ZusammensetzungWirkstoffe
Ixazomib als Ixazomibzitrat.
Hilfsstoffe
Kapselinhalt: Mikrokristalline Cellulose, Magnesiumstearat, Talkum.
Kapselhülle: Gelatine, Titandioxid (E171), Eisenoxid (E172) rot (Ninlaro 2,3 mg und 4,0 mg), gelb (Ninlaro 4,0 mg) bzw. schwarz (Ninlaro 3,0 mg).
Drucktinte: Schellack, Propylenglycol, Kaliumhydroxid, schwarzes Eisenoxid (E172), Ammoniaklösung 30%.
Indikationen/AnwendungsmöglichkeitenNINLARO in Kombination mit Lenalidomid und Dexamethason ist für die Behandlung von erwachsenen Patienten mit multiplem Myelom indiziert,
·die mindestens eine vorangegangene Therapie erhalten haben und Hochrisikomerkmale aufweisen, oder
·die mindestens zwei vorangegangene Therapien erhalten haben.
NINLARO ist ausserhalb kontrollierter klinischer Studien nicht für die Erhaltungstherapie beim multiplen Myelom empfohlen.
Siehe Abschnitt «Eigenschaften/Wirkungen».
Dosierung/AnwendungDie Behandlung muss unter der Aufsicht von Ärzten mit Erfahrung in der Behandlung des multiplen Myeloms begonnen und überwacht werden.
Dosierung
NINLARO in Kombination mit Lenalidomid und Dexamethason
Die empfohlene Anfangsdosis NINLARO beträgt 4 mg (eine Hartkapsel) oral einmal wöchentlich an den Tagen 1, 8 und 15 eines 28-tägigen Behandlungszyklus.
Die empfohlene Anfangsdosis Lenalidomid beträgt 25 mg einmal täglich an den Tagen 1 bis 21 eines 28-tägigen Behandlungszyklus.
Die empfohlene Anfangsdosis Dexamethason beträgt 40 mg an den Tagen 1, 8, 15 und 22 eines 28-tägigen Behandlungszyklus.
Dosierungsschema: NINLARO in Kombination mit Lenalidomid und Dexamethason
+ Arzneimitteleinnahme
|
28-tägiger Zyklus (4-Wochen-Zyklus)
| |
|
Woche 1
|
Woche 2
|
Woche 3
|
Woche 4
| |
|
Tag
1
|
Tage
2 bis 7
|
Tag
8
|
Tage
9 bis 14
|
Tag
15
|
Tage
16 bis 21
|
Tag
22
|
Tage
23 bis 28
| |
NINLARO
|
+
|
|
+
|
|
+
|
|
|
| |
Lenalidomid
|
+
|
+ Täglich
|
+
|
+ Täglich
|
+
|
+ Täglich
|
|
| |
Dexamethason
|
+
|
|
+
|
|
+
|
|
+
|
|
Für zusätzliche Informationen zu Lenalidomid und Dexamethason, siehe entsprechende Arzneimittelinformation.
Vor Beginn eines neuen Behandlungszyklus:
·Die absolute Neutrophilenanzahl muss ≥1'000/mm3 betragen
·Die Thrombozytenanzahl muss ≥75'000/mm3 betragen
·Nicht-hämatologische Toxizitäten müssen sich nach Ermessen des Arztes allgemein bis zum Ausgangszustand des Patienten oder ≤ Grad 1 rückbilden.
Die Behandlung ist bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Toxizität fortzusetzen.
Verspätete oder vergessene Dosis
Erfolgte die Einnahme einer Dosis NINLARO verspätet oder wurde sie vergessen, ist die Dosis nur einzunehmen, wenn die nächste geplante Dosis in ≥72 Stunden erfolgt. Eine vergessene Dosis darf nicht innerhalb von 72 Stunden vor der nächsten geplanten Dosis eingenommen werden. Es darf keine doppelte Dosis eingenommen werden, um eine vergessene Dosis nachzuholen.
Erbricht ein Patient nach der Einnahme einer Dosis, darf der Patient die Dosis nicht erneut einnehmen, sondern sollte die Einnahme zum Zeitpunkt der nächsten Dosis fortsetzen.
Dosisanpassungen
Die Reduktionsschritte der NINLARO-Dosis sind in Tabelle 1 dargestellt. Richtlinien zur Dosisanpassung sind in Tabelle 2 aufgeführt.
Tabelle 1: Reduktionsschritte der NINLARO-Dosis
|
Empfohlene Anfangsdosis*
|
Erste Reduktion auf
|
Zweite Reduktion auf
|
Absetzen
| |
4 mg
|
3 mg
|
2,3 mg
|
* Empfohlene Anfangsdosis 3 mg bei Patienten mit mässiger oder schwerer Leberinsuffizienz, schwerer Niereninsuffizienz oder terminaler dialysepflichtiger Nierenerkrankung (ESRD).
Für NINLARO und Lenalidomid wird eine alternierende Dosisänderung aufgrund überlappender Toxizitäten in Form von Thrombozytopenie, Neutropenie und Rash empfohlen. Bei diesen Toxizitäten ist der erste Dosisänderungsschritt die Aussetzung oder Reduktion von Lenalidomid. Bezüglich der Dosisreduktionsschritte bei diesen Toxizitäten, siehe Fachinformation Lenalidomid, Rubrik «Dosierung/Anwendung».
Tabelle 2: Richtlinien zur Dosisanpassung von NINLARO in Kombination mit Lenalidomid und Dexamethason
|
Hämatologische Toxizitäten
|
Empfohlene Massnahmen
| |
Thrombozytopenie (Thrombozytenzahl)
| |
Thrombozytenzahl <30'000/mm3
|
·NINLARO und Lenalidomid aussetzen, bis Thrombozytenzahl ≥30'000/mm3.
·Bei Normalisierung mit Lenalidomid in der nächst niedrigeren Dosierung gemäss der entsprechenden Fachinformation sowie mit NINLARO in der letzten Dosierung fortfahren.
·Sinkt die Thrombozytenzahl erneut <30'000/mm3, NINLARO und Lenalidomid aussetzen, bis die Thrombozytenzahl ≥30'000/mm3.
·Bei Normalisierung mit NINLARO in der nächst niedrigeren Dosierung sowie mit Lenalidomid in der letzten Dosierung fortfahren.*
| |
Neutropenie (absolute Neutrophilenzahl)
| |
Absolute Neutrophilenzahl <500/mm3
|
·NINLARO und Lenalidomid aussetzen, bis die absolute Neutrophilenzahl ≥500/mm3 ist.
·Gemäss den klinischen Leitlinien kann auch das Hinzufügen von G-CSF in Betracht gezogen werden.
·Bei Normalisierung mit Lenalidomid in der nächst niedrigeren Dosierung gemäss der entsprechenden Fachinformation sowie mit NINLARO in der letzten Dosierung fortfahren.
·Sinkt die absolute Neutrophilenzahl erneut <500/mm3, NINLARO und Lenalidomid aussetzen, bis die absolute Neutrophilenzahl ≥500/mm3 ist.
·Bei Normalisierung mit NINLARO in der nächst niedrigeren Dosierung sowie mit Lenalidomid in der letzten Dosierung fortfahren.*
| |
Nicht-hämatologische Toxizitäten
|
Empfohlene Massnahmen
| |
Rash
| |
Grad† 2 oder 3
|
·Lenalidomid aussetzen, bis Rash sich auf ≤ Grad 1 zurückbildet.
·Bei Normalisierung mit Lenalidomid in der nächst niedrigeren Dosierung gemäss der entsprechenden Fachinformation fortfahren.
·Tritt erneut Rash Grad 2 oder 3 auf, NINLARO und Lenalidomid aussetzen, bis Rash sich auf ≤ Grad 1 zurückbildet.
·Bei Normalisierung mit NINLARO in der nächst niedrigeren Dosierung sowie mit Lenalidomid in der letzten Dosierung fortfahren.*
| |
Grad 4
|
Behandlung unterbrechen.
| |
Andere Nicht-hämatologische Toxizitäten
| |
Periphere Neuropathie Grad 1 mit Schmerzen oder periphere Neuropathie Grad 2
|
·NINLARO aussetzen, bis die periphere Neuropathie sich auf ≤ Grad 1 ohne Schmerzen oder bis zum Ausgangszustand des Patienten zurückbildet.
·Bei Normalisierung mit NINLARO in der letzten Dosierung fortfahren.
| |
Periphere Neuropathie Grad 2 mit Schmerzen oder periphere Neuropathie Grad 3
Weitere nicht-hämatologische Toxizitäten Grad 3 oder 4
|
·NINLARO aussetzen. Toxizitäten müssen sich nach Ermessen des Arztes/der Ärztin allgemein bis zum Ausgangszustand des Patienten oder ≤ Grad 1 rückbilden, bevor die Behandlung mit NINLARO fortgesetzt wird.
·Bei Normalisierung mit NINLARO in der nächst niedrigeren Dosierung fortfahren.
| |
Periphere Neuropathie Grad 4
|
Behandlung unterbrechen.
|
* Bei weiteren Ereignissen Dosisanpassung von Lenalidomid und NINLARO alternieren
† Einstufung basierend auf Toxizitätskriterien des Nationalen Krebsinstituts der USA (CTCAE) Version 4.03
Begleitmedikation
Bei Patienten, die mit Ixazomib behandelt werden, sollte eine antivirale Prophylaxe in Betracht gezogen werden, um das Risiko einer Herpes-Zoster-Reaktivierung zu reduzieren. Bei Patienten, die eine antivirale Prophylaxe erhielten, traten in Studien mit Ixazomib seltener Herpes-Zoster-Infektionen auf als bei Patienten, bei denen keine solche Prophylaxe durchgeführt wurde.
Spezielle Dosierungsanweisungen
Ältere Patienten
Bei Patienten über 65 Jahren ist basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse keine Dosisanpassung von NINLARO erforderlich. In Studien zu NINLARO gab es keine klinisch signifikanten Unterschiede hinsichtlich Sicherheit und Wirksamkeit zwischen Patienten unter 65 Jahren und Patienten über 65 Jahren.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von NINLARO bei Kindern und Jugendlichen unter 18 Jahren ist nicht gezeigt. Es liegen keine Daten vor.
Patienten mit Leberfunktionsstörung
Bei Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin ≤ obere Normgrenze (ULN) und Aspartataminotransferase (AST) >ULN oder Gesamtbilirubin >1-1,5 x ULN und beliebige AST) ist basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse keine Dosisanpassung von NINLARO erforderlich. Bei Patienten mit mässiger (Gesamtbilirubin >1,5-3 x ULN) oder schwerer (Gesamtbilirubin >3 x ULN) Leberfunktionsstörung wird basierend auf den Ergebnissen einer pharmakokinetischen Studie (siehe Rubrik «Pharmakokinetik») eine niedrigere Anfangsdosis (3 mg) empfohlen.
Patienten mit Nierenfunktionsstörung
Bei Patienten mit leichter oder mässiger Nierenfunktionsstörung (Kreatinin-Clearance ≥30 ml/Min) ist basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse keine Dosisanpassung von NINLARO erforderlich. Bei Patienten mit schwerer Nierenfunktionsstörung (Kreatinin-Clearance <30 ml/Min) oder terminaler dialysepflichtiger Nierenerkrankung (ESRD) wird basierend auf den Ergebnissen einer pharmakokinetischen Studie eine geringere Anfangsdosis (3 mg) empfohlen. NINLARO ist nicht dialysierbar und kann daher unabhängig von der Dialysezeit eingenommen werden (siehe Rubrik «Pharmakokinetik»).
Für Dosierungsempfehlungen bei Patienten mit Nierenfunktionsstörungen, siehe auch Fachinformation Lenalidomid.
Ethnische Herkunft
Die Sicherheit und Wirksamkeit mit einer Anfangsdosis von 4 mg Ixazomib in Kombination mit Lenalidomid und Dexamethason wurde in einer globalen klinischen Studie mit asiatischen Patienten nachgewiesen. Bei kaukasischen und asiatischen Patienten gab es eine Überschneidung der AUC von Ixazomib; jedoch war die mittlere AUC bei asiatischen Patienten um 35% erhöht. Eine genaue Beobachtung dieser Patienten wird empfohlen. Siehe Abschnitt «Dosisanpassungen».
Art der Anwendung
Oral.
NINLARO sollte jeweils etwa zur gleichen Tageszeit an den Tagen 1, 8 und 15 mindestens 1 Stunde vor oder mindestens 2 Stunden nach der Mahlzeit eingenommen werden (siehe Rubrik «Pharmakokinetik»). Die Hartkapsel muss vollständig und mit Wasser geschluckt werden. Die Hartkapsel darf nicht zerdrückt, zerkaut oder geöffnet werden (siehe Rubrik «Sonstige Hinweise, Hinweise für die Handhabung»).
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenThrombozytopenie
Thrombozytopenien wurden unter NINLARO beobachtet (siehe Rubrik «Unerwünschte Wirkungen»). Der Thrombozyten-Nadir trat dabei üblicherweise zwischen den Tagen 14-21 des sich wiederholenden 28-tägigen Zyklus auf. Zu Beginn des nächsten Zyklus war der Wert auf die Ausgangszahl zurückgekehrt. Die Thrombozytopenie führte nicht zu einem Anstieg von hämorrhagischen Ereignissen oder von Thrombozytentransfusionen.
Während der Behandlung mit NINLARO ist die Thrombozytenzahl mindestens monatlich zu kontrollieren. Eine häufigere Überwachung sollte gemäss Fachinformation von Lenalidomid während der ersten drei Zyklen in Erwägung gezogen werden. Eine Thrombozytopenie kann mittels Dosisanpassungen (siehe Rubrik «Dosierung/Anwendung») und Thrombozytentransfusionen entsprechend medizinischer Standardleitlinien gesteuert werden.
Gastrointestinale Toxizitäten
Über Übelkeit, Erbrechen und Diarrhoe wurde unter NINLARO berichtet (siehe Rubrik «Unerwünschte Wirkungen»). Gelegentlich war die Anwendung von Antiemetika und Antidiarrhoika sowie unterstützender Massnahmen erforderlich. Die Dosis sollte bei schweren Symptomen (Grad 3-4) angepasst werden (siehe Rubrik «Dosierung/Anwendung»).
Periphere Neuropathie
Bei Patienten, die mit NINLARO behandelt wurden, wurden Fälle von peripherer Neuropathie berichtet (siehe Rubrik «Unerwünschte Wirkungen»). Die Patienten sollten auf Symptome einer Neuropathie überwacht werden. Bei Patienten mit neu auftretender oder sich verschlechternder peripherer Neuropathie ist möglicherweise eine Dosisanpassung erforderlich (siehe Rubrik «Dosierung/Anwendung»).
Periphere Ödeme
Unter Therapie mit NINLARO wurden periphere Ödeme berichtet (siehe Rubrik «Unerwünschte Wirkungen»). Die zugrundeliegenden Ursachen sollten untersucht und bei Bedarf unterstützende Massnahmen ergriffen werden. Die Dosierung von Dexamethason sollte entsprechend der Fachinformation bzw. die Dosierung von NINLARO entsprechend den Dosierungsvorschriften für den Fall des Auftretens von Symptomen vom Grad 3 oder 4 angepasst werden (siehe Rubrik «Dosierung/Anwendung»).
Hautreaktionen
Rash wurde unter NINLARO berichtet (siehe Rubrik «Unerwünschte Wirkungen»). Rash sollte mit unterstützender Therapie, oder bei Grad 2 und höher, mit einer Dosisanpassung behandelt werden (siehe Rubrik «Dosierung/Anwendung»). Es wurde auch über das Stevens-Johnson-Syndrom und die toxische epidermale Nekrolyse bei NINLARO berichtet, einschliesslich tödlicher Fälle (siehe Rubrik «Unerwünschte Wirkungen»). Wenn das Stevens-Johnson-Syndrom oder die toxische epidermale Nekrolyse auftritt, muss NINLARO abgesetzt werden.
Schwangerschaft
Basierend auf seinem Wirkmechanismus und den Ergebnissen von Tierstudien ist davon auszugehen, dass Ixazomib zu fetalen Schäden führen kann, wenn es Schwangeren verabreicht wird. Gebärfähige Frauen sollten angehalten werden, während der Behandlung mit Ixazomib eine Schwangerschaft zu vermeiden. Falls Ixazomib während der Schwangerschaft angewendet wird oder die Patientin während der Behandlung mit Ixazomib schwanger wird, sollte die Patientin über die potenziellen Gefahren für den Fötus in Kenntnis gesetzt werden. Gebärfähige Frauen sind anzuweisen, während der Behandlung mit Ixazomib und für 90 Tage nach der letzten Dosis eine wirksame Verhütungsmethode anzuwenden. Frauen, die hormonelle Kontrazeptiva anwenden, sollten zusätzlich eine Barrieremethode zur Empfängnisverhütung benutzen. (Siehe Abschnitte «Schwangerschaft, Stillzeit» und «Präklinische Daten»).
Starke CYP3A-Induktoren
Starke Induktoren können die Wirksamkeit von NINLARO verringern. Daher sollte die gleichzeitige Anwendung von starken CYP3A-Induktoren wie Carbamazepin, Phenytoin, Rifampicin und Johanniskraut (Hypericum perforatum) vermieden werden (siehe «Interaktionen» und «Pharmakokinetik»). Falls die gleichzeitige Anwendung eines starken CYP3A-Induktors unvermeidlich ist, sollten die Patienten engmaschig auf eine gute Krankheitskontrolle überwacht werden.
InteraktionenStarke CYP3A-Induktoren
Die gleichzeitige Anwendung starker CYP3A-Induktoren und NINLARO wird nicht empfohlen. Bei gleichzeitiger Verabreichung von NINLARO und Rifampin nahm die Cmax von Ixazomib um 54% und die AUC um 74% ab.
Starke CYP3A Inhibitoren
Die gleichzeitige Verabreichung von NINLARO und Clarithromycin führte nicht zu einer klinisch bedeutsamen Änderung der systemischen Exposition von Ixazomib. Die Cmax von Ixazomib nahm um 4% ab, die AUC um 11% zu. In einer separaten Studie mit Ketoconazol, bei der ein fixed-sequence Design angewendet wurde, wurden bei der gemeinsamen Verabreichung mit Ketoconazol in der zweiten Periode höhere Expositionswerte für Ixazomib festgestellt. Die tatsächliche Auswirkung von Ketoconazol wurde nicht bewertet, da er durch einen Periodeneffekt verfälscht wurde. Aus diesen Gründen ist bei der gemeinsamen Verabreichung von starken CYP3A-Hemmern mit NINLARO Vorsicht geboten.
Die gleichzeitige Verabreichung von NINLARO und Clarithrhomycin kann hingegen ohne Dosisanpassung erfolgen.
Starke CYP1A2 Inhibitoren
Bei gleichzeitiger Anwendung starker CYP1A2-Inhibitoren ist keine Dosisanpassung von NINLARO erforderlich. Die gleichzeitige Verabreichung von NINLARO und starken CYP1A2-Inhibitoren führte basierend auf den Ergebnissen einer pharmakokinetischen Populationsanalyse nicht zu einer klinisch bedeutsamen Änderung der systemischen Exposition von Ixazomib.
Wirkung von NINLARO auf andere Arzneimittel
Es wird nicht erwartet, dass NINLARO aufgrund CYP-Inhibition oder -Induktion zu Arzneimittelinteraktionen führt.
Ixazomib ist kein reversibler oder zeitabhängiger Inhibitor von CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6 oder 3A4/5. Ixazomib induzierte keine CYP1A2-, CYP2B6-, und CYP3A4/5-Aktivität oder entsprechende immunreaktive Proteinspiegel.
Transporterbasierte Interaktionen
Es wird nicht erwartet, dass NINLARO transportervermittelte Interaktionen zwischen Arzneimitteln hervorruft. Ixazomib ist ein Low-Affinity-Substrat von P-gp. Ixazomib ist kein Substrat von BCRP, MRP2 und hepatischen OATPs. Ixazomib ist kein Inhibitor von P-gp, BCRP, MRP2, OATP1B1, OATP1B3, dem organischen Kationentransporter (OCT)2 oder den organischen Anionentransportern (OAT)1, OAT3, MATE1 oder MATE2-K.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Humandaten hinsichtlich des potenziellen Effekts von Ixazomib auf eine Schwangerschaft oder die Entwicklung des Embryos oder Fötus vor. In tierexperimentellen Studien zur embryonalen und fötalen Entwicklung hat sich jedoch gezeigt, dass Ixazomib einen potenziell letalen Effekt bei Embryonen oder Föten haben kann (siehe Rubrik «Präklinische Daten»).
Während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine Verhütungsmethode anwenden, wird NINLARO nicht empfohlen.
Männliche und weibliche Patienten im fortpflanzungsfähigen Alter müssen während der Behandlung und 90 Tage nach der Behandlung wirksame Verhütungsmethoden anwenden.
Wenn Ixazomib zusammen mit Dexamethason, das als schwacher bis moderater Induktor von CYP3A4 sowie weiterer Enzyme und Transportproteine bekannt ist, verabreicht wird, dann ist das Risiko einer reduzierten Wirksamkeit oraler Kontrazeptiva in Erwägung zu ziehen. Frauen, die orale hormonelle Kontrazeptiva anwenden, sollten zusätzlich eine Barrieremethode zur Empfängnisverhütung benutzen.
Stillzeit
Es ist nicht bekannt, ob NINLARO oder seine Metaboliten in die Muttermilch übergehen.
Ein Risiko für Neugeborene/Säuglinge kann nicht ausgeschlossen werden.
Es wird empfohlen, den Nutzen des Stillens für das Kind gegenüber dem Nutzen der Therapie für die Mutter abzuwägen und dementsprechend muss entweder das Stillen unterbrochen oder die Behandlung mit NINLARO abgesetzt bzw. darauf verzichtet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs liegen keine Daten zu den Auswirkungen auf die Verkehrstüchtigkeit und das Bedienen von Maschinen vor.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die Sicherheitspopulation der randomisierten, doppelblinden, Placebo-kontrollierten klinischen Phase-3-Studie umfasste 720 Patienten mit rezidivierendem und/oder refraktärem multiplem Myelom, die NINLARO in Kombination mit Lenalidomid und Dexamethason (NINLARO-Regime; N=361) oder Placebo in Kombination mit Lenalidomid und Dexamethason (Placebo-Regime; N=359) erhielten.
Die am häufigsten berichteten unerwünschten Wirkungen (≥20%) unter dem NINLARO-Regime umfassten Diarrhoe, Thrombozytopenie, Verstopfung, periphere Neuropathie, Übelkeit, Infektion der oberen Atemwege, periphere Ödeme, Rückenschmerzen, Rash, Erbrechen und Bronchitis. Zu den bei ≥2% der Patienten beobachteten schwerwiegenden unerwünschten Wirkungen zählten Diarrhoe (3%), Thrombozytopenie (2%) und Bronchitis (2%).
Unerwünschte Wirkungen
Die nachfolgenden unerwünschten Wirkungen sind geordnet nach Systemorganklassen und Häufigkeit aufgelistet. Die Häufigkeiten sind wie folgt definiert: «Sehr häufig» (≥1/10), «häufig» (<1/10, ≥1/100), «gelegentlich» (<1/100, ≥1/1000), «selten» (<1/1000, ≥1/10'000), «sehr selten» (<1/10'000); nicht bekannt (kann anhand der verfügbaren Daten nicht abgeschätzt werden).
Unerwünschte Wirkungen bei Patienten, die mit NINLARO in Kombination mit Lenalidomid und Dexamethason behandelt wurden (alle Schweregrade, Grad 3 und Grad 4).
Infektionen und parasitäre Erkrankungen
Sehr häufig (alle Grade): Infektion der oberen Atemwege (27%), Bronchitis (22%).
Häufig (alle Grade): Herpes Zoster.
Häufig (Grad 3): Infektion der oberen Atemwege, Bronchitis.
Gelegentlich (Grad 3): Herpes Zoster.
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig (alle Grade): Thrombozytopenie* (37%).
Sehr häufig (Grad 3): Thrombozytopenie* (13%).
Häufig (Grad 4): Thrombozytopenie*.
Erkrankungen des Immunsystems
Selten (alle Grade): anaphylaktische Reaktion†.
Sehr selten (Grad 3): anaphylaktische Reaktion†.
Sehr selten (Grad 4): anaphylaktische Reaktion†.
Erkrankungen des Nervensystems
Sehr häufig (alle Grade): periphere Neuropathien* (32%).
Häufig (Grad 3): periphere Neuropathien*.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig (alle Grade): Diarrhoe (52%), Verstopfung (35%), Übelkeit (32%), Erbrechen (26%).
Sehr häufig (Grad 3): Diarrhoe (10%)
Häufig (Grad 3): Übelkeit, Erbrechen.
Gelegentlich (Grad 3): Verstopfung.
Erkrankungen der Haut und des Unterhautzellgewebes
Sehr häufig (alle Grade): Rash* (27%).
Häufig (Grad 3): Rash*.
Selten (alle Grade): Stevens-Johnson-Syndrom†, toxische epidermale Nekrolyse†, Angioödem†.
Selten (Grad 3): Stevens-Johnson-Syndrom†, Angioödem†.
Selten (Grad 4): toxische epidermale Nekrolyse†.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig (alle Grade): Rückenschmerzen (27%).
Gelegentlich (Grad 3): Rückenschmerzen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig (alle Grade): periphere Ödeme (27%).
Häufig (Grad 3): periphere Ödeme.
* Stellt eine Zusammenfassung bevorzugter Begriffe dar.
† Ausserhalb der Phase-3-Studie berichtet.
Beschreibung ausgewählter unerwünschter Wirkungen
Rash
Rash trat bei 27% der Patienten unter dem NINLARO-Regime auf. Die meisten Rash-Ereignisse entsprachen Grad 1 oder Grad 2. Rash Grad 3 wurde bei 3% der Patienten unter dem NINLARO-Regime beobachtet. Es wurden keine Grad-4-Ereignisse von Rash beobachtet. Schwere unerwünschte Reaktionen von Rash wurden bei <1% der Patienten im NINLARO-Regime beobachtet. Die am häufigsten beobachteten Rashtypen waren makulopapulöse und makulare Exantheme. Rash führte bei <1% der Patienten zu einem Absetzen eines oder mehrerer der drei Arzneimittel.
Ausserhalb der Phase-3-Studie wurde selten das folgende schwerwiegende unerwünschte Ereignis, für welches die Kausalität nicht gezeigt ist, beobachtet: akute febrile neutrophile Dermatose (Sweet's Syndrom). Es wurde auch selten über das Stevens-Johnson-Syndrom bei NINLARO berichtet, darunter ein Todesfall.
Dermale Ereignisse wurden unter Lenalidomid und Dexamethason beobachtet.
Periphere Neuropathie
Die meisten unerwünschten Reaktionen einer peripheren Neuropathie waren vom Grad 1 (18% im NINLARO- und 16% im Placebo-Regime) und vom Grad 2 (11% im NINLARO- und 6% im Placebo-Regime). Unerwünschte Reaktionen einer peripheren Neuropathie vom Grad 3 wurden bei 2% in beiden Regimen beobachtet; es kam zu keinen Grad-4-Ereignissen oder schwerwiegenden unerwünschten Wirkungen.
Die am häufigsten berichtete Reaktion war eine periphere sensorische Neuropathie (24% im NINLARO- und 17% im Placebo-Regime). Periphere motorische Neuropathien wurden in beiden Regimen gelegentlich beobachtet (<1%). Periphere Neuropathien führten bei 4% der Patienten im NINLARO- und <1% der Patienten im Placebo-Regime zu einem Absetzen eines oder mehrerer der drei Arzneimittel.
Thrombozytopenie
Zwei Prozent der Patienten sowohl im NINLARO- als auch im Placebo-Regime wiesen während der Behandlung eine Thrombozytenzahl ≤10'000/mm3 auf. Weniger als 1% der Patienten unter beiden Behandlungen wiesen während der Behandlung eine Thrombozytenzahl ≤5'000/mm3 auf. Die Thrombozytopenie führte bei 2% der Patienten im NINLARO- und 3% der Patienten im Placebo-Regime zu einem Absetzen eines oder mehrerer der drei Arzneimittel.
Gastrointestinale Toxizitäten
Diarrhoe führte bei 3% der Patienten im NINLARO- und 2% der Patienten im Placebo-Regime zu einem Absetzen eines oder mehrerer der drei Arzneimittel.
Periphere Ödeme
Periphere Ödeme wurden bei 27% der Patienten im NINLARO-Regime und 21% der Patienten im Placebo-Regime berichtet. Die meisten unerwünschten Reaktionen in Form peripherer Ödeme waren vom Grad 1 (17% im NINLARO- und 14% im Placebo-Regime) und vom Grad 2 (7% im NINLARO- und 6% im Placebo-Regime).
Periphere Ödeme vom Grad 3 wurden bei 2% der Patienten im NINLARO-Regime und 1% der Patienten im Placebo-Regime berichtet. Es wurden keine peripheren Ödeme vom Grad 4 berichtet. Periphere Ödeme führten bei <1% der Patienten in beiden Regimen zum Absetzen eines oder mehrerer der drei Arzneimittel.
Sonstige unerwünschte Wirkungen
Ausserhalb der Phase-3-Studie wurden selten folgende schwerwiegende unerwünschte Ereignisse, für welche die Kausalität nicht gezeigt ist, beobachtet: transverse Myelitis, posteriores reversibles Enzephalopathie-Syndrom, Tumorlysesyndrom, und thrombotische thrombozytopenische Purpura.
Unerwünschte Wirkungen nach Markteinführung
Klinisch bedeutsame unerwünschte Wirkungen sind hier aufgeführt, wenn sie oben nicht beschrieben wurden.
Thrombotische Mikroangiopathie
Es wurden Fälle von thrombotischer Mikroangiopathie (TMA), einschliesslich thrombotischthrombozytopenischer Purpura (TTP), bei Patienten berichtet, die Ixazomib erhielten. Einige dieser Fälle verliefen tödlich. Es sollte eine Überwachung auf Anzeichen und Symptome von TMA erfolgen. Bei Verdacht dieser Diagnose ist Ixazomib abzusetzen und die Patienten sind auf eine mögliche TMA hin zu untersuchen. Bei Ausschluss der Diagnose TMA kann die Verabreichung von Ixazomib wieder aufgenommen werden. Die Sicherheit einer erneuten Ixazomibtherapie bei Patienten, bei denen zuvor eine TMA aufgetreten ist, ist nicht bekannt.
Hepatotoxizität
Bei mit Ixazomib behandelten Patienten wurden gelegentlich arzneimittelbedingte Leberschäden (Drug-induced liver injury), hepatozelluläre Schädigungen, hepatische Steatose, cholestatische Hepatitis und Hepatotoxizität gemeldet (siehe «Unerwünschte Wirkungen»). Die Leberenzymwerte sollten regelmässig überwacht und die Dosis sollte bei Symptomen der Grade 3 oder 4 angepasst werden (siehe «Dosierung/Anwendung»).
Posteriores reversibles Enzephalopathie-Syndrom (PRES)
Posteriores reversibles Enzephalopathie-Syndrom (PRES) trat bei Patienten unter der Behandlung mit Ixazomib auf. PRES ist eine seltene, reversible, neurologische Störung, die sich mit epileptischen Anfällen, Bluthochdruck, Kopfschmerzen, Bewusstseinsveränderungen und Sehstörungen zeigen kann. Eine Tomographie des Gehirns, vorzugsweise Magnetresonanztomographie, wird für die Bestätigung der Diagnose verwendet. Wenn Patienten PRES entwickeln, ist die Behandlung mit Ixazomib abzubrechen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurde über Überdosierung bei Patienten die NINLARO einnahmen berichtet. Die Symptome bei Überdosierung stimmen im Allgemeinen mit den bekannten Risiken von NINLARO überein (siehe «Unerwünschte Wirkungen»). Berichte über versehentliche Überdosierungen wurden mit schwerwiegenden unerwünschten Ereignissen wie schwerer Übelkeit, Aspirationspneumonie, Multiorganversagen und Tod in Verbindung gebracht.
Es ist kein spezifisches Antidot für eine Überdosierung von NINLARO bekannt. Im Falle einer Überdosierung ist der Patient engmaschig auf unerwünschte Reaktionen zu überwachen und es sind geeignete unterstützende Massnahmen zu ergreifen.
Das Gesundheitspersonal sollte Patienten und Pflegepersonal anweisen, jeweils nur eine Dosis NINLARO auf einmal und nur im vorgeschriebenen Intervall einzunehmen (eine Hartkapsel, einmal pro Woche, an den Tagen 1, 8 und 15 jedes 28-tägigen Zyklus). Die Bedeutung der sorgfältigen Befolgung aller Dosierungsanweisungen sollte mit Patienten, die die Behandlung beginnen, besprochen werden.
Eigenschaften/WirkungenATC-Code
L01XG03
Wirkungsmechanismus
Ixazomibzitrat, eine Prodrug, ist ein Wirkstoff, der unter physiologischen Bedingungen schnell in seine biologisch aktive Form, Ixazomib, hydrolysiert. Die chemische Bezeichnung von Ixazomibzitrat lautet 1,3,2- Dioxaborolan-4,4-Diessigsäure, 2-[(1R)-1-[[2-[(2,5-Dichlorobenzoyl)amino]acetyl]amino]-3- methylbutyl]-5-oxo-.
Ixazomib ist ein oraler, hochselektiver und reversibler Proteasom-Inhibitor. Ixazomib bindet und hemmt bevorzugt die Chymotrypsin-ähnliche Aktivität der Beta-5-Untereinheit des 20S-Proteasoms.
Ixazomib induzierte Apoptose einzelner Tumorzelltypen in vitro. Ixazomib zeigte in vitro eine Zytotoxizität gegenüber Myelomzellen von Patienten, bei denen es nach mehreren vorangehenden Therapien, einschliesslich Bortezomib, Lenalidomid und Dexamethason, zu einem Rezidiv kam. Die Kombination von Ixazomib und Lenalidomid zeigte bei mehreren multiplen Myelom-Zelllinien eine synergistische zytotoxische Wirkung. In vivo zeigte Ixazomib eine antitumorale Aktivität bei verschiedenen Tumor-Xenograft-Modellen einschliesslich multiplen Myelom-Modellen.
Ixazomib stört auch die Knochenmark-Mikroumgebung. In vitro hemmte Ixazomib die Proliferation multipler Myelomzellen in Co-Kultur mit Knochenmark-Stromazellen. Ixazomib zeigte eine antiangiogene Wirkung in einem in-vitro-Kapillarenausbildungstest. Ixazomib förderte in vitro die Osteoblastogenese und Osteoblastenaktivität und hemmte die Osteoklastogenese und Osteoklastenresorption. Zudem beugte Ixazomib dem Knochenverlust in einem in-vivo-Mausmodell des multiplen Myeloms vor.
Pharmakodynamik
Kardiale Elektrophysiologie
Bei klinisch relevanten Expositionen verlängert NINLARO, basierend auf den Ergebnissen einer pharmakokinetisch-pharmakodynamischen Analyse der Daten von 245 Patienten, das QTc-Intervall nicht. Bei der 4-mg-Dosis wird die mittlere QTcF-Veränderung gegenüber der Baseline auf Grundlage der modellbasierten Analyse auf 0,07 mSek geschätzt (90% Kl; -0,22, 0,36).
Es gab keinen erkennbaren Zusammenhang zwischen der Ixazomib-Konzentration und dem RR-Intervall, was auf keine klinisch bedeutsame Wirkung von NINLARO auf die Herzfrequenz hindeutet.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von NINLARO in Kombination mit Lenalidomid und Dexamethason wurde in einer internationalen randomisierten, doppelblinden, Placebo-kontrollierten, multizentrischen Überlegenheitsstudie der Phase 3 bei Patienten mit rezidivierendem und/oder refraktärem multiplem Myelom untersucht, die mindestens eine vorangehende Therapie erhalten hatten.
Insgesamt wurden 722 Patienten (Intent-to-treat [ITT]-Population) in einem 1:1-Verhältnis randomisiert und erhielten entweder die Kombination aus NINLARO, Lenalidomid und Dexamethason (N=360; NINLARO-Regime) oder Placebo, Lenalidomid und Dexamethason (N=362; Placebo-Regime) bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Toxizität. Die Randomisierung wurde entsprechend der Anzahl vorangegangener Therapielinien (1 gegenüber 2 oder 3), dem Internationalen Myelom-Staging-System (ISS) (Stadium I oder II gegenüber III) und vorangegangenen Therapien mit einem Proteasom-Inhibitor (exponiert oder naiv) stratifiziert.
Die in die Studie eingeschlossenen Patienten hatten ein multiples Myelom, das über Paraprotein im Serum und im Urin oder Bestimmungen der freien Leichtketten messbar war und deren Erkrankung refraktär einschliesslich primär refraktär (d. h., keinerlei Ansprechen auf vorhergehende Therapien) war, bei denen es nach einer vorangegangenen Behandlung zu einem Rezidiv kam oder bei denen es zu einem Rezidiv kam und die Erkrankung refraktär auf alle vorangegangenen Therapien war.
Auch Patienten, die die Therapien vor der Krankheitsprogression wechselten, wurden eingeschlossen, ebenso Patienten mit kontrollierten kardiovaskulären Erkrankungen. Von der Phase-3-Studie ausgeschlossen waren Patienten, die refraktär auf Lenalidomid oder Proteasom-Inhibitoren waren oder mehr als drei vorangegangene Therapien erhalten haben. Da zu diesen Patienten nur begrenzte Daten vorliegen, wird vor Beginn der Behandlung mit NINLARO eine sorgfältige Nutzen-Risiko-Abwägung empfohlen.
Entsprechend der Arzneimittelinformation von Lenalidomid wurde für alle Patienten in beiden Behandlungsgruppen eine Thromboseprophylaxe empfohlen. Begleitmedikationen wie Antiemetika, antivirale Arzneimittel und Antihistaminika wurden den Patienten nach Ermessen des Arztes als Prophylaxe und/oder zur Behandlung von Symptomen verabreicht.
Die Patienten erhielten NINLARO 4 mg oder Placebo an den Tagen 1, 8 und 15 plus Lenalidomid (25 mg) an den Tagen 1 bis 21 und Dexamethason (40 mg) an den Tagen 1, 8, 15 und 22 eines 28-tägigen Zyklus. Patienten mit Nierenfunktionsstörung erhielten eine Anfangsdosis Lenalidomid gemäss der entsprechenden Arzneimittelinformation. Die Behandlung wurde bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Toxizität fortgesetzt.
Die demographischen Ausgangsdaten und die Krankheitscharakteristika wurden gleich gewichtet und waren zwischen den Studiengruppen vergleichbar. Das mediane Alter lag bei 66 Jahren (Bereich: 38-91 Jahre); 58% der Patienten waren älter als 65 Jahre. 57% der Patienten waren männlich. 85% der Studienpopulation waren kaukasisch, 9% asiatisch und 2% schwarz. 93% der Patienten hatten einen ECOG-Performance-Status von 0-1 und 12% befanden sich zu Studienbeginn im ISS-Stadium III (N=90). 25% der Patienten hatten eine Kreatinin-Clearance <60 ml/min. Bei 23% der Patienten lag ein Leichtkettenmyelom vor und bei 12% der Patienten war die Krankheit nur mit einem freien Leichtkettentest nachweisbar. 43% der Gesamtpopulation (N=309) waren Patienten mit erhöhtem zytogenetischem Risiko (Hochrisiko [del(17), t(4;14), t(14;16)] oder 1q-Amplifikation [1q21]). 19% (N=137) wiesen hochrisikobehaftete zytogenetische Anomalitäten und 10% (N=69) die Deletion del(17) auf. Die Patienten erhielten ein bis drei vorangegangene Therapielinien (median: 1) einschliesslich einer früheren Behandlung mit Bortezomib (69%), Carfilzomib (<1%), Thalidomid (45%), Lenalidomid (12%) und Melphalan (81%). Bei 57% der Patienten war zuvor eine Stammzelltransplantation durchgeführt worden. Bei 77% der Patienten kam es nach vorangegangenen Therapielinien zu einem Rezidiv und 11% der Patienten waren refraktär auf vorangegangene Therapielinien. Primäre Refraktärität (d.h. das beste Ansprechen bei allen vorherigen Therapielinien war eine stabile Erkrankung oder Krankheitsprogression) wurde bei 6% der Patienten dokumentiert.
Primärer Endpunkt war das progressionsfreie Überleben (PFS) gemäss des Konsens der International Myeloma Working Group 2011 (IMWG) über einheitliche Responsekriterien nach Prüfung durch ein verblindetes unabhängiges Expertenkomitee (IRC) basierend auf zentralen Laborergebnissen. Das Ansprechen wurde alle 4 Wochen bis zur Krankheitsprogression überprüft.
Bei der primären und abschliessenden Analyse für die statistische Prüfung (mediane Nachbeobachtungszeit von 14,7 Monaten und mediane Zyklusanzahl 13) wurden für das NINLARO-Behandlungsregime signifikant bessere Ergebnisse mit einem Anstieg des medianen PFS um etwa 6 Monate belegt. Im NINLARO-Regime traten 129 (36%) und im Placebo-Regime 157 (43%) PFS-Ereignisse auf. Das mediane PFS betrug im NINLARO-Regime 20,6 Monate und im Placebo-Regime 14,7 Monate (HR=0,74 [95%-KI (0,587, 0,939)], p-Wert=0,012).
Hinweis: Die Hazard Ratio basiert auf dem geschichteten proportionalen Hazard-Regressionsmodell nach Cox; eine Hazard Ratio von weniger als 1 deutet auf eine Überlegenheit der NINLARO-Therapie hin. Der p-Wert basiert auf dem geschichtetem Log-Rank-Test.
Die Verbesserung des PFS im NINLARO-Regime wurde durch Verbesserungen der Gesamtansprechrate gestützt.
Eine geplante Interimsanalyse des Gesamtüberlebens (OS) wurde nach einer medianen Nachbeobachtungszeit von 23 Monaten mit 35% der für die finale OS-Analyse erforderlichen Anzahl Todesfälle in der ITT-Population durchgeführt. Im NINLARO-Arm waren 81 Todesfälle, im Placebo-Arm 90 Todesfälle aufgetreten. Das mediane OS wurde in keinem der beiden Behandlungsarme erreicht. Gleichzeitig wurde eine nichtinferentielle exploratorische PFS-Analyse durchgeführt. Das geschätzte mediane PFS in der ITT-Population betrug im NINLARO-Arm 20 Monate und im Placebo-Arm 15,9 Monate (HR=0,82 [95%-KI (0,67, 1,0)]).
Bei der finalen OS-Analyse nach einer medianen Nachbeobachtungszeit von ca. 85 Monaten betrug das mediane OS in der ITT-Population 53,6 Monate für Patienten im NINLARO-Regime und 51,6 Monate für Patienten im Placebo-Regime (HR=0,94 [95%-KI (0,78, 1,13)]).
Bei der OS-Interimsanalyse kam es in der Untergruppe der Hochrisikopatienten, deren multiples Myelom zentral in einem CLIA-zertifizierten Labor auf eine Deletion des Chromosoms 17 (del[17]) untersucht wurde, zu weniger Todesfällen bei Patienten mit del(17) im NINLARO-Arm (11,1%) als bei Patienten im Placebo-Arm (27,3%). Bei der finalen OS-Analyse betrug das mediane OS in dieser Subpopulation 42,2 Monate für Patienten im NINLARO-Regime und 29,4 Monate für Patienten im Placebo-Regime (HR=0,92 [95%-KI (0,52, 1,63)]).
Bei Patienten mit einem hohen zytogenetischen Risiko betrug das mediane OS in der finalen Analyse 46,9 Monate im NINLARO-Regime und 30,9 Monate im Placebo-Regime (HR=0,87 [95%-KI (0,58, 1,31)]) und bei Patienten mit erhöhtem zytogenetischem Risiko betrug das mediane OS 44,6 Monate im NINLARO-Regime und 33,4 Monate im Placebo-Regime (HR=0,86 [95%-KI (0,66, 1,12)]).
Da es sich beim multiplen Myelom um eine heterogene Erkrankung handelt, kann der Nutzen in verschiedenen Untergruppen variieren (siehe Abbildung 1).
Abbildung 1: Forest-Plot des progressionsfreien Überlebens in Untergruppen
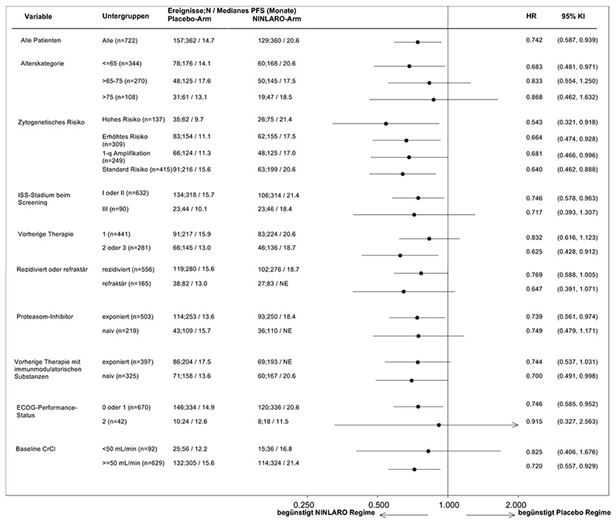
Indizierte Population
Es wurde eine Analyse zur Ermittlung der klinischen Wirksamkeitsergebnisse anhand festgelegter zytogenetischer und stratifizierter klinisch-ungünstiger Risikomerkmale durchgeführt. In der ITT-Population wurde eine stärkere Verbesserung der Wirksamkeitsergebnisse bei 70% der Patienten (N=505, «indizierte Population») beobachtet, die mindestens eine vorangegangene Therapie erhalten haben und Hochrisikomerkmale aufwiesen (definiert als erhöhtes zytogenetisches Risiko [Hochrisiko (del[17], t[4;14], t[14;16]) oder 1q21] oder ISS-Stadium III) oder die mindestens zwei vorangegangene Therapien erhalten haben. Bei der Primäranalyse traten im NINLARO-Arm 98 (39%) PFS-Ereignisse und im Placebo-Arm 122 (49%) PFS-Ereignisse auf. Das mediane PFS betrug im NINLARO-Arm 18,4 Monate und im Placebo-Arm 12,2 Monate (HR=0,681 [95%-KI (0,520, 0,892)], p-Wert=0,005).
Die Verbesserung des PFS im NINLARO-Arm wurde durch Verbesserungen der Gesamtansprechrate in der indizierten Population gestützt.
Zum Zeitpunkt der geplanten OS-Interimsanalyse waren in der indizierten Population im NINLARO-Arm 58 Todesfälle und im Placebo-Arm 76 Todesfälle aufgetreten. Das mediane OS wurde im NINLARO-Arm nicht erreicht und betrug im Placebo-Arm 30,9 Monate (HR=0,706 [95%-KI (0,499, 0,997)]). Die Ergebnisse der nichtinferentiellen exploratorischen PFS-Analyse zeigten ein geschätztes medianes PFS in der indizierten Population von 19,1 Monaten im NINLARO-Arm und von 12,6 Monaten im Placebo-Arm (HR=0,728 [95%-KI (0,573, 0,924)]). Ergebnisse stimmen mit der Schlussfolgerung der primären PFS-Analyse überein, dass ein positiver Behandlungseffekt besteht.
Zum Zeitpunkt der abschliessenden OS-Analyse gab es in der indizierten Population 181 Todesfälle im NINLARO-Regime und 179 Todesfälle im Placebo-Regime. Das mediane OS betrug 50,4 Monate im NINLARO-Regime und 42,0 Monate im Placebo-Regime (HR=0,912 [95%-KI (0,74, 1,125)]).
Pädiatrie
Die Europäische Arzneimittelagentur hat die Verpflichtung, die Ergebnisse der Studien zu NINLARO bei allen Untergruppen der pädiatrischen Population vorzulegen, aufgehoben (siehe Rubrik «Dosierung/Anwendung, Kinder und Jugendliche»).
PharmakokinetikAbsorption
Nach der oralen Anwendung wurden Spitzenplasmakonzentrationen bei Ixazomib ungefähr eine Stunde nach der Gabe erreicht. Die durchschnittliche absolute Bioverfügbarkeit beträgt 58%. Die Ixazomib AUC steigt dosisproportional über einen Dosisbereich von 0,2-10,6 mg an.
Bei Einnahme mit einer Mahlzeit mit hohem Fettgehalt nahm die AUC von Ixazomib um 28% im Vergleich zur Verabreichung auf nüchternen Magen ab (siehe Rubrik «Dosierung/Anwendung»).
Distribution
Ixazomib wird zu 99% an Plasmaproteine gebunden und verteilt sich in die Erythrozyten mit einem Blut-zu-Plasma-AUC-Verhältnis von 10. Das Steady-State-Verteilungsvolumen beträgt 543 l.
Metabolismus
Keine Daten vorhanden.
Elimination
Ixazomib weist ein multiexponenzielles Dispositionsprofil auf. Basierend auf einer pharmakokinetischen (PK) Populationsanalyse lag die systemische Clearance (CL) bei etwa 1,86 l/Std, mit einer interindividuellen Variabilität von 44%. Die Halbwertszeit der (terminalen) Alpha-, Beta- und Gamma-Dispositionsphasen des pharmakokinetischen Profils betrug 0,272 Stunden, 17,8 Stunden und 9,5 Tage.
Bei wöchentlicher oraler Gabe wurde an Tag 15 eine etwa zweifache Akkumulation der AUC beobachtet.
Biotransformation
Nach oraler Verabreichung einer radioaktiv markierten Dosis bestanden 70% des drug-related Materials im Plasma aus Ixazomib. Ixazomib wird zu mehreren deboronierten Metaboliten verstoffwechselt, die pharmakologisch inaktiv sind und im Urin ausgeschieden werden. Die Metabolisierung über mehrere CYP-Enzyme und Nicht-CYP-Proteine ist wahrscheinlich der wichtigste Clearance-Mechanismus für Ixazomib. Bei klinisch relevanten Ixazomib-Konzentrationen weisen In-vitro-Studien mit humanen cDNA-exprimierten Cytochrom-P450-Isozymen darauf hin, dass kein spezifisches CYP-Isozym vorwiegend zur Ixazomib-Metabolisierung beiträgt und Nicht-CYP-Proteine an der gesamten Metabolisierung beteiligt sind.
Ausscheidung
Nach Verabreichung einer oralen Einzeldosis von 14C-Ixazomib an fünf Patienten mit fortgeschrittenem Karzinom wurden 62% der applizierten Radioaktivität über den Urin und 22% über den Stuhl ausgeschieden. Der Anteil an unverändertem Ixazomib betrug <3,5% der im Urin nachgewiesenen, verabreichten Dosis.
Kinetik spezieller Patientengruppen
Leberfunktionsstörung
Die Pharmakokinetik von Ixazomib ist bei Patienten mit normaler Leberfunktion und Patienten mit leichter Leberfunktionsstörung (Gesamtbilirubin ≤ ULN und AST >ULN oder Gesamtbilirubin >1-1,5 x ULN und beliebige AST) basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse vergleichbar.
Die Pharmakokinetik von Ixazomib wurde bei Patienten mit normaler Leberfunktion mit 4 mg (N=12), mässiger Leberfunktionsstörung mit 2,3 mg (Gesamtbilirubin >1,5-3 x ULN, N=13) oder schwerer Leberfunktionsstörung mit 1,5 mg (Gesamtbilirubin >3 x ULN, N=18) untersucht. Die ungebundene dosisnormierte AUC war bei Patienten mit mässiger oder schwerer Leberfunktionsstörung gegenüber Patienten mit normaler Leberfunktion um 27% erhöht (siehe Rubrik «Dosierung/Anwendung»).
Nierenfunktionsstörung
Die Pharmakokinetik von Ixazomib ist bei Patienten mit normaler Nierenfunktion und Patienten mit leichter oder mässiger Nierenfunktionsstörung (Kreatinin- Clearance ≥30 ml/Min) basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse vergleichbar.
Die Pharmakokinetik von Ixazomib wurde mit einer Dosis von 3 mg bei Patienten mit normaler Nierenfunktion (Kreatinin-Clearance ≥90 ml/Min, N=18), schwerer Nierenfunktionsstörung (Kreatinin-Clearance <30 ml/Min, N=14), oder dialysepflichtiger Nierenerkrankung im Endstadium (ESRD) (N=6) untersucht. Die ungebundene AUC war bei Patienten mit schwerer Nierenfunktionsstörung oder dialysepflichtiger ESRD gegenüber Patienten mit normaler Nierenfunktion um 38% erhöht. Die während der Hämodialyse gemessenen Prä- und Postdialysekonzentrationen waren vergleichbar, was darauf hindeutet, dass Ixazomib nicht dialysierbar ist (siehe Rubrik «Dosierung/Anwendung»).
Alter, Geschlecht
Es gab basierend auf den Ergebnissen einer pharmakokinetischen (PK) Populationsanalyse keine klinisch bedeutsamen Auswirkungen von Alter (23-91 Jahre), Geschlecht oder der Körperoberfläche (1,2-2,7 m2) auf die Clearance von Ixazomib.
Präklinische DatenKarzinogenese und Mutagenese
Ixazomib war nicht mutagen in einem bakteriellen Rückmutationstest (Ames-Test) und nicht klastogen in einem Mikronukleustest am Knochenmark von Mäusen. Ixazomib zeigte sich als positiv in einem in-vitro-Klastogenitätstest an humanen peripheren Blutlymphozyten. Andererseits war Ixazomib negativ in einem in-vivo-Comet-Assay an Mäusen, bei dem die anteilige Tail-DNA in Magen und Leber gemessen wurde. Die Evidenzlage lässt deshalb darauf schliessen, dass NINLARO nicht mit einem genotoxischen Risiko behaftet ist. Es wurden keine Karzinogenitätsstudien mit NINLARO durchgeführt.
Reproduktive und embryofetale Entwicklung
Studien zur Entwicklungstoxizität an Ratten und Kaninchen ergaben keine direkte embryofetale Toxizität unterhalb maternalen toxischen Ixazomib-Dosen. In Dosisfindungsstudien an trächtigen Ratten (0,6 mg/kg; 3,6 mg/m2) und Kaninchen (1,0 mg/kg; 12 mg/m2) kam es zu einem reduzierten Körpergewicht der Feten, einer Tendenz zu geringerer fetaler Überlebensfähigkeit und/oder höheren Postimplantationsverlusten; diese Ergebnisse wurden jedoch in den definitiven Studien nicht eindeutig reproduziert und wurden nur bei maternalen toxischen Dosen beobachtet (Dosen, die zu einer Reduzierung von Körpergewicht und/oder Nahrungsaufnahme führten). In der definitiven Studie an Kaninchen wurden bei Dosen ≥0,3 mg/kg (3,6 mg/m2) vermehrt Veränderungen/Anomalitäten des fetalen Skeletts (untere Wirbel, Anzahl lumbaler Wirbel und vollständige überzählige Rippen) beobachtet, die ebenfalls mit der maternalen Toxizität in Verbindung gebracht wurden. Bei Dosen, die auch maternal toxisch sind, ist Ixazomib als embryofetotoxisch zu bezeichnen. Eine Dosis von 0,1 mg/kg (1,2 mg/m2) führte nicht zu einer maternalen Toxizität oder hatte embryofetale Wirkungen zur Folge.
Studien zur Fertilität und frühen embryonalen Entwicklung sowie zur prä- und postnatalen Toxizität wurden mit Ixazomib nicht durchgeführt. In den allgemeinen Toxizitätsstudien wurde jedoch eine Beurteilung der reproduktiven Gewebe vorgenommen. In Studien mit einer Dauer von bis zu sechs Monaten an Ratten und bis zu neun Monaten an Hunden zeigten sich keine Auswirkungen der Behandlung mit Ixazomib auf die männlichen oder weiblichen Reproduktionsorgane.
Toxikologische und/oder pharmakologische Eigenschaften bei Tieren
In multizyklischen allgemeinen Toxizitätsstudien an Ratten und Hunden zählten der Gastrointestinaltrakt (GI-Trakt), die Lymphgewebe und das Nervensystem zu den wichtigsten Zielorganen. Die GI-Befunde umfassten Erbrechen und/oder Diarrhoe, einen Anstieg der Leukozyten und mikroskopische Veränderungen (Entzündung, epitheliale Hyperplasie, neutrophile Infiltration, Einzelzellnekrose und Erosion/Ulzeration). Die Toxizität auf das Lymphsystem war gekennzeichnet durch lymphoide Depletion/Nekrose (einschliesslich Knochenmark), neutrophile Infiltration und Einzelzellnekrose. Auswirkungen auf das Nervensystem traten vorwiegend bei Hunden bei Dosen ≥0,1 mg/kg (2 mg/m2) auf und umfassten mikroskopische Befunde minimaler bis leichter neuronaler Degeneration der sympathischen, spinalen-, peripheren autonomen (Speicheldrüse) und Endorganganglien und minimale sekundäre axonale Degenerationen der peripheren Nerven und des aufsteigenden Trakts in den Hintersäulen des Rückenmarks (Neurotoxizität). In der neun Monate (10 Zyklen umfassenden) Studie an Hunden, in der das Dosierungsschema dem klinischen Schema (28-tägiger Zyklus) entsprach, waren die mikroskopischen neuronalen Wirkungen allgemein minimal und wurden nur bei 0,2 mg/kg (4 mg/m2; AUC168 1940 h*ng/ml) beobachtet. Die meisten Zielorganbefunde bildeten sich nach Behandlungsabbruch teilweise bis vollständig zurück. Ausgenommen hiervon waren die neuronalen Befunde der lumbalen Spinalganglien und der dorsalen Säule. Eine fehlende fortschreitende neuronale Degeneration in den peripheren Ganglien und nur sekundäre periphere Veränderungen der Nervenfasern und Axone stehen für eine fehlende persistente Toxizität.
Tierstudien zufolge passiert Ixazomib die Blut-Hirn-Schranke nicht. Zudem zeigten präklinische Studien und Untersuchungen zur Sicherheitspharmakologie in vitro (an hERG-Kanälen) und in vivo, dass Ixazomib keine Auswirkungen auf die Herz-Kreislauffunktion hat.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Für Kinder unzugänglich aufbewahren. Nicht über 30°C, vor Feuchtigkeit geschützt lagern. Nicht einfrieren.
Die Hartkapsel sollte erst unmittelbar vor der Anwendung entnommen werden.
Hinweise für die Handhabung
NINLARO ist zytotoxisch. Die Hartkapseln dürfen nicht geöffnet oder zerbrochen werden. Ein direkter Kontakt mit dem Kapselinhalt ist zu vermeiden. Bei Zerbrechen einer Hartkapsel Staubaufwirbelung bei der Säuberung vermeiden. Sollte es zu einem Kontakt kommen, gründlich mit Seife und Wasser abwaschen.
Nicht verwendete Arzneimittel oder Abfallmaterialien sind entsprechend den lokalen Anforderungen zu beseitigen.
Zulassungsnummer65959 (Swissmedic).
PackungenNINLARO 4 mg: Bündelpackung mit 3 einzelnen Blister-Walletpackungen mit je einer Hartkapsel (A).
NINLARO 3 mg: Bündelpackung mit 3 einzelnen Blister-Walletpackungen mit je einer Hartkapsel (A).
NINLARO 2.3 mg: Bündelpackung mit 3 einzelnen Blister-Walletpackungen mit je einer Hartkapsel (A).
ZulassungsinhaberinTakeda Pharma AG, 8152 Opfikon
Stand der InformationFebruar 2024
|