ZusammensetzungWirkstoffe
Nintedanib (als Nintedanib-Esilat).
Hilfsstoffe
·Kapselinhalt: mittelkettige Triglyceride, Hartfett, Lecithin (Soja)(E322).
·Kapselhülle: Gelatine, Glycerol (85 %), Titandioxid (E171), Eisenoxid gelb (E172), Eisenoxid rot (E172).
Indikationen/AnwendungsmöglichkeitenOfev ist bei Erwachsenen indiziert für die Behandlung von:
·der idiopathischen Lungenfibrose (IPF)
·chronischen fibrosierenden interstitiellen Lungenerkrankungen (ILDs) mit einem progressiven Phänotyp (siehe Rubrik «Klinische Wirksamkeit»)
·der mit systemischer Sklerose assoziierten interstitiellen Lungenerkrankung (SSc-ILD)
Dosierung/AnwendungDie Behandlung sollte von einem Arzt eingeleitet werden, der Erfahrung mit der Diagnostik und Therapie der Erkrankungen, für die Ofev indiziert ist, hat.
Die empfohlene Dosis von Ofev beträgt 150 mg zweimal täglich im Abstand von etwa 12 Stunden.
Ofev Kapseln sollen im Ganzen mit Wasser und zu Nahrung eingenommen werden. Die Kapseln dürfen nicht zerkaut werden.
Ofev Kapseln können mit einer kleinen Menge (Teelöffel) kalter oder zimmerwarmer weicher Nahrung, wie Apfelmus oder Schokoladenpudding, eingenommen werden und müssen sofort unzerkaut geschluckt werden, damit die Kapsel unversehrt bleibt.
Wenn eine Dosis Ofev ausgelassen wurde, die Behandlung am nächsten geplanten Einnahmezeitpunkt in empfohlener Dosierung fortsetzen. Wenn eine Dosis ausgelassen wurde, sollte der Patient keine zusätzliche Dosis erhalten. Die empfohlene maximale Tagesdosis von 300 mg sollte nicht überschritten werden.
Die Kapseln dürfen nicht geöffnet oder zerkleinert werden. Beim Kontakt mit dem Inhalt einer Kapsel müssen die Hände sofort und gründlich gewaschen werden.
Dosierungsanpassungen
Massnahmen bei Nebenwirkungen (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen») von Ofev können zusätzlich zu einer symptomatischen Therapie (sofern angezeigt), eine Dosisreduktion oder eine vorübergehende Unterbrechung der Behandlung umfassen, bis die jeweilige Nebenwirkung abgeklungen ist oder auf einen Grad abgenommen hat, der eine Fortsetzung der Therapie erlaubt. Die Behandlung mit Ofev kann in voller Dosis (150 mg zweimal täglich) oder in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen werden. Verträgt der Patient eine Dosis von 100 mg zweimal täglich nicht, so sollte die Behandlung mit Ofev abgesetzt werden.
Bei Unterbrechung der Behandlung aufgrund eines Transaminasenanstiegs (AST oder ALT) auf mehr als das 3-fache der Obergrenze des Normbereichs (OGN) kann die Behandlung mit Ofev nach Normalisierung der Transaminasenwerte in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen und anschliessend wieder auf die volle Dosis (150 mg zweimal täglich) erhöht werden. Bei einem Anstieg der AST oder ALT auf mehr als das 5-fache der OGN oder bei einem Anstieg auf mehr als das 3-fache der OGN mit gleichzeitig bestehenden Befunden oder Symptomen einer schweren Leberschädigung ist Ofev abzusetzen (siehe «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen»).
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktionsstörungen
Weniger als 1 % einer Einzeldosis Nintedanib wird über die Nieren ausgeschieden (siehe «Pharmakokinetik»). Bei Patienten mit leichter bis mittelschwerer Einschränkung der Nierenfunktion ist keine Anpassung der Anfangsdosis erforderlich. Die Sicherheit, Wirksamkeit und Pharmakokinetik von Nintedanib bei Patienten mit schwerer Einschränkung der Nierenfunktion wurden nicht untersucht (CrCL <30 ml/min).
Patienten mit Leberfunktionsstörungen
Nintedanib wird vorwiegend über die Galle/die Fäzes ausgeschieden (> 90 %; siehe «Pharmakokinetik»). Die Exposition stieg bei Patienten mit Einschränkung der Leberfunktion (Child-Pugh-Klasse A, Child-Pugh-Klasse B) an. Bei Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) beträgt die empfohlene Ofev-Dosis 100 mg zweimal täglich im Abstand von jeweils etwa 12 Stunden. Bei Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) sollte eine Unterbrechung oder ein Absetzen der Behandlung, um unerwünschte Reaktionen behandeln zu können, in Erwägung gezogen werden.
Die Sicherheit und Wirksamkeit von Nintedanib bei Patienten mit Einschränkung der Leberfunktion vom Grad Child Pugh B oder C wurde nicht untersucht. Die Anwendung von Ofev bei Patienten mit mittelschwerer (Child-Pugh-Klasse B) oder schwerer (Child-Pugh-Klasse C) Einschränkung der Leberfunktion wird nicht empfohlen (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Kinder und Jugendliche
Nintedanib sollte nicht von Kindern und Jugendlichen unter 18 Jahren eingenommen werden.
Ältere Patienten (≥65 Jahre)
Bei älteren Patienten wurden hinsichtlich der allgemeinen Sicherheit und Wirksamkeit keine Unterschiede zu Patienten unter 65 Jahren beobachtet. Es ist keine Dosisanpassung auf Grundlage des Lebensalters des Patienten erforderlich (siehe «Pharmakokinetik»).
KontraindikationenOfev ist bei Patienten mit bekannter Überempfindlichkeit gegen Nintedanib, Erdnüsse, Soja oder einen der sonstigen Bestandteile kontraindiziert.
Ofev ist in der Schwangerschaft kontraindiziert (siehe «Schwangerschaft /Stillzeit» und «Präklinische Daten»).
Warnhinweise und VorsichtsmassnahmenErkrankungen des Gastrointestinaltrakts
Diarrhö
In den klinischen Studien (siehe «Klinische Wirksamkeit») war Diarrhö das am häufigsten genannte gastrointestinale Ereignis. Bei den meisten Patienten war Diarrhö leicht bis mittelschwer ausgeprägt und trat in den ersten 3 Behandlungsmonaten auf.
In den INPULSIS-Studien (IPF-Patienten) wurde eine Diarrhö bei 62,4% der mit Ofev versus 18,4% der mit Placebo behandelten Patienten berichtet. Durchfall führte bei 10,7% der Patienten zu einer Dosisreduktion von Ofev versus bei keinem mit Placebo behandelten Patienten und bei 4,4% der Patienten zum Abbruch der Behandlung mit Ofev versus 0,2% der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Diarrhö bei 66,9 % der mit Ofev versus 23,9 % der mit Placebo behandelten Patienten berichtet. Diarrhö führte bei 16,0 % der Patienten zu einer Dosisreduktion von Ofev versus 0,9% bei mit Placebo behandelten Patienten und bei 5,7 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3% bei mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Diarrhö bei 75,7 % der mit Ofev versus 31,6% der mit Placebo behandelten Patienten berichtet. Durchfall führte bei 22,2% der Patienten zu einer Dosisreduktion von Ofev versus 1,0% bei mit Placebo behandelten Patienten und bei 6,9% der Patienten zum Abbruch der Behandlung versus 0,3% bei mit Placebo-behandelten Patienten (siehe Abschnitt «Unerwünschte Wirkungen»).
Diarrhö kann zu Dehydrierung mit oder ohne Elektrolytstörungen führen, was eine Nierenfunktionsstörung zur Folge haben kann.
Eine Diarrhö sollte bei den ersten Anzeichen mit angemessener Hydrierung und Antidiarrhoika wie beispielsweise Loperamid behandelt werden. Gegebenenfalls ist eine Dosisreduktion oder eine Unterbrechung der Behandlung erforderlich. Die Behandlung mit Ofev kann in verringerter Dosis (100 mg zweimal täglich) oder in voller Dosis (150 mg zweimal täglich) wieder aufgenommen werden. Wenn eine schwere Diarrhö trotz symptomatischer Therapie persistiert, sollte die Behandlung mit Ofev abgebrochen werden.
Übelkeit und Erbrechen
Übelkeit und Erbrechen sind häufig genannte unerwünschte Ereignisse (siehe «Unerwünschte Wirkungen») und meist leicht bis mittelschwer ausgeprägt.
In den INPULSIS-Studien (IPF-Patienten) wurde Übelkeit bei 24,5 % der mit Ofev versus 6,6 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 1,7 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 2,0 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Übelkeit bei 28,9 % der mit Ofev versus 9,4 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 3,3 % der Patienten zu einer Dosisreduktion von Ofev versus 0,6 % bei mit Placebo behandelten Patienten und bei 0,3 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3 % bei mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Übelkeit bei 31,6 % der mit Ofev versus 13,5 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 2,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 2,1 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In den INPULSIS-Studien (IPF-Patienten) wurde Erbrechen bei 11,6 % der mit Ofev versus 2,6 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 1,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 0,8 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Erbrechen bei 18,4 % der mit Ofev versus 5,1 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 2,4 % der Patienten zu einer Dosisreduktion von Ofev versus 0,9 % bei mit Placebo behandelten Patienten und bei 0,9 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Erbrechen bei 24,7 % der mit Ofev versus 10,4 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 2,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 1,4 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3 % bei mit Placebo behandelten Patienten.
Erbrechen kann zu Dehydrierung mit oder ohne Elektrolytstörungen führen, was eine Nierenfunktionsstörung zur Folge haben kann.
Wenn die Symptome trotz geeigneter supportiver Massnahmen (einschliesslich einer antiemetischen Therapie) persistieren, kann eine Dosisreduktion oder Unterbrechung der Behandlung erforderlich sein. Die Behandlung kann in verringerter Dosis (100 mg zweimal täglich) oder in voller Dosis (150 mg zweimal täglich) wieder aufgenommen werden. Bei persistierenden schweren Symptomen sollte die Behandlung mit Ofev abgesetzt werden.
Leberfunktion
Die Sicherheit und Wirksamkeit von Ofev bei Patienten mit mittelschwerer (Child-Pugh-Klasse B) oder schwerer (Child-Pugh-Klasse C) Einschränkung der Leberfunktion wurde nicht untersucht. Daher wird die Anwendung von Ofev bei diesen Patienten nicht empfohlen.
Aufgrund der erhöhten Exposition kann das Risiko für unerwünschte Ereignisse bei Patienten mit leichter Einschränkung der Leberfunktion (Child-Pugh-Klasse A) erhöht sein. Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) sollten mit einer reduzierten Ofev-Dosis behandelt werden (siehe «Dosierung / Anwendung» und «Pharmakokinetik»).
Fälle von arzneimittelinduzierten Leberschäden wurden unter der Behandlung mit Nintedanib beobachtet. In der Marktbeobachtung wurden nicht-schwerwiegende und schwerwiegende Fälle von arzneimittelinduzierten Leberschäden gemeldet, darunter auch schwere Leberschäden mit tödlichem Ausgang.
Die meisten unerwünschten hepatischen Ereignisse traten innerhalb der ersten drei Behandlungsmonate auf. Daher sollten die hepatischen Transaminasen und Bilirubin-Konzentrationen zu Beginn der Behandlung mit Ofev, in regelmässigen Abständen während der ersten drei Monate der Behandlung und anschliessend periodisch (z.B. bei jedem Termin des Patienten) oder wenn klinisch indiziert kontrolliert werden.
Erhöhungen der Leberenzymwerte (ALT, AST, ALKP, Gammaglutamyltransferase (GGT)) und des Bilirubinwerts waren nach Dosisreduktion oder Behandlungsunterbrechung in den meisten Fällen reversibel. Erhöhte ALT- und/oder AST-Werte, die auf mindestens das 3-Fache der OGN anstiegen, wurden bei bis zu 5,0%, 7,8% bzw. 4,9 % der mit Ofev behandelten Patienten in den INPULSIS (IPF-Patienten) -, INBUILD (Patienten mit progressiven fibrosierenden ILDs) - bzw. SENSCIS (SSc-ILD-Patienten) -Studien beobachtet, während es bei Patienten, die Placebo erhielten, insgesamt weniger als 2% waren.
Bei einem Anstieg der Transaminasen (AST oder ALT) auf das mehr als 3-fache der Obergrenze des Normbereichs (OGN) sollte die Behandlung mit Ofev unterbrochen oder die Dosis auf 100 mg zweimal täglich verringert werden. Ausserdem sollte der Patient engmaschig überwacht werden und alternative Ursachen für den Anstieg der Leberenzyme sind zu untersuchen. Nach Normalisierung der Transaminasen kann die Behandlung mit Ofev in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen und anschliessend auf die volle Dosis (150 mg zweimal täglich) erhöht werden. Bestehen parallel zu einem Anstieg von Leberwerten klinische Zeichen einer Leberschädigung wie beispielsweise eine Gelbsucht, oder steigen die Transaminasen (AST oder ALT) auf das mehr als 5-fache der OGN an, so ist die Behandlung mit Ofev endgültig abzusetzen (siehe «Dosierung/Anwendung»).
Bei Patienten mit geringem Körpergewicht (< 65 kg), Asiaten und Frauen besteht ein höheres Risiko für Leberenzymerhöhungen.
Die Nintedanib-Exposition erhöhte sich linear mit dem Alter der Patienten. Dies könnte auch zu einem Anstieg des Risikos für Leberenzymerhöhungen führen (siehe Abschnitt «Pharmakokinetik»).
Bei Patienten mit diesen Risikofaktoren wird eine engmaschige Überwachung empfohlen.
Nierenfunktion
Bei der Anwendung von Nintedanib wurde über Fälle von Nierenfunktionsstörungen bzw. Nierenversagen berichtet, von denen einige tödlich verliefen (siehe «Unerwünschte Wirkungen»).
Während der Therapie mit Nintedanib sollten die Patienten überwacht werden, insbesondere solche Patienten, die Risikofaktoren für eine Nierenfunktionsstörung bzw. ein Nierenversagen aufweisen. Bei einer Nierenfunktionsstörung bzw. einem Nierenversagen ist eine Anpassung der Therapie in Erwägung zu ziehen (siehe «Dosierungsanpassungen»).
Blutungen
Angesichts seines Wirkmechanismus (Hemmung des vaskulären endothelialen Wachstumsfaktorrezeptors (vascular endothelial growth factor receptor, VEGFR)) kann Ofev mit einem erhöhten Blutungsrisiko verbunden sein.
In klinischen Studien mit Ofev war die Häufigkeit von Blutungsereignissen bei Patienten unter Ofev etwas höher bzw. zwischen den Behandlungsgruppen vergleichbar (10,3% versus Placebo 7,8% in den INPULSIS-Studien (IPF-Patienten); 11,1 % versus Placebo 12,7% in der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs),11,1 % versus Placebo 8,3% in der SENSCIS-Studie (SSc-ILD-Patienten)).
Nicht schwerwiegende Epistaxis stellte das häufigste Blutungsereignis dar.
Die Häufigkeit von schwerwiegenden Blutungsereignissen war in den beiden Behandlungsgruppen gering (Ofev 1,3 % versus Placebo 1,4 % in INPULSIS (IPF-Patienten); Ofev 0,9 % versus Placebo 1,5 % in INBUILD (Patienten mit progressiven fibrosierenden ILDs); Ofev 1,4 % versus Placebo 0,7 % in SENSCIS (SSc-ILD-Patienten))
An den klinischen-Studien nahmen keine Patienten mit bekanntem Blutungsrisiko wie solche mit angeborener Blutungsneigung oder Patienten, die eine Antikoagulation in voller Dosis erhalten, teil. Nach Markteinführung wurden Fälle von Blutungen berichtet, darunter schwerwiegende und tödliche (einschliesslich Patienten mit oder ohne Behandlung mit Antikoagulanzien oder anderen Arzneimitteln, die Blutungen hervorrufen könnten). Daher sollten Patienten mit zusätzlichen Risikofaktoren nur dann mit Ofev behandelt werden, wenn der erwartete Nutzen das potenzielle Risiko überwiegt.
Arterielle thromboembolische Ereignisse
Patienten mit kürzlichem Myokardinfarkt oder Schlaganfall waren von den klinischen Studien ausgeschlossen.
In den klinischen Studien wurden arterielle thromboembolische Ereignisse mit geringer Häufigkeit beschrieben (Ofev 2,5 % versus Placebo 0,7 % in INPULSIS (IPF-Patienten); Ofev 0,9 % versus Placebo 0,9 % in INBUILD (Patienten mit progressiven fibrosierenden ILDs); Ofev 0,7 % versus Placebo 0,7 % in SENSCIS (SSc-ILD-Patienten)). In den INPULSIS-Studien wurden bei einem höheren Prozentsatz der Patienten in der Ofev-Gruppe (1,6%) als der Patienten in der Placebogruppe (0,5 %) Myokardinfarkte beschrieben, während unerwünschte Ereignisse, die eine ischämische Herzkrankheit widerspiegeln, in der Ofev- und Placebogruppe ausgeglichen waren. In der INBUILD- und der SENSCIS-Studie wurde eine geringe Häufigkeit von Myokardinfarkten beobachtet: Ofev 0,9 % versus Placebo 0,9 % in INBUILD; Ofev 0 % versus Placebo 0,7 % in SENSCIS.
Bei Patienten mit erhöhtem kardiovaskulärem Risiko, wie beispielsweise solchen mit bekannter koronarer Herzkrankheit, mit Vorsicht anwenden. Bei Patienten, bei denen Befunde oder Symptome einer akuten Myokardischämie auftreten, ist eine Unterbrechung der Behandlung in Betracht zu ziehen.
Aneurysmen und Arteriendissektionen
Die Verwendung von VEGF-Signalweg-Hemmern bei Patienten mit oder ohne Hypertonie kann die Entstehung von Aneurysmen und/oder Arteriendissektionen begünstigen. Vor Beginn der Behandlung mit Ofev sollte dieses Risiko bei Patienten mit Risikofaktoren wie Hypertonie oder Aneurysmen in der Vorgeschichte sorgfältig abgewogen werden.
Venöse Thromboembolien
In den klinischen Studien wurde bei den mit Nintedanib behandelten Patienten kein erhöhtes Risiko für venöse Thromboembolien beobachtet. Angesichts des Wirkmechanismus von Nintedanib könnte ein erhöhtes Risiko für thromboembolische Ereignisse bestehen.
Gastrointestinale Perforationen und ischämische Kolitis
Mit Nintedanib behandelte Patienten können aufgrund des Wirkmechanismus des Arzneimittels ein erhöhtes Risiko für gastrointestinale Perforationen aufweisen. In den INPULSIS-Studien (IPF-Patienten) wurde bei 0,3 % der mit Ofev und bei keinem (0) der mit Placebo behandelten Patienten eine gastrointestinale Perforation beschrieben. In der SENSCIS-Studie (SSc-ILD-Patienten) und der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurden in beiden Behandlungsgruppen keine Fälle von gastrointestinaler Perforation berichtet.
Nach Markteinführung wurden Fälle von gastrointestinalen Perforationen und ischämischer Kolitis, darunter auch tödliche, berichtet. Bei Patienten mit kürzlichem bauchchirurgischem Eingriff, kürzlich aufgetretener Perforation eines Hohlorgans, peptischen Ulzera in der Anamnese, Divertikulose oder bei begleitender Anwendung von Corticosteroiden oder NSAR ist besondere Vorsicht geboten. Bei Auftreten einer gastrointestinalen Perforation oder ischämischen Kolitis ist die Behandlung mit Ofev endgültig abzusetzen.
Patienten mit bekanntem Risiko für eine gastrointestinale Perforation oder ischämischer Kolitis dürfen nur mit Ofev behandelt werden, wenn der erwartete Nutzen das potenzielle Risiko überwiegt.
Proteinurie im nephrotischen Bereich und thrombotische Mikroangiopathie
Nach dem Inverkehrbringen wurden sehr wenige Fälle einer Proteinurie im nephrotischen Bereich mit oder ohne Beeinträchtigung der Nierenfunktion gemeldet. In einzelnen Fällen waren die histologischen Befunde mit einer glomerulären Mikroangiopathie mit oder ohne Nierenthrombus vereinbar. Nach dem Absetzen von Ofev waren die Symptome reversibel, wobei in einigen Fällen eine anhaltende Proteinurie beobachtet wurde. Bei Patienten, die Anzeichen oder Symptome eines nephrotischen Syndroms entwickeln, sollte eine Unterbrechung der Behandlung erwogen werden.
Die Anwendung von VEGF-Signalweg-Hemmern wurde mit einer thrombotischen Mikroangiopathie (TMA) assoziiert; einschliesslich sehr weniger Fallberichte für Nintedanib. Sollten bei einem Patienten, der Nintedanib erhält, mit einer TMA in Verbindung stehende Laborwerte oder klinische Befunde auftreten, ist die Behandlung mit Nintedanib abzusetzen und eine eingehende Untersuchung auf TMA durchzuführen.
Posteriores reversibles Enzephalopathie-Syndrom (PRES)
Es wurden sehr wenige Fälle eines posterioren reversiblen Enzephalopathie-Syndroms (PRES) nach der Marktzulassung berichtet.
PRES ist ein neurologisches Syndrom, das mit Kopfschmerzen, visuellen Störungen, Krampfanfällen, Lethargie, Verwirrtheit und anderen neurologischen Störungen einhergehen kann.
Eine leichte bis schwere Hypertonie kann auftreten. Zur Bestätigung der Diagnose eines PRES ist eine Magnetresonanztomographie erforderlich.
Die Behandlung mit Nintedanib muss bei Verdacht auf PRES abgesetzt werden.
Es ist nicht bekannt, wie die Sicherheit im Falle einer erneuten Aufnahme der Nintedanib-Therapie bei Patienten mit zuvor aufgetretenem PRES einzuschätzen ist.
Wundheilungsstörungen
In den klinischen Studien wurde keine erhöhte Inzidenz von Wundheilungsstörungen beobachtet. Nintedanib kann angesichts seines Wirkmechanismus die Wundheilung beeinträchtigen. Es wurden keine Studien durchgeführt, die eigens die Auswirkungen von Nintedanib auf die Wundheilung untersuchten. Daher sollte die Behandlung mit Ofev erst dann begonnen – bzw. bei perioperativer Unterbrechung der Behandlung wieder aufgenommen – werden, wenn nach klinischem Ermessen eine ausreichende Wundheilung erfolgt ist.
Sojalecithin
Ofev Weichkapseln enthalten Sojalecithin (siehe «Kontraindikationen»).
InteraktionenStudien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Einfluss anderer Wirkstoffe auf Nintedanib:
P-Glycoprotein (P-gp) und CYP3A4 Inhibitoren und Induktoren
Nintedanib ist ein Substrat von P-gp (siehe «Pharmakokinetik») und zu einem geringen Teil auch von CYP3A4. Die gemeinsame Verabreichung mit dem starken P-gp-Inhibitor und CYP3A4 Inhibitor Ketoconazol bewirkte einen Anstieg der AUC von Nintedanib auf das 1,61-fache und der Cmax auf das 1,83-fache.
Starke P-gp-Inhibitoren (z.B. Ketoconazol oder Erythromycin) können bei gemeinsamer Verabreichung mit Ofev die Nintedanib-Exposition erhöhen. In solchen Fällen ist eine engmaschige Überwachung der Verträglichkeit von Nintedanib geboten. Nebenwirkungen können eine Unterbrechung der Behandlung mit Ofev, eine Dosisreduktion oder ein Absetzen von Ofev erforderlich machen (siehe «Dosierung/Anwendung»).
Bei gleichzeitiger Verabreichung mit dem starken P-gp- und CYP3A4 Induktor Rifampicin nahm die AUC von Nintedanib auf 50,3 % und die Cmax auf 60,3 % des bei alleiniger Verabreichung von Nintedanib gemessenen Wertes ab.
Starke P-gp-Induktoren (z.B. Rifampicin, Carbamazepin, Phenytoin und Johanniskraut) können die Nintedanib-Exposition verringern. Die Auswahl einer anderen Begleitmedikation ohne oder mit minimalem Potential für eine P-gp-Induktion ist zu erwägen.
Nahrung
Ofev soll zusammen mit Nahrung eingenommen werden um die Bioverfügbarkeit zu erhöhen (siehe «Pharmakokinetik»).
pH-Wert
Die Löslichkeit von Nintedanib ist stark vom pH-Wert abhängig und sinkt bei einem pH-Wert >3. In klinischen Studien hatte die gleichzeitige Einnahme von Medikamenten, die zu einer Erhöhung des Magen pH-Wertes führen, bei Einnahme von Nintedanib mit Nahrung keinen Einfluss auf die Ctrough Werte von Nintedanib. Bei nüchterner Einnahme könnte die gleichzeitige Einnahme von Medikamenten, die zu einer Erhöhung des Magen pH-Wertes führen, die Bioverfügbarkeit von Nintedanib reduzieren. Nintedanib sollte daher mit Nahrung eingenommen werden.
Rauchen
Rauchen steht mit einer reduzierten Nintedanib Exposition im Zusammenhang, was zu einer veränderten Wirksamkeit führen könnte. Patienten sollten angehalten werden vor Beginn einer Therapie mit Ofev mit dem Rauchen aufzuhören oder das Rauchen während der Therapie mit Ofev zu reduzieren.
In-vitro-Untersuchungen zufolge ist Nintedanib kein Substrat von OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, BCRP oder MRP-2. In-vitro-Studien zeigten, dass Nintedanib ein Substrat von OCT-1 ist. Es wird davon ausgegangen, dass diese Beobachtungen geringe klinische Relevanz haben.
Einfluss von Nintedanib auf andere Wirkstoffe:
Transporter
In vitro wurde eine schwache hemmende Wirkung auf OCT-1, BCRP und P-gp beobachtet. Die klinische Relevanz wurde nicht untersucht, wird aber als gering betrachtet.
In-vitro-Untersuchungen zufolge ist Nintedanib kein Inhibitor von OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, oder MRP-2.
Der Metabolit BIBF 1202 war in vitro ein schwacher Hemmer von OATP-1B1, OATP-1B3, OATP-2B1, OCT-1 und BCRP. Die klinische Relevanz wurde nicht untersucht, wird aber als gering betrachtet.
Cytochrom-(CYP-)Enzyme
Die Biotransformation von Nintedanib erfolgt nur in geringem Masse über CYP-Enzyme. Nintedanib und seine Metaboliten, die freie Säure BIBF 1202 und ihr Glucuronid BIBF-1202-Glucuronid, hatten in präklinischen Studien weder einen hemmenden, noch einen induzierenden Einfluss auf CYP-Enzyme (siehe «Pharmakokinetik»). Die Wahrscheinlichkeit für Arzneimittelwechselwirkungen aufgrund von Beeinflussung von CYP-Stoffwechselwegen durch Nintedanib wird daher als gering eingeschätzt.
Gemeinsame Verabreichung mit anderen Arzneimitteln
Begleittherapie mit Pirfenidon:
Ausgehend von den Ergebnissen einer speziellen Pharmakokinetik (PK)-Studie, gibt es keine Hinweise auf eine relevante pharmakokinetische Interaktion zwischen Nintedanib und Pirfenidon, wenn diese Wirkstoffe in Kombination verabreicht werden.
Begleittherapie mit Bosentan
In einer speziellen Pharmakokinetik (PK)-Studie wurde die gleichzeitige Behandlung von Ofev mit Bosentan an gesunden Freiwilligen untersucht. Die Studienteilnehmer erhielten eine Einzeldosis von 150 mg Ofev vor und nach einer Mehrfachgabe von 125 mg Bosentan zweimal täglich im Steady State. Die adjustierten Verhältnisse der geometrischen Mittelwerte (90% Konfidenzintervall [KI]) lagen bei 103% (86% - 124%) bzw. 99% (91% - 107%) für die Cmax bzw. AUC0-tz von Nintedanib (n=13), was darauf hinweist, dass die gleichzeitige Gabe von Nintedanib mit Bosentan die Pharmakokinetik von Nintedanib nicht veränderte.
Begleittherapie mit oralen hormonellen Kontrazeptiva
Die gleichzeitige Gabe von Nintedanib und oralen hormonellen Kontrazeptiva hatte keinen nennenswerten Einfluss auf die Pharmakokinetik der oralen hormonellen Kontrazeptiva.
In einer speziellen PK-Studie erhielten weibliche Patienten mit SSc-ILD eine Einzeldosis einer Kombination aus 30 µg Ethinylestradiol und 150 µg Levonorgestrel vor und nach der zweimal täglichen Gabe von 150 mg Nintedanib über mindestens 10 Tage. Die adjustierten Verhältnisse der geometrischen Mittelwerte (90%-KI) betrugen 117% (108%-127%; Cmax) bzw. 101% (93%-111%; AUC0–tz) für Ethinylestradiol und 101% (90%-113%; Cmax) bzw. 96% (91%-102%; AUC0–tz) für Levonorgestrel (n = 15), was darauf hindeutet, dass die gleichzeitige Gabe von Nintedanib keine nennenswerten Auswirkungen auf die Plasmaspiegel von Ethinylestradiol und Levonorgestrel hat.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Daten zur Anwendung von Ofev bei Schwangeren vor, allerdings zeigten präklinische tierexperimentelle Studien eine Reproduktionstoxizität des Arzneimittels (siehe «Präklinische Daten»). Da Nintedanib auch beim Menschen den Fötus schädigen kann, darf es in der Schwangerschaft nicht angewendet werden.
Vor Beginn der Behandlung mit Ofev ist ein Schwangerschaftstest vorzunehmen, der während der Behandlung nach Bedarf zu wiederholen ist.
Weibliche Patienten sind darauf hinzuweisen, dass sie ihren Arzt oder Apotheker informieren müssen, wenn sie während der Behandlung mit Ofev schwanger werden.
Wird eine Patientin während der Behandlung mit Ofev schwanger, ist die Behandlung abzusetzen und die Patientin sollte auf die potenziellen Risiken für den Fötus hingewiesen werden.
Empfängnisverhütung
Nintedanib kann den Fötus schädigen (siehe «Präklinische Daten»). Mit Ofev behandelte Frauen, die schwanger werden können, sollten darauf hingewiesen werden, dass sie während der Behandlung mit Ofev nicht schwanger werden sollen und dass zu Beginn, während der Behandlung und bis mindestens 3 Monate nach der letzten Dosis Ofev hoch wirksame Verhütungsmethoden anzuwenden sind. Nintedanib hat keinen nennenswerten Einfluss auf die Plasmaspiegel von Ethinylestradiol und Levonorgestrel (siehe «Interaktionen»). Die Wirksamkeit oraler hormoneller Kontrazeptiva kann bei Erbrechen und/oder Diarrhoe sowie anderen Zuständen mit beeinträchtigter Resorption vermindert sein. Frauen, die orale hormonelle Kontrazeptiva einnehmen und bei denen solche Zustände auftreten, sollte geraten werden, eine andere sehr zuverlässige Verhütungsmethode anzuwenden.
Stillzeit
Es liegen keine Daten zur Ausscheidung von Nintedanib und seinen Metaboliten in die Muttermilch vor.
In präklinischen Studien wurden bei säugenden Ratten geringe Mengen an Nintedanib und seinen Metaboliten (≤0,5 % der verabreichten Dosis) in der Milch nachgewiesen.
Ein Risiko für das Neugeborene / Kind kann nicht ausgeschlossen werden. Das Stillen soll während der Behandlung mit Ofev unterbrochen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Die Patienten sind darauf hinzuweisen, dass sie während einer Behandlung mit Ofev beim Führen eines Fahrzeugs und beim Bedienen von Maschinen vorsichtig sein sollten.
Unerwünschte WirkungenDie Sicherheit von Ofev wurde in klinischen Studien bei mehr als 1'000 Patienten mit IPF untersucht, von denen mehr als 200 Ofev mehr als 2 Jahre lang erhielten.
Ofev wurde in drei 52-wöchigen randomisierten, placebokontrollierten Doppelblind-Studien bei Patienten mit IPF untersucht. In der Phase-II-Studie (TOMORROW) und den Phase-III-Studien (INPULSIS-1 und INPULSIS-2) erhielten 723 Patienten mit IPF Ofev 150 mg zweimal täglich und 508 Patienten Placebo. Die mediane Dauer der Exposition betrug bei mit Ofev behandelten Patienten 10 Monate und bei mit Placebo behandelten Patienten 11 Monate. Die Studienteilnehmer waren zwischen 42 und 89 Jahre alt (medianes Alter von 67 Jahren). Die meisten Patienten waren Männer (79 %) und kaukasischer Abstammung (60 %).
Ofev wurde zudem in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie (INBUILD) untersucht, in der die Behandlung mit Ofev 150 mg zweimal täglich (n=332) mit Placebo (n=331) an 663 Patienten mit anderen chronischen fibrosierenden ILDs mit einem progressiven Phänotyp verglichen wurde. Während die Patienten mindestens 52 Wochen behandelt wurden, dauerte die Behandlung bei einzelnen Patienten bis zu 27 Monate. Die mediane Expositionsdauer betrug 16 Monate bei mit Ofev behandelten Patienten und 17 Monate bei Patienten, die Placebo erhielten. Das Alter der Studienteilnehmer reichte von 27 bis 87 Jahren (medianes Alter: 67 Jahre). Die meisten Patienten waren männlich (54 %). Die Patienten waren überwiegend Kaukasier (74 %) oder Asiaten (25 %).
Ofev wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie (SENSCIS) untersucht, in der 576 Patienten mit SSc-ILD zweimal täglich 150 mg Ofev (n=288) oder Placebo (n=288) erhielten. Während die Patienten mindestens 52 Wochen behandelt wurden, dauerte die Behandlung bei einzelnen Patienten bis zu 100 Wochen. Die mediane Expositionsdauer betrug 15 Monate bei mit Ofev behandelten Patienten und 16 Monate bei Patienten, die Placebo erhielten. Das Alter der Probanden reichte von 20 bis 79 Jahre (medianes Alter: 55 Jahre). Die meisten Patienten waren weiblich (75 %). Die Patienten waren überwiegend Kaukasier (67 %), Asiaten (25 %) oder hatten eine schwarze Hautfarbe (6 %). 49 % der Patienten erhielten zu Studienbeginn eine stabile Therapie mit Mycophenolat.
Die häufigsten im Zusammenhang mit der Anwendung von Ofev beschriebenen Nebenwirkungen beinhalteten Diarrhö, Übelkeit und Erbrechen, abdominelle Schmerzen, Appetitabnahme, Gewichtsabnahme, Blutungen, Erhöhung der Leberenzyme und Hautausschlag.
Das Sicherheitsprofil bei mit Ofev behandelten Patienten war vergleichbar, und zwar unabhängig davon, ob sie zu Studienbeginn Mycophenolat erhielten oder nicht.
Der Abschnitt «Warnhinweise und Vorsichtsmassnahmen» enthält Hinweise zum Vorgehen bei ausgewählten Nebenwirkungen.
Bei den Nebenwirkungshäufigkeiten werden folgende Häufigkeitskategorien zugrunde gelegt: «sehr häufig» (≥1/10), «häufig» (< 1/10, ≥1/100), «gelegentlich» (< 1/100, ≥1/1000), «selten» (< 1/1000, ≥1/10'000), «sehr selten» (< 1/10'000).
Idiopathische Lungenfibrose (IPF)
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Häufig: Appetitverlust, Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Häufigkeit nicht bekannt: Posteriores reversibles Enzephalopathie-Syndrom (PRES).
Herzerkrankungen
Gelegentlich: Myokardinfarkt.
Gefässerkrankungen
Häufig: Blutungena.
Gelegentlich: Hypertonieb.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (53,6%), Übelkeit (19,1%), abdominelle Schmerzenc (10,2%).
Häufig: Erbrechen.
Gelegentlich: Pankreatitis, gastrointestinale Perforation, Kolitis.
Häufigkeit unbekannt: Ischämische Kolitis (Beobachtung nach Markteinführung)
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (10,5%).
Häufig: Erhöhung der Alaninaminotransferase (ALT), Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT).
Gelegentlich: Erhöhung der Alkalischen Phosphatase (ALP) im Blut, Hyperbilirubinämie, Arzneimittelbedingter Leberschaden.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag.
Gelegentlich: Pruritus, Alopezie.
Erkrankungen der Nieren und Harnwege
Gelegentlich: Proteinurie.
Häufigkeit nicht bekannt: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
a Nach Markteinführung wurden nicht schwerwiegende und schwerwiegende Blutungsereignisse, darunter tödliche, beobachtet, die mit den Erfahrungen aus klinischen Studien übereinstimmen. Die häufigsten Blutungsereignisse umfassten rektale Blutung/Hämatochezie, Nasenbluten sowie Bluterguss. Tödliche Ereignisse betrafen gastrointestinale, intrakranielle und pulmonale Blutungen.sowie DIC.
b Umfasst Hypertonie, Blutdruck erhöht, hypertensive Krise und hypertensive Kardiomyopathie.
c Umfasst Abdominalschmerz, Schmerzen Oberbauch, Schmerzen Unterbauch, gastrointestinale Schmerzen, abdominaler Druckschmerz.
Andere chronische fibrotisierende interstitielle Lungenerkrankungen (ILD) mit einem progressiven Phänotyp
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Appetitverlust (11,1%).
Häufig: Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Häufigkeit nicht bekannt: Posteriores reversibles Enzephalopathie-Syndrom (PRES).
Herzerkrankungen
Gelegentlich: Myokardinfarkt.
Gefässerkrankungen
Häufig: Hypertonie, Blutungen.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (59%), Übelkeit (24%), abdominelle Schmerzen (11%), Erbrechen (12%).
Gelegentlich: Pankreatitis, Kolitis.
Häufigkeit nicht bekannt: gastrointestinale Perforation, ischämische Kolitis (Beobachtung nach Markteinführung).
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (18,4%), Erhöhung der Alaninaminotransferase (ALT) (10,8%).
Häufig: Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT), Erhöhung der Alkalischen Phosphatase (ALP) im Blut, Arzneimittelbedingter Leberschaden.
Gelegentlich: Hyperbilirubinämie.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag.
Gelegentlich: Pruritus, Alopezie.
Erkrankungen der Nieren und Harnwege
Gelegentlich: Proteinurie.
Häufigkeit nicht bekannt: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Häufig: Appetitverlust, Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Häufigkeit nicht bekannt: Posteriores reversibles Enzephalopathie-Syndrom (PRES).
Herzerkrankungen
Häufigkeit nicht bekannt: Myokardinfarkt.
Gefässerkrankungen
Häufig: Hypertonie, Blutungen.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (68,4%), Übelkeit (24,7%), abdominelle Schmerzen (11,8%), Erbrechen (17,7%).
Gelegentlich: Kolitis.
Häufigkeit nicht bekannt: Pankreatitis, gastrointestinale Perforation, ischämische Kolitis (Beobachtung nach Markteinführung).
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (11,1%).
Häufig: Erhöhung der Alaninaminotransferase (ALT), Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT), Erhöhung der Alkalischen Phosphatase (ALP) im Blut.
Gelegentlich: Arzneimittelbedingter Leberschaden.
Häufigkeit nicht bekannt: Hyperbilirubinämie.
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Hautausschlag, Pruritus.
Häufigkeit nicht bekannt: Alopezie.
Erkrankungen der Nieren und Harnwege
Gelegentlich: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Häufigkeit nicht bekannt: Proteinurie.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs gibt kein spezifisches Antidot und keine spezifische Behandlung für eine Überdosierung mit Ofev. Die höchste in Phase-I-Studien verabreichte Nintedanib-Einzeldosis betrug 450 mg einmal täglich. Darüber hinaus erhielten 2 Patienten bis zu acht Tage lang eine Überdosierung mit maximal 600 mg zweimal täglich. Die beobachteten unerwünschten Ereignisse standen im Einklang mit dem bekannten Sicherheitsprofil von Nintedanib, d.h. mit erhöhten Leberenzymen und gastrointestinalen Symptomen. Beide Patienten erholten sich von diesen Nebenwirkungen.
In den INPULSIS-Studien (IPF-Patienten) erhielt ein Patient versehentlich insgesamt 21 Tage lang eine Dosis von 600 mg pro Tag. In der Phase der fehlerhaften Dosierung trat ein nicht-schwerwiegendes unerwünschtes Ereignis (Nasopharyngitis) auf und klang wieder ab. Es traten keine weiteren beschriebenen Ereignisse auf.
Im Fall einer Überdosierung sollte die Behandlung unterbrochen werden und es sollten geeignete allgemeine supportive Massnahmen eingeleitet werden.
Eigenschaften/WirkungenATC-Code
L01EX09
Wirkungsmechanismus
Nintedanib ist ein Tyrosinkinaseinhibitor vom Typ der kleinen Moleküle. Zu den inhibierten Tyrosinkinasen gehören die Rezeptoren Platelet-Derived Growth Factor-Receptor (PDGFR) α und β, Fibroblastenwachstumsfaktorrezeptor (FGFR) 1-3 und Vaskulärer Endothelialer Wachstumsfaktorrezeptor (VEGFR) 1-3. Nintedanib bindet kompetitiv an die ATP-Bindungstasche dieser Rezeptoren und blockiert die intrazelluläre Signalgebung, die für die Proliferation, Migration und Transformation von Fibroblasten – die essentielle Pathomechanismen der IPF sind – entscheidend ist. Darüber hinaus hemmt Nintedanib die Kinasen Flt-3, Lck, Lyn und Src.
Pharmakodynamik
Die Aktivierung der FGFR- und PDGFR-Signalkaskaden ist ein entscheidender Faktor für die Proliferation und Migration von Lungenfibroblasten/Myofibroblasten, den kennzeichnenden Zellen der Pathologie der idiopathischen Lungenfibrose. Die potenzielle Bedeutung der Hemmung von VEGFR für die Pathologie der IPF wurde noch nicht vollständig aufgeklärt. Es wird angenommen, dass Nintedanib auf molekularer Ebene die FGFR- und PDGFR-Signalkaskaden hemmt, die die Proliferation und Migration von Lungenfibroblasten vermitteln, indem es an die Adenosintriphosphat-(ATP)Bindungstasche der intrazellulären Rezeptorkinasedomäne bindet und auf diese Weise die Kreuzaktivierung (über Autophosphorylierung der Rezeptor-Homodimere) beeinträchtigt.
In vitro hemmt Nintedanib seine Zielrezeptoren in Konzentrationen im niedrigen nanomolaren Bereich. An Lungenfibroblasten von Patienten mit IPF hemmte Nintedanib die durch PDGF, FGF und VEGF stimulierte Proliferation mit EC50-Werten von 11 nmol/l, 5,5 nmol/l bzw. unter 1 nmol/l. In Konzentrationen zwischen 100 und 1'000 nmol/l hemmte Nintedanib darüber hinaus die durch PDGF, FGF und VEGF stimulierte Fibroblastenmigration und die TGF-β2-induzierte Transformation von Fibroblasten in Myofibroblasten. Darüber hinaus wird angenommen, dass die antiinflammatorische Aktivität von Nintedanib die fibrotische Stimulation begrenzt, indem sie profibrotische Mediatoren wie IL-1β und IL-6 reduziert. Es wurde noch nicht geklärt, welchen Anteil die antiangiogene Aktivität von Nintedanib am Wirkmechanismus des Arzneimittels bei fibrotischen Lungenerkrankungen hat. In In-vivo-Studien hatte Nintedanib eine starke antifibrotische und antiinflammatorische Aktivität.
Klinische Wirksamkeit
Idiopathische Lungenfibrose (IPF)
Die klinische Wirksamkeit von Nintedanib wurde in einer Phase-II-Studie (TOMORROW) und zwei Phase-III-Studien (INPULSIS-1 und INPULSIS-2) bei 1231 Patienten mit IPF untersucht. Bei den genannten Studien handelte es sich um randomisierte, doppelblinde, placebokontrollierte Studien, in denen eine 52-wöchige Behandlung mit Ofev 150 mg zweimal täglich mit Placebo verglichen wurde.
Die Studien INPULSIS-1 und INPULSIS-2 hatten ein identisches Studiendesign und das Design der TOMORROW-Studie war sehr ähnlich. Die Patienten wurden in einem Verhältnis von 3:2 (in der TOMORROW-Studie 1:1) randomisiert einer 52-wöchigen Behandlung mit Ofev 150 mg oder Placebo, jeweils zweimal täglich, zugeteilt. Die TOMORROW-Studie besass darüber hinaus weitere Behandlungsarme (50 mg pro Tag, 50 mg zweimal täglich und 100 mg zweimal täglich), die hier nicht weiter besprochen werden.
Primärer Endpunkt war die jährliche Abnahme der forcierten Vitalkapazität (FVC). Die Änderung des Gesamtscores im Saint George's Respiratory Questionnaire (SGRQ) zwischen Studienbeginn und Woche 52 und die Zeit bis zur ersten akuten IPF-Exazerbation waren wichtige sekundäre Endpunkte der INPULSIS-Studien und sekundäre Endpunkte der TOMORROW-Studie.
Jährliche Abnahme der FVC
Die jährliche Abnahme der FVC (in ml) war bei mit Nintedanib behandelten Patienten signifikant geringer ausgeprägt als bei mit Placebo behandelten Patienten. Die Wirkung der Behandlung war in allen 3 Studien einheitlich. Tabelle 1 enthält die Ergebnisse der Einzelstudien und der gepoolten INPULSIS-Analyse.
Tabelle 1: Jährliche Abnahme der FVC (ml) in den Studien TOMORROW, INPULSIS-1 und INPULSIS-2 sowie der gepoolten INPULSIS-Analyse – behandelte Population2
|
|
TOMORROW
|
INPULSIS-1
|
INPULSIS-2
|
INPULSIS-1 und INPULSIS-2
gepoolt
| |
|
Placebo
|
Ofev 150 mg zweimal täglich
|
Placebo
|
Ofev 150 mg zweimal täglich
|
Placebo
|
Ofev 150 mg zweimal täglich
|
Placebo
|
Ofev 150 mg zweimal täglich
| |
Anzahl der ausgewerteten Patienten
|
83
|
84
|
204
|
309
|
219
|
329
|
423
|
638
| |
Rate 1,2 (SE) der Abnahme über 52 Wochen
|
-190 (36)
|
-60 (39)
|
−239,9 (18,71)
|
−114,7 (15,33)
|
−207,3 (19,31)
|
−113,6 (15,73)
|
−223,5 (13,45)
|
−113,6(10,98)
| |
Vergleich vs. Placebo
| |
Differenz1
|
|
131
|
|
125,3
|
|
93,7
|
|
109,9
| |
95%-KI
|
|
(27; 235)
|
|
(77,7; 172,8)
|
|
(44,8; 142,7)
|
|
(75,9; 144,0)
| |
p-Wert
|
|
0,01363
|
|
<0,0001
|
|
0,0002
|
|
<0,0001
| |
1
Berechnung auf Grundlage eines Regressionsmodells mit Zufallskoeffizienten.
2 Randomisierte Population in der TOMORROW-Studie; behandelte Population in INPULSIS-1, INPULSIS-2 und den gepoolten INPULSIS-Daten
3 Nominaler p-Wert.
|
Alle präspezifizierten Sensitivitätsanalysen bestätigten die Robustheit der Wirkung von Nintedanib im Sinne einer Reduktion der jährlichen Abnahme der FVC. Darüber hinaus wurden vergleichbare Auswirkungen auf andere Endpunkte der Lungenfunktion wie beispielsweise die Änderung der FVC in Woche 52 gegenüber dem Ausgangwert und die FVC-Responder-Analyse beobachtet, was die Wirkungen von Nintedanib im Sinne einer Verlangsamung des Fortschreitens der Erkrankung untermauert. Abbildung 1 zeigt den Verlauf der Änderung gegenüber dem Ausgangswert über die Zeit in beiden Behandlungsgruppen (auf Grundlage der gepoolten Analyse der Studien INPULSIS-1 und INPULSIS-2).
Abbildung 1: Mittlere (SEM) beobachtete Änderung der FVC gegenüber dem Ausgangswert (ml) über die Zeit, Studien INPULSIS-1 und INPULSIS-2 (gepoolte Daten)
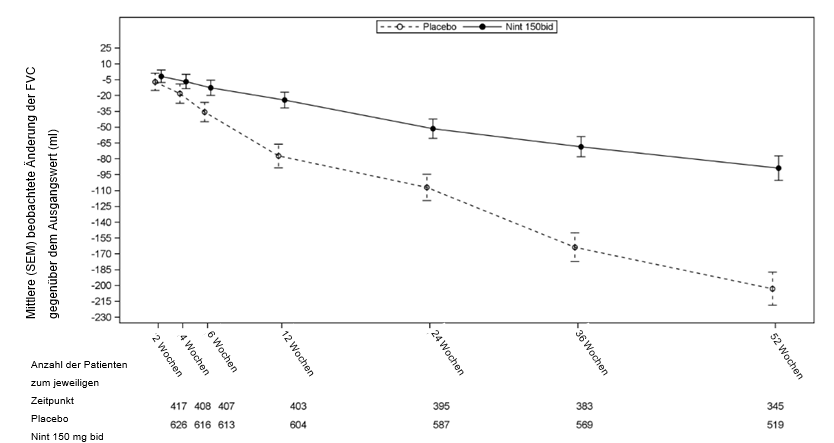
bid = zweimal täglich, SEM = Standardfehler des Mittelwerts
Änderung im SGRQ-Gesamtscore in Woche 52 gegenüber dem Ausgangswert
Der Gesamtscore im St. George's Respiratory Questionnaire (SGRQ) ist ein Mass für die gesundheitsbezogene Lebensqualität (HRQoL) und wurde in Woche 52 ausgewertet. In der TOMORROW-Studie betrug die geschätzte mittlere Änderung im SGRQ-Gesamtscore zwischen Studienbeginn (Baseline) und Woche 52 unter Placebo 5,46, was auf eine Verschlechterung der gesundheitsbezogenen Lebensqualität hinweist, und unter Nintedanib -0,66, was eine stabile gesundheitsbezogene Lebensqualität anzeigt. Die geschätzte mittlere Differenz zwischen Nintedanib und Placebo betrug -6,12 (95%-KI: -10,57; -1,67; p = 0,0071).
In der Studie INPULSIS-2 wiesen die mit Placebo behandelten Patienten einen ausgeprägteren Anstieg des SGRQ-Gesamtscores gegenüber dem Ausgangswert auf als die mit Nintedanib 150 mg zweimal täglich behandelten Patienten. Die Verschlechterung der HRQoL war in der Nintedanib-Gruppe geringer ausgeprägt. Die Differenz zwischen den Behandlungsgruppen war statistisch signifikant (-2,69; 95%-KI: -4,95; -0,43; p = 0,0197).
In der Studie INPULSIS-1 fiel der Anstieg des SGRQ-Gesamtscores in Woche 52 gegenüber dem Ausgangswert in der Nintedanib- und Placebogruppe vergleichbar aus (Differenz zwischen den Behandlungsgruppen: -0,05; 95%-KI: -2,50; 2,40; p = 0,9657). In der gepoolten Analyse der INPULSIS-Studien war die geschätzte mittlere Änderung im SGRQ-Gesamtscore zwischen Ausgangswert und Woche 52 in der Nintedanib-Gruppe (3,53) geringer als in der Placebogruppe (4,96). Die Differenz zwischen den Behandlungen betrug -1,43 (95%-KI: -3,09; 0,23; p = 0,0923). Insgesamt ist der über den SGRQ-Gesamtscore beurteilte Einfluss von Nintedanib auf die gesundheitsbezogene Lebensqualität moderat und weist auf eine geringer ausgeprägte Verschlechterung hin als unter Placebo.
Zeit bis zur ersten akuten IPF-Exazerbation
In den Studien TOMORROW und INPULSIS-2 war das Risiko für eine erste akute IPF-Exazerbation über den Zeitraum von 52 Wochen bei mit Nintedanib behandelten Patienten signifikant geringer als bei mit Placebo behandelten Patienten (Hazard Ratio [HR]: 0,16; 95%-KI: 0,04; 0,71; p = 0,0054) bzw. (HR: 0,38; 95%-KI: 0,19; 0,77; p = 0,005). In der Studie INPULSIS-1 gab es keine Differenz zwischen den Behandlungsgruppen (HR: 1,15; 95%-KI: 0,54; 2,42; p = 0,6728).
Überlebensanalyse
In der präspezifizierten gepoolten Analyse der Überlebensdaten der INPULSIS-Studien fiel die Gesamtmortalität über 52 Wochen in der Nintedanib-Gruppe (5,5 %) numerisch niedriger aus als in der Placebogruppe (7,8 %). Die Analyse der Zeit bis zum Tod ergab eine HR von 0,70 (95%-KI: 0,43; 1,12; p = 0,1399). Die Ergebnisse für alle Überlebensendpunkte (wie Mortalität während der Behandlung und respiratorische Mortalität) zeigten einheitlich numerische Differenzen zugunsten von Nintedanib, die jedoch keine statistische Signifikanz erreichten.
Langzeitbehandlung mit Ofev bei Patienten mit IPF (INPULSIS-ON)
In eine unverblindete Verlängerungsstudie zu Ofev wurden 734 Patienten mit IPF aufgenommen. Einige Patienten wurden für mehr als 5 Jahre mit Ofev behandelt. Patienten, welche die 52-wöchige Behandlungsphase in einer INPULSIS-Studie abgeschlossen haben, erhielten im Rahmen der Verlängerungsstudie INPULSIS-ON eine unverblindete Behandlung mit Ofev. Die mediane Expositionsdauer für Patienten, die sowohl in einer INPULSIS-Studie als auch in der INPULSIS-ON-Studie mit Ofev behandelt wurden, betrug 44,7 Monate (Bereich 11,9–68,3). Die adjustierte jährliche Rate der FVC-Abnahme über 192 Wochen lag bei −135,1 (5,8) ml/Jahr bei allen behandelten Patienten und deckte sich mit der jährlichen Rate der FVC-Abnahme bei Patienten, die in den INPULSIS-Studien der Phase III mit Ofev behandelt wurden (−113,6 ml pro Jahr). Das Profil unerwünschter Ereignisse von Ofev in der INPULSIS-ON-Studie war dem in den INPULSIS-Studien der Phase III ähnlich.
IPF-Patienten mit fortgeschrittener Beeinträchtigung der Lungenfunktion (INSTAGE)
Eine doppelblinde, randomisierte Parallelgruppenstudie zur Beurteilung der Wirksamkeit und Sicherheit von Ofev in Kombination mit oralem Sildenafil im Vergleich zur Behandlung mit Ofev allein bei 273 Patienten mit IPF und fortgeschrittener Beeinträchtigung der Lungenfunktion (DLCO < 35 % vorhergesagt) über 24 Wochen.
Die FVC-Abnahme bei Patienten, die mit Ofev allein behandelt wurden, stimmte mit der FVC-Abnahme bei Patienten mit weniger fortgeschrittenen Erkrankungen überein, die in den INPULSIS-Studien der Phase III mit Ofev behandelt wurden. Die Zugabe von Sildenafil zu Ofev brachte keinen signifikanten Nutzen in Bezug auf die Lebensqualität im Vergleich zu Ofev allein. Das Sicherheits- und Verträglichkeitsprofil von Ofev bei IPF-Patienten mit fortgeschrittener Beeinträchtigung der Lungenfunktion deckte sich mit den Profilen, die in den INPULSIS-Studien beobachtet wurden. Das Profil unerwünschter Ereignisse der Kombination aus Ofev und Sildenafil stimmte mit dem bekannten Sicherheitsprofil der einzelnen Komponenten überein, ohne einen Anstieg von schwerwiegenden oder tödlichen unerwünschten Ereignissen im Vergleich zu Ofev allein.
Weitere Daten aus der Phase-IV-Studie INJOURNEY mit zweimal täglicher Gabe von 150 mg Ofev und zusätzlich Pirfenidon:
In einer exploratorischen, unverblindeten, randomisierten Studie wurde die gemeinsame Gabe von 150 mg Nintedanib zweimal täglich plus Pirfenidon (Titration auf 801 mg dreimal täglich) im Vergleich zur alleinigen Gabe von 150 mg Nintedanib zweimal täglich bei 105 Patienten über 12 Wochen untersucht. Primärer Endpunkt war der Prozentsatz an Patienten mit unerwünschten gastrointestinalen Ereignissen von Studienbeginn bis Woche 12. Gastrointestinale unerwünschte Ereignisse traten häufig auf, was mit dem belegten Sicherheitsprofil für jede der beiden Substanzen übereinstimmt. Diarrhoe, Übelkeit und Erbrechen waren die häufigsten unerwünschten Ereignisse, die bei 20 (37,7%) gegenüber 16 (31,4%), 22 (41,5%) gegenüber 6 (11,8%) bzw. 15 (28,3%) gegenüber 6 (11,8%) der Patienten nach Behandlung mit Pirfenidon zusätzlich zu Nintedanib gegenüber Nintedanib allein gemeldet wurden.
Die mittleren (SE) absoluten Änderungen der FVC gegenüber den Ausgangswerte nach 12 Wochen waren −13,3 (17,4) mL in Patienten, welche mit Pirfenidon zusätzlich zu Nintedanib behandelt wurden (n=48) gegenüber −40,9 (31,4) mL in Patienten, welche mit Nintedanib allein behandelt wurden (n=44).
Chronisch fibrosierende Interstitielle Lungenerkrankungen (ILD) mit einem progressiven Phänotyp
Die klinische Wirksamkeit von OFEV bei Patienten mit chronisch fibrosierender ILD mit einem progressiven Phänotyp wurde in einer randomisierten, doppelblinden, Placebo-kontrollierten Phase 3 Studie (INBUILD) untersucht.
Insgesamt wurden n=663 Patienten 1:1 randomisiert. Sie erhielten entweder OFEV 2x150mg pro Tag oder Placebo für mindestens 52 Wochen.
Die Randomisierung wurde stratifiziert gemäss Fibrosemuster in der hochauflösenden Computertomographie (HRCT). N=412 Patienten mit einem UIP (usual interstitial pneumonia)-artigen Fibrosemuster und n=251 Patienten mit anderen Fibrosemustern wurden randomisiert. Für die Studienauswertung wurden 2 co-primäre Populationen definiert: die Gesamtpopulation und die Population mit UIP-artigem Fibrosemuster gemäss HRCT.
Der primäre Studienendpunkt war die jährliche Abnahme der FVC (ml) über 52 Wochen. Weitere Studienendpunkte waren beispielsweise die Zeit bis zur ersten akuten ILD Exazerbation oder zum Tod oder die Zeit bis zum Tod.
Patienten mit der Diagnose einer chronisch fibrosierenden ILD wurden für die Studie berücksichtigt, wenn sie im HRCT eine relevante Fibrosierung aufwiesen (>10% Fibrosierung) und klinische Merkmale einer Krankheitsprogression (definiert als Abnahme der FVC ≥10%, Abnahme der FVC zwischen 5 und 10% mit Verschlechterung in Krankheitssymptomen ODER Bildgebung oder Verschlechterung in Krankheitssymptomen UND Bildgebung) über die vorausgehenden 24 Monate trotz adäquater Behandlung aufwiesen. Die FVC musste ≥45% predicted und die DLCO zwischen 30% und 80% predicted betragen.
Von einer Teilnahme an der Studie ausgeschlossen waren Patienten mit
·IPF, relevanter Bronchokonstriktion (z.B. FEV1/FVC <0,7 vor Bronchodilatation) oder signifikanter pulmonaler Hypertonie
·Erhöhungen der Transaminasen oder des Bilirubins >1,5 ULN
·bekanntem Blutungsrisiko, voller Antikoagulation, kürzlichem Myokardinfarkt oder Schlaganfall
·vorausgegangener Behandlung mit Nintedanib, Pirfenidon, Azathioprin, Cyclosporin, Mycophenolat, Tacrolimus, oralen Kortikoiden >20mg/Tag oder oralen Kortikoiden+Azathioprin+N-Acetylcystein innert 4 Wochen vor Randomisierung*
·vorausgegangener Behandlung mit Cyclophosphamid innert 8 Wochen vor Randomisierung*
·vorausgegangener Behandlung mit Rituximab innert 6 Monaten vor Randomisierung*
* der Einsatz der genannten Medikamente war ab 6 Monaten nach Studienbeginn wieder gestattet, soweit klinisch indiziert
Die Studienteilnehmer waren im Wesentlichen Kaukasier (74%) und Asiaten (25%). 54% der Patienten waren männlich, 49% hatten niemals geraucht, das mittlere Alter betrug 66 Jahre und die FVC betrug im Mittel 69% predicted. Die zugrundeliegenden ILDs waren Hypersensitivitätspneumonien (26%), autoimmun-assoziierte ILDs (26%), idiopathische nicht-spezifische interstitielle Pneumonie (19%), unklassifizierbare idiopathische interstitielle Pneumonie (17%) und andere ILDs (12%).
Jährliche Abnahme der FVC (ml)
Über 52 Wochen wurde eine statistisch signifikante Reduktion in der Abnahme der FVC unter der Behandlung mit OFEV versus Placebo beobachtet. Der Unterschied von OFEV zu Placebo betrug 107ml (Tabelle 2).
Tabelle 2: Jährliche Abnahme der FVC (ml, primärer Studienendpunkt INBUILD Studie)
|
|
Gesamtpopulation
|
Subpopulation
UIP-artig
|
Subpopulation
Andere Fibrosemuster in der HRCT
| |
|
OFEV
|
Placebo
|
OFEV
|
Placebo
|
OFEV
|
Placebo
| |
Anzahl der ausgewerteten Patienten
|
332
|
331
|
206
|
206
|
126
|
125
| |
Ratea (SE) der Abnahme über 52 Wochen
|
-80,8
|
-187,8
|
-82,9
|
-211,1
|
-79,0
|
-154,2
| |
Vergleich vs Placebo
Differenza
|
107,0
|
128,2
|
75,2*
| |
95%-KI
|
(65,4; 148.5)
|
(70,8; 185,6)
|
(15,5; 135,0)*
| |
p-Wert
|
< 0,0001
|
< 0,0001
|
| |
* Ein auf der Subpopulation mit anderen Fibrosemustern in der HRCT beruhender Vergleich wurde nicht in die multiple Testprozedur aufgenommen. Die hier gezeigten Werte dienen beschreibenden Zwecken.
a Basierend auf einem Regressionsmodell mit zufälligen Koeffizienten und den festen kategorialen Effekten von Behandlung, HRCT-Muster, den festen kontinuierlichen Effekten von Zeit, FVC-Ausgangswert (ml) und einschliesslich der Interaktion zwischen Therapie und Zeit und der Interaktion zwischen Ausgangswert und Zeit.
|
Abbildung 2 zeigt die zeitliche Entwicklung der FVC-Veränderung gegenüber Studienbeginn in den Behandlungsgruppen.
Abbildung 2. Mittlere (SEM) beobachtete Veränderung der FVC (ml) gegenüber Studienbeginn über 52 Wochen
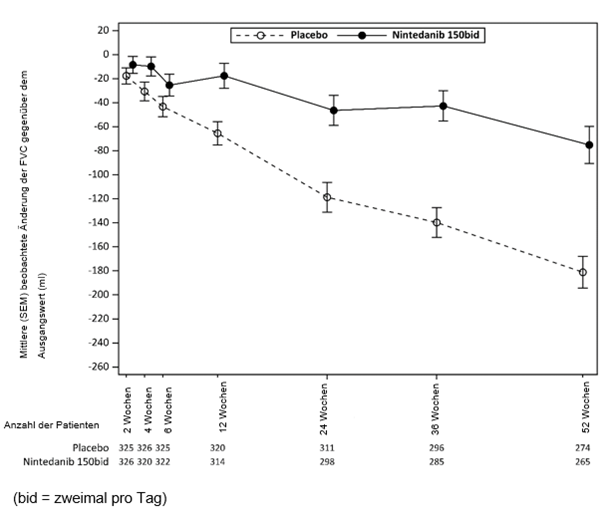
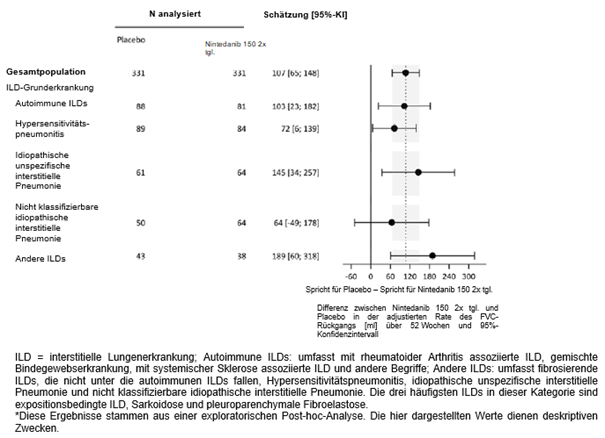
Eine post-hoc durchgeführte exploratorische Analyse nach ILD-Diagnose wurde durchgeführt (Abbildung 3). Insgesamt wurde ein konsistenter Behandlungseffekt in den unterschiedlichen ILD-Diagnosen gezeigt.
Abbildung 3: Jährliche Rate des FVC-Rückgangs (ml) über 52 Wochen basierend auf ILD-Grunddiagnose in Studie 5*
Prozentuale Abnahme der FVC
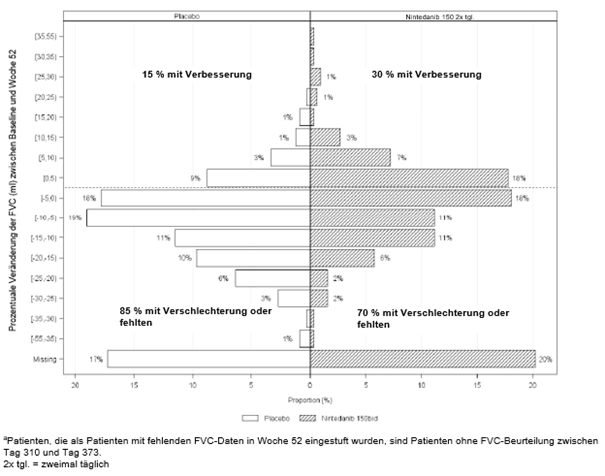
Abbildung 4 zeigt die prozentuale Veränderung der FVC (ml) von Baseline bis Woche 52. Für die Mehrzahl der Patienten fiel die Abnahme der Lungenfunktion unter OFEV geringer aus als unter Placebo.
Abbildung 4: Histogramm der prozentualen Veränderung der FVC (ml) von Baseline bis Woche 52 nach Behandlung mit prozentualen Inkrementen oder Dekrementen in 5er-Schritten (Studie 5)a
Zeit bis zur ersten akuten ILD-Exazerbation oder zum Tod
Das Risiko einer ersten akuten ILD-Exazerbation oder des Todes verminderte sich in der Ofev-Gruppe gegenüber der Placebogruppe über 52 Wochen (HR Ofev versus Placebo: 0,80; 95%-KI: 0,48; 1,34) oder über die gesamte Studiendauer (HR Ofev versus Placebo: 0,67; 95%-KI: 0,46; 0,98), was einer Reduktion des Risikos für eine erste akute ILD-Exazerbation oder den Tod um 33 % bei Patienten unter Ofev im Vergleich zu Placebo entspricht.
Überlebensanalyse
In der INBUILD-Studie wurden die Überlebensdaten unter Ofev im Vergleich zu Placebo ausgewertet, um den primären Endpunkt (FVC) zu untermauern. Die Mortalität jeglicher Ursache wurde über die gesamte Studiendauer und den verfügbaren Nachbeobachtungszeitraum beurteilt, unabhängig von der Todesursache und davon, ob die Behandlung weitergeführt wurde oder nicht. Die Analyse ergab keinen statistisch signifikanten Unterschied über 52 Wochen (HR OFEV versus Placebo 0,94: 95%-KI 0,47; 1,86) bzw. über die gesamte Studiendauer (HR OFEV versus Placebo 0,78; 95%-KI 0,5; 1,21).
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Die klinische Wirksamkeit von Nintedanib wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie (SENSCIS) bei Patienten mit SSc-ILD untersucht.
Insgesamt wurden 580 Patienten in einem Verhältnis von 1:1 randomisiert um mindestens 52Wochen entweder Ofev 150 mg zweimal täglich oder Placebo zu erhalten. Die Randomisierung wurde nach Anti-Topoisomerase-Antikörper-(ATA)-Status stratifiziert. Einzelne Patienten setzten die verblindete Studienbehandlung bis zu 100 Wochen lang fort.
Die Diagnose der Patienten mit SSc-ILD beruhte auf den Klassifikationskriterien für SSc des American College of Rheumatology / der European League Against Rheumatism, wenn das Auftreten der Erkrankung (erstes Nicht-Raynaud-Symptom) weniger als 7 Jahre zurücklag und eine mindestens 10 %ige Lungenfibrose vorlag, ermittelt anhand eines in den vorausgegangenen 12 Monaten durchgeführten hochauflösenden Computertomografie (HRCT)-Scans.
Die FVC musste bei den Patienten mindestens 40 % des geschätzten Wertes betragen, während die DLCO 30-89 % des geschätzten Wertes betragen musste. Patienten mit relevanten Atemwegsobstruktionen (d.h. FEV1/FVC vor Bronchodilatation unter 0,7) oder einer vorherigen oder anstehenden hämatopoetischen Stammzelltransplantation durften nicht an der Studie teilnehmen. Weitere Ausschlusskriterien waren erhöhte ALT-, AST- oder Bilirubin-Werte (>1,5xOGN), ein erhöhtes Risiko für Blutungen (einschliesslich therapeutische Antikoagulation), kürzlich erfolgter Myokardinfarkt oder Schlaganfall, eine signifikante pulmonale Hypertonie, Geschwüre an mindestens drei Fingerkuppen, eine schwere Fingernekrose mit erforderlicher Hospitalisierung in der Anamnese sowie eine sklerodermale renale Krise in der Anamnese. Patienten wurden ebenfalls ausgeschlossen, wenn sie andere Prüfmedikamente erhielten, bereits mit Nintedanib oder Pirfenidon behandelt worden waren und 8 Wochen vor Randomisierung Azathioprin oder 6 Monate vor Randomisierung Cyclophosphamid oder Cyclosporin A erhalten hatten. Eine stabile Therapie mit Mycophenolat oder Methotrexat sowie Prednison ≤10 mg/Tag oder Äquivalent war zulässig.
Primärer Endpunkt war die jährliche Abnahme der forcierten Vitalkapazität (FVC) über 52 Wochen.
Wichtige sekundäre Endpunkte waren die absolute Veränderung des modifizierten Rodnan Skin Score (mRSS) in Woche 52 gegenüber dem Ausgangswert, die absolute Veränderung des Gesamtscores im Saint George's Respiratory Questionnaire (SGRQ) in Woche 52 gegenüber dem Ausgangswert sowie die Mortalität während der gesamten Studie.
75,2 % der Patienten waren weiblich. Die Patienten waren überwiegend Kaukasier (67 %), Asiaten (25 %) oder hatten eine schwarze Hautfarbe (6 %). Das mittlere (Standardabweichung [SD], Min.-Max.) Alter betrug 54,0 (12,2, 20–79) Jahre. Insgesamt litten 51,9 % der Patienten an einer diffusen kutanen systemischen Sklerose (SSc) und 48,1 % an einer limitierten kutanen SSc. Die mittlere (SD) Zeit seit dem ersten Auftreten eines Nicht-Raynaud-Symptoms betrug 3,49 (1,7) Jahre. 49,0 % der Patienten erhielten zu Studienbeginn eine stabile Therapie mit Mycophenolat (46.5% Mycophenolat-Mofetil, 1.9% Mycophenolat-Natrium, 0.5% Mycophenolsäure) und das bei Patienten mit bzw. ohne Mycophenolat beobachtete Sicherheitsprofil war vergleichbar.
Jährliche Abnahme der FVC
Die jährliche Abnahme der FVC (in ml) über 52 Wochen wurde bei mit Ofev behandelten Patienten um 41,0 ml im Vergleich zu Placebo signifikant reduziert (siehe Tabelle 3), was einem relativen Behandlungseffekt von 43,8% entspricht.
Tabelle 3: Jährliche Abnahme der FVC (ml) über 52 Wochen
|
|
Placebo
|
Ofev
150 mg zweimal täglich
| |
Anzahl der ausgewerteten Patienten
|
288
|
287
| |
Rate1 (SE) der Abnahme über 52 Wochen
|
-93,3 (13,5)
|
-52,4 (13,8)
| |
Vergleich vs. Placebo
| |
Differenz1
|
|
41,0
| |
95%-KI
|
|
(2,9, 79,0)
| |
p-Wert
|
|
<0,05
| |
1
Basierend auf einer Regression mit zufälligen Koeffizienten, adjustiert für Behandlung, Geschlecht, Grösse, Alter, ATA status, FVC-Ausgangswert, FVC*Zeit und Behandlung*Zeit
|
Eine explorative Analyse der Daten bis zu 100 Wochen (maximale Behandlungsdauer in der SENSCIS-Studie) wies darauf hin, dass die Wirkung unter einer Behandlung mit Ofev auf die Verlangsamung der Progression der SSc-ILD über 52 Wochen hinaus anhielt.
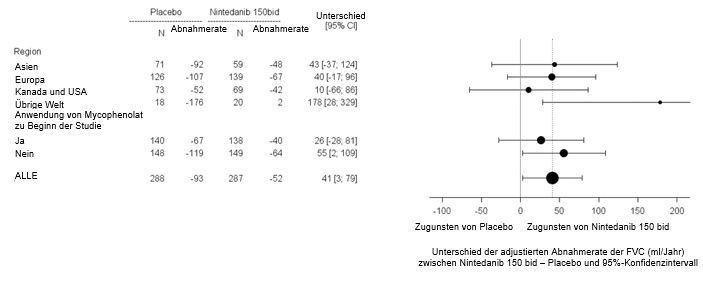
In zwei vorgegebenen Subgruppen-Wirksamkeitsanalysen wurde der mittlere Behandlungsunterschied in Bezug auf die Abnahme der FVC in Woche 52 bei den Patienten nach Region und Anwendung von Mycophenolat untersucht (siehe Abbildung 5).
Abbildung 5: Subgruppenanalyse des mittleren Behandlungsunterschiedes in Bezug auf die Abnahme der FVC (ml) in Woche 52 nach Region und Anwendung von Mycophenolat (SENSCIS)
Prozentuale Änderung der forcierten Vitalkapazität ab Studienbeginn
In Abbildung 6 wird für die SENSCIS Studie die prozentuale Änderung der FVC in ml in Woche 52 gegenüber dem Ausgangswert gezeigt. Die Verschlechterung der Lungenfunktion war bei der Mehrheit der Patienten unter Ofev geringer ausgeprägt als unter Placebo.
Abbildung 6: Histogramm der prozentualen Änderung der FVC (ml) in Woche 52 gegenüber dem Ausgangswert je nach Behandlung und prozentualer Zunahme oder Abnahme um 5 (SENSCIS)a
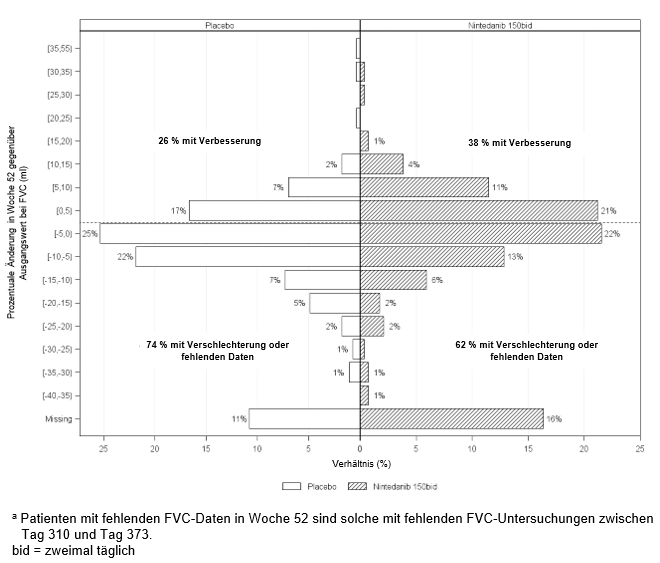
Abbildung 7: Mittlere (SEM) beobachtete Änderung der FVC gegenüber dem Ausgangswert (ml) über 52 Wochen
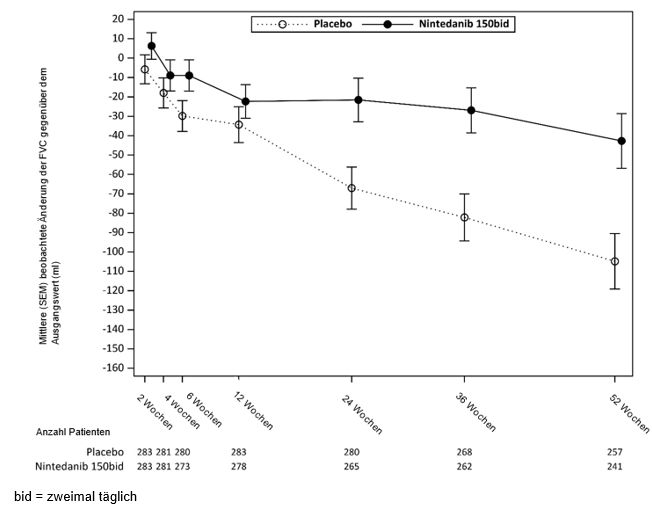
Tabelle 4: Jährliche Abnahme der FVC (% des Sollwerts) über 52 Wochen
|
|
Placebo
|
Ofev
150 mg zweimal täglich
| |
Anzahl der ausgewerteten Patienten
|
288
|
287
| |
Rate1 (SE) der Abnahme über 52 Wochen
|
-2,6 (0,4)
|
-1,4 (0,4)
| |
Vergleich vs. Placebo
| |
Differenz1
|
|
1,15
| |
95%-KI
|
|
(0,09, 2,21)
| |
p-Wert
|
|
<0,05
| |
1
Basierend auf einer Regression mit zufälligen Koeffizienten und den festen kategorialen Effekten von Behandlung, ATA-Status, den festen kontinuierlichen Effekten von Zeit, FVC-Ausgangswert [% des Sollwerts] und einschliesslich der Interaktion zwischen Therapie und Zeit und der Interaktion zwischen Ausgangswert und Zeit. Als zufälliger Effekt wurden patientenspezifisch Intercept und Zeit angegeben. Die Messfehler-Varianz (within-patient error) innerhalb eines Patienten wurde durch eine unstrukturierte Varianz-Kovarianz-Matrix modelliert. Die interindividuelle Variabilität wurde durch eine Varianz-Kovarianz-Matrix der Varianzkomponenten modelliert.
|
Änderung des modifizierten Rodnan Skin Scores (mRSS) in Woche 52 gegenüber dem Ausgangswert
Die adjustierte mittlere absolute Veränderung des mRSS in Woche 52 gegenüber dem Ausgangswert war zwischen der Ofev-Gruppe (-2,17 (95%-KI -2,69, -1,65)) und der Placebogruppe (-1,96 (95%-KI -2,48, -1,45)) vergleichbar. Die adjustierte mittlere Differenz zwischen den Behandlungsgruppen betrug -0,21 (95%-KI -0,94, 0,53; p = 0,5785).
Änderung des Gesamtscores im St. George's Respiratory Questionnaire (SGRQ) in Woche 52 gegenüber dem Ausgangswert
Die adjustierte mittlere absolute Veränderung des SGRQ-Gesamtscores in Woche 52 gegenüber dem Ausgangswert war zwischen der Ofev-Gruppe (0,81 (95%-KI -0,92, 2,55)) und der Placebogruppe (-0,88 (95%-KI -2,58, 0,82)) vergleichbar. Die adjustierte mittlere Differenz zwischen den Behandlungsgruppen betrug 1,69 (95%-KI -0,73, 4,12; p = 0,1711).
Überlebensanalyse
Über die gesamte Studie war die Mortalität zwischen der Ofev-Gruppe (N = 10; 3,5 %) und der Placebogruppe (N = 9; 3,1 %) vergleichbar. Über die gesamte Studie ergab die Analyse der Zeit bis zum Tod eine HR von 1,16 (95%-KI 0,47, 2,84; p = 0,7535).
Wirkung auf das QT-Intervall
In einer speziellen Studie zu Patienten mit Nierenzellkarzinom zeigten QT/QTc-Messungen, dass eine orale Nintedanib Einzeldosis zu 200 mg und mehrmalige orale Nintedanib-Dosen zu 200 mg zweimal täglich für 15 Tage keine Verlängerung des QTcF-Intervalls zur Folge hatten. Eine vollständige Thorough-QT-Study wurde mit Nintedanib nicht durchgeführt.
Studien mit Kindern und Jugendlichen
Nintedanib sollte bei Kindern und Jugendlichen Patienten mit chronisch fibrosierenden interstitiellen Lungenerkrankungen (ILDs) nicht angewendet werden.
PharmakokinetikDie pharmakokinetischen Eigenschaften von Nintedanib waren bei gesunden Probanden, Patienten mit IPF, SSc-ILD, ILDs und Patienten mit Krebserkrankung vergleichbar.
Die Pharmakokinetik von Nintedanib ist über die Zeit linear. Es wurde eine Dosisproportionalität mit steigender Nintedanib-Exposition bei steigenden Dosen nachgewiesen (Dosisspanne von 50 bis 450 mg einmal täglich und 150 bis 300 mg zweimal täglich). Die Akkumulation betrug nach mehrmaliger Verabreichung bei IPF Patienten in Bezug auf AUCτ das 1,76-fache. Steady-State-Plasmakonzentrationen wurden nach 1-wöchiger Verabreichung erreicht. Die Nintedanib-Talspiegel blieben über mehr als ein Jahr stabil. Die Pharmakokinetik von Nintedanib hat eine moderate bis hohe interindividuelle Variabilität (Variationskoeffizient für pharmakokinetische Standardparameter im Bereich von 30 % bis 70 %). Die intraindividuelle Variabilität ist niedrig bis moderat (Variationskoeffizient unter 40 %).
Absorption
Maximale Nintedanib-Plasmakonzentrationen wurden nach oraler Einnahme von Weichgelatinekapseln zusammen mit einer Mahlzeit innerhalb von etwa 2 4 Stunden erreicht (Spanne: 0,5 8 h). Die absolute Bioverfügbarkeit einer 100-mg-Dosis betrug bei gesunden Probanden 4,69 % (90%-KI: 3,62-6,08). Resorption und Bioverfügbarkeit werden durch den Einfluss von Transportern (P-gp), und einen substantiellen First-Pass-Metabolismus und evtl. durch schlechte Löslichkeit von Nintedanib bei neutralem pH-Wert verringert.
Nach Nahrungsaufnahme stieg die Nintedanib-Exposition gegenüber der Verabreichung im Nüchternzustand um etwa 20 % an (KI: 95,3-152,5 %) und die Resorption verlief verzögert (mediane tmax im Nüchternzustand: 2,00 h; nach Nahrungsaufnahme: 3,98 h).
Die In-vitro Studie hat gezeigt, dass wenn die Kapsel (alle Dosisstärken) bis 15 Minuten in Apfelmus oder Schokoladepudding verweilt, kein relevanter Einfluss auf die Integrität der Kapsel sowie auf deren Wirkstofffreisetzung, die potenziellen Abbauprodukte und die Menge an Wirkstoff in der Kapsel gemessen werden kann, und deshalb auch kein Einfluss auf die klinische Wirksamkeit zu erwarten ist.
Distribution
Nintedanib folgt einer zumindest biphasischen Verteilungskinetik. Nach intravenöser Infusion wurde ein hohes Verteilungsvolumen beobachtet (Vss: 1050 Liter).
Die Proteinbindung von Nintedanib in menschlichem Plasma war in vitro hoch (97,8 %). Es wird angenommen, dass die Bindung vorwiegend an Serumalbumin erfolgt. Nintedanib ist vorwiegend im Plasma verteilt mit einem Blut-Plasma-Verhältnis von 0,869.
Metabolismus
Hauptmetabolisierungsweg von Nintedanib ist eine hydrolytische Spaltung durch Esterasen, die zur Bildung der freien Säure BIBF 1202 führt. Anschliessend wird BIBF 1202 durch Enzyme (namentlich UGT 1A1, UGT 1A7, UGT 1A8 und UGT 1A10) in BIBF-1202-Glucuronid umgewandelt.
Nur ein kleiner Teil der Biotransformation von Nintedanib verläuft über CYP-Enzyme, vorwiegend über CYP 3A4. In der ADME-Studie entfiel der wesentliche Anteil der Radioaktivität im menschlichen Plasma auf Nintedanib (24 %), BIBF 1202 (32 %) und BIBF-1202-Glucuronid (30 %). In der ADME-Studie konnten die CYP-abhängigen Hauptmetaboliten im menschlichen Plasma nicht nachgewiesen werden. In vitro macht der CYP-abhängige Metabolismus etwa 5 % des Umsatzes aus, während unter den gegebenen experimentellen Bedingungen etwa 25 % auf die Esterspaltung entfielen.
Elimination
Die Gesamtplasmaclearance war nach intravenöser Infusion hoch (CL: 1390 ml/min). Innerhalb von 48 Stunden wurden nach oraler Gabe etwa 0,05 % und nach intravenöser Gabe etwa 1,4 % der Dosis als unveränderter Wirkstoff im Urin ausgeschieden. Die renale Clearance betrug 20 ml/min.
Ausscheidung
Nach oraler Verabreichung von [14C]-Nintedanib war der Haupteliminationsweg für arzneimittelgebundene Radioaktivität die Exkretion über Fäzes/Galle (93,4% der Dosis). Der grösste Teil von Ofev wurde in Form von BIBF 1202 über die Fäzes ausgeschieden (58% der Dosis); 20% wurden als Muttersubstanz und weniger als 7 % in Form von unterschiedlichen weniger wichtigen Metaboliten ausgeschieden. Der Beitrag der renalen Exkretion an der Gesamtclearance war niedrig (0,649% der Dosis). Die Wiederfindung wurde innerhalb von 4 Tagen nach der Verabreichung als vollständig bewertet (mehr als 90%). Die terminale Halbwertzeit von Nintedanib betrug 10-15 Stunden.
Beziehung Exposition/Ansprechen
An den Daten der Phase-II-Studie zur IPF durchgeführte explorative Analysen zur Beziehung zwischen Pharmakokinetik und unerwünschten Ereignissen zeigten, dass eine höhere Nintedanib-Exposition tendenziell mit einem Anstieg von Leberenzymen verbunden war (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Kinetik spezieller Patientengruppen, Intrinsische und extrinsische Faktoren
Den Ergebnissen einer populationspharmakokinetischen Analyse zu Patienten mit IPF und nicht-kleinzelligem Bronchialkarzinom (NSCLC) (N = 1191) sowie deskriptiven Untersuchungen zufolge hatten die Faktoren Geschlecht (für das Körpergewicht korrigiert), leichte und mittelschwere Einschränkung der Nierenfunktion (auf Grundlage der Creatinin-Clearance geschätzt), Alkoholkonsum und P-gp-Genotyp keinen Einfluss auf die Nintedanib-Exposition. Die populationspharmakokinetische Analyse zeigte einen moderaten Einfluss der weiter unten beschriebenen Faktoren Lebensalter, Körpergewicht und ethnische Abstammung auf die Nintedanib-Exposition. Vor dem Hintergrund der beobachteten hohen interindividuellen Variabilität kann aufgrund der moderaten Effekte dieser Kovariablen keine Empfehlung für eine Dosisanpassung gegeben werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Lebensalter
Die Nintedanib-Exposition stieg mit dem Alter linear an. Die AUCτ,ss fiel bei einem 45 Jahre alten Patienten (5. Perzentil) um 16 % niedriger und bei einem 76 Jahre alten Patienten (95. Perzentil) um 13 % höher aus als bei einem Patienten mit einem Alter von 62 Jahren (medianes Alter). Die Alterspanne der Analyse betrug 29 bis 85 Jahre und etwa 5 % der Population war älter als 75 Jahre.
Es wurden keine Studien zu Kindern und Jugendlichen durchgeführt.
Körpergewicht
Es wurde eine inverse Korrelation zwischen dem Körpergewicht und der Nintedanib-Exposition beobachtet. Die AUCτ,ss fiel bei einem 50 kg schweren Patienten (5. Perzentil) um 25 % höher und bei einem 100 kg schweren Patienten (95. Perzentil) um 19 % niedriger aus als bei einem Patienten mit einem Körpergewicht von 71,5 kg (medianes Körpergewicht).
Ethnische Abstammung
Der Populationsmittelwert der Nintedanib-Exposition war bei Chinesen, Taiwanern und Indern um 33-50 % höher und bei japanischen Patienten um 16 % höher, während er bei Koreanern um 16-22 % niedriger war als bei Patienten kaukasischer Abstammung (für das Körpergewicht korrigiert). Daten zu schwarzen Patienten liegen nur sehr begrenzt vor, bewegen sich jedoch im gleichen Bereich wie bei Patienten kaukasischer Abstammung.
Raucher
In der populationspharmakokinetischen Analyse fiel die Nintedanib-Exposition bei aktuellen Rauchern um 21 % niedriger aus als bei ehemaligen Rauchern und lebenslangen Nichtrauchern. Ein Effekt dieses Ausmasses rechtfertigt keine Dosisanpassung.
Eingeschränkte Leberfunktion
In einer speziellen Phase-I-Studie mit Einzeldosis und verglichen mit gesunden Teilnehmern war die Nintedanib-Exposition basierend auf der Cmax und der AUC bei Freiwilligen mit leichter Einschränkung der Leberfunktion (Child-Pugh-Klasse A; 90%-KI 1,3 – 3,7 für Cmax bzw. 1,2 – 3,8 für AUC) um das 2,2-Fache erhöht. Bei Freiwilligen mit moderater Einschränkung der Leberfunktion (Child-Pugh-Klasse B) war, verglichen mit gesunden Freiwilligen, die Exposition um das 7,6-Fache basierend auf der Cmax (90%-KI 4,4 – 13,2) bzw. um das 8,7-Fache (90%-KI 5,7 – 13,1) basierend auf der AUC erhöht. Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) wurden nicht untersucht.
Eingeschränkte Nierenfunktion
Die Pharmakokinetik von Nintedanib wurde nicht in einer speziellen PK Studie in Patienten mit eingeschränkter Nierenfunktion untersucht. Den Ergebnissen der populationspharmakokinetischen Analyse zufolge, hatte eine leichte (CrCl: 60 bis 90 ml/min) oder moderate (CrCl: 30 bis 60 ml/min) Einschränkung der Nierenfunktion keinen Einfluss auf die Nintedanib-Exposition. Es liegen nur begrenzt Daten zu Patienten mit schwerer Einschränkung der Nierenfunktion vor (CrCl unterhalb von 30 ml/min).
Präklinische DatenAllgemeine Toxikologie
Untersuchungen zur Einzeldosis-Toxizität an Ratten und Mäusen weisen auf ein niedriges Potential für eine akute Toxizität von Nintedanib hin. In Untersuchungen zur Toxizität bei wiederholter Gabe an Ratten zeigten die meisten Nebenwirkungen (z.B. Verdickung der Epiphysenfuge, Läsionen an den Schneidezähnen) einen Zusammenhang mit dem Wirkmechanismus von Nintedanib (d.h. VEGFR-2-Hemmung). Derartige Veränderungen sind von anderen VEGFR-2-Inhibitoren bekannt und können als Klasseneffekte betrachtet werden.
In Toxizitätsstudien bei wiederholter Gabe an Nicht-Nagetier-Spezies wurden Diarrhö und Erbrechen, begleitet von verringerter Futteraufnahme und Gewichtsabnahme beobachtet.
Bei Ratten, Hunden und Langschwanzmakaken (Cynomolgus-Affen) gab es keine Hinweise auf einen Anstieg von Leberenzymen. Lediglich bei Rhesus-Affen wurden leichte Anstiege der Leberenzyme beobachtet, die nicht auf schwerwiegende Nebenwirkungen wie eine Diarrhö zurückzuführen waren.
Mutagenität/ Karzinogenität
Untersuchungen zur Genotoxizität zeigten kein mutagenes Potential von Nintedanib.
Die 2-Jahres-Studien zur Kanzerogenität an Mäusen und Ratten lieferten keine Evidenz für ein kanzerogenes Potential von Nintedanib.
Reproduktionstoxizität
Bei Ratten wurden bei einer Exposition, die etwa 3,6- bis 7,2-mal niedriger als die maximal empfohlene Humandosis (MRHD) von 150 mg zweimal täglich war, embryofetale Letalität und teratogene Wirkungen beobachtet. Bei einer Exposition, die etwa 12- bis 18-mal niedriger als die MRHD war, wurden geringfügige Wirkungen auf die Entwicklung des Achsenskeletts und die Entwicklung der grossen Arterien festgestellt.
Bei Kaninchen wurden bei einer Exposition in Höhe des etwa 3-fachen der MRHD embryofetale Letalität und teratogene Wirkungen beobachtet, während bei einer geringeren Exposition in Höhe der MRHD von 150 mg zweimal täglich bereits eine weniger eindeutige Beeinträchtigung der embryofetalen Entwicklung des Achsenskeletts und des Herzens festgestellt wurde.
Eine Studie an Ratten zur männlichen Fertilität und frühen embryonalen Entwicklung bis zur Implantation zeigte keinen Einfluss auf die männliche Reproduktionsrate oder Fertilität bei Expositionen in Höhe des etwa 3-fachen der MRHD.
Bei Ratten wurden geringe Mengen an radioaktiv markiertem Nintedanib und/oder seinen Metaboliten in die Milch ausgeschieden (≤0,5 % der verabreichten Dosis).
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Bei Raumtemperatur (15-25°C) lagern. In der Originalverpackung aufbewahrt, um den Inhalt vor Feuchtigkeit zu schützen.
Zulassungsnummer65330 (Swissmedic)
PackungenOfev 100 mg: Packungen zu 60 Kapseln [B].
Ofev 150 mg: Packungen zu 60 Kapseln [B].
ZulassungsinhaberinBoehringer Ingelheim (Schweiz) GmbH, Basel.
Stand der InformationMärz 2025
|