ZusammensetzungWirkstoffe
Rurioctocogum alfa pegolum (Factor VIII coagulationis humanus (ADNr)). Der Ausgangsstoff, rekombinanter Blutgerinnungsfaktor VIII, wird in Ovarienzellen des chinesischen Hamsters (CHO) hergestellt.
Hilfsstoffe
Trometamolum, Calcii chloridum dihydricum, Mannitolum, Natrii chloridum (entspricht 10.4 mg Natrium pro Durchstechflasche), Trehalosum dihydricum, Histidinum, Polysorbatum 80, L-Glutathionum
Lösungsmittel: Aqua ad iniectabile
Indikationen/AnwendungsmöglichkeitenBehandlung und Prophylaxe von Blutungen bei Patienten mit Hämophilie A (angeborener Faktor VIII-Mangel).
ADYNOVI enthält keine pharmakologisch wirksamen Mengen des von Willebrand-Faktors und ist daher nicht zur Behandlung des von Willebrand-Jürgens-Syndroms geeignet.
Dosierung/AnwendungDie Behandlung muss unter Überwachung eines Arztes erfolgen, der mit der Behandlung der Hämophilie vertraut ist.
Übliche Dosierung
Die Dosis und Dauer der Substitutionstherapie richten sich nach dem Schweregrad des Faktor VIII-Mangels, nach dem Ort und dem Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die Zahl der Einheiten des Faktors VIII wird in Internationalen Einheiten (I.E.) angegeben, entsprechend dem WHO-Standard für Faktor VIII-Präparate. Die Faktor VIII-Aktivität im Plasma wird entweder als Prozentsatz (relativ zur Aktivität normalen menschlichen Plasmas) oder in I.E. (relativ zum internationalen Standard für Faktor VIII im Plasma) angegeben.
Eine Internationale Einheit (I.E.) der Faktor VIII-Aktivität entspricht der Menge an Faktor VIII in einem ml normalem menschlichen Plasma.
Bedarfsbehandlung
Die Berechnung der erforderlichen Faktor-VIII-Dosis basiert auf dem empirischen Befund, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor VIII-Aktivität im Plasma um 2 I.E./dl erhöht.
Die erforderliche Dosis wird mit folgender Formel berechnet:
Erforderliche Einheiten (I.E.) = Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (%) x 0.5
Die Dosis und Häufigkeit der Verabreichung sollen immer entsprechend der klinischen Wirksamkeit des Präparates im Einzelfall angepasst werden.
Bei folgenden hämorrhagischen Ereignissen soll die Faktor VIII-Aktivität im entsprechenden Zeitraum nicht unter die angegebenen Plasmaspiegel (in % der Norm oder in I.E./dl) sinken.
Die folgende Tabelle 1 enthält Richtwerte für die Dosierung bei Blutungen und chirurgischen Eingriffen:
Bei schweren oder lebensbedrohlichen Blutungen ist es besonders wichtig, die Therapie möglichst sorgfältig zu überwachen.
Tabelle 1
|
Grad der Blutung / Art des chirurgischen Eingriffes
|
Erforderlicher Faktor VIII Plasmaspiegel (% oder I.E./dl)
|
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage)
| |
Blutung
| |
Gelenkblutung im Frühstadium, Muskelblutungen oder Blutungen im Mund
|
20 – 40
|
Injektion alle 12 – 24 Stunden für mind. 1 Tag, bis die Blutung – angezeigt durch das Verschwinden der Schmerzen – steht oder Heilung erreicht ist.
| |
Ausgeprägtere Gelenkblutung, Muskelblutung oder Hämatom
|
30 – 60
|
Injektion alle 12 – 24 Stunden für 3 – 4 Tage oder länger wiederholen, bis die Schmerzen und die akute Beeinträchtigung beseitigt sind.
| |
Lebensbedrohliche Blutungen
|
60 – 100
|
Injektion alle 8 – 24 Stunden wiederholen bis die Gefahr für den Patienten vorüber ist.
| |
Chirurgische Eingriffe
| |
Kleinere Eingriffe
Einschliesslich Zahnextraktion
|
30 – 60
|
Alle 12 Stunden für mind. 1 Tag bis die Wundheilung erreicht ist.
| |
Grössere Eingriffe
|
80 – 100
(prä- und postoperativ)
|
Injektion alle 8 – 24 Stunden bis zu angemessener Wundheilung wiederholen, dann Therapie für noch mind. 7 Tage fortsetzen, um eine Faktor VIII-Aktivität von 30 – 60% (I.E./dl) aufrecht zu erhalten.
|
Prophylaxe
ADYNOVI wird weniger häufig als rekombinanter Faktor VIII verabreicht. Die empfohlene Dosis für ADYNOVI ist 40-50 I.E./kg Körpergewicht zweimal wöchentlich bei Jugendlichen und Erwachsenen (≥12 Jahre) und 40-60 I.E./kg Körpergewicht zweimal wöchentlich bei Kindern (< 12 Jahren). Verabreichen Sie ADYNOVI am nächsten geplanten Infusionstag und passen Sie die Häufigkeit entsprechend an, um einen maximalen Schutz für die körperliche Aktivität zu bieten.
Individualisierte Dosierung
Bis zu 80 I.E./kg können verabreicht werden, um einen Faktor VIII-Zieltalspiegel ≥1% zu erhalten. Die Dosis ist unter Berücksichtigung der klinischen Antwort des Patienten anzupassen.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Spezielle Dosierungsanweisungen
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit für Kinder, Jugendliche und Erwachsene war in den klinischen Studien (Bedarfstherapie, Prophylaxe) vergleichbar.
Pharmakokinetische Untersuchungen haben bei Kindern unter 12 Jahren eine höhere Clearance, eine kürzere Halbwertszeit und eine niedrigere inkrementelle Recovery des Faktors VIII im Vergleich zu Erwachsenen gezeigt.
Art der Anwendung
ADYNOVI wird intravenös verabreicht. Wenn es nicht durch medizinisches Personal verabreicht werden soll, ist vorher ein entsprechendes Training erforderlich.
Die Verabreichungsgeschwindigkeit soll sich nach dem Befinden des Patienten richten, wobei eine maximale Injektionsrate von 10 ml/min nicht überschritten werden sollte.
Anweisungen zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt «Hinweise für die Handhabung».
KontraindikationenBekannte lebensbedrohliche Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, gegenüber ADYNOVI, gegenüber der Ausgangssubstanz ADVATE, gegenüber Maus- oder Hamsterproteinen, sowie anderen Bestandteilen von ADYNOVI.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeitsreaktionen
Allergische Überempfindlichkeitsreaktionen sind mit ADYNOVI möglich. Es sind Fälle von allergischen Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, nach Anwendung von rekombinanten Blutgerinnungsfaktor VIII-Präparaten, einschliesslich ADYNOVI und dessen Ausgangssubstanz ADVATE bekannt. Wenn Überempfindlichkeitsreaktionen auftreten, sollte die Verabreichung sofort abgebrochen und eine geeignete Behandlung eingeleitet werden.
Patienten sollen über die frühen Anzeichen einer Überempfindlichkeitsreaktion einschliesslich Ausschlag, generalisierte Urtikaria, Engegefühl in der Brust, Giemen, Hypotonie und Anaphylaxie aufgeklärt sein.
Im Falle eines Schocks sollte die medizinische Standardschocktherapie durchgeführt werden.
In klinischen Studien wurde über keine Fälle von Überempfindlichkeitsreaktionen mit ADYNOVI berichtet.
Inhibitoren
Die Bildung von neutralisierenden Antikörpern (Inhibitoren) gegen Faktor VIII ist eine bekannte Komplikation bei der Behandlung von Patienten mit Hämophilie A. Diese Inhibitoren sind stets gegen die prokoagulatorische Aktivität von Faktor VIII gerichtete IgG-Immunglobuline, die in Bethesda-Einheiten (B.E.) pro ml Plasma mittels modifiziertem Assay quantifiziert werden.
Das Risiko, Inhibitoren zu entwickeln, korreliert mit dem Ausmass der Exposition gegenüber dem Faktor VIII, wobei das Risiko innerhalb der ersten 20 Expositionstage am grössten ist. In seltenen Fällen können sich Inhibitoren nach den ersten 100 Expositionstagen bilden.
Bei vorbehandelten Patienten (PTPs) mit mehr als 100 Expositionstagen und anamnestisch bekannter Inhibitorentwicklung wurde, nach Umstellung von einem rekombinanten Faktor VIII-Präparat auf ein anderes, das Wiederauftreten von (niedrigtitrigen) Inhibitoren beobachtet.
Daher wird empfohlen, alle Patienten nach jeder Umstellung auf ein anderes Präparat sorgfältig auf die Inzidenz von Inhibitoren zu überwachen.
Ganz allgemein sollten alle Patienten, die mit Blutgerinnungsfaktor VIII behandelt werden, sorgfältig klinisch und mit geeigneten Labortests hinsichtlich der Entwicklung von Inhibitoren überwacht werden. Wenn der erwartete Faktor-VIII-Spiegel nicht erreicht wird oder die Blutung nicht durch die Verabreichung einer geeigneten Dosis gestillt werden kann, sollte der Patient auf Faktor-VIII-Hemmkörper untersucht werden. Bei Patienten mit hohen Inhibitorspiegeln kann die Faktor-VIII-Ersatztherapie unwirksam sein und es sollten andere Therapiemöglichkeiten in Betracht gezogen werden. Die Behandlung solcher Patienten sollte durch Ärzte erfolgen, die Erfahrung mit der Pflege der Hämophile und von Inhibitoren gegen Faktor VIII haben.
Katheterbedingte Komplikationen bei der Behandlung
Falls ein zentralvenöser Zugang erforderlich sein sollte, ist auf Komplikationen, z.B. lokale Infektionen, Bakteriämie und Katheterthrombose zu achten.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natriumfrei».
InteraktionenEs wurden keine Untersuchungen zur Wechselwirkung von ADYNOVI mit anderen Arzneimitteln durchgeführt.
Schwangerschaft, StillzeitReproduktionsstudien an Tieren wurden mit ADYNOVI nicht durchgeführt. Aufgrund des seltenen Auftretens der Hämophilie A bei Frauen liegen über die Anwendung von ADYNOVI während der Schwangerschaft und Stillzeit keine Erfahrungen vor. Deshalb muss in der Schwangerschaft und Stillzeit der Nutzen der ADYNOVI Behandlung gegen das mögliche Risiko für Mutter und Kind sorgfältig abgewogen werden und ADYNOVI darf nur bei eindeutiger Indikationsstellung angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs liegt keine Information über die Wirkung von ADYNOVI auf die Fahrtüchtigkeit und das Bedienen von Maschinen vor.
Unerwünschte WirkungenÜberempfindlichkeits- oder allergische Reaktionen (welche Angioödeme, Brennen und Stechen an der Infusionsstelle, Frösteln, Hitzegefühl, generalisierte Urtikaria, Kopfschmerzen, Ausschlag, Hypotonie, Lethargie, Übelkeit, nervöse Unruhe, Tachykardie, Engegefühl in der Brust, Parästhesie, Erbrechen, Giemen einschliessen können) wurden selten nach Behandlung mit Faktor VIII beobachtet und können in manchen Fällen zu schweren anaphylaktischen Reaktionen (einschliesslich Schock) führen.
Patienten mit Hämophilie A können neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Bei Auftreten solcher Inhibitoren kann sich dieser Zustand in einem unzureichenden klinischen Ansprechen manifestieren. In diesem Fall wird empfohlen, Kontakt mit einem auf Hämophilie spezialisierten Zentrum aufzunehmen.
Die Sicherheit von ADYNOVI wurde in 6 multizentrischen, prospektiven, offenen klinischen Studien und in 1 laufenden Studie an 365 vorbehandelten oder zuvor unbehandelten Patienten mit schwerer Hämophilie die mindestens eine Dosis ADYNOVI erhielten, untersucht.
In klinischen Studien wurde über Kopfschmerzen als häufige unerwünschte Wirkung (≥1% der Patienten) berichtet.
Die Häufigkeit wurde ermittelt unter Verwendung der folgenden Kriterien: sehr häufig (≥10%), häufig (≥1% bis <10%), gelegentlich (≥0.1% bis<1%), selten (≥0.01% bis <0.1%), sehr selten (<0.01%)
In klinischen Studien wurden die folgenden Nebenwirkungen beschrieben:
Erkrankungen des Gastrointestinaltrakts
Häufig: Diarrhoe, Übelkeit
Augenerkrankungen:
Gelegentlich: Okuläre Hyperämie
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeit
Erkrankungen des Nervensystem
Sehr häufig: Kopfschmerzen (11.2%)
Häufig: Schwindelgefühl
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Ausschlag
Gelegentlich: Ausschlag mit Juckreiz
Häufig: Urtikaria
Gefässerkrankungen
Gelegentlich: Hitzewallung
Untersuchungen:
Gelegentlich: Eosinophilenzahl erhöht
Untersuchungen Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen:
Gelegentlich: Reaktion im Zusammenhang mit einer Infusion
Unerwünschte Wirkungen nach Markteinführung
Erkrankungen des Immunsystems
Nicht bekannt: Anaphylaktische Reaktionen
Beschreibung ausgewählter Nebenwirkungen
Inhibitorenentwickung
Die Bildung neutralisierender Antikörper (Inhibitoren) gegen Faktor VIII kann nach der Verabreichung von ADYNOVI auftreten.
Keiner der Patienten, die an einer oder mehreren der 6 abgeschlossenen Studien mit vorbehandelten Patienten (PTPs) teilnahmen, entwickelte persistierende neutralisierende (inhibitorische) Antikörper gegen FVIII von ≥0.6 B.E./ml (basierend auf dem modifizierten Nijmegen-Bethesda-Assay). Ein Patient entwickelte während einer personalisierten Prophylaxe mit einem FVIII-Zielwert von 8-12% vorübergehend FVIII-Inhibitoren an der niedrigsten positiven Nachweisgrenze (0.6 BU).
Aus einer laufenden Studie mit nicht vorbehandelten Patienten im Alter von < 6 Jahren mit schwerer Hämophilie A liegen vorläufige Berichte über 9 Fälle einer FVIII-Inhibitor-Entwicklung in Verbindung mit der ADYNOVI-Behandlung vor.
Die Erkennung von Antikörpern die auf Faktor VIII reaktiv sind, hängt stark von verschiedenen Faktoren ab, einschliesslich der Sensitivität und Spezifität des Tests, Handhabung der Proben, Zeitpunkt der Probenentnahme, gleichzeitig verabreiche Arzneimittel und der zugrundeliegenden Erkrankung.
Klasseneffekte
Unerwünschte Wirkungen, die mit der Ausgangsubstanz assoziiert werden, schliessen ein: Anaphylaktische Reaktion, Überempfindlichkeit, Faktor VIII-Hemmung.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurde über keine Symptome durch Überdosierung mit rekombinantem Blutgerinnungsfaktor VIII vom Menschen berichtet.
Eigenschaften/WirkungenATC-Code
B02BD02 (Gerinnungsfaktor VIII)
Wirkungsmechanismus
Der Faktor-VIII/von-Willebrand-Faktor-Komplex besteht aus zwei Molekülen (Faktor VIII und von-Willebrand-Faktor) mit unterschiedlichen physiologischen Funktionen.
Wird einem Hämophilie Patienten Faktor VIII injiziert, so bindet sich dieser im Blutkreislauf an den endogenen von-Willebrand-Faktor. Der aktivierte Faktor VIII wirkt als Cofaktor für den aktivierten Faktor IX und beschleunigt die Bildung von aktiviertem Faktor X aus Faktor X. Der aktivierte Faktor X wandelt Prothrombin in Thrombin um.
Dieses setzt dann Fibrin aus Fibrinogen frei und die Gerinnselbildung kann erfolgen. Hämophilie A ist eine geschlechtsgebundene, erbliche Störung (verbunden mit dem X-Chromosom) der Blutgerinnung aufgrund erniedrigter Faktor VIII-Spiegel. Dies führt, entweder spontan oder in Folge unfallbedingter oder chirurgischer Traumata, zu starken Blutungen in Gelenken, Muskeln oder inneren Organen. Die Faktor VIII-Plasmaspiegel werden durch die Substitutionstherapie erhöht, wodurch eine vorübergehende Korrektur des Faktor VIII-Mangels und der Blutungsneigung erfolgt.
ADYNOVI (pegylierter rekombinanter menschlicher Faktor VIII) enthält den ADVATE Wirkstoff (Octocogum alfa, rekombinanter menschlicher Faktor VIII) in voller Länge welches aus 2332 Aminosäuren (Molekulargewicht (MW) ca. 280 kD) besteht und mit Polyethylenglycol (PEG) (MW 20 kD) konjugiert wurde.
Die therapeutische Aktivität von ADYNOVI wird von ADVATE abgeleitet. ADVATE wird mittels rekombinanter DNA-Technologie aus Ovarialzellen des chinesischen Hamsters (CHO) hergestellt.
Das ADVATE Molekül wird dann kovalent mit PEG konjugiert, welches die Lysinreste als Zielgruppe anstrebt. Die funktionelle Gruppe PEG wird an den ADVATE Wirkstoff konjugiert, um die Plasmahalbwertszeit durch Verminderung der LRP-1 Rezeptorvermittelten Clearance des Faktor VIII Moleküls zu erhöhen.
Pharmakodynamik
Klinische Wirksamkeit
Die klinische Wirksamkeit von ADYNOVI wurde in einer prospektiven, multizentrischen open-label-Phase 2/3-Studie gezeigt. 137 vorbehandelte Patienten im Alter von 12 bis 65 Jahren erhielten ADYNOVI entweder prophylaktisch (zweimal wöchentlich 40-50 I.E./kg; n = 120) oder zur Bedarfsbehandlung von Blutungen (10-60 I.E./kg; n = 17). Alle Patienten litten an schwerer Hämophilie A (Faktor VIII-Spiegel <1%). Das mittlere Dosierungsintervall betrug 3.6 Tage. Bei 91 von 98 Patienten, die vor Studienbeginn prophylaktisch behandelt wurden, konnte die Dosierungsfrequenz verringert werden (um median 33.7%).
Die mediane jährliche totale Blutungsrate in der prophylaktischen Behandlungsgruppe betrug 1.9 (min. 0.0, max. 18.4), für spontane Blutungen und Gelenkblutungen 0.0 (spontane Blutungen: min. 0.0, max. 18.2; Gelenkblutungen: min. 0.0, max. 13.1). 40% der Patienten hatten keine Blutungsepisode.
Insgesamt wurden 518 Blutungsepisoden mit ADYNOVI behandelt, davon 361 Blutungen in der Akutbehandlungsgruppe und 157 bei prophylaktisch behandelten Patienten. Die Blutungen wurden in 95.9% mit 1 bis 2 ADYNOVI Injektionen behandelt.
In 96.1% aller behandelten Blutungsepisoden wurde die hämostatische Wirksamkeit als exzellent oder gut eingestuft.
Die Sicherheit und Wirksamkeit von ADYNOVI bei perioperativer Anwendung wurde in einer prospektiven, open-label, unkontrollierten, multizentrischen Phase-3-Studie an vorbehandelten männlichen Patienten mit schwerer Hämophilie A evaluiert. In die Analyse wurden insgesamt 15 Operationen (11 grosse und 4 kleine Operationen) bei 15 Patienten (im Alter von 19 bis 52 Jahren) eingeschlossen. Die perioperative hämostatische Wirksamkeit wurde bei allen 15 Eingriffen mit exzellent bewertet (erwarteter Blutverlust geringer oder vergleichbar zu einem Patienten ohne Hämophilie bei gleicher Operation).
Sicherheit und Wirksamkeit bei pädiatrischen Patienten
Patienten < 12 Jahre
In der pädiatrischen Studie wurden insgesamt 66 vorbehandelte Patienten mit schwerer Hämophilie A (32 waren < 6 Jahre alt und 34 waren 6 bis < 12 Jahre alt) aufgenommen.
Die prophylaktische Dosierung betrug 40 bis 60 I.E./kg ADYNOVI 2x wöchentlich.
Die mediane jährliche totale Blutungsrate betrug 2.0 (IQR: 3.9) für die 65 Patienten in der per-protocol Population und die mediane Blutungsrate betrug 0 (IQR: 1.9) sowohl für spontane Blutungen als auch für Gelenksblutungen. Bei 37% der Patienten traten keine Blutungsepisoden, bei 72% traten keine Gelenksblutungen und bei 66% traten keine spontanen Blutungsepisoden unter Prophylaxe auf.
Von den 70 beobachteten Blutungsepisoden, konnten in der pädiatrischen Studie 82.9% mit einer Infusion und 91.4% mit einer oder zwei Infusionen kontrolliert werden.
Die Kontrolle der Blutungen wurde in 90% der Blutungsepisoden als exzellent oder gut bewertet.
Patienten < 6 Jahre
Die Sicherheit, Immunogenität und hämostatische Wirksamkeit von ADYNOVI wurden in einer prospektiven, multizentrischen, offenen Phase-3-Studie an zuvor unbehandelten Patienten < 6 Jahren mit schwerer Hämophilie A (Faktor VIII < 1%) evaluiert. Die Patienten erhielten entweder eine Bedarfsbehandlung in einer Dosis von 10 bis 50 I.E./kg, bis zu 80 I.E./kg oder eine prophylaktische Behandlung mit einer mindestens einmal wöchentlichen Dosis von 25 bis 50 I.E./kg, die auf 80 I.E./kg erhöht werden konnte.
Die Zwischenauswertung umfasste 59 Patienten, die mindestens eine Dosis erhalten hatten. Von den 59 behandelten Patienten wurden 52 in die Primäranalyse der Bildung von Inhibitoren eingeschlossen, darunter 10 Patienten, bei denen es zur Bildung von Inhibitoren kam (19.2%). Auf der Basis eines mittleren generalisierten linearen Modells (GLM) mit negativer Binomialverteilung betrug der Punktschätzer (95-%-KI) der jährlichen Gesamtblutungsrate für die Gruppen unter Bedarfsbehandlung und unter prophylaktischer Behandlung 3.206 (1.632–6.297) bzw. 3.199 (2.026–5.049).
Bei den meisten Blutungen war lediglich eine Infusion erforderlich (77.7%). Bei Blutungsstillung wurde die hämostatische Wirksamkeit von den Patienten oder Betreuungspersonen als exzellent (32.7%) oder gut (27.1%) bewertet. Es ist zu beachten, dass die hämostatische Wirksamkeit bei der Blutungsstillung bei 36.8% der Blutungen nicht bewertet wurde. Diese Zwischenergebnisse bestätigen die Sicherheit und Wirksamkeit, die in den Studien mit pädiatrischen, jugendlichen und erwachsenen vorbehandelten Patienten gezeigt wurden.
Langzeitprophylaxe bei pädiatrischen und erwachsenen Patienten
Die Langzeit-Sicherheit und -Wirksamkeit von ADYNOVI zur Prophylaxe und Bedarfsbehandlung von Blutungsepisoden wurde bei 216 pädiatrischen und erwachsenen PTPs mit schwerer Hämophilie A untersucht, die entweder zuvor an anderen ADYNOVI-Studien teilgenommen hatten oder bisher ADYNOVI-unbehandelt waren. Die Teilnehmer der behandelten Population erhielten zweimal wöchentlich eine festgelegte Dosis von 40 bis 50 I.E./kg (Teilnehmer im Alter ≥12 Jahre) bzw. 40 bis 60 I.E./kg (Teilnehmer im Alter < 12 Jahre). Die Dosis wurde auf bis zu 80 I.E./kg zweimal wöchentlich angepasst, falls dies erforderlich war, um einen FVIII-Talspiegel von > 1% aufrecht zu erhalten. Teilnehmer, die sich für ein personalisiertes (pharmakokinetisch individualisiertes) Prophylaxeschema entschieden, erhielten mindestens zweimal wöchentlich Dosen von bis zu 80 I.E./kg pro Infusion, die auf FVIII-Talspiegel von ≥3% abzielten. Teilnehmer im Alter von ≥12 Jahren, die ein Regime mit zweimal wöchentlicher Gabe erhielten und in einem Zeitraum von 6 konsekutiven Monaten keinerlei (null) Spontanblutungen hatten, konnten auf eine Prophylaxe mit feststehender Dosis alle 5 Tage wechseln. Hatten diese Probanden weitere 6 Monate lang keinerlei (null) Spontanblutungen, so konnten sie ein Regime mit Gabe alle 7 Tage erhalten. Insgesamt betrug die langfristige Exposition im Mittel (SA) 195.4 (101.57) Prophylaxe-Expositionstage (ED) je Proband. In Tabelle 2 ist die ABR nach Prophylaxeschema, Blutungsstelle und Ätiologie dargestellt.
|
Tabelle 2: Annualisierte Blutungsrate (ABR) nach Prophylaxeschema (ITT-Population)
| |
Blutungslokalisation und Ätiologie
|
Zweimal wöchentlich
(n = 186)
|
Alle 5 Tage
(n = 56)
|
Alle 7 Tage
(n = 15)
|
PK-basierta
(n = 25)
| |
Mittelwert
[Punktschätzung – 95%-Konfidenzintervall]
| |
Gesamt
|
2.2 [1.85-2.69]
|
2.1 [1.54-2.86]
|
2.7 [1.44-5.20]
|
2.6 [1.70-4.08]
| |
Gelenk
|
1.2 [0.96-1.58]
|
1.1 [0.81-1.55]
|
2.0 [0.90-4.62]
|
1.4 [0.91-2.17]
| |
Spontan
|
1.2 [0.92-1.56]
|
1.3 [0.87-2.01]
|
1.8 [0.78-4.06]
|
1.0 [0.54-1.71]
| |
Traumatisch
|
1.0 [0.83-1.28]
|
0.7 [0.45-0.99]
|
0.9 [0.41-1.91]
|
1.6 [1.03-2.50]
| |
Punktschätzung und 95%-Konfidenzintervalle aus einem generalisierten linearen Modell entsprechend einer negativen Binomialverteilung mit logarithmischer Linkfunktion.
Teilnehmer, die Dosen aus mehreren Schemata erhielten, sind in Zusammenfassungen für mehrere Schemata berücksichtigt.
n = Anzahl der in die Analyse eingeschlossenen Patienten
a Angestrebte Talwerte für die FVIII-Aktivität von ≥3% des Normwerts
|
Die hämostatische Langzeitwirkung wurde bei 910 mit ADYNOVI behandelten Blutungsepisoden untersucht und bei 88.5% der Blutungsepisoden als ausgezeichnet oder gut eingestuft. Über alle Altersklassen hinweg wurden sowohl für das Dosierungsschema mit feststehender Dosis als auch für die PK-basierte Dosierung > 85% der Blutungsbehandlungen als ausgezeichnet oder gut eingestuft. Die meisten Blutungsepisoden wurden mit einer (74.0%) oder zwei (15.4%) Infusionen kontrolliert. Insgesamt hatten während einer Langzeitexposition von im Mittel (SA) 195.4 (101.57) Prophylaxe-Expositionstagen pro Patient (Median von 208,5 ED) 16.7% (36/216) der Patienten in diesem erweiterten Zeitraum keine Blutungsepisoden.
Personalisierte Prophylaxe, PROPEL-Studie bei Jugendlichen und Erwachsenen
Die Sicherheit und Wirksamkeit von ADYNOVI wurden in einer prospektiven, randomisierten, offenen, multizentrischen Studie mit 121 (115 randomisierten) jugendlichen (Alter 12-18 Jahre) und erwachsenen PTPs mit schwerer Hämophilie A über einen 12monatigen Behandlungszeitraum untersucht. In der Studie wurden 2 PK-gelenkte prophylaktische Dosierungsschemata von ADYNOVI verglichen, die bei zweimal wöchentlicher Gabe auf einen Faktor-VIII-Talspiegel von 1 bis 3% (n = 57) bzw. bei einer Gabe jeden zweiten Tag (n = 58) von 8 bis 12% abzielten. Der Vergleich erfolgte durch die Bewertung des Anteils an Patienten, die im zweiten 6monatigen Studienzeitraum eine Gesamt-ABR von 0 erreichten.
Im Durchschnitt wurden im Arm mit dem angestrebten Talspiegel von 1 bis 3% bzw. im Arm mit dem angestrebten Talspiegel von 8 bis 12% prophylaktische Dosen von jeweils 3866.1 I.E./kg pro Jahr (mittlere [SA] Infusionen/Woche = 2.3 [0.58]) bzw. 7532.8 I.E./kg pro Jahr (mittlere [SA] Infusionen/Woche = 3.6 [1.18]) verabreicht. Nach Dosisanpassung in den ersten 6 Monaten der Prophylaxe lagen die medianen Talspiegel im zweiten 6-Monatszeitraum (basierend auf dem einstufigen Gerinnungsassay und am Ende des geplanten Infusionsintervalls berechnet) im Arm mit dem angestrebten Talspiegel von 1 bis 3% zwischen 2.10 I.E./dl und 3.00 I.E./dl und im Arm mit dem angestrebten Talspiegel von 8 bis 12% zwischen 10.70 I.E./dl und 11.70 I.E./dl. Die Dosierung war in beiden Prophylaxeschemata somit generell geeignet, um die gewünschten FVIII-Talspiegel zu erreichen und aufrecht zu halten.
Der primäre Endpunkt der Studie, der Anteil der Probanden, die während des zweiten 6-Monatszeitraums eine Gesamt-ABR von 0 hatten, wurde in der ITT-Patientenpopulation nicht erreicht (p = 0.0545), aber in der Per-Protokoll-Population (p = 0.0154).
Insgesamt wurden 242 Blutungsepisoden bei 66 Teilnehmern mit ADYNOVI behandelt, davon 155 Blutungen bei 40 Teilnehmern im Talspiegel-Arm von 1 bis 3% und 87 Blutungen bei 26 Teilnehmern im Talspiegel-Arm von 8 bis 12%. Die meisten Blutungen (86.0%, 208/242) wurden mit 1 oder 2 Infusionen kontrolliert. Bei 84.7% (205/242) der Blutungsfälle wurde die Blutungskontrolle nach Ende der Blutungsepisode als ausgezeichnet oder gut eingestuft.
PharmakokinetikDie Pharmakokinetik von ADYNOVI wurde in einer Crossover Studie mit ADVATE in 26 Patienten (18 Erwachsene und 8 Jugendliche) und in 22 Patienten (16 Erwachsene und 6 Jugendliche) nach einer 6 monatigen Behandlung mit ADYNOVI ermittelt. Für beide Präparate wurde eine Einzeldosis von 45 ± 5 IE/kg gebraucht. Die PK Parameter, aufgelistet pro Altergruppe (Erwachsene und Jugendliche) in den unterstehenden Tabellen, basieren auf Faktor-VIII-Plasmaaktivität gemessen mittels den Einstufen-Gerinnungstests.
ADYNOVI hat eine um das 1.4 bis 1.5-fach verlängerte Halbwertszeit, wie mittels des Einstufen-Gerinnungstests und chromogenen Assays bestimmt. Eine Erhöhung in AUC und einer Verminderung der Clearance verglichen mit dem Ausgangswirkstoff ADVATE wurde ebenfalls beobachtet. Die Incremental Recovery war mit beiden Präparaten vergleichbar.
Die Änderungen der pharmakokinetischen Parameter waren in beiden Gruppen, Erwachsene und Jugendliche, sowie zwischen dem Einstufen-Gerinnungstest und dem chromogenic substrate Assay, ähnlich.
|
Pharmakokinetische Parameter bei Erwachsenen (18 Jahre und älter)
| |
PK-Parameter (Mittel ± SD)
|
ADVATE bei Initialdosis
n = 18
|
ADYNOVI bei Initialdosis
n = 18
|
ADYNOVI ≥50 EDs
n = 16
| |
Terminale Halbwertszeit (h)
|
10.83 ± 2.08
|
14.69 ± 3.79
|
16.39 ± 5.28
| |
MRT (h)
|
13.41 ± 3.00
|
20.27 ± 5.23
|
21.09 ± 4.73
| |
Clearance (dl/kg*h)
|
0.0388 ± 0.0124
|
0.0227 ± 0.0084
|
0.0237 ± 0.0077
| |
Incremental-Recovery (I.E./dl/I.E./kg)*
|
2.57 ± 0.43
|
2.66 ± 0.68
|
2.33 ± 0.55
| |
AUC0-Inf (I.E. * h/dl)
|
1286 ± 390
|
2264 ± 729
|
2062 ± 575
| |
Vss (dl/kg)
|
0.50 ± 0.11
|
0.43 ± 0.11
|
0.49 ± 0.17
| |
Cmax (I.E./dl)
|
117 ± 20
|
122 ± 29
|
105 ± 25
| |
Tmax (h)
|
0.033 ± 0.19
|
0.46 ± 0.29
|
0.38 ± 0.18
|
|
Pharmakokinetische Parameter bei Jugendlichen (12 bis unter 18 Jahren)
| |
PK-Parameter (Mittel ± SD)
|
ADVATE bei Initialdosis
(95% CI)
n = 8
|
ADYNOVI bei Initialdosis
(95% CI)
n = 8
|
ADYNOVI ≥50 EDs
(95% CI)
n = 6
| |
Terminale Halbwertszeit (h)
|
9.45 ± 2.45
|
13.43 ± 4.05
|
15.06 ± 4.08
| |
MRT (h)
|
11.63 ± 2.94
|
17.96 ± 5.49
|
19.47 ± 5.32
| |
Clearance (dl/kg*h)
|
0.0607 ± 0.0305
|
0.0387 ± 0.0331
|
0.0275 ± 0.0096
| |
Incremental-Recovery (I.E./dl/I.E./kg)*
|
1.94 ± 0.52
|
2.12 ± 0.60
|
2.22 ± 0.88
| |
AUC0-Inf (I.E. * h/dl)
|
902 ± 400
|
1642 ± 752
|
1868 ± 807
| |
Vss (dl/kg)
|
0.67 ± 0.31
|
0.56 ± 0.18
|
0.51 ± 0.13
| |
Cmax (I.E./dl)
|
89 ± 29
|
95 ± 25
|
100 ± 42
| |
Tmax (h)
|
0.21 ± 0.04
|
0.26 ± 0.10
|
0.71 ± 1.16
|
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Die pharmakokinetischen Parameter welche anhand der Daten von 39 Patienten unter 18 Jahren (intent-to-treat Analyse) berechnet wurden, sind für 14 Kinder (2 bis < 6 Jahre), für 17 ältere Kinder (6 bis < 12 Jahre) und für 8 Jugendliche (12 bis < 18 Jahre) verfügbar (siehe untenstehende Tabellen).
Im Vergleich zu Erwachsenen war bei Kindern unter 12 Jahren die mittlere Clearance von ADYNOVI bezogen auf das Körpergewicht höher und die mittlere Halbwertszeit kürzer.
Eine höhere Dosierung kann bei Kindern unter 12 Jahren erforderlich sein.
|
Pharmakokinetische Parameter bei pädiatrischen Patienten unter Verwendung des Einstufen-Gerinnungstests
| |
PK-Parameter (Mittel ± SD)
|
Pädiatrische Studie
|
Pivotale Studie bei Jugendlichen und Erwachsenen
| |
< 6 Jahre
n = 14
|
6 bis < 12 Jahre
n = 17
|
12 - < 18 Jahre
n = 8
| |
Design
|
PK Population mit «Sparse Sampling»a
|
Individuelle PK mit «Full Sampling»b
| |
Terminale Halbwertszeit (h)
|
11.8 ± 2.43
|
12.4 ± 1.67
|
13.43 ± 4.05
| |
MRT (h)
|
17.0 ± 3.50
|
17.8 ± 2.42
|
17.96 ± 5.49
| |
Clearance (ml/kg*h)
|
3.53 ± 1.29
|
3.11 ± 0.76
|
3.87 ± 3.31
(2.73 ± 0.93)*
| |
Incremental-Recovery (I.E./dl/I.E./kg)*
|
nac
(1.89 ± 0.49)
|
nac
(1.95 ± 0.47)
|
2.12 ± 0.60
| |
AUC0-Inf (I.E. * h/dl)
|
1947 ± 757
|
2012 ± 45
|
1642 ± 752
| |
Vss (dl/kg)
|
56 ± 0.12
|
0.54 ± 0.09
|
0.56 ± 0.18
| |
Cmax (I.E./dl)
|
nac
(115 ± 30)
|
nac
(115 ± 33)
|
95 ± 25
| |
Tmax (h)
|
d
|
d
|
0.26 ± 0.10
|
* Geschätzter Mittelwert und SD berechnet ohne einen Patienten, dessen geschätzte Clearance 11.8 ml/(kg·h) betrug. Median aller Patienten beträgt 2.78 ml/(kg·h).
a Populations PK-Modell mit 3 post-Infusions Proben basierend auf randomisierten drawing schedule.
b Individuelle PK mit 12 post-Infusions Proben.
c NA, nicht anwendbar, da incremental Recovery und Cmax bei Kindern durch eine individuelle PK bestimmt wurden. In Klammern die Resultate für die incremental Recovery und Cmax, welche durch individuelle PK bestimmt wurden.
d Tmax konnte nicht für Patienten in der pädiatrischen Studie berechnet werden da nur eine Probe innerhalb der ersten 3 Stunden nach Infusion entnommen wurde (15-30 Minuten post-Infusion)
|
Pharmakokinetische Parameter bei pädiatrischen Patienten unter Verwendung des chromogenen Tests
| |
PK-Parameter (Mittel ± SD)
|
Pädiatrische Studie
|
Pivotale Studie bei Jugendlichen und Erwachsenen
| |
< 6 Jahre
n = 14
|
6 bis < 12 Jahre
n = 17
|
12 - < 18 Jahre
n = 8
| |
Design
|
PK Population mit «Sparse Sampling»a
|
Individuelle PK mit «Full Sampling»b
| |
Terminale Halbwertszeit (h)
|
13.0 ± 8.74
|
11.9 ± 2.58
|
13.80 ± 4.00
| |
MRT (h)
|
18.7 ± 12.6
|
17.2 ± 3.72
|
17.73 ± 5.44
| |
Clearance (ml/kg*h)
|
|
|
3.41 ± 3.14
| |
Incremental-Recovery (I.E./dl/I.E./kg)*
|
nac
1.90 ± 0.27
|
nac
2.20 ± 0.38
|
2.60 ± 0.69
| |
AUC0-Inf (I.E. * h/dl)
|
2190 ± 1590
|
2260 ± 514
|
1900 ± 841
| |
Vss (dl/kg)
|
0.91 ± 0.12
|
1.33 ± 0.23
|
0.49 ± 0.20
| |
Cmax (I.E./dl)
|
nac
117 ± 16
|
nac
130 ± 24
|
117 ± 28
| |
Tmax (h)
|
d
|
d
|
0.26 ± 0.14
|
a Populations PK-Modell mit 3 post-Infusions Proben basierend auf randomisierten drawing schedule.
b Individuelle PK mit 12 post-infusions Proben.
c NA, nicht anwendbar, da incremental Recovery und Cmax bei Kindern durch eine individuelle PK bestimmt wurden. In Klammern die Resultate für die incremental Recovery und Cmax, welche durch individuelle PK bestimmt wurden.
d Tmax konnte nicht für Patienten in der pädiatrischen Studie berechnet werden da nur eine Probe innerhalb der ersten 3 Stunden nach Infusion entnommen wurde (15-30 Minuten post-Infusion)
Die pharmakokinetischen Daten zeigen, dass ADYNOVI eine verlängerte zirkulierende Halbwertszeit hat.
Präklinische DatenSicherheitspharmakologie
Studien zur Sicherheitspharmakologie haben keine Anzeichen eines thrombogenen Potentials oder von unerwünschten Wirkungen auf die respiratorischen oder kardiovaskulären Funktionen gezeigt.
Langzeittoxizität (bzw. Toxizität bei wiederholter Verabreichung)
Einzel- und wiederholte Dosierungen zeigten keine Anzeichen von Toxizität von ADYNOVI bei Labortieren (Mäuse, Ratten, Kaninchen und Javaneraffen).
Wiederholte Verabreichung des 20 kDa PEG-Teils zeigte keine Anzeichen von Toxizität, bei der höchsten getesteten Dosis von 65 mg/kg.
Mutagenität und Karzinogenität
Es wurden keine Studien mit dem Wirkstoff von ADYNOVI durchgeführt um das mutagene oder karzinogene Potential zu beurteilen.
Reproduktionstoxizität
Es wurden keine toxikologische Tierstudien zur Reproduktion oder Entwicklung mit ADYNOVI durchgeführt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf ADYNOVI nicht mit anderen Arzneimitteln gemischt werden.
Zur Rekonstitution nur das beigepackte sterilisierte Wasser für Injektionszwecke und das beigepackte Verabreichungsset verwenden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Chemische und physikalische in-use Stabilität wurde für 3 Stunden bei 25 °C gezeigt. Aus mikrobiologischen Gründen sollte die gebrauchsfertige Zubereitung unmittelbar nach Rekonstitution verwendet werden. Falls dies nicht möglich ist, liegen Aufbrauchsfristen und Lagerbedingungen in der Verantwortung des Anwenders, ausser wenn die Rekonstitution unter kontrollierten und validierten aseptischen Bedingungen erfolgte. Nach der Rekonstitution nicht kühl stellen.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern. Nicht einfrieren. Innerhalb des aufgedruckten Verfallsdatums darf das Produkt für den ambulanten Einsatz einmalig aus dem Kühlschrank entnommen werden und dann maximal 3 Monate vor Gebrauch unterhalb 30 °C gelagert werden. Nach dem Entnehmen aus dem Kühlschrank darf ADYNOVI nicht erneut im Kühlschrank gelagert werden, sondern ist zu vernichten.
ADYNOVI mit BAXJECT II Gerät: Die Pulverdurchstechflasche im Umkarton vor Licht geschützt aufbewahren.
ADYNOVI mit BAXJECT III System: Den verschlossenen Blister in der Originalverpackung vor Licht geschützt aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Die rekonstituierte Lösung sollte einer Sichtprüfung auf Fremdkörper und/oder Verfärbungen unterzogen werden. Nach der Rekonstitution ist die Lösung klar oder leicht opaleszierend.
Verwenden Sie keine Lösungen, die trüb sind oder Ablagerungen enthalten.
Nach der Rekonstitution ist die Lösung klar, farblos und frei von Fremdkörpern. Sie hat einen pH-Wert zwischen 6.7 und 7.3. Die Osmolalität beträgt ≥380 mOsmol/kg.
ADYNOVI wird entweder mit einem BAXJECT II Gerät zur Rekonstitution oder mit einem gebrauchsfertigem BAXJECT III System in einem verschlossenen Blister (das Lyophilisat und das Wasser für Injektionszwecke sind vormontiert mit einem System zur Rekonstitution) geliefert.
Rekonstitution mit dem BAXJECT II Gerät
Zur Rekonstitution nur das beigepackte sterilisierte Wasser für Injektionszwecke und das beigepackte Gerät zur Rekonstitution verwenden.
1.Achten Sie auf aseptische Arbeitsweise und auf die Verwendung einer flachen Arbeitsfläche während der Rekonstitution.
2.Bringen Sie die Durchstechflasche mit ADYNOVI, sowie die Glasflasche mit dem Lösungsmittel (Aqua ad iniectabile) auf Raumtemperatur.
3.Schutzkappen von dem Fläschchen mit ADYNOVI und Lösungsmittel entfernen.
4.Gummistopfen mit Alkoholtupfern reinigen und diese vor dem Gebrauch trocknen lassen.
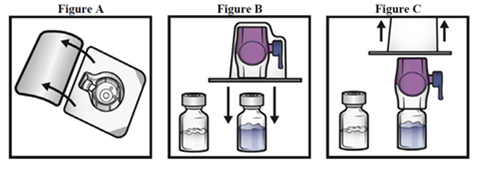
5.Öffnen Sie die Packung des BAXJECT II Gerätes durch Abziehen der Folie, ohne dabei das Innere zu berühren (Fig. A). Nehmen Sie das Gerät nicht aus der Packung.
6.Drehen Sie die Packung um und drücken Sie nach unten, um den klaren Plastikspike vollständig durch den Stopfen der Lösungsmittel-Durchstechflasche zu stechen (Fig. B).
7.Fassen Sie die Verpackung des BAXJECT II Gerätes an den Seiten und ziehen Sie sie vom Gerät herunter (Bild Fig. C). Die blaue Kappe nicht vom BAXJECT II Gerät entfernen. Den freiliegenden lila Plastikspike nicht berühren.
8.Drehen Sie das System so herum, dass die Lösungsmittel-Durchstechflasche nach oben zeigt. Den lila Plastikspike schnell in den ADYNOVI-Durchstechflaschen-Stopfen drücken, in dem man ihn direkt herunter drückt (Fig. D). Durch das Vakuum wird das Lösungsmittel in die ADYNOVI-Durchstechflasche gezogen.
9.Vorsichtig schwenken, bis ADYNOVI komplett gelöst ist. Nach Rekonstitution nicht kühl stellen.
Rekonstitution mit dem BAXJECT III System
Nicht verwenden, falls die Schutzfolie des Blisters nicht vollständig versiegelt ist.
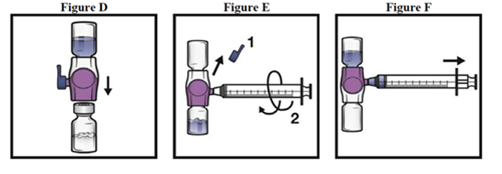
1.Den verschlossenen Blister (enthält Fläschchen mit Lyophilisat und Aqua ad iniectabile vormontiert mit einem System zur Rekonstitution) auf 15 – 25 °C bringen.
2.Hände sorgfältig mit Seife und warmen Wasser waschen.
3.Die ADYNOVI Blisterpackung öffnen, indem die Schutzfolie abgezogen wird. Das BAXJECT III System aus dem Blister entnehmen.
4.Das ADYNOVI auf eine flache Oberfläche stellen, so dass sich das Lösungsmittelfläschchen oben befindet (Fig. 1). Das Lösungsmittelfläschchen hat einen blauen Streifen. Die blaue Schutzkappe nicht entfernen, bis dies in einem späteren Schritt angewiesen wird.
5.Während Sie mit einer Hand das ADYNOVI im BAXJECT III System halten, drücken Sie mit der anderen Hand fest auf das Lösungsmittelfläschchen, bis das System vollständig zusammengedrückt ist und das Lösungsmittel in das ADYNOVI Fläschchen hinunterfliesst (Fig. 2). Kippen Sie das System nicht um, bis der Transfer abgeschlossen ist.
6.Überprüfen Sie, ob der Transfer des Lösungsmittels abgeschlossen ist. Vorsichtig schwenken bis ADYNOVI vollständig gelöst ist, da sonst wirksame Substanz durch den Filter zurückgehalten wird. Das Präparat löst sich schnell auf (normalerweise in weniger als 1 Minute). Nach der Rekonstitution sollte die Lösung klar, farblos und frei von fremden Partikeln sein.
Verabreichung
Vor der Verabreichung immer auf Schwebeteilchen oder Verfärbung überprüfen. Die Lösung sollte klar oder farblos sein. Lösungen, die Ablagerungen oder Verfärbungen aufweisen, nicht verwenden.
Das rekonstituierte Produkt muss sofort verwendet werden aber auf keinen Fall später als 3 Stunden nach der Rekonstitution.
1.Entfernen Sie die blaue Schutzkappe vom BAXJECT II / BAXJECT III Gerät. Schliessen Sie die Spritze an das BAXJECT II / BAXJECT III Gerät an (Fig. E). Ziehen Sie keine Luft auf.
2.Drehen Sie das System einmal herum – die ADYNOVI-Glasflasche ist nun oben. Überführen Sie nun das Faktor-Konzentrat in die Spritze indem sie den Kolben langsam zurückziehen (Fig. F).
3.Nehmen Sie die Spritze ab, setzen Sie eine passende Nadel auf und beginnen Sie mit der intravenösen Injektion. Wenn einem Patienten mehr als eine Durchstechflasche ADYNOVI injiziert werden muss, kann für diese Durchstechflaschen dieselbe Spritze verwendet werden.Es ist zu beachten, dass der BAXJECT II nur für die Verwendung von einer einzigen Durchstechflasche ADYNOVI und Lösungsmittel vorgesehen ist.Deshalb ist es nötig für eine weitere Rekonstitution und für ein weiteres Aufziehen der Spritze ein zweites BAXJECT II Gerät zu verwenden.
4.ADYNOVI über 5 oder weniger Minuten verabreichen. Die maximale Injektionsrate beträgt 10 ml/min.
Zulassungsnummer65953 (Swissmedic)
PackungenPackungen mit BAXJECT II Gerät
Jede Packung ADYNOVI enthält: 1 Durchstechflasche Lyophilisat, 1 Durchstechflasche mit Wasser für Injektionszwecke als Lösungsmittel, 1 BAXJECT II zur Auflösung
Packungen mit BAXJECT III System
Jede Packung ADYNOVI enthält: ein gebrauchsfertiges BAXJECT III System in einem verschlossenen Blister (das Lyophilisat und das Wasser für Injektionszwecke sind vormontiert mit dem System zur Rekonstitution)
ADYNOVI 250 [B] Lyophilisat zur Injektion mit 250 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADYNOVI 250 [B] Lyophilisat zur Injektion mit 250 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 500 [B] Lyophilisat zur Injektion mit 500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADYNOVI 500 [B] Lyophilisat zur Injektion mit 500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 750 [B] Lyophilisat zur Injektion mit 750 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADYNOVI 750 [B] Lyophilisat zur Injektion mit 750 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 1000 [B] Lyophilisat zur Injektion mit 1000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADYNOVI 1000 [B] Lyophilisat zur Injektion mit 1000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 1500 [B] Lyophilisat zur Injektion mit 1500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 2 ml Wasser für Injektionszwecke.
ADYNOVI 1500 [B] Lyophilisat zur Injektion mit 1500 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 2000 [B] Lyophilisat zur Injektion mit 2000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
ADYNOVI 3000 [B] Lyophilisat zur Injektion mit 3000 I.E. rekombinantem Faktor VIII pro Durchstechflasche und eine Durchstechflasche mit 5 ml Wasser für Injektionszwecke.
Mit jeder Packung ADYNOVI wird ein Gerätesatz mitgeliefert:
1 Mini-Infusionsset
1 sterile 10 ml Einmalspritze
2 Alkoholtupfer
2 Pflaster
ZulassungsinhaberinTakeda Pharma AG, 8152 Opfikon
Stand der InformationFebruar 2024.
|