ZusammensetzungWirkstoffe
INN = Lonoctocog alfa.
Rekombinanter Blutgerinnungsfaktor VIII, einkettig (rVIII-SingleChain), der in Ovarienzellen des chinesischen Hamsters (CHO) hergestellt wird.
Hilfsstoffe
Pulver: L-Histidin, Polysorbat 80, Calciumchlorid-Dihydrat, Natriumchlorid (entspricht 0,7 mmol bzw. 16,1 mg Natrium pro 500 I.E. und 1000 I.E. Durchstechflasche und 1,4 mmol bzw. 32,3 mg pro 2000 I.E. Durchstechflasche), Saccharose.
Lösungsmittel: Wasser für Injektionszwecke.
Indikationen/AnwendungsmöglichkeitenBehandlung und Prophylaxe von Blutungen bei vorbehandelten Patienten mit Hämophilie A (angeborener Faktor-VIII-Mangel).
AFSTYLA ist nicht zur Behandlung von von-Willebrand-Krankheit (vWD) indiziert.
Dosierung/AnwendungDosierung für Erwachsene und Kinder
Die Behandlung mit AFSTYLA ist unter Aufsicht eines in der Behandlung von Hämophilie erfahrenen Arztes einzuleiten.
Die Entscheidung hinsichtlich einer häuslichen Anwendung des Produkts zur Behandlung von Blutungen beziehungsweise zur Prophylaxe von Blutungen bei Patienten mit Hämophilie A muss individuell für jeden Patienten durch den behandelnden Arzt getroffen werden. Der behandelnde Arzt muss gewährleisten, dass eine entsprechende Unterweisung des Patienten erfolgt und die Anwendung in regelmässigen Intervallen überprüft wird.
Dosierung
Die Dosierung und die Dauer der Behandlung richten sich nach dem Schweregrad des Faktor-VIII-Mangels, dem Ort und Ausmass der Blutung und dem klinischen Zustand des Patienten.
Die Anzahl der verabreichten Einheiten des Faktors VIII wird in Internationalen Einheiten (I.E.) angegeben, die sich auf den derzeitigen WHO-Standard für Faktor-VIII-Produkte beziehen. Die Faktor-VIII-Aktivität in Plasma wird entweder als Prozentsatz (bezogen auf das normale Humanplasma) oder in Internationalen Einheiten (bezogen auf den Internationalen Standard für Faktor VIII im Plasma) angegeben.
Auf jedem Flaschenetikett von AFSTYLA wird die Faktor-VIII-Aktivität in Internationalen Einheiten (I.E.) angegeben. Eine I.E. entspricht der Aktivität des Faktors VIII, der in einem Milliliter normalen Humanplasma enthalten ist.
Die Bestimmung der ausgewiesenen Aktivität erfolgt unter Verwendung eines chromogenen Substrat-Tests.
Die Überwachung der Faktor-VIII-Plasmaspiegel kann entweder mit einem chromogenen Substrat-Test oder einem One-Stage-Gerinnungstest erfolgen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Bedarfsbehandlung
Die Berechnung der erforderlichen Faktor-VIII-Dosis basiert auf dem empirischen Befund, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Faktor-VIII-Aktivität im Plasma um 2 I.E./dl erhöht. Der erwartete In-vivo-Maximalanstieg des Faktor-VIII-Spiegels, ausgedrückt als I.E./dl (oder % des Normalwerts) wird über die folgende Formel geschätzt:
Geschätzter Anstieg von Faktor VIII (I.E./dl oder % des Normalwerts) = [Gesamtdosis (I.E.)/Körpergewicht (kg)] x 2 (I.E./dl pro I.E./kg)
Die erforderliche Dosis zum Erreichen des gewünschten In-vivo-Maximalanstiegs des Faktor-VIII-Spiegels kann nach folgender Formel berechnet werden:
Dosis (I.E.) = Körpergewicht (kg) x gewünschter Faktor-VIII-Anstieg (I.E./dl oder % des Normalwerts) x 0,5 (I.E./kg pro I.E./dl)
Die Dosierung und die Häufigkeit der Anwendung sollten sich stets an der klinischen Wirksamkeit im Einzelfall orientieren.
Die folgende Tabelle dient als Empfehlung für die Dosierung von AFSTYLA zur Kontrolle und Vermeidung von Blutungsereignissen. Der Faktor-VIII-Spiegel sollte im Zielbereich oder oberhalb des Zielbereichs erhalten werden.
|
Grad der Blutung / Art des chirurgischen Eingriffs
|
Erforderlicher Faktor-VIII-Spiegel (% oder I.E./dl)
|
Häufigkeit der Dosierung (Stunden) / Behandlungsdauer (Tage)
| |
Blutung
| |
Beginnende Hämarthrose, Muskelblutungen oder Blutungen in der Mundhöhle
|
20 bis 40
|
Wiederholung der Injektion alle 12 bis 24 Stunden, bis die Blutung gestillt ist.
| |
Ausgedehntere Hämarthrosen, Muskelblutungen oder Hämatome
|
30 bis 60
|
Wiederholung der Injektion alle 12 bis 24 Stunden, bis die Blutung gestillt ist.
| |
Lebensbedrohliche Blutungen
|
60 bis 100
|
Wiederholung der Injektion alle 8 bis 24 Stunden, bis die Blutung gestillt ist.
| |
Chirurgische Eingriffe
| |
Kleinere Eingriffe
einschliesslich Zahnextraktion
|
30 bis 60
|
Wiederholung der Injektion alle 24 Stunden für mind. 1 Tag, bis die Wundheilung erzielt ist.
| |
Grössere Eingriffe
|
80 bis 100
(prä- und
postoperativ)
|
Wiederholung der Injektion alle 8 bis 24 Stunden bis zu einer angemessenen Wundheilung, dann Fortsetzung der Therapie um mindestens weitere 7 Tage zur Erhaltung einer Faktor-VIII-Aktivität von 30 bis 60 % (I.E./dl).
|
Prophylaxe
Die empfohlene Anfangsdosierung beträgt 20 bis 50 I.E./kg AFSTYLA 2- bis 3-mal wöchentlich.
Die Dosierung soll entsprechend dem Ansprechen des Patienten angepasst werden.
Zuvor unbehandelte Patienten (PUPs)
Insgesamt entsprach das Sicherheits- und Wirksamkeitsprofil von AFSTYLA bei PUPs dem Nutzen-Risiko-Profil, das bei den zuvor behandelten Patienten (previously treated patients, PTPs) berichtet wurde, mit Ausnahme des Auftretens von neutralisierenden Antikörpern, die ein anerkanntes Risiko einer FVIII-Substitutionstherapie in dieser Patientenpopulation darstellen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Ältere Patienten
Bei klinischen Studien von AFSTYLA waren keine Probanden im Alter von über 65 Jahren eingeschlossen.
Kinder und Jugendliche
Die empfohlene Anfangsdosierung bei Kindern (im Alter von 0 bis zu 12 Jahren) beträgt 30 bis 50 I.E./kg AFSTYLA 2- bis 3-mal wöchentlich. Eine häufigere oder höhere Dosierung kann für Kinder unter 12 Jahren erforderlich sein, da eine höhere Clearance in dieser Altersgruppe zu berücksichtigen ist.
Für Jugendliche im Alter von über 12 Jahren ist die empfohlene Dosierung dieselbe wie für Erwachsene.
Derzeit verfügbare Daten sind in Abschnitt «Pharmakokinetik» beschrieben.
Überwachung auf Inhibitoren
Patienten sollten auf die Entwicklung von Faktor-VIII-Inhibitoren hin überwacht werden, siehe auch Abschnitt «Warnhinweise und Vorsichtsmassnahmen».
Art der Anwendung
Infusionsgeschwindigkeit
Zum intravenösen Gebrauch.
Für Anweisungen zur Rekonstitution des Arzneimittels vor der Verabreichung siehe «Hinweise für die Handhabung» im Abschnitt «Sonstige Hinweise». Das rekonstituierte Präparat sollte langsam und mit einer für den Patienten angenehmen Geschwindigkeit injiziert werden.
Der Patient sollte auf unmittelbare Reaktionen hin beobachtet werden. Im Fall des Auftretens einer Reaktion, die mit der Verabreichung von AFSTYLA in Verbindung stehen könnte, ist abhängig von dem klinischen Zustand des Patienten die Injektionsgeschwindigkeit zu verringern oder die Anwendung zu beenden (siehe auch «Warnhinweise und Vorsichtsmassnahmen»).
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
KontraindikationenAFSTYLA ist bei Patienten kontraindiziert, die lebensbedrohliche Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, auf den Wirkstoff, auf einen der Hilfsstoffe gemäss Zusammensetzung oder auf Hamsterproteine erfahren haben.
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeit
Allergische Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, sind bei AFSTYLA möglich. Patienten sollten über die frühen Anzeichen von Überempfindlichkeitsreaktionen, wie z.B. quaddelartiger Hautausschlag, generalisierte Nesselsucht, Engegefühl in der Brust, Stridor, Hypotonie und Pruritus, aufgeklärt werden. Die Verabreichung ist sofort abzubrechen und eine angemessene Behandlung ist einzuleiten, wenn Überempfindlichkeitsreaktionen auftreten.
Den Patienten ist anzuraten, die Anwendung von AFSTYLA zu unterbrechen und ihren Arzt zu kontaktieren.
Bei Patienten mit früheren Überempfindlichkeitsreaktionen kann eine Vorbehandlung mit Antihistaminika in Betracht gezogen werden.
Neutralisierende Antikörper
Die Entstehung von neutralisierenden Antikörpern (Inhibitoren) gegen Faktor VIII wurde nach Verabreichung von Faktor-VIII-Produkten, einschliesslich AFSTYLA, festgestellt.
Patienten sollten durch geeignete klinische und im Labor durchgeführte Untersuchungen auf die Entwicklung von neutralisierenden Antikörpern (Inhibitoren) beobachtet werden. Falls nach Verabreichung von AFSTYLA der erwartete Faktor-VIII-Plasmawirkspiegel nicht erreicht oder die Blutung nicht gestillt werden kann, sollte das Vorhandensein eines Inhibitors (neutralisierenden Antikörpers) vermutet werden.
Falls ein Patient einen Inhibitor entwickelt, sollte ein Hämophilie-Zentrum kontaktiert werden.
Ein Bethesda-Inhibitortest ist durchzuführen, wenn die erwarteten Faktor-VIII-Plasmaspiegel nicht erreicht werden oder die Blutung mit der erwarteten Dosis von AFSTYLA nicht gestillt werden kann. Zur Meldung von Inhibitorspiegeln sind Bethesda-Einheiten (B.E.) zu verwenden.
Labortests zur Überwachung
Die Plasmaaktivität des Faktors-VIII sollte unter der Verwendung des chromogenen Substrat- oder des One-Stage-Gerinnungs-Assays bei AFSTYLA-behandelten Patienten überwacht werden.
Wirksamkeitsergebnisse einer breit angelegten, klinischen Pivotalstudie bestätigten, dass die Ergebnisse des chromogenen Substrat-Tests das klinische hämostatische Potential am genauesten wiedergeben. Sofern verfügbar, sollte daher der chromogene Substrat-Test zur Bestimmung der Faktor-VIII-Aktivität in Patientenproben verwendet werden. Wird die One-Stage-Methode zur Bestimmung der Faktor-VIII-Aktivität in Patientenproben verwendet, ist zu beachten, dass die Ergebnisse des One-Stage-Tests etwa 45 % unter denen des chromogenen Substrat-Tests liegen und durch Multiplikation mit Faktor 2 entsprechend angeglichen werden können (s. oben).
Katheter-assoziierte Komplikationen
Wenn ein zentralvenöser Katheter erforderlich ist, sollte das Risiko von Katheter-assoziierten Komplikationen einschliesslich lokaler Infektionen, Bakteriämie und Katheter-assoziierten Thrombosen beachtet werden.
Kinder und Jugendliche
Die aufgeführten Warnhinweise und Vorsichtsmassnahmen gelten für Erwachsene und Kinder gleichermassen.
AFSTYLA 500 I.E. und 1000 I.E. enthalten weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, das heisst, es ist nahezu «natriumfrei».
AFSTYLA 2000 I.E. enthält ≤1,4 mmol (32,3 mg) Natrium pro Durchstechflasche, entsprechend 1,6 % der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenEs sind keine Wechselwirkungen von AFSTYLA mit anderen Arzneimitteln gemeldet worden.
Schwangerschaft, StillzeitSchwangerschaft
Es wurden keine Studien zur Entwicklungs- und Reproduktionstoxizität am Tier mit AFSTYLA durchgeführt. Es ist nicht bekannt, ob AFSTYLA bei Verabreichung an schwangere Frauen zur Schädigung des Fötus führen oder die Reproduktion beeinträchtigen kann. Daher sollte AFSTYLA während der Schwangerschaft nur bei eindeutiger Indikationsstellung angewendet werden.
Stillzeit
Es ist nicht bekannt, ob AFSTYLA in die menschliche Muttermilch übergeht. Da viele Arzneistoffe in die menschliche Muttermilch übergehen, sollte AFSTYLA nicht bei stillenden Müttern angewendet werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenAFSTYLA hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Überempfindlichkeit oder allergische Reaktionen, die Angioödem, Brennen und Stechen an der Injektionsstelle, Schüttelfrost, Hitzegefühl, generalisierte Nesselsucht, Kopfschmerz, quaddelartiger Hautausschlag, Hypotonie, Lethargie, Übelkeit, Unruhe, Tachykardie, Engegefühl in der Brust, Kribbelgefühl, Erbrechen oder Stridor einschliessen können, wurden bei Anwendung von Faktor-VIII-Produkten selten beobachtet. In einigen Fällen entwickelten sich diese Reaktionen zu schwerer Anaphylaxie (einschliesslich Schock) (siehe auch «Warnhinweise und Vorsichtsmassnahmen»). Überempfindlichkeitsreaktionen wurden in klinischen Studien von AFSTYLA beobachtet (siehe ADR-Tabelle unten), jedoch wurden keine anaphylaktischen Reaktionen gemeldet.
Patienten mit Hämophilie A können neutralisierende Antikörper (Inhibitoren) gegen Faktor VIII entwickeln. Wenn solche Inhibitoren auftreten, manifestiert sich der Zustand als unzureichende klinische Antwort. Für diese Fälle wird empfohlenen, ein spezialisiertes Hämophilie-Zentrum zu kontaktieren. In klinischen Studien mit AFSTYLA wurden keine solchen Reaktionen festgestellt. Die Bildung von neutralisierenden Antikörpern (Inhibitoren) gegen Faktor VIII wurde in der klinischen Studie an zuvor unbehandelten Patienten mit AFSTYLA beobachtet (siehe nachstehende Auflistung der unerwünschten Arzneimittelwirkungen).
Auflistung der unerwünschten Arzneimittelwirkungen
Die unten aufgeführte Liste entspricht der MedDRA-Systemorganklassifizierung (SOC und bevorzugte Begriffe).
Die Häufigkeiten der unerwünschten Wirkungen wurde pro Patient, basierend auf Daten aus abgeschlossenen klinischen Studien berechnet und nach folgender Konvention kategorisiert: häufig (≥1/100 bis <1/10), gelegentlich (≥1/1000 bis <1/100), selten (≥1/10'000 bis <1/1000) und sehr selten (<1/10'000), nicht bekannt (auf Grundlage der verfügbaren Daten nicht abschätzbar).
Erkrankungen des Blutes und des Lymphsystems:
Gelegentlich (bei zuvor behandelten Patienten, PTPs), Häufigkeit der Faktor-VIII-Inhibition basiert auf der Klassenhäufigkeit für FVIII-Produkte), sehr häufig (bei zuvor unbehandelten Patienten (previously untreated patients, PUPs): Faktor-VIII-Inhibition.
Erkrankungen des Immunsystems:
Häufig: Überempfindlichkeit.
Erkrankungen des Nervensystems:
Häufig: Schwindelgefühl, Parästhesie.
Erkrankungen der Haut und des Unterhautzellgewebes:
Häufig: Ausschlag.
Gelegentlich: Erythem, Pruritus.
Allgemeine Störungen und Beschwerden am Verabreichungsort:
Häufig: Fieber.
Gelegentlich: Schmerzen an der Injektionsstelle, Schüttelfrost, Wärmegefühl.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung mit AFSTYLA berichtet.
Von einem Patienten wurde berichtet, dass er mehr als das Zweifache der verschriebenen Dosis erhalten hatte. Es wurden keine mit dieser Überdosierung in Zusammenhang stehenden Nebenwirkungen berichtet.
Eigenschaften/WirkungenATC-Code
B02BD02
Pharmakotherapeutische Gruppe: Antihämorrhagika: Blutgerinnungsfaktor VIII.
Wirkungsmechanismus
AFSTYLA (INN: Lonoctocog alfa) enthält ein rekombinantes Protein, das den fehlenden Faktor VIII, der zu einer wirkungsvollen Blutstillung benötigt wird, ersetzt. Lonoctocog alfa ist ein rekombinanter Faktor VIII, der einkettig vorliegt und bei dem ein Grossteil der bei dem Faktor-VIII-Wildtyp voller Länge vorkommenden B-Domäne entfernt wurde. Nach Aktivierung enthält das Lonoctocog-alfa-Molekül eine Aminosäuresequenz, die identisch mit der des Faktors VIIIa ist, der aus dem endogenen Faktor VIII voller Länge gebildet wird. Ausserdem führt der einzelkettige Aufbau zu einer hohen Bindungsaffinität von Lonoctocog alfa zum von-Willebrand-Faktor.
Pharmakodynamik
Hämophilie A ist eine x-chromosomal vererbbare Erkrankung der Blutgerinnung, verursacht durch erniedrigte Faktor-VIII-Spiegel und führt zu starken Blutungen in die Gelenke, Muskeln oder inneren Organe, entweder spontan oder als Folge von unfallbedingten oder chirurgischen Verletzungen. Durch die Substitutionstherapie wird der Plasmaspiegel von Faktor VIII erhöht und somit eine zeitlich begrenzte Korrektur des Faktor-VIII-Mangels und eine Korrektur der Blutungsneigung erreicht.
Klinische Wirksamkeit
Offene Studien an zuvor behandelten Patienten (previously treated patients, PTPs): Studie 1001 an Jugendlichen und Erwachsenen, Studie 3002 an einer pädiatrischen Population
Die Pharmakokinetik, Sicherheit und klinische Wirksamkeit von AFSTYLA wurde in einer Phase I/III-Studie mit Erwachsenen/Jugendlichen und einer Phase III Studie mit Kindern untersucht. In den Studien wurde die Pharmakokinetik von AFSTYLA charakterisiert und die blutstillende Wirksamkeit in der Kontrolle von Blutungen und in der Routineprophylaxe zur Vermeidung von Blutungen bestimmt. In der Phase I/III Studie mit Erwachsenen/Jugendlichen wurde auch die blutstillende Wirksamkeit in der perioperativen Prophylaxe bei Patienten untersucht, bei denen chirurgische Eingriffe vorgenommen wurden.
An der Phase I/III Studie mit Erwachsenen/Jugendlichen nahmen 175 männliche Patienten mit schwerer Hämophilie A (<1 % endogene FVIII Aktivität) teil, die zuvor schon eine Behandlung erfahren hatten. Die Patienten waren zwischen 12 und 65 Jahre alt; darunter befanden sich 14 Jugendliche im Alter von mindestens 12 bis unter 18 Jahren. Von den 175 Studienteilnehmern erhielten 174 mindestens 1 Dosis AFSTYLA und 173 (99 %) waren für die Wirksamkeitsstudie auswertbar. 161 Patienten (92,5 %) schlossen die Studie ab. 120 (69,0 %) der Studienteilnehmer wurden mindestens 50 Behandlungstagen unterworfen, 52 (29,9 %) dieser Patienten erhielten mindestens 100 Behandlungstage. Die Patienten erhielten insgesamt 14'592 Injektionen, der Median lag bei 67 Injektionen, entsprechend einer Spannbreite von 1 bis 395 Injektionen pro Patient.
An der Phase-III-Studie mit Kindern nahmen 84 männliche Patienten mit schwerer Hämophilie A teil, 35 der Patienten waren im Alter zwischen 0 und weniger als 6 Jahren, und 49 zwischen 6 und 12 Jahren. Von den 84 teilnehmenden Patienten erhielten alle mindestens eine Dosis AFSTYLA und 83 (99 %) waren für die Wirksamkeitsstudie auswertbar. 65 (77,4 %) der Studienteilnehmer wurden mindestens 50 Behandlungstagen unterworfen, 8 (9,5 %) dieser Patienten erhielten mindestens 100 Behandlungstage. Die Patienten erhielten insgesamt 5'313 Injektionen, der Median lag bei 59 Injektionen, entsprechend einer Spannbreite von 4 bis 145 Injektionen pro Patient.
Kontrolle und Vermeidung von Blutungen
In der Studie mit Erwachsenen/Jugendlichen wurden 848 Blutungsepisoden, mehrheitlich Gelenksblutungen, mit AFSTYLA behandelt und 835 wurden bezüglich der Wirksamkeit durch den klinischen Prüfer ausgewertet.
Der Median der Injektionsdosen für die Behandlung der Blutungsepisoden lag bei 31,7 I.E./kg. 686 (81 %) dieser gesamthaft 848 Blutungsepisoden wurden mit einer einzigen Dosis AFSTYLA unter Kontrolle gebracht, weitere 107 (13 %) mit 2 Injektionsdosen. 55 (6 %) der 848 Blutungsepisoden benötigten 3 oder mehr Injektionen.
Die hämostatische Wirksamkeit wurde durch den klinischen Prüfer für 94 % der Blutungsepisoden als ausgezeichnet oder gut eingestuft.
In der pädiatrischen Studie wurden gesamthaft 347 Blutungsepisoden, mehrheitlich Gelenksblutungen, mit AFSTYLA behandelt und bezüglich der Wirksamkeit durch den klinischen Prüfer ausgewertet.
Der Median der Injektionsdosen für die Behandlung der Blutungsepisoden lag bei 27,3 I.E./kg. 298 (86 %) dieser gesamthaft 347 Blutungsepisoden wurden mit einer einzigen Dosis AFSTYLA unter Kontrolle gebracht, weitere 34 (10 %) mit 2 Injektionsdosen. 15 (4 %) der 347 Blutungsepisoden benötigten 3 oder mehr Injektionen.
Die hämostatische Wirksamkeit wurde durch den klinischen Prüfer für 96 % der Blutungsepisoden als ausgezeichnet oder gut eingestuft.
Routineprophylaxe
In der Studie mit Erwachsenen/Jugendlichen und der pädiatrischen Studie wurde das Dosierungsschema für die Routineprophylaxe von dem klinischen Prüfer bestimmt. Dies geschah unter Berücksichtigung des FVIII-Behandlungsschemas, dem der jeweilige Patient vor Studienbeginn folgte, und unter Berücksichtigung seines Blutungsphänotyps.
In der Studie mit Erwachsenen/Jugendlichen erhielten 54 % der insgesamt 146 Prophylaxe-Patienten 3-mal wöchentlich AFSTYLA. 32 % erhielten AFSTYLA 2-mal wöchentlich. 6 % der Prophylaxe-Patienten erhielten alle 2 Tage AFSTYLA und 8 % der Prophylaxe-Patienten folgten einem anderen Dosierungsschema.
Unter Routineprophylaxe traten bei 63 (43 %) der 146 Patienten keine Blutungsepisoden auf.
Der Median der jährlichen Blutungsrate (ABR) unter Routineprophylaxe lag bei 1,14 (Interquartilbereich 0,0 bis 4,2) und war somit statistisch signifikant niedriger (p <0,0001) als bei Patienten unter Bedarfsbehandlung (Median der ABR 19,64, Interquartilbereich 6,2 bis 46,5). Die ABR von Patienten, die 3-mal wöchentlich behandelt wurden (Median der ABR 1,53, Interquartilbereich 0,0 bis 4.,4) war vergleichbar mit der ABR der Patienten, die 2-mal wöchentlich behandelt wurden (Median der ABR 0,00, Interquartilbereich 0,00 – 3,3). Die jährliche spontane Blutungsrate (AsBR) war bei diesen Patienten identisch, mit einem Median der AsBR von 0,00 sowohl bei 3-mal wöchentlicher Behandlung (Interquartilbereich 0,00 bis 3,5) als auch bei 2-mal wöchentlicher Behandlung (Interquartilbereich 0,00 bis 1,1).
Der Median der verschriebenen Dosis für Patienten, die 3-mal wöchentlich behandelt wurden, lag bei 30 I.E./kg pro Injektion, und bei 35 I.E./kg für Patienten, die 2-mal wöchentlich behandelt wurden.
In der pädiatrischen Studie erhielten 54 % der insgesamt 80 Prophylaxe-Patienten 2-mal wöchentlich AFSTYLA. 30 % der Prophylaxe-Patienten erhielten AFSTYLA 3-mal wöchentlich. 4 % der Prophylaxe-Patienten erhielten alle 2 Tage AFSTYLA und 12 % der Prophylaxe-Patienten folgten einem anderen Dosierungsschema.
Unter Routineprophylaxe traten bei 21 (26 %) der 80 Patienten keine Blutungsepisoden auf.
Die ABR lag bei Prophylaxe-Patienten bei 3,69 (Interquartilbereich 0,00 bis 7,20) mit einem Median der ABR von 2,30 (Interquartilbereich 0,00 bis 11,58) für Patienten, die 3-mal wöchentlich behandelt wurden und einem Median der ABR von 4,37 (Interquartilbereich 2,31 bis 7,24) für Patienten, die 2-mal wöchentlich behandelt wurden. Die jährliche spontane Blutungsrate (AsBR) war bei diesen Patienten identisch, der Median der AsBR bei 3-mal wöchentlicher Behandlung sowie 2-mal wöchentlicher Behandlung lag bei 0,00 (Interquartilbereich 0,00 bis 3,03 und 0,00 bis 2,08).
Der Median der verschriebenen Dosis für Patienten, die 3-mal wöchentlich behandelt wurden, lag bei 32 I.E./kg pro Injektion, und bei 35 I.E./kg für Patienten, die 2-mal wöchentlich behandelt wurden.
Perioperativen Prophylaxe (chirurgische Prophylaxe)
13 der Patienten, die an der Studie mit Erwachsenen/Jugendlichen teilnahmen, wurden insgesamt 16 chirurgischen Eingriffen unterzogen. Bei diesen Eingriffen handelte es sich um eine Extraktion von Weisheitszähnen, Bauchwandhernien-Operation, ein Ersatz des Ellenbogengelenks, fünfmal Ersetzen des Kniegelenks, eine Entfernung der Gallenblase und Dehnung der Achillessehne kombiniert mit Richten, drei Zirkumzisionen und ein Osteosynthese-Verfahren am rechten Fussgelenk mit offener Reposition und interner Fixation und anschliessender Entfernung dieser Fixation. Insgesamt wurde die hämostatische Wirksamkeit von AFSTYLA in 15 von 16 Eingriffen von den klinischen Prüfern als ausgezeichnet und in 1 von 16 Eingriffen (ein Ersatz des Kniegelenks) als gut eingestuft. Der Median des FVIII-Verbrauchs am Operations-Tag lag bei 89,4 I.E./kg (Spannbreite 40,5 – 108,6 I.E./kg).
Kinder, Jugendliche und Erwachsene (Verlängerungsstudie 3001)
In die Studie 3001 wurden 222 PTPs (67 Teilnehmende <12 Jahre) aufgenommen, darunter 204 PTPs aus den Lead-in-Studien 1001 und 3002. Die mittlere (SD) Anzahl Expositionstage (Eds) bei den PTPs in dieser Studie betrug 341,9 (135,48). Insgesamt erreichten 212 Teilnehmende (95,5 %) >100 Eds. Kein Patient entwickelte einen Inhibitor oder erlebte eine anaphylaktische Reaktion. Es wurden keine neuen Sicherheitsbedenken festgestellt in dieser Verlängerungsstudie. Die Ergebnisse zur individualisierten Prophylaxe und zur Behandlung von Blutungen waren vergleichbar mit denen früherer Studien.
Individualisierte Prophylaxe: Von den 211 prophylaktisch behandelten Patienten (Median der ABR 1,21 [Interquartilbereich: 0,33; 3,03]) wurden 85 (40 %) einem Zweimal-wöchentlich-Schema und 97 (46 %) einem Dreimal-wöchentlich-Schema zugewiesen. Patienten unter 2- und 3-mal wöchentlicher prophylaktischer Behandlung erhielten Dosen von im Median 35 bzw. 32 I.E./kg pro Injektion mit einem jährlichen Verbrauch von im Median 4,603 I.E./kg pro Jahr über alle Prophylaxeschemata hinweg.
Behandlung von Blutungen: Von den 2413 behandelten Blutungsereignissen, die in Studie 3001 auftraten, wurden 86,3 % mit 2 oder weniger Injektionen unter Kontrolle gebracht.
Zuvor unbehandelte Patienten (PUPs)
Pädiatrische Population <12 Jahre (Studie 3001)
In die Studie wurden insgesamt 24 PUPs mit einem Altersmedian von 1,0 Jahren (Bereich: 0 bis 5 Jahre) aufgenommen. Die Studienteilnehmenden erreichten insgesamt 5909 Eds mit rVIII-SingleChain.
Individualisierte Prophylaxe: Insgesamt 23 PUPs erhielten das prophylaktische Behandlungsschema während der Studie (11 nach Wechsel von der Bedarfsbehandlung). Die ABR betrug im Median 1,84 (Bereich: 0,0 bis 23,6), die AsBR im Median 0,88 (Bereich: 0,0 bis 19,7).
Behandlung von Blutungen: Von den 315 behandelten Blutungsereignissen, wurden 88,9 % mit 2 oder weniger Injektionen unter Kontrolle gebracht. Die zur Behandlung eines Blutungsereignisses verwendete Dosis lag im Median bei 66,2 I.E./kg.
Insgesamt stimmte das Nutzen-Risiko-Profil von AFSTYLA bei PUPs mit dem bei PTPs berichteten überein, mit Ausnahme des Auftretens von neutralisierenden Antikörpern, die ein anerkanntes Risiko einer FVIII-Substitutionstherapie bei PUPs darstellen.
Daten zur Immuntoleranzinduktion (ITI) wurden von Patienten mit Hämophilie A erhoben, die Inhibitoren gegen FVIII entwickelten. Während der klinischen Studie an PUPs erhielten 11 Patienten eine ITI-Behandlung mit AFSTYLA, und 9 Patienten erreichten eine Immuntoleranz, die durch ein negatives Ergebnis im Hemmkörper-Test bei Abschluss der Studie belegt wurde.
Zu beachten ist, dass die jährliche Blutungsrate (ABR) zwischen verschiedenen Faktorkonzentraten und verschiedenen klinischen Studien nicht vergleichbar ist.
PharmakokinetikErwachsene
Die Pharmakokinetik (PK) von AFSTYLA wurde bei 81 erwachsenen Probanden nach intravenöser Injektion einer Einzeldosis von 50 I.E./kg untersucht.
Die PK-Parameter stützen sich auf die durch den chromogenen Substrat-Test gemessene Plasmafaktor-VIII-Aktivität. Das 3 bis 6 Monate nach der initialen PK-Bewertung erhaltene PK-Profil war mit dem nach der ersten Dosis erhaltenen Profil vergleichbar.
Pharmakokinetische Parameter (Arithmetisches Mittel, CV %) nach einer Einzelinjektion von 50 I.E./kg AFSTYLA – Chromogener Substrat-Test:
|
PK-Parameter
|
rVIII-SingleChain
50 I.E./kg
(N = 81)
| |
IR (I.E./dl)/(I.E./kg)
|
2,00 (20,8)
| |
Cmax (I.E./dl)
|
106 (18,1)
| |
AUC0-inf (I.E.*h/dl)
|
1960 (33,1)
| |
t½ (h)
|
14,2 (26,0)
| |
MRT (h)
|
20,4 (25,8)
| |
CL (ml/h/kg)
|
2,90 (34,4)
| |
Vss (ml/kg)
|
55,2 (20,8)
|
IR = Inkrementelle Recovery, aufgenommen 30 Minuten nach Injektion; Cmax = maximale Konzentration; AUC0-inf = Fläche unter der Faktor-VIII-Aktivität-Zeit-Kurve, extrapoliert bis unendlich; t½ = Halbwertszeit; MRT = mittlere Verweildauer; CL = Körpergewicht-korrigierte Clearance; Vss = Körpergewicht-korrigiertes Verteilungsvolumen im Steady-State.
Kinder und Jugendliche
Die Pharmakokinetik (PK) von AFSTYLA wurde an 10 Jugendlichen (Alter 12 bis < 18 Jahre) und 39 Kindern (Alter 0 bis < 12 Jahre) nach intravenöser Injektion einer Einzeldosis von 50 I.E./kg untersucht.
Die PK-Parameter stützen sich auf die durch den chromogenen Substrat-Test gemessene Plasmafaktor-VIII-Aktivität.
Vergleich der pharmakokinetischen Parameter je nach Altersklasse (Arithmetisches Mittel, CV %) nach einer Einzelinjektion von 50 I.E./kg AFSTYLA – Chromogener Substrat-Test:
|
PK-Parameter
|
0 bis < 6 Jahre
(N = 20)
|
6 bis < 12 Jahre
(N = 19)
|
12 bis < 18 Jahre
(N = 10)
| |
IR (I.E./dl)/(I.E./kg)
|
1,60 (21,1)
|
1,66 (19,7)
|
1,69 (24,8)
| |
Cmax (I.E./dl)
|
80,2 (20,6)
|
83,5 (19,5)
|
89,7 (24,8)
| |
AUC0-inf (I.E.*h/dl)
|
1080 (31,0)
|
1170 (26,3)
|
1540 (36,5)
| |
t½ (h)
|
10,4 (28,7)
|
10,2 (19,4)
|
14,3 (33,3)
| |
MRT (h)
|
12,4 (25,0)
|
12,3 (16,8)
|
20,0 (32,2)
| |
CL (ml/h/kg)
|
5,07 (29,6)
|
4,63 (29,5)
|
3,80 (46,9)
| |
Vss (ml/kg)
|
71,0 (11,8)
|
67,1 (22,3)
|
68,5 (29,9)
|
IR = Inkrementelle Recovery, aufgenommen 30 Minuten nach Injektion bei Probanden im Alter von 12 bis < 18 Jahren und 60 Minuten nach Injektion bei Probanden im Alter von 1 bis < 12 Jahren; Cmax = maximale Konzentration; AUC0-inf = Fläche unter der Faktor-VIII-Aktivität-Zeit-Kurve extrapoliert bis unendlich; t½ = Halbwertszeit; MRT = mittlere Verweildauer; CL = Körpergewicht-korrigierte Clearance; Vss = Körpergewicht-korrigiertes Verteilungsvolumen im Steady-State
Absorption
Keine Angaben.
Distribution
Keine Angaben.
Metabolismus
Keine Angaben.
Elimination
Keine Angaben.
Präklinische DatenPräklinische Daten, die auf der Grundlage von konventionellen Studien zur Sicherheitspharmakologie, Toxizitätsstudien mit einzelnen und wiederholten Dosisgaben und Prüfungen zur lokalen Verträglichkeit und Thrombogenität erhoben wurden, zeigen kein spezielles Risiko für den Menschen.
Es wurden keine Studien zur Untersuchung der Genotoxizität, Karzinogenität und Reproduktionstoxizität mit dem Wirkstoff von AFSTYLA durchgeführt.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln, Verdünnungs- oder Lösungsmitteln gemischt werden mit Ausnahme der unter «Zusammensetzung» angegebenen Lösungsmittel.
Haltbarkeit
AFSTYLA darf nur bis zu dem auf Etikette und Faltschachtel mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Die chemische und physikalische Anwendungsstabilität nach Rekonstitution wurde für 48 Stunden bei Raumtemperatur (unter 25 °C) nachgewiesen. Aus mikrobiologischer Sicht sollte das Produkt sofort verwendet werden. Falls es nicht sofort angewendet wird, sollte eine Aufbewahrung von 4 Stunden bei Raumtemperatur (bis max. 25 °C) nicht überschritten werden.
Besondere Lagerungshinweise
Im Kühlschrank (2 – 8 °C) lagern.
Nicht einfrieren.
Ausser Reichweite von Kindern aufbewahren.
Den Behälter in der geschlossenen Faltschachtel aufbewahren, um den Inhalt vor Licht zu schützen.
AFSTYLA kann bei Raumtemperatur, die 25 °C nicht übersteigen darf, einmalig für einen Zeitraum von bis zu 3 Monaten innerhalb der auf dem Karton und dem Flaschenetikett angegebenen Haltbarkeitsfrist aufbewahrt werden.
Hinweise für die Handhabung
Allgemeine Hinweise
·Die Lösung sollte nahezu farblos, klar oder leicht opaleszent sein. Nach Filtration/Entnahme (siehe unten) sollte das rekonstituierte Produkt vor der Verabreichung visuell auf das Vorhandensein von Partikeln und Verfärbung überprüft werden.
·Keine Lösungen verwenden, die sichtbar trüb sind oder noch Flocken oder Partikel enthalten.
·Die Rekonstitution und Entnahme müssen unter aseptischen Bedingungen erfolgen.
Zubereitung
Das Lösungsmittel auf Raumtemperatur (15 – 25 °C) bringen. Die Schutzkappen der AFSTYLA- und Lösungsmittelflaschen entfernen, die Stopfen mit antiseptischer Lösung behandeln und vor dem Öffnen der Mix2Vial™-Verpackung trocknen lassen.
|
|
1
|
1. Öffnen Sie die Mix2Vial Packung durch Abziehen des Deckpapiers. Das Mix2Vial dabei nicht aus dem Blister entnehmen!
| |
|
2
|
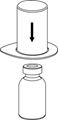
2. Die Lösungsmittelflasche auf eine ebene, saubere Oberfläche stellen und festhalten. Das Mix2Vial zusammen mit dem Blister greifen und den Dorn des blauen Adapterendes gerade nach unten durch den Stopfen der Lösungsmittelflasche drücken.
| |
|
3
|
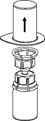
3. Den Blister vorsichtig aus dem Mix2Vial Set entfernen, indem er am Siegelrand festgehalten und senkrecht nach oben gezogen wird. Stellen Sie sicher, dass nur der Blister und nicht das Mix2Vial Set entfernt wird.
| |
|
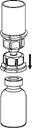
4
|
4. Die AFSTYLA-Flasche auf eine ebene und feste Oberfläche stellen. Die Lösungsmittelflasche mit dem aufgesetzten Mix2Vial Set herumdrehen und den Dorn des transparenten Adapterendes senkrecht nach unten in den Stopfen der AFSTYLA-Flasche einstechen. Das Lösungsmittel läuft automatisch in die AFSTYLA-Flasche über.
| |
|
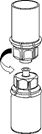
5
|
5. Mit der einen Hand die AFSTYLA-Seite und mit der anderen Hand die Lösungsmittelseite des Mix2Vial Set greifen und das Set vorsichtig gegen den Uhrzeigersinn auseinanderschrauben, um eine übermässige Schaumbildung beim Auflösen des Produktes zu vermeiden.
Die Lösungsmittelflasche mit dem aufgesetzten blauen Mix2Vial Adapter entsorgen.
| |
|
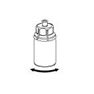
6
|
6. Die AFSTYLA-Flasche mit aufgesetztem transparentem Adapter vorsichtig schwenken, bis das Produkt vollständig aufgelöst ist. Nicht schütteln!
| |
|
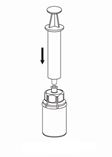
7
|
7. Luft in eine leere, sterile Spritze aufziehen. Die AFSTYLA-Flasche aufrecht halten, die Spritze durch Aufschrauben im Uhrzeigersinn mit dem Luer-Lock-Anschluss des Mix2Vial verbinden und die Luft in die AFSTYLA-Flasche injizieren.
|
Entnahme und Anwendung
|
|

8
|
8. Den Stempel der Spritze gedrückt halten, das gesamte System herumdrehen und das Produkt durch langsames Zurückziehen der Kolbenstange in die Spritze aufziehen.
| |
|

9
|
9. Nachdem die Lösung vollständig in die Spritze überführt ist, den Spritzenzylinder fassen (dabei die Kolbenstange nach unten gerichtet lassen) und den transparenten Mix2Vial Set Adapter durch Abdrehen gegen den Uhrzeigersinn von der Spritze lösen.
|
Zur Injektion von AFSTYLA wird die Verwendung des mitgelieferten Sets zur Anwendung empfohlen, da als Folge von Adsorption von Faktor VIII an die innere Oberfläche einiger Injektionsvorrichtungen ein Behandlungsmisserfolg auftreten kann.
Es muss darauf geachtet werden, dass kein Blut in die mit Produkt gefüllte Spritze gelangt, da das Risiko besteht, dass das Blut in der Spritze koaguliert und dadurch dem Patienten Fibringerinnsel verabreicht werden könnten.
Die AFSTYLA-Lösung darf nicht verdünnt werden.
Die rekonstituierte Lösung sollte über eine getrennte Injektions-/Infusionsleitung durch langsame intravenöse Injektion mit einer für den Patienten angenehmen Geschwindigkeit injiziert werden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist den lokalen Bestimmungen gemäss zu entsorgen.
Zulassungsnummer66030 (Swissmedic)
PackungenJe 1 Flasche zu:
500 I.E. Pulver und Lösungsmittel (2,5 ml) (B)
1000 I.E. Pulver und Lösungsmittel (2,5 ml) (B)
2000 I.E. Pulver und Lösungsmittel (5 ml) (B)
Je:
1 Filter Transfer Set 20/20
1 Verabreichungsset (beigepackter Zusatzkarton):
1 Venenpunktionsbesteck
2 Alkoholtupfer
1 nicht steriles Pflaster
1 Einmalspritze
ZulassungsinhaberinCSL Behring Lengnau AG, Lengnau
Stand der InformationSeptember 2023
|