ZusammensetzungWirkstoffe
Liraglutidum
Hilfsstoffe
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabilia.
Das Arzneimittel enthält 0.367 mg/ml Natrium.
Indikationen/AnwendungsmöglichkeitenSaxenda wird als Ergänzung zu einer kalorienreduzierten Ernährung und verstärkter körperlicher Aktivität zur Gewichtsregulierung angewendet bei:
·Erwachsenen Patienten mit einem Ausgangs-Body-Mass-Index (BMI) von
·≥ 30 kg/m² (adipös) oder
·≥ 27 kg/m² falls zusätzliche gewichtsbedingte Begleiterkrankungen (Prädiabetes oder Diabetes mellitus Typ 2, arterielle Hypertonie oder Dyslipidämie) vorliegen.
Falls die Patienten nach 12-wöchiger Behandlung mit einer Dosis von 3.0 mg/Tag nicht mindestens 5 % ihres Körpergewichts verloren haben, ist Saxenda abzusetzen.
·Jugendlichen ab 12 Jahren mit einem Körpergewicht ≥60 kg und einer Adipositas gemäss den dafür international akzeptierten Grenzwerten (entspricht einem BMI ≥30 kg/m2 bei Erwachsenen)*.
*BMI-Grenzwerte der IOTF für Fettleibigkeit nach Geschlecht für Jugendliche von 12–18 Jahren.
|
Alter
(Jahre)
|
Body Mass Index
30 kg/m2
| |
Männlich
|
Weiblich
| |
12
|
26.02
|
26.67
| |
12.5
|
26.43
|
27.24
| |
13
|
26.84
|
27.76
| |
13.5
|
27.25
|
28.20
| |
14
|
27.63
|
28.57
| |
14.5
|
27.98
|
28.87
| |
15
|
28.30
|
29.11
| |
15.5
|
28.60
|
29.29
| |
16
|
28.88
|
29.43
| |
16.5
|
29.14
|
29.56
| |
17
|
29.41
|
29.69
| |
17.5
|
29.70
|
29.84
| |
18
|
30.00-
|
30.00
|
Die Behandlung mit Saxenda sollte unterbrochen und neu beurteilt werden, wenn sich der BMI oder der BMI-Z-Wert der Patienten nach Anwendung von 3.0 mg/Tag oder der maximal verträglichen Dosis über 12 Wochen nicht um mindestens 4 % verbessert hat.
Dosierung/AnwendungÜbliche Dosierung
Die Anfangsdosis beträgt 0.6 mg einmal pro Tag. Die Dosis sollte schrittweise auf maximal 3.0 mg einmal pro Tag erhöht werden. Zur Verbesserung der gastrointestinalen Verträglichkeit sollte dies in Abstufungen von 0.6 mg jeweils im Abstand von mindestens einer Woche erfolgen (siehe Tabelle 1). Wird die Dosissteigerung auf die nächste Dosisstufe in zwei aufeinanderfolgenden Wochen nicht vertragen, ist ein Abbruch der Behandlung in Betracht zu ziehen. Höhere Tagesdosen als 3.0 mg werden nicht empfohlen.
Dosisanpassung/Titration
Tabelle 1. Dosiseskalationsschema
|
|
Dosis
|
Wochen
| |
Dosiseskalation
4 Wochen
|
0.6 mg
|
1
| |
1.2 mg
|
1
| |
1.8 mg
|
1
| |
2.4 mg
|
1
| |
Erhaltungsdosis
|
3.0 mg
|
Patienten mit Typ 2 Diabetes mellitus
Saxenda ist zur Behandlung von Diabetes mellitus Typ 2 nicht indiziert. Saxenda darf nicht in Kombination mit einem anderen GLP-1-Rezeptor-Agonisten angewendet werden.
Wenn die Behandlung mit Saxenda begonnen wird, ist eine Dosisreduktion von gleichzeitig angewendetem Insulin oder Insulinsekretagoga (wie Sulfonylharnstoffe) zu erwägen, um das Risiko einer Hypoglykämie zu senken. Eine Selbstkontrolle des Blutzuckers durch den Patienten ist notwendig, um die Dosis von Insulin oder lnsulinsekretagoga anzupassen.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter oder mittelschwerer Einschränkung der Leberfunktion wird keine Dosisanpassung empfohlen. Saxenda wird nicht zur Anwendung bei Patienten mit schwerer Leberfunktionsstörung empfohlen und muss bei Patienten mit leichter oder mittelschwerer Einschränkung der Leberfunktion mit Vorsicht angewendet werden (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter oder mittelschwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance ≥30 ml/min) ist keine Dosisanpassung erforderlich. Die klinische Erfahrung mit solchen Patienten ist allerdings limitiert, Saxenda sollte in dieser Population mit Vorsicht angewendet werden. Saxenda wird nicht zur Anwendung bei Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance < 30 ml/min), einschliesslich Patienten mit terminaler Niereninsuffizienz, empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen» und «Pharmakokinetik»).
Ältere Patienten (≥65 Jahre alt)
Eine Dosisanpassung bei älteren Menschen ist nicht erforderlich. Bei Patienten ≥75 Jahre sind die therapeutischen Erfahrungen begrenzt und die Anwendung wird bei diesen Patienten nicht empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Kinder und Jugendliche
Die Anwendung von Saxenda bei Kindern unterhalb 12 Jahren oder bei Jugendlichen mit einem Körpergewicht von 60 kg oder weniger wird aufgrund fehlender Daten nicht empfohlen (siehe «Eigenschaften/Wirkungen»).
Bei Jugendlichen im Alter von 12 bis zu 18 Jahren sollte eine ähnliche Dosissteigerung wie bei Erwachsenen angewendet werden (siehe Tabelle 1). Die Dosis sollte bis auf 3.0 mg (Erhaltungsdosis) erhöht werden oder bis die maximal verträgliche Dosis erreicht ist. Tägliche Dosen über 3.0 mg werden nicht empfohlen.
Art der Anwendung
Saxenda ist nur für die subkutane Anwendung bestimmt. Es darf nicht intravenös oder intramuskulär angewendet werden.
Saxenda wird einmal täglich zu einem beliebigen Zeitpunkt und unabhängig von den Mahlzeiten gegeben. Die Injektion sollte in Abdomen, Oberschenkel oder Oberarm erfolgen. Die Einstichstelle ist bei jeder Injektion innerhalb derselben Körperregion zu wechseln, um das Risiko einer kutanen Amyloidose zu reduzieren (siehe «Unerwünschte Wirkungen»).
Die Injektionsstelle und der Zeitpunkt der Gabe können ohne Dosisanpassung geändert werden. Saxenda sollte jedoch vorzugsweise in etwa zum gleichen Tageszeitpunkt injiziert werden, sobald der geeignetste Tageszeitpunkt gewählt wurde.
Saxenda darf nicht mit anderen injizierbaren Arzneimitteln gemischt werden (z.B. Insuline).
Wird eine Dosis vergessen und es sind weniger als 12 Stunden seit dem normalen Anwendungszeitpunkt vergangen, sollte der Patient die Dosis so bald wie möglich nachholen. Verbleiben weniger als 12 Stunden bis zur nächsten Dosis, sollte der Patient die vergessene Dosis nicht nachholen, sondern mit der nächsten Dosis zu seinem gewohnten einmal täglichen Dosierungsschema zurückkehren. In diesem Fall sollte keine Extra-Dosis gespritzt oder die nächste Dosis erhöht werden, um die vergessene Dosis auszugleichen. Weitere Hinweise zur Handhabung, siehe «Sonstige Hinweise». Eine genaue Gebrauchsanweisung ist in der Patienteninformation integriert.
KontraindikationenÜberempfindlichkeit gegen Liraglutide oder einen der in «Zusammensetzung» genannten sonstigen Bestandteile.
Warnhinweise und VorsichtsmassnahmenBei Patienten mit Diabetes mellitus darf Saxenda nicht als Ersatz für Insulin angewendet werden. Es liegen Berichte über diabetische Ketoazidose bei insulinabhängigen Patienten nach raschem Absetzen oder einer schnellen Dosisreduktion von Insulin vor (siehe «Dosierung/Anwendung»).
Es gibt keine klinischen Erfahrungen bei Patienten mit Herzinsuffizienz des New York Heart Association (NYHA)-Stadiums IV, daher wird die Anwendung von Liraglutide bei diesen Patienten nicht empfohlen. Erfahrungen für Patienten mit Herzinsuffizienz NYHA I-III basieren auf der LEADER Studie, in welcher diese mit Liraglutide in einer maximalen Tagesdosis von 1.8 mg behandelt wurden.
Die Sicherheit und Wirksamkeit von Liraglutide zur Gewichtsregulierung sind nicht erwiesen bei Patienten:
·im Alter von 75 Jahren und mehr
·die mit anderen Produkten zur Gewichtsregulierung behandelt werden
·mit einer Adipositas als Folge endokrinologischer Störungen oder Essstörungen, wie Bulimie oder Binge Eating, oder der Behandlung mit Arzneimitteln, die eine Gewichtszunahme verursachen können
·mit schwerer Einschränkung der Nierenfunktion
·mit schwerer Einschränkung der Leberfunktion.
Die Anwendung wird bei diesen Patienten nicht empfohlen (siehe «Dosierung/Anwendung»).
Da Liraglutide nicht zur Gewichtsregulierung bei Patienten mit leichter oder mittelschwerer Einschränkung der Leberfunktion oder der Nierenfunktion untersucht wurde, muss es bei diesen Patienten mit Vorsicht angewendet werden (siehe «Dosierung/Anwendung» und «Pharmakokinetik»).
Aspiration im Zusammenhang mit einer Vollnarkose oder tiefer Sedierung
Bei Patienten, die GLP-1-Rezeptor-Agonisten erhielten und die sich einer Vollnarkose oder einer tiefen Sedierung unterzogen, wurden Fälle von pulmonaler Aspiration berichtet, trotz der berichteten Einhaltung der präoperativen Nüchternempfehlungen. Daher sollte vor der Durchführung von Eingriffen unter Vollnarkose oder tiefer Sedierung das erhöhte Risiko für Restmageninhalt aufgrund einer verzögerten Magenentleerung berücksichtigt werden.
Entzündliche Darmerkrankung und diabetische Gastroparese
Bei Patienten mit entzündlichen Darmkrankheiten und diabetischer Gastroparese liegen nur begrenzte Erfahrungen vor. Die Anwendung von Liraglutide wird bei diesen Patienten nicht empfohlen, da sie mit vorübergehenden gastrointestinalen Nebenwirkungen, einschliesslich Übelkeit, Erbrechen und Durchfall, verbunden ist.
Pankreatitis
Akute Pankreatitis wurde unter der Anwendung von GLP-1-Rezeptor Agonisten beobachtet. Patienten sollten über die charakteristischen Symptome einer akuten Pankreatitis informiert werden. Wird eine Pankreatitis vermutet, ist Liraglutide abzusetzen; falls eine akute Pankreatitis bestätigt wird, ist die Behandlung mit Liraglutide nicht wieder aufzunehmen.
Eine isolierte Erhöhung der Pankreasenzyme unter der Behandlung mit Saxenda (ohne charakteristische Symptomatik) manifestiert nicht zwingend eine akute Pankreatitis (siehe «Unerwünschte Wirkungen»).
Cholelithiasis und Cholezystitis
In klinischen Studien zur Gewichtsregulierung wurde bei Patienten, die mit Liraglutide behandelt wurden, ein häufigeres Auftreten von Cholelithiasis und Cholezystitis beobachtet als bei den mit Placebo behandelten Patienten. Die Tatsache, dass starker Gewichtsverlust mit einem erhöhten Risiko für Cholelithiasis und dadurch auch für Cholezystitis einhergehen kann, erklärte nur teilweise das häufigere Auftreten mit Liraglutide. Cholelithiasis und Cholezystitis können eine stationäre Behandlung und eine Cholezystektomie erforderlich machen. Patienten sollten über die charakteristischen Symptome von Cholelithiasis und Cholezystitis informiert werden.
Schilddrüsenerkrankungen
In klinischen Studien bei Patienten mit Typ 2 Diabetes wurde über unerwünschte Ereignisse in Zusammenhang mit der Schilddrüse wie Struma insbesondere bei Patienten mit bestehender Schilddrüsenerkrankung, berichtet. Liraglutide sollte deshalb bei Patienten mit Schilddrüsenerkrankungen mit Vorsicht angewendet werden.
Herzfrequenz
Liraglutide erhöht die Herzfrequenz (siehe «Eigenschaften/Wirkungen»). Die Herzfrequenz sollte gemäss der gängigen klinischen Praxis in regelmässigen Abständen überprüft werden. Patienten sollten über die charakteristischen Symptome einer erhöhten Herzfrequenz (Palpitationen oder gefühltes Herzrasen im Ruhezustand) informiert werden. Bei Patienten, bei denen es zu einer anhaltenden, klinisch relevanten Erhöhung der Herzfrequenz kommt, sollte Liraglutide abgesetzt werden.
Dehydrierung
Bei Patienten, die mit GLP-1-Rezeptor-Agonisten behandelt wurden, wurde über Anzeichen und Symptome von Dehydrierung einschliesslich Beeinträchtigung der Nierenfunktion und akutem Nierenversagen berichtet. Patienten, die mit Liraglutide behandelt werden, müssen auf das potenzielle Dehydrierungs-Risiko im Zusammenhang mit gastrointestinalen Nebenwirkungen hingewiesen werden und Vorkehrungen gegen Flüssigkeitsverluste treffen.
Hypoglykämie bei Patienten mit Diabetes mellitus Typ 2
Patienten mit Diabetes mellitus Typ 2, die Liraglutide in Kombination mit Insulinsekretagoga, z.B. einem Sulfonylharnstoff, oder mit Insulin erhalten, können ein erhöhtes Risiko für eine Hypoglykämie haben. Das Risiko einer Hypoglykämie kann durch Reduktion der Dosis des Insulins und/oder des Sekretagogums gesenkt werden.
Kinder und Jugendliche
Bei mit Liraglutide behandelten Jugendlichen (≥12 Jahre alt) wurden Episoden einer klinisch relevanten Hypoglykämie berichtet. Die Patienten sollten über die charakteristischen Symptome von Hypoglykämie und geeignete Gegenmassnahmen informiert werden.
Sonstige Bestandteile
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Dosis, d.h. es ist nahezu «natriumfrei».
InteraktionenPharmakokinetische Interaktionen
In-vitro-Beurteilung von Arzneimittel-Wechselwirkungen mit Liraglutide
Das Potential von Liraglutide zur Beteiligung an pharmakokinetischen Wechselwirkungen mit anderen Wirkstoffen, die mit Cytochrom P450 (CYP) und der Bindung an Plasmaproteine in Zusammenhang stehen, hat sich als sehr gering erwiesen.
In-vivo-Beurteilung von Arzneimittel-Wechselwirkungen mit Liraglutide
Die durch Liraglutide verursachte geringe Verzögerung der Magenentleerung könnte die Resorption gleichzeitig oral angewendeter Arzneimittel beeinflussen. Interaktionsstudien zeigten keine klinisch relevante Verzögerung der Resorption, und daher ist keine Dosisanpassung erforderlich.
Die Interaktionsstudien wurden mit 1.8 mg Liraglutide durchgeführt. Die Wirkung auf die Geschwindigkeit der Magenentleerung war bei 1.8 mg und 3.0 mg Liraglutide gleich (Paracetamol AUC0-300 min).
Einige mit Liraglutide behandelte Patienten berichteten von mindestens einer schweren Durchfall-Episode. Diarrhö kann die Resorption von gleichzeitig oral gegebener Arzneimitteln beeinträchtigen.
Wirkung von Saxenda auf andere Arzneimittel
Warfarin und andere Cumarin-Derivate
Es wurde keine Interaktionsstudie durchgeführt. Klinisch relevante Wechselwirkungen mit Wirkstoffen wie Warfarin, die eine geringe Löslichkeit oder einen engen therapeutischen Bereich haben, können nicht ausgeschlossen werden. Bei Patienten, die mit Warfarin oder anderen Cumarin-Derivaten behandelt werden, wird zu Beginn der Behandlung mit Liraglutide eine häufigere Überwachung der INR (International Normalized Ratio) empfohlen.
Paracetamol (Acetaminophen)
Nach einer Einzeldosis von 1'000 mg Paracetamol führte Liraglutide nicht zu einer Änderung der Gesamtexposition von Paracetamol. Die Cmax von Paracetamol war um 31 % verringert, die mittlere tmax war um bis zu 15 Min. verzögert. Bei begleitender Anwendung von Paracetamol ist keine Dosisanpassung erforderlich.
Atorvastatin
Nach Gabe einer Einzeldosis von 40 mg Atorvastatin führte Liraglutide nicht zu einer Änderung der Gesamtexposition von Atorvastatin. Es ist deshalb keine Dosisanpassung von Atorvastatin erforderlich, wenn es gemeinsam mit Liraglutide gegeben wird. Mit Liraglutide war die Cmax von Atorvastatin um 38 % verringert, die mittlere tmax war um 1 bis 3 Stunden verzögert.
Griseofulvin
Nach Gabe einer Einzeldosis von 500 mg Griseofulvin führte Liraglutide nicht zu einer Änderung der Gesamtexposition von Griseofulvin. Die Cmax von Griseofulvin erhöhte sich um 37 %, während die mittlere tmax unverändert blieb. Dosisanpassungen von Griseofulvin und anderen Präparaten mit geringer Löslichkeit und hoher Permeabilität sind nicht erforderlich.
Digoxin
Die Gabe von Liraglutide zusammen mit einer Einzeldosis von 1 mg Digoxin führte zu einer Verringerung der AUC von Digoxin um 16 %; die Cmax nahm um 31 % ab. Die mittlere tmax von Digoxin war um 1 bis 1.5 Stunden verzögert. Ausgehend von diesen Ergebnissen ist keine Dosisanpassung von Digoxin erforderlich.
Lisinopril
Die Gabe von Liraglutide zusammen mit einer Einzeldosis von 20 mg Lisinopril führte zu einer Verringerung der AUC von Lisinopril um 15 %; die Cmax nahm um 27 % ab. Mit Liraglutide war die mittlere tmax von Lisinopril um 6 bis 8 Stunden verzögert. Ausgehend von diesen Ergebnissen ist keine Dosisanpassung von Lisinopril erforderlich.
Orale Kontrazeptiva
Nach Gabe einer Einzeldosis eines oralen Kontrazeptivums senkte Liraglutide die Cmax von Ethinylestradiol und Levonorgestrel um 12 % bzw. 13 %. Die tmax war mit Liraglutide bei beiden Wirkstoffen um 1.5 Stunden verzögert. Es gab keine klinisch relevante Auswirkung auf die Gesamtexposition von Ethinylestradiol oder Levonorgestrel. Folglich ist zu erwarten, dass die kontrazeptive Wirkung bei gleichzeitiger Gabe von Liraglutide nicht beeinträchtigt wird.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen nur begrenzte Daten für die Anwendung von Liraglutide bei schwangeren Frauen vor. In tierexperimentellen Studien fand sich eine Reproduktionstoxizität (siehe «Präklinische Daten»). Das potenzielle Risiko für den Menschen ist nicht bekannt.
Saxenda sollte während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich. Möchte eine Patientin schwanger werden oder tritt eine Schwangerschaft ein, sollte die Behandlung mit Saxenda abgebrochen werden.
Stillzeit
Es ist nicht bekannt, ob Liraglutide in die Muttermilch übergeht. Tierexperimentelle Studien haben gezeigt, dass der Übergang von Liraglutide und strukturell eng verwandten Metaboliten in die Muttermilch gering ist. Präklinische Studien zeigten in Zusammenhang mit der Behandlung eine Abnahme des neonatalen Wachstums von gesäugten Ratten (siehe «Präklinische Daten»). Aufgrund mangelnder Erfahrung soll Saxenda nicht in der Stillzeit angewendet werden.
Fertilität
Abgesehen von einer leichten Reduktion der Implantationsrate zeigten tierexperimentelle Studien bezüglich Fertilität keine unmittelbar schädlichen Effekte (siehe «Präklinische Daten»). Es gibt keine hinreichenden klinischen Daten zur Beeinflussung der Fertilität beim Menschen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenSaxenda hat keinen oder einen zu vernachlässigenden Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheit von Saxenda wurde in 5 doppelblinden, placebokontrollierten klinischen Studien, an denen 5'813 erwachsene Patienten mit Übergewicht oder Adipositas teilnahmen, die mindestens eine gewichtsbedingte Begleiterkrankung aufwiesen, beurteilt. Insgesamt waren die während der Behandlung mit Saxenda am häufigsten berichteten Nebenwirkungen gastrointestinale Nebenwirkungen (siehe «Beschreibung ausgewählter Nebenwirkungen»).
Tabellarische Auflistung der Nebenwirkungen
In Tabelle 2 sind Nebenwirkungen aufgeführt, die in kontrollierten Studien der Phase 2 und Phase 3 mit Erwachsenen berichtet wurden und von Postmarketing Berichten. Die Nebenwirkungen sind nach Systemorganklassen und Häufigkeit aufgeführt. Die Häufigkeit der Nebenwirkungen basieren auf den gepoolten Daten der Phase 2 und Phase 3 Studien.
Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1'000, < 1/100), selten (≥1/10'000, < 1/1'000), sehr selten (< 1/10'000), nicht bekannt (Häufigkeit auf Grundlage der bekannten Daten nicht abschätzbar).
Innerhalb der Häufigkeitsbereiche werden die Nebenwirkungen in absteigender Reihenfolge bezüglich ihres Schweregrades angegeben.
Tabelle 2. Aus kontrollierten Phase-2 und Phase-3-Studien mit Erwachsenen berichtete Nebenwirkungen und aus Postmarketing Berichten
|
Systemorganklassen gemäss MedDRA
|
Häufigkeit
|
Unerwünschte Wirkung
| |
Erkrankungen des Immunsystems
|
Selten
|
Anaphylaktische Reaktion
| |
Stoffwechsel- und Ernährungsstörungen
|
Häufig
|
Hypoglykämie*
| |
Gelegentlich
|
Dehydrierung
| |
Psychiatrische Erkrankungen
|
Häufig
|
Schlaflosigkeit**
| |
Erkrankungen des Nervensystems
|
Sehr häufig
|
Kopfschmerzen
| |
Häufig
|
Schwindel
Geschmacksstörung
| |
Herzerkrankungen
|
Gelegentlich
|
Tachykardie
| |
Erkrankungen des Gastrointestinaltrakts
|
Sehr häufig
|
Übelkeit
Erbrechen
Durchfall
Obstipation
| |
Häufig
|
Mundtrockenheit
Dyspepsie
Gastritis
Gastroösophageale Refluxkrankheit
Oberbauchschmerzen
Flatulenz
Aufstossen
Abdominelles Spannungsgefühl
| |
Gelegentlich
|
Verzögerte Magenentleerung
Pankreatitis***
| |
Nicht bekannt
|
Darmobstruktion†a
| |
Leber- und Gallenerkrankungen
|
Häufig
|
Cholelithiasis***
| |
Gelegentlich
|
Cholezystitis***
| |
Untersuchungen
|
Häufig
|
Erhöhte Lipase
Erhöhte Amylase
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Häufig
|
Hautausschlag
| |
Gelegentlich
|
Urtikaria
| |
Erkrankungen der Nieren und Harnwege
|
Selten
|
Akutes Nierenversagen
Beeinträchtigung der Nierenfunktion
| |
Nicht bekannt
|
Kutane Amyloidose†
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Häufig
|
Reaktionen an der Injektionsstelle
Asthenie
Erschöpfung
| |
Gelegentlich
|
Unwohlsein
|
*Hypoglykämie (basierend auf Symptomen, die von den Patienten selbst berichtet, anhand von Blutzuckermessungen jedoch nicht bestätigt wurden) berichtet bei Patienten ohne Diabetes mellitus Typ 2, die mit Saxenda in Kombination mit Diät und körperlicher Aktivität behandelt wurden. Siehe Abschnitt «Beschreibung ausgewählter Nebenwirkungen» für weitere Informationen.
**Schlaflosigkeit wurde hauptsächlich während der ersten 3 Behandlungsmonate beobachtet.
***Siehe «Warnhinweise und Vorsichtsmassnahmen».
†Arzneimittelnebenwirkung aus Meldungen nach Markteinführung.
a) Die zusammengefasste Bezeichnung umfasst die unerwünschten Ereignisse Darmobstruktion, Ileus und Dünndarmobstruktion.
Beschreibung ausgewählter Nebenwirkungen
Gutartige, bösartige und unspezifische Neubildungen (einschl. Zysten und Polypen)
Gelegentlich – Brustkrebs
In den klinischen Studien zu Saxenda wurde Brustkrebs bei 17 (0.7 %) von 2'379 mit Saxenda behandelten Frauen berichtet, im Vergleich zu 3 (0.2 %) von 1'300 mit Placebo behandelten Frauen, einschliesslich invasiver Krebs (13 mit Saxenda und 2 mit Placebo behandelte Frauen) und duktales Karzinom in situ (4 mit Saxenda und 1 mit Placebo behandelte Frau). Die Mehrheit der Krebserkrankungen waren Östrogen- und Progresteron-Rezeptor-positiv. Es gab zu wenige Fälle, um ermitteln zu können, ob bei diesen Fällen ein Zusammenhang mit Saxenda bestand. Ausserdem liegen keine ausreichenden Daten vor, um zu ermitteln, ob Saxenda Auswirkungen auf bereits bestehende Brustneoplasien hat.
Gelegentlich – Papillärer Schilddrüsenkrebs
In den klinischen Studien zu Saxenda wurde papillärer Schilddrüsenkrebs bei 8 (0.2 %) von 3'291 mit Saxenda behandelten Patienten berichtet, im Vergleich zu keinen Fällen bei den 1'843 mit Placebo behandelten Patienten. Vier dieser papillären Schilddrüsenkarzinome hatten einen maximalen Durchmesser von weniger als 1 cm, und 4 wurden in Gewebeproben nach Thyroidektomie diagnostiziert, die durch vor der Saxenda-Behandlung identifizierte Befunde veranlasst wurden.
Gelegentlich – Kolorektale Neoplasmen
In den klinischen Studien zu Saxenda wurden benigne kolorektale Neoplasmen (zumeist Kolonadenome) bei 20 (0.6 %) von 3'291 mit Saxenda behandelten Patienten berichtet, im Vergleich zu 7 (0.4 %) von 1'843 mit Placebo behandelten Patienten. Sechs Fälle von malignem kolorektalem Karzinom wurden bei 5 der mit Saxenda behandelten Patienten (0.2 %) berichtet und einer bei den mit Placebo behandelten Patienten (0.1 %).
Erkrankungen des Immunsystems
Selten – Anaphylaktische Reaktionen
Einige Fälle anaphylaktischer Reaktionen mit Symptomen wie niedrigem Blutdruck, Herzklopfen, Atemnot und Ödemen wurden bei der Anwendung von Liraglutide nach der Markteinführung gemeldet. Anaphylaktische Reaktionen können potenziell lebensbedrohlich sein. Besteht der Verdacht auf eine anaphylaktische Reaktion, ist Liraglutide abzusetzen und die Behandlung nicht wieder aufzunehmen (siehe «Kontraindikationen»).
Stoffwechsel- und Ernährungsstörungen
Sehr häufig – Hypoglykämie bei Patienten mit Typ 2 Diabetes mellitus (23 %)
In einer klinischen Studie mit übergewichtigen oder adipösen Patienten mit Diabetes mellitus Typ 2, die mit Saxenda in Kombination mit Diät und körperlicher Aktivität behandelt wurden, wurden schwere hypoglykämische Ereignisse (für die der Patient Fremdhilfe benötigte) von 0.7 % der mit Saxenda behandelten Patienten berichtet, aber nur bei Patienten die gleichzeitig auch mit Sulfonylharnstoff behandelt wurden. Darüber hinaus wurde bei diesen Patienten dokumentierte symptomatische Hypoglykämien von 43.6 % der mit Saxenda behandelten Patienten und von 27.3 % der mit Placebo behandelten Patienten berichtet. Von den nicht gleichzeitig mit Sulfonylharnstoff behandelten Patienten berichteten 15.7 % der mit Saxenda behandelten Patienten und 7.6 % der mit Placebo behandelten Patienten über dokumentierte symptomatische hypoglykämische Ereignisse (definiert durch einen Plasmaglucosewert von ≤3.9 mmol/l begleitet von Symptomen).
Häufig – Hypoglykämie bei Patienten ohne Typ 2 Diabetes mellitus
In klinischen Studien mit übergewichtigen oder adipösen Patienten ohne Diabetes mellitus Typ 2, die mit Saxenda in Kombination mit Diät und körperlicher Aktivität behandelt wurden, wurden keine schweren hypoglykämischen Ereignisse (für die der Patient Fremdhilfe benötigt hätte) berichtet. Mit einer Hypoglykämie kompatible Symptome wurden von 1.6 % der mit Saxenda behandelten Patienten und von 1.1 % der mit Placebo behandelten Patienten berichtet; diese Ereignisse wurden jedoch nicht durch Blutzuckermessungen bestätigt. Die meisten dieser Ereignisse waren leichter Natur.
Sehr häufig – Hypoglykämie bei mit Insulin behandelten Patienten mit Typ 2 Diabetes mellitus
In einer klinischen Studie mit übergewichtigen oder adipösen Patienten mit Typ 2 Diabetes mellitus, die mit Insulin und Saxenda in Kombination mit Diät und körperlicher Aktivität und mit bis zu zwei OAD's behandelt wurden, wurden 1.5% schwere Hypoglykämien (es wurde Fremdhilfe benötigt) unter Saxenda gemeldet. In dieser Studie wurden 47.2% symptomatische Hypoglykämien (definiert durch einen Plasmaglukosewert von ≤3.9 mmol/l begleitet von Symptomen) unter Saxenda und 51.8 % unter Placebo gemeldet. In der Untergruppe, die zusätzlich mit Sulfonylharnstoff behandelt wurden, berichteten 60.9 % der Patienten unter Saxenda und 60 % der Patienten unter Placebo dokumentierte symptomatische Hypoglykämie-Ereignisse.
Herzerkrankungen
Gelegentlich – Tachykardie
In klinischen Studien wurde eine Tachykardie bei 0.6 % der Patienten, die mit Saxenda behandelt wurden, und bei 0.1 % der mit Placebo behandelten Patienten beobachtet. Die meisten dieser Ereignisse waren leichter oder mittelschwerer Natur. Die Ereignisse traten nur isoliert auf und die meisten klangen im Laufe der Behandlung mit Saxenda ab.
Häufig – Hypotonie
Nebenwirkungen im Zusammenhang mit Hypotonie (das heisst, Berichte von Hypotonie, orthostatischer Hypotonie, Kreislaufkollaps und vermindertem Blutdruck) wurden in den klinischen Studien zu Saxenda häufiger bei Saxenda (1.1 %) berichtet als bei Placebo (0.5 %). Verminderungen des systolischen Blutdrucks auf weniger als 80 mmHg wurden bei 4 (0.1 %) der mit Saxenda behandelten Patienten beobachtet, im Vergleich zu keinem bei den mit Placebo behandelten Patienten. Bei einem der mit Saxenda behandelten Patienten ist Hypotonie im Zusammenhang mit gastrointestinalen Nebenwirkungen und Niereninsuffizienz aufgetreten.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig – Übelkeit (39.3 %), Erbrechen (15.7 %), Durchfall (20.9 %), Obstipation (19.4 %)
Häufig – Mundtrockenheit, Dyspepsie, Gastritis, gastroösophageale Refluxkrankheit, Oberbauchschmerzen, Flatulenz, Aufstossen, abdominelles Spannungsgefühl
Gelegentlich – Verzögerte Magenentleerung
Die meisten Episoden von gastrointestinalen Ereignissen waren leicht bis mittelschwer, vorübergehend und führten grösstenteils nicht zum Abbruch der Behandlung. Die Reaktionen traten in der Regel in den ersten Behandlungswochen auf und nahmen bei Fortsetzung der Behandlung innerhalb weniger Tage oder Wochen ab.
Bei Patienten im Alter von ≥65 Jahre können bei Behandlung mit Saxenda häufiger gastrointestinale Beschwerden auftreten.
Patienten mit leichter oder mittelschwerer Einschränkung der Nierenfunktion (Kreatinin-Clearance ≥30 ml/min) können unter der Behandlung mit Saxenda häufiger gastrointestinale Beschwerden haben.
Erkrankungen der Nieren und Harnwege
Selten – Akutes Nierenversagen
Bei Patienten, die mit GLP-1-Rezeptor-Agonisten behandelt wurden, wurde über akutes Nierenversagen berichtet. Die meisten der berichteten Ereignisse traten bei Patienten auf, bei denen es zu Übelkeit, Erbrechen oder Durchfall mit anschliessender Volumendepletion gekommen war («Warnhinweise und Vorsichtsmassnahmen»).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig – Reaktionen an der Injektionsstelle
Reaktionen an der Injektionsstelle wurden bei mit Saxenda behandelten Patienten berichtet. Diese Reaktionen waren in der Regel leicht und vorübergehend und die meisten verschwanden im Laufe der Behandlung.
Unerwünschte Wirkungen nach Markteinführung
Die folgenden Nebenwirkungen wurden bei der Anwendung von Liraglutide, dem Wirkstoff von Saxenda, nach Erteilung der Zulassung berichtet. Weil diese Nebenwirkungen freiwillig von einer Population mit unbestimmter Grösse berichtet werden, ist es nicht immer möglich, zuverlässig ihre Häufigkeit abzuschätzen oder einen kausalen Zusammenhang mit der Wirkstoffexposition zu begründen.
Neoplasmen: Medulläres Schilddrüsenkarzinom.
Erkrankungen des Gastrointestinaltrakts: Akute Pankreatitis, hämorrhagische und nekrotisierende Pankreatitis, mitunter mit Todesfolge.
Stoffwechsel und Ernährungsstörungen: Dehydrierung infolge von Übelkeit, Erbrechen und Diarrhöe.
Erkrankungen der Nieren und Harnwege: Anstieg des Serumkreatinins, akute Niereninsuffizienz oder Verschlechterung bei chronischer Niereninsuffizienz, erforderte mitunter eine Hämodialyse.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Allergische Reaktionen: Hautausschlag und Pruritus.
Erkrankungen des Immunsystems: Angioödem und anaphylaktische Reaktionen.
Leber- und Gallenerkrankungen: Anstiege von Leberenzymkonzentrationen, Hyperbilirubinämie, Cholestase, Hepatitis.
Kinder und Jugendliche
In einer klinischen Studie, die bei Jugendlichen von 12 bis 18 Jahren mit Adipositas durchgeführt wurde, wurden 125 Patienten über einen Zeitraum von 56 Wochen mit Saxenda behandelt.
Die Häufigkeit, Art und Schwere der Nebenwirkungen war insgesamt mit den bei den Erwachsenen beobachteten Nebenwirkungen vergleichbar. Erbrechen trat bei Jugendlichen im Vergleich zu Erwachsenen doppelt so häufig auf.
Der Anteil der Patienten, die über mindestens eine Episode einer klinisch relevanten Hypoglykämie berichteten, war unter Liraglutide (1.6 %) höher als bei den mit Placebo behandelten Patienten (0.8 %). Während der Studie traten keine schweren hypoglykämischen Episoden auf.
Es wurden keine Auswirkungen auf das Wachstum und die pubertäre Entwicklung nach einer 56-wöchigen Behandlung festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien und bei der Anwendung von Liraglutide nach Markteinführung wurden Überdosierungen von bis zu 72 mg (24-Fache der für die Gewichtsregulierung empfohlenen Dosis) berichtet.
Anzeichen und Symptome
Die berichteten Ereignisse schliessen schwere Übelkeit, starkes Erbrechen und Durchfall ein, welches auch die zu erwartenden Symptome einer Überdosierung von Liraglutide sind. Es wurden Fälle schwerer Hypoglykämien berichtet. Alle Patienten erholten sich komplikationslos.
Behandlung
Im Fall einer Überdosierung ist eine angemessene unterstützende Behandlung entsprechend den klinischen Anzeichen und Symptomen des Patienten einzuleiten. Der Patient muss bezüglich klinischer Anzeichen von Dehydrierung beobachtet werden und der Blutzuckerspiegel muss überwacht werden.
Eigenschaften/WirkungenATC-Code
A10BJ02
Wirkungsmechanismus
Liraglutide ist ein acyliertes Analogon des humanen GLP-1 (Glucagon-like Peptid-1) mit einer 97-%-igen Aminosäurensequenz-Homologie zum endogenen humanen GLP-1. Liraglutide bindet an den GLP-1-Rezeptor (GLP-1R) und aktiviert diesen.
GLP-1 ist ein physiologischer Regulator des Appetits und der Nahrungsaufnahme, doch der genaue Wirkmechanismus ist noch nicht vollständig bekannt. In tierexperimentellen Studien führte die periphere Verabreichung zu einer Aufnahme von Liraglutide in bestimmten Hirnregionen, die mit der Appetitregulierung assoziiert sind, wo Liraglutide über die spezifische Aktivierung von GLP-1R zu einem Anstieg der wichtigsten Sättigungssignale und einer Abnahme der wichtigsten Hungersignale führte und damit zu einem geringeren Körpergewicht.
GLP-1-Rezeptoren werden an spezifischen Orten des Herzens, des Gefässsystems, des Immunsystems und der Nieren exprimiert. Human- bzw. Tierstudien belegen, dass diese Rezeptoren kardiovaskuläre Wirkungen von Liraglutide vermitteln können, einschliesslich verminderter Entzündung und der verzögerten Progression einer Atherosklerose.
Pharmakodynamik
Liraglutide reduziert das Körpergewicht beim Menschen hauptsächlich durch eine Abnahme der Fettmasse, wobei der relative Verlust an viszeralem Fett grösser ist als der Verlust an subkutanem Fett. Liraglutide reguliert den Appetit durch eine Steigerung des Völle- und Sättigungsgefühls und eine Reduzierung des Hungergefühls und des Wunsches nach Nahrungsverzehr und führt so zu einer geringeren Nahrungsaufnahme. Liraglutide erhöht im Vergleich zu Placebo nicht den Energieverbrauch.
Liraglutide stimuliert die Insulinsekretion und senkt die Glucagonsekretion in einem glucoseabhängigen Mechanismus, was zu einer Senkung des postprandialen und des Nüchternblutzuckers führt. Die blutzuckersenkende Wirkung ist bei Patienten mit Prädiabetes und Diabetes stärker ausgeprägt als bei Patienten mit Blutzuckerwerten im Normbereich. Klinische Studien legen nahe, dass Liraglutide die Betazellfunktion verbessert und unterstützt. Dabei wurden Messungen wie das HOMA-B und das Verhältnis von Proinsulin zu Insulin zugrunde gelegt.
Klinische Wirksamkeit
Die Wirksamkeit und die Sicherheit von Liraglutide für die Gewichtsregulierung in Verbindung mit einer verminderten Kalorienzufuhr und verstärkter körperlicher Aktivität wurden in vier randomisierten, doppelblinden, Placebo-kontrollierten Phase-3-Studien untersucht, an denen insgesamt 5'358 Patienten teilnahmen.
Studie 1 (SCALE Obesity & Pre-Diabetes – 1839): Insgesamt wurden 3'731 Patienten mit Adipositas (BMI ≥30 kg/m2) oder Übergewicht (BMI ≥27 kg/m2) mit Dyslipidämie und/oder Hypertonie, nach ihrem Prädiabetes-Stadium zum Zeitpunkt der Einschlussuntersuchung sowie nach ihrem BMI bei Studienbeginn (≥30 kg/m² oder < 30 kg/m²), stratifiziert. Alle 3'731 randomisierten Patienten erhielten eine 56-wöchige Behandlung, und 2'254 randomisierte Patienten, mit einem vorhandenen Prädiabetes zum Zeitpunkt der Einschlussuntersuchung, erhielten eine 160-wöchige Behandlung. Auf beide Behandlungszeiträume folgte eine 12-wöchige Nachbeobachtungszeit ohne Arzneimittel/Placebo. Hintergrundtherapie für alle Patienten war eine Lebensstilintervention in Form von einer energiereduzierten Diät sowie einer Beratung hinsichtlich körperlicher Aktivität.
Im 56-wöchigen Teil der Studie 1 wurde der Gewichtsverlust bei allen 3'731 randomisierten Patienten bewertet (2'590 Patienten schlossen die Studie ab).
Im 160-wöchigen Teil der Studie 1 wurde die Zeit bis zum Auftreten eines Diabetes mellitus Typ 2 in den 2'254 randomisierten Patienten mit einem bereits vorhandenen Prädiabetes bewertet (1'128 Patienten schlossen die Studie ab).
Studie 2 (SCALE Diabetes – 1922): Eine 56-wöchige Studie zur Bewertung des Gewichtsverlusts als Primärendpunkt bei 846 randomisierten adipösen und übergewichtigen Patienten (628 Patienten schlossen die Studie ab) mit unzureichend kontrolliertem Diabetes mellitus Typ 2 (HbA1c-Bereich 7–10 %). Die Standardtherapie bei Studienbeginn war entweder ausschliesslich Diät und körperliche Aktivität, Metformin, ein Sulfonylharnstoff oder ein Glitazon, jeweils als Einzelwirkstoff oder in einer Kombination hiervon.
Studie 3 (SCALE Maintenance – 1923): Eine 56-wöchige Studie zur Bewertung der Erhaltung des Körpergewichts und des Gewichtsverlusts als Primärendpunkt bei 422 randomisierten adipösen und übergewichtigen Patienten (305 Patienten schlossen die Studie ab) mit Hypertonie oder Dyslipidämie nach einer vorangegangenen Gewichtsabnahme von ≥5 % infolge einer kalorienarmen Diät.
Studie 4 (SCALE Sleep Apnoe – 3970): Eine 32-wöchige Studie zur Bewertung des Schweregrads der Schlafapnoe als Primärendpunkt und des Gewichtsverlusts als Sekundärendpunkt bei 359 randomisierten adipösen Patienten (276 Patienten schlossen die Studie ab) mit mittelschwerer oder schwerer obstruktiver Schlafapnoe.
Körpergewicht
Die Daten zu Gewichtsabnahme, Therapie-Respondern, Zeitverlauf und kumulativer Verteilung der Gewichtsveränderung (%) für die Studien 1–3 sind in der Tabelle 3 und den Abbildungen 1, 2, 3 und 4 dargestellt.
Tabelle 3. Änderungen gegenüber dem Ausgangswert bei Körpergewicht in Woche 56 und 160 – Studien 1, 2 und 3
|
|
Studie 1 (Woche 56)
|
Studie 1 (Woche 160)
|
Studie 2
|
Studie 3
| |
|
Saxenda
n=2'437
|
Placebo
n=1'225
|
Saxenda (N=1'472)
|
Placebo (N=738)
|
Saxenda
n=412
|
Placebo
n=211
|
Saxenda
n=207
|
Placebo
n=206
| |
Körpergewicht
| |
Ausgangswert im Mittel, kg (SA)
|
106.3
(21.2)
|
106.3
(21.7)
|
107.6 (21.6)
|
108.0 (21.8)
|
105.6
(21.9)
|
106.7
(21.2)
|
100.7 (20.8)
|
98.9 (21.2)
| |
Änderung gegenüber Ausgangswert, %
|
-8.0
|
-2.6
|
-6.2
|
-1.8
|
-5.9
|
-2.0
|
-6.3
|
-0.2
| |
Saxenda gegenüber
Placebo,
% (95 % KI)
|
-5.4*
(-5.8; -5.0)
|
-4.3** (-4.9; -3.7)
|
-4.0**
(-4.8; -3.1)
|
-6.1**
(-7.5; -4.6)
| |
Änderung gegenüber Ausgangswert, kg
|
-8.4
|
-2.8
|
-6.5
|
-2.0
|
-6.2
|
-2.2
|
-6.0
|
-0.2
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
-5.6**
(-6.0; -5.1)
|
-4.6** (-5.3; -3.9)
|
-4.1**
(-5.0; -3.1)
|
-5.9**
(-7.3; -4.4)
| |
% der Patienten mit
≥5 % Gewichtsabnahme
|
63.5
|
26.6
|
49.6
|
23.4
|
49.8
|
13.5
|
50.7
|
21.3
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
4.8**
(4.1; 5.6)
|
3.2** (2.6; 3.9)
|
6.4**
(4.1; 10.0)
|
3.8**
(2.4; 6.0)
| |
% der Patienten mit
> 10 % Gewichtsabnahme
|
32.8
|
10.1
|
24.4
|
9.5
|
22.9
|
4.2
|
27.4
|
6.8
| |
Saxenda gegenüber
Placebo,
kg (95 % KI)
|
4.3**
(3.5; 5.3)
|
3.1** (2.3; 4.1)
|
6.8**
(3.4; 13.8)
|
5.1**
(2.7; 9.7)
|
Gesamtgruppe (FAS=Full Analysis Set). Für Körpergewicht sind die Ausgangswerte Mittelwerte, Änderungen gegenüber den Ausgangswerten in Woche 56 und Woche 160 sind geschätzte Mittelwerte (LSMeans = kleinste Fehlerquadrate) und Behandlungsunterschiede in Woche 56 und Woche 160 sind geschätzte Behandlungsunterschiede. Für die Anteile der Patienten (%), die ≥5/> 10 % Körpergewicht verloren haben, wurden geschätzte Odds-Verhältnisse verwendet. Fehlende Werte nach Studienbeginn wurden unter Verwendung der Last Observation Carried Forward (LOCF) berechnet. * p < 0.05. ** p < 0.0001. SA=Standardabweichung. KI=Konfidenzintervall.
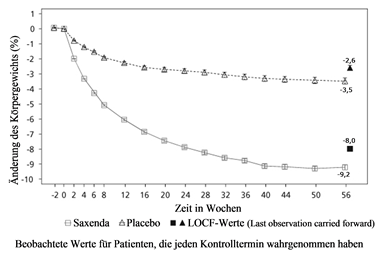
Abbildung 1. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 1 (0–56 Wochen) gegenüber dem Ausgangswert
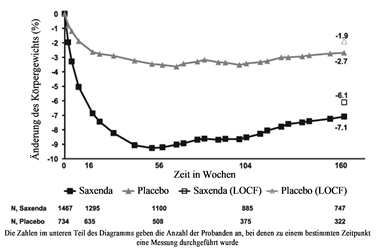
Abbildung 2. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 1 (0–160 Wochen) gegenüber dem Ausgangswert
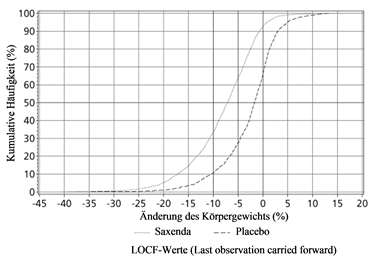
Abbildung 3. Kumulative Verteilung der Gewichtsänderung (%) nach 56 Behandlungswochen in Studie 1
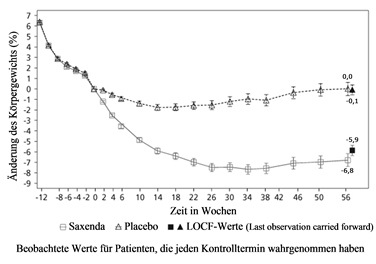
Abbildung 4. Änderung des Körpergewichts (%) im Zeitverlauf in Studie 3 gegenüber der Randomisierung (Woche 0)
Vor Woche 0 bestand die Behandlung der Patienten nur aus kalorienarmer Diät und körperlicher Aktivität. In Woche 0 wurden die Patienten randomisiert der Behandlungsgruppe mit Saxenda oder Placebo zugeteilt.
Gewichtsabnahme nach 12-wöchiger Behandlung mit Liraglutide (3.0 mg)
Als «Early Responders» wurden die Patienten definiert, die nach 12-wöchiger Therapie mit der Behandlungsdosis von Liraglutide (4 Wochen Dosissteigerung und 12 Wochen Behandlungsdosis) eine Gewichtsabnahme von ≥5 % erzielten. In Studie 1 waren 67.5 % «Early Responders». In Studie 2 waren es 50.4 % der Patienten. Bei Fortsetzung der Behandlung mit Liraglutide erzielen voraussichtlich 86.2 % dieser «Early Responders» nach 1 Jahr Behandlung eine Gewichtsabnahme von ≥5 % und 51 % erzielen voraussichtlich eine Gewichtsabnahme von ≥10 %. Die voraussichtliche durchschnittliche Gewichtsabnahme bei den «Early Responders», die 1 Jahr Behandlung durchlaufen, beträgt 11.2 % ihres Ausgangskörpergewichts (9.7 % bei Männern und 11.6 % bei Frauen). In der Gruppe von Patienten, die nach 12-wöchiger Therapie mit 3.0 mg Liraglutide pro Tag eine Gewichtsabnahme von < 5 % erreicht haben, erreichen noch 6.6 % der Patienten eine Gewichtsabnahme von ≥10 % nach 1 Jahr.
Blutzucker und kardiometabolische Parameter
Daten zum Blutzucker und kardiometabolischen Parametern in den Studien 1 und 2 sind in der Tabelle 4 dargestellt.
Tabelle 4. Änderungen nach 56 Wochen (Studien 1 und 2) und 160 Wochen (Studie 1) gegenüber dem Ausgangswert bei Blutzucker und kardiometabolischen Parametern
|
|
Saxenda
(n=2'437)
|
Placebo
(n=1'225)
|
Saxenda gegenüber Placebo
| |
Studie 1
(Woche 56)
|
Ausgangswert
|
Änderung
|
Ausgangswert
|
Änderung
|
| |
HbA1c, %
|
5.6
|
-0.3
|
5.6
|
-0.1
|
-0.23**
(0.25; -0.21)
| |
NPG, mmol/l
|
5.3
|
-0.4
|
5.3
|
-0.01
|
-0.38**
(0.42; -0.35)
| |
Systolischer Blutdruck, mmHg
|
123.0
|
-4.3
|
123.3
|
-1.5
|
-2.8**
(-3.6; -2.1)
| |
Diastolischer Blutdruck, mmHg
|
78.7
|
-2.7
|
78.9
|
-1.8
|
-0.9*
(-1.4; - 0.4)
| |
Taillenumfang, cm
|
115.0
|
-8.2
|
114.5
|
-4.0
|
-4.2**
(-4.7; -3.7)
| |
|
Saxenda (n=1'472)
|
Placebo
(n=738)
|
Saxenda gegenüber Placebo
| |
Studie 1
(Woche 160)
|
Ausgangswert
|
Änderung
|
Ausgangswert
|
Änderung
|
| |
HbA1c, %
|
5.75
|
-0.35
|
5.74
|
-0.14
|
-0.21**
(-0.24; -0.18)
| |
NPG, mmol/l
|
5,50
|
-0,37
|
5,46
|
0,04
|
-0.41**
(-0.46; -0.36)
| |
Systolischer Blutdruck, mmHg
|
124.80
|
-3.24
|
125.01
|
-0.44
|
-2.80**
(-3.81; -1.79)
| |
Diastolischer Blutdruck, mmHg
|
79.40
|
-2.36
|
79.83
|
-1.74
|
-0.62
(-1.33; 0.09)
| |
Taillenumfang, cm
|
116.64
|
-6.88
|
116.74
|
-3.35
|
-3.53**
(-4.23; -2.83)
| |
Studie 2
|
(n=412)
|
(n=211)
|
| |
HbA1c, %
|
7.9
|
-1.3
|
7.9
|
-0.4
|
-0.9**
(-1.1; -0.8)
| |
NPG, mmol/l
|
8.8
|
-1.9
|
8.6
|
-0.1
|
-1.8**
(-2.1; -1.4)
| |
Systolischer Blutdruck, mmHg
|
128.9
|
-3.0
|
129.2
|
-0.4
|
-2.6*
(-4.6; - 0.6)
| |
Diastolischer Blutdruck, mmHg
|
79.0
|
-1.0
|
79.3
|
-0.6
|
-0.4
(1.7; 1.0)
| |
Taillenumfang, cm
|
118.1
|
-6.0
|
117.3
|
-2.8
|
-3.2**
(-4.2; -2.2)
| |
Bei der statistischen Analyse der glykämischen und kardiometabolischen Parameter wurde das multiple Testen nicht berücksichtigt, und somit müssen die Ergebnisse als statistisch nicht-konfirmatorisch betrachtet werden. Gesamtgruppe (FAS=Full Analysis Set). Für HbA1c, NPG, Blutdruck und Taillenumfang sind die Ausgangswerte Mittelwerte, Änderungen gegenüber den Ausgangswerten in Woche 56 und Woche 160 sind geschätzte Mittelwerte (LSMeans) und Behandlungsunterschiede in Woche 56 und Woche 160 sind geschätzte Behandlungsunterschiede. Fehlende Werte nach Studienbeginn wurden unter Verwendung der Last Observation Carried Forward (LOCF) berechnet. * p < 0.05. ** p < 0.0001.
SA=Standardabweichung. KI=Konfidenzintervall.
|
Immunogenität
Mit Saxenda behandelte Patienten können Anti-Liraglutide-Antikörper entwickeln. Bei einer Untersuchung nach Behandlungsbeginn wurden bei 42 (2.8 %) von 1'505 mit Saxenda behandelten Patienten Anti-Liraglutide-Antikörper nachgewiesen. Antikörper, die in einem in-vitro-Test eine neutralisierende Wirkung hatten, traten bei 18 (1.2 %) von 1'505 mit Saxenda behandelten Patienten auf. Das Vorhandensein von Antikörpern könnte im Zusammenhang mit einem vermehrten Auftreten von Reaktionen an der Injektionsstelle und Berichten von niedrigen Blutzuckerspiegeln stehen. In klinischen Studien wurden diese Ereignisse üblicherweise als leicht eingestuft und verschwanden im weiteren Behandlungsverlauf wieder.
Der Nachweis der Antikörperbildung ist stark abhängig von der Empfindlichkeit und Spezifität des Tests. Ausserdem kann die beobachtete Inzidenz von nachgewiesenen Antikörpern (einschliesslich neutralisierender Antikörper) in einem Test durch verschiedene Faktoren beeinflusst werden, zu denen die Testmethodik, die Probenhandhabung, der zeitliche Ablauf der Probenentnahme, die Begleitmedikation sowie Grunderkrankungen zählen. Aus diesen Gründen kann das Auftreten von Antikörpern gegen Saxenda nicht direkt mit dem Auftreten von Antikörpern anderer Produkte verglichen werden.
Kardiovaskuläre Bewertung
Schwere, unerwünschte kardiovaskuläre Ereignisse (MACE) wurden von einer externen unabhängigen Expertengruppe beurteilt und als nicht-tödlicher Myokardinfarkt, nicht-tödlicher Schlaganfall und kardiovaskulärer Tod klassiert. In allen Studien mit Saxenda traten 6 MACE bei Patienten, die mit Liraglutide behandelt wurden, und 10 MACE bei mit Placebo behandelten Patienten auf. Die Hazard Ratio und 95 % KI ist 0.33 [0.12–0.90] für Liraglutide gegenüber Placebo. In klinischen Phase-3-Studien wurde bei Behandlung mit Liraglutide eine mittlere Erhöhung der Herzfrequenz gegenüber dem Ausgangswert in Höhe von 2.5 Schlägen pro Minute beobachtet (in allen Studien zwischen 1.6 und 3.6 Schläge pro Minute), welche nach etwa 6 Wochen ihr Maximum erreichte und nach Absetzen von Liraglutide reversibel war (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results (LEADER) Studie untersuchte die Häufigkeit schwerer kardiovaskulärer Ereignisse (MACE: kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt, nicht-tödlicher Schlaganfall) in 9'340 Patienten mit erhöhten Diabetes mellitus Typ 2 Diabetes und einem erhöhten kardiovaskulären Risiko. Randomisiert (1:1) wurden die Patienten zusätzlich zur Standardtherapie entweder mit bis zu 1.8 mg Liraglutide täglich (4'668) oder Placebo (4'672) behandelt (mediane Behandlungsdauer zirka 3.5 Jahre). Primärer Endpunkt war die Zeit bis zum ersten Auftreten eines MACE. Dieser wurde durch Liraglutide in der untersuchten Dosierung signifikant reduziert (Hazard Ratio 0.87 [0.78; 0.97] 95 % KI).
Kinder und Jugendliche
In einer doppelblinden Studie, in der die Wirksamkeit und Sicherheit von Saxenda gegenüber Placebo hinsichtlich des Gewichtsverlusts bei Jugendlichen ab 12 Jahren mit Adipositas untersucht wurde, war Saxenda gegenüber Placebo in der Reduktion des Körpergewichtes nach 56 Wochen überlegen (gemessen anhand des BMI-SDS; siehe Tabelle 5).
Ein grösserer Anteil der Patienten erreichten eine ≥5 % und ≥10 % BMI-Reduktion mit Saxenda als mit Placebo sowie eine grössere mittlere BMI-Abnahme und Körpergewichtsabnahme (siehe Tabelle 5).
Die Veränderung der Körperzusammensetzung wurde nicht untersucht.
Alle Patienten sollten engmaschig überwacht werden (siehe auch Abbruchregel unter «Indikation»).
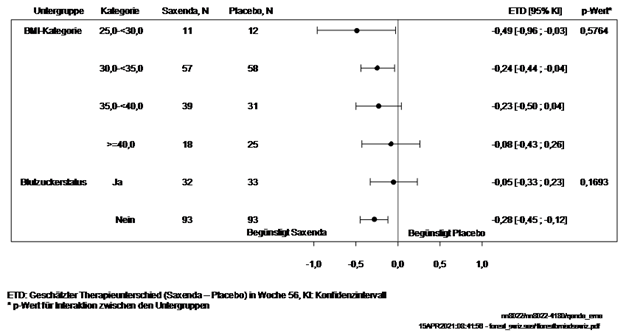
Abbildung 5.
Der Forest-Plot zeigt die geschätzten Unterschiede (Saxenda vs. Placebo) in der Änderung des BMI-SDS von Studienbeginn bis Woche 56 in Abhängigkeit vom BMI Ausgangswert ([kg/m2]: ≥25 -<30, ≥30 - <35, ≥35 - <40 und ≥40) und vom glykämischen Status zu Beginn der Studie (Normoglykämie vs. Prädiabetes oder Diabetes Typ 2) inklusive der der p-Werte für die Interaktionstests.
Nach einer 26-wöchigen Nachbeobachtungszeitraum ohne die Studienmedikation bzw. Placebo wurde in der Saxenda Gruppe gegenüber der Placebo-Gruppe eine erneute Gewichtszunahme beobachtet (siehe Tabelle 5):
Tabelle 5. NN8022-4180 Änderung des Körpergewichts und des BMI in Woche 56 und Änderung des BMI-SDS von Woche 56 bis Woche 82
|
|
Saxenda (N=125)
|
Placebo (N=126)
|
Saxenda vs.
Placebo
| |
BMI-SDS
|
|
|
| |
Ausgangswert, BMI-SDS (SD)
|
3.14 (0.65)
|
3.20 (0.77)
|
| |
Mittlere Änderung in Woche 56 (95%-KI)
|
-0.23
|
-0.00
|
-0.22*
(-0.37; -0.08)
| |
Woche 56, BMI-SDS (SD)
|
2.88 (0.94)
|
3.14 (0.98)
|
| |
Mittlere Änderung von Woche 56 bis Woche 82, BMI-SDS (95%-KI)
|
0.22
|
0.07
|
0.15**
(0.07; 0.23)
| |
Körpergewicht
|
|
|
| |
Ausgangswert, kg (SD)
|
99.3 (19.7)
|
102.2 (21,6)
|
-
| |
Mittlere Änderung in Woche 56, % (95%-KI)
|
-2.65
|
2.37
|
-5.01**
(-7.63; -2.39)
| |
Mittlere Änderung in Woche 56, kg (95%-KI)
|
-2.,26
|
2.25
|
-4.50**
(-7.17; -1.84)
| |
BMI
|
|
|
| |
Ausgangswert, kg/m2 (SD)
|
35.3 (5.1)
|
35.8 (5.7)
|
-
| |
Mittlere Änderung in Woche 56, kg/m2 (95%-KI)
|
-1.39
|
0.19
|
-1.58**
(-2.47; -0.69)
| |
Anteil der Patienten mit einer Reduzierung des BMI-Ausgangswertes von ≥5% in Woche 56, % (95%-KI)
|
43.25
|
18.73
|
3.31**
(1.78; 6.16)
| |
Anteil der Patienten mit einer Reduzierung des BMI-Ausgangswertes von ≥10% in Woche 56, % (95%-KI)
|
26.08
|
8.11
|
4.00**
(1.81; 8.83)
| |
Gesamtgruppe (FAS=Full Analysis Set). Für BMI-SDS, Körpergewicht und BMI sind die Ausgangswerte Mittelwerte, die Änderungen gegenüber den Ausgangswerten in Woche 56 sind geschätzte Mittelwerte (LSMeans) und die Behandlungskontraste in Woche 56 sind geschätzte Behandlungsunterschiede. Die BMI-SDS-Werte in Woche 56 sind Mittelwerte, die Änderungen von Woche 56 bis Woche 82 sind geschätzte Mittelwerte (LSMeans) und die Behandlungskontraste in Woche 82 sind geschätzte Behandlungsunterschiede. Für den Anteil der Patienten, bei denen sich der BMI-Ausgangswert um ≥5%/≥10% reduziert hat, wird die geschätzte Odds Ratio angegeben. Fehlende Beobachtungen wurden mittels multipler Imputation (Jump-to-Reference-Ansatz, x100) basierend auf der Placebo-Gruppe vervollständigt.
*p<0.01, **p<0.001. KI=Konfidenzintervall. SD=Standardabweichung.
|
Basierend auf der Verträglichkeit wurde die Dosis bei 103 Patienten (82.4 %) bis auf 3.0 mg, bei 11 Patienten (8.8 %) bis auf 2.4 mg, bei 4 Patienten (3.2 %) bis auf 1.8 mg, bei 4 Patienten (3.2 %) bis auf 1.2 mg erhöht und 3 Patienten (2.4 %) blieben auf 0.6 mg.
Die Wirksamkeit und Sicherheit von Saxenda bei pädiatrischen Patienten mit Prader-Willi-Syndrom und Adipositas wurde in einer 16-wöchigen doppelblinden Studie (Teil A) mit anschliessender 36-wöchiger unverblindeter (open label) Phase untersucht. Im Teil A wurden insgesamt 32 Jugendliche im Alter von ≥12 bis <18 Jahren (Tanner-Stadium 2-5) randomisiert mit Liraglutid 3 mg (n=20) bzw. Placebo (n=12) behandelt, von denen 18 bzw. 12 die doppelblinde Phase bis Woche 16 sowie 17 bzw. 12 die unverblindete Extensionsphase bis Woche 52 abschlossen. Im Teil B wurden insgesamt 24 Kinder zwischen ≥6 bis <12 Jahren (Tanner-Stadium unter 2) randomisiert mit Liraglutid 3 mg (n=17) bzw. Placebo (n=7) behandelt, von denen 16 bzw. 7 die doppelblinde Phase bis Woche 16 sowie 14 bzw. 7 die unverblindete Extensionsphase bis Woche 52 abschlossen.
Patienten mit einem Körpergewicht von weniger als 45 kg starteten die Dosiseskalation mit einer niedrigeren Dosis von 0.3 mg anstelle von 0.6 mg und erhöhten die maximale Dosis auf 2.4 mg.
Die geschätzten Änderungen des mittleren BMI-SDS in den beiden Behandlungsarmen waren sowohl in Woche 16 (Differenz Liraglutid 3 mg vs. Placebo [95% KI]: -0,07 [-0,23, 0,09] im Teil A und -0,06 [-1.06, 0.93] im Teil B) als auch in Woche 52 (Differenz Liraglutid 3 mg vs. Placebo [95% KI]: -0,14 [-0.62, 0.34] im Teil A und -0.07 [-0.89, 0.76] im Teil B) vergleichbar.
PharmakokinetikAbsorption
Die Resorption von Liraglutide nach subkutaner Gabe war langsam, Maximalkonzentrationen wurden ungefähr 11 Stunden nach der Injektion erreicht. Nach Anwendung von 3.0 mg Liraglutide bei adipösen Patienten (BMI 30–40 kg/m2) erreichte die durchschnittliche Steady-State-Konzentration (AUCτ/24) von Liraglutide etwa 31 nmol/l. Die Liraglutide-Exposition erhöhte sich im Dosisbereich von 0.6 mg bis 3.0 mg proportional zur Dosis. Die absolute Bioverfügbarkeit von Liraglutide nach subkutaner Gabe liegt bei ungefähr 55 %.
Distribution
Das mittlere scheinbare Verteilungsvolumen nach subkutaner Gabe von 3.0 mg Liraglutide beträgt 20–25 l (bei einer Person, die etwa 100 kg wiegt). Liraglutide ist stark an Plasmaproteine gebunden (> 98 %).
Metabolismus
In den 24 Stunden nach Gabe einer Einzeldosis von [3H]-Liraglutide bei gesunden Probanden war intaktes Liraglutide die Hauptkomponente im Plasma. Zwei Metaboliten wurden nachgewiesen (≤9 % und ≤5 % der gesamten Radioaktivitätsexposition im Plasma).
Elimination
Liraglutide wird auf ähnliche Weise wie grosse Proteine endogen metabolisiert, ohne einen bestimmten Haupteliminationsweg. Nach einer Dosis [3H]-Liraglutide wurde kein intaktes Liraglutide in Urin oder Fäzes nachgewiesen. Nur ein geringer Teil der eingesetzten Radioaktivität wurde als Liraglutide-verwandte Metabolite in Urin oder Fäzes ausgeschieden (6 % bzw. 5 %). Die Radioaktivität in Urin und Fäzes wurde hauptsächlich in den ersten 6–8 Tagen ausgeschieden und stimmte jeweils mit den drei Nebenmetaboliten überein.
Die mittlere Clearance nach subkutaner Gabe von Liraglutide beträgt ungefähr 0.9–1.4 l/h mit einer Eliminationshalbwertszeit von ca. 13 Stunden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
In einer Einzeldosis-Studie (0.75 mg) wurde die Pharmakokinetik von Liraglutide bei Patienten mit unterschiedlichen Graden einer Leberfunktionsstörung beurteilt. Verglichen mit gesunden Probanden war die Liraglutide-Exposition bei Patienten mit leichter bis mittelschwerer Leberfunktionsstörung um 13–23 % vermindert. Bei Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Score > 9) war die Exposition deutlich geringer (44 %).
Nierenfunktionsstörungen
In einer Einzeldosis-Studie (0.75 mg) war bei Patienten mit Niereninsuffizienz die Liraglutide-Exposition im Vergleich zu Personen mit normaler Nierenfunktion reduziert. Bei Patienten mit leichter (Kreatinin-Clearance, CrCl 50–80 ml/min), mittelschwerer (CrCl 30-50 ml/min) und schwerer (CrCl < 30 ml/min) Nierenfunktionsstörung und bei dialysepflichtigen Patienten mit einer Nierenerkrankung im Endstadium war die Liraglutide-Exposition um 33 %, 14 %, 27 % bzw. 26 % vermindert.
Ältere Patienten
Ausgehend von Ergebnissen einer populationspharmakokinetischen Analyse von Daten übergewichtiger und adipöser Patienten (18 bis 82 Jahre) hat das Alter keine klinisch relevante Auswirkung auf die Pharmakokinetik von Liraglutide. Eine Dosisanpassung ist bei älteren Menschen nicht erforderlich.
Kinder und Jugendliche
Es wurde für Liraglutide Populationsanalysen mit Daten aus pädiatrischen klinischen Studien durchgeführt.
Die pharmakokinetischen Eigenschaften von Liraglutide 3.0 mg wurden in klinischen Studien bei Jugendlichen mit Adipositas im Alter von 12 bis 18 Jahren bewertet (134 Patienten, Körpergewicht 62–178 kg). Die Liraglutide-Exposition bei Jugendlichen (12–18 Jahre) war mit der bei Erwachsenen mit Adipositas vergleichbar.
Die pharmakokinetischen Eigenschaften wurden auch in einer Studie zur klinischen Pharmakologie bei Kindern mit Adipositas im Alter von 7–11 Jahren (13 Patienten, Körpergewicht von 54–87 kg) bewertet. Die Exposition in Verbindung mit 3.0 mg Liraglutide wurde nach der Korrektur aufgrund des Körpergewichts bei Kindern im Alter von 7–11 Jahren und Erwachsenen als vergleichbar eingeschätzt.
Geschlecht
Ausgehend von Ergebnissen populationspharmakokinetischer Datenanalysen haben Frauen eine um 24 % niedrigere gewichtskorrigierte Clearance von Liraglutide als Männer. Ausgehend von den Expositions-Wirkungs-Daten ist keine geschlechtsspezifische Dosisanpassung erforderlich.
Ethnische Zugehörigkeit
Ausgehend von Ergebnissen populationspharmakokinetischer Analysen von Daten übergewichtiger und adipöser weisser, schwarzer, asiatischer und lateinamerikanischer/nicht-lateinamerikanischer Patienten hat die ethnische Zugehörigkeit keine klinisch relevante Auswirkung auf die Pharmakokinetik von Liraglutide.
Körpergewicht
Die Exposition von Liraglutide nimmt mit zunehmendem Ausgangskörpergewicht ab. Nach Beurteilung der Expositions-Wirkungs-Daten der klinischen Studien ermöglichte die Tagesdosis von 3.0 mg Liraglutide eine angemessene systemische Exposition in einem Körpergewichtsbereich von 60–234 kg. Bei Patienten mit einem Körpergewicht > 234 kg wurde die Liraglutide-Exposition nicht untersucht.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe oder Genotoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Karzinogenität
Bei einer zweijährigen Karzinogenitätsstudie bei Ratten und Mäusen traten nichtletale C-Zelltumoren der Schilddrüse auf. Bei Ratten wurde ein No Observed Adverse Effect Level (NOAEL) nicht beobachtet. Bei Affen, die 20 Monate lang behandelt wurden, wurden diese Tumoren nicht beobachtet. Diese Befunde bei Nagetieren werden durch einen nichtgenotoxischen, spezifisch durch den GLP-1-Rezeptor vermittelten Mechanismus verursacht, für den Nager besonders empfänglich sind. Die Relevanz für den Menschen ist wahrscheinlich gering, kann jedoch nicht komplett ausgeschlossen werden. Im Zusammenhang mit der Behandlung wurden keine anderen Tumoren festgestellt.
Reproduktionstoxizität
Tierexperimentelle Studien zeigten keine direkt schädigende Wirkung hinsichtlich Fertilität, aber bei der höchsten Dosis eine leicht erhöhte Embryonensterblichkeit in frühen Stadien. Eine Anwendung von Liraglutide während des mittleren Abschnitts der Tragzeit führte zu einer Reduktion des mütterlichen Gewichts und des Fötuswachstums mit nicht eindeutigen Auswirkungen auf die Rippen von Ratten und Skelettveränderungen bei Kaninchen. Unter Einwirkung von Liraglutide war bei Ratten das neonatale Wachstum reduziert. In der Gruppe mit der höchsten Dosis hielt dieser Effekt in der Zeit nach dem Absetzen an. Es ist nicht bekannt, ob das verminderte Wachstum der Jungtiere durch eine geringere Milchaufnahme aufgrund einer direkten GLP-1-Wirkung oder durch geringere Milchproduktion der Muttertiere aufgrund einer verminderten Kalorienaufnahme verursacht wird.
Juvenile Studien
Bei juvenilen männlichen und weiblichen Ratten verursachte Liraglutide bei klinisch relevanten Expositionen eine verzögerte Geschlechtsreife. Diese Verzögerungen hatten weder Auswirkung auf die Fertilität und die Fortpflanzungsfähigkeit beider Geschlechter noch auf die Fähigkeit der Weibchen, eine Trächtigkeit auszutragen.
Sonstige HinweiseInkompatibilitäten
Werden Substanzen zu Saxenda hinzugefügt, können diese zu einer Degradation von Liraglutide führen. Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach dem erstmaligen Gebrauch des Saxenda-Pens kann das Produkt 1 Monat bei Raumtemperatur (nicht über 30°C) oder im Kühlschrank (2–8°C) aufbewahrt werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Im Kühlschrank (2–8°C) lagern.
Nicht einfrieren.
Nicht in der Nähe des Gefrierfachs lagern.
Verschlusskappe aufgesetzt lassen, um den Inhalt vor Licht zu schützen.
Hinweise für die Handhabung
Die Lösung darf nicht verwendet werden, wenn sie nicht klar und farblos oder nahezu farblos aussieht.
Einmal gefrorenes Saxenda darf nicht mehr verwendet werden.
Der Pen ist für die Verwendung mit NovoFine® oder NovoTwist® Einwegnadeln mit einer Länge von bis zu 8 mm und einem minimalen Aussendurchmesser von 32 G vorgesehen.
Nadeln sind nicht enthalten.
Der Patient ist anzuweisen, die Injektionsnadel nach jeder Injektion zu entsorgen und den Pen ohne aufgeschraubte Injektionsnadel zu lagern. Dies beugt Kontamination, Infektion und Austreten von Flüssigkeit vor. Ausserdem wird dadurch eine genaue Dosierung sichergestellt.
Zulassungsnummer65899 (Swissmedic)
PackungenPackungen zu 3 und 5 Fertigpens. [B]
Jeder Pen enthält 3 ml Lösung und ermöglicht die Abgabe von Dosen zu 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg und 3.0 mg.
ZulassungsinhaberinNovo Nordisk Pharma AG, Kloten
Domizil: Zürich
Stand der InformationApril 2025
|