierten klinischen Studiena
|
Monotherapie (26 Wochen)
| |
|
Invokana
|
Placebo
(N=192)
| |
100 mg
(N=195)
|
300 mg
(N=197)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,06
|
8,01
|
7,97
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-0,77
|
-1,03
|
0,14
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-0,91b
(-1,09; -0,73)
|
-1,16b
(-1,34; -0,99)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
44,5b
|
62,4b
|
20,6
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,57
|
9,57
|
9,20
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,51
|
-1,94
|
0,46
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-1,97b
(-2,34; -1,60)
|
-2,41b
(-2,78; -2,03)
|
N/Ac
| |
2-Stunden postprandiale Glukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
13,87
|
14,10
|
12,74
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-2,38
|
-3,27
|
0,29
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-2,67b
(-3,28; -2,05)
|
-3,55b
(-4,17; -2,94)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
85,9
|
86,9
|
87,5
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-2,8
|
-3,9
|
-0,6
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-2,2b
(-2,9; -1,6)
|
-3,3b
(-4,0; -2,6)
|
N/Ac
|
|
Duale Therapie mit Metformin (26 Wochen)
| |
|
Invokana + Metformin
|
Placebo +
Metformin
(N=183)
| |
100 mg
(N=368)
|
300 mg
(N=367)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
7,94
|
7,95
|
7,96
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-0,79
|
-0,94
|
-0,17
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-0,62b
(-0,76; -0,48)
|
-0,77b
(-0,91; -0,64)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
45,5
|
57,8
|
29,8
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,36
|
9,59
|
9,12
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,52
|
-2,10
|
0,14
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-1,65b
(-1,99; -1,32)
|
-2,23b
(-2,57; -1,90)
|
N/Ac
| |
2-Stunden postprandiale Glukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
14,30
|
14,54
|
13,81
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-2,66
|
-3,17
|
-0,55
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-2,12b
(-2,73; -1,51)
|
-2,62b
(-3,24; -2,01)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
88,7
|
85,4
|
86,7
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-3,7
|
-4,2
|
-1,2
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-2,5b
(-3,1; -1,9)
|
-2,9b
(-3,5; -2,3)
|
N/Ac
|
|
Dreifache Therapie mit Metformin und Sulfonylharnstoff (26 Wochen)
| |
|
Invokana + Metformin und
Sulfonylharnstoff
|
Placebo + Metformin
und Sulfonylharnstoff
(N=156)
| |
100 mg
(N=157)
|
300 mg
(N=156)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,13
|
8,13
|
8,12
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-0,85
|
-1,06
|
-0,13
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-0,71b
(-0,90; -0,52)
|
-0,92b
(-1,11; -0,73)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
43,2b
|
56,6b
|
18,0
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,60
|
9,34
|
9,42
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,01
|
-1,69
|
0,23
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-1,24b
(-1,75; -0,73)
|
-1,92b
(-2,43; -1,41)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
93,5
|
93,5
|
90,8
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-2,1
|
-2,6
|
-0,7
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-1,4b
(-2,1;-0,7)
|
-2,0b
(-2,7;-1,3)
|
N/Ac
|
|
Zusatztherapie zu Insulind (18 Wochen)
| |
|
Invokana + Insulin
|
Placebo + Insulin
(N=565)
| |
100 mg
(N=566)
|
300 mg
(N=587)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,33
|
8,27
|
8,20
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-0,63
|
-0,72
|
0,01
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-0,65b
(-0,73; -0,56)
|
-0,73b
(-0,82; -0,65)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
19,8
|
24,7
|
7,7
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,43
|
9,33
|
9,38
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,03
|
-1,39
|
0,22
| |
Unterschied zu Placebo (LS-Mittelwert) (95% KI)
|
-1,25b
(-1,55; -0,96)
|
-1,61b
(-1,90; -1,31)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
96,9
|
96,7
|
97,7
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,8
|
-2,3
|
0,1
| |
Unterschied zu Placebo (LS-Mittelwert) (97.5% KI)
|
-1,9b
(-2,2; -1,5)
|
-2,4b
(-2,8; -2,0)
|
N/Ac
|
a Intenttotreat-Population unter Anwendung der letzten Beobachtung in der Studie vor Einsatz einer glykämischen Rettungstherapie.
b p<0.001 im Vergleich zu Placebo.
c Nicht anwendbar.
d Invokana als Zusatztherapie zu Insulin (mit oder ohne andere antihyperglykämische Arzneimittel).
Zusatztherapie: Canagliflozin in Kombination mit Metformin und einem Dipeptidylpeptidase-4-Inhibitor
Canagliflozin wurde als Add-on bei Patienten, die unter vorheriger Behandlung mit Metformin und Sitagliptin keine adäquate glykämische Kontrolle erzielten, untersucht und dabei gemäss Titrationsplan (Anfangsdosis 100 mg; Erhöhung auf 300 mg nach 6 Wochen bei Patienten mit unzureichender glykämischer Kontrolle, angemessener eGFR und guter Verträglichkeit für 100 mg Canagliflozin) dosiert.
Bei diesen Patienten verbesserte sich die glykämische Kontrolle (siehe Tabelle 6) nach Gabe von Canagliflozin im Vergleich zu Placebo. Zusätzlich reduzierte Canagliflozin gegenüber Placebo das Körpergewicht und den systolischen Blutdruck.
Tabelle 6: Ergebnisse aus der 26-wöchigen placebokontrollierten klinischen Studie mit Canagliflozin in Kombination mit Metformin und Sitagliptin*
|
Wirksamkeitsparameter
|
Placebo +
Metformin und Sitagliptin
(N=106)
|
Canagliflozin +
Metformin und Sitagliptin
(N=107)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,38
|
8,53
| |
Abweichung vom Ausgangswert (angepasster Mittelwert)
|
-0,01
|
-0,91
| |
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)†
|
|
-0,89‡
(-1,19; -0,59)
| |
Patienten (%), die ein HbA1c <7% erreichten
|
12
|
32§
| |
Nüchtern-Plasmaglukose (mg/dl)
| |
Ausgangswert (Mittelwert)
|
180
|
186
| |
Abweichung vom Ausgangswert (angepasster Mittelwert)
|
-3
|
-30
| |
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)†
|
|
-27‡
(-40; -14)
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
89,9
|
93,8
| |
% Abweichung vom Ausgangswert (angepasster Mittelwert)
|
-1,6
|
-3,4
| |
Unterschied zu Placebo (angepasster Mittelwert) (95% KI)†
|
|
-1,8‡
(-2,7; -0,9)
|
* Intent-to-treat-Population
† Der angepasste Mittelwert und das KI wurden mit einem Mischmodell für Messwiederholungen berechnet
‡ p<0,001
§ p<0,01
Aktiv-kontrollierte Studien
Invokana wurde als duale Therapie (zusammen mit Metformin) mit Glimepirid sowie als dreifache Therapie (zusammen mit Metformin und Sulfonylharnstoff) mit Sitagliptin verglichen (Tabelle 7).
Tabelle 7: Wirksamkeitsresultate aus den aktiv-kontrollierten Studiena
|
Im Vergleich zu Glimepirid als duale Therapie zusammen mit Metformin (52 Wochen)
| |
|
Invokana + Metformin
|
Glimepirid (titriert)
+ Metformin
(N=482)
| |
100 mg
(N=483)
|
300 mg
(N=485)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
7,78
|
7,79
|
7,83
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-0,82
|
-0,93
|
-0,81
| |
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI)
|
-0,01b
(−0,11; 0,09)
|
-0,12b
(−0,22; −0,02)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
53,6
|
60,1
|
55,8
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,18
|
9,09
|
9,20
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,35
|
-1,52
|
-1,02
| |
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI)
|
-0,33
(−0,56; -0,11)
|
-0,51
(−0,73; −0,28)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
86,8
|
86,6
|
86,6
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-4,2
|
-4,7
|
1,0
| |
Unterschied zu Glimepirid (LS-Mittelwert) (95% KI)
|
-5,2d
(−5,7; −4,7)
|
-5,7d
(−6,2; −5,1)
|
N/Ac
|
|
Im Vergleich zu Sitagliptin als Dreifachtherapie zusammen mit Metformin und Sulfonylharnstoff (52 Wochen)
| |
|
Invokana 300 mg +
Metformin und
Sulfonylharnstoff
(N=377)
|
Sitagliptin 100 mg +
Metformin und
Sulfonylharnstoff
(N=378)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,12
|
8,13
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,03
|
-0,66
| |
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI)
|
-0,37e
(-0,50; -0,25)
|
N/Ac
| |
Patienten (%), die ein HbA1c <7% erreichten
|
47,6
|
35,3
| |
Nüchtern-Plasmaglukose (mmol/l)
| |
Ausgangswert (Mittelwert)
|
9,42
|
9,09
| |
Abweichung vom Ausgangswert (LS-Mittelwert)
|
-1,66
|
-0,32
| |
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI)
|
-1,34
(-1,66; -1,01)
|
N/Ac
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
87,6
|
89,6
| |
% Abweichung vom Ausgangswert (LS-Mittelwert)
|
-2,5
|
0,3
| |
Unterschied zu Sitagliptin (LS-Mittelwert) (95% KI)
|
-2,8d
(-3,3; -2,2)
|
N/Ac
|
a Intenttotreat-Population LOCF in der Studie vor Einsatz einer glykämischen Rettungstherapie.
b Invokana + Metformin wird gegenüber Glimepirid+Metformin als nicht unterlegen angesehen, da die obere Grenze des Konfidenzintervalls kleiner ist als die vordefinierte Nicht-Unterlegenheitsgrenze von <0,3%.
c Nicht anwendbar.
d p<0.01.
e Invokana+Metformin+Sulfonylharnstoff wird gegenüber Sitagliptin+Metformin+Sulfonylharnstoff als nicht unterlegen angesehen, da die obere Grenze des Konfidenzintervalls kleiner ist als die vordefinierte Nicht-Unterlegenheitsgrenze von <0,3%.
Canagliflozin in Kombination mit Metformin als Initialtherapie
Die Kombination von Canagliflozin und Metformin wurde bei nicht vorbehandelten Patienten mit Diabetes mellitus Typ 2 untersucht, welche mit Ernährungsmassnahmen und Bewegung allein keine adäquate glykämische Kontrolle erzielen konnten. Die initiale Kombinationstherapie (Canagliflozin 100 mg bzw. Canagliflozin 300 mg mit Metformin XR) war der jeweiligen Monotherapien mit Canagliflozin (100 mg oder 300 mg) und der Metformin-Monotherapie hinsichtlich der Verbesserung des HbA1c überlegen (siehe Tabelle 8).
Tabelle 8: Ergebnisse aus der 26-wöchigen aktiv-kontrollierten klinischen Studie mit Canagliflozin in Kombination mit Metformin als Initialtherapie*
|
Wirksamkeitsparameter
|
Metformin XR
(N=237)
|
Canagliflozin 100 mg
(N=237)
|
Canagliflozin 300 mg
(N=238)
|
Canagliflozin 100 mg +
Metformin XR
(N=237)
|
Canagliflozin 300 mg +
Metformin XR
(N=237)
| |
HbA1c (%)
| |
Ausgangswert (Mittelwert)
|
8,81
|
8,78
|
8,77
|
8,83
|
8,90
| |
Abweichung vom Ausgangswert (angepasster Mittelwert)
|
-1,30
|
-1,37
|
-1,42
|
-1,77
|
-1,78
| |
Unterschied zu 100 mg Canagliflozin (angepasster Mittelwert) (95% KI) †
|
|
|
|
-0,40‡
(-0,59, -0,21)
|
| |
Unterschied zu 300 mg Canagliflozin (angepasster Mittelwert) (95% KI) †
|
|
|
|
|
-0,36‡
(-0,56, -0,17)
| |
Unterschied zu Metformin XR (angepasster Mittelwert) (95% KI) †
|
|
-0,06‡
(-0,26, 0,13)
|
-0,11‡
(-0,31, 0,08)
|
-0,46‡
(-0,66, -0,27)
|
-0,48‡
(-0,67, -0,28)
| |
Patienten (%), die ein HbA1c <7% erreichten
|
43
|
39
|
43
|
50§§
|
57§§
| |
Körpergewicht
| |
Ausgangswert (Mittelwert) in kg
|
92,1
|
90,3
|
93,0
|
88,3
|
91,5
| |
% Abweichung vom Ausgangswert (angepasster Mittelwert)
|
-2,1
|
-3,0
|
-3,9
|
-3,5
|
-4,2
| |
Unterschied zu Metformin XR (angepasster Mittelwert) (95% KI)†
|
|
-0,9§§
(-1,6, -0,2)
|
-1,8§
(-2,6, -1,1)
|
-1,4‡
(-2,1, -0,6)
|
-2,1‡
(-2,9, -1,4)
|
* Intent-to-treat-Population
† Least squares mean (LS-Mittelwert), angepasst bzgl. Covariaten einschliesslich Ausgangswert und Stratifizierungsfaktor
‡ Angepasst p=0,001
§ Angepasst p<0,01
§§ Angepasst p<0,05
Spezifische Populationen
In drei Studien an spezifischen Populationen (ältere Patienten, Patienten mit eGFR von 30 bis <50 ml/min/1,73 m2 und Patienten mit kardiovaskulärer Erkrankung oder einem erhöhten Risiko für eine solche) wurde Invokana der aktuellen Diabetesbehandlung der Patienten (Diät, Monotherapie, Kombinationstherapie) hinzugefügt.
Ältere Patienten
An einer doppelblinden, Placebo-kontrollierten Studie über 26 Wochen nahmen insgesamt 714 Patienten im Alter von ≥55 bis ≤80 Jahren (davon 227 Patienten zwischen 65 und <75 Jahren und 46 Patienten zwischen 75 und <85 Jahren) mit ungenügender glykämischer Kontrolle unter der aktuellen Diabetes-Behandlung (entweder Diät und körperliche Betätigung alleine oder in Kombination mit oralen oder parenteralen antihyperglykämischen Wirkstoffen) teil. Unter Invokana 100 mg bzw. 300 mg wurden jeweils im Vergleich zu Placebo statistisch signifikante (p<0,001) Veränderungen des HbA1c gegenüber dem Ausgangswert von -0,57% und -0,70% beobachtet. Darüber hinaus kam es zu einer statistisch signifikanten Senkung der FPG und ein höherer Prozentsatz der Patienten erreichte ein HbA1c von <7,0%, verglichen mit Placebo (siehe «Dosierung/Anwendung» und «Unerwünschte Wirkungen»).
Patienten mit einer eGFR von 45 bis <60 ml/min/1,73 m2
In einer gepoolten Analyse der Daten von Patienten (n=722) mit einer Ausgangs-eGFR von 45 ml/min/1,73 m2 bis <60 ml/min/1,73 m2 bewirkte Canagliflozin verglichen mit dem Placebo eine Reduzierung des HbA1c um -0,47% unter Canagliflozin 100 mg. Patienten mit einer Ausgangs-eGFR von 45 ml/min/1,73 m2 bis <60 ml/min/1,73 m2 zeigten unter Canagliflozin 100 mg eine mittlere Abnahme des Körpergewichts um -1,8% verglichen mit Placebo.
Eine Mehrheit der Patienten mit einer Ausgangs-eGFR von 45 bis <60 ml/min/1,73 m2 wurde mit Insulin und/oder Sulfonylharnstoffen therapiert (85% [614/722]). In dieser Analyse von gepoolten Daten hatten 27,3% der Patienten unter Placebo ≥1 Hypoglykämie, versus 39,8% unter 100 mg Invokana und 41,8% unter Invokana 300 mg. Diese erhöhte Inzidenz von Hypoglykämien ist erklärbar durch die gleichzeitige Behandlung mit Insulin und/oder Sulfonylharnstoffen (siehe «Unerwünschte Wirkungen»).
Blutdruck
In den Placebo-kontrollierten Studien wurden relativ zu Placebo folgende mittlere Senkungen des systolischen Blutdrucks beobachtet: unter Invokana 100 mg -3,9 mmHg, unter Invokana 300 mg -5,3 mmHg und unter Placebo -0,1 mmHg. In der gleichen Population war die Wirkung auf den diastolischen Blutdruck geringer ausgeprägt, mit mittleren Veränderungen von -2,1 mmHg unter Invokana 100 mg, -2,5 mmHg unter Invokana 300 mg und -0,3 mmHg unter Placebo. Es kam zu keinen merklichen Veränderungen der Herzfrequenz.
Kardiovaskuläre Ergebnisse im CANVAS-Programm
Die Auswirkung von Canagliflozin auf das kardiovaskuläre Risiko bei Erwachsenen mit Typ-2-Diabetes mellitus und manifestierter kardiovaskulärer (KV) Erkrankung oder mit KV-Risiko (zwei oder mehr KV-Risikofaktoren) wurde im CANVAS-Programm (Studie CANVAS und Studie CANVAS-R) untersucht. Dabei handelte es sich um multizentrische, multinationale, randomisierte, doppelblinde Studien mit Parallelgruppen und ähnlichen Ein- und Ausschlusskriterien und Patientenkollektiven. In den Studien wurde das Risiko des Auftretens eines gravierenden unerwünschten kardiovaskulären Ereignisses (MACE; definiert als Tod infolge eines kardiovaskulären Ereignisses, nicht-tödlicher Myokardinfarkt oder nicht-tödlicher Schlaganfall) zwischen Canagliflozin und einem Placebo vor dem Hintergrund einer Standardbehandlung gegen Diabetes und atherosklerotische kardiovaskuläre Erkrankung verglichen.
In der CANVAS-Studie wurden die Patienten im Zufallsverfahren 1:1:1 randomisiert und der Behandlung mit Canagliflozin 100 mg, Canagliflozin 300 mg oder einem passenden Placebo zugeteilt. In der Studie CANVAS-R wurden die Patienten im Zufallsverfahren 1:1 randomisiert und der Behandlung mit Canagliflozin 100 mg oder einem passenden Placebo zugeteilt. Nach Ermessen des Prüfarztes war nach Woche 13 je nach Verträglichkeit und glykämischem Bedarf eine Dosiserhöhung auf 300 mg zulässig. Zur Sicherstellung, dass die Teilnehmer dem Versorgungsstandard entsprechend eine Behandlung gegen Diabetes und Atherosklerose erhielten, war eine Anpassung der entsprechenden Begleittherapien nach Ermessen des Prüfarztes zulässig.
Es wurden insgesamt 10'134 Patienten (4'327 in der CANVAS-Studie und 5'807 in der Studie CANVAS-R; insgesamt wurden 4'344 im Zufallsverfahren dem Placebo zugewiesen und 5'790 der Behandlung mit Canagliflozin) über 149 Wochen behandelt (Mittelwert; in der CANVAS-Studie betrug die Behandlungsdauer 223 Wochen und in der CANVAS-R 94 Wochen). Bei 99,6% der Teilnehmer beider Studien wurde der Vitalstatus erfasst. Ungefähr 78% der Studienteilnehmer waren Kaukasier, 13% waren Asiaten und 3% waren Schwarze. Das mittlere Alter betrug 63 Jahre und ungefähr 64% waren männlich.
Alle Patienten in der Studie hatten zum Screening-Zeitpunkt einen unzureichend kontrollierten Typ-2-Diabetes mellitus (HbA1c ≥7,0% bis ≤10,5%). Der mittlere HbA1c zum Studienbeginn betrug 8,2% und die mittlere Diabetesdauer betrug 13,5 Jahre. Bei ungefähr 31%, 21% und 18% lag eine Vorgeschichte mit Neuropathie, Retinopathie bzw. Nephropathie vor. Die Nierenfunktion zum Studienbeginn war bei 80% der Patienten normal oder leicht beeinträchtigt und bei 20% der Patienten moderat beeinträchtigt (mittlere eGFR: 77 ml/min/1,73 m2). Zum Studienbeginn standen die Patienten unter Behandlung mit mindestens einer Antidiabetesmedikation, einschliesslich Metformin (77%), Insulin (50%) und Sulfonylharnstoff (43%).
Sechsundsechzig Prozent der Teilnehmer hatten eine manifestierte kardiovaskuläre Erkrankung in der Vorgeschichte, davon hatten 56% eine Koronarerkrankung, 19% eine zerebrovaskuläre Erkrankung und 21% eine periphere Gefässerkrankung; 14% hatten eine Herzinsuffizienz in der Vorgeschichte. Zum Studienbeginn betrug der mittlere systolische Blutdruck 137 mmHg und der mittlere diastolische Blutdruck 78 mmHg, der mittlere LDL-Wert betrug 89 mg/dl, der mittlere HDL-Wert 46 mg/dl und die mittlere Urin-Albumin-Kreatinin-Ratio (UACR) 115 mg/g. Ungefähr 80% der Patienten standen zum Studienbeginn unter Behandlung mit Renin-Angiotensin-System-Hemmern, 54% unter Behandlung mit Betablockern, 13% unter Behandlung mit Schleifendiuretika, 36% unter Behandlung mit anderen Diuretika, 75% unter Behandlung mit Statinen und 74% unter Behandlung mit Thrombozytenaggregationshemmern (überwiegend Acetylsalicylsäure).
Der primäre Endpunkt im CANVAS-Programm war die Dauer des Zeitraums bis zum ersten Auftreten eines MACE. Die MACE-HR bei Patienten unter Behandlung mit Canagliflozin im Vergleich zu denen in der Placebogruppe und das dazugehörige 95%-KI wurden mit einem stetigen Cox-Regressionsmodell mit Stratifizierung nach Studie und manifestierter kardiovaskulärer Erkrankung geschätzt.
Canagliflozin bewirkte eine signifikante Reduktion des Risikos des Erstauftretens des primären kombinierten Endpunktes MACE (HR: 0,86; 95%-KI 0,75, 0,97), zu welcher jede einzelne MACE-Komponente beitrug. Die Ergebnisse für die beiden empfohlenen Canagliflozin-Dosierungen (100 mg und 300 mg) waren konsistent mit dem Ergebnis für die gepoolten Daten. Die Wirksamkeit von Canagliflozin in Bezug auf MACE unterschied sich abhängig davon, ob Patienten zu Studienbeginn bereits eine manifeste kardiovaskuläre Erkrankung (HR: 0,82; 95%-KI 0,72, 0,95) oder nur kardiovaskuläre Risikofaktoren (HR: 0,98; 95%-KI 0,74, 1,30) aufwiesen.
Es gab 2'011 Patienten mit einer eGFR von 30 bis <60 ml/min/1,73 m2. Die MACE-Ergebnisse in dieser Teilgruppe korrelierten mit den Gesamtbefunden.
Tabelle 9: Effekt der Behandlung für den primären kombinierten Endpunkt und seinen Komponenten
|
|
Placebo
N=4347
Ereignisrate pro 1000 Patientenjahre
|
Canagliflozin
N=5795
Ereignisrate pro 1000 Patientenjahre
|
Hazard Ratio
(95% KI)
| |
Kombination aus Tod infolge eines kardiovaskulären Ereignisses, nicht-tödlicher Myokardinfarkt, oder nicht-tödlicher Schlaganfall (Dauer des Zeitraums bis zum ersten Auftreten; Intent-to-Treat-Analysegruppe)1
|
31,48
|
26,93
|
0,86 (0,75-0,97)
| |
Tod infolge eines kardiovaskulären Ereignisses
|
12,84
|
11,60
|
0,87 (0,72-1,06)
| |
nicht-tödlicher Myokardinfarkt
|
11,61
|
9,74
|
0,85 (0,69-1,05)
| |
nicht-tödlicher Schlaganfall
|
8,39
|
7,12
|
0,90 (0,71-1,15)
|
1 P Wert für die Überlegenheit (zweiseitig) = 0,0158
Ergebnisse nach Subgruppen Vorliegen oder Abwesenheit einer manifestierten kardiovaskulären Erkrankung:
Die primäre Wirksamkeitsanalyse basierte auf der Gesamtpopulation. Eine vordefinierte Subgruppenanalyse für Patienten mit bzw. ohne manifeste kardiovaskuläre Vorerkrankung erbrachte zwar keinen eindeutigen Hinweis auf eine Heterogenität der Wirksamkeit in diesen beiden Gruppen (Interaktion p-Wert = 0,1803); allerdings war nur bei Patienten mit manifester Vorerkrankung eine klinisch relevante Reduktion des kardiovaskulären Risikos (HR: 0,82; 95% KI 0,72, 0,95) zu beobachten, nicht bei Patienten ohne manifeste Vorerkrankung (HR: 0,98; 95% KI: 0,74, 1,30).
Ausgehend von dem Kaplan-Meier-Plot des ersten Auftretens eines MACE war eine Reduzierung des MACE in der Canagliflozin-Gruppe bereits in Woche 26 festzustellen und blieb für den Rest der Studie erhalten.
Gesamtmortalität:
In der kombinierten Canagliflozin-Gruppe betrug die HR für die Gesamtmortalität gegenüber Placebo 0,87 (0,74; 1,01).
Stationär behandelte Herzinsuffizienz
Canagliflozin reduzierte im Vergleich mit Placebo das Risiko für eine Herzinsuffizienz, die einen Krankenhausaufenthalt erforderte (HR: 0,67; 95% KI [0,52; 0,87]).
Renale Endpunkte im CANVAS Programm
Im CANVAS-Programm lag die HR für die Zeit bis zur ersten bestätigten Nephropathie (Verdoppelung des Serum-Kreatinins, Notwendigkeit einer Nierenersatztherapie, Nierentod) bei 0,53 (95% CI: 0,33; 0,84) für Canagliflozin (1,5 Ereignisse pro 1000 Patientenjahre) im Vergleich zu Placebo (2,8 Ereignisse pro 1000 Patientenjahre). Zusätzlich reduzierte Canagliflozin das Fortschreiten der Albuminurie bei Patienten mit einer Normo- oder Mikroalbuminurie vor Behandlungsbeginn um 25,8% gegenüber 29,2% unter Placebo (HR: 0,73; 95% KI: 0,67, 0,79). Obwohl die Behandlung mit ACEi und/oder ARB im CANVAS-Programm kein Einschlusskriterium darstellte, nahmen 80% der Teilnehmer zum Zeitpunkt der Randomisierung einen ACEi und/oder ARB ein.
Nierenbezogene Therapieergebnisse in der CREDENCE-Studie
Die Wirkung von Canagliflozin 100 mg auf die renalen Ereignisse bei Erwachsenen mit Diabetes mellitus Typ 2 und diabetischer Nierenerkrankung (DKD) mit einer geschätzten glomerulären Filtrationsrate (eGFR) von 30 bis <90 ml/min/1,73 m2 oder einer CrCl von 30 bis <90 ml/min und Albuminurie (>300 bis 5'000 mg/g Kreatinin) wurde in der Studie «Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation Trial» (CREDENCE) bewertet. Dabei handelte es sich um eine multizentrische, internationale, randomisierte (1:1), doppelblinde, ereignisgesteuerte, placebokontrollierte Parallelgruppenstudie. Insgesamt 4'401 Probanden wurden randomisiert, um den ITT (Intent-To-Treat) Analysedatensatz zu bilden, welcher für die wichtigsten Wirksamkeitsanalysen verwendet wurde, während 4'397 behandelte Probanden für die Bewertung der Sicherheit zur Verfügung standen. In der CREDENCE-Studie wurde das Risiko des Auftretens einer DKD (definiert als zusammengesetzter Endpunkt aus terminaler Niereninsuffizienz, Verdopplung des Serumkreatinins und Tod durch renale oder kardiovaskuläre Ursachen) bei Gabe von Canagliflozin 100 mg bzw. eines Placebos vor dem Hintergrund einer Standardbehandlung gegen DKD (einschliesslich Angiotensinkonversionsenzym-Hemmer [ACE-Hemmer] und Angiotensin-Rezeptorblocker [ARB]) untersucht und verglichen.
In der CREDENCE-Studie wurden die Probanden entweder mit Canagliflozin 100 mg oder Placebo, stratifiziert nach der Screening-eGFR (30 bis <45, 45 bis <60, 60 bis <90 ml/min/1,73 m2) oder der CrCl (30 bis <45, 45<60, 60<90 ml/min) bis zum Beginn einer Dialyse oder bis zur Nierentransplantation behandelt.
Insgesamt wurden 4'397 Probanden behandelt, die mittlere Expositionsdauer betrug 115 Wochen. Der Vitalstatus wurde während der Studie von 99,9% der Probanden erhoben. Das Durchschnittsalter betrug 63 Jahre, 66% waren Männer.
Der mittlere HbA1c-Ausgangswert betrug 8,3%, der mediane Ausgangswert des Albumin-Kreatinin-Quotienten im Urin lag bei 927 mg/g. Die zu Studienbeginn am häufigsten verwendeten antihyperglykämischen Wirkstoffe waren Insulin (65,5%), Biguanid-Derivate (57,8%) und Sulfonylharnstoffe (28,8%). Beinahe alle Probanden (99,9%) erhielten zum Zeitpunkt der Randomisierung ACE-Hemmer oder ARB. Rund 92% der Probanden erhielten zu Studienbeginn eine kardiovaskuläre Therapie (abgesehen von ACE-Hemmern und ARB), etwa 60% wurden mit einem antithrombotischen Wirkstoff (einschliesslich Acetylsalicylsäure) und 69% mit Statinen behandelt.
Zu Studienbeginn betrug die mittlere eGFR 56,2 ml/min/1,73 m2 (rund 60% der Studienpopulation hatten eine eGFR von <60 ml/min/1,73 m2 ). Die Probanden litten durchschnittlich seit rund 16 Jahren an Diabetes. Der Anteil der Probanden mit einer kardiovaskulären Erkrankung in der Vergangenheit betrug 50,4%; bei 14,8% war in der Vergangenheit Herzinsuffizienz aufgetreten. Zusätzlich zur diabetischen Nierenerkrankung wiesen etwa 64% der Population mindestens eine weitere mikrovaskuläre Komplikation auf.
HRs und zugehörige 95%-KI für den primären und die sekundären Endpunkte wurden mittels eines stratifizierten Proportional-Hazards-Regressionsmodell nach Cox geschätzt (mit der Therapie als der erklärenden Variable und stratifiziert nach der Screening-eGFR [30 bis <45, 45 bis <60, 60 bis <90 ml/min/1,73 m2] oder CrCl [30 bis <45, 45 bis <60, 60 bis <90 ml/min].
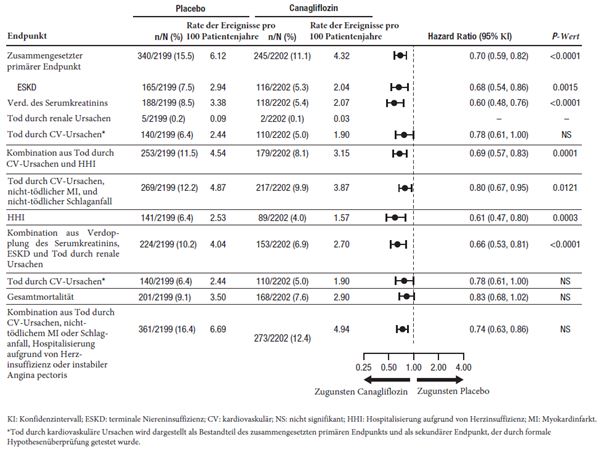
Der zusammengesetzte primäre Endpunkt der CREDENCE-Studie bestand aus dem Zeitraum bis zum/zur a) ersten Auftreten einer terminalen Niereninsuffizienz (ESKD; definiert durch einen Abfall der eGFR unter 15 ml/min/1,73 m2 bzw. der CrCl unter 15 ml/min) oder Beginn einer chronischen Dialyse oder einer Nierentransplantation, b) Verdopplung des Serumkreatinins, c) Tod durch renale Ursachen, oder d) Tod durch kardiovaskuläre Ursachen.
Canagliflozin 100 mg verringerte das Risiko des ersten Auftretens des zusammengesetzten primären Endpunkts (ESKD, Verdopplung des Serumkreatinins und Tod durch renale oder kardiovaskuläre Ursachen) signifikant [p <0,0001; HR: 0,70; 95%-KI: 0,59, 0,82] (siehe Abbildung 1). Der Behandlungseffekt war in allen Subgruppen vergleichbar, darunter in den drei aufgrund der eGFR definierten Subgruppen und bei Probanden mit einer kardiovaskulären Erkrankung in der Anamnese oder ohne diese.
Basierend auf dem Kaplan-Meier-Plot der Zeit bis zum ersten Auftreten des primären zusammengesetzten Endpunkts war die Wirkung auf die Behandlung von 100 mg Canagliflozin ab Woche 52 erkennbar und blieb bis zum Ende der Studie erhalten.
Die Effekte von Canagliflozin 100 mg auf sekundäre Endpunkte sind in Abbildung 1 zusammengefasst.
Abbildung 1: Behandlungseffekt im Hinblick auf die zusammengesetzten primären und sekundären Endpunkte und ihre Bestandteile
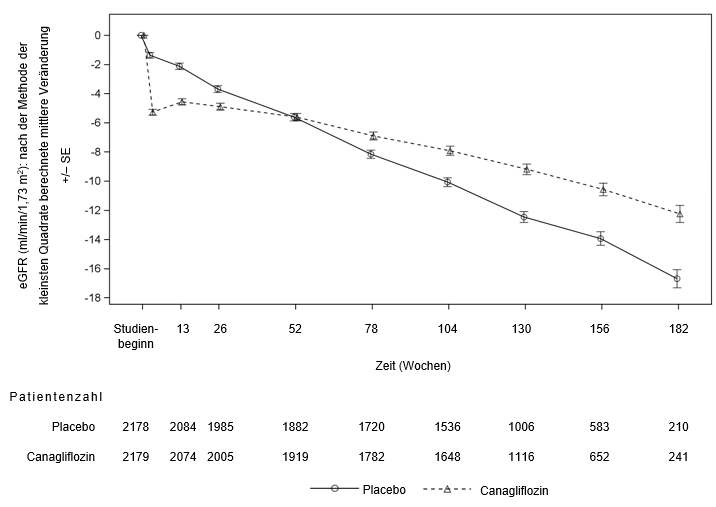
Wie in Abbildung 2 dargestellt, sank die eGFR bei placebobehandelten Patienten allmählich auf progressive und lineare Weise; in der Canagliflozin-Gruppe dagegen war eine abrupte Verringerung in Woche 3 zu beobachten, die sich danach mit der Zeit abschwächte; die nach der Methode der kleinsten Quadrate errechnete mittlere Verringerung der eGFR war nach Woche 52 in der Canagliflozin-Gruppe geringer als in der Placebogruppe, der Behandlungseffekt blieb bis zum Behandlungsende erhalten.
Abbildung 2: Nach der Methode der kleinsten Quadrate berechnete mittlere Veränderung des eGFR-Ausgangswerts im zeitlichen Verlauf (Analyseset der weiterhin behandelten Patienten)
In der CREDENCE-Studie war die Inzidenzrate nierenbezogener unerwünschter Ereignisse in der Canagliflozin-100-mg-Gruppe geringer als in der Placebogruppe (57 pro 1'000 Patientenjahre in der Canagliflozin- bzw. 79 pro 1'000 Patientenjahre in der Placebogruppe).
|