Eigenschaften/WirkungenATC-Code
L01XG03
Wirkungsmechanismus
Ixazomibzitrat, eine Prodrug, ist ein Wirkstoff, der unter physiologischen Bedingungen schnell in seine biologisch aktive Form, Ixazomib, hydrolysiert. Die chemische Bezeichnung von Ixazomibzitrat lautet 1,3,2- Dioxaborolan-4,4-Diessigsäure, 2-[(1R)-1-[[2-[(2,5-Dichlorobenzoyl)amino]acetyl]amino]-3- methylbutyl]-5-oxo-.
Ixazomib ist ein oraler, hochselektiver und reversibler Proteasom-Inhibitor. Ixazomib bindet und hemmt bevorzugt die Chymotrypsin-ähnliche Aktivität der Beta-5-Untereinheit des 20S-Proteasoms.
Ixazomib induzierte Apoptose einzelner Tumorzelltypen in vitro. Ixazomib zeigte in vitro eine Zytotoxizität gegenüber Myelomzellen von Patienten, bei denen es nach mehreren vorangehenden Therapien, einschliesslich Bortezomib, Lenalidomid und Dexamethason, zu einem Rezidiv kam. Die Kombination von Ixazomib und Lenalidomid zeigte bei mehreren multiplen Myelom-Zelllinien eine synergistische zytotoxische Wirkung. In vivo zeigte Ixazomib eine antitumorale Aktivität bei verschiedenen Tumor-Xenograft-Modellen einschliesslich multiplen Myelom-Modellen.
Ixazomib stört auch die Knochenmark-Mikroumgebung. In vitro hemmte Ixazomib die Proliferation multipler Myelomzellen in Co-Kultur mit Knochenmark-Stromazellen. Ixazomib zeigte eine antiangiogene Wirkung in einem in-vitro-Kapillarenausbildungstest. Ixazomib förderte in vitro die Osteoblastogenese und Osteoblastenaktivität und hemmte die Osteoklastogenese und Osteoklastenresorption. Zudem beugte Ixazomib dem Knochenverlust in einem in-vivo-Mausmodell des multiplen Myeloms vor.
Pharmakodynamik
Kardiale Elektrophysiologie
Bei klinisch relevanten Expositionen verlängert NINLARO, basierend auf den Ergebnissen einer pharmakokinetisch-pharmakodynamischen Analyse der Daten von 245 Patienten, das QTc-Intervall nicht. Bei der 4-mg-Dosis wird die mittlere QTcF-Veränderung gegenüber der Baseline auf Grundlage der modellbasierten Analyse auf 0,07 mSek geschätzt (90% Kl; -0,22, 0,36).
Es gab keinen erkennbaren Zusammenhang zwischen der Ixazomib-Konzentration und dem RR-Intervall, was auf keine klinisch bedeutsame Wirkung von NINLARO auf die Herzfrequenz hindeutet.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von NINLARO in Kombination mit Lenalidomid und Dexamethason wurde in einer internationalen randomisierten, doppelblinden, Placebo-kontrollierten, multizentrischen Überlegenheitsstudie der Phase 3 bei Patienten mit rezidivierendem und/oder refraktärem multiplem Myelom untersucht, die mindestens eine vorangehende Therapie erhalten hatten.
Insgesamt wurden 722 Patienten (Intent-to-treat [ITT]-Population) in einem 1:1-Verhältnis randomisiert und erhielten entweder die Kombination aus NINLARO, Lenalidomid und Dexamethason (N=360; NINLARO-Regime) oder Placebo, Lenalidomid und Dexamethason (N=362; Placebo-Regime) bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Toxizität. Die Randomisierung wurde entsprechend der Anzahl vorangegangener Therapielinien (1 gegenüber 2 oder 3), dem Internationalen Myelom-Staging-System (ISS) (Stadium I oder II gegenüber III) und vorangegangenen Therapien mit einem Proteasom-Inhibitor (exponiert oder naiv) stratifiziert.
Die in die Studie eingeschlossenen Patienten hatten ein multiples Myelom, das über Paraprotein im Serum und im Urin oder Bestimmungen der freien Leichtketten messbar war und deren Erkrankung refraktär einschliesslich primär refraktär (d. h., keinerlei Ansprechen auf vorhergehende Therapien) war, bei denen es nach einer vorangegangenen Behandlung zu einem Rezidiv kam oder bei denen es zu einem Rezidiv kam und die Erkrankung refraktär auf alle vorangegangenen Therapien war.
Auch Patienten, die die Therapien vor der Krankheitsprogression wechselten, wurden eingeschlossen, ebenso Patienten mit kontrollierten kardiovaskulären Erkrankungen. Von der Phase-3-Studie ausgeschlossen waren Patienten, die refraktär auf Lenalidomid oder Proteasom-Inhibitoren waren oder mehr als drei vorangegangene Therapien erhalten haben. Da zu diesen Patienten nur begrenzte Daten vorliegen, wird vor Beginn der Behandlung mit NINLARO eine sorgfältige Nutzen-Risiko-Abwägung empfohlen.
Entsprechend der Arzneimittelinformation von Lenalidomid wurde für alle Patienten in beiden Behandlungsgruppen eine Thromboseprophylaxe empfohlen. Begleitmedikationen wie Antiemetika, antivirale Arzneimittel und Antihistaminika wurden den Patienten nach Ermessen des Arztes als Prophylaxe und/oder zur Behandlung von Symptomen verabreicht.
Die Patienten erhielten NINLARO 4 mg oder Placebo an den Tagen 1, 8 und 15 plus Lenalidomid (25 mg) an den Tagen 1 bis 21 und Dexamethason (40 mg) an den Tagen 1, 8, 15 und 22 eines 28-tägigen Zyklus. Patienten mit Nierenfunktionsstörung erhielten eine Anfangsdosis Lenalidomid gemäss der entsprechenden Arzneimittelinformation. Die Behandlung wurde bis zum Fortschreiten der Krankheit oder bis zum Auftreten inakzeptabler Toxizität fortgesetzt.
Die demographischen Ausgangsdaten und die Krankheitscharakteristika wurden gleich gewichtet und waren zwischen den Studiengruppen vergleichbar. Das mediane Alter lag bei 66 Jahren (Bereich: 38-91 Jahre); 58% der Patienten waren älter als 65 Jahre. 57% der Patienten waren männlich. 85% der Studienpopulation waren kaukasisch, 9% asiatisch und 2% schwarz. 93% der Patienten hatten einen ECOG-Performance-Status von 0-1 und 12% befanden sich zu Studienbeginn im ISS-Stadium III (N=90). 25% der Patienten hatten eine Kreatinin-Clearance <60 ml/min. Bei 23% der Patienten lag ein Leichtkettenmyelom vor und bei 12% der Patienten war die Krankheit nur mit einem freien Leichtkettentest nachweisbar. 43% der Gesamtpopulation (N=309) waren Patienten mit erhöhtem zytogenetischem Risiko (Hochrisiko [del(17), t(4;14), t(14;16)] oder 1q-Amplifikation [1q21]). 19% (N=137) wiesen hochrisikobehaftete zytogenetische Anomalitäten und 10% (N=69) die Deletion del(17) auf. Die Patienten erhielten ein bis drei vorangegangene Therapielinien (median: 1) einschliesslich einer früheren Behandlung mit Bortezomib (69%), Carfilzomib (<1%), Thalidomid (45%), Lenalidomid (12%) und Melphalan (81%). Bei 57% der Patienten war zuvor eine Stammzelltransplantation durchgeführt worden. Bei 77% der Patienten kam es nach vorangegangenen Therapielinien zu einem Rezidiv und 11% der Patienten waren refraktär auf vorangegangene Therapielinien. Primäre Refraktärität (d.h. das beste Ansprechen bei allen vorherigen Therapielinien war eine stabile Erkrankung oder Krankheitsprogression) wurde bei 6% der Patienten dokumentiert.
Primärer Endpunkt war das progressionsfreie Überleben (PFS) gemäss des Konsens der International Myeloma Working Group 2011 (IMWG) über einheitliche Responsekriterien nach Prüfung durch ein verblindetes unabhängiges Expertenkomitee (IRC) basierend auf zentralen Laborergebnissen. Das Ansprechen wurde alle 4 Wochen bis zur Krankheitsprogression überprüft.
Bei der primären und abschliessenden Analyse für die statistische Prüfung (mediane Nachbeobachtungszeit von 14,7 Monaten und mediane Zyklusanzahl 13) wurden für das NINLARO-Behandlungsregime signifikant bessere Ergebnisse mit einem Anstieg des medianen PFS um etwa 6 Monate belegt. Im NINLARO-Regime traten 129 (36%) und im Placebo-Regime 157 (43%) PFS-Ereignisse auf. Das mediane PFS betrug im NINLARO-Regime 20,6 Monate und im Placebo-Regime 14,7 Monate (HR=0,74 [95%-KI (0,587, 0,939)], p-Wert=0,012).
Hinweis: Die Hazard Ratio basiert auf dem geschichteten proportionalen Hazard-Regressionsmodell nach Cox; eine Hazard Ratio von weniger als 1 deutet auf eine Überlegenheit der NINLARO-Therapie hin. Der p-Wert basiert auf dem geschichtetem Log-Rank-Test.
Die Verbesserung des PFS im NINLARO-Regime wurde durch Verbesserungen der Gesamtansprechrate gestützt.
Eine geplante Interimsanalyse des Gesamtüberlebens (OS) wurde nach einer medianen Nachbeobachtungszeit von 23 Monaten mit 35% der für die finale OS-Analyse erforderlichen Anzahl Todesfälle in der ITT-Population durchgeführt. Im NINLARO-Arm waren 81 Todesfälle, im Placebo-Arm 90 Todesfälle aufgetreten. Das mediane OS wurde in keinem der beiden Behandlungsarme erreicht. Gleichzeitig wurde eine nichtinferentielle exploratorische PFS-Analyse durchgeführt. Das geschätzte mediane PFS in der ITT-Population betrug im NINLARO-Arm 20 Monate und im Placebo-Arm 15,9 Monate (HR=0,82 [95%-KI (0,67, 1,0)]).
Bei der finalen OS-Analyse nach einer medianen Nachbeobachtungszeit von ca. 85 Monaten betrug das mediane OS in der ITT-Population 53,6 Monate für Patienten im NINLARO-Regime und 51,6 Monate für Patienten im Placebo-Regime (HR=0,94 [95%-KI (0,78, 1,13)]).
Bei der OS-Interimsanalyse kam es in der Untergruppe der Hochrisikopatienten, deren multiples Myelom zentral in einem CLIA-zertifizierten Labor auf eine Deletion des Chromosoms 17 (del[17]) untersucht wurde, zu weniger Todesfällen bei Patienten mit del(17) im NINLARO-Arm (11,1%) als bei Patienten im Placebo-Arm (27,3%). Bei der finalen OS-Analyse betrug das mediane OS in dieser Subpopulation 42,2 Monate für Patienten im NINLARO-Regime und 29,4 Monate für Patienten im Placebo-Regime (HR=0,92 [95%-KI (0,52, 1,63)]).
Bei Patienten mit einem hohen zytogenetischen Risiko betrug das mediane OS in der finalen Analyse 46,9 Monate im NINLARO-Regime und 30,9 Monate im Placebo-Regime (HR=0,87 [95%-KI (0,58, 1,31)]) und bei Patienten mit erhöhtem zytogenetischem Risiko betrug das mediane OS 44,6 Monate im NINLARO-Regime und 33,4 Monate im Placebo-Regime (HR=0,86 [95%-KI (0,66, 1,12)]).
Da es sich beim multiplen Myelom um eine heterogene Erkrankung handelt, kann der Nutzen in verschiedenen Untergruppen variieren (siehe Abbildung 1).
Abbildung 1: Forest-Plot des progressionsfreien Überlebens in Untergruppen
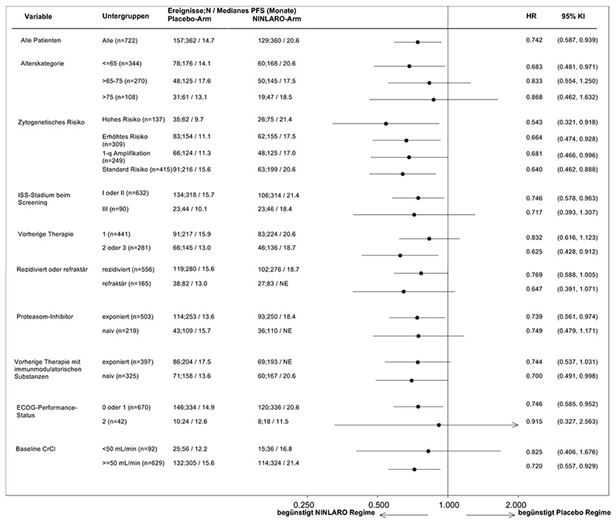
Indizierte Population
Es wurde eine Analyse zur Ermittlung der klinischen Wirksamkeitsergebnisse anhand festgelegter zytogenetischer und stratifizierter klinisch-ungünstiger Risikomerkmale durchgeführt. In der ITT-Population wurde eine stärkere Verbesserung der Wirksamkeitsergebnisse bei 70% der Patienten (N=505, «indizierte Population») beobachtet, die mindestens eine vorangegangene Therapie erhalten haben und Hochrisikomerkmale aufwiesen (definiert als erhöhtes zytogenetisches Risiko [Hochrisiko (del[17], t[4;14], t[14;16]) oder 1q21] oder ISS-Stadium III) oder die mindestens zwei vorangegangene Therapien erhalten haben. Bei der Primäranalyse traten im NINLARO-Arm 98 (39%) PFS-Ereignisse und im Placebo-Arm 122 (49%) PFS-Ereignisse auf. Das mediane PFS betrug im NINLARO-Arm 18,4 Monate und im Placebo-Arm 12,2 Monate (HR=0,681 [95%-KI (0,520, 0,892)], p-Wert=0,005).
Die Verbesserung des PFS im NINLARO-Arm wurde durch Verbesserungen der Gesamtansprechrate in der indizierten Population gestützt.
Zum Zeitpunkt der geplanten OS-Interimsanalyse waren in der indizierten Population im NINLARO-Arm 58 Todesfälle und im Placebo-Arm 76 Todesfälle aufgetreten. Das mediane OS wurde im NINLARO-Arm nicht erreicht und betrug im Placebo-Arm 30,9 Monate (HR=0,706 [95%-KI (0,499, 0,997)]). Die Ergebnisse der nichtinferentiellen exploratorischen PFS-Analyse zeigten ein geschätztes medianes PFS in der indizierten Population von 19,1 Monaten im NINLARO-Arm und von 12,6 Monaten im Placebo-Arm (HR=0,728 [95%-KI (0,573, 0,924)]). Ergebnisse stimmen mit der Schlussfolgerung der primären PFS-Analyse überein, dass ein positiver Behandlungseffekt besteht.
Zum Zeitpunkt der abschliessenden OS-Analyse gab es in der indizierten Population 181 Todesfälle im NINLARO-Regime und 179 Todesfälle im Placebo-Regime. Das mediane OS betrug 50,4 Monate im NINLARO-Regime und 42,0 Monate im Placebo-Regime (HR=0,912 [95%-KI (0,74, 1,125)]).
Pädiatrie
Die Europäische Arzneimittelagentur hat die Verpflichtung, die Ergebnisse der Studien zu NINLARO bei allen Untergruppen der pädiatrischen Population vorzulegen, aufgehoben (siehe Rubrik «Dosierung/Anwendung, Kinder und Jugendliche»).
|