Eigenschaften/WirkungenATC-Code
L04AA37
Wirkungsmechanismus
Baricitinib ist ein selektiver und reversibler Inhibitor von Janus-Kinasen (JAK), mit höherer Selektivität für JAK 1 und JAK 2. In Tests mit isolierten Enzymen hemmte Baricitinib die Aktivität JAK1, JAK2, Tyrosin Kinase 2 und JAK3 mit IC50 Werten von 5.9, 5.7, 53 bzw. > 400 nM.
Die Janus-Kinasen sind Enzyme, die intrazelluläre Signale von Rezeptoren auf der Zelloberfläche für verschiedene Zytokine und Wachstumsfaktoren, die an der Hämatopoese, der Entzündung und der Immunfunktion beteiligt sind, weiterleiten. Innerhalb der intrazellulären Signalkaskade werden STATs (signal transducers and activators of transcription) von JAKs phosphoryliert und aktiviert, welches die Genexpression in der Zelle aktiviert. Baricitinib moduliert diese Signalkaskade durch die Hemmung der enzymatischen Aktivität von JAK1 und JAK2, welches die Phosphorylierung und Aktivierung der STATs vermindert.
Baricitinib wurde als Inhibitor der NAK (numb-associated kinase) identifiziert, mit hoher Affinität für AP2-assoziierte Proteinkinase 1 (AAK1) - 8.2 nM, BIKE - 20 nM und GAK - 120 nM. Speziell die NAKs AAK1 und GAK sind mit dem Eindringen von SARS-CoV-2 (COVID-19) in die menschlichen Zellen assoziiert.
Pharmakodynamik
Hemmung der IL-6 induzierten STAT3 Phosphorylierung
Die Gabe von Baricitinib führte zu einer dosisabhängigen Hemmung der IL-6-induzierten STAT3 Phosphorylierung im Vollblut gesunder Probanden, mit maximaler Hemmung 2 Stunden nach der Gabe und Rückkehr zu den ungefähren Ausgangswerten innerhalb von 24 Stunden. Ähnliche Grade der Inhibition wurden nach Verwendung von IL-6 oder Thrombopoietin (TPO) als Stimulus beobachtet.
Immunoglobuline
Nach Beginn der Behandlung mit Olumiant nahmen die mittleren Serumwerte von IgG, IgM und IgA bis Woche 12 ab und blieben bis mindestens Woche 104 stabil. Bei den meisten Patienten erfolgte die Veränderung der Immunglobuline innerhalb des Normalbereichs.
Anstiege der IgG-Antikörper gegen die S1/S2-Antigene von SARS-CoV-2 wurden in einer begrenzten Stichprobe von mittelschwer bis schwer erkrankten hospitalisierten COVID-19 Patienten, die mit Baricitinib behandelt wurden, beobachtet.
Lymphozyten
Die mittlere absolute Lymphozytenzahl stieg eine Woche nach Beginn der Behandlung mit Olumiant, ging bis Woche 24 auf die Ausgangswerte zurück und blieb dann über mindestens 104 Wochen stabil. Bei den meisten Patienten erfolgte die Veränderung der Lymphozytenzahlen innerhalb des Normalbereichs.
Thrombozyten
Klinisch wurden unter Baricitinib gegenüber Placebo häufiger Thrombozytosen beobachtet (siehe «Unerwünschte Wirkungen, Beschreibung ausgewählter Nebenwirkungen, Thrombozytose»).
C-reaktives Protein
Bei Patienten mit rheumatoider Arthritis wurden schon eine Woche nach Beginn der Behandlung mit Olumiant Abnahmen des C-reaktiven Proteins (CRP) im Serum beobachtet und während der Therapie beibehalten.
Haut
In einem mit proinflammatorischen Zytokinen behandelten Modell der menschlichen Haut (d.h. IL-4, IL-13, IL-31) reduzierte Baricitinibdie pSTAT3-Expression der epidermalen Keratinozyten und erhöhte die Expression von Filaggrin.
Biomarker für COVID-19
Baricitinib senkt die Werte von Zytokinen und Biomarkern, die mit COVID-19 in Zusammenhang stehen, darunter IL-6, IFN-γ, MCP-3, CXCL10, IL-10, MCP-2, CCL19, PTX3 und IL-27. Darüber stiegen die Werte für Marker, die bei mittelschwer bis schwer erkrankten COVID-19-Patienten verringert sind, durch die Anwendung von Baricitinib; dies betrifft CCL17, GDF2 und SCF.
Klinische Wirksamkeit
Rheumatoide Arthritis
Die Wirksamkeit und Sicherheit von Olumiant einmal täglich wurde in 4 Phase III pivotalen, randomisierten, doppelblinden, multizentrischen Studien untersucht, bei Patienten mit mittelschwerer bis schwerer aktiver rheumatoider Arthritis, diagnostiziert gemäss den Kriterien des ACR/EULAR 2010 (siehe Tabelle 1). Eingeschlossen werden konnten Patienten über 18 Jahre. Zu Studienbeginn mussten mindestens 6 berührungsempfindliche und 6 geschwollene Gelenke vorhanden sein. Alle Patienten, die diese Studien abgeschlossen hatten, konnten an einer anschliessenden Langzeitstudie mit bis zu 4 Jahren fortgesetzter Behandlung teilnehmen.
Tabelle 1. Zusammenfassung der klinischen Studien
|
Name der Studie (Dauer)
|
Population
(Gesamt-Anzahl)
|
Therapiearme
|
Zusammenfassung der wesentlichen Studienziele
| |
RA-BEGIN
(52 Wochen)
|
MTX-naiv1
(584)
|
Olumiant 4 mg QD
Olumiant 4 mg QD + MTX
MTX
|
Primärer Endpunkt: ACR20 in Woche 24
Körperliche Funktion (HAQ-DI)
Radiologische Progression (mTSS)
Niedrige Krankheitsaktivität und Remission (SDAI)
| |
RA-BEAM
(52 Wochen)
|
MTX-IR2
(1305)
|
Olumiant 4 mg QD
Adalimumab 40 mg SC Q2W
Placebo
Alle Patienten erhielten eine Basistherapie mit MTX
|
Primärer Endpunkt: ACR20 in Woche 12
Körperliche Funktion (HAQ-DI)
Radiologische Progression (mTSS)
Niedrige Krankheitsaktivität und Remission (SDAI)
Morgendliche Gelenksteifigkeit
| |
RA-BUILD
(24 Wochen)
|
cDMARD-IR3
(684)
|
Olumiant 4 mg QD
Olumiant 2 mg QD
Placebo
Patienten erhielten eine Basistherapie mit cDMARDs, wenn bei stabiler Therapie mit cDMARD zu Studienbeginn
|
Primärer Endpunkt: ACR20 in Woche 12
Körperliche Funktion (HAQ-DI)
Niedrige Krankheitsaktivität und Remission (SDAI)
Radiologische Progression (mTSS)
Morgendliche Gelenksteifigkeit
| |
RA-BEACON
(24 Wochen)
|
TNF-IR4
(527)
|
Olumiant 4 mg QD
Olumiant 2 mg QD
Placebo
Alle Patienten erhielten eine Basistherapie mit cDMARDs
|
Primärer Endpunkt: ACR20 in Woche 12
Körperliche Funktion (HAQ-DI)
Niedrige Krankheitsaktivität und Remission (SDAI)
|
QD = einmal täglich; Q2W = einmal alle 2 Wochen; SC = Subkutan.
1 Patienten, die weniger als 3 Dosierungen Methotrexat (MTX) erhalten haben; naiv gegenüber anderen konventionellen (cDMARDs) oder biologischen (bDMARDs) krankheitsmodifizierenden Antirheumatika.
2 Patienten mit unzureichendem Ansprechen auf MTX (+/- andere cDMARDs); naiv gegenüber Biologika.
3 Patienten mit unzureichendem Ansprechen auf oder Unverträglichkeit von ≥1 cDMARDs; naiv gegenüber Biologika.
4 Patienten mit unzureichendem Ansprechen auf oder Unverträglichkeit von ≥1 bDMARDs, darunter mindestens ein TNF Inhibitor.
Klinisches Ansprechen
In den 3 Placebo kontrollierten Studien erreichte ein statistisch signifikant grösserer Anteil der Patienten unter Olumiant 4 mg einmal täglich ein ACR20, ACR50 und ACR70 Ansprechen nach 12 und 24 Wochen im Vergleich zu Placebo (Tabelle 2). Die Zeit bis zum Eintritt der Wirkung war kurz über die Messwerte hinweg mit einem signifikant grösserem Ansprechen bereits in Woche 1. Anhaltende, dauerhafte Ansprechraten wurden beobachtet, die ACR20/50/70 Ansprechen blieben über mindestens 2 Jahre erhalten, einschliesslich in der anschliessenden Langzeitstudie.
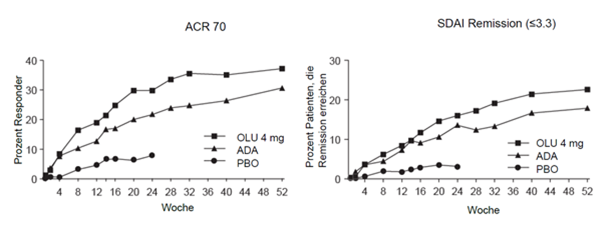
In der aktiv kontrollierten Studie RA-BEAM erreichte ein statistisch signifikant grösserer Anteil der mit Olumiant behandelten Patienten ein ACR20/50/70 Ansprechen nach 12 Wochen im Vergleich zu Adalimumab und die Unterschiede blieben über 52 Wochen erhalten (Abbildung 1). Radiologisch zeigten sowohl Olumiant als auch Adalimumab statistisch signifikante Effekte auf die Hemmung der strukturellen Gelenkschäden im Vergleich zu Placebo; Adalimumab schnitt gegenüber Olumiant numerisch etwas besser ab (siehe unten «Radiologisches Ansprechen»).
Die Behandlung mit Olumiant 4 mg, allein oder in Kombination mit cDMARDs, führte zu einer signifikanten Verbesserung aller einzelnen ACR Komponenten, einschliesslich der Anzahl berührungsempfindlicher und geschwollener Gelenke, der Gesamteinschätzung durch Patient und Arzt, des Health Assessment Questionnaire-Disability Index (HAQ-DI), der Schmerzbeurteilung und des C-reaktiven Proteins (CRP) im Vergleich zu Placebo oder MTX Monotherapie. In der Studie RA-BEAM führte die Behandlung mit Olumiant zu einer signifikanten Verbesserung der Gesamteinschätzung durch Patient und Arzt, des Health Assessment Questionnaire-Disability Index (HAQ-DI), der Schmerzbeurteilung und des C-reaktiven Proteins (CRP) in Woche 12, 24 und 52 im Vergleich zu Adalimumab.
Remission und niedrige Krankheitsaktivität
Olumiant 4 mg zeigte im Vergleich zu Placebo oder MTX eine statistisch signifikante Verbesserung hinsichtlich des Anteils von Patienten, der eine Remission definiert als SDAI ≤3.3 und CDAI ≤2.8 erreichte, in den Wochen 12 und 24 (Tabelle 2). In der Studie RA-BEAM, waren Olumiant und Adalimumab im Vergleich zu Placebo beide überlegen, gemessen anhand des SDAI ≤3.3 in Woche 12 und die Unterschiede wurden bis Woche 52 beibehalten (Abbildung 1).
In allen 4 Studien zeigte Olumiant 4 mg eine signifikante Verbesserung im Vergleich zu Placebo oder MTX hinsichtlich des Anteils von Patienten, die eine niedrige Krankheitsaktivität oder Remission erreichten (DAS28-ESR oder DAS28-hsCRP ≤3.2 und DAS28-ESR oder DAS28-hsCRP < 2.6) in Woche 12 und 24.
Einschliesslich der Daten aus einer anschliessenden Langzeitstudie wurden die Raten für Remission und geringe Krankheitsaktivität über mindestens 2 Jahre beibehalten.
Tabelle 2: Ansprechen, Remission und körperliche Funktion
|
Studie
|
RA-BEGIN
MTX-naiv Patienten
|
RA-BEAM
MTX-IR Patienten
|
RA-BUILD
cDMARD-IR Patienten
|
RA-BEACON
TNF-IR Patienten
| |
Therapie-Gruppe
|
MTX
|
OLU
4 mg
|
OLU
4 mg
+ MTX
|
PBO
|
OLU
4 mg
|
ADA
40 mg Q2W
|
PBO
|
OLU
2 mg
|
OLU 4 mg
|
PBO
|
OLU 2 mg
|
OLU
4 mg
| |
N
|
210
|
159
|
215
|
488
|
487
|
330
|
228
|
229
|
227
|
176
|
174
|
177
| |
ACR20:
| |
Woche 12
|
59 %
|
79 %***
|
77 %***
|
40 %
|
70 %***†
|
61 %***
|
39 %
|
66 %***
|
62 %***
|
27 %
|
49 %***
|
55 %***
| |
Woche 24
|
62 %
|
77 %**
|
78 %***
|
37 %
|
74 %***†
|
66 %***
|
42 %
|
61 %***
|
65 %***
|
27 %
|
45 %***
|
46 %***
| |
Woche 52
|
56 %
|
73 %***
|
73 %***
|
|
71 %††
|
62 %
|
|
|
|
|
|
| |
ACR50:
| |
Woche 12
|
33 %
|
55 %***
|
60 %***
|
17 %
|
45 %***††
|
35 %***
|
13 %
|
33 %***
|
34 %***
|
8 %
|
20 %**
|
28 %***
| |
Woche 24
|
43 %
|
60 %**
|
63 %***
|
19 %
|
51 %***
|
45 %***
|
21 %
|
41 %***
|
44 %***
|
13 %
|
23 %*
|
29 %***
| |
Woche 52
|
38 %
|
57 %***
|
62 %***
|
|
56 %†
|
47 %
|
|
|
|
|
|
| |
ACR70:
| |
Woche 12
|
16 %
|
31 %***
|
34 %***
|
5 %
|
19 %***†
|
13 %***
|
3 %
|
18 %***
|
18 %***
|
2 %
|
13 %***
|
11 %**
| |
Woche 24
|
21 %
|
42 %***
|
40 %***
|
8 %
|
30 %***†
|
22 %***
|
8 %
|
25 %***
|
24 %***
|
3 %
|
13 %***
|
17 %***
| |
Woche 52
|
25 %
|
42 %***
|
46 %***
|
|
37 %
|
31 %
|
|
|
|
|
|
| |
DAS28-hsCRP ≤3.2:
| |
Woche 12
|
30 %
|
47 %***
|
56 %***
|
14 %
|
44 %***††
|
35 %***
|
17 %
|
36 %***
|
39 %***
|
9 %
|
24 %***
|
32 %***
| |
Woche 24
|
38 %
|
57 %***
|
60 %***
|
19 %
|
52 %***
|
48 %***
|
24 %
|
46 %***
|
52 %***
|
11 %
|
20 %*
|
33 %***
| |
Woche 52
|
38 %
|
57 %***
|
63 %***
|
|
56 %†
|
48 %
|
|
|
|
|
|
| |
DAS28-ESR ≤3.2:
| |
Woche 12
|
15 %
|
21 %
|
34 %***
|
7 %
|
24 %***
|
21 %***
|
7 %
|
21 %***
|
22 %***
|
4 %
|
13 %**
|
12 %**
| |
Woche 24
|
23 %
|
36 %**
|
39 %***
|
10 %
|
32 %***
|
34 %***
|
10 %
|
29 %***
|
32 %***
|
7 %
|
11 %
|
17 %**
| |
Woche 52
|
27 %
|
36 %
|
45 %***
|
|
39 %
|
36 %
|
|
|
|
|
|
| |
SDAI ≤3.3:
| |
Woche 12
|
6 %
|
14 %*
|
20 %***
|
2 %
|
8 %***
|
7 %***
|
1 %
|
9 %***
|
9 %***
|
2 %
|
2 %
|
5 %
| |
Woche 24
|
10 %
|
22 %**
|
23 %**
|
3 %
|
16 %***
|
14 %***
|
4 %
|
17 %***
|
15 %***
|
2 %
|
5 %
|
9 %**
| |
Woche 52
|
13 %
|
25 %**
|
30 %***
|
|
23 %
|
18 %
|
|
|
|
|
|
| |
CDAI ≤2.8:
| |
Woche 12
|
7 %
|
14 %*
|
19 %***
|
2 %
|
8 %***
|
7 %**
|
2 %
|
10 %***
|
9 %***
|
2 %
|
3 %
|
6 %
| |
Woche 24
|
11 %
|
21 %**
|
22 %**
|
4 %
|
16 %***
|
12 %***
|
4 %
|
15 %***
|
15 %***
|
3 %
|
5 %
|
9 %*
| |
Woche 52
|
16 %
|
25 %*
|
28 %**
|
|
22 %
|
18 %
|
|
|
|
|
|
| |
HAQ-DI (Veränderung gegenüber dem Ausgangswert:):
| |
Woche 12
|
-0.61
|
-0.92***
|
-0.98***
|
-0.34
|
-0.66***††
|
-0.56***
|
-0.36
|
-0.57***
|
-0.56***
|
-0.17
|
-0.37***
|
-0.41***
| |
Woche 24
|
-0.74
|
-1.04***
|
-1.03***
|
-0.35
|
-0.75***††
|
-0.63***
|
-0.38
|
-0.62***
|
-0.62***
|
-0.15
|
-0.38***
|
-0.43***
| |
Woche 52
|
-0.71
|
-0.99***
|
-1.06***
|
|
-0.77††
|
-0.66
|
|
|
|
|
|
|
ADA = Adalimumab; MTX = Methotrexat; OLU = Olumiant; PBO = Placebo
* p ≤0.05; ** p ≤0.01; *** p ≤0.001 vs. Placebo (vs. MTX in Studie RA-BEGIN)
† p ≤0.05; †† p ≤0.01; ††† p ≤0.001 vs. Adalimumab
Abbildung 1. ACR70 und SDAI Remission, Studie RA-BEAM
Radiologisches Ansprechen
Die Wirkungen von Olumiant auf die Progression struktureller Gelenkschäden wurden in den Studien RA-BEGIN (Hauptstudienziel), RA-BEAM (Hauptstudienziel) und RA-BUILD (exploratives Studienziel) radiologisch gemessen und beurteilt unter Verwendung des modifizierten Total Sharp Score (mTSS) und seiner Komponenten, der Erosionen (Erosions-Score) und der Gelenksspaltverengung (Joint Space Narrowing Score).
Im RA-BEAM zeigten sowohl Olumiant 4 mg als auch Adalimumab statistisch signifikante Effekte auf die Hemmung der strukturellen Gelenkschäden im Vergleich zu Placebo (Tabelle 3). Auswertungen der Erosion und der Gelenksspaltverengung (Joint Space Narrowing Score) entsprachen den Gesamtwerten. Der Anteil der Patienten ohne radiologische Progression (mTSS Veränderung ≤0) war signifikant grösser unter Olumiant 4 mg im Vergleich zu Placebo nach 24 und 52 Wochen.
Tabelle 3. Radiologische Veränderungen
|
Studie
|
RA-BEGIN
MTX-naive Patienten
|
RA-BEAM
MTX-IR Patienten
|
RA-BUILD °
cDMARD-IR Patienten
| |
Therapie-Gruppe
|
MTX
|
OLU 4 mg
|
OLU 4 mg
+ MTX
|
PBOa
|
OLU 4 mg
|
ADA 40 mg Q2W
|
PBO
|
OLU 2 mg
|
OLU 4 mg
| |
N
|
210
|
159
|
215
|
488
|
487
|
330
|
228
|
229
|
227
| |
modifizierter Total Sharp Score, mittlere Veränderung gegenüber dem Ausgangswert:
| |
Woche 16
|
|
|
|
0.69
|
0.35***
|
0.28***
|
|
|
| |
Woche 24
|
0.61
|
0.39
|
0.29*
|
0.90
|
0.41***
|
0.33***
|
0.70
|
0.33*
|
0.15**
| |
Woche 52
|
1.02
|
0.80
|
0.40**
|
1.80
|
0.71***
|
0.60***
|
|
|
| |
Erosions-Score, mittlere Veränderung gegenüber dem Ausgangswert:
| |
Woche 16
|
|
|
|
0.50
|
0.25***
|
0.21***
|
|
|
| |
Woche 24
|
0.47
|
0.33
|
0.26*
|
0.61
|
0.29***
|
0.24***
|
0.47
|
0.30
|
0.11**
| |
Woche 52
|
0.81
|
0.55
|
0.34**
|
1.23
|
0.51***
|
0.42***
|
|
|
| |
Joint Space Narrowing Score, mittlere Veränderung gegenüber dem Ausgangswert:
| |
Woche 16
|
|
|
|
0.20
|
0.11
|
0.08*
|
|
|
| |
Woche 24
|
0.14
|
0.06
|
0.03
|
0.29
|
0.12**
|
0.10**
|
0.23
|
0.03*
|
0.04*
| |
Woche 52
|
0.21
|
0.25
|
0.06
|
0.58
|
0.21***
|
0.19**
|
|
|
| |
Anteil der Patienten ohne radiologischen Progressb:
| |
Woche 16
|
|
|
|
72 %
|
81 %**
|
82 %**
|
|
|
| |
Woche 24
|
68 %
|
76 %
|
81 %**
|
70 %
|
81 %***
|
83 %***
|
74 %
|
72 %*
|
80 %**
| |
Woche 52
|
66 %
|
69 %
|
80 %**
|
70 %
|
79 %**
|
81 %**
|
|
|
|
ADA = Adalimumab; MTX = Methotrexat; OLU = Olumiant; PBO = Placebo
a Placebo Daten in Woche 52 stammen aus der linearen Extrapolation.
b Kein Progress definiert als Veränderung des mTSS ≤0.
* p ≤0.05; ** p ≤0.01; *** p ≤0.001 vs. Placebo (vs. MTX in Studie RA-BEGIN)
° exploratives Studienziel in RA-BUILD
Ansprechen der körperlichen Funktion und gesundheitsbezogene Ergebnisse
Die Therapie mit Olumiant 4 mg allein oder in Kombination mit cDMARDs zeigte eine signifikante Verbesserung im Vergleich zu allen Komparatoren (Placebo, MTX, Adalimumab) hinsichtlich der Verbesserung der körperlichen Funktion in Woche 12, 24 und 52, gemessen anhand des HAQ-DI (Tabelle 2). Verbesserungen wurden bereits in Woche 1 beobachtet, in den Studien RA-BEGIN und RA-BEAM blieben sie bis Woche 52 erhalten. In der Studie RA-BEAM erreichten 67 % der Patienten unter Olumiant 4 mg mindestens einen klinisch bedeutsamen Unterschied (Abnahme des HAQ-DI Score um ≥0.30) in Woche 24 im Vergleich zu 37 % unter Placebo und 60 % unter Adalimumab (p < 0.001 und p = 0.049 bzw.). Die Verbesserung mit Olumiant blieb mindestens 2 Jahre bestehen.
Die Therapie mit Olumiant 4 mg allein oder in Kombination mit cDMARDs zeigte eine signifikante Verbesserung im Vergleich zu allen Komparatoren (Placebo, MTX, Adalimumab) hinsichtlich der Schmerzreduktion in Woche 12, 24 und 52, gemessen anhand einer 0-100 visuellen Analogskala. Eine statistisch signifikante Schmerzreduktion wurde bereits in Woche 1 festgestellt und in den Studien RA-BEGIN und RA-BEAM blieb diese über bis zu 52 Wochen erhalten. In der Studie RA-BEAM betrug die mittlere Veränderung des Schmerzwertes unter Olumiant 4 mg ab Ausgangswert -33.6 in Woche 24 im Vergleich zu -17.5 unter Placebo und -28.8 in der Adalimumab Behandlungsgruppe (p < 0.001 und p = 0.004 bzw.).
In den Studien RA-BEAM und RA-BUILD wurden Dauer und Schweregrad der morgendlichen Gelenksteifigkeit mit Hilfe eines täglichen Patiententagebuchs über 12 Wochen beurteilt. In Studie RA-BEAM zeigten Patienten unter Olumiant nach 12 Wochen eine mediane Dauer der morgendlichen Gelenksteifigkeit von 27 Minuten im Vergleich zu 60 Minuten unter Placebo und 37 Minuten unter Adalimumab (p < 0.001 und p = 0.024 bzw.). In Studie RA-BEAM zeigten Patienten nach 12 Wochen unter Olumiant einen mittleren Schweregrad der morgendlichen Gelenkssteifigkeit von 3.0 im Vergleich zu 4.1 unter Placebo und 3.5 unter Adalimumab, gemessen anhand einer numerischen Beurteilungsskala von 0 bis 10 mit 0 für keine Gelenkssteifigkeit (p < 0.001 und p = 0.002 bzw.). In der Studie RA-BUILD wurden ähnliche Ergebnisse versus Placebo beobachtet.
In allen Studien berichteten Patienten unter Olumiant Verbesserungen der von Patienten berichteten Lebensqualität gemessen anhand von Short Form (36) Health Survey (SF-36) Physical Component Score, Müdigkeit gemessen anhand des Functional Assessment of Chronic Illness Therapy-Fatigue Score (FACIT-F) und der Arbeitsproduktivität gemessen anhand des Work Productivity and Activity Impairment Questionnaire: Rheumatoid arthritis (WPAI-RA).
Olumiant 4 mg vs. 2 mg
In klinischen Studien, die Dosierungen von 2 mg und 4 mg Olumiant einmal täglich beinhalteten (RA-BUILD und RA-BEACON), wurde die Wirksamkeit auf Zeichen und Symptome bei beiden Dosierungen gezeigt. Unter der 4mg Dosis gegenüber 2 mg wurde ein schnellerer Wirkungseintritt mit einem numerisch höheren Anteil an Patienten, die eine niedrige Krankheitsaktivität und eine geringere Progression der radiologischen Gelenkschäden aufwiesen, festgestellt. Die Unterschiede wurden vorallem bei Patienten der bDMARD-IR Population (RA-BEACON) beobachtet, jedoch kaum bei Patienten, welche vorher noch nie mit einem Biologikum behandelt worden waren.
In einer langfristigen Verlängerungsstudie wurden Patienten aus den Studien RA-BEAM, RA-BUILD und RA-BEACON, die eine anhaltende niedrige Krankheitsaktivität oder Remission erreicht hatten (CDAI ≤10) nach mindestens 15 Monaten Behandlung mit Olumiant 4 mg einmal täglich doppelblind erneut 1:1 randomisiert um mit 4 mg einmal täglich fortzusetzen oder auf 2 mg einmal täglich zu reduzieren.
Die Mehrzahl der Patienten behielt auch nach Reduktion auf 2 mg eine niedrige Krankheitsaktivität oder Remission bei:
·In Woche 12: 234/251 Patienten (93 %) unter Fortsetzung von 4 mg, und 207/251 (82 %) nach Reduktion auf 2 mg (p ≤0.001).
·In Woche 24: 163/191 Patienten (85 %) unter Fortsetzung von 4 mg, und 144/189 (76 %) nach Reduktion auf 2 mg (p ≤0.05).
·In Woche 48: 57/73 Patienten (78 %) unter Fortsetzung von 4 mg, und 51/86 (59 %) nach Reduktion auf 2 mg (p ≤0.05).
Die Mehrzahl der Patienten, die nach der Dosisreduktion den Status einer niedrigen Krankheitsaktivität oder Remission verloren hatten, konnten die Krankheitskontrolle nach Rückkehr auf 4 mg wiedererlangen.
Atopische Dermatitis
Die Wirksamkeit und Sicherheit von Olumiant als Monotherapie oder in Kombination mit topischen Kortikosteroiden (TCS) wurde in 3 randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien mit einer Dauer von 16 Wochen (BREEZE-AD1, -AD2 und -AD7) untersucht. Die Studien umfassten 1568 Patienten mit mittelschwerer bis schwerer atopischer Dermatitis, definiert durch den Score Investigator's Global Assessment (IGA) ≥3, einen Score Eczema Area und Severity Index (EASI) ≥16 und einen Body Surface Area (Körperoberfläche, BSA) Anteil von ≥10 %. Die teilnahmeberechtigten Patienten waren über 18 Jahre alt und hatten zuvor ein unzureichendes Ansprechen oder eine Unverträglichkeit gegenüber topischen Arzneimitteln. Die Patienten durften eine Rescue-Therapie (einschliesslich topischer oder systemischer Therapie) erhalten, galten dann aber ab diesem Zeitpunkt als Non-Responder. Alle Patienten, die diese Studien abgeschlossen hatten, waren berechtigt, sich für eine Langzeitverlängerungsstudie (BREEZE AD--3) für bis zu vier Jahre Dauerbehandlung einzuschreiben.
Die randomisierte, doppelblinde, placebokontrollierte Phase-III-Studie BREEZE-AD4 untersuchte die Wirksamkeit von Baricitinib in Kombination mit topischen Kortikosteroiden bei Patienten mit mittelschwerer bis schwerer AD mit Therapieversagen, Unverträglichkeit oder Kontraindikation für eine orale Ciclosporin-Behandlung.
Tabelle 4. Zusammenfassung der klinischen Studien
|
Name der Studie (Dauer)
|
Anzahl der behandelten Patienten (N)
|
Hintergrundbehandlunga
|
Behandlungsgruppen (QD)
|
Ergebnismessungen
| |
BREEZE AD-1
(16 Wochen)
|
624
|
Keine
|
OLU 4 mg
OLU 2 mg
OLU 1 mg Placebo
|
·Primärer Endpunkt: IGA 0 oder 1b in Woche 16
·Verbesserung von ≥50%, 75 % oder 90 % beim Eczema Area and Severity Index gegenüber Ausgangswert (Baseline) (EASI 50, 75, 90)
·Verbesserung von ≥75 % auf der Skala SCORing Atopic Dermatitis (SCORAD)
·Itch Numerical Rating Scale (NRS) ≥4 Punkte Verbesserung
·Auswirkung des Juckreizes auf den Schlaf, gemessen an der Atopic Dermatitis Sleep Scale (ADSS)
·Schweregrad der Hautschmerzen gemessen an der Skin Pain Numerical Rating Scale (NRS)
·Patient-Oriented Eczema Measure (POEM)
·Dermatologischer Lebensqualität-Index (DLQI)
·Hospital Anxiety and Depression Scale (HADS)
| |
BREEZE AD-2
(16 Wochen)
|
615
|
Keine
| |
BREEZE AD-7
(16 Wochen)
|
329
|
TCS;
TCI nach Bedarf
|
OLU 4 mg
OLU 2 mg Placebo
| |
BREEZE AD-4
(bis zu 200 Wochen)
|
462
|
TCS;
TCI nach Bedarf
|
OLU 4 mg
OLU 2 mg
OLU 1 mg
Placebo
|
OLU=Olumiant; QD = einmal täglich; TCI = Topical Calcineurin Inhibitor; TCS = Topical Kortikosteroid
a Patienten verwendeten Linderungsmittel während der gesamten Studie.
b Investigators Global Assessment Score von 0 («erscheinungsfrei») oder 1 («fast erscheinungsfrei») mit einer Verringerung von ≥2 Punkten auf einer 5-Punkte-Schweregrad-Skala von 0 bis 4.
Ausgangs-Charakteristika
In den Monotherapiestudien (BREEZE-AD1 und BREEZE-AD2) lag das mittlere Alter über alle Behandlungsgruppen hinweg bei 35.2, das mittlere Gewicht bei 73.3 kg, 37.7 % waren weiblich, 63.5 % kaukasisch, 30 % asiatisch und 0.2 % mit schwarzer Hautfarbe. In diesen Studien hatten 54 % der Patienten einen Baseline-IGA-Score von 3 (mittelschwere AD), 46 % einen Baseline-IGA-Score von 4 (schwere AD), und 59.9 % der Patienten hatten zuvor eine systemische Behandlung für die atopische Dermatitis erhalten. Der mittlere Baseline-EASI-Score betrug 32.2, mittlere Baseline-BSA-Score 52.3, wöchentliche Durchschnittswert gemäss Pruritus-NRS bei Baseline 6.6, mittlere Baseline-SCORAD-Score 67.8, mittlere Baseline-POEM 20.6, mittlere Baseline-DLQI 14.0, mittlere Baseline-HADS Depression Score 5.0 und mittlere Baseline-HADS Anxiety Score 6.1.
In der Kombination-TCS-Studie (BREEZE-AD7) lag das mittlere Alter über alle Behandlungsgruppen hinweg bei 33.8, das mittlere Gewicht bei 72.9 kg, 34.3 % waren weiblich, 45.6 % kaukasisch und 51.1 % asiatisch. In dieser Studie hatten 54.9 % der Patienten einen Baseline-IGA-Score von 3, 45.1 % einen Baseline-IGA-Score von 4 und 66.4 % der Patienten hatten zuvor eine systemische Behandlung erhalten. Der mittlere Baseline-EASI-Score betrug 29.6, mittlere Baseline-BSA-Score 50.3, Durchschnittswert gemäss Pruritus-NRS bei Baseline 7.1, mittlere Baseline-SCORAD-Score 67.2, mittlere Baseline-POEM 21.1, mittlere Baseline-DLQI 14.9, mittlere Baseline-HADS Depression Score 5.5 und mittlere Baseline-HADS Anxiety Score 6.6.
In der Kombination-TCS-Studie (BREEZE-AD4) lag das mittlere Alter über alle Behandlungsgruppen hinweg bei 38.2, das mittlere Gewicht bei 75.5 kg, 35.9 % waren weiblich, 77.8 % kaukasisch und 19.2 % asiatisch. In dieser Studie hatten 48.7 % der Patienten einen Baseline-IGA-Score von 3, 51.3% einen Baseline-IGA-Score von 4 und 78.8 % der Patienten hatten zuvor eine systemische Behandlung erhalten. Der mittlere Baseline-EASI-Score betrug 31.8, mittlere Baseline-BSA-Score 51.8, Durchschnittswert gemäss Pruritus-NRS bei Baseline 6.8, mittlere Baseline-SCORAD-Score 68.8, mittlere Baseline-POEM 21.2, mittlere Baseline-DLQI 14.0, mittlere Baseline-HADS Depression Score 5.0 und mittlere Baseline-HADS Anxiety Score 6.2.
Klinisches Ansprechen
16-wöchige Monotherapie-Studien (BREEZE-AD1 und BREEZE-AD2)
In BREEZE-AD1 und BREEZE-AD2 erreichte ein signifikant höherer Anteil von Patienten, die auf Baricitinib 4 mg randomisiert waren, ein IGA-Ansprechen von 0 oder 1, EASI-75 oder eine Verbesserung von ≥4 Punkten auf der Itch NRS im Vergleich zu Placebo in Woche 16 (Tabelle 5).
Ein signifikant höherer Anteil von Patienten, die auf Baricitinib 4 mg randomisiert waren, erreichte eine rasche Verbesserung auf der Itch NRS im Vergleich zu Placebo (definiert als ≥4 Punkte Verbesserung innerhalb der ersten Behandlungswoche; p <0.001).
Die Abbildungen 2 und 3 zeigen jeweils die mittlere prozentuale Veränderung gegenüber dem Ausgangswert in EASI und der Anteil von Patienten mit mindestens 4 Punkte Verbesserung auf der Itch NRS Die Ergebnisse für den placebokontrollierten Zeitraum werden bis Woche 16 angezeigt, wobei Langzeitergebnisse bis Woche 68 und 32 für die Abbildungen 2 bzw. 3 verfügbar sind.
Die Behandlungseffekte in den Untergruppen (Gewicht, Alter, Geschlecht, ethnischer Abstammung, Schweregrad der Erkrankung und frühere Behandlung, einschliesslich Immunsuppressiva) in BREEZE-AD1 und BREEZE-AD2 waren mit den Ergebnissen in der gesamten Studienpopulation konsistent.
Tabelle 5. Wirksamkeit der Baricitinib-Monotherapie in Woche 16 (FASa)
|
Studie
|
BREEZE-AD1
|
BREEZE-AD2
| |
Behandlungsgruppe
|
PBO
|
OLU
2 mg
|
OLU
4 mg
|
PBO
|
OLU
2 mg
|
OLU
4 mg
| |
N
|
N = 249
|
N = 123
|
N = 125
|
N = 244
|
N = 123
|
N = 123
| |
IGA 0 oder 1,
% Ansprechendeb, c
|
4.8 %
|
11.4 %*
|
16.8 %***
|
4.5 %
|
10.6 %*
|
13.8 %***
| |
EASI-75,
% Ansprechendec
|
8.8 %
|
18.7 %**
|
24.8 %***
|
6.1 %
|
17.9 %***
|
21.1 %***
| |
SCORAD75,
% Ansprechendec
|
1.2 %
|
7.3 %+
|
10.4 %***
|
1.6 %
|
7.3 %**
|
11.4 %***
| |
Itch NRS (≥4 Punkte Verbesserung), % Ansprechendec,d
|
7.2 %
|
12.0+ %
|
21.5 %***
|
4.7 %
|
15.1 %**
|
18.7 %***
|
OLU = Olumiant; PBO = Placebo
*p ≤0.05; **p ≤0.01; ***p ≤0.001 +nicht signifikant, teils aufgrund Testhierarchie, vs. Placebo.
a Full analysis set (FAS, Gesamtanalyse-Datensatz), einschliesslich aller randomisierten Patienten.
b Ansprechender war definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verringerung von ≥2 Punkten auf der IGA-Skala von 0-4.
c Non-Responder Imputation: Patienten mit Rescue-Therapie, wurden dauerhaft von der Studienmedikation abgesetzt. Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
d Die Ergebnisse wurden in einer Untergruppe von Patienten gezeigt, die für eine Beurteilung in Frage kommen (Patienten mit Itch NRS ≥4 zu Studienbeginn).
Abbildung 2. Mittlere prozentuale Veränderung gegenüber dem Ausgangswert in EASI in BREEZE-AD1 und BREEZE-AD2 (FAS)a mit Langzeitdaten in BREEZE-AD3b, c
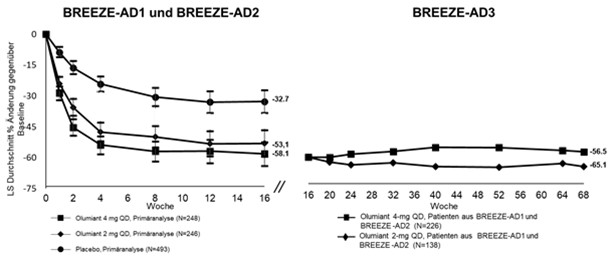
a Full analysis set (FAS) einschliesslich aller randomisierten Patienten. Daten, die nach der Rescue-Therapie oder nach einem dauerhaften Absetzen der Studienmedikation gesammelt wurden, galten als fehlend. LS-Mittelwerte stammen aus MMRM-Analysen.
b Modified last observation carried forward (mLOCF) wurde verwendet. Die mLOCF-Imputationstechnik ersetzt fehlende Daten durch die letzte nicht fehlende Bewertung nach der Baseline.
c In BREEZE-AD3 wird die Patientenprobengrösse für Olumiant 2 mg reduziert, da Non-Responder auf diese Dosis in BREEZE-AD1 und BREEZE-AD2 auf die 2-mg- oder 4-mg-Dosis 1: 1 re-randomisiert wurden. Responder und Partial Responder blieben auf der gleichen Dosis.
Hinweis: Fehlerbalken repräsentieren 95% CI.
Abbildung 3. Anteil von Patienten mit mindestens 4 Punkte Verbesserung auf der Itch NRS in BREEZE-AD1 and BREEZE-AD2 a mit Langzeitdaten in BREEZE-AD3b
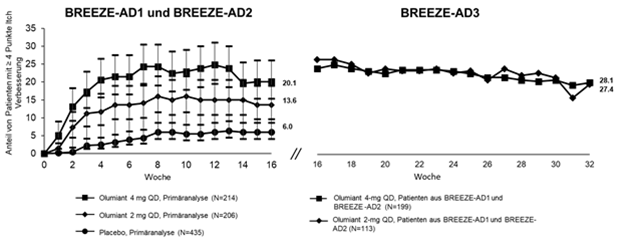
a Die Ergebnisse wurden in einer Untergruppe von Patienten gezeigt, die für eine Beurteilung in Frage kommen (Patienten mit Itch NRS ≥4 zu Studienbeginn). Patienten mit Rescue-Therapie wurden dauerhaft von der Studienmedikation abgesetzt. Patienten mit fehlenden Daten wurden als Non-Responder gewertet (Non Responder Imputation).
b In BREEZE-AD3 wird die Patientenprobengrösse für Olumiant 2 mg reduziert, da Non-Responder auf diese Dosis in BREEZE-AD1 und BREEZE-AD2 auf die 2-mg- oder 4-mg-Dosis 1: 1 re-randomisiert wurden. Responder und Partial Responder blieben auf der gleichen Dosis. Patienten, die dauerhaft von der Studienmedikation abgesetzt wurden oder mit fehlenden Daten wurden als Non-Responder gewertet (Non Responder Imputation).
Hinweis: Fehlerbalken repräsentieren 95% CI. Der beobachtete prozentuale Anstieg in BREEZE-AD3 ist darauf zurückzuführen, dass Patienten, die in BREEZE-AD1 und -AD2 mit TCS gerettet wurden, nicht als Non-Responder angesehen wurden.
16wöchige Kombination TCS-Studie (BREEZE-AD7)
In BREEZE-AD7 erreichte ein signifikant höherer Anteil von Patienten, die auf Baricitinib 4 mg + TCS randomisiert waren, ein IGA-Ansprechen von 0 oder 1, EASI-75 oder eine Verbesserung von ≥4 Punkten auf der Itch NRS im Vergleich zu Placebo in Woche 16 (Tabelle 6).
Ein signifikant höherer Anteil von Patienten, die auf Baricitinib 4 mg randomisiert waren, erreichte eine rasche Verbesserung auf der Itch NRS im Vergleich zu Placebo (definiert als ≥4 Punkte Verbesserung bereits ab Woche 2; p <0.001).
Die Abbildungen 4 und 5 zeigen jeweils die mittlere prozentuale Veränderung gegenüber dem Ausgangswert in EASI und der Anteil von Patienten mit mindestens 4 Punkte Verbesserung auf der Itch NRS. Die Ergebnisse für den placebokontrollierten Zeitraum werden bis Woche 16 angezeigt, wobei Langzeitergebnisse bis Woche 68 und 32 für die Abbildungen 4 bzw. 5 verfügbar sind.
Die Behandlungseffekte in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Abstammung, Schweregrad der Erkrankung und frühere Behandlung, einschliesslich Immunsuppressiva) in BREEZE-AD7 waren mit den Ergebnissen in der gesamten Studienpopulation konsistent.
Tabelle 6. Wirksamkeit von Baricitinib in Kombination mit TCSa in Woche 16 (FAS)b
|
Studie
|
BREEZE- AD7
| |
Behandlungsgruppe
|
PBOa
|
OLU 2 mg a
|
OLU 4 mg a
| |
N
|
109
|
109
|
111
| |
IGA 0 oder 1,
% Ansprechendec, d
|
14.7 %
|
23.9 %
|
30.6 %**
| |
EASI-75,
% Ansprechended
|
22.9 %
|
43.1 %+
|
47.7 %***
| |
SCORAD75,
% Ansprechended
|
7.3 %
|
11.0 %+
|
18.0 %+
| |
Itch NRS (≥4 Punkte Verbesserung), % Ansprechende d, e
|
20.2 %
|
38.1 %+
|
44.0 %***
|
OLU = Olumiant; PBO = Placebo
*p ≤0.05; **p ≤0.01; ***p ≤0.001 vs. +nicht signifikant, teils aufgrund Testhierarchie Placebo.
a Alle Patienten erhielten eine topische Kortikosteroidtherapie im Hintergrund und durften topische Calcineurinhemmer verwenden.
b Full Analysis Set (FAS) einschliesslich aller randomisierten Patienten.
c Ansprechender war definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei») oder «fast erscheinungsfrei») mit einer Verringerung von ≥2 Punkten auf einer IGA-Skala von 0-4.
d Non-Responder Imputation: Patienten mit Rescue-Therapie, wurden dauerhaft von der Studienmedikation abgesetzt. Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
e Die Ergebnisse wurden in einer Untergruppe von Patienten gezeigt, die für eine Beurteilung in Frage kommen (Patienten mit Itch NRS ≥4 zu Studienbeginn).
Abbildung 4. Mittlere prozentuale Veränderung gegenüber dem Ausgangswert in EASI in BREEZE-AD7 (FAS) a mit Langzeitdaten in BREEZE-AD3b
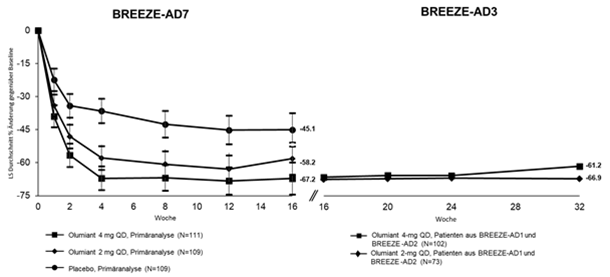
a Full Analysis Set (FAS) einschliesslich aller randomisierten Patienten. Daten, die nach der Rescue-Therapie oder nach einem dauerhaften Absetzen der Studienmedikation gesammelt wurden, gelten als fehlend. LS-Mittelwerte stammen aus MMRM-Analysen.
b Modified last observation carried forward (mLOCF) wurde verwendet. Die mLOCF-Imputationstechnik ersetzt fehlende Daten durch die letzte nicht fehlende Bewertung nach der Baseline.
Hinweis: Fehlerbalken repräsentieren 95% CI
Abbildung 5. Anteil von Patienten mit mindestens 4 Punkte Verbesserung auf der Itch NRS in BREEZE-AD7 a mit Langzeitdaten in BREEZE-AD3 b
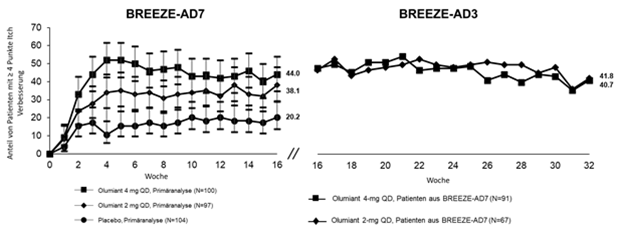
a Die Ergebnisse wurden in einer Untergruppe von Patienten gezeigt, die für eine Beurteilung in Frage kommen (Patienten mit Itch NRS ≥4 zu Studienbeginn). Patienten mit Rescue-Therapiewurden dauerhaft von der Studienmedikation abgesetzt. Patienten mit fehlenden Daten wurden als Non-Responder gewertet (Non Responder Imputation).
b In BREEZE-AD3 wird die Patientenprobengrösse für Olumiant 2 mg reduziert, da Non-Responder auf diese Dosis in BREEZE-AD7 auf die 2-mg- oder 4-mg-Dosis 1:1 re-randomisiert wurden. Responder und Partial Responder blieben auf der gleichen Dosis. Patienten, die dauerhaft von der Studienmedikation abgesetzt wurden oder mit fehlenden Daten wurden als Non-Responder gewertet (Non Responder Imputation).
Hinweis: Fehlerbalken repräsentieren 95% CI
Aufrechterhaltung des Ansprechens
Um die Aufrechterhaltung des Ansprechens zu bewerten, waren 1373 Patienten, die 16 Wochen lang mit Baricitinib in BREEZE-AD1 (N=541), und BREEZE-AD2 (N=540) und BREEZE-AD7 (N =292) behandelt wurden, berechtigt, sich für eine Langzeitverlängerungsstudie BREEZE-AD3 einzuschreiben. Daten sind bis zu 68 Wochen kumulativer Behandlung für Patienten aus BREEZE AD1 und BREEZE AD2 und bis zu 32 Wochen kumulativer Behandlung für Patienten aus BREEZE AD7 verfügbar. Es wurde in den Baricitinib-Armen ein anhaltender Unterschied zu Placebo beobachtet. Zwischen der höheren 4 mg und der 2 mg Dosis zeigten sich konsistent numerische Unterschiede zuungunsten der höheren Dosis von 4 mg (siehe Abbildung 6).
Abbildung 6. IGA 0 or 1 Persistenz für Baricitinib 4 mg und 2 mg in BREEZE-AD3 bis zu Woche 68a
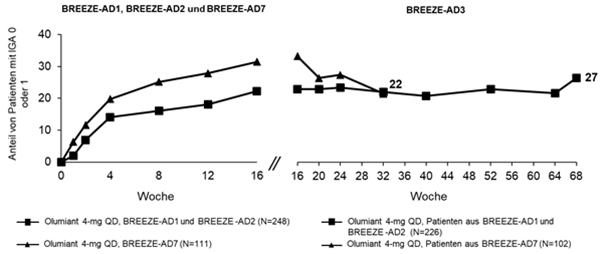

a Non-Responder Imputation wurde verwendet. Patienten, die dauerhaft von der Studienmedikation abgesetzt wurden oder mit fehlenden Daten wurden als Non-Responder gewertet.
Bei Patienten, die in der Langzeit-Verlängerungsstudie BREEZE-AD3 kontinuierlich mit Olumiant 2 mg oder 4 mg einmal täglich behandelt wurden und in Woche 52 eine erscheinungsfreie Haut oder eine anhaltende minimale Erkrankung (d.h. IGA 0 oder 1) erreicht hatten, wurde ein IGA-Ansprechen von 0 oder 1 bis Woche 200 bei 60 % bzw. 70 % der Patienten, die Olumiant 2 mg bzw. 4 mg erhielten, beobachtet.
Klinisches Ansprechen bei Patienten, mit Versagen der Ciclosporin-Behandlung oder die diese nicht vertragen oder die eine Kontraindikation für die Ciclosporin-Behandlung haben (BREEZE-AD4-Studie)
Insgesamt wurden 462 Patienten eingeschlossen, die entweder ein Therapieversagen hatten (n = 173) oder eine Unverträglichkeit (n = 75) oder eine Kontraindikation (n = 126) für orale Ciclosporin hatten.
Der primäre Endpunkt war der Anteil der Patienten, die in Woche 16 EASI-75 erreichten, und wurde in der Barictinib 4 mg Behandlungsgruppe erreicht. Endpunkte in Woche 24 sind in Tabelle 7 zusammengefasst.
Tabelle 7. Wirkung von Baricitinib in Kombination mit TCS in Woche 24 in BREEZE-AD4 (FAS)b
|
Studie
|
BREEZE- AD4
| |
Behandlungsgruppe
|
PBOa
|
OLU 2 mga
|
OLU 4 mga
| |
N
|
93
|
185
|
92
| |
EASI-75,
% Ansprechendec
|
15.1 %
|
27.0 %
|
28.3 %*
| |
EASI, LS Durchschnitt % Änderung gegenüber Baseline (SE) d
|
-45.10
(4.24)
|
-57.24 **
(2.80)
|
-58.46 ***
(4.02)
| |
IGA 0 oder 1,
% Ansprechendec, e
|
12.9 %
|
18.9 %
|
13.0 %*
| |
Itch NRS, LS Durchschnitt % Änderung gegenüber Baseline (SE) d,f
|
-15.35
(5.35)
|
-30.11 **
(3.50)
|
-33.16 **
(4.97)
| |
Änderung bei DLQI, Durchschnitt (SE)d
|
-4.90 (0.74)
|
-6.81
(0.49)
|
-7.65 **
(0.71)
|
SE = standard error (Standardfehler). *p ≤0.05; **p ≤0.01; ***p ≤0.001, +nicht signifikant, teils aufgrund Testhierarchievs Placebo
Hinweis: IGA (0,1) und EASI-75 sind gated Endpunkte. Alle anderen Endpunkte sind nicht gated.
a Alle Patienten erhielten eine topische Kortikosteroidtherapie im Hintergrund und durften topische Calcineurinhemmer verwenden.
b Full analysis set (FAS, Gesamtanalyse-Datensatz), einschliesslich aller randomisierten Patienten.
c Non-Responder Imputation: Patienten mit Rescue-Therapie wurden dauerhaft von der Studienmedikation abgesetzt. Patienten mit fehlenden Daten wurden als Non-Responder gewertet.
d Daten, die nach der Rescue-Therapie oder nach einem dauerhaften Absetzen der Studienmedikation gesammelt wurden, galten als fehlend. LS-Mittelwerte stammen aus MMRM-Analysen, die mehrere Imputationen für fehlende Daten beinhalten.
e Ansprechender war definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verringerung von ≥2 Punkten auf der IGA-Skala von 0-4.
f Der auswertbare Analysensatz umfasst Patienten mit nicht-null NRS zu Studienbeginn.
Dosisreduktion
In der Langzeitverlängerungsstudie BREEZE AD3 wurden Patienten, die mit Olumiant 4 mg einmal täglich eine erscheinungsfreie Haut oder eine anhaltende minimale oder milde Erkrankung (IGA 0, 1 oder 2) erreichten, doppelblind re-randomisiert in Woche 52, um die Therapie mit 4 mg einmal täglich fortzusetzen oder die Dosis auf 2 mg einmal täglich zu reduzieren. Bei Patienten mit IGA 0 oder 1 in Woche 52, die die Dosis auf 2 mg reduzierten, blieb bei 51% ein IGA 0 oder 1 Ansprechen und bei 67% ein EASI-75-Ansprechen bis zu 200 Wochen erhalten. Die Mehrheit der Patienten, die nach der Dosisreduktion ihre milde Krankheitsaktivität oder ihren erscheinungsfreien Hautstatus verloren hatten, erreichte nach Rückkehr zu Olumiant 4 mg die Krankheitskontrolle wieder.
Lebensqualität/Patientenberichtete Ergebnisse
Während des 16-wöchigen placebokontrollierten Zeitraums zeigten Patienten, die mit 2 mg und 4 mg Baricitinib behandelt wurden, Verbesserungen bei Juckreiz, Schlaf (ADSS-Scores), Hautschmerzen und Lebensqualität (DLQI-Scores) im Vergleich zu Patienten, die mit Placebo randomisiert wurden.
Alopecia areata
Die Wirksamkeit und Sicherheit von Baricitinib einmal täglich wurde in einer adaptiven Phase-II/III-Studie (BRAVE-AA1) und einer Phase-III-Studie (BRAVE-AA2) untersucht. Beide waren randomisierte, doppelblinde, placebokontrollierte Studien über 36 Wochen mit Verlängerungsphasen bis zu 200 Wochen. In beiden Studien wurden die Patienten auf Placebo, 2 mg oder 4 mg Baricitinib im Verhältnis 2:2:3 randomisiert. Teilnahmeberechtigt waren Erwachsene im Alter zwischen 18 und 60 Jahren bei männlichen Patienten und zwischen 18 und 70 Jahren bei weiblichen Patienten mit einer aktuellen Episode von mehr als 6 Monaten schwerer Alopecia areata (Haarausfall, der ≥50 % der Kopfhaut umfasst). Patienten mit einer aktuellen Episode von mehr als 8 Jahren waren nicht teilnahmeberechtigt, es sei denn, es wurden in den letzten 8 Jahren Episoden von erneutem Wachstum der Haare auf den betroffenen Bereichen der Kopfhaut beobachtet. Patienten mit chronischer viraler Hepatitis und/oder aktiven Infektionen waren von den Studien ausgeschlossen.
Die einzigen erlaubten begleitenden AA-Therapien waren Finasterid (oder andere 5-alpha-Reduktase-Hemmer), orales oder topisches Minoxidil und ophthalmische Bimatoprost-Lösung für die Wimpern, wenn bei Studienbeginn eine stabile Dosis vorhanden war.
Primärer Endpunkt beider Studien war der Anteil der Probanden, die in Woche 36 einen SALT-Score ≤20 (mindestens 80 % der Kopfhaut behaart) erreichten. Zusätzlich bewerteten beide Studien den durch Patienten mit einer 5-Punkte-Skala beurteilten Haarausfall an der Kopfhaut (Scalp Hair Assessment PRO™) und den durch den Studienarzt mit einer 4-Punkte-Skala (ClinRO Measure for Eyebrow Hair Loss™, ClinRO Measure for Eyelash Hair Loss™) beurteilten Haarausfall an Augenbrauen und Wimpern.
Ausgangs-Charakteristika
Der Phase-III-Teil der BRAVE-AA1-Studie und die Phase-III-Studie BRAVE-AA2 umfassten 1200 erwachsene Patienten. Über alle Behandlungsgruppen hinweg lag das mittlere Alter bei 37.5 Jahren, 61 % der Patienten waren Frauen. Die mittlere Dauer der AA ab Beginn und die mittlere Dauer der aktuellen Episode von Haarausfall betrugen 12.2 bzw. 3.9 Jahre. Der mediane SALT-Score über die Studien hinweg betrug 96, und bei etwa 44 % der Patienten bestand eine AA universalis. Über die Studien hinweg wiesen 69 % der Patienten zu Studienbeginn einen relevanten oder vollständigen Haarausfall der Augenbrauen auf, und 58 % einen relevanten oder vollständigen Haarausfall der Wimpern, gemessen anhand eines ClinRO Measures for Eyebrow and Eyelash-Scores von 2 oder 3. Ungefähr 90 % der Patienten hatten vor Studienbeginn mindestens eine AA-Behandlung erhalten und 50 % mindestens ein systemisches Immunsuppressivum. Die Anwendung zugelassener begleitender AA-Behandlungen während der Studien wurde nur von 4.3 % der Patienten berichtet.
Klinisches Ansprechen
In beiden Studien erreichte ein signifikant grösserer Anteil der Patienten, die auf Baricitinib 4 mg einmal täglich randomisiert wurden, einen SALT ≤20 in Woche 36 im Vergleich zu Placebo, dies begann bereits in Woche 8 der Studie BRAVE-AA1 und Woche 12 der Studie BRAVE-AA2. Eine übereinstimmende Wirksamkeit zeigte sich über einige der wichtigsten sekundären Endpunkte hinweg (Tabelle 8). Abbildung 7 zeigt den Anteil der Patienten, die SALT ≤20 vom Ausgangswert bis Woche 36 erreichten.
Die Behandlungseffekte in den Subgruppen (Geschlecht, Alter, Gewicht, geschätzte glomeruläre Filtrationsrate (eGFR), Abstammung, geografische Region, Schwere der Erkrankung, Dauer der aktuellen AA-Episode) stimmten in Woche 36 mit den Ergebnissen in der gesamten Studienpopulation überein.
Tabelle 8. Wirksamkeit von Baricitinib bis Woche 36 (FASa)
|
Studie
|
BRAVE-AA1
|
BRAVE-AA2
| |
Behandlungsgruppe
|
PBO
N=189
|
BARI 2 mg
N=184
|
BARI 4 mg
N=281
|
PBO
N=156
|
BARI 2 mg
N=156
|
BARI 4 mg
N=234
| |
SALT ≤20 in Woche 36
|
5.3%
|
21.7%**
|
35.2%**
|
2.6%
|
17.3%**
|
32.5%**
| |
SALT ≤20in Woche 24
|
4.8%
|
11.4%**
|
26.7%**
|
1.3%
|
10.9%**
|
28.2%**
| |
Scalp Hair Assessment PRO 0 oder 1 in Woche 36 mit ≥2 Punkte Verbesserung gegenüber dem Ausgangswert b
|
5.0%
|
16.0%**
|
33.1%**
|
4.0%
|
16.1%**
|
34.4%**
| |
ClinRO Measure for Eyebrow Hair Loss 0 oder 1 in Woche 36 mit ≥2 Punkte Verbesserung gegenüber dem Ausgangswert c
|
3.2%
|
19.1%**
|
31.4%**
|
4.5%
|
11.5%*
|
34.8%**
| |
ClinRO Measure for Eyelash Hair Loss 0 oder 1 in Woche 36 mit ≥2 Punkte Verbesserung gegenüber dem Ausgangswert d
|
3.1%
|
13.5%*
|
33.5%**
|
5.6%
|
10.1%
|
34.3%**
|
BARI = Baricitinib; PBO = Placebo
* statistisch signifikant vs. Placebo ohne Anpassung für Multiplizität; ** statistisch signifikant im Vergleich zu Placebo mit Anpassung für Multiplizität.
a Full Analysis Set (FAS, Gesamtpopulation), umfasst alle randomisierten Patienten.
b 0 = Kein Haarausfall, 1 = Haarausfall in einem begrenzten Bereich der Kopfhaut (1% bis 20%). Bei Patienten mit einem Scalp Hair PRO-Score ≥3 zu Studienbeginn (n=181, 175 bzw. 275 für BRAVE-AA1 und n=151, 149 bzw. 215 für BRAVE-AA2)
c 0 = vollständig abdeckende Augenbrauen ohne Bereiche mit Haarausfall, 1 = Minimale Lücken in den Augenbrauenhaaren bei gleichmässiger Verteilung. Bei Patienten mit ClinRO Measure for Eyebrow Hair loss Score ≥2 zu Studienbeginn (n=124, 136 bzw. 188 für BRAVE-AA1 und n = 112, 104 bzw. 161 für BRAVE-AA2)
d 0 = Wimpern bilden eine durchgehende Linie entlang der Augenlider beider Augen, 1 = Es gibt minimale Lücken, die Wimpern stehen in gleichmässigen Abständen entlang der Augenlider beider Augen. Bei Patienten mit ClinRO Measure for Eyelash Hair loss Score ≥2 zu Studienbeginn (n=96, 111 bzw. 167 für BRAVE-AA1 und n=90, 89 bzw. 140 für BRAVE-AA2)
Abbildung 7. Anteil der Patienten mit SALT ≤20
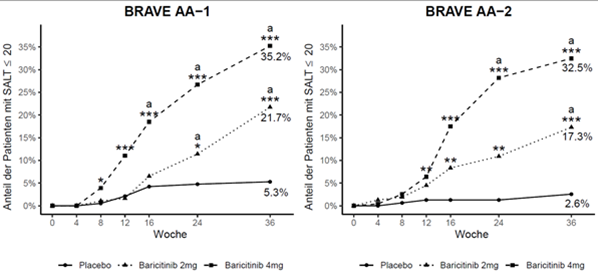
aStatistisch signifikant nach Adjustierung für Multiplizität.
* p-Wert für Baricitinib versus Placebo ≤0.05; **p- Wert für Baricitinib versus Placebo ≤0.01; ***p- Wert für Baricitinib versus Placebo ≤0.001.
Wirksamkeit bis Woche 52
Der Anteil der mit Baricitinib behandelten Patienten, die einen SALT ≤20 erreichten, stieg nach Woche 36 weiter an und erreichte 39.0 % der Patienten unter Baricitinib 4 mg in Woche 52. Die Ergebnisse für die Subpopulationen bezüglich Schweregrad der Erkrankung zu Studienbeginn und Episodendauer in Woche 52 stimmten mit denen überein, die in Woche 36 beobachtet wurden, und mit den Ergebnissen in der gesamten Studienpopulation.
Dosisreduktion
In der Studie BRAVE-AA2 wurden Patienten, die seit der initialen Randomisierung einmal täglich 4 mg Baricitinib erhalten hatten und in Woche 52 SALT ≤20 erreichten doppelblind re-randomisiert, um die Therapie mit 4 mg einmal täglich fortzusetzen oder die Dosis auf 2 mg einmal täglich zu reduzieren. Die Ergebnisse zeigen, dass 96 % der Patienten, die weiterhin Baricitinib 4 mg erhielten, und 74 % der Patienten, die auf Baricitinib 2 mg re-randomisiert wurden, ihr Ansprechen in Woche 76 aufrechterhielten.
COVID-19
In einer randomisierten, doppelblinden, Placebo-kontrollierten klinischen Studie (ACTT-2) wurde Baricitinib 4 mg einmal täglich + Remdesivir im Vergleich zu Placebo + Remdesivir in 1033 hospitalisierten erwachsenen Patienten mit COVID-19 untersucht.
Die folgende 8-Punkte-Ordnungsskala (Ordinal Score, OS) des NIAID (National Institute of Allergy and Infectious Diseases) wurde verwendet, um den Schweregrad der Grunderkrankung zu klassifizieren:
8. Tod;
7. Hospitalisiert, mit invasiver mechanischer Beatmung oder ECMO (Extrakorporaler Membranoxygenierung);
6. Hospitalisiert, mit nicht-invasiver Beatmung or «High-Flow» Sauerstofftherapie;
5. Hospitalisiert, mit «Low-Flow» Sauerstoffsupplementation;
4. Hospitalisiert, ohne «Low-Flow» Sauerstoffsupplementation, aber mit medizinischer Behandlung (COVID-19 bezogen oder anderweitig);
3. Hospitalisiert, ohne «Low-Flow» Sauerstoffsupplementation - nicht länger einer medizinischen Behandlung bedürfend;
2. Nicht hospitalisiert, mit Aktivitätseinschränkungen und/oder Sauerstoffsupplementation zu Hause;
1. Nicht hospitalisiert, ohne Aktivitätseinschränkungen.
Die Studie umfasste 14% Patienten mit OS 4, 55% Patienten mit OS 5, 21% Patienten mit OS 6 und 11 % Patienten mit OS 7.
Der primäre klinische Endpunkt war die Zeit bis zur Genesung innerhalb von 29 Tagen nach der Randomisierung, wobei diese als Erreichen der OS Kategorie 1, 2 oder 3 definiert war.
Die Patienten wurden 1:1 randomisiert, nach dem Schweregrad der Erkrankung bei Aufnahme in die Studie stratifiziert, einer Behandlung mit Baricitinib + Remdesivir (n=515) oder Placebo + Remdesivir (n=518) zugewiesen. Die Patienten wurden nach folgendem Schema behandelt:
·Baricitinib 4 mg oder Placebo einmal täglich (oral) über 14 Tage oder bis zur Entlassung aus dem Krankenhaus
·Remdesivir 200 mg am Tag 1, gefolgt von 100 mg einmal täglich (mittels intravenöser Infusion) an den darauffolgenden Tagen über eine gesamte Behandlungsdauer von 10 Tagen oder bis zur Entlassung aus dem Krankenhaus.
Das mittlere Alter der Patienten bei Studienbeginn lag bei 55 Jahren, wobei 30 % der Patienten 65 Jahre oder älter waren. 63 % der Patienten waren männlich, 48 % waren kaukasisch, 15 % waren schwarz und 10 % waren asiatisch. 83% der Patienten wiesen Komorbiditäten auf, wobei Adipositas (56 %), Bluthochdruck (52 %) und Typ-2-Diabetes (37 %) die häufigsten Komorbiditäten waren. Demografische und krankheitsbezogene Merkmale waren in der Kombinationsgruppe und der Placebogruppe ausgewogen.
Was die Gesamtpopulation (ITT) betrifft, so betrug die mediane Zeit bis zur Genesung 7 Tage bei Baricitinib + Remdesivir, im Vergleich zu 8 Tagen bei Placebo + Remdesivir [Hazard Ratio: 1.15 (95 % KI 1.00, 1.31); p=0.047]. Am ausgeprägtesten war der klinische Nutzen von Baricitinib + Remdesivir bei Patienten, die eine «Low-Flow»-Sauerstoffsupplementation (OS 5), eine nichtinvasive Beatmung oder eine «High-Flow»-Sauerstofftherapie (OS 6) benötigten (siehe Tabelle 9). Bei Patienten, die eine invasive mechanische Beatmung oder ECMO (OS 7) benötigten, war die mediane Zeit bis zur Genesung nicht einschätzbar. Bei nicht sauerstoffpflichtigen Patienten (OS 4) gab es keinen offensichtlichen Vorteil bei der medianen Zeit bis zur Genesung mit Baricitinib + Remdesivir (5 Tage) im Vergleich zu Remdesivir (4 Tage) (Recovery Rate Ratio 0.88 [95 % KI 0.62–1.23]).
Tabelle 9. Genesungs-Ergebnisse nach ordinalem Score bei Studienbeginn für sauerstoffpflichtige Patienten – Studie ACTT-2a
|
|
Ordinaler Score bei Studienbeginn – ITT Population
| |
|
5
|
6
|
7
| |
|
«Low-Flow»- Sauerstoff
|
Nichtinvasive Beatmung oder «High-Flow»-Sauerstoff
|
Invasive mechanische Beatmung/ECMO
| |
|
BARI
+ RDV
(n=288)
|
PBO
+ RDV
(n=276)
|
BARI
+ RDV
(n=103)
|
PBO
+ RDV
(n=113)
|
BARI
+ RDV
(n=54)
|
PBO
+ RDV
(n=57)
| |
Anzahl der Genesenen
|
262
|
243
|
82
|
73
|
22
|
21
| |
Mediane Zeit bis zur Genesung
Tage (95 % KI)
|
5
(5, 6)
|
6
(5, 6)
|
10
(9, 13)
|
18
(13, 21)
|
NE
(25, NE)
|
NE
(26, NE)
| |
Recovery Rate Ratio
Tagea (95 % KI)
|
1.17
(0.98, 1.39)
|
1.51
(1.10, 2.08)
|
1.08
(0.59, 1.97)
|
ECMO = Extracorpora Membranoxygenation, ITT = Intention to Treat, RDV = Remdesivir, PBO = placebo, BARI= baricitinib
a Recovery Rate Ratio berechnet nach dem stratifizierten Cox-Modell. Eine Recovery Rate Ratio >1 zeigt einen Vorteil für Baricitinib + Remdesivir an. NE = nicht einschätzbar
Die Mortalität am Tag 29 in der Gesamtpopulation betrug 4.7 % (n=24/515) in der Baricitinib/Remdesivir-Gruppe im Vergleich zu 7.1 % (n=37/518) in der Placebo/Remdesivir-Gruppe (Hazard Ratio: 0.65; [95 % KI 0.39 bis 1.09]; p=0.102). Der klinische Nutzen von Baricitinib war am ausgeprägtesten bei Patienten, die eine «Low-Flow»-Sauerstoffsupplementation, eine nichtinvasive Beatmung oder eine «High-Flow»-Sauerstofftherapie benötigten (siehe Tabelle 10):
Tabelle 10 . Mortalitätsergebnisse am Tag 29 nach ordinalem Score bei Studienbeginn für sauerstoffpflichtige Patienten– Studie ACTT-2
|
|
Ordinaler Score bei Studienbeginn – ITT Populationc
| |
|
5
|
6
|
7
| |
|
«Low-Flow»-Sauerstoff
|
Nichtinvasive Beatmung
oder «High-Flow»-Sauerstoff
|
Invasive mechanische Beatmung/ECMO
| |
|
BARI
+ RDV
(n=288)
|
PBO
+ RDV
(n=276)
|
BARI
+ RDV
(n= 103)
|
PBO
+ RDV
(n=113)
|
BARI
+ RDV
(n=54)
|
PBO
+ RDV
(n=57)
| |
Mortalität am Tag 29 – N (%)a
|
5
(1.9%)
|
12
(4.7%)
|
7
(7.5%)
|
13
(13.0%)
|
12 (23.1%)
|
12
(22.6%)
| |
Hazard Ratiob
(95 % KI)
|
0.4
(0.14, 1.14)
|
0.55
(0.22, 1.38)
|
1.00 (0.45, 2.22)
|
a Die Prozentangaben basieren auf der Kaplan-Meier-Methode.
b Die Hazard Ratios für die Subgruppen der ordinalen Skala bei Studienbeginn stammen aus unstratifizierten Cox-Proportional-Hazard-Modellen.
c ITT = Intention-To-Treat Population = alle randomisierten Patienten.
Subgruppenanalysen zeigten für Patienten unter 40 Jahren und für Patienten ohne Komorbiditäten eine längere Zeit bis zur Genesung, wenn sie mit Baricitinib+Remdesivir statt mit Remdesivir allein behandelt waren, aber ihre Genesungschance (Recovery Rate Ratio) lag etwas über der bei alleiniger Remdesivirbehandlung:
·Patienten unter 40 Jahren (n=162/1033): 6 versus 5 Tage; Recovery Rate Ratio (95 % KI): 1.01 (0.74-1.38)
·Patienten ohne Komorbiditäten (n=138/1033): 7 versus 6 Tage; Recovery Rate Ratio (95 % KI): 1.11 (0.79-1.55)
Subgruppenanalysen zeigten für weibliche Patienten und für nicht adipöse Patienten keinen Unterschied in der Zeit bis zur Genesung, wenn sie mit Baricitinib+Remdesivir statt mit Remdesivir allein behandelt waren, aber ihre Genesungschance (Recovery Rate Ratio) lag etwas über der bei alleiniger Remdesivirbehandlung:
·Weibliche Patienten (n=316/1033): 7 versus 7 Tage; Recovery Rate Ratio (95 % KI): 1.06 (0.85-1.32)
·Nicht-adipöse Patienten (n=362/1033): 9 versus 9 Tage; Recovery Rate Ratio (95 % KI): 1.07 (0.87-1.32)
|