ZusammensetzungWirkstoffe
Simoctocog alfa (rekombinanter humaner Blutgerinnungsfaktor VIII).
Hilfsstoffe
Saccharose, Natriumchlorid, Calciumchlorid-Dihydrat, Argininhydrochlorid, Natriumcitrat-Dihydrat, Poloxamer 188 (entspricht einem Gesamtnatriumgehalt von 18,4 mg/vial).
Lösungsmittel: Wasser für Injektionszwecke.
Indikationen/AnwendungsmöglichkeitenBehandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor VIII-Mangel).
Nuwiq enthält keine pharmakologisch wirksamen Mengen des von-Willebrand-Faktors und ist daher nicht zur Behandlung des von Willebrand-Jürgens-Syndroms geeignet.
Dosierung/AnwendungDie Behandlung muss unter der Aufsicht eines in der Behandlung der Hämophilie erfahrenen Arztes erfolgen.
Übliche Dosierung
Die Dosis und Dauer der Substitutionstherapie richtet sich nach dem Schweregrad des Faktor VIII-Mangels, dem Ort und Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die Anzahl der verabreichten Faktor VIII-Einheiten wird in Internationalen Einheiten (IE) angegeben, bezogen auf den aktuellen WHO-Standard für Faktor VIII-Produkte. Die Faktor VIII-Aktivität im Plasma wird entweder in Prozent (bezogen auf humanes Normalplasma) oder in Internationalen Einheiten (bezogen auf einen internationalen Faktor VIII-Standard im Plasma) angegeben. Sowohl der einstufige Gerinnungstest als auch der chromogene Test eignen sich für die Messung der FVIII-Aktivität im Plasma. Für die klinische Anwendung und Aktivitätsvergleich mit anderen rekombinanten und plasmatischen FVIII Präparaten ist die Angabe des einstufigen Gerinnungstests üblich.
Eine Internationale Einheit (IE) Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem Milliliter humanem Normalplasma.
Bedarfstherapie
Die Berechnung der erforderlichen Faktor VIII-Dosis basiert auf dem empirischen Ergebnis, dass eine Internationale Einheit (IE) Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma im Durchschnitt um 2% der normalen Aktivität oder 2 IE/dl erhöht. Die erforderliche Dosis wird anhand der folgenden Formel ermittelt:

Die zu verabreichende Menge und die Häufigkeit der Verabreichung sollten sich immer nach der klinischen Wirksamkeit im Einzelfall richten.
Im Falle der folgenden Blutungsereignisse sollte die Faktor VIII-Aktivität nicht unter die angegebene Plasmaaktivität (in % des Normalwerts oder IE/dl) im entsprechenden Zeitraum fallen. Die Angaben in der nachstehenden Tabelle können als Dosierungsrichtwerte bei Blutungsepisoden und chirurgischen Eingriffen verwendet werden:
|
Schweregrad der Blutung/
Art des chirurgischen Eingriffs
|
Erforderlicher Faktor VIII-Spiegel (%) (IE/dl)
|
Häufigkeit der Anwendung (Stunden)/Dauer der Therapie (Tage)
| |
Blutung
| |
Beginnende Gelenkblutungen, Muskelblutungen oder Blutungen im Mundbereich
|
20–40
|
Alle 12 bis 24 Stunden wiederholen. Mindestens 1 Tag, bis die durch Schmerzen erkennbare Blutung gestillt ist oder eine Heilung erreicht ist.
| |
Grössere Gelenkblutungen, Muskelblutung oder Hämatome
|
30–60
|
Infusion alle 12 bis 24 Stunden wiederholen, über 3 bis 4 Tage oder länger wiederholen, bis die Schmerzen und die akuten Beeinträchtigungen aufhören.
| |
Lebensbedrohliche Blutungen
|
60–100
|
Infusion alle 8 bis 24 Stunden wiederholen, bis der Patient ausser Gefahr ist.
| |
Chirurgischer Eingriff
| |
Kleinere Eingriffe
einschliesslich Zahnextraktion
|
30–60
|
Alle 24 h, mindestens 1 Tag, bis eine Heilung eintritt.
| |
Grössere Eingriffe
|
80–100
(prä- und postoperativ)
|
Infusion alle 8-24 h wiederholen, bis eine angemessene Wundheilung erzielt ist. Dann die Therapie für mindestens 7 Tage weiterführen, um eine Faktor-VIII-Aktivität von 30% bis 60% (IE/dl) aufrechtzuerhalten.
|
Prophylaxe
Übliche Dosen zur Langzeitprophylaxe von Blutungen bei Patienten mit schwerer Hämophilie A sind 20 bis 40 IE Faktor VIII pro kg Körpergewicht in Abständen von 2 bis 3 Tagen. In manchen Fällen, insbesondere bei jüngeren Patienten, können kürzere Dosierungsintervalle oder höhere Dosen erforderlich sein.
Im Laufe der Behandlung ist es ratsam, eine angemessene Kontrolle des Faktor VIII-Spiegels durchzuführen, um die zu verabreichende Dosis und die Häufigkeit der wiederholten Infusionen entsprechend festzulegen. Vor allem bei grossen chirurgischen Eingriffen ist eine genaue Kontrolle der Substitutionstherapie mit Hilfe der Gerinnungsanalyse (Faktor VIII-Aktivität im Plasma) unerlässlich. Die Antwort auf die Gabe von Faktor VIII, gemessen als In-Vivo-Recovery und die Halbwertszeit können von Patient zu Patient variieren.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Ältere Patienten
Es liegen nur wenige Erfahrungen mit älteren Patienten vor.
Kinder und Jugendliche
Zur langfristigen Vorbeugung von Blutungen bei unter 12 jährigen Patienten wird eine Dosierung von 30 – 50 IE/kg FVIII alle 2 Tage oder dreimal wöchentlich empfohlen.
Zuvor unbehandelte Patienten
Die Sicherheit und Wirksamkeit von Nuwiq bei zuvor unbehandelten Patienten wurde in einer prospektiven klinischen Studie untersucht.
Verabreichungsschema
Es wird empfohlen, nicht mehr als 4 ml pro Minute zu verabreichen.
Art der Anwendung
Zur intravenösen Anwendung.
Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt «Sonstige Hinweise – Hinweise für die Handhabung».
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder den Hilfsstoffen gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeit
Wie bei jedem intravenösen Proteinprodukt sind allergische Überempfindlichkeitsreaktionen möglich. Nuwiq enthält neben Faktor VIII Spuren anderer menschlicher Proteine aus den Wirtszellen. Wenn Überempfindlichkeitssymptome auftreten, sollten die Patienten angewiesen werden, das Arzneimittel sofort abzusetzen und ihren Arzt zu kontaktieren. Patienten sollten über Frühzeichen von Überempfindlichkeitsreaktionen aufgeklärt werden, wie zum Beispiel Nesselausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Keuchen, Hypotonie und anaphylaktischer Schock.
Im Falle eines Schocks sind die medizinischen Standards für die Behandlung von Schockzuständen zu befolgen.
Inhibitoren
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Hämophilie-A-Patienten. Diese Antikörper sind normalerweise IgG-Immunglobuline, die gegen die prokoagulatorische Aktivität des Faktors VIII gerichtet sind. Die Messung erfolgt mit Hilfe des modifizierten Bethesda-Tests und wird in Bethesda-Einheiten (B.E.) pro Milliliter Plasma ausgedrückt. Das Risiko, Antikörper zu entwickeln, korreliert mit dem Ausmass der Exposition gegenüber dem Faktor VIII, wobei das Risiko innerhalb der ersten 20 Expositionstage (EDs) am grössten ist. In seltenen Fällen können sich Antikörper nach den ersten 100 EDs bilden.
Bei vorbehandelten Patienten mit mehr als 100 EDs, die in der Vergangenheit bereits Antikörper entwickelt hatten, wurden beim Wechsel von einem Faktor VIII-Produkt zu einem anderen Fälle wiederkehrender (niedrigtitriger) Antikörper beobachtet. Daher wird empfohlen, Patienten nach jedem Produktwechsel sorgfältig auf das Auftreten eines Antikörpers hin zu überwachen.
Die Immunogenität von Nuwiq wurde bei zuvor unbehandelten Patienten mit schwerer Hämophilie A (<1% FVIII:C) in einer prospektiven offenen klinischen Studie untersucht. Von 105 Patienten, die Nuwiq erhielten und nach Beginn der Behandlung mindestens einmal auf neutralisierende Antikörper überprüft wurden, entwickelten 17 Patienten (16,2%) neutralisierende Antikörper mit hohem Titer und 11 Patienten (10,5%) neutralisierende Antikörper mit niedrigem Titer (5 davon waren transiente Antikörper, die durch Fortsetzung der prophylaktischen Behandlung ohne Änderung der Dosierung eliminiert wurden). Von den 28 Patienten, die einen Antikörper entwickelten, hatten 25 weniger als 20 EDs vor dem Nachweis. Die mittleren Zeiten bis zur Entwicklung von Antikörper mit hohem Titer bzw. niedrigem Titer betrugen 9,0 EDs (Bereich 4–24 EDs) bzw. 12,0 EDs (Bereich 6–34 EDs). Bei Patienten mit Non-Null F8 Gen Mutationen wurden keine neutralisierenden Antikörper nachgewiesen.
Im Allgemeinen sollten alle Patienten, die mit Blutgerinnungsfaktor VIII behandelt werden, durch sorgfältige klinische Beobachtung und Labortests auf die Entwicklung eines Antikörpers hin überwacht werden. Wenn die erwarteten Faktor VIII-Spiegel im Plasma nicht erreicht werden oder die Blutung nicht durch eine angemessene Dosis unter Kontrolle gebracht werden kann, sollte auf das Vorhandensein von Faktor VIII-Antikörper getestet werden. Bei Patienten mit einem hohen Antikörperspiegel ist eine Faktor VIII-Therapie möglicherweise nicht wirksam und es sollten andere Behandlungsoptionen, wie z.B. Immuntoleranzinduktion (ITI) in Betracht gezogen werden. Die Behandlung solcher Patienten sollte von Ärzten geleitet werden, die im Umgang mit Hämophilie und Faktor VIII-Antikörper Erfahrung haben.
Kardiovaskuläre Ereignisse
Bei Patienten mit bestehenden kardiovaskulären Risikofaktoren kann eine Substitutionstherapie mit Faktor VIII das kardiovaskuläre Risiko erhöhen.
Katheter-assoziierte Komplikationen
Wenn ein zentraler Venenkatheter (ZVK) erforderlich ist, sollte das Risiko ZVK-assoziierter Komplikationen, einschliesslich lokaler Infektionen, Bakteriämie und Thrombose an der Katheterstelle berücksichtigt werden.
Es wird dringend empfohlen, bei jeder Verabreichung von Nuwiq den Namen und die Chargennummer des Produkts zu dokumentieren, damit jederzeit ein Zusammenhang zwischen Patient und der Produktcharge hergestellt werden kann.
Kinder und Jugendliche
Die aufgelisteten Warnungen und Vorsichtsmassnahmen gelten für Erwachsene und Kinder gleichermassen.
Hinweis zu den sonstigen Bestandteilen (Natriumgehalt)
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natrium-frei».
Allerdings besteht die Möglichkeit, dass ein Patient, je nach Körpergewicht und Dosierung, mehr als eine Durchstechflasche erhält (siehe Rubrik «Darreichungsform und Wirkstoffmenge pro Einheit» für den Gehalt pro Durchstechflasche). Dies muss von Patienten, die eine natriumkontrollierte Diät erhalten, berücksichtigt werden.
InteraktionenEs wurden keine Interaktionsstudien zu Wechselwirkungen von Nuwiq durchgeführt.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung bei Schwangeren.
Es liegen keine hinreichenden tierexperimentellen Studien zur Auswirkung auf Schwangerschaft, Embryonalentwicklung, Entwicklung des Föten und/oder die postnatale Entwicklung vor. Das potentielle Risiko für den Menschen ist nicht bekannt.
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Nuwiq bei Schwangeren vor. Daher sollte Nuwiq während der Schwangerschaft nur dann angewandt werden, wenn dies unbedingt erforderlich ist.
Stillzeit
Aufgrund des seltenen Auftretens von Hämophilie A bei Frauen liegen keine Erfahrungen über die Anwendung von Nuwiq bei stillenden Müttern vor. Daher sollte Nuwiq während der Stillzeit nur dann angewandt werden, wenn dies unbedingt erforderlich ist.
Fertilität
Es liegen keine Daten zur Beeinflussung der Fertilität vor.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenNuwiq hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
In seltenen Fällen wurden bei Faktor VIII-Präparaten Überempfindlichkeits- oder allergische Reaktionen (wie z.B. Angioödem, Brennen und Stechen an der Infusionsstelle, Schüttelfrost, Gesichtsrötung und Hitzegefühl (Flushing), Kopfschmerzen, Nesselausschlag, Hypotonie, Lethargie, Übelkeit, Hautausschlag, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbeln, Urtikaria einschliesslich generalisierter Urtikaria, Erbrechen, Keuchen) beobachtet. In einigen Fällen können sich diese Symptome/Reaktionen zur schweren Anaphylaxie (einschliesslich Schock) entwickeln.
Patienten mit Hämophilie A können neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Falls derartige Antikörper auftreten, äussert sich dies in Form einer unzureichenden klinischen Wirksamkeit. In solchen Fällen wird die Kontaktaufnahme mit einem spezialisierten Hämophiliezentrum empfohlen.
Tabellarische Auflistung von Nebenwirkungen
In klinischen Studien mit Nuwiq an zuvor behandelten Kindern (2 bis 11 Jahre, n = 58), Jugendlichen (12 bis 17 Jahre, n = 3) und erwachsenen Patienten (n = 186) mit schwerer Hämophilie A wurden bei 9 Patienten (5 Erwachsenen, 4 Kindern) insgesamt 15 unerwünschte Arzneimittelwirkungen (UAWs) (11 bei Erwachsenen, 4 bei Kindern) berichtet. In einer klinischen Studie mit 108 vorher unbehandelten Patienten (im Alter zwischen 0-146 Monaten) mit schwerer Hämophilie A wurden folgende UAWs berichtet: neutralisierende Antikörper gegen Faktor VIII (Inhibitoren) bei 28 Patienten, Pyrexie bei 21 Patienten, Überempfindlichkeit bei 10 Patienten, Anämie bei 2 Patienten, und hämorrhagische Anämie bei einem Patienten.
Die nachfolgende Tabelle 1 entspricht der MedDRA-Systemorganklassifizierung (SOC und Preferred Term-Level).
Die Häufigkeiten wurden gemäss folgender Konvention beurteilt: sehr häufig (≥1/10); häufig (≥1/100 bis <1/10); gelegentlich (≥1/1'000 bis <1/100); selten (≥1/10'000 bis <1/1'000); sehr selten (<1/10'000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar).
Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 1. Häufigkeit des Auftretens von unerwünschten Arzneimittelwirkungen (UAWs) pro Patient in klinischen Studien
|
MedDRA-Systemorganklasse
|
Unerwünschte Wirkungen
|
Häufigkeit
| |
Blut- und Lymphsystem
Störungen
|
Anämie
|
gelegentlich*
| |
Antikörper gegen Faktor VIII
|
gelegentlich (bei zuvor behandelten Patienten)#
sehr häufig (bei nicht vorbehandelten Patienten)#
| |
Hämorrhagische Anämie
|
gelegentlich*
| |
Erkrankungen des Immunsystems
|
Überempfindlichkeit
|
häufig*
| |
Erkrankungen des Nervensystems
|
Schwindelgefühl;
Kopfschmerzen;
Parästhesien
|
gelegentlich*
| |
Erkrankungen des Ohrs und des Labyrinths
|
Schwindel
|
gelegentlich*
| |
Atemwege, Brustkorb und mediastinale Störungen
|
Dyspnoe
|
gelegentlich*
| |
Erkrankungen des Gastrointestinaltrakts
|
Mundtrockenheit
|
gelegentlich*
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
Rückenschmerzen
|
gelegentlich*
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Fieber
|
häufig*
| |
Brustkorbschmerz;
Entzündung an der Injektionsstelle;
Schmerzen an der Injektionsstelle;
Unwohlsein
|
gelegentlich*
| |
Untersuchungen
|
Nicht-neutralisierende Antikörper gegen Faktor VIII (bei zuvor behandelten Patienten)
|
gelegentlich*
|
* Berechnet als Patienten mit UAWs pro Gesamtzahl von 355 Studienpatienten, davon 247 zuvor behandelte Patienten und 108 nicht vorbehandelten Patienten.
# Die Häufigkeit basiert auf Studien mit allen FVIII-Produkten, die Patienten mit schwerer Hämophilie A einschlossen.
Beschreibung ausgewählter Nebenwirkungen
Ein nicht-neutralisierender Antikörper gegen Faktor VIII wurde bei einem erwachsenen Patienten festgestellt (siehe Tabelle 1). Die Probe wurde vom Zentrallabor in 8 Verdünnungen getestet. Nur bei Verdünnungsfaktor 1 war das Ergebnis positiv und der Antikörpertiter war sehr niedrig. Eine inhibitorische Aktivität, gemäss modifiziertem Bethesda-Test, wurde bei diesem Patienten nicht festgestellt. Die klinische Wirksamkeit und die In-vivo-Recovery von Nuwiq waren bei diesem Patienten nicht beeinträchtigt.
Kinder und Jugendliche
Es wird davon ausgegangen, dass die Häufigkeit, die Art und der Schweregrad von Nebenwirkungen bei Kindern die gleichen sind, wie bei Erwachsenen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/WirkungenATC-Code
B02BD02
Wirkungsmechanismus
Simoctocog alfa [humaner Blutgerinnungsfaktor VIII (rDNA)] ist ein gereinigtes Protein, bestehend aus 1440 Aminosäuren. Die Aminosäuresequenz ist vergleichbar mit der 90- und 80-kDa-Form des humanen Plasma-Faktor-VIII (d.h., die B-Domäne ist entfernt). Nuwiq wird mittels rekombinanter DNS-Technologie in genetisch veränderten, menschlichen, embryonalen Nierenzellen (HEK) der Zelllinie HEK-293F hergestellt. Materialien menschlichen oder tierischen Ursprunges werden weder während der Herstellung noch dem fertigen Produkt hinzugefügt.
Hämophilie A ist eine X chromosomalgebundene, erbliche Störung der Blutgerinnung, aufgrund erniedrigter Faktor VIII:C -Spiegel. Als Folge treten starke Blutungen in Gelenken, Muskeln oder inneren Organen auf. Diese können spontan oder als Folge von Unfällen oder chirurgischen Eingriffen entstehen. Die Substitutionstherapie hebt den Plasmaspiegel des Faktor VIII an und ermöglicht so eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung.
Pharmakodynamik
Der Faktor VIII/von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit verschiedenen physiologischen Funktionen. Wird einem Hämophilie-A-Patienten Faktor VIII injiziert, so bindet dieser im Blutkreislauf an den von-Willebrand-Faktor. Aktivierter Faktor VIII wirkt als Kofaktor für aktivierten Faktor IX und beschleunigt die Umwandlung von Faktor X in aktivierten Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um. Thrombin wandelt dann Fibrinogen in Fibrin um und führt so zur Bildung eines Gerinnsels.
Klinische Wirksamkeit
Die Immunogenität von Nuwiq wurde in klinischen Studien bei 135 zuvor behandelten Patienten mit schwerer Hämophilie A untersucht (74 Erwachsene sowie 61 Kinder). In 4 Patienten konnten nicht-neutralisierende Faktor VIII Antikörper nachgewiesen werden. 3 von den 4 Patienten hatten bereits einen nicht-neutralisierenden Antikörper vor der Verabreichung von Nuwiq. Ein nicht-neutralisierender Antikörper gegen Faktor VIII wurde bei einem erwachsenen Patienten festgestellt. Die Probe wurde vom Zentrallabor in 8 Verdünnungen getestet. Nur bei Verdünnungsfaktor 1 war das Ergebnis positiv und der Antikörpertiter war sehr niedrig. Eine inhibitorische Aktivität, gemäss modifiziertem Bethesda-Test, wurde bei diesem Patienten nicht festgestellt. Die klinische Wirksamkeit und die In-vivo-Recovery von Nuwiq waren bei diesem Patienten nicht beeinträchtigt.
In einer klinischen Studie mit 32 erwachsenen Patienten mit schwerer Hämophilie A lag der mittlere Verbrauch von Nuwiq zur Prophylaxe bei 468,7 IE/kg/Monat. Die mittlere Dosis zur Behandlung von Episoden mit Durchbruchsblutungen betrug 33,0 IE/kg bei diesen Patienten unter prophylaktischer Behandlung. In einer anderen klinischen Studie erhielten 22 erwachsene Patienten eine Bedarfsbehandlung. Insgesamt wurden 986 Blutungsepisoden mit einer mittleren Dosis von 30,9 IE/kg behandelt. Im Allgemeinen waren für leichte Blutungen etwas niedrigere und für schwerere Blutungen bis zu dreifach höhere mittlere Dosen erforderlich.
Individualisierte Prophylaxe: Die individualisierte PK-basierte Prophylaxe wurde in 66 erwachsenen PTPs mit schwerer Hämophilie A untersucht. Nach einer 1-3-monatigen Standard-Prophylaxe-Phase (jeden zweiten Tag oder 3-mal wöchentliche Dosierung), konnten 44 (67%) Patienten auf der Grundlage ihrer PK-Bewertung auf ein Dosierschema umgestellt werden, und 40 absolvierten die 6 Monate Prophylaxe nach dem empfohlenen kalkulierten Dosierungs- und Behandlungsschema. Von diesen Patienten wurden 34 (85%) zweimal wöchentlich oder seltener behandelt. Bei 33 (82,5 %) Patienten traten keine Blutungen auf, und 36 (90,0 %) Patienten hatten keine spontanen Blutungen. Die annualisierte Blutungsrate (ABR; Mittelwert ± SD) betrug 1,2 ± 3,9 und die mittlere Dosis ± SD lag bei 52,2 ± 12,2 I.E./kg pro Injektion bzw. 99,7 ± 25,6 I.E./kg pro Woche.
Es gilt zu beachten, dass die ABR zwischen verschiedenen Faktorkonzentraten und zwischen verschiedenen klinischen Studien nicht vergleichbar ist.
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Die Daten wurden bei 29 zuvor behandelten Kindern zwischen 2 und 5 Jahren, 31 Kindern zwischen 6 und 12 Jahren und einem Jugendlichen von 14 Jahren erhoben. Die mittlere Dosis pro prophylaktischer Infusion lag bei 37,8 IE/kg. Zwanzig Patienten verwendeten mittlere Dosen von mehr als 45 IE/kg. Der mittlere Verbrauch von Nuwiq zur Prophylaxe pro Monat lag bei 521,9 IE/kg. Für die Behandlung von Blutungen bei Kindern war eine höhere Dosis Nuwiq (43,9 IE/kg) erforderlich als bei Erwachsenen (33,0 IE/kg) und eine höhere mittlere Dosis war für die Behandlung von moderaten bis schweren als für leichte Blutungen (78,2 IE/kg vs. 41,7 IE/kg) erforderlich. Bei jüngeren Kindern waren im Allgemeinen höhere mittlere Dosen erforderlich (6-12 Jahre: 43,9 IE/kg; 2-5 Jahre: 52,6 IE/kg). Diese Daten wurden durch die Langzeitbeobachtung von 49 dieser Kinder, die für einen zusätzlichen Medianzeitraum von ca. 30 Monaten behandelt wurden (Bereich von 9,5 bis 52 Monaten), bestätigt; in diesem Zeitraum hatten 45% der Kinder keine spontanen Blutungen.
PharmakokinetikAbsorption
Nicht zutreffend.
Distribution
FVIII verteilt sich im Plasma.
Metabolismus
Nicht zutreffend.
Elimination
Siehe Tabellen 2 – 4.
Tabelle 2. Pharmakokinetik-(PK)-Parameter für Nuwiq (Dosis: 50 IE/kg) bei erwachsenen vorbehandelten Patienten (im Alter von 18-65 Jahren) mit schwerer Hämophilie A (n = 20)
|
PK-Parameter
|
Chromogener Test
|
Einstufiger Gerinnungstest
| |
Mittelwert ± SD
|
Mittelwert ± SD
| |
AUCnorm (h*IE/ml/(IE/kg))
|
0,39 ± 0,14
|
0,37 ± 0,11
| |
T½ (h)
|
14,7 ± 10,4
|
17,0 ± 11,8
| |
IVR (%/IE/kg)
|
2,5 ± 0,4
|
2,2 ± 0,3
| |
CL (ml/h/kg)
|
3,0 ± 1,2
|
2,9 ± 1,0
|
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Kinetik spezieller Patientengruppen
Gewichtsangepasste Untergruppen
Bei den in der PK Studie eingeschlossenen erwachsenen präadipösen (BMI 25-30 kg/m2) und adipösen Patienten (BMI >30 kg/m2) war die Clearance höher als bei den Normalgewichtigen.
Kinder und Jugendliche
Tabelle 3. PK-Parameter für Nuwiq (Dosis: 50 IE/kg) bei vorbehandelten Kindern im Alter von 6 bis 12 Jahren mit schwerer Hämophilie A (n = 12)
|
PK-Parameter
|
Chromogener Test
|
Einstufiger Gerinnungstest
| |
Mittelwert ± SD
|
Mittelwert ± SD
| |
AUCnorm (h*IE/ml/(IE/kg))
|
0,25 ± 0,1
|
0,26 ± 0,1
| |
T½(h)
|
10,0 ± 1,9
|
13,1 ± 2,6
| |
IVR (%/IE/kg)
|
1,9 ± 0,4
|
1,6 ± 0,4
| |
CL (ml/h/kg)
|
4,3 ± 1,2
|
4,1 ± 0,9
|
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Tabelle 4. PK-Parameter für Nuwiq (Dosis: 50 IE/kg) bei vorbehandelten Kindern im Alter von 2 bis 5 Jahren mit schwerer Hämophilie A (n = 13)
|
PK-Parameter
|
Chromogener Test
|
Einstufiger Gerinnungstest
| |
Mittelwert ± SD
|
Mittelwert ± SD
| |
AUCnorm (h*IE/ml/(IE/kg))
|
0,22 ± 0,1
|
0,22 ± 0,1
| |
T½(h)
|
9,5 ± 3,3
|
11,9 ± 5,4
| |
IVR (%/IE/kg)
|
1,9 ± 0,3
|
1,6 ± 0,2
| |
CL (ml/h/kg)
|
5,4 ± 2,4
|
5,4 ±2,3
|
AUC = Fläche unter der Kurve (FVIII:C), T½ = terminale Halbwertszeit,
IVR = inkrementelle In-Vivo-Recovery, CL = Clearance, SD = Standardabweichung
Wie aus der Literatur bekannt, waren bei jüngeren Kindern Recovery und Halbwertszeit niedriger und die Clearance höher als bei Erwachsenen, was teilweise am bekanntermassen höheren Plasmavolumen pro Kilogramm Körpergewicht bei jüngeren Patienten liegen könnte.
Präklinische DatenToxikologische Studien zeigten, dass die lokale intravenöse Verabreichung und systemische Exposition von Labortieren (Ratten und Cynomolgus-Affen) gut vertragen wurde. Die Bedeutung der nach wiederholter Simoctocog alfa-Gabe in Affen beobachteten erniedrigten Thymusgewichte ist unklar.
Aufgrund der Immunreaktion auf heterologe Proteine bei allen nicht-humanen Säugetierarten wurden mit Nuwiq keine spezifischen Studien mit wiederholter Verabreichung über einen längeren Zeitraum durchgeführt (wie z.B. Studien zu Reproduktionstoxizität, chronischer Toxizität und Kanzerogenität).
Es wurden keine Studien zum mutagenen Potential von Nuwiq durchgeführt.
Ex-Vivo-Untersuchungen mit Hilfe eines kommerziellen Test-Kits zur Quantifizierung der T-Zell-Antwort auf Proteintherapeutika zeigen eine zu vergleichbaren Präparaten mindestens gleichermassen ausgeprägte Immunogenität an.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Es dürfen nur die mitgelieferten Infusionssets verwendet werden, da Therapieversagen als Folge einer Adsorption von humanem Gerinnungsfaktor VIII an der inneren Oberfläche mancher Infusionssets auftreten kann.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «Verw. bis/EXP» bezeichneten Datum verwendet werden.
Während der Haltbarkeitsfrist kann das Präparat für einen Zeitraum von nicht mehr als 1 Monat bei Raumtemperatur (bis zu 25 °C) aufbewahrt werden. Sobald das Präparat aus dem Kühlschrank genommen wird, darf es nicht wieder in diesen zurückgelegt werden. Vermerken Sie bitte den Beginn der Aufbewahrung bei Raumtemperatur auf der Packung. Die Durchstechflasche muss in der Originalverpackung zum Schutz vor Licht gelagert werden.
Nach der Rekonstitution
Aus mikrobiologischer Sicht sollte das gebrauchsfertige Arzneimittel sofort nach Rekonstitution verwendet werden. Geschieht das nicht, so ist das rekonstituierte Präparat bei einer Lagertemperatur von ≤25 °C innerhalb von 3 Stunden zu verwenden. Das rekonstituierte Präparat muss in der Durchstechflasche aufbewahrt werden.
Nicht verwendete Präparate, die länger als 3 Stunden bei Raumtemperatur gelagert wurden, müssen entsorgt werden.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern.
Nicht einfrieren.
In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zu den Lagerungsbedingungen für das rekonstituierte Präparat siehe unter «Sonstige Hinweise: Haltbarkeit – Nach der Rekonstitution».
Entsorgung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Hinweise für die Handhabung
Das Pulver darf nur mit dem mitgelieferten Lösungsmittel (2,5 ml Wasser für Injektionszwecke) und unter Verwendung des mitgelieferten Injektionssets rekonstituiert werden. Die Durchstechflasche sollte vorsichtig geschwenkt werden, bis das gesamte Pulver aufgelöst ist. Nach der Rekonstitution sollte die Lösung wieder in die Spritze aufgezogen werden.
Das rekonstituierte Arzneimittel muss vor der Verabreichung per Sichtkontrolle auf Partikel und Verfärbungen hin geprüft werden. Das rekonstituierte Arzneimittel ist eine klare, farblose Lösung, die frei von Fremdkörpern ist und einen pH-Wert zwischen 6,5 und 7,5 aufweist. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen enthalten.
Rekonstitution:
1.Bringen Sie die Lösungsmittelspritze (mit Wasser für Injektionszwecke) und das Pulver in der verschlossenen Durchstechflasche vor der Anwendung auf Zimmertemperatur. Sie können dazu die Behälter in den Händen halten, bis sie sich so warm wie Ihre Hände anfühlen. Wenden Sie keine andere Methode zum Aufwärmen der Durchstechflasche und der Fertigspritze an. Diese Temperatur muss während der Rekonstitution beibehalten werden.
2.Entfernen Sie die Flipp-off-Kappe aus Plastik von der Durchstechflasche mit dem Pulver, so dass der mittlere Teil des Gummistopfens sichtbar wird. Entfernen Sie nicht den grauen Stopfen oder den Metallring (Bördelkappe) am oberen Ende der Durchstechflasche.

3.Wischen Sie den Gummistopfen der Durchstechflasche mit einem Alkoholtupfer ab. Lassen Sie den Alkohol trocknen.
4.Ziehen Sie die Schutzfolie von der Verpackung des Durchstechflaschen-Adapters ab. Nehmen Sie den Adapter nicht aus seiner Verpackung.

5.Stellen Sie die Durchstechflasche mit dem Pulver auf eine ebene Unterlage und halten Sie sie fest. Nehmen Sie den Adapter mit seiner Verpackung und platzieren Sie den Durchstechflaschen-Adapter mittig über dem Gummistopfen der Durchstechflasche mit dem Pulver. Drücken Sie die Adapterverpackung mit dem Adapter kräftig auf, bis der Adapterdorn den Gummistopfen durchdringt. Dabei rastet der Adapter auf der Durchstechflasche ein.

6.Ziehen Sie die Schutzfolie von der Verpackung der Fertigspritze ab. Halten Sie den Spritzenstempel am Ende und berühren Sie nicht den Schaft. Stecken Sie das Ende des Spritzenstempels mit dem Gewinde auf den Kolben der Lösungsmittelspritze. Drehen Sie den Stempel im Uhrzeigersinn, bis Sie einen leichten Widerstand spüren.

7.Brechen Sie die Spritzenkappe aus Plastik von der Lösungsmittelspritze an der Perforation ab. Berühren Sie nicht die Innenseite der Kappe oder der Spritzenspitze. Wird die Lösung nicht sofort verwendet, verschliessen Sie die gefüllte Spritze mit der Spritzenkappe zur Aufbewahrung.
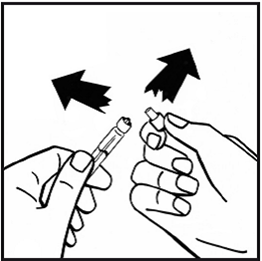
8.Entfernen Sie die Adapterverpackung und entsorgen Sie diese.
9.Verbinden Sie die Lösungsmittelspritze fest mit dem Durchstechflaschen-Adapter, indem Sie sie im Uhrzeigersinn drehen, bis ein Widerstand spürbar wird.

10.Injizieren Sie langsam das gesamte Lösungsmittel in die Durchstechflasche mit dem Pulver, indem Sie den Spritzenstempel nach unten drücken.
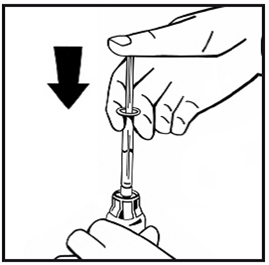
11.Zum Auflösen des Pulvers schwenken Sie die Durchstechflasche einige Male leicht im Kreis, ohne dabei die Spritze zu entfernen. Nicht schütteln. Warten Sie bis sich das Pulver vollständig aufgelöst hat.
12.Prüfen Sie die fertige Lösung vor der Verabreichung visuell auf Partikel. Die Lösung sollte klar und farblos sein und frei von sichtbaren Partikeln. Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen enthalten.
13.Drehen Sie die mit der Spritze verbundene Durchstechflasche auf den Kopf und ziehen Sie die fertige Lösung langsam in die Spritze auf. Stellen Sie sicher, dass der gesamte Inhalt der Durchstechflasche in die Spritze überführt wird.
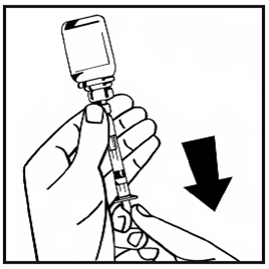
14.Ziehen Sie die gefüllte Spritze mit einer Drehbewegung gegen den Uhrzeigersinn aus dem Durchstechflaschen-Adapter und verwerfen Sie die leere Durchstechflasche.
15.Die Lösung steht nun zur sofortigen Verwendung bereit. Nicht im Kühlschrank lagern.
16.Reinigen Sie die vorgesehene Injektionsstelle mit einem der mitgelieferten Alkoholtupfer.
17.Verbinden Sie das mitgelieferte Infusionsset mit der Spritze. Führen Sie die Nadel der Flügelkanüle in die ausgewählte Vene ein. Wenn Sie die Vene vor der Punktion gestaut haben, damit Sie sie besser sehen können, müssen Sie die Stauung öffnen, bevor Sie mit der Injektion der Lösung beginnen. Es darf kein Blut in die Spritze gelangen, da dies zur Bildung von Blutgerinnseln führen könnte.
18.Injizieren Sie die Lösung langsam in die Vene. Die Injektionsgeschwindigkeit sollte höchstens 4 ml pro Minute betragen.
Wenn Sie mehr als eine Durchstechflasche mit Pulver für eine Behandlung benötigen, können Sie dieselbe Flügelkanüle verwenden. Der Durchstechflaschen-Adapter und die Spritze sind nur für den einmaligen Gebrauch bestimmt.
Zulassungsnummer65551 (Swissmedic).
PackungenNuwiq ist in den Packungsgrössen zu je 1 Durchstechflasche mit 250 IE (100 IE/ml), 500 IE (200 IE/ml), 1000 IE (400 IE/ml) und 2000 IE (800 IE/ml) erhältlich.
Jede Packung enthält:
1 Durchstechflasche mit Pulver mit 250/ 500/ 1000/ 2000 Internationalen Einheiten Simoctocog alfa, 1 Fertigspritze mit 2,5 ml Wasser für Injektionszwecke, 1 Durchstechflaschenadapter, 1 Injektionsnadel (Butterfly), 2 Alkoholtupfer
Abgabekategorie B
ZulassungsinhaberinOctapharma AG,
8853 Lachen
Stand der InformationNovember 2023
|