ZusammensetzungWirkstoffe
Patiromer (als Patiromer Sorbitol Calcium (Patiromer Sorbitex Calcium)).
Hilfsstoffe
Xanthangummi.
Indikationen/AnwendungsmöglichkeitenVeltassa ist für die Behandlung einer Hyperkaliämie bei Erwachsenen indiziert.
Dosierung/AnwendungTherapieeinleitung
Die empfohlene Anfangsdosis Veltassa liegt bei mindestens 8,4 g Patiromer einmal täglich.
Im Allgemeinen sollte der zeitliche Abstand zwischen der Einnahme von Veltassa und anderen oral einzunehmenden Arzneimitteln 3 Stunden betragen. Bei bestimmten Arzneimitteln ist kein Abstand erforderlich (siehe Abschnitt Interaktionen).
Aufgrund des verzögerten Wirkungseintritts sollte Veltassa keine Notfallbehandlung einer lebensbedrohlichen Hyperkaliämie ersetzen.
Dosisanpassung und Erhaltungstherapie
Die Tagesdosis Veltassa kann je nach Serumkalium und gewünschtem Zielbereich angepasst werden. Die Tagesdosis kann in wöchentlichen Intervallen um jeweils 8,4 g erhöht oder gesenkt werden, je nachdem was zum Erreichen des Zielbereichs erforderlich ist. Die maximale Dosis beträgt 25,2 g täglich. Um die gewünschte Dosis zu erreichen, können mehrere Beutel verwendet werden. Wenn das Serumkalium unter den gewünschten Zielbereich fällt, sollte die Dosis Veltassa reduziert oder die Behandlung abgebrochen werden.
Wenn die Einnahme einer Dosis Veltassa vergessen wird, sollte diese Dosis möglichst bald noch am selben Tag eingenommen werden. Die vergessene Dosis sollte nicht mit der nächsten Dosis zusammen eingenommen werden.
Spezielle Dosierungsanweisungen
Dialysepatienten
Es liegen nur begrenzte Daten zur Anwendung von Veltassa bei Dialysepatienten vor. In klinischen Studien wurden keine speziellen Richtlinien für die Dosierung und Verabreichung bei diesen sechs Patienten angewandt.
Ältere Patienten
Von der Gesamtanzahl an Patienten, die im Rahmen von klinischen Studien mit Veltassa behandelt wurden, waren 1307 (61,2 %) Patienten 65 Jahre und älter, wovon 500 (23,4 %) 75 Jahre und älter waren. Im Rahmen dieser Studien wurden keine speziellen Richtlinien für die Dosierung bei diesen Patienten angewandt.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Veltassa bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Zum Einnehmen. Veltassa sollte folgendermassen mit Wasser oder anderen Flüssigkeiten oder den unten aufgeführten weichen Nahrungsmitteln vermischt und zu einer Suspension einheitlicher Konsistenz verrührt werden:
Die komplette Dosis Veltassa sollte zunächst in ein Glas mit ca. 40 ml Wasser (3 Esslöffel) gegeben und verrührt werden. Danach weitere ca. 40 ml Wasser hinzufügen und die Suspension erneut gründlich verrühren. Das Pulver löst sich nicht auf. Zum Erreichen der gewünschten Konsistenz kann bei Bedarf noch mehr Wasser hinzugefügt werden.
Die Mischung sollte sofort eingenommen werden. Wenn nach der Einnahme Pulverrückstände im Glas verbleiben, Wasser nachfüllen, umrühren und sofort austrinken. Dieser Vorgang kann so oft wiederholt werden, bis die gesamte Dosis eingenommen wurde.
Die folgenden Flüssigkeiten oder weichen Nahrungsmittel können anstelle von Wasser zum Herstellen der Mischung nach denselben, oben beschriebenen Schritten verwendet werden: Apfelsaft, Cranberrysaft, Ananassaft, Orangensaft, Traubensaft, Birnensaft, Aprikosennektar, Pfirsichnektar, Joghurt, Milch, Verdickungsmittel, Apfelmus, Vanille- und Schokoladenpudding.
Der Kaliumgehalt der für die Herstellung der Mischung verwendeten Flüssigkeiten oder weichen Nahrungsmittel sollte als Teil der für jeden Patienten individuell empfohlenen Kaliumzufuhr mit der Nahrung berücksichtigt werden.
Veltassa kann zu einer Mahlzeit oder unabhängig davon eingenommen werden. Veltassa sollte nicht (z.B. in der Mikrowelle) erhitzt oder mit erhitzten Lebensmitteln oder Flüssigkeiten vermischt werden. Veltassa sollte nicht in trockenem Zustand eingenommen werden.
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenNiedrige Magnesiumwerte
Patiromer kann zu niedrigen Magnesiumwerten führen, da es Magnesium im Darm bindet. In klinischen Studien wiesen 7,1 % der mit Veltassa behandelten Patienten Magnesiumwerte < 1,4 mg/dl auf. 0,3 % der Patienten entwickelten ein Serummagnesium < 1,0 mg/dl. Der mittlere Abfall des Serummagnesiums trat zu Beginn der Behandlung mit Veltassa auf und betrug ≤0,118 mg/dl eine Woche nach der Einleitung der Behandlung mit Veltassa. Das Serummagnesium sollte nach Einleitung der Behandlung mit Veltassa einen Monat lang überwacht werden und die Überwachung sollte im Falle eines Abfalls des Serummagnesiumspiegels fortgesetzt werden. Die Gabe eines Magnesiumpräparats sollte bei Patienten, die während der Behandlung mit Veltassa ein niedriges Serummagnesium entwickeln, in Erwägung gezogen werden.
Erkrankungen des Gastrointestinaltrakts
Patienten mit Darmverschluss oder einer grösseren gastrointestinalen Operation, diabetischen Gastroparese, schweren Erkrankungen des Gastrointestinaltrakts oder Schluckbeschwerden in der Anamnese wurden nicht in die klinischen Studien aufgenommen. Diese Patienten sollten nicht mit Veltassa behandelt werden.
Absetzen von Veltassa
Wird die Einnahme von Veltassa abgebrochen, kann das Serumkalium wieder ansteigen, vor allem wenn weiterhin eine Behandlung mit RAAS-Hemmern erfolgt. Die Patienten müssen angewiesen werden, die Therapie nicht ohne vorherige Absprache mit ihrem Arzt abzubrechen. Ein Anstieg des Serumkaliums kann bereits 2 Tage nach der letzten Veltassa-Dosis eintreten.
Serumkaliumspiegel
Das Serumkalium sollte bei klinischer Indikation überwacht werden, u.a. wenn Veränderungen bei Arzneimitteln vorgenommen werden, die einen Einfluss auf die Serumkaliumkonzentration haben (z.B. RAAS-Hemmer oder Diuretika), und nach Anpassung der Veltassa-Dosierung.
Sorbitol
Veltassa enthält Sorbitol als Bestandteil des Gegenionkomplexes. Der Sorbitolgehalt entspricht ca. 4 g (10,4 kcal) pro 8,4 g Patiromer. Patienten mit einer hereditären Fruktoseintoleranz (HFI) dürfen dieses Arzneimittel nicht einnehmen / erhalten.
Fluor
Veltassa enthält Fluor. Bei Patienten mit schwerer chronischer Niereninsuffizienz sollte jegliche Zufuhr von Fluor eingeschränkt werden.
Informationen zu Calcium
Veltassa enthält Calcium als Bestandteil des Gegenionkomplexes. Das Calcium wird teilweise freigesetzt und ein Teil davon möglicherweise resorbiert. Im Rahmen von klinischen Studien mit einer Laufzeit von bis zu einem Jahr wurde jedoch keine Veränderung des Serumcalciumspiegels beobachtet. Bei Risikopatienten sollte der Arzt Nutzen und Risiken einer Verabreichung dieses Arzneimittels sorgfältig abwägen.
InteraktionenVeltassa besitzt die Eigenschaft, einige oral verabreichte Arzneimittel zu binden, wodurch sich deren gastrointestinale Resorption vermindern könnte. Da Patiromer nicht vom Körper resorbiert oder metabolisiert wird, sind die Auswirkungen auf die Wirkungsweise anderer Arzneimittel begrenzt.
Tabelle 1 zeigt die auf Interaktionen mit Veltassa getesteten Arzneimittel und die Empfehlungen für die Verabreichung dieser Arzneimittel zusammen mit Veltassa. Aus Gründen der Vorsicht sollte die Einnahme von Patiromer im Abstand von mindestens 3 Stunden zu den nicht aufgelisteten, oral verabreichten Arzneimitteln erfolgen.
Tabelle 1: Empfehlungen für die Verabreichung von auf Interaktionen mit Veltassa getesteten Arzneimitteln
|
Arzneimittel
|
Klinische Empfehlung
|
Grundlage für die Empfehlung
| |
Angiotensinkonversionsenzymhemmer (ACE)
| |
Benazepril, Captopril, Enalapril, Fosinopril, Lisinopril, Perindopril, Quinapril, Ramipril, Trandolapril
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Antiotensin-II-Rezeptorblocker (ARB)
| |
Telmisartan
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vitro: Bindung beobachtet
| |
Azilsartan, Candesartan, Irbesartan, Losartan, Olmesartan, Valsartan
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
β-Adrenozeptorblocker (Betablocker)
| |
Bisoprolol, Carvedilol, Nebivolol
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vitro: Bindung beobachtet
| |
Metoprolol
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Schleifendiuretika
| |
Furosemid
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Bumetanid, Torasemid
|
In vitro: keine Interaktion beobachtet
| |
Mineralokortikoid-Rezeptorantagonisten (MRA)
| |
Eplerenon, Finerenon, Spironolacton
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Neprilysin-Inhibitoren
| |
Sacubitril
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Natrium-Glucose-Cotransporter-2 (SGLT-2)-Inhibitoren
| |
Canagliflozin, Dapagliflozin, Empagliflozin
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Antibiotika
| |
Ciprofloxacin
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vivo: Interaktion beobachtet, jedoch nicht bei 3-stündigem Abstand
| |
Trimethoprim
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Amoxicillin, Cephalexin
|
In vitro: keine Interaktion beobachtet
| |
Antikoagulatien
| |
Warfarin
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Apixaban, Rivaroxaban
|
In vitro: keine Interaktion beobachtet
| |
Arzneimittel zur Senkung des PTH-Werts und Schilddrüsenpräparate
| |
Levothyroxin
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vivo: Interaktion beobachtet, jedoch nicht bei 3-stündigem Abstand
| |
Cinacalcet
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Antithrombotika
| |
Clopidogrel
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Acetylsalicylsäure
|
In vitro: keine Interaktion beobachtet
| |
Antidiabetika
| |
Metformin
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vivo: Interaktion beobachtet, jedoch nicht bei 3-stündigem Abstand
| |
Glipizid
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Calciumkanalblocker
| |
Amlodipin, Verapamil
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Immunsuppressiva
| |
Mycophenolat-Mofetil
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vitro: Bindung beobachtet
| |
Tacrolimus
|
Kein Abstand zu Veltassa erforderlich
|
In vitro: keine Interaktion beobachtet
| |
Andere
| |
Chinidin
|
Anwendung mit mindestens 3-stündigem Abstand zu Veltassa
|
In vitro: Bindung beobachtet
| |
Lithium
|
Kein Abstand zu Veltassa erforderlich
|
In vivo: keine Interkation beobachtet
| |
Allopurinol, Atorvastatin, Digoxin, Phenytoin, Riboflavin, Sevelamer
|
In vitro: keine Interaktion beobachtet
|
Wirkung von Nahrungsmitteln auf Veltassa
Veltassa kann zu einer Mahlzeit oder unabhängig davon eingenommen werden. In einer Open-Label-Studie wurden 114 Patienten mit Hyperkaliämie randomisiert und nahmen Veltassa einmal täglich zu einer Mahlzeit oder unabhängig davon ein. Das Serumkalium am Ende der Behandlung, die Differenz zum Baseline-Serumkalium und die mittlere Veltassa-Dosis waren in beiden Gruppen vergleichbar.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen bisher keine klinischen Daten für die Anwendung von Veltassa bei Schwangeren vor. Tierexperimentelle Studien geben keine Hinweise auf eine direkte oder indirekte Toxizität mit Auswirkung auf Schwangerschaft, Embryonalentwicklung, Entwicklung des Fötus und/oder postnatale Entwicklung.
Aus Gründen der Vorsicht sollte die Anwendung von Veltassa während der Schwangerschaft unterbleiben.
Stillzeit
Es werden keine Auswirkungen auf gestillte Neugeborene/Säuglinge erwartet, da die systemische Exposition der stillenden Mutter gegenüber Patiromer vernachlässigbar ist. Bei der Entscheidung, ob abgestillt oder die Patiromer-Therapie beendet/abgesetzt werden sollte, ist der Nutzen des Stillens für das Kind gegen den Nutzen der Therapie für die Frau abzuwägen.
Fertilität
Es liegen keine Daten zu den Auswirkungen von Veltassa auf die Fertilität beim Menschen vor. Tierexperimentelle Studien zeigten keine Auswirkungen auf die Fortpflanzungsfähigkeit oder Fertilität (siehe Abschnitt Präklinische Daten).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenVeltassa hat keinen oder einen zu vernachlässigenden Einfluss auf die Fahrtüchtigkeit und das Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Das aktuelle Sicherheitsprofil von Veltassa basiert auf den Daten von 2135 Patienten, die an klinischen Studien teilnahmen, sowie auf Erfahrungen nach der Markteinführung.
Bei einem Grossteil der gemeldeten unerwünschten Wirkungen (UWs) handelte es sich um Hypomagnesiämie und gastrointestinale Störungen, wobei am häufigsten von Obstipation, Diarrhö, Abdominalschmerzen, Übelkeit, Flatulenz, Erbrechen und Überempfindlichkeit berichtet wurde. Die gastrointestinalen Störungen waren im Allgemeinen von leichter bis mittelschwerer Ausprägung, schienen dosisunabhängig zu sein, verschwanden in der Regel spontan oder nach Behandlung und wurden in keinem Fall als schwerwiegend bezeichnet. Zu den Überempfindlichkeitsreaktionen gehörten Hautausschlag, Urtikaria, Schwellungen in der Mundhöhle und an den Lippen und waren leicht bis mittelschwer.
Hinweis: Diarrhö ist ein Sammelbegriff für Durchfall und häufigen Darmentleerungen. Abdominalschmerz ist ein Sammelbegriff, der die bevorzugten Begriffe abdominale Beschwerden, Abdominalschmerz und abdominaler Oberbauchschmerz umfasst. Obstipation ist ein Sammelbegriff, der die bevorzugten Begriffe Obstipation und harter Stuhl zusammenfasst.
Liste der unerwünschten Wirkungen
Die unerwünschten Wirkungen sind nach MedDRA-Systemorganklassen und Häufigkeit gemäss folgender Konvention geordnet:
«sehr häufig» (≥1/10)
«häufig» (≥1/100, <1/10),
«gelegentlich» (≥1/1'000, <1/100)
«selten» (≥1/10'000, <1/1'000)
«sehr selten» (<1/10'000)
«nicht bekannt» (kann aus den verfügbaren Daten nicht abgeschätzt werden)
Erkrankungen des Immunsystems
Nicht bekannt: Überempfindlichkeit.
Stoffwechsel- und Ernährungsstörungen
Häufig: Hypomagnesiämie.
Erkrankungen des Gastrointestinaltrakts
Häufig: Obstipation, Diarrhö, Abdominalschmerzen, Übelkeit, Flatulenz.
Gelegentlich: Erbrechen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEine Dosis Veltassa von mehr als 50,4 g Patiromer pro Tag wurde nicht untersucht. Da eine übermässige Dosierung von Veltassa zu einer Hypokaliämie führen kann, sollte der Serumkaliumspiegel überwacht werden. Wenn festgestellt wird, dass eine medizinische Intervention erforderlich ist, können angemessene Massnahmen zur Wiederherstellung eines normalen Serumkaliums erwogen werden.
Eigenschaften/WirkungenATC-Code
V03AE09
Pharmakotherapeutische Gruppe: Behandlung von Hyperkaliämie.
Wirkungsmechanismus
Veltassa ist ein nicht resorbierbares Kationenaustauschpolymer, das einen Calciumsorbitolkomplex als Gegenion enthält.
Veltassa erhöht die fäkale Kaliumausscheidung durch die Bindung von Kalium im Lumen des Gastrointestinaltrakts. Die Kaliumbindung lässt die Konzentration des freien Kaliums im Gastrointestinallumen absinken und führt so zu einer Verringerung des Serumkaliums.
Pharmakodynamik
Veltassa bindet Kalium nachweislich in vitro und in vivo in Tiermodellen.
In einer Studie der Phase I mit gesunden freiwilligen Erwachsenen (6 bis 8 Probanden pro Gruppe) hat Veltassa (0 g bis 50,4 g Patiromer täglich) bei dreimal täglicher Gabe innerhalb von 8 Stunden zu einem dosisabhängigen Anstieg der fäkalen Ausscheidung von Kalium geführt. Eine entsprechende dosisabhängige Verringerung der Kaliumausscheidung im Urin ohne Veränderung des Serumkaliums konnte ebenfalls beobachtet werden. Im Vergleich zum Placebo senkte Veltassa bei einer Dosis von 25,2 g bzw. 50,4 g täglich signifikant die durchschnittliche tägliche Kaliumausscheidung im Urin.
In einer offenen Cross-over-Studie der Phase I mit wiederholter Dosis bei 12 gesunden Freiwilligen wurde die Tagesdosis von 25,2 g Patiromer entsprechend der randomisierten Zuordnung zu einem Dosierungsschema einmal täglich, zweimal täglich oder dreimal täglich über einen Zeitraum von 6 Tagen oral verabreicht. Bei diesen drei Dosierungsschemata wurde ein signifikanter Anstieg der durchschnittlichen täglichen fäkalen Kaliumausscheidung und gleichzeitig eine Verringerung der durchschnittlichen täglichen Kaliumausscheidung im Urin beobachtet. Für den Bereich dieser drei Dosierungsschemata lag der mittlere Anstieg der fäkalen Kaliumausscheidung zwischen 1.283 mg/Tag und 1.550 mg/Tag und die mittlere Verringerung der Kaliumausscheidung im Urin zwischen 1.438 mg/Tag und 1.534 mg/Tag. Es wurde kein signifikanter Unterschied zwischen diesen Dosierungsschemata in Bezug auf die durchschnittliche tägliche Ausscheidung im Stuhl oder im Urin festgestellt, und zwar weder im gesamten noch im paarweisen Vergleich. Die tägliche Calciumausscheidung über den Urin stieg gegenüber der Baseline um 53 mg/Tag, 66 mg/Tag bzw. 73 mg/Tag bei Dosierung gemäss der Schemata einmal täglich, zweimal täglich bzw. dreimal täglich.
In einer nicht kontrollierten Open-Label-Studie haben 25 Patienten mit Hyperkaliämie (durchschnittliches Baseline-Serumkalium 5,9 mEq/l) und einer chronischen Niereninsuffizienz über einen Zeitraum von 3 Tagen eine kontrollierte Kaliumdiät eingehalten und dann 16,8 g Patiromer täglich (auf mehrere Gaben verteilt) innerhalb von 2 Tagen bei fortgesetzter Kaliumdiät erhalten. 7 Stunden nach der ersten Dosis konnte eine statistisch signifikante Verringerung des Serumkaliums (-0,2 mEq/l) beobachtet werden. Während der 48-stündigen Behandlung sanken die Kaliumwerte weiter ab (-0,8 mEq/l, 48 Stunden nach der ersten Dosis). Die Kaliumspiegel blieben nach der letzten Dosis 24 Stunden lang stabil, anschliessend stiegen sie während eines 4-tägigen Beobachtungszeitraums nach Absetzen von Veltassa wieder an.
Abbildung 1: Wirkungseintritt – Mittleres (95%-KI) Serumkalium (mEq/l) im zeitlichen Verlauf
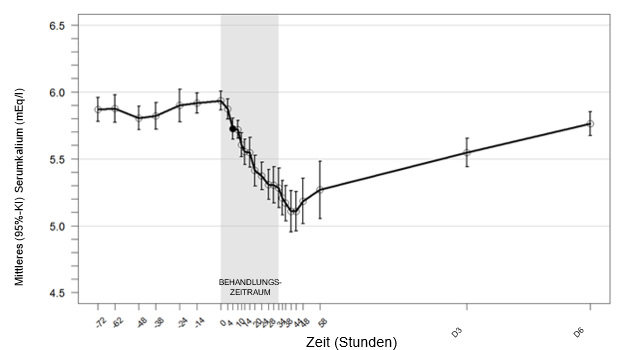
* Der gefüllte Kreis zeigt die Stunde an, in der die erste statistisch signifikante Verringerung identifiziert wurde. Eine durchschnittliche Verringerung um 0,8 mEq/l wurde nach 48 Stunden beobachtet (p < 0,001).
Anmerkung: Stunde -72 bis 0 = Phase vor der Aufnahme in die Studie mit kontrollierter Kaliumdiät; Stunde 0 bis 58 = Phase der stationären Behandlung mit 16,8 g Veltassa täglich, auf mehrere Gaben verteilt; Stunde 58 bis Tag 6 = ambulante Nachsorgephase.
Klinische Wirksamkeit
Die kaliumsenkende Wirkung von Patiromer wurde in 5 klinischen Studien nachgewiesen, an denen Patienten mit Hyperkaliämie oder normokaliämische Patienten teilnahmen, die RAAS-Hemmer erhielten und somit ein erhöhtes Risiko für eine Hyperkaliämie aufwiesen. Die Wirkung von Patiromer bei hyperkaliämischen Patienten mit chronischer Nierenerkrankung, die RAAS-Hemmer erhielten, wurde in einer Einfachblindstudie mit anschliessendem randomisiertem Absetzungsteil untersucht. Es wurde eine offene, 52-wöchige Studie mit hyperkaliämischen Patienten mit chronischer Nierenerkrankung, nicht kontrolliertem Bluthochdruck und Diabetes mellitus Typ 2, die RAAS-Hemmer erhielten, durchgeführt. In zwei randomisierten, doppelblinden, placebokontrollierten Studien wurde Patiromer bei normokaliämischen Patienten mit Herzinsuffizienz oder chronischer Nierenerkrankung und resistentem Bluthochdruck bzw. während gleichzeitiger Einleitung einer Behandlung mit RAAS-Hemmer (Spironolacton) untersucht. In einer doppelblinden, placebokontrollierten Studie wurden Patienten mit Herzinsuffizienz und reduzierter Ejektionsfraktion eingeschlossen, die eine Hyperkaliämie oder eine kürzlich aufgetretene Hyperkaliämie aufwiesen und in der Lage waren, zugelassene Dosen von RAAS-Hemmern anzuwenden und beizubehalten und eine Normokaliämie zu erreichen oder beizubehalten, während sie Patiromer einnahmen. Anschliessend wurde in einem randomisierten Absetzungsverfahren die Wirkung von Patiromer auf den Serumkaliumspiegel untersucht.
Insgesamt nahmen an den 5 Studien 1918 Patienten teil, die mindestens eine Dosis Patiromer erhielten. Davon litten 64,6 % an einer chronischen Nierenerkrankung mit einer eGFR < 60 ml/min/1,73 m2, 54,3 % an Diabetes mellitus und 77,9 % an einer Herzinsuffizienz.
Studie 1 (OPAL-HK)
Zweiteilige, einfachblinde, randomisierte Absetzungsstudie mit Patienten mit chronischer Niereninsuffizienz und Hyperkaliämie, die im Rahmen der Studie mit einer gleichbleibenden Dosierung wenigstens einen RAAS-Hemmer erhielten.
Die Sicherheit und Wirksamkeit von Veltassa wurden in einer zweiteiligen, einfachblinden, randomisierten Absetzungsstudie nachgewiesen, in der diese Behandlung an Patienten mit chronischer Niereninsuffizienz und Hyperkaliämie mit einer gleichbleibenden Dosierung von wenigstens einem RAAS-Hemmer (d.h. Angiotensinkonversionsenzymhemmer [ACE-Hemmer], Angiotensin-II-Rezeptorblocker [ARB] oder Mineralokortikoid-Rezeptorantagonisten [MRA]) untersucht wurde.
In Teil A wurden 243 Patienten 4 Wochen lang mit Veltassa behandelt. Patienten mit einem Baseline-Serumkalium von 5,1 mEq/l bis < 5,5 mEq/l erhielten eine Anfangsdosis Veltassa von 8,4 g Patiromer pro Tag (auf mehrere Gaben verteilt) und Patienten mit einem Baseline-Serumkalium von 5,5 mEq/l bis < 6,5 mEq/l erhielten eine Anfangsdosis von 16,8 g Patiromer pro Tag (auf mehrere Gaben verteilt). Die Dosis Veltassa wurde bedarfsgerecht je nach Kaliumwert angepasst, beginnend an Tag 3 sowie anschliessend an den jeweiligen Wochenterminen (Woche 1, 2 und 3) bis zum Ende des 4-wöchigen Behandlungszeitraums, mit der Vorgabe, das Serumkalium im Zielbereich (3,8 mEq/l bis < 5,1 mEq/l) zu halten.
Das mittlere Alter der Patienten lag bei 64 Jahren, 58 % der Patienten waren Männer und 98 % Weisse. Ca. 97 % der Patienten litten an Bluthochdruck, 57 % an Diabetes mellitus Typ 2 und 42 % an Herzinsuffizienz.
Die mittlere Veltassa-Tagesdosis betrug 13 g bzw. 21 g bei Patienten mit einem Serumkalium von 5,1 mEq/l bis < 5,5 mEq/l bzw. 5,5 mEq/l bis < 6,5 mEq/l.
Das mittlere Baseline-Serumkalium betrug 5,58 mEq/l und die durchschnittliche Veränderung des Serumkaliums zwischen dem Termin zur Aufnahme in die Studie und Woche 4 von Teil A lag bei -1,01 (0,031) mEq/l, 95%-KI: [-1,07; -0,95] (siehe Abbildung 2); diese durchschnittliche Verringerung des Serumkaliums war statistisch signifikant. In Bezug auf den sekundären Endpunkt von Teil A lag das Serumkalium von 76 % der Patienten (95%-KI: 70 %; 81 %) in Woche 4 von Teil A im Zielbereich von 3,8 mEq/L bis < 5,1 mEq/l.
Abbildung 2: Mittleres, geschätztes (95%-KI) Serumkalium (mEq/l), im Zentrallabor bestimmt, im zeitlichen Verlauf
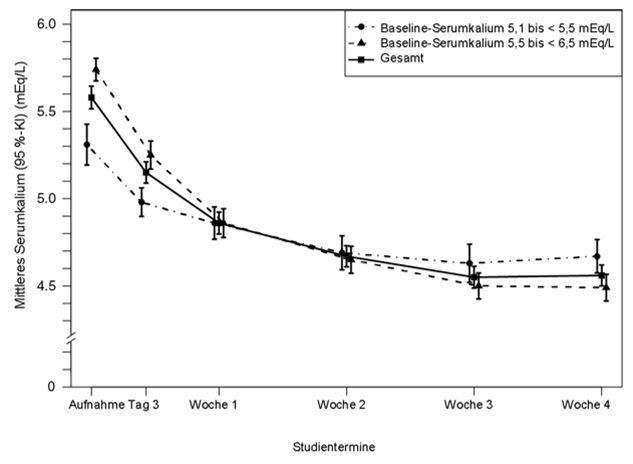
In Teil B wurden 107 Patienten mit einem Baseline-Serumkalium von 5,5 mEq/l bis < 6,5 mEq/l in Teil A, deren Serumkalium in Woche 4 von Teil A im Zielbereich (3,8 mEq/l bis < 5,1 mEq/l) lag und die noch immer wenigstens einen RAAS-Hemmer erhielten, randomisiert und setzten entweder die Veltassa-Behandlung fort oder erhielten über einen Zeitraum von 8 Wochen ein Placebo, um zu untersuchen, wie sich das Absetzen der Veltassa-Behandlung auf das Serumkalium auswirkt. Dabei betrug die mittlere Tagesdosis bei den Patienten der Veltassa-Gruppe zu Beginn von Teil B und im Verlauf von Teil B jeweils 21 g.
Der primäre Endpunkt von Teil B war die Veränderung des Serumkaliums vom Baseline-Wert in Teil B bis zum ersten Termin, an dem das Serumkalium des Patienten zum ersten Mal ausserhalb des Bereichs von 3,8 mEq/l bis < 5,5 mEq/l lag, oder bis Woche 4 von Teil B, sofern das Serumkalium des Patienten in diesem Bereich blieb. In Teil B stieg das Serumkalium bei Patienten unter Placebo um 0,72 mEq/l, 95%-KI [0,46, 0,99] an, wohingegen das Serumkalium bei Patienten unverändert blieb, die weiterhin mit Veltassa behandelt wurden.
Der prozentuale Anteil der Patienten, die im Verlauf von Teil B ein Serumkalium ≥5,1 mEq/l entwickelten, war in der Placebo-Gruppe höher (91 % [95%-KI: 83 %; 99 %]) als in der Veltassa-Gruppe (43 % [95%-KI: 30 %; 56 %]). Der prozentuale Anteil der Patienten, die im Verlauf von Teil B ein Serumkalium ≥5,5 mEq/l entwickelten, war in der Placebo-Gruppe höher (60 % [95%-KI: 47 %; 74 %]) als in der Veltassa-Gruppe (15 % [95%-KI: 6 %; 24 %]).
Zweiundfünfzig Prozent (52 %) der Patienten, die ein Placebo erhielten, setzten die Behandlung mit dem RAAS-Hemmer wegen einer wiederkehrenden Hyperkaliämie ab, im Vergleich zu 5 % der Patienten, die mit Veltassa behandelt wurden.
Studie 2 (AMETHYST-DN)
Die Wirkung der Veltassa-Behandlung wurde bis zu 52 Wochen lang in einer Open-Label-Studie an 304 Patienten mit Diabetes mellitus Typ 2 und chronischer Niereninsuffizienz sowie Hyperkaliämie untersucht, die eine gleichbleibende Dosis eines RAAS-Hemmers erhielten. Die Verringerung des Serumkaliums unter Veltassa konnte in der 1-jährigen Langzeitbehandlung aufrechterhalten werden. Bei Patienten mit einem Baseline-Serumkalium von > 5,0 mEq/l bis 5,5 mEq/l und einer Anfangsdosis von 8,4 g Patiromer pro Tag (auf mehrere Gaben verteilt) betrug die mittlere Tagesdosis 14 g und bei Patienten mit einem Baseline-Serumkalium von > 5,5 mEq/l bis < 6,0 mEq/l und einer Anfangsdosis von 16,8 g Patiromer pro Tag (auf mehrere Gaben verteilt) betrug die mittlere Tagesdosis 20 g während der gesamten Studie.
Abbildung 3: Mittleres Serumkalium (95%-KI) im zeitlichen Verlauf
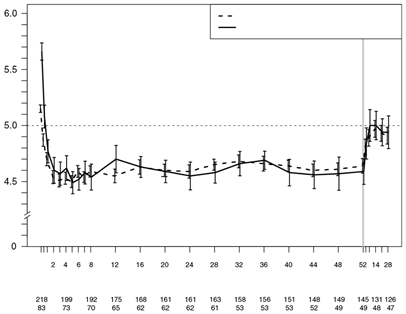
Studie 3 (PEARL-HF)
Die Eigenschaft von Veltassa, eine gleichzeitige Spironolacton-Therapie zu ermöglichen, wurde in einer randomisierten, doppelblinden, placebokontrollierten Studie an Patienten mit Herzinsuffizienz untersucht, bei denen die Gabe von MRA klinisch angezeigt war. Die Patienten begannen die Einnahme von Spironolacton mit 25 mg/Tag zeitgleich zu ihrer randomisierten Behandlung (Veltassa 12,6 g zweimal täglich oder Placebo) und steigerten ihre Dosis auf 50 mg/Tag nach Tag 14, sofern das Serumkalium > 3,5 mEq/l bzw. ≤5,1 mEq/l betrug. Von den 105 Patienten, die randomisiert wurden und die Studienmedikation (Veltassa 56; Placebo 49) erhielten, lag das mittlere Alter bei 68,3 Jahren, 60,6 % waren Männer, 97,1 % Weisse und die mittlere eGFR betrug 81,3 ml/min. Das mittlere Baseline-Serumkalium betrug 4,71 mEq/l bei Veltassa und 4,68 mEq/l beim Placebo.
Der primäre Wirksamkeitsendpunkt, die Veränderung des Serumkaliums von der Baseline bis zum Ende des 28-tägigen Behandlungszeitraums, war in der Veltassa-Gruppe (p < 0,001) signifikant geringer (LSQ-Mittelwert [SEM]: -0,21 [0,07] mEq/l) als in der Placebo-Gruppe (LSQ-Mittelwert [SEM]: +0,23 [0,07] mEq/l). In der Veltassa-Gruppe war die Anzahl der Patienten, deren Serumkaliumwerte > 5,5 mEq/l (7,3 % vs. 24,5 %; p = 0,027) betrug, weiterhin geringer und die Anzahl der Patienten, die Spironolacton 50 mg/Tag erhielten, war deutlich höher (90,9 % vs. 73,5 %, p = 0,022).
Studie 4 (AMBER)
Eine weitere randomisierte, doppelblinde, placebokontrollierte Studie untersuchte den Effekt von Patiromer auf die Verträglichkeit einer Spironolacton-Behandlung bei Patienten mit therapieresistentem Bluthochdruck und CKD. Dabei wurden die Patienten zusätzlich zu Spironolacton (25 mg/Tag; Erhöhung auf 50 mg/Tag ab Woche 3 bei Patienten mit einem systolischen Blutdruck ≥120 mmHg und einem Serumkalium von ≤5,1 mEq/l) randomisiert mit Patiromer oder Placebo behandelt. Zur Aufrechterhaltung eines Serumkaliumspiegels zwischen ≥4,0 mEq/l und ≤5,1 mEq/l konnte die Patiromer/Placebo-Dosis wöchentlich von der Startdosis 8,4 g/Tag bis auf maximal 25,2 g/Tag gesteigert werden. Insgesamt wurden 295 Patienten (Patiromer 147; Placebo 148) mit einem mittleren Alter von 68,1 Jahren und einer mittleren eGFR von 35,73 ml/min/1,73 m² randomisiert, stratifiziert nach Serumkalium zu Studienbeginn (mittlerer Spiegel 4,71 mEq/l im Patiromer-Arm und 4,68 mEq/l in der Placebo-Kontrolle) und Vorgeschichte eines Diabetes mellitus (Typ 1 oder Typ 2). Primärer Endpunkt war der Anteil an Patienten, welche die Spironolacton-Behandlung bis zum Ende der Studie (12 Wochen) fortführten. Dieser Anteil war in der Patiromer-Gruppe gegenüber der Placebo-Gruppe signifikant erhöht (85,7 % vs. 66,2 %; p < 0,0001). Die Patienten im Patiromer-Arm erhielten dabei öfter eine Spironolacton-Dosis von 50 mg/Tag (69,4 % versus 51,4 % in der Placebo-Kontrolle) und zeigten seltener Serumkaliumwerte ≥5,5 mEq/l als die (35,4 % vs. 64,2 % in der Placebo-Kontrolle; p < 0,001).
Studie 5 (DIAMOND)
Die Wirkung von Patiromer auf das Serumkalium wurde in einer doppelblinden, placebokontrollierten, randomisierten Absetzungs-Parallelgruppenstudie bei Patienten untersucht, die zur Behandlung der Herzinsuffizienz zugelassene Dosen von RAAS-Hemmern erhielten und während der Einnahme von Patiromer eine Normokaliämie beibehielten oder erreichten [39]. Vor der Randomisierung waren die Patienten entweder hyperkaliämisch, während sie RAAS-Hemmer erhielten, oder normokaliämisch mit einer Vorgeschichte von Hyperkaliämie während der Einnahme von RAAS-Hemmern in den letzten 12 Monaten, die zu einer Dosisverringerung der RAAS-Hemmer oder zum Absetzen von RAAS-Hemmern führte. Patiromer wurde bei allen Patienten mit einer Dosis von 8,4 g pro Tag eingeleitet und wöchentlich bis zu einer maximalen Dosis von 25,2 g pro Tag titriert, um den Serumkaliumspiegel ≥4,0 mEq/l und ≤5,0 mEq/l zu halten. Hyperkaliämische Patienten erhielten Patiromer direkt zu Beginn; normokaliämische Patienten erhielten Patiromer in Woche 1 oder später. Parallel dazu wurde die Medikation des RAAS-Hemmers optimiert, einschliesslich der Einleitung, Dosiseskalation und Erhaltung von mindestens 50 % der vorgegebenen ACEi/ARB/ARNI-Zieldosen und 100 % der vorgegebenen MRA- Zieldosen. Die Patienten wurden, sobald diese RAAS-Hemmer-Zieldosen erreicht waren und sie mindestens eine Woche lang stabile Serumkaliumwerte von ≥4,0 mEq/l und ≤5,0 mEq/l aufwiesen, entweder zu Patiromer oder Placebo randomisiert.
Von den 1168 Patienten, bei denen Patiromer eingeleitet wurde, schlossen 130 den Run-in-Abschnitt nicht ab. Von den 1038 Patienten, die den Run-in-Abschnitt abschlossen, wurden 878 (85 %) randomisiert (Patiromer: 439, Placebo: 439). Das mittlere Alter der randomisierten Patienten lag bei 66,9 Jahren, 72,9 % waren Männer, 97,9 % Weisse, und die mittlere eGFR betrug 63,0 ml/min/1,73 m2. Zum Zeitpunkt der Randomisierung betrug der mittlere Baseline-Serumkaliumwert 4,72 mEq/l.
Alle Teilnehmer hatten eine Vorgeschichte von Hyperkaliämie. Insgesamt waren 40,3 % der Teilnehmer bei Studienbeginn hyperkaliämisch und 59,7 % normokaliämisch, hatten jedoch eine Vorgeschichte von Hyperkaliämie in den letzten 12 Monaten.
Der primäre Wirksamkeitsendpunkt war die Differenz der adjustierten mittleren Veränderung (Standardfehler [SE]) des Serumkaliumspiegels von Baseline, die mit Hilfe eines gemischten Modells für Messwiederholungen (MMRM) analysiert wurde. Die Differenz der adjustierten mittleren Veränderungen gegenüber Baseline betrug -0,097 mEq/l (95%-KI: -0,128; -0,067). Die mittlere Veränderung des Serum-K+ vom Baseline-Wert war in der Patiromer-Gruppe (0,029 [0,019] mEq/l) statistisch signifikant niedriger (p < 0,001) als in der Placebo-Gruppe (0,127 [0,019] mEq/l).
Die Zeit bis zum ersten Hyperkaliämie-Ereignis (definiert als Serumkaliumwert > 5,5 mEq/l) war für Patiromer im Vergleich zu Placebo signifikant verkürzt (p = 0,006), mit einer Hazard Ratio von 0,63 (95%-KI: 0,45; 0,87).
Der Anteil der Patienten, bei denen die MRA-Medikation (Spironolacton oder Eplerenon) unter die Zieldosis reduziert wurde, betrug 13,9 % in der Patiromer-Gruppe gegenüber 18,9 % in der Placebo-Gruppe. Die Hazard Ratio für die Zeit bis zur ersten MRA-Dosisreduktion von Patiromer gegenüber Placebo betrug 0,62 (95%-KI: 0,45; 0,87), und der Behandlungsunterschied war statistisch signifikant (p = 0,006).
In den vordefinierten Untergruppen eGFR ml/min/1,73 m2, ml/min/1,73 m2 mit Post-hoc-Analyse der eGFR </≥30 ml/min/1,73 m2 ermöglichte Patiromer den Einsatz wirksamer RAAS-Hemmer-Dosen, kontrollierte das Serum-K+ und minimierte das Hyperkaliämie-Risiko bei Patienten mit HFrEF und leichter bis schwerer chronischer Nierenerkrankung.
Insgesamt trat bei 31,2 % der Patienten in der Patiromer-Gruppe und bei 45,1 % der Patienten in der Placebo-Gruppe mindestens ein vom Prüfarzt gemeldetes Hyperkaliämie-Ereignis mit einem Serumkaliumwert von > 5,5 mEq/l auf. Das Verhältnis der annualisierten Ereignisrate für Patiromer betrug im Vergleich zu Placebo 0,658 (95%-KI: 0,534; 0,810), was statistisch signifikant war (p < 0,001).
Für zwei hierarchisch getestete Sekundärendpunkte wurde ein unangepasster Win-Ratio-Ansatz verwendet: ein Ansatz, für den zusammengesetzte Hyperkaliämie-bedingte harte Endpunkte verwendet wurden, darunter Tod aus kardiovaskulärer Ursache, Krankenhausaufenthalte aufgrund von kardiovaskulären Erkrankungen und drei Grade von Hyperkaliämie-Ereignissen, die eine Win-Ratio von 1,526 (95%-KI:1,231; 1,906; p < 0,001) aufwiesen, was für eine Patiromer-Behandlung spricht, und einer, bei dem ein neuer Score für die Anwendung von RAAS-Hemmern (0–8 Punkte) verwendet wurde, der eine günstige Quote für das Erreichen eines höheren Scores für die Anwendung von RAAS-Hemmern für mit Patiromer behandelte Patienten im Vergleich zur Placebo-Gruppe zeigt, mit einer Win-Ratio von 1,248 (95%-KI: 1,003; 1,564; p = 0,048).
PharmakokinetikAbsorption
Veltassa wirkt, indem Kalium im Gastrointestinaltrakt gebunden wird, deshalb ist die Serumkonzentration für die Wirksamkeit nicht relevant. Aufgrund der Unlöslichkeit und der fehlenden Resorbierbarkeit von Veltassa können zahlreiche klassische pharmakokinetische Untersuchungen nicht durchgeführt werden.
Im Rahmen von ADME-Studien (Absorption, Distribution, Metabolismus, Elimination) mit radioaktiver Markierung von Ratten und Hunden wurde Patiromer nicht systemisch resorbiert.
Distribution
Quantitative Ganzkörper-Autoradiographie-Analysen bei Ratten zeigten, dass die Radioaktivität auf den Gastrointestinaltrakt beschränkt war und keine Radioaktivität in anderen Geweben oder Organen nachgewiesen werden konnte.
Metabolismus
Nicht zutreffend aufgrund der Unlöslichkeit und der fehlenden Absorption von Veltassa.
Elimination
Basierend auf den ADME-Studien mit radioaktiver Markierung von Ratten und Hunden wurde Patiromer in den Fäzes ausgeschieden.
Präklinische DatenBasierend auf konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, Reproduktions- und Entwicklungstoxizität, lassen die nicht klinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Patiromer zeigte im Rückmutationstest (Ames-Test), in Chromosomenaberrations- oder Ratten-Mikronukleus-Tests keine Genotoxizität.
Studien zur Karzinogenität wurden nicht durchgeführt.
Patiromer hat bei einer bis zu 10-fachen Dosis der empfohlenen Höchstdosis für Menschen keine Veränderungen der Fertilität bei männlichen oder weiblichen Ratten gezeigt. Patiromer hat auch nach Gabe an trächtige Ratten mit einer Dosis von bis zu 6g/kg keine schädlichen Auswirkungen auf die Entwicklung des Embryos und des Fötus gezeigt, was eine Sicherheitsmarge ergibt, die das 12-fache der empfohlenen Höchstdosis für Menschen beträgt, und wurde trächtigen Kaninchen mit einer Dosis von bis zu 3 g/kg verabreicht, was eine Sicherheitsmarge ergibt, die das 6-fache der empfohlenen Höchstdosis für Menschen beträgt.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
Veltassa darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Kühl transportieren und lagern (2 °C - 8 °C).
Nicht Temperaturen über 40 °C aussetzen.
Patienten können Veltassa bei Temperaturen unter 25 °C bis zu 6 Monate lang aufbewahren.
Veltassa nach Ablauf des Verfallsdatums auf dem Beutel nicht verwenden.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer66411 (Swissmedic)
PackungenVeltassa ist in Beuteln mit 8,4 g oder 16,8 g Patiromer (als Patiromer Sorbitol Calcium (Patiromer Sorbitex Calcium)) erhältlich.
Packungen mit 30 Beuteln. [B]
ZulassungsinhaberinVifor Fresenius Medical Care Renal Pharma Ltd.
St. Gallen
Stand der InformationMai 2024
|