Eigenschaften/WirkungenATC-Code
D11AH05
Wirkungsmechanismus
Dupilumab ist ein rekombinanter, humaner, monoklonaler IgG4-Antikörper, der die Signalwege von Interleukin 4 (IL-4) und Interleukin 13 (IL-13) hemmt. Dupilumab hemmt den IL-4-Signalweg über den Typ-I-Rezeptor (IL-4Rα/γc) und die Signalwege von IL-4 und IL-13 über den Typ-II-Rezeptor (IL-4Rα/IL-13Rα).
IL-4 und IL-13 sind Zytokine im Zusammenhang mit entzündlichen Erkrankungen vom Typ 2 wie atopische Dermatitis und Asthma. Durch Hemmung des IL-4-/IL-13-Signalwegs mit Dupilumab kommt es zu einer Verringerung mehrerer Mediatoren der Typ-2-Inflammation.
Dupilumab normalisiert das RNA-Expressionsprofil der Ösophagusbiopsate, einschliesslich der Gene, die mit Typ-2-Inflammationen, Eosinophilen und mit sonstigen an der eosinophilen Ösophagitis beteiligten biologischen Prozessen (wie Zellproliferation, Barrierefunktion, Fibrose und Remodeling) in Verbindung stehen.
Pharmakodynamik
In klinischen Studien zu atopischer Dermatitis war die Behandlung mit Dupilumab mit einer Senkung der Konzentration von Biomarkern der Immunität Typ 2 wie thymusaktivitätsreguliertes Chemokin (TARC/CCL17), dem Gesamt-IgE im Serum sowie dem allergenspezifischen IgE im Serum gegenüber den Ausgangswerten assoziiert. Unter der Behandlung mit Dupilumab wurde bei Erwachsenen und Jugendlichen mit atopischer Dermatitis eine Abnahme der Laktatdehydrogenase (LDH) beobachtet.
Während der Behandlung kommt es durch die Präsenz von Dupilumab zu einem Anstieg des IgG4-Spiegels. Die Auswirkungen einer Langzeittherapie mit einem monoklonalen IgG4-Antikörper wurden noch nicht ausreichend untersucht.
In klinischen Studien zu Asthma kam es unter der Behandlung mit Dupilumab im Vergleich zu Placebo zu einer deutlichen Verringerung der FeNO-Werte und der Konzentrationen an Eotaxin-3, Gesamt-IgE, allergenspezifischem IgE, TARC und Periostin im Blutkreislauf. Diese Verringerungen von Biomarkern der Typ-2-Inflammation waren für die Behandlungsschemata mit 200 mg Q2W und 300 mg Q2W vergleichbar. Diese Marker waren nach 2-wöchiger Behandlung nahezu nicht nachweisbar, mit Ausnahme von IgE, das einen langsameren Rückgang zeigte. Diese Wirkungen hielten über die gesamte Behandlungsdauer an.
Bei COPD-Patienten verringerte die Behandlung mit Dupilumab gegenüber Placebo die Typ-2-Biomarker, insbesondere FeNO und Gesamt-IgE. Ein Rückgang von FeNO wurde bereits in Woche 4 beobachtet. Diese Auswirkungen auf die Typ-2-Biomarker blieben während der gesamten Behandlung mit Dupilumab erhalten.
Klinische Wirksamkeit
1) Atopische Dermatitis
Klinische Wirksamkeit und Sicherheit bei atopischer Dermatitis bei Erwachsenen
Die Wirksamkeit und Sicherheit von Dupilumab als Monotherapie und mit einer begleitenden Behandlung mit topischen Kortikosteroiden wurden in drei randomisierten, placebokontrollierten, doppelblinden Zulassungsstudien (SOLO 1, SOLO 2 und CHRONOS) untersucht. Eingeschlossen waren 2'119 Patienten ab 18 Jahren mit einer mittelschweren bis schweren atopischen Dermatitis (AD), definiert durch einen Investigator's Global Assessment-Score (IGA) ≥3, einen EASI-Score (Eczema Area and Severity Index) ≥16 und eine betroffene Körperoberfläche von 10 % oder mehr. Die für diese drei Studien geeigneten und darin eingeschlossenen Patienten hatten vorher nur unzureichend auf eine topische Behandlung angesprochen.
In den drei Studien erhielten die Patienten 1) eine Anfangsdosis von 600 mg Dupilumab (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg einmal alle zwei Wochen (Q2W), 2) eine Anfangsdosis von 600 mg Dupilumab an Tag 1, gefolgt von 300 mg einmal wöchentlich (QW) oder 3) ein entsprechendes Placebo. Dupilumab wurde in allen Studien als subkutane (s. c.) Injektion verabreicht. Um als unerträglich empfundene Symptome der atopischen Dermatitis zu lindern, konnten die Patienten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten (u.a. topische Steroide mit höherer Wirksamkeit oder systemische Immunsuppressiva). Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In die Studie SOLO 1 wurden 671 Patienten eingeschlossen (224 in die Placebo-Gruppe, 224 in die Gruppe mit Dupilumab 300 mg Q2W und 223 in die Gruppe mit Dupilumab 300 mg QW). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie SOLO 2 wurden 708 Patienten eingeschlossen (236 in die Placebo-Gruppe, 233 in die Gruppe mit Dupilumab 300 mg Q2W und 239 in die Gruppe mit Dupilumab 300 mg QW). Der Behandlungszeitraum betrug 16 Wochen.
In die Studie CHRONOS wurden 740 Patienten eingeschlossen (315 in die Gruppe Placebo + TCS, 106 in die Gruppe mit Dupilumab 300 mg Q2W + TCS und 319 in die Gruppe mit Dupilumab 300 mg QW + TCS). Der Behandlungszeitraum betrug 52 Wochen. Die Patienten erhielten ab Baseline Dupilumab oder ein Placebo sowie eine begleitende Therapie mit TCS gemäss standardisiertem Behandlungsschema. Die Patienten konnten auch topische Calcineurin-Inhibitoren (TCI) erhalten.
Primäre Endpunkte:
Die koprimären Endpunkte aller drei Zulassungsstudien waren der Anteil der Patienten, bei denen sich der Wert auf einer IGA-Skala (von 0 bis 4) von der Baseline bis Woche 16 um ≥2 Punkte auf dann 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») verbesserte, sowie der Anteil der Patienten, deren EASI-Score sich von der Baseline bis Woche 16 um mindestens 75 % (EASI-75) verbesserte. Die weiteren untersuchten Resultate umfassten den Anteil der Patienten mit einer EASI-Verbesserung von mindestens 50 % (EASI-50) bzw. 90 % (EASI-90), einer Verringerung des Juckreizes, gemessen an der numerischen Bewertungsskala für Pruritus (Peak Pruritus Numerical Rating Scale [NRS]), und einer prozentualen Veränderung des SCORAD (SCORing Atopic Dermatitis) von der Baseline bis Woche 16. Zusätzliche sekundäre Endpunkte umfassten die mittlere Veränderung des POEM (Patient-Oriented Eczema Measure), des DLQI (Dermatology Life Quality Index) sowie des HADS-Werts (Hospital Anxiety and Depression Scale) von der Baseline bis Woche 16. In der CHRONOS-Studie wurde die Wirksamkeit auch in Woche 52 bewertet.
Patientencharakteristika bei der Baseline:
In allen Behandlungsgruppen der Monotherapie-Studien (SOLO 1 und SOLO 2) lag das mittlere Alter bei 38,3 Jahren und das mittlere Gewicht bei 76,9 kg. 42,1 % der Studienteilnehmer waren Frauen, 68,1 % Weisse, 21,8 % Asiaten und 6,8 % Schwarze. In diesen Studien hatten 51,6 % der Patienten einen Baseline-IGA-Score von 3 (mittelschwere AD), 48,3 % einen Baseline-IGA-Score von 4 (schwere AD) und 32,4 % der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,0, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,4, der mittlere SCORAD 67,8, der mittlere POEM 20,5, der mittlere DLQI 15,0 und der mittlere HADS-Gesamtwert 13,3.
In allen Behandlungsgruppen der Studie mit begleitender TCS-Therapie (CHRONOS) lag das mittlere Alter bei 37,1 Jahren und das mittlere Gewicht bei 74,5 kg. 39,7 % der Studienteilnehmer waren Frauen, 66,2 % Weisse, 27,2 % Asiaten und 4,6 % Schwarze. In dieser Studie hatten 53,1 % der Patienten einen Baseline-IGA-Score von 3, 46,9 % einen Baseline-IGA-Score von 4, und 33,6 % der Patienten wurden in der Vergangenheit mit systemischen Immunsuppressiva behandelt. Bei Behandlungsbeginn betrug der mittlere EASI-Score 32,5, der wöchentliche Durchschnittswert gemäss Pruritus NRS 7,3, der mittlere SCORAD 66,4, der mittlere POEM 20,1, der mittlere DLQI 14,5 und der mittlere HADS-Gesamtwert 12,7.
Klinische Wirksamkeit: 16-wöchige Monotherapie-Studien (SOLO 1 und SOLO 2)
Im Vergleich zu Placebo erreichte in den Studien SOLO 1 und SOLO 2 von der Baseline bis Woche 16 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupilumab zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um > 4 Punkte gemäss Pruritus NRS (siehe Tabelle 6).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupilumab erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um ≥4 Punkte bereits in Woche 2; p < 0,01); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat parallel zu einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 1 und Abbildung 2 stellen die mittlere prozentuale Veränderung der EASI- bzw. Pruritus-NRS-Scores von der Baseline bis Woche 16 dar.
Tabelle 6: Wirksamkeitsergebnisse der Dupilumab-Monotherapie in Woche 16
|
|
SOLO 1 (FSA)a
|
SOLO 2 (FSA)a
| |
|
Placebo
|
Dupilumab
300 mg Q2W
|
Placebo
|
Dupilumab
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
| |
IGA 0 oder 1b,
% der Responderc
|
10,3 %
|
37,9 %e
|
8,5 %
|
36,1 %e
| |
EASI-50,
% der Responderc
|
24,6 %
|
68,8 %e
|
22,0 %
|
65,2 %e
| |
EASI-75,
% der Responderc
|
14,7 %
|
51,3 %e
|
11,9 %
|
44,2 %e
| |
EASI-90,
% der Responderc
|
7,6 %
|
35,7 %e
|
7,2 %
|
30,0 %e
| |
Anzahl Patienten mit Pruritus-NRS-Wert bei Baseline ≥ 4
|
212
|
213
|
221
|
225
| |
Pruritus gemäss NRS (Verbesserung ≥4 Punkte)
% der Responderc,d
|
12,3 %
|
40,8 %e
|
9,5 %
|
36,0 %e
|
a Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
b Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
c Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
d In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, in den Dupilumab-Gruppen signifikant höher als in der Placebo-Gruppe (p < 0,01).
e p < 0,0001
Abbildung 1: Mittlere prozentuale Veränderung des EASI-Scores gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
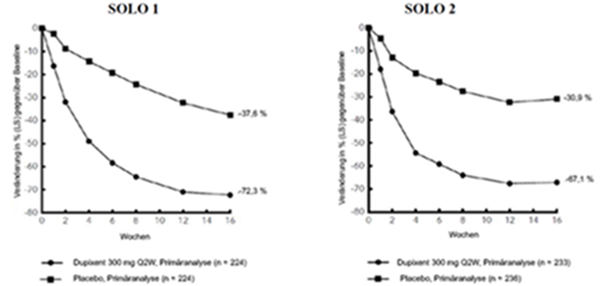
Abbildung 2: Mittlere prozentuale Veränderung gemäss Pruritus NRS gegenüber der Baseline in den Studien SOLO 1a und SOLO 2a (FSA)b
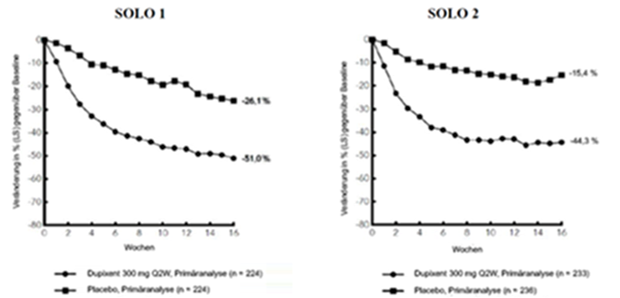
LS = Least Squares
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten.
In den Studien SOLO 1 und SOLO 2 stimmte die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Basistherapie einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinische Wirksamkeit: 52-wöchige Studie mit begleitender Therapie mit TCS (CHRONOS)
Im Vergleich zu Placebo + TCS erreichte in der Studie CHRONOS von der Baseline bis Woche 16 und Woche 52 ein signifikant höherer Anteil an Patienten, denen randomisiert Dupilumab 300 mg Q2W + TCS zugewiesen worden war, einen IGA-Score von 0 oder 1, den EASI-75 und/oder eine Verbesserung um > 4 Punkte gemäss Pruritus NRS (siehe Tabelle 7).
Im Vergleich zu Placebo + TCS erreichte ein signifikant höherer Anteil an Patienten, die randomisiert Dupilumab + TCS erhielten, eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um > 4 Punkte bereits in Woche 2; p < 0,05); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus NRS weiterhin an. Die Verbesserung gemäss Pruritus NRS trat im Zusammenhang mit einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 3 und Abbildung 4 stellen die mittlere prozentuale Veränderung der EASI- bzw. NRS-Scores in der CHRONOS-Studie von der Baseline bis Woche 52 dar.
Tabelle 7: Wirksamkeitsergebnisse von Dupilumab mit begleitenden TCSa in Woche 16 und Woche 52 in der CHRONOS-Studie
|
|
Woche 16 (FSA)b
|
Woche 52 (FSA Woche 52)b
| |
|
Placebo
+
TCS
|
Dupilumab
300 mg
Q2W + TCS
|
Placebo
+
TCS
|
Dupilumab
300 mg
Q2W + TCS
| |
Randomisierte Patienten
|
315
|
106
|
264
|
89
| |
IGA 0 oder 1c,
% der Responderd
|
12,4 %
|
38,7 %f
|
12,5 %
|
36,0 %f
| |
EASI-50,
% der Responderd
|
37,5 %
|
80,2 %f
|
29,9 %
|
78,7 %f
| |
EASI-75,
% der Responderd
|
23,2 %
|
68,9 %f
|
21,6 %
|
65,2 %f
| |
EASI-90,
% der Responderd
|
11,1 %
|
39,6 %f
|
15,5 %
|
50,6 %f
| |
Anzahl Patienten mit Pruritus NRS-Wert bei Baseline ≥4
|
299
|
102
|
249
|
86
| |
Pruritus NRS-Wert
(Verbesserung ≥4 Punkte),
% der Responderd,e
|
19,7 %
|
58,8 %f
|
12,9 %
|
51,2 %f
|
a Alle Patienten erhielten eine Basistherapie mit TCS. Die Patienten konnten topische Calcineurin-Inhibitoren anwenden.
b Die Full-Sample-Analyse (FSA) umfasst alle randomisierten Patienten. Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
c Als Responder angesehen wurden Patienten mit einem IGA-Score von 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei») mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
d Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
e In Woche 2 war der Anteil der Patienten, bei denen eine Verbesserung gemäss Pruritus NRS um ≥4 Punkte festzustellen war, unter Dupilumab signifikant höher als unter Placebo (p < 0,05).
f p < 0,0001
Abbildung 3: Mittlere Veränderung (in %) des EASI-Score gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
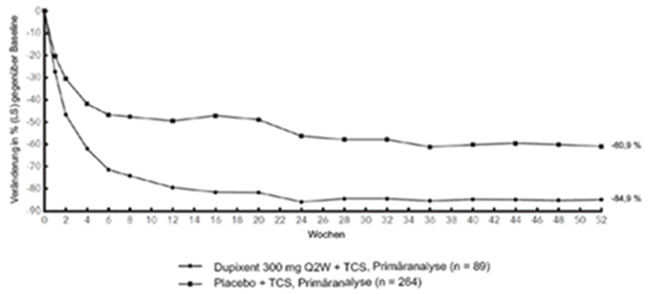
Abbildung 4: Mittlere Veränderung (in %) gemäss Pruritus NRS gegenüber der Baseline in der CHRONOS-Studiea (FSA in Woche 52)b
CHRONOS
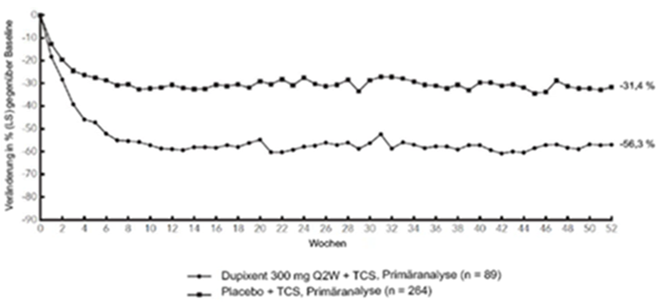
LS = Least Squares [Methode der kleinsten Quadrate]
a In den primären Analysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die FSA in Woche 52 umfasst alle Patienten, die mindestens ein Jahr vor dem Enddatum der Primäranalyse randomisiert wurden.
In der CHRONOS-Studie stimmte die Wirksamkeit der Behandlung in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Basistherapie einschliesslich Immunsuppressiva) mit den Ergebnissen der gesamten Studienpopulation überein.
Klinische Wirksamkeit: Patienten, die unter einer Ciclosporin-Behandlung unzureichend eingestellt waren, eine Unverträglichkeit gegenüber Ciclosporin aufwiesen oder für die diese Behandlung medizinisch nicht indiziert war (CAFE-Studie)
Im Rahmen der CAFE-Studie wurde die Wirksamkeit von Dupilumab mit begleitender TCS-Therapie gegenüber Placebo innerhalb eines 16-wöchigen Behandlungszeitraums bewertet. Bei den Studienteilnehmern handelte es sich um erwachsene Patienten mit schwerer atopischer Dermatitis, die unter einem oralen Ciclosporin unzureichend eingestellt waren, dieses nicht vertrugen oder für die diese Behandlung derzeit kontraindiziert oder medizinisch nicht angezeigt ist.
Insgesamt wurden 325 Patienten eingeschlossen, von denen 210 bereits in der Vergangenheit mit Ciclosporin behandelt wurden, während 115 Patienten noch nie Ciclosporin erhalten hatten oder eine Ciclosporin-Behandlung bei ihnen medizinisch nicht angezeigt war. Das mittlere Alter lag bei 38,4 Jahren, 38,8 % der Patienten waren Frauen. Bei Behandlungsbeginn betrug der mittlere EASI-Score 33,1, die durchschnittliche betroffene Körperoberfläche 55,7, der wöchentliche Durchschnittswert gemäss Pruritus NRS 6,4, der mittlere SCORAD 67,2 und der mittlere DLQI 13,8.
Primärer Endpunkt war der Anteil der Patienten mit einem EASI-75 in Woche 16.
In Tabelle 8 sind sowohl die primären als auch die sekundären Endpunkte der 16-wöchigen CAFE-Studie zusammengefasst.
Tabelle 8: Ergebnisse der primären und sekundären Endpunkte der CAFE-Studie
|
|
Placebo + TCS
|
Dupilumab
300 mg Q2W + TCS
| |
Randomisierte Patienten
|
108
|
107
| |
EASI-75, Responder (in %)
|
29,6 %
|
62,6 %
| |
EASI, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-46,6
(2,76)
|
-79,8
(2,59)
| |
Wert gemäss Pruritus NRS, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-25,4 % (3,39)
|
-53,9 % (3,14)
| |
SCORAD, mittlere prozentuale Veränderung (LS) gegenüber den Ausgangswerten (+/– SE)
|
-29,5 % (2,55)
|
-62,4 % (2,48)
| |
DLQI, mittlere Veränderung (LS) gegenüber den Ausgangswerten (SE)
|
-4,5
(0,49)
|
-9,5
(0,46)
|
In der Patientenuntergruppe der 52-wöchigen CHRONOS-Studie, die der Studienpopulation der CAFE-Studie ähnelte, erreichten bis Woche 16 69,6 % der mit Dupilumab 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen, während es bei den mit Placebo behandelten Patienten 18,0 % waren. Bis Woche 52 erreichten 52,4 % der mit Dupilumab 300 mg Q2W behandelten Patienten ein EASI-75-Ansprechen, gegenüber 18,6 % in der Placebo-Gruppe. In dieser Untergruppe lag die mittlere prozentuale Veränderung des Pruritus-NRS-Werts von der Baseline bis Woche 16 bei –51,4 % unter Dupilumab 300 mg Q2W und bei –30,2 % unter Placebo beziehungsweise bis Woche 52 bei –54,8 % in der Gruppe mit Dupilumab 300 mg Q2W und bei –30,9 % in der Placebo-Gruppe.
Aufrechterhaltung und Dauer des Ansprechens (Studie SOLO CONTINUE)
Um Aufrechterhaltung und Dauer des Ansprechens zu untersuchen, wurden die Studienteilnehmer, die in den Studien SOLO 1 und SOLO 2 16 Wochen lang mit Dupilumab behandelt wurden und einen IGA-Wert von 0 oder 1 oder ein EASI-75-Ansprechen erreichten, im Rahmen der Studie SOLO CONTINUE erneut randomisiert. Diese Studie umfasste eine zusätzliche 36-wöchige Behandlung mit Dupilumab oder Placebo, sodass sich die Gesamtdauer der Studienbehandlung auf 52 Wochen belief. Die Beurteilung der Endpunkte erfolgte in Woche 51 oder 52.
Die koprimären Endpunkte waren der Unterschied zwischen der Baseline (Woche 0) und Woche 36 gemessen an der prozentualen Veränderung des EASI-Scores gegenüber der Baseline der Studien SOLO 1 und SOLO 2, sowie der prozentuale Anteil an Patienten mit einem EASI-75-Ansprechen in Woche 36 bei Patienten, die bereits bei Behandlungsbeginn ein EASI-75-Ansprechen hatten.
Bei Patienten, die dasselbe Behandlungsschema beibehielten, mit dem sie in den Studien SOLO 1 und SOLO 2 behandelt worden waren (300 mg Q2W oder 300 mg QW), konnte ein optimaler Erhaltungseffekt des klinischen Ansprechens nachgewiesen werden, wohingegen sich die Wirksamkeit bei anderen Dosierungsschemata dosisabhängig verringerte.
Sowohl die primären als auch die sekundären Endpunkte der 52-wöchigen Studie SOLO CONTINUE sind in Tabelle 9 zusammengefasst.
Tabelle 9: Ergebnisse der primären und sekundären Endpunkte der Studie SOLO CONTINUE
|
|
Placebo
|
Dupilumab 300 mg
| |
|
n = 83
|
Q8W
n = 84
|
Q2W/QW
n = 169
| |
Koprimäre Endpunkte
| |
Mittlerer Unterschied (LS) (+/– SE) zwischen der Baseline und Woche 36 in prozentualer EASI-Veränderung gegenüber der Baseline der Vorläufer-Studien
|
21,7
(3,13)
|
6,8***
(2,43)
|
0,1***
(1,74)
| |
Prozentualer Anteil an Patienten mit EASI-75-Ansprechen in Woche 36 bei Patienten mit EASI-75-Ansprechen bei der Baseline (n [%])
|
24/79
(30,4 %)
|
45/82*
(54,9 %)
|
116/162***
(71,6 %)
| |
Wichtige sekundäre Endpunkte:
| |
Prozentualer Anteil an Patienten, deren IGA-Wert in Woche 36 um nicht mehr als 1 Punkt vom Baseline-Wert abwich, unter den Patienten mit IGA 0 oder 1 bei der Baseline (n [%])
|
18/63
(28,6)
|
32/64†
(50,0)
|
89/126***
(70,6)
| |
Prozentualer Anteil an Patienten mit einem IGA-Wert von 0 oder 1 in Woche 36 unter den Patienten mit IGA von 0 oder 1 bei der Baseline (n [%])
|
9/63
(14,3)
|
21/64†
(32,8)
|
68/126***
(54,0)
| |
Prozentualer Anteil an Patienten, deren maximaler Pruritus-NRS-Wert von der Baseline bis Woche 35 um ≥3 Punkte anstieg, unter den Patienten mit einem maximalen Baseline-Pruritus-NRS-Wert ≤7 (n [%])
|
56/80
(70,0)
|
45/81
(55,6)
|
57/168***
(33,9)
|
† p < 0,05; *p < 0,01; ***p ≤0,0001
In der Studie SOLO CONTINUE wurde bei verlängerten Dosierungsintervallen eine Tendenz zu einer behandlungsbedingten stärkeren Produktion von Antikörpern gegen den Wirkstoff (Anti-Drug Antibodies, ADA) beobachtet.
Behandlungsbedingte ADA-Reaktionen: QW: 1,2 %; Q2W: 4,3 %; Q4W: 6,0 %; Q8W: 11,7 %. ADA-Reaktionen, die länger als 12 Wochen andauerten: QW: 0,0 %; Q2W: 1,4 %; Q4W: 0,0 %; Q8W: 2,6 %.
Lebensqualität / von Patienten berichtete Ergebnisse (Patient Reported Outcomes, PRO)
In den Studien SOLO 1 und SOLO 2 wurde mit Dupilumab im Vergleich zu Placebo gemäss der POEM-, der DLQI- und der HADS-Gesamtbewertung jeweils eine signifikante Verbesserung der Symptome der atopischen Dermatitis (AD), der gesundheitsbezogenen Lebensqualität sowie der Angst- und Depressionssymptome erzielt.
In der CHRONOS-Studie wurde mit Dupilumab im Vergleich zu Placebo gemäss der POEM- und der DLQI-Gesamtbewertung nach 16 Wochen jeweils eine signifikante Verbesserung der Symptome der AD und der gesundheitsbezogenen Lebensqualität erzielt.
In den Studien SOLO 1 und SOLO 2 waren die mittleren Veränderungen (± SE) der Least Squares der POEM-, der DLQI- und der HADS-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p < 0,0001 für alle Vergleiche ausser HADS in der Studie SOLO 2, p < 0,001), siehe Tabelle 10.
In der Studie CHRONOS waren die mittleren Veränderungen (± SE) der Least Squares der POEM- und der DLQI-Gesamtbewertung von der Baseline bis Woche 16 bei Patienten, die Dupilumab erhielten, signifikant höher als bei Placebo-Patienten (p < 0,0001 für alle Vergleiche), siehe Tabelle 10.
Tabelle 10: Zusätzliche Ergebnisse der sekundären Endpunkte der Dupilumab-Behandlung mit oder ohne TCS in Woche 16
|
|
Monotherapie in Woche 16
|
Begleitende Therapie mit TCS in Woche 16
| |
|
SOLO 1
|
SOLO 2
|
CHRONOS
| |
|
Placebo
|
Dupilumab
300 mg Q2W
|
Placebo
|
Dupilumab
300 mg Q2W
|
Placebo
|
Dupilumab
300 mg Q2W
| |
Randomisierte Patienten
|
224
|
224
|
236
|
233
|
315
|
106
| |
POEM, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,1
(0,67)
|
-11,6a
(0,49)
|
-3,3
(0,55)
|
-10,2a
(0,49)
|
-5,3
(0,41)
|
-12,7a
(0,64)
| |
DLQI, mittlere Veränderung (LS) gegenüber der Baseline (SE)
|
-5,3
(0,50)
|
-9,3a
(0,40)
|
-3,6
(0,50)
|
-9,3a
(0,38)
|
-5,8
(0,34)
|
-10,0a
(0,50)
|
LS = Least Squares [Methode der kleinsten Quadrate]; SE = Standard Error [Standardfehler]
a p < 0,0001
Klinische Wirksamkeit und Sicherheit bei Jugendlichen mit atopischer Dermatitis:
Die Wirksamkeit und Sicherheit einer Monotherapie mit Dupilumab bei Jugendlichen wurden in einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (AD-1526) bei 251 jugendlichen Patienten von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis (AD) beurteilt. Diese wurde definiert als ein Wert ≥3 gemäss IGA (Investigator's Global Assessment) bei der Gesamtbeurteilung der AD-Läsionen auf einer Schweregrad-Skala von 0 bis 4, ein Wert ≥16 gemäss EASI (Eczema Area and Severity Index) auf einer Skala von 0 bis 72 und eine betroffene Körperoberfläche (Body Surface Area, BSA) von mindestens ≥10 %. Patienten, die für die Studie geeignet waren und in diese eingeschlossen wurden, hatten zuvor nur unzureichend auf eine topische Medikation angesprochen.
Die Patienten erhielten:
1.eine Anfangsdosis von 400 mg Dupilumab (zwei Injektionen zu je 200 mg) an Tag 1, gefolgt von 200 mg einmal alle zwei Wochen (Q2W), bei einem Ausgangsgewicht von < 60 kg oder eine Anfangsdosis von 600 mg Dupilumab (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg Q2W, bei einem Ausgangsgewicht von ≥60 kg,
2.eine Anfangsdosis von 600 mg Dupilumab (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg alle 4 Wochen (Q4W), unabhängig vom Ausgangsgewichtoder
3.ein entsprechendes Placebo.
Dupilumab wurde als subkutane (s. c.) Injektion verabreicht. Um als unerträglich empfundene Symptome der atopischen Dermatitis zu lindern, konnten die Patienten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten. Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In dieser Studie betrug das mittlere Alter 14,5 Jahre, das mediane Körpergewicht 59,4 kg, 41,0 % der Patienten waren weiblich, 62,5 % Weisse, 15,1 % Asiaten und 12,0 % Schwarze. Zu Studienbeginn hatten 46,2 % der Patienten einen Baseline-IGA-Wert von 3 (mittelschwere AD), 53,8 % einen Baseline-IGA-Wert von 4 (schwere AD), die mittlere BSA-Beteiligung betrug 56,5 %, und 42,4 % der Patienten hatten in der Vergangenheit systemische Immunsuppressiva erhalten. Bei der Aufnahme in die Studie betrug der mittlere EASI-Wert 35,5, der wöchentliche Durchschnittswert gemäss Pruritus NRS (Numeral Rating Scale) 7,6, der mittlere SCORAD(SCORing Atopic Dermatitis)-Wert 70,3, der mittlere POEM(Patient Oriented Eczema Measure)-Wert 21,0 und der mittlere CDLQI(Children Dermatology Life Quality Index)-Wert 13,6. Insgesamt lag bei 92,0 % der Patienten mindestens eine allergische Begleiterkrankung vor; 65,6 % hatten allergische Rhinitis, 53,6 % Asthma und 60,8 % Nahrungsmittelallergien.
Koprimäre Endpunkte waren der Anteil der Patienten mit IGA 0 oder 1 mit einer Verbesserung um mindestens 2 Punkte und der Anteil der Patienten mit EASI-75 (EASI-Verbesserung um mindestens 75 %) zwischen Baseline und Woche 16. Die weiteren beurteilten Endpunkte umfassten den Anteil der Patienten mit EASI-50 bzw. EASI-90 (EASI-Verbesserung von mindestens 50 % bzw. 90 % gegenüber Baseline), die Verringerung des Juckreizes gemäss Peak-Pruritus-NRS und die prozentuale Veränderung auf der SCORAD-Skala zwischen Baseline und Woche 16. Zusätzliche sekundäre Endpunkte beinhalteten die mittlere Veränderung des POEM- und CDLQI-Scores zwischen Baseline und Woche 16.
Klinische Wirksamkeit
Die Wirksamkeitsergebnisse aus Woche 16 der Studie für atopische Dermatitis bei Jugendlichen sind in Tabelle 11 angegeben.
Tabelle 11: Wirksamkeitsergebnisse der Dupilumab-Therapie aus Woche 16 der Studie für atopische Dermatitis bei Jugendlichen (FAS)
|
|
AD-1526 (FAS)a
| |
|
Placebo
|
Dupilumab 200 mg (< 60 kg) und 300 mg (≥ 60 kg) Q2W
| |
Randomisierte Patienten
|
85a
|
82a
| |
IGA 0 oder 1b, % Responderc
|
2,4 %
|
24,4 %
| |
EASI-50, % Responderc
|
12,9 %
|
61,0 %
| |
EASI-75, % Responderc
|
8,2 %
|
41,5 %
| |
EASI-90, % Responderc
|
2,4 %
|
23,2 %
| |
EASI, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-23,6 %
(5,49)
|
-65,9 %
(3,99)
| |
SCORAD, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-17,6 %
(3,76)
|
-51,6 %
(3,23)
| |
Pruritus NRS, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-19,0 %
(4,09)
|
-47,9 %
(3,43)
| |
Pruritus NRS (> 4 Punkte Verbesserung), % Responderc
|
4,8 %
|
36,6 %
| |
BSA, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-11,7 %
(2,72)
|
-30,1 %
(2,34)
| |
CDLQI, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-5,1
(0,62)
|
-8,5
(0,50)
| |
CDLQI (≥6 Punkte Verbesserung), % Responder
|
19,7 %
|
60,6 %
| |
POEM, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-3,8
(0,96)
|
-10,1
(0,76)
| |
POEM (≥ 6 Punkte Verbesserung), % Responder
|
9,5 %
|
63,4 %
|
a Die FAS(Full Analysis Set [Gesamtanalyse-Datensatz])-Population umfasst alle randomisierten Patienten.
b Responder wurde definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei»), mit einer Verbesserung um ≥2 Punkte auf einer von 0 bis 4 reichenden IGA-Skala.
c Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft (58,8 % im Placebo-Arm und 20,7 % im Dupixent-Arm).
Alle p-Werte < 0,0001.
Ein grösserer Anteil der in die Placebo-Gruppe randomisierten Patienten benötigte eine Rescue-Therapie (topische Kortikosteroide, systemische Kortikosteroide oder systemische nichtsteroidale Immunsuppressiva) im Vergleich zur Dupilumab-Gruppe (58,8 % bzw. 20,7 %).
Im Vergleich zu Placebo erreichte ein signifikant höherer Anteil der in die Dupilumab-Gruppe randomisierten Patienten eine schnelle Verbesserung gemäss Pruritus NRS (definiert als Verbesserung um > 4 Punkte bereits in Woche 4; nominaler p-Wert < 0,001); im Laufe des Behandlungszeitraums stieg der Patientenanteil mit einem Ansprechen gemäss Pruritus-NRS weiterhin an (siehe Abbildung 5 unten). Die Verbesserung gemäss Pruritus NRS trat im Zusammenhang mit einer Verbesserung der objektiven Anzeichen der atopischen Dermatitis auf.
Abbildung 5: Anteil der Jugendlichen mit einer Verbesserung um ≥4 Punkte gemäss Pruritus NRS in der AD-1526-Studiea (FAS)b
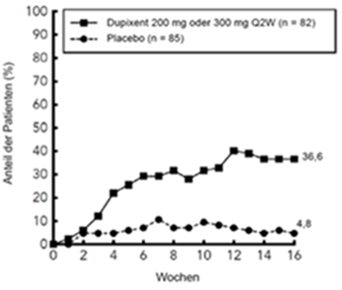
a In den Primäranalysen der Wirksamkeitsendpunkte wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die FAS(Full Analysis Set [Gesamtanalyse-Datensatz])-Population umfasst alle randomisierten Patienten.
Im Vergleich zu Placebo wurde in der Dupilumab-Gruppe in Woche 16 eine signifikante Verbesserung der von Patienten angegebenen Symptome verzeichnet. Dies galt auch für die Auswirkungen der AD auf die Schlafqualität und die gesundheitsbezogene Lebensqualität gemäss den POEM-, SCORAD- und CDLQI-Werten.
Die langfristige Wirksamkeit von Dupilumab bei Jugendlichen mit mittelschwerer bis schwerer AD, die an den vorherigen klinischen Studien mit Dupilumab teilgenommen hatten, wurde in einer unverblindeten Verlängerungsstudie (AD-1434) untersucht. Wirksamkeitsdaten aus dieser Studie weisen darauf hin, dass der in Woche 16 erzielte klinische Nutzen bis einschliesslich Woche 52 anhielt.
Klinische Wirksamkeit und Sicherheit bei Kindern (6 bis 11 Jahre) mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit der Anwendung von Dupilumab in Kombination mit TCS bei pädiatrischen Patienten wurden in einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (AD-1652) mit 367 Patienten im Alter von 6 bis 11 Jahren untersucht. Atopische Dermatitis wurde dabei definiert als ein Wert von 4 gemäss IGA (Skala von 0 bis 4), ein Wert ≥21 gemäss EASI (Skala von 0 bis 72) und eine betroffene Körperoberfläche (Body Surface Area, BSA) von ≥15 %. Patienten, die für die Studie geeignet waren und in diese eingeschlossen wurden, hatten zuvor nur unzureichend auf eine topische Medikation angesprochen. Die Rekrutierung erfolgte stratifiziert nach Ausgangsgewicht (< 30 kg; ≥30 kg).
Patienten in der Gruppe Dupilumab Q4W + TCS erhielten unabhängig vom Gewicht eine Anfangsdosis von 600 mg an Tag 1, gefolgt von 300 mg Q4W von Woche 4 bis Woche 12. Patienten in der Gruppe Dupilumab Q2W + TCS mit einem Ausgangsgewicht von < 30 kg erhielten eine Anfangsdosis von 200 mg an Tag 1, gefolgt von 100 mg Q2W von Woche 2 bis Woche 14; Patienten mit einem Ausgangsgewicht von ≥30 kg erhielten eine Anfangsdosis von 400 mg an Tag 1, gefolgt von 200 mg Q2W von Woche 2 bis Woche 14. Die Patienten konnten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten. Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In der Studie AD-1652 betrug das mittlere Alter 8,5 Jahre, das mediane Körpergewicht 29,8 kg, 50 % der Patienten waren weiblich, 69,2 % Weisse, 16,7 % Schwarze und 7,6 % Asiaten. Zu Beginn betrug die mittlere BSA-Beteiligung 57,6 %, und 16,9 % hatten in der Vergangenheit bereits systemische nichtsteroidale Immunsuppressiva erhalten. Zu Beginn betrug der mittlere EASI-Score 37,9, der wöchentliche Durchschnittswert des täglich stärksten Juckreizes lag bei 7,8 auf einer Skala von 0 bis 10, der mittlere SCORAD bei der Baseline betrug 73,6, der mittlere POEM 20,9 und der mittlere CDLQI 15,1. Insgesamt lag bei 91,7 % der Patienten mindestens eine allergische Begleiterkrankung vor; 64,4 % hatten Nahrungsmittelallergien, 62,7 % andere Allergien, 60,2 % allergische Rhinitis und 46,7 % Asthma.
Primärer Endpunkt war der Anteil der Patienten mit IGA 0 («erscheinungsfrei») oder 1 («fast erscheinungsfrei») in Woche 16. Die weiteren untersuchten Resultate umfassten den Anteil der Patienten mit EASI-75 bzw. EASI-90 (EASI-Verbesserung von mindestens 75 % bzw. 90 % gegenüber Baseline), die prozentuale Veränderung des EASI-Scores zwischen Baseline und Woche 16 sowie die Verringerung des Juckreizes gemäss Pruritus NRS (Verbesserung um ≥4 Punkte). Zusätzliche sekundäre Endpunkte beinhalteten die mittlere Veränderung der POEM- und CDLQI-Scores zwischen Baseline und Woche 16.
Tabelle 12 zeigt die nach Ausgangsgewicht stratifizierten Ergebnisse für die zugelassenen Dosierungsschemata.
Tabelle 12: Wirksamkeitsergebnisse von Dupixent mit begleitenden TCS in der Studie AD-1652 in Woche 16 (FAS)a
|
|
Dupixent
300 mg Q4Wd
+ TCS
(n = 61)
|
Placebo
+ TCS
(n = 61)
|
Dupixent
200 mg Q2We
+ TCS
(n = 59)
|
Placebo
+ TCS
(n = 62)
| |
< 30 kg
|
< 30 kg
|
≥30 kg
|
≥30 kg
| |
IGA 0 oder 1 b, % Responderc
|
29,5 %
|
13,1 %
|
39,0 %
|
9,7 %
| |
EASI-50, % Responderc
|
95,1 %
|
42,6 %
|
86,4 %
|
43,5 %
| |
EASI-75, % Responderc
|
75,4 %
|
27,9 %
|
74,6 %
|
25,8 %
| |
EASI-90, % Responderc
|
45,9 %
|
6,6 %
|
35,6 %
|
8,1 %
| |
EASI, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-84,3 %
(3,08)
|
-49,1 %
(3,30)
|
-80,4 %
(3,61)
|
-48,3 %
(3,63)
| |
SCORAD, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-65,3 %
(2,87)
|
-28,9 %
(3,05)
|
-62,7 %
(3,14)
|
-30,7 %
(3,28)
| |
Pruritus NRS, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-55,1 %
(3,94)
|
-27,0 %
(4,24)
|
-58,2 %
(4,01)
|
-25,0 %
(3,95)
| |
Pruritus NRS (≥ 4 Punkte Verbesserung), % Responderc
|
54,1 %
|
11,7 %
|
61,4 %
|
12,9 %
| |
CDLQI, mittlere LS-Veränderung gegenüber Baseline (+/- SE)
|
-11,5
(0,69)
|
-7,2
(0,76)
|
-9,8
(0,63)
|
-5,6
(0,66)
| |
CDLQI (≥ 6 Punkte Verbesserung), % Responder
|
81,8 %
|
48,3 %
|
80,8 %
|
35,8 %
| |
POEM, mittlere LS-Veränderung gegenüber Baseline (+/- SE)
|
-14,0
(0,95)
|
-5,9
(1,04)
|
-13,6
(0,90)
|
-4,7
(0,91)
|
a Die FAS-(Full Analysis Set [Gesamtanalyse-Datensatz])-Population umfasst alle randomisierten Patienten.
b Responder wurde definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei»).
c Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
d An Tag 1 erhielten die Patienten 600 mg Dupilumab.
e An Tag 1 erhielten die Patienten 200 mg (Ausgangsgewicht < 30 kg) oder 400 mg (Ausgangsgewicht ≥ 30 kg) Dupixent.
Im Vergleich zu Placebo + TCS erreichte ein grösserer Anteil an Patienten, die randomisiert Dupilumab + TCS erhielten, eine Verbesserung gemäss Pruritus NRS (definiert als Verbesserung ≥4 Punkte in Woche 16). Siehe Abbildung 6.
Abbildung 6: Anteil der pädiatrischen Patienten mit einer Verbesserung um ≥4 Punkte auf der Pruritus NRS in Woche 16 der Studie AD-1652a (FAS)b
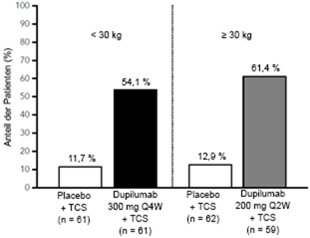
a In den Primäranalysen der Wirksamkeitsparameter wurden Patienten, die eine Rescue-Therapie erhielten oder bei denen Daten fehlten, als Non-Responder eingestuft.
b Die FAS(Full Analysis Set [Gesamtanalyse-Datensatz])-Population umfasst alle randomisierten Patienten.
Im Vergleich zu Placebo wurde in der Dupilumab-Gruppe in Woche 16 eine signifikante Verbesserung der von Patienten angegebenen Symptome verzeichnet. Dies galt auch für die Auswirkungen der AD auf die Schlafqualität und die gesundheitsbezogene Lebensqualität gemäss den POEM-, SCORAD- und CDLQI-Werten.
Die langfristige Wirksamkeit von Dupilumab + TCS bei pädiatrischen Patienten mit atopischer Dermatitis, die an den vorherigen klinischen Studien mit Dupilumab + TCS teilgenommen hatten, wurde in einer unverblindeten Verlängerungsstudie (AD-1434) untersucht. In dieser Studie zeigten die in Woche 52 verfügbaren nicht kontrollierten Daten zu 45 Patienten, dass der Anteil der Patienten mit klinisch bedeutsamen IGA- oder EASI-Verbesserungen im Laufe der Zeit stabil blieb.
Klinische Wirksamkeit und Sicherheit bei Kindern (6 Monate bis 5 Jahre) mit atopischer Dermatitis
Die Wirksamkeit und Sicherheit der Anwendung von Dupilumab in Kombination mit TCS bei pädiatrischen Patienten wurden in einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (AD-1539) bei 162 Patienten im Alter von 6 Monaten bis 5 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis untersucht. Mittelschwere bis schwere atopische Dermatitis wurde dabei definiert als ein Wert von ≥3 gemäss IGA (Skala von 0 bis 4), ein Wert ≥16 gemäss EASI (Skala von 0 bis 72) und eine betroffene Körperoberfläche (Body Surface Area, BSA) von ≥10 %. Patienten, die für die Studie geeignet waren und in diese eingeschlossen wurden, hatten zuvor nur unzureichend auf eine topische Medikation angesprochen. Die Rekrutierung erfolgte stratifiziert nach Ausgangsgewicht (≥5 bis < 15 kg und ≥15 bis < 30 kg).
Patienten in der Gruppe Dupilumab Q4W + TCS mit einem Ausgangsgewicht von ≥5 bis < 15 kg erhielten eine Anfangsdosis von 200 mg an Tag 1, gefolgt von 200 mg Q4W von Woche 4 bis Woche 12; Patienten mit einem Ausgangsgewicht von ≥15 bis < 30 kg erhielten eine Anfangsdosis von 300 mg an Tag 1, gefolgt von 300 mg Q4W von Woche 4 bis Woche 12. Die Patienten konnten nach Ermessen des Prüfarztes eine Rescue-Therapie erhalten. Patienten, die eine Rescue-Therapie erhielten, wurden als Non-Responder eingestuft.
In der Studie AD-1539 betrug das mittlere Alter 3,8 Jahre, das mediane Körpergewicht 16,5 kg, 38,9 % der Patienten waren weiblich, 68,5 % Weisse, 18,5 % Schwarze und 6,2 % Asiaten. Zu Beginn betrug der mittlere EASI-Score 34,1, 22,8 % hatten einen IGA-Wert von 3 und 77,2 % einen IGA-Wert von 4.
Primärer Endpunkt war der Anteil der Patienten mit IGA 0 («erscheinungsfrei») oder 1 («fast erscheinungsfrei») in Woche 16 und die wichtigsten sekundären Endpunkte umfassten den Anteil der Patienten mit EASI-75 (EASI-Verbesserung von mindestens 75 % gegenüber Baseline).
Klinisches Ansprechen
Die Wirksamkeitsergebnisse der Woche 16 für die Studie AD-1539 sind in Tabelle 13 dargestellt.
Tabelle 13: Wirksamkeitsergebnisse von Dupixent mit begleitenden TCS in der Studie AD-1539 in Woche 16 (FAS)a bei pädiatrischen Patienten im Alter von 6 Monaten bis 5 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis
|
|
Dupixent
200 mg (5 bis < 15 kg) oder 300 mg (15 bis < 30 kg) Q4Wd
+ TCS
(n = 83)a
|
Placebo
+ TCS
(n = 79)a
| |
IGA 0 oder 1b,c
|
27,7 %
|
3,9 %
| |
EASI-50, % Responderc
|
68,7 %
|
20,2 %
| |
EASI-75c
|
53,0 %
|
10,7 %
| |
EASI-90c
|
25,3 %
|
2,8 %
| |
EASI, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-70,0 %e
(4,85)
|
-19,6 %
(5,13)
| |
SCORAD, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-54,7 %e
(3,39)
|
-16,2 %
(3,54)
| |
Pruritus-NRS, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)
|
-49,4 %e
(5,03)
|
-2,2 %
(5,22)
| |
Pruritus-NRS (≥ 4 Punkte Verbesserung), % Responderc
|
48,1 %
|
8,9 %
| |
Schlafqualität NRS, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)*
|
2,0e
(0,25)
|
0,3
(0,26)
| |
Hautschmerzen NRS, mittlere LS-Veränderung in % gegenüber Baseline (+/- SE)*
|
-3,9e
(0,30)
|
-0,6
(0,30)
| |
POEM, mittlere LS-Veränderung gegenüber Baseline (+/- SE)*
|
-12,9e
(0,89)
|
-3,8
(0,92)
|
a Die FAS(Full Analysis Set [Gesamtanalyse-Datensatz])-Population umfasst alle randomisierten Patienten.
b Responder wurde definiert als ein Patient mit IGA 0 oder 1 («erscheinungsfrei» oder «fast erscheinungsfrei»).
c Patienten, die eine Rescue-Therapie erhielten (62,8 % und 19 % in der Placebo- bzw. Dupixent-Gruppe) oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
d An Tag 1 erhielten die Patienten 200 mg (5 bis < 15 kg) oder 300 mg (15 bis < 30 kg) Dupixent
e Werte von p < 0,0001
*Von Pflegeperson berichtetes Ergebnis
Im Vergleich zu Placebo + TCS erreichte ein grösserer Anteil an Patienten, die randomisiert Dupilumab + TCS erhielten, eine Verbesserung gemäss Pruritus-NRS (definiert als Verbesserung ≥ 4 Punkte ab Woche 3, nominaler p-Wert < 0,005), und der Anteil der Patienten, die gemäss Pruritus NRS ansprachen, stieg während der gesamten Behandlung weiter an (siehe Abbildung 7).
Abbildung 7: Anteil der pädiatrischen Patienten im Alter von 6 Monaten bis 5 Jahren mit einer Verbesserung um ≥4 Punkte auf der Pruritus NRS in Woche 16 der Studie AD-1539a (FAS)b
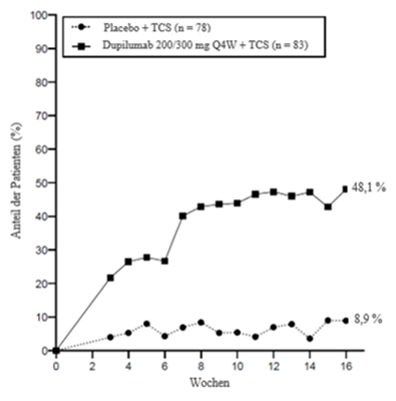
Die langfristige Wirksamkeit und Sicherheit von Dupilumab + TCS bei pädiatrischen Patienten mit mittelschwerer bis schwerer atopischer Dermatitis, die an den vorherigen klinischen Studien mit Dupilumab + TCS teilgenommen hatten, wurden in einer unverblindeten Verlängerungsstudie (AD-1434) untersucht. In dieser Studie zeigen die Wirksamkeitsdaten, dass die in Woche 16 erreichten klinischen Verbesserungen bis Woche 52 anhalten. Das Sicherheitsprofil von Dupilumab bei den Patienten, die bis Woche 52 nachbeobachtet wurden, war vergleichbar mit dem Sicherheitsprofil, das in Woche 16 in der Studie AD-1539 beobachtet wurde.
Atopische Dermatitis der Hände und Füsse
Die Wirksamkeit und Sicherheit von Dupilumab wurden in einer 16-wöchigen multizentrischen, randomisierten, doppelblinden, placebokontrollierten Parallelgruppenstudie (AD-1924) mit 106 erwachsenen und 27 pädiatrischen Patienten im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis der Hände und Füsse, definiert durch einen IGA-Score (Hände und Füsse) ≥3 (auf einer Skala von 0 bis 4) und einen Score für den maximalen Pruritus ≥4 (auf einer Skala von 0 bis 10) auf der numerischen Bewertungsskala für den maximalen Pruritus (NRS) der Hände und Füsse, untersucht. Die geeigneten Patienten hatten zuvor nur unzureichend auf die topische Behandlung der atopischen Dermatitis der Hände und Füsse angesprochen oder haben diese nicht vertragen.
Zu Studienbeginn waren 38 % der Patienten männlich, 80 % waren weiss, 72 % der Patienten hatten bei Studieneinschluss einen IGA-Score (Hände und Füsse) von 3 (mittelschwere atopische Dermatitis der Hände und Füsse) und 28 % der Patienten hatten bei Studieneinschluss einen IGA-Score (Hände und Füsse) von 4 (schwere atopische Dermatitis der Hände und Füsse). Der wöchentliche Durchschnittswert gemäss Pruritus NRS der Hände und Füsse betrug bei Studieneinschluss 7,1.
Der primäre Endpunkt war der Anteil der Patienten mit einem IGA-Score für Hände und Füsse von 0 («erscheinungsfrei») oder 1 («fast erscheinungsfrei») in der Woche 16. Der wichtigste sekundäre Endpunkt war die Verringerung des Juckreizes gemessen an der numerischen Bewertungsskala für den maximalen Pruritus der Hände und Füsse (Verbesserung um ≥4 Punkte). Die anderen Patienten-berichtete Endpunkte umfassten Hautschmerzen NRS (0–10) der Hände und Füsse, Schlafqualität NRS (0–10), den Wert für die Lebensqualität im Fragebogen zu Handekzem (0–117) (QoLHEQ) sowie den Score für die Arbeitsproduktivität und die Beeinträchtigung von Aktivitäten (WPAI) (0–100 %).
Der Anteil an Patienten mit einem IGA (Hände und Füsse) von 0 («erscheinungsfrei») oder 1 («fast erscheinungsfrei») in der Woche 16 betrug 40,3 % bei Dupilumab und 16,7 % bei Placebo (Behandlungsunterschied 23,6 %; 95 %-KI: 8,84; 38,42; p-Wert = 0,0030). Der Anteil an Patienten mit Verbesserung (Verringerung) des wöchentlichen Durchschnittswerts gemäss Pruritus NRS der Hände und Füsse ≥4 in der Woche 16 betrug 52,2 % bei Dupilumab und 13,6 % bei Placebo (Behandlungsunterschied 38,6 %; 95 %-KI: 24,06; 53,15; p-Wert = 0,0001).
In der Dupilumab-Gruppe wurden grössere Verbesserungen des Hautschmerzen NRS für Hände und Füsse, des Schlafqualität NRS, des QoLHEQ-Scores für die Arbeitsproduktivität und die Beeinträchtigung von Aktivitäten zwischen Studieneinschluss und Woche 16 im Vergleich zur Placebo-Gruppe beobachtet (mittlere LS- Veränderung von Dupilumab im Vergleich zum Placebo: -4,66 vs. -1,93 (p < 0,0001); 0,88 vs. -0,00 (p < 0,05); -40,28 vs. -16,18 (p < 0,0001); -38,57 % vs. -22,83 % (nominaler p-Wert < 0,001) und -36,39 % vs. -21,26 % (nominaler p-Wert < 0,001)).
2) Asthma
Klinische Wirksamkeit und Sicherheit bei Erwachsenen und Jugendlichen mit Asthma:
Das Entwicklungsprogramm zu Asthma beinhaltete drei randomisierte, doppelblinde, placebokontrollierte, multizentrische Parallelgruppenstudien (DRI12544, QUEST und VENTURE) mit 24- bis 52-wöchiger Behandlungsdauer, in die insgesamt 2'888 Patienten (ab 12 Jahren) eingeschlossen wurden.
Die Patienten wurden unabhängig von ihren Eosinophilen-(EOS-)Werten im Blut bzw. ihrer FeNO- oder IgE-Konzentration aufgenommen.
In den Studien DRI12544 und QUEST umfassten die präspezifizierten Subgruppenanalysen EOS-Werte im Blut von ≥150 und ≥300 Zellen/μl.
Bei DRI12544 handelte es sich um eine 24-wöchige Dosisfindungsstudie, in die 776 Patienten (ab 18 Jahren) eingeschlossen wurden. Dupilumab wurde bei erwachsenen Patienten mit mittelschwerem bis schwerem Asthma, die ein mittel- bis hochdosiertes inhalatives Kortikosteroid und ein langwirksames Beta-2-Sympathomimetikum anwendeten, im Vergleich zu Placebo beurteilt. Der primäre Endpunkt war die Veränderung des FEV1 (Einsekundenkapazität, forciertes Exspirationsvolumen (1 s)) (Liter) zwischen dem Ausgangswert und Woche 12 bei Patienten mit einem Eosinophilen-Ausgangswert ≥300 Zellen/µl. Die schweren Asthmaexazerbationsereignisse während des 24-wöchigen placebokontrollierten Behandlungszeitraums wurden ebenfalls für einen einjährigen Zeitraum bestimmt. Die Auswertung der Ergebnisse erfolgte für die Gesamtpopulation (ohne Einschränkung des Mindestausgangswerts für Eosinophile) und die Subgruppen, die auf dem Eosinophilen-Ausgangswert im Blut beruhten.
Bei QUEST handelte es sich um eine 52-wöchige konfirmatorische Studie, in die 1'902 Patienten (ab 12 Jahren) eingeschlossen wurden. Dupilumab im Vergleich zu Placebo wurde bei 107 jugendlichen und 1'795 erwachsenen Patienten mit persistierendem Asthma, die ein mittel- bis hochdosiertes inhalatives Kortikosteroid (ICS) und ein zweites Arzneimittel als Basistherapie anwendeten, beurteilt. Patienten, die ein drittes Arzneimittel als Basistherapie benötigten, durften ebenfalls an dieser Studie teilnehmen. Nach der Randomisierung erhielten die Patienten entweder 200 mg (n = 631) oder 300 mg (n = 633) Dupilumab alle zwei Wochen (oder das entsprechende Placebo alle zwei Wochen entweder 200 mg (n = 317) oder 300 mg (n = 321)) nach einer Anfangsdosis von 400 mg bzw. 600 mg bzw. Placebo. Die primären Endpunkte waren die Jahresrate schwerer Exazerbationsereignisse während des 52-wöchigen placebokontrollierten Zeitraums und die Veränderung des FEV1 vor Anwendung eines Bronchodilatators zwischen der Baseline und Woche 12 in der Gesamtpopulation (ohne Einschränkung des Mindestausgangswerts für Eosinophile).
Die zusätzlichen sekundären Endpunkte umfassten die Jahresrate schwerer Exazerbationsereignisse und FEV1 in den Subgruppen, die auf dem Eosinophilen-Ausgangswert im Blut beruhten.
Bei VENTURE handelte es sich um eine 24-wöchige Studie zur Reduktion von oralen Kortikosteroiden (OCS) bei 210 Asthmapatienten ohne Einschränkung der Biomarker-Ausgangswerte für die Typ-2-Inflammation, die täglich orale Kortikosteroide zusätzlich zur regelmässigen Anwendung hochdosierter inhalativer Kortikosteroide plus ein weiteres Arzneimittel als Basistherapie benötigten. Nach Optimierung der OCS-Dosis während des Screeningzeitraums erhielten die Patienten 300 mg Dupilumab (n = 103) oder Placebo (n = 107) einmal alle zwei Wochen über 24 Wochen, nach einer Anfangsdosis von 600 mg oder Placebo. Während der Studie erhielten die Patienten weiter ihre bestehende Asthmamedikation als Basistherapie, die OCS-Dosis wurde jedoch während der OCS-Reduktionsphase (Woche 4–20) alle 4 Wochen verringert, solange die Asthmakontrolle aufrechterhalten werden konnte. Der primäre Endpunkt war die prozentuale Verringerung der oralen Kortikosteroiddosis, beurteilt in der Gesamtpopulation, beruhend auf einem Vergleich der oralen Kortikosteroiddosis in den Wochen 20 bis 24, bei der die Asthmakontrolle aufrechterhalten wurde, mit der zuvor (bei Studienbeginn) optimierten Dosis oraler Kortikosteroide in der Gesamtpopulation (ohne Einschränkung des Mindestausgangswerts für Eosinophile). Weitere sekundäre Endpunkte umfassten die Jahresrate schwerer Exazerbationsereignisse im Behandlungszeitraum und den Prozentsatz der Responder bei dem ACQ-5- und AQLQ-Score (S).
Die demografischen Angaben und Baseline-Charakteristika bzgl. Asthma sind für diese 3 Studien in Tabelle 14 dargestellt.
Tabelle 14: Demografische Angaben und Baseline-Charakteristika in den Asthma-Studien
|
Parameter
|
DRI12544
(n = 776)
|
QUEST
(n = 1'902)
|
VENTURE
(n = 210)
| |
Mittleres Alter in Jahren (SD)
|
48,6 (13,0)
|
47,9 (15,3)
|
51,3 (12,6)
| |
% weiblich
|
63,1
|
62,9
|
60,5
| |
% weiss
|
78,2
|
82,9
|
93,8
| |
Dauer des Asthmas (Jahre), Mittelwert ± SD
|
22,03 (15,42)
|
20,94 (15,36)
|
19,95 (13,90)
| |
Nie geraucht, (%)
|
77,4
|
80,7
|
80,5
| |
Mittlere Anzahl der Exazerbationen im vorherigen Jahr ± SD
|
2,17 (2,14)
|
2,09 (2,15)
|
2,09 (2,16)
| |
Anwendung hochdosierter ICS (%)a
|
49,5
|
51,5
|
88,6
| |
FEV1 (l) vor Verabreichung bei der Baseline ± SD
|
1,84 (0,54)
|
1,78 (0,60)
|
1,58 (0,57)
| |
Mittlere prozentuelle Veränderung des FEV1 bei der Baseline (%) (± SD)
|
60,77 (10,72)
|
58,43 (13,52)
|
52,18 (15,18)
| |
% Reversibilität (± SD)
|
26,85 (15,43)
|
26,29 (21,73)
|
19,47 (23,25)
| |
Mittlerer ACQ-5-Score (± SD)
|
2,74 (0,81)
|
2,76 (0,77)
|
2,50 (1,16)
| |
Mittlerer AQLQ-Score (± SD)
|
4,02 (1,09)
|
4,29 (1,05)
|
4,35 (1,17)
| |
Atopische Krankheiten in der Anamnese% insgesamt
(AD %, NP %, AR %)
|
72,9
(8,0, 10,6, 61,7)
|
77,7
(10,3, 12,7, 68,6)
|
72,4
(7,6, 21,0, 55,7)
| |
Mittelwert FeNO in ppb (± SD)
|
39,10 (35,09)
|
34,97 (32,85)
|
37,61 (31,38)
| |
% Patienten mit FeNO ppb
| |
≥ 25
|
49,9
|
49,6
|
54,3
| |
≥ 50
|
21,6
|
20,5
|
25,2
| |
Mittelwert Gesamt-IgE, I.E./ml (± SD)
|
435,05 (753,88)
|
432,40 (746,66)
|
430,58 (775,96)
| |
Mittlere Eosinophilenzahl bei der Baseline (± SD) in Zellen/µl
|
350 (430)
|
360 (370)
|
350 (310)
| |
% Patienten mit EOS
| |
≥ 150 Zellen/µl
|
77,8
|
71,4
|
71,4
| |
≥ 300 Zellen/µl
|
41,9
|
43,7
|
42,4
|
ICS = inhalatives Kortikosteroid; FEV1 = Einsekundenkapazität (forciertes Exspirationsvolumen (1 s));
ACQ-5 = Fragebogen zur Asthmakontrolle (Asthma Control Questionnaire-5); AQLQ = Fragebogen zur Lebensqualität bei Asthma (Asthma Quality of Life Questionnaire); AD = atopische Dermatitis; NP = Nasenpolypen; AR = allergische Rhinitis; FeNO = fraktioniertes exhaliertes Stickstoffmonoxid; EOS = Eosinophilenzahl im Blut
a Die Population, die in Studien zur Therapie von Asthma mit Dupilumab behandelt wurde, umfasste Patienten mit mittleren und hochdosierten ICS. Die mittlere ICS-Dosis war definiert als gleichwertig zu 500 μg Fluticason oder Äquivalent pro Tag.
Exazerbationen
Die Studien DRI12544 und QUEST untersuchten die Häufigkeit schwerer Asthmaexazerbationen, definiert als eine Verschlechterung des Asthmas, die die Anwendung systemischer Kortikosteroide über mindestens 3 Tage, eine Hospitalisierung oder einen Besuch der Notfallstation aufgrund eines Asthmas, das systemische Kortikosteroide erfordert, notwendig macht.
In der Primäranalysenpopulation (Patienten mit einem Eosinophilen-Ausgangswert ≥300 Zellen/µl in der Studie DRI12544 und der Gesamtpopulation in der Studie QUEST) kam es bei den Teilnehmern, die Dupixent 200 mg oder 300 mg Q2W erhielten, zu einem signifikativen Rückgang der Rate schwerer Asthmaexazerbationen im Vergleich zu Placebo. In der Gesamtpopulation der Studie QUEST lag die Rate schwerer Exazerbationen bei 0,46 für Dupixent 200 mg Q2W bzw. 0,52 für Dupixent 300 mg Q2W, unter Placebo bei 0,87 bzw. 0,97.
Die Rate schwerer Exazerbationen im Vergleich zu Placebo lag bei 0,52 (95 %-KI: 0,41, 0,66) und 0,54 (95 %-KI: 0,43, 0,68) für Dupixent 200 mg Q2W bzw. 300 mg Q2W. Die Ergebnisse für die Patienten mit einem Eosinophilen-Ausgangswert im Blut ≥150 Zellen/µl und ≥300 Zellen/µl in den Studien DRI12544 und QUEST sind in Tabelle 15 dargestellt.
Die Responderquote nach Eosinophilen-Ausgangswert für die Studie QUEST ist in Abbildung 8 angegeben. Die präspezifizierten Subgruppenanalysen der Studien DRI12544 und QUEST belegten stärkere Verringerungen von schweren Exazerbationen bei Teilnehmern mit höheren Eosinophilen-Ausgangswerten im Blut. In der Studie QUEST war die Reduzierung von Exazerbationen in der Subgruppe mit einem Eosinophilen-Ausgangswert im Blut ≥150 Zellen/µl signifikant. Bei Patienten mit Eosinophilen-Werten im Blut < 150 Zellen/µl war die Rate schwerer Exazerbationen unter Dupixent und Placebo vergleichbar.
In der Studie QUEST lag die Rate schwerer Exazerbationen, die zu Hospitalisierungen und/oder Besuchen der Notfallstation führten, im Vergleich zu Placebo bei 0,53 (95 %-KI: 0,28, 1,03) und 0,74 (95 %-KI: 0,32, 1,70) unter Dupixent 200 mg bzw. 300 mg Q2W.
Tabelle 15: Rate schwerer Exazerbationen in DRI12544 und QUEST
|
Behandlung
|
EOS-Wert im Blut
| |
|
≥ 150 Zellen/µl
|
≥ 300 Zellen/µl
| |
Exazerbationen pro Jahr
|
% Reduktion
|
Exazerbationen pro Jahr
|
%
Reduktion
| |
N
|
Rate
(95 %-KI)
|
Ratenverhältnis (95 %-KI)
|
N
|
Rate
(95 %-KI)
|
Ratenverhältnis (95 %-KI)
| |
Schwere Exazerbationen gesamt
| |
DRI12544 Studie
| |
Dupilumab 200 mg Q2W
|
120
|
0,29
(0,16, 0,53)
|
0,28a
(0,14, 0,55)
|
72 %
|
65
|
0,30
(0,13, 0,68)
|
0,29c
(0,11, 0,76)
|
71 %
| |
Dupilumab 300 mg
Q2W
|
129
|
0,28
(0,16, 0,50)
|
0,27b
(0,14, 0,52)
|
73 %
|
64
|
0,20
(0,08, 0,52)
|
0,19d
(0,07, 0,56)
|
81 %
| |
Placebo
|
127
|
1,05
(0,69, 1,60)
|
|
|
68
|
1,04
(0,57, 1,90)
|
|
| |
QUEST Studie
| |
Dupilumab 200 mg
Q2W
|
437
|
0,45
(0,37, 0,54)
|
0,44e
(0,34,0,58)
|
56 %
|
264
|
0,37
(0,29, 0,48)
|
0,34e
(0,24,0,48)
|
66 %
| |
Placebo
|
232
|
1,01
(0,81, 1,25)
|
|
|
148
|
1,08
(0,85, 1,38)
|
|
| |
Dupilumab 300 mg
Q2W
|
452
|
0,43
(0,36, 0,53)
|
0,40 e
(0,31,0,53)
|
60 %
|
277
|
0,40
(0,32, 0,51)
|
0,33e
(0,23,0,45)
|
67 %
| |
Placebo
|
237
|
1,08
(0,88, 1,33)
|
|
|
142
|
1,24
(0,97, 1,57)
|
|
|
Abbildung 8: Relatives Risiko in Bezug auf die jährliche Rate schwerer Exazerbationen nach Eosinophilen-Ausgangswerten im Blut (Zellen/µl) in QUEST

In der Studie QUEST verging bei Patienten unter Dupixent mehr Zeit bis zur ersten Exazerbation als bei Patienten unter Placebo (Abbildung 9).
Abbildung 9: Kaplan-Meier-Kurve der Zeitspanne bis zur ersten schweren Exazerbation bei Patienten mit Eosinophilen-Ausgangswerten im Blut ≥300 Zellen/µl (QUEST)a
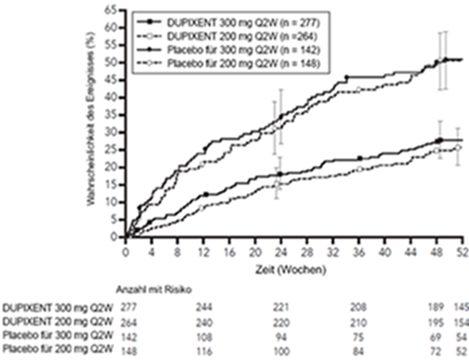
a Zum Zeitpunkt des Schliessens der Datenbank hatten noch nicht alle Patienten Woche 52 beendet.
Lungenfunktion
Klinisch signifikante Zunahmen des FEV1 vor Anwendung eines Bronchodilatators wurden in Woche 12 der Studien DRI12544 und QUEST in den Primäranalysenpopulationen (Patienten mit einem Eosinophilen-Ausgangswert ≥300 Zellen/µl in der Studie DRI12544 und der Gesamtpopulation in der Studie QUEST) beobachtet. In der Gesamtpopulation der Studie QUEST lag die mittlere Veränderung des FEV1 (l) gegenüber dem Ausgangswert bei 0,32 l (21%) und 0,34 l (23 %) für Dupixent 200 mg Q2W bzw. 300 mg Q2W im Vergleich zur durchschnittlichen Veränderung unter Placebo von 0,18 l (12 %) bzw. 0,21 l (14 %).
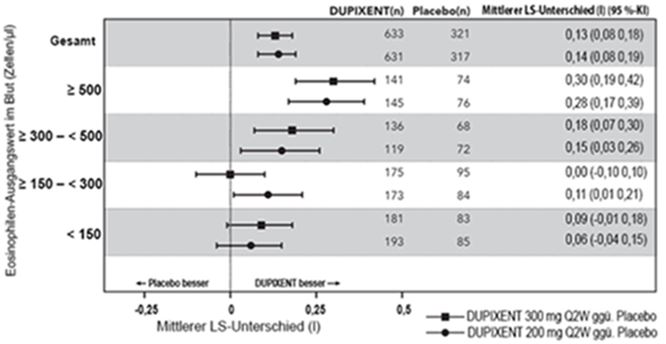
Der mittlere Behandlungsunterschied gegenüber Placebo betrug 0,14 l (95 %-KI: 0,08, 0,19) bzw. 0,13 l (95 %-KI: 0,08, 0,18) für Dupixent 200 mg Q2W bzw. 300 mg Q2W. Die Ergebnisse bei den Patienten mit einem Eosinophilen-Ausgangswert im Blut ≥150 Zellen/µl und ≥300 Zellen/µl in den Studien DRI12544 und QUEST sind in Tabelle 16 aufgeführt. Die Verbesserungen des FEV1 je nach Eosinophilen-Ausgangswert für die Studie QUEST sind in Abbildung 10 dargestellt. Die Subgruppenanalyse der Studien DRI12544 und QUEST ergab eine grössere Verbesserung bei den Teilnehmern mit einem höheren Eosinophilen-Ausgangswert.
Tabelle 16: Mittlere Veränderung des FEV1 vor Anwendung eines Bronchodilatators gegenüber der Baseline in Woche 12 in DRI12544 und QUEST
|
Behandlung
|
EOS-Ausgangswert im Blut
| |
|
≥150 Zellen/µl
|
≥300 Zellen/µl
| |
n
|
Mittlere LS-Veränderung
gegenüber der
Baseline
l (%)
|
Mittlerer LS-Unterschied
vs.
Placebo (95 %-
KI)
|
n
|
Mittlere LS-Veränderung
gegenüber der
Baseline
l (%)
|
Mittlerer LS-Unterschied
vs.
Placebo
(95 %-KI)
| |
Studie DRI12544
| |
Dupilumab 200 mg Q2W
|
120
|
0,32 (18,25)
|
0,23a
(0,13, 0,33)
|
65
|
0,43 (25,9)
|
0,26c
(0,11, 0,40)
| |
Dupilumab 300 mg Q2W
|
129
|
0,26 (17,1)
|
0,18b
(0,08, 0,27)
|
64
|
0,39 (25,8)
|
0,21d
(0,06, 0,36)
| |
Placebo
|
127
|
0,09 (4,36)
|
|
68
|
0,18 (10,2)
|
| |
Studie QUEST
| |
Dupilumab 200 mg Q2W
|
437
|
0,36 (23,6)
|
0,17e
(0,11, 0,23)
|
264
|
0,43 (29,0)
|
0,21e
(0,13, 0,29)
| |
Placebo
|
232
|
0,18 (12,4)
|
|
148
|
0,21 (15,6)
|
| |
Dupilumab 300 mg Q2W
|
452
|
0,37 (25,3)
|
0,15e
(0,09, 0,21)
|
277
|
0,47 (32,5)
|
0,24e
(0,16, 0,32)
| |
Placebo
|
237
|
0,22 (14,2)
|
|
142
|
0,22 (14,4)
|
|
a p-Wert < 0,0001, b p-Wert = 0,0004, c p-Wert = 0,0008, d p-Wert = 0,0063, e p-Wert < 0,0001
Abbildung 10: Mittlerer LS-Unterschied im Vergleich zu Placebo bzgl. FEV1 vor Anwendung eines Bronchodilatators gegenüber der Baseline in Woche 12 der QUEST-Studie nach Eosinophilen-Ausgangswert im Blut (Zellen/µl) in QUEST
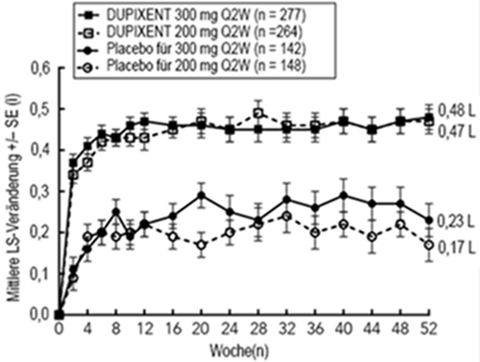
Die mittlere Veränderung des FEV1 im Zeitverlauf in der QUEST-Studie ist in Abbildung 11 dargestellt.
Abbildung 11: Mittlere Veränderung des FEV1 (l) vor Anwendung eines Bronchodilatators gegenüber der Baseline im Zeitverlauf bei Patienten mit einem Eosinophilen-Ausgangswert ≥300 Zellen/µl (QUEST)
Weitere sekundäre Endpunkte:
Ergebnisse zur Lebensqualität / Patienten-berichtete Endpunkte bei Asthma
Endpunkte bezüglich ACQ-5 und AQLQ(S) wurden in der QUEST-Studie nach 52 Wochen analysiert. Die Responderrate war definiert als eine Verbesserung des Wertes um mindestens 0,5 Punkte (Skalenbereich 0–6 für ACQ-5 und 1–7 für AQLQ(S)).
·Die ACQ-5-Responderrate für Dupilumab 200 mg und 300 mg Q2W in der Gesamtpopulation betrug 69 % gegenüber 62 % unter Placebo (Odds Ratio 1,37; 95 %-KI: 1,01, 1,86) bzw. 69 % gegenüber 63 % unter Placebo (Odds Ratio 1,28; 95 %-KI: 0,94, 1,73). Die AQLQ(S)-Responderrate belief sich auf 62 % gegenüber 54 % unter Placebo (Odds Ratio 1,61; 95 %-KI: 1,17, 2,21) bzw. 62 % gegenüber 57 % unter Placebo (Odds Ratio 1,33; 95 %-KI: 0,98, 1,81).
·Die ACQ-5-Responderrate für Dupilumab 200 mg und 300 mg Q2W bei Patienten mit einem Eosinophilen-Ausgangswert ≥300 Zellen/μl betrug 75 % gegenüber 67 % unter Placebo (Odds Ratio: 1,46; 95 %-KI: 0,90, 2,35) bzw. 71% gegenüber 64 % unter Placebo (Odds Ratio: 1,39; 95 %-KI: 0,88, 2,19). Die AQLQ(S)-Responderrate belief sich auf 71 % gegenüber 55 % unter Placebo (Odds Ratio: 2,02; 95 %-KI: 1,24, 3,32) bzw. 65 % gegenüber 55 % unter Placebo (Odds Ratio: 1,79; 95 %-KI: 1,13, 2,85).
Studie zur Reduktion oraler Kortikosteroide (VENTURE)
In der VENTURE-Studie wurde die Wirkung von Dupilumab in Bezug auf die Reduktion der Anwendung oraler Kortikosteroide für die Erhaltungstherapie untersucht. Bei der Baseline lag die im Mittel angewendete Dosis oraler Kortikosteroide bei 12 mg in der Placebo-Gruppe und 11 mg in der Gruppe, die Dupilumab erhielt. Primärer Endpunkt war die Reduktionsrate der Enddosis oraler Kortikosteroide gegenüber der Baseline in Woche 24, bei der die Asthmakontrolle aufrechterhalten wurde.
Patienten, die mit Dupilumab behandelt wurden, erzielten im Vergleich zu Patienten unter Placebo eine stärkere Reduktion der täglichen Dosis oraler Kortikosteroide unter Aufrechterhaltung der Asthmakontrolle. Die durchschnittliche Reduktionsrate der täglichen OCS-Dosis gegenüber Baseline lag bei Patienten, die Dupilumab erhielten, bei 70 % (median 100 %) (95 %-KI: 60 %, 80 %) im Vergleich zu 42 % (median 50 %) bei Patienten unter Placebo (95 %-KI: 33 %, 51 %). Bei 82 (80 %) der Patienten unter Dupilumab vs. 57 (53 %) der Patienten unter Placebo wurden Reduktionen der OCS-Dosis von mindestens 50 % beobachtet. Der Anteil der Patienten mit einer durchschnittlichen Enddosis unter 5 mg in Woche 24 lag für Dupilumab bei 72 % und für Placebo bei 37 % (Odds Ratio 4,48; 95 %-KI: 2,39, 8,39). Insgesamt 54 (52 %) Patienten, die Dupilumab erhielten, im Vergleich zu 31 (29 %) Patienten in der Placebo-Gruppe erzielten eine Reduktion ihrer OCS-Dosis von 100 %.
In dieser 24-wöchigen Studie traten bei Patienten, die mit Dupilumab behandelt wurden, weniger Asthmaexazerbationen (definiert als vorübergehende Erhöhung der Dosis oraler Kortikosteroide über mindestens 3 Tage) auf als bei Patienten, die Placebo erhielten (annualisierte Rate 0,65 und 1,60 für die Dupilumab- bzw. Placebo-Gruppe; Odds Ratio 0,41 [95 %-KI 0,26, 0,63]), und die Verbesserung des FEV1 vor Anwendung eines Bronchodilatators zwischen der Baseline und Woche 24 war bei Patienten, die Dupilumab erhielten, grösser als unter Placebo (mittlere LS-Veränderung für Dupilumab versus Placebo von 0,22 l [95 %-KI: 0,09 bis 0,34 l]). Die Auswirkungen auf die Lungenfunktion, die Anwendung oraler Steroide und die Verringerung von Exazerbationen fielen, unabhängig von den Eosinophilen-Ausgangswerten im Blut, ähnlich aus. Die Fragebögen ACQ-5 und AQLQ(S) wurden in der VENTURE-Studie ebenfalls beurteilt und ergaben ähnliche Verbesserungen, wie sie in der QUEST-Studie festgestellt wurden.
Langzeit-Verlängerungsstudie (TRAVERSE)
Die Langzeitsicherheit von Dupilumab wurde in der unverblindeten Verlängerungsstudie (TRAVERSE) bei 2'193 Erwachsenen und 89 Jugendlichen mit mittelschwerem bis schwerem Asthma untersucht; darunter 185 Erwachsene mit oral steroidpflichtigem Asthma, die an früheren klinischen Studien mit Dupilumab (DRI12544, QUEST und VENTURE) teilgenommen hatten (siehe «Unerwünschte Wirkungen»). Die als sekundärer Endpunkt gemessene Wirksamkeit war mit den in den Zulassungsstudien beobachteten Ergebnissen vergleichbar und hielt bis zu 96 Wochen an. Bei Erwachsenen mit oral steroidpflichtigem Asthma wurde trotz Dosisreduktion bzw. Absetzen des oralen Kortikosteroids eine anhaltendende Verringerung der Exazerbationen sowie eine Verbesserung der Lungenfunktion während bis zu 96 Wochen beobachtet.
Klinische Wirksamkeit und Sicherheit bei Kindern (6 bis 11 Jahre) mit Asthma:
Die Wirksamkeit und Sicherheit von Dupilumab bei pädiatrischen Patienten wurden in einer 52-wöchigen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (VOYAGE) mit 408 Patienten im Alter von 6 bis 11 Jahren mit mittelschwerem bis schwerem Asthma untersucht, die ein mittel- oder hochdosiertes inhalatives Kortikosteroid (ICS) und ein weiteres Arzneimittel zur Erhaltungstherapie oder hochdosiertes ICS allein erhielten. Die Patienten erhielten randomisiert Dupilumab (n = 273) oder ein entsprechendes Placebo (n = 135) alle zwei Wochen jeweils auf Grundlage ihres Körpergewichts von ≤30 kg oder > 30 kg.
Der primäre Endpunkt war die jährliche Rate schwerer Exazerbationsereignisse während des 52-wöchigen placebokontrollierten Zeitraums, und der sekundäre Endpunkt war die prozentuale Veränderung des vorhergesagten FEV1 in Woche 12 vor Anwendung eines Bronchodilatators im Vergleich zur Baseline. Weitere sekundäre Endpunkte waren die mittleren Veränderungen gegenüber der Baseline und die Responderraten bei ACQ-7-IA und PAQLQ(S)-IA.
Die demografischen Daten und Baseline-Charakteristika der VOYAGE-Studie sind in Tabelle 17 dargestellt.
Tabelle 17: Demografische Angaben und Baseline-Charakteristika in der VOYAGE-Studie
|
Parameter
|
ITT
(n = 408)
| |
Mittleres Alter in Jahren (SD)
|
8,9 (1,6)
| |
% weiblich
|
35,8
| |
% weiss
|
88,2
| |
Mittleres Körpergewicht (kg)
|
35,91
| |
Mittlere Anzahl der Exazerbationen im vorherigen Jahr ± SD
|
2,44 (2,18)
| |
ICS-Dosis (%)
|
| |
Mittel
|
55,1
| |
Hoch
|
44,1
| |
FEV1 (l) vor Anwendung bei Baseline (± SD)
|
1,48 (0,41)
| |
Mittlerer Prozentsatz der vorhergesagten FEV1 (%) (± SD)
|
78,07 (14,72)
| |
Mittlere Reversibilität in % (± SD)
|
19,58 (20,76)
| |
Mittlerer ACQ-7-IA-Score (± SD)
|
2,13 (0,73)
| |
Mittlerer PAQLQ(S)-IA-Score (± SD)
|
4,91 (1,13)
| |
Atopische Krankheiten in der Anamnese % insgesamt
(AD %, AR %)
|
92,4
(36,3, 81,9)
| |
Median Gesamt-IgE I.E./ml ± SD)
|
792,28 (1093,46)
| |
Mittelwert FeNO in ppb (± SD)
|
27,71 (23,84)
| |
% Patienten mit FeNO ppb ≥20
|
49,7
| |
Mittlere Eosinophilenzahl bei Baseline (± SD) in Zellen/µl
|
500 (400)
| |
% Patienten mit EOS
| |
≥150 Zellen/µl
|
81,1
| |
≥300 Zellen/µl
|
63,5
|
Exazerbationen waren definiert als eine Verschlechterung des Asthmas, welche die Anwendung systemischer Kortikosteroide über mindestens 3 Tage, eine Hospitalisierung oder einen Besuch der Notfallstation aufgrund eines Asthmas, das systemische Kortikosteroide erfordert, notwendig macht. In der Population mit Typ-2-Inflammation (Eosinophilenzahl im Blut ≥150 Zellen/µl oder FeNO ≥20 ppb) und in der Population mit einer Eosinophilenzahl im Blut ≥300 Zellen/µl bei Baseline führte Dupilumab zu einer signifikanten Verringerung der jährlichen Rate schwerer Asthmaexazerbationsereignisse während des 52-wöchigen Behandlungszeitraums im Vergleich zu Placebo. Subgruppenanalysen zur Wirksamkeit von Dupilumab, die auf dem Eosinophilen-Ausgangswert oder dem FeNO-Ausgangswert beruhten, zeigten ähnliche Ergebnisse wie die klinischen Studien mit jugendlichen Patienten (12 bis 17 Jahre) und erwachsenen Patienten (siehe oben). Bei Patienten mit einer Eosinophilenzahl im Blut < 150 Zellen/µl bei der Baseline oder einem FeNO-Wert < 20 ppb war die jährliche Rate der beobachteten schweren Asthmaexazerbationsereignisse bei Dupilumab und Placebo vergleichbar. Die Wirksamkeitsergebnisse der VOYAGE-Studie sind in Tabelle 18 dargestellt.
Tabelle 18: Rate schwerer Exazerbationen, mittlere Veränderung des FEV1 gegenüber Baseline
|
Behandlung
|
EOS ≥150 Zellen/µl oder FeNO ≥20 ppb
|
EOS
≥300 Zellen/µl
| |
Jährliche Rate schwerer Exazerbationen innerhalb von 52 Wochen
| |
|
N
|
Rate
(95 %-KI)
|
Rate
(95 %-KI)
|
N
|
Rate
(95 %-KI)
|
Rate
(95 %-KI)
| |
Dupixent
100 mg alle 2 Wochen (< 30 kg)/
200 mg alle 2 Wochen (≥ 30 kg)
|
236
|
0,305
(0,223, 0,416)
|
0,407
(0,274, 0,605)
|
175
|
0,235
(0,160, 0,345)
|
0,353
(0,222, 0,562)
| |
Placebo
|
114
|
0,748
(0,542, 1,034)
|
|
84
|
0,665
(0,467, 0,949)
|
| |
Mittlere prozentuale Veränderung des vorhergesagten FEV1 in Woche 12 gegenüber Baseline
| |
|
N
|
Mittlere LS-Veränderungen gegenüber Baseline in % des vorhergesagten FEV1
|
N
|
Mittlere LS-Veränderungen gegenüber Baseline in % des vorhergesagten FEV1
| |
Dupixent
100 mg alle 2 Wochen (< 30 kg)/
200 mg alle 2 Wochen (≥30 kg)
|
229
|
10,53
|
168
|
10,15
| |
Placebo
|
110
|
5,32
|
80
|
4,83
|
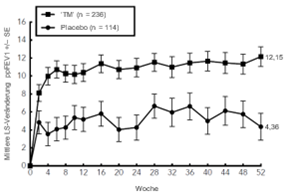
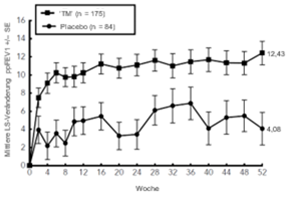
In Woche 12 wurden signifikante prozentuale Verbesserungen des vorhergesagten FEV1 vor Anwendung des Bronchodilatators beobachtet.
Signifikante prozentuale Verbesserungen des vorhergesagten FEV1 wurden bereits in Woche 2 beobachtet und hielten bis Woche 52 der VOYAGE-Studie an.
Die prozentualen Verbesserungen des vorhergesagten FEV1 im Zeitverlauf der VOYAGE-Studie sind in Abbildung 12 dargestellt.
Abbildung 12: Mittlere prozentuale Veränderung des vorhergesagten FEV1 gegenüber Baseline vor Anwendung eines Bronchodilatators (Liter) im Zeitverlauf der VOYAGE-Studie (Ausgangswert Eosinophilenzahl im Blut ≥150 Zellen/µl oder FeNO ≥20 ppb, Ausgangswert Eosinophilenzahl ≥300 Zellen/µl)
|
Ausgangswert Eosinophilenzahl im Blut ≥150 Zellen/µl oder FeNO ≥20 ppb
|
Ausgangswert Eosinophilenzahl im Blut ≥300 Zellen/µl
| |
|
|
|
|
Verbesserungen wurden bei ACQ-7-IA und PAQLQ(S)-IA in Woche 24 beobachtet und hielten bis Woche 52 an. Die Responderraten in den Populationen mit einer Eosinophilenzahl im Blut ≥150 Zellen/µl oder einem FeNO ≥20 ppb und mit einer Eosinophilenzahl im Blut ≥150 Zellen/µl waren verglichen mit Placebo in Woche 24 höher bei ACQ-7-IA und PAQLQ(S)-IA. Die Responderrate war definiert als eine Verbesserung des Wertes um mindestens 0,5 in der Skala (Skalenbereich von 0–6 bei ACQ-7-IA und von 1–7 bei PAQLQ(S)-IA). In der Subgruppe der Patienten mit einer Eosinophilenzahl im Blut ≥300 Zellen/µl als Ausgangswert war bei Dupixent in Woche 24 der Anteil der Responder bei ACQ-7-IA höher (80,6% gegenüber 64,3% bei Placebo) bei einer Odds Ratio (OR) von 2,79 (95 %-KI: 1,43; 5,44), ebenso der Anteil der Responder bei PAQLQ(S)-IA (72,8% gegenüber 63,0% bei Placebo) mit einer OR von 1,84 (95 %-KI: 0,92; 3,65).
Langzeit-Verlängerungsstudie (EXCURSION)
Die Wirksamkeit von Dupilumab, die als sekundärer Endpunkt bestimmt wurde, wurde bei 365 Kindern (im Alter von 6 bis 11 Jahren) mit Asthma in der Langzeit-Verlängerungsstudie (EXCURSION) untersucht. Es wurde eine dauerhafte Verringerung der Exazerbationen, die eine Hospitalisierung und/oder einen Besuch der Notfallstation erforderten, sowie eine Verringerung der Exposition gegenüber oralen systemischen Kortikosteroiden beobachtet. Anhaltende Verbesserungen der Lungenfunktion wurden für mehrere Parameter beobachtet, darunter der Prozentsatz des vorhergesagten FEV1, der Prozentsatz der vorhergesagten FVC, das Verhältnis von FEV1 zu FVC und der Prozentsatz des vorhergesagten FEF 25–75 %. Zudem erreichten und/oder behielten 75 % der Patienten eine normale Lungenfunktion mit einer FEV1 vor Anwendung eines Bronchodilatators in Prozent des vorhergesagten Wertes > 80 % am Ende der EXCURSION-Studie. Die Wirksamkeit wurde über eine kumulative Behandlungsdauer von bis zu 104 Wochen aufrechterhalten (VOYAGE und EXCURSION).
3) Klinische Wirksamkeit bei chronischer Rhinosinusitis mit Nasenpolypen (CRSwNP)
Das Entwicklungsprogramm zur chronischen Rhinosinusitis mit Nasenpolypen (CRSwNP) beinhaltete zwei randomisierte, doppelblinde, multizentrische, placebokontrollierte Parallelgruppenstudien (SINUS-24 und SINUS-52) mit 724 Patienten ab 18 Jahren, eingestellt auf intranasale Kortikosteroide (INCS) als Basistherapie. Diese Studien schlossen Patienten ein, die trotz vorherigem operativem Eingriff an den Nasennebenhöhlen oder vorheriger Behandlung mit systemischen Kortikosteroiden in den vergangenen 2 Jahren unter einer schweren CRSwNP litten bzw. die für eine Therapie mit systemischen Kortikosteroiden in den vergangenen 2 Jahren nicht in Frage kamen. Es lag im Ermessen des Prüfarztes, Notfallmedikation mit systemischen Kortikosteroiden oder einen operativen Eingriff während der Studien zu veranlassen.
In der SINUS-24-Studie wurden insgesamt 276 Patienten randomisiert und erhielten für 24 Wochen entweder 300 mg Dupilumab (n = 143) oder Placebo (n = 133) alle zwei Wochen. In der SINUS-52-Studie wurden 448 Patienten randomisiert und erhielten entweder für 52 Wochen 300 mg Dupilumab (n = 150) alle zwei Wochen oder bis Woche 24 300 mg Dupilumab (n = 145) alle zwei Wochen, gefolgt von 300 mg Dupilumab alle 4 Wochen bis Woche 52, oder Placebo (n = 153). Alle Patienten zeigten eine Verschattung der Nasennebenhöhlen im Lund-MacKay-Gesamt-CT-Score (LMK), und 73 % bis 90 % der Patienten wiesen eine Verschattung aller Nasennebenhöhlen auf. Patienten wurden stratifiziert anhand ihrer vorherigen operativen Eingriffe und ihres komorbiden Asthmas / Analgetika-Intoleranz-Syndroms (nonsteroidal anti-inflammatory drug exacerbated respiratory disease, NSAID-ERD).
Die koprimären Wirksamkeitsendpunkte waren die Veränderung des bilateralen endoskopischen Nasenpolypenscores (NPS), bewertet von zentral verblindeten Prüfern, zwischen dem Ausgangswert und dem Wert in Woche 24 sowie die Veränderung des über 28 Tage gemittelten Scores der nasalen Kongestion/Obstruktion (NC), täglich ermittelt durch Patienten anhand von Tagebüchern, zwischen dem Ausgangswert und dem Wert in Woche 24. Bei dem NPS wurden Polypen auf jeder Seite der Nase auf einer kategorialen Skala beurteilt (0 = keine Polypen; 1 = kleine Polypen im mittleren Nasengang, die nicht weiter als bis zur unteren Grenze der mittleren Nasenmuschel reichen; 2 = Polypen, die weiter als bis zur unteren Grenze der mittleren Nasenmuschel reichen; 3 = grosse Polypen, die bis zur unteren Grenze der unteren Nasenmuschel reichen oder Polypen medial der mittleren Nasenmuschel; 4 = grosse Polypen, die die untere Nasenhöhle komplett blockieren). Der Gesamtscore war die Summe der rechten und linken Scores. Die nasale Kongestion wurde von den Teilnehmern täglich anhand einer kategorialen Schweregrad-Skala von 0 bis 3 beurteilt (0 = keine Symptome; 1 = leichte Symptome; 2 = mittelschwere Symptome; 3 = schwere Symptome).
In beiden Studien umfassten die sekundären Hauptendpunkte in Woche 24 Veränderungen gegenüber dem Ausgangwert bei: Lund-MacKay-Gesamt-CT-Score (LMK), Gesamtsymptom-Score (total symptom score, TSS), Riechtest UPSIT (University of Pennsylvania smell identification test), täglicher Verlust des Geruchssinns und dem aus 22 Fragen bestehenden Sino-Nasal-Outcome-Test (22-item Sino-Nasal Outcome Test, SNOT-22). Anhand der gepoolten Daten der beiden Studien wurde die Verringerung des Anteils der Patienten beurteilt, die im Notfall systemische Kortikosteroide anwendeten und/oder sich einem operativen Eingriff an den Nasennebenhöhlen unterzogen, sowie die Verbesserung des FEV1 in der Asthma-Subgruppe. Zu den weiteren sekundären Endpunkten gehörte der aus 6 Fragen bestehende Fragebogen zur Asthmakontrolle (Asthma Control Questionnaire, ACQ-6) in der Subgruppe mit komorbidem Asthma.
Die demografischen Angaben und Baseline-Charakteristika für diese zwei Studien sind in Tabelle 19 dargestellt.
Tabelle 19: Demografische Angaben und Baseline-Charakteristika in den CRSwNP-Studien
|
Parameter
|
SINUS-24
(n = 276)
|
SINUS-52
(n = 448)
| |
Mittleres Alter in Jahren (SD)
|
50,49 (13,39)
|
51,95 (12,45)
| |
% Männlich
|
57,2
|
62,3
| |
Mittlere Dauer der CRSwNP (Jahre) (SD)
|
11,11 (9,16)
|
10,94 (9,63)
| |
Patienten mit ≥1 vorherigen operativen Eingriff (%)
|
71,7
|
58,3
| |
Patienten mit Anwendung systemischer Kortikosteroide in den vergangenen 2 Jahren (%)
|
64,9
|
80,1
| |
Mittlerer bilateraler endoskopischer NPSa (SD), Bereich: 0–8
|
5,75 (1,28)
|
6,10 (1,21)
| |
Mittlerer nasaler Kongestion (NC)-Scorea (SD), Bereich: 0–3
|
2,35 (0,57)
|
2,43 (0,59)
| |
Mittlerer Lund-MacKay-Gesamt-CT-Scorea (SD), Bereich: 0–24
|
19,03 (4,44)
|
17,96 (3,76)
| |
Mittlerer Riechtest UPSIT-Scorea (SD), Bereich: 0–40
|
14,56 (2,71)
|
13,61 (8,02)
| |
Mittlerer Verlust des Geruchssinn-Scorea, (SD), Bereich: 0–3
|
2,71 (0,54)
|
2,75 (0,52)
| |
Mittlerer SNOT-22-Gesamtscorea (SD), Bereich: 0–110
|
49,40 (20,20)
|
51,86 (20,90)
| |
Mittelwert Rhinosinusitis-Schweregrad-Skalaa (VAS) (SD), 0–10 cm
|
7,68 (2,05)
|
8,00 (2,08)
| |
Mittlere Eosinophilenzahl im Blut (Zellen/µl) (SD)
|
437 (333)
|
431 (353)
| |
Mittleres Gesamt-IgE I.E./ml (SD)
|
211,97 (275,73)
|
239,84
(341,53)
| |
Atopische Krankheiten in der Anamnese (mit Typ-2- Inflammation)
% insgesamt
|
75,4 %
|
82,4 %
| |
Asthma (%)
|
58,3
|
59,6
| |
Mittlere FEV1 (l) (SD)
|
2,69
(0,96)
|
2,57
(0,83)
| |
Mittlerer Prozentsatz des vorhergesagten FEV1 (%) (SD)
|
85,30
(20,23)
|
83,39
(17,72)
| |
Mittlerer ACQ-6-Scorea (SD)
|
1,62
(1,14)
|
1,58
(1,09)
| |
NSAID-ERD (%)
|
30,4
|
26,8
|
a Höhere Scores weisen auf eine stärkere Ausprägung der Erkrankung hin, ausser bei UPSIT, bei dem höhere Scores auf eine geringere Ausprägung der Erkrankung hinweisen; SD = Standardabweichung; NPS = Nasenpolypenscore; UPSIT = Riechtest (University of Pennsylvania smell identification test); SNOT-22 = Sino-Nasal-Outcome-Test mit 22 Fragen (22-item Sino-Nasal Outcome Test); VAS = visuelle Analogskala (visual analogue scale); FEV1 = Einsekundenkapazität (forciertes Exspirationsvolumen (1 s)); ACQ-6 = Fragebogen zur Asthmakontrolle (Asthma Control Questionnaire-6); NSAID-ERD = Aspirin/Analgetika-Intoleranz-Syndrom (nonsteroidal anti-inflammatory drug exacerbated respiratory disease).
Klinisches Ansprechen (SINUS-24 und SINUS-52)
Die Ergebnisse der primären und sekundären Endpunkte der CRSwNP-Studien sind in Tabelle 20 dargestellt.
Tabelle 20: Ergebnisse der primären und sekundären Endpunkte in den CRSwNP-Studien
|
|
SINUS-24
|
SINUS-52
| |
|
Placebo
(n = 133)
|
Dupilumab
300 mg
Q2W
(n = 143)
|
Mittlerer LS-Unterschied vs. Placebo (95 %-KI)
|
Placebo
(n = 153)
|
Dupilumab
300 mg
Q2W
(n = 295)
|
Mittlerer LS-Unterschied vs. Placebo (95 %-KI)
| |
Primäre Endpunkte in Woche 24
| |
Scores
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
| |
NPS
|
5,86
|
0,17
|
5,64
|
-1,89
|
-2,06
(-2,43; -1,69)
|
5,96
|
0,10
|
6,18
|
-1,71
|
-1,80
(-2,10; -1,51)
| |
NC
|
2,45
|
-0,45
|
2,26
|
-1,34
|
-0,89
(-1,07; -0,71)
|
2,38
|
-0,38
|
2,46
|
-1,25
|
-0,87
(-1,03; -0,71)
| |
Sekundäre Hauptendpunkte in Woche 24
| |
Scores
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
Mittlere Baseline
|
Mittlere
LS-Veränderung
|
| |
LMK-Score
|
19,55
|
-0,74
|
18,55
|
-8,18
|
-7,44
(-8,35; -6,53)
|
17,65
|
-0,09
|
18,12
|
-5,21
|
-5,13
(-5,80; -4,46)
| |
Gesamtsymptom-Score (TSS)
|
7,28
|
-1,17
|
6,82
|
-3,77
|
-2,61
(-3,04; -2,17)
|
7,08
|
-1,00
|
7,30
|
-3,45
|
-2,44
(-2,87; -2,02)
| |
UPSIT
|
14,44
|
0,70
|
14,68
|
11,26
|
10,56
(8,79; 12,34)
|
13,78
|
-0,81
|
13,53
|
9,71
|
10,52
(8,98; 12,07)
| |
Verlust des Geruchssinns
|
2,73
|
-0,29
|
2,70
|
-1,41
|
-1,12
(-1,31; -0,93)
|
2,72
|
-0,23
|
2,77
|
-1,21
|
-0,98
(-1,15; -0,81)
| |
SNOT-22
|
50,87
|
-9,31
|
48,0
|
-30,43
|
-21,12
(-25,17; -17,06)
|
53,48
|
-10,40
|
51,02
|
-27,77
|
-17,36
(-20,87; -13,85)
| |
VAS
|
7,96
|
-1,34
|
7,42
|
-4,54
|
-3,20
(-3,79; -2,60)
|
7,98
|
-1,39
|
8,01
|
-4,32
|
-2,93
(-3,45, -2,40)
|
Eine Verringerung des Scores bedeutet eine Verbesserung, ausser bei UPSIT, bei dem ein Anstieg eine Verbesserung bedeutet.
Der Gesamtsymptom-Score (TSS) ist ein zusammengesetzter Schweregrad-Score, bestehend aus der Summe der täglichen Symptome von NC, Verlust des Geruchssinns und anteriorer/posteriorer Rhinorrhö.
NC = nasale Kongestion; NPS = Nasenpolypenscore; LMK = Lund-MacKay-Gesamt-CT-Score; UPSIT = Riechtest (University of Pennsylvania smell identification test); SNOT-22 = Sino-Nasal-Outcome-Test mit 22 Fragen (22-item Sino-Nasal Outcome Test); TSS = Gesamtsymptom-Score (total symptom score); VAS = visuelle Analogskala für Rhinosinusitis (visual analogue scale) (alle p-Werte < 0,0001, nominal für VAS)
Die Ergebnisse der SINUS-52-Studie in Woche 52 sind in Tabelle 21 dargestellt.
Tabelle 21: Ergebnisse zur Wirksamkeit in Woche 52 der SINUS-52-Studie
|
|
Placebo
(n = 153)
|
Dupilumab
300 mg Q2W
(n = 150)
|
Mittlerer LS-Unterschied vs. Placebo (95 %-KI)
|
Dupilumab
300 mg Q2W–Q4W
(n = 145)
|
Mittlerer LS-Unterschied vs. Placebo (95 %-KI)
| |
Mittlere Baseline
|
Mittlere LS-Veränderung
|
Mittlere Baseline
|
Mittlere LS-Veränderung
|
Mittlere Baseline
|
Mittlere LS-Veränderung
| |
NPS
|
5,96
|
0,15
|
6,07
|
-2,24
|
-2,40
(-2,77; -2,02)
|
6,29
|
-2,06
|
-2,21
(-2,59; -1,83)
| |
NC
|
2,38
|
-0,37
|
2,48
|
-1,35
|
-0,98
(-1,17; -0,79)
|
2,44
|
-1,48
|
-1,10
(-1,29; -0,91)
| |
LMK-Score
|
17,65
|
0,11
|
18,42
|
-6,83
|
-6,94
(-7,87; -6,01)
|
17,81
|
-5,60
|
-5,71
(-6,64; -4,77)
| |
Gesamtsymptom-Score (TSS)
|
7,08
|
-0,94
|
7,31
|
-3,79
|
-2,85
(-3,35; -2,35)
|
7,28
|
-4,16
|
-3,22
(-3,73; -2,72)
| |
UPSIT
|
13,78
|
-0,77
|
13,46
|
9,53
|
10,30
(8,50; 12,10)
|
13,60
|
9,99
|
10,76
(8,95; 12,57)
| |
Verlust des Geruchssinns
|
2,72
|
-0,19
|
2,81
|
-1,29
|
-1,10
(-1,31, -0,89)
|
2,73
|
-1,49
|
-1,30
(-1,51; -1,09)
| |
SNOT-22
|
53,48
|
-8,88
|
50,16
|
-29,84
|
-20,96
(-25,03; -16,89)
|
51,89
|
-30,52
|
-21,65
(-25,71; -17,58)
| |
VAS
|
7,98
|
-0,93
|
8,24
|
-4,74
|
-3,81
(-4,46; -3,17)
|
7,78
|
-4,39
|
-3,46
(-4,10, -2,81)
|
Eine Verringerung des Scores bedeutet eine Verbesserung, ausser bei UPSIT, bei dem ein Anstieg eine Verbesserung bedeutet.
Der Gesamtsymptom-Score (TSS) ist ein zusammengesetzter Schweregrad-Score, bestehend aus der Summe der täglichen Symptome von NC, Verlust des Geruchssinns und anteriorer/posteriorer Rhinorrhö.
NC = nasale Kongestion; NPS = Nasenpolypenscore; LMK = Lund-MacKay-Gesamt-CT-Score; UPSIT = Riechtest (University of Pennsylvania smell identification test); SNOT-22 = Sino-Nasal-Outcome-Test mit 22 Fragen (22-item Sino-Nasal Outcome Test); TSS = Gesamtsymptom-Score (total symptom score); VAS = visuelle Analogskala für Rhinosinusitis (visual analogue scale). (alle p-Werte < 0,0001)
Eine statistisch signifikante und klinisch bedeutsame Wirksamkeit wurde in der SINUS-24-Studie hinsichtlich der Verbesserung bei dem bilateralen endoskopischen NPS-Score in Woche 24 beobachtet. Im Zeitraum nach der Behandlung, als Patienten kein Dupilumab erhielten, nahm der Behandlungseffekt mit der Zeit ab (siehe Abbildung 13a). Ähnliche Verbesserungen bei dem bilateralen endoskopischen NPS-Score wurden ebenfalls in der SINUS-52-Studie sowohl in Woche 24 als auch in Woche 52 mit einer progressiven Verbesserung im Laufe der Zeit beobachtet (siehe Abbildung 13b).
Abbildung 13: Mittlere LS-Veränderung gegenüber Baseline bei dem bilateralen Nasenpolypenscore (NPS) in SINUS-24 und SINUS-52 (ITT-Population)
|
Abbildung 13a. SINUS-24
|
Abbildung 13b. SINUS-52
| |
|
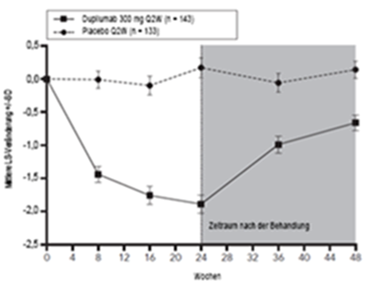
|
|
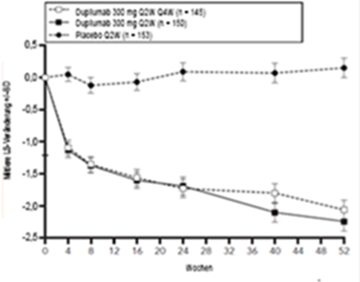
|
In beiden Studien wurden bereits bei der ersten Beurteilung in Woche 4 signifikante Verbesserungen bei der NC und bei dem Schweregrad des täglichen Verlusts des Geruchssinns beobachtet. Der mittlere LS-Unterschied bei der NC in Woche 4 lag in der Dupilumab-Gruppe im Vergleich zur Placebo-Gruppe bei -0,41 (95 %-KI: -0,52; -0,30) in SINUS-24 und -0,37 (95 %-KI: -0,46; -0,27) in SINUS-52. Der mittlere LS-Unterschied bei dem Verlust des Geruchssinns in Woche 4 lag in der Dupilumab-Gruppe im Vergleich zur Placebo-Gruppe bei -0,34 (95 %-KI: -0,44; -0,25) in SINUS-24 und -0,31 (95 %-KI: -0,41; -0,22) in SINUS-52.
Eine Verringerung im Anteil der Patienten mit Anosmie wurde in den SINUS-24- und SINUS-52-Studien beobachtet. Zu Beginn litten 74 % bis 79 % der Patienten an Anosmie, was sich in Woche 24 auf 24 % in der SINUS-24-Studie und 30 % in der SINUS-52-Studie verringerte, während es unter Placebo keine Veränderung gab. Eine Verbesserung im nasalen maximalen inspiratorischen Atemstrom (nasal peak inspiratory flow, NPIF) wurde in SINUS-24 und SINUS-52 in Woche 24 beobachtet. Der mittlere LS-Unterschied in der Dupilumab-Gruppe betrug 40,4 l/min (95 %-KI: 30,4; 50,4), während er in der Placebo-Gruppe 36,6 l/min (95 %-KI: 28,0; 45,3) betrug.
Bei den Patienten mit einem Rhinosinusitis-VAS-Score > 7 zu Studienbeginn erreichte in Woche 24 ein grösserer Anteil der Patienten in der Dupilumab-Gruppe einen VAS-Score ≤7 als in der Placebo-Gruppe (83,3 % versus 39,4 % in SINUS-24 und 75,0 % versus 39,3 % in SINUS-52).
In der präspezifizierten, für Multiplizität adjustierten gepoolten Analyse der beiden Studien führte die Behandlung mit Dupilumab im Vergleich mit Placebo zu einer signifikanten Verringerung bei der Anwendung von systemischen Kortikosteroiden und der Notwendigkeit eines operativen Eingriffs an den Nasennebenhöhlen (HR von 0,24; 95 %-KI: 0,17; 0,35) (siehe Abbildung 14). Der Anteil der Patienten, die systemische Kortikosteroide benötigten, verringerte sich um 74 % (HR von 0,26; 95 %-KI: 0,18; 0,38). Die Gesamtzahl der jährlichen Anwendungen systemischer Kortikosteroide wurde um 75 % verringert (RR von 0,25; 95 %-KI: 0,17; 0,37). Die mittlere individuelle jährlich verschriebene Gesamtdosis von systemischen Kortikosteroiden (in mg) während des Behandlungszeitraums war 71 % geringer in der gepoolten Dupilumab-Gruppe als in der gepoolten Placebo-Gruppe (60,5 [531,3] mg versus 209,5 [497,2] mg). Der Anteil der Patienten, die einen operativen Eingriff benötigten, verringerte sich um 83 % (HR von 0,17; 95 %-KI: 0,07; 0,46).
Abbildung 14: Kaplan-Meier-Kurve der Zeitspanne bis zur ersten Anwendung systemischer Kortikosteroide und/oder eines operativen Eingriffs an den Nasennebenhöhlen während des Behandlungszeitraums (ITT-Population) [SINUS-24 und SINUS-52 gepoolt]
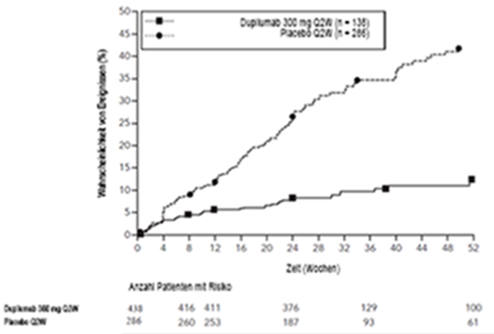
Die Auswirkungen von Dupilumab auf die primären Endpunkte NPS und nasale Kongestion sowie den sekundären Hauptendpunkt Lund-MacKay-Gesamt-CT-Score (LMK) waren vergleichbar bei Patienten mit und ohne vorherigen operativen Eingriff.
Bei Patienten mit komorbidem Asthma wurden in Woche 24, unabhängig vom Eosinophilen-Ausgangswert im Blut, signifikante Verbesserungen bei FEV1 und ACQ-6 beobachtet. Die gepoolte mittlere LS-Veränderung des FEV1 im Vergleich zum Ausgangswert lag in Woche 24 für Dupilumab 300 mg alle 2 Wochen (Q2W) bei 0,14 l, während sie bei Placebo bei -0,07 l lag, was einem Unterschied von 0,21 l entspricht (95 %-KI: 0,13; 0,29). Darüber hinaus wurden im Vergleich zu den Ausgangswerten Verbesserungen des FEV1 ab der ersten Beurteilung nach Studienbeginn in Woche 8 in SINUS-24 und in Woche 4 in SINUS-52 beobachtet. Verbesserungen im ACQ-6 bei Patienten mit komorbidem Asthma wurden in beiden Studien beobachtet. Ein Ansprechen war definiert als eine Verbesserung des Scores um 0,5 oder mehr. Der mittlere LS-Unterschied in Woche 24 betrug in der Dupilumab-Gruppe im Vergleich zu Placebo -0,76 (95 %-KI: -1,00 bis -0,51) in SINUS-24 und -0,94 (95 %-KI: -1,19; -0,69) in SINUS-52.
Die Responderrate bei ACQ-6 betrug in Woche 24 bei Dupilumab 300 mg alle 2 Wochen (Q2W) in der SINUS-24-Studie 56 %, bei Placebo 28 % (Odds Ratio 3,17; 95 %-KI: 1,65; 6,09). Die Ansprechrate bei ACQ-6 betrug in Woche 52 bei Dupilumab 300 mg alle 2 Wochen (Q2W) in der SINUS-52-Studie 46 %, bei Placebo 14 % (Odds Ratio 7,02; 95 %-KI: 3,10; 15,90).
Bei Patienten mit NSAID-ERD stimmten die Auswirkungen von Dupilumab auf die primären Endpunkte NPS und NC sowie den sekundären Hauptendpunkt Lund-MacKay-Gesamt-CT-Score (LMK) mit denen überein, die in der CRSwNP-Gesamtpopulation beobachtet wurden.
4) Klinische Wirksamkeit bei Prurigo nodularis (PN)
Das Entwicklungsprogramm zu Prurigo nodularis (PN) beinhaltete zwei 24-wöchige, randomisierte, doppelblinde, placebokontrollierte, multizentrische Parallelgruppenstudien (PRIME und PRIME2) mit 311 Patienten ab 18 Jahren mit mittelschwerer bis schwerer PN, definiert als schwerer Pruritus (WI- NRS ≥7 auf einer Skala von 0 bis 10) und mindestens 20 noduläre Läsionen, deren Erkrankung mit verschreibungspflichtigen topischen Therapien nicht ausreichend kontrolliert werden konnte oder wenn derartige Therapien nicht empfohlen wurden. PRIME und PRIME2 untersuchten neben der Wirkung von Dupilumab auf die Verbesserung des Juckreizes auch dessen Wirkung auf PN-Läsionen, den Dermatologischer-Lebensqualitäts-Index (Dermatology Life Quality Index, DLQI), die Skala zur Erfassung von Angst und Depression (Hospital Anxiety and Depression Scale, HADS) sowie auf Hautschmerzen.
In diesen beiden Studien erhielten Patienten entweder subkutan 600 mg Dupilumab (zwei Injektionen zu je 300 mg) an Tag 1, gefolgt von 300 mg alle 2 Wochen (Q2W) für 24 Wochen, oder ein entsprechendes Placebo. Patienten, die eine stabile Behandlung mit schwach oder mittelstark wirksamen TCS oder TCI erhielten, konnten die begleitende Anwendung topischer Steroide fortsetzen. Stark oder sehr stark wirksame TCS oder TCI waren simultan als Rescue-Therapie zugelassen.
In diesen Studien lag das mittlere Alter bei 49,5 Jahren und das mediane Gewicht bei 71,3 kg. 65,3 % der Patienten waren Frauen, 56,6 % Weisse, 6,1 % Schwarze und 34,1 % Asiaten. Zu Studienbeginn lag der mittlere WI-NRS-Wert bei 8,5; 66,3 % hatten 20 bis 100 Knoten (mittelschwer), 33,7 % hatten mehr als 100 Knoten (schwer), 99,7 % waren zuvor topisch behandelt worden, 12,5 % hatten zuvor systemische Kortikosteroide erhalten, 20,6 % hatten zuvor systemische nichtsteroidale Immunsuppressiva erhalten und 4,5 % hatten zuvor Gabapentinoide erhalten. Zu Studienbeginn nahmen 11 % der Patienten stabile Dosen von Antidepressiva ein und wurden angewiesen, diese Arzneimittel während der Studie weiter einzunehmen. 43,4 % wiesen in ihrer Anamnese eine Atopie auf (definiert als atopische Dermatitis in der Anamnese, allergische Rhinitis/Rhinokonjunktivitis, Asthma oder Nahrungsmittelallergie). Der WI-NRS-Wert besteht aus einem Einzelwert, bewertet anhand einer Skala von 0 («kein Juckreiz) bis 10 («schlimmster vorstellbarer Juckreiz»). Teilnehmer wurden gebeten, die Intensität ihres schlimmsten Pruritus-Ereignisses (Juckreiz) innerhalb der letzten 24 Stunden anhand dieser Skala zu bewerten. Der IGA PN-S ist ein Instrument zur Erfassung der ungefähren Anzahl an Knoten, das eine Einteilung in fünf Schweregrade von 0 (frei) bis 4 (schwer) vornimmt.
Der primäre Wirksamkeitsendpunkt war der Anteil an Patienten mit Verbesserung (Reduktion) auf der WI-NRS um ≥4. Wichtige sekundäre Endpunkte waren unter anderem der Anteil an Studienteilnehmern mit einem IGA PN-S von 0 oder 1 (entspricht 0 bis 5 Knoten).
Die Wirksamkeitsergebnisse für PRIME und PRIME 2 sind in Tabelle 22 dargestellt.
Tabelle 22: Ergebnisse der primären und sekundären Endpunkte in den Studien PRIME und PRIME 2
|
|
PRIME
|
PRIME2
| |
|
Placebo
(N=76)
|
Dupilumab 300 mg Q2W
(N=75)
|
Unterschied (95 %-KI) Dupilumab vs. Placebo
|
Placebo
(N=82)
|
Dupilumab 300 mg Q2W
(N=78)
|
Unterschied (95 %-KI) Dupilumab vs. Placebo
| |
Anteil Patienten mit Verbesserung (Reduktion) auf WI-NRS um ≥4 Punkte gegenüber der Baseline in Woche 24 (primärer Endpunkt in PRIME)b
|
18,4 %
|
60,0 %
|
42,7 %
(27,76; 57,72)
|
19,5 %
|
57,7 %
|
42,6 %
(29,06; 56,08)
| |
Anteil Patienten mit Verbesserung (Reduktion) auf WI-NRS um ≥4 Punkte gegenüber der Baseline in Woche 12 (primärer Endpunkt in PRIME2)b
|
15,8 %a
|
44,0 %a
|
29,2 %
(14,49; 43,81)a
|
22,0 %
|
37,2 %
|
16,8 %
(2,34; 31,16)
| |
Anteil Patienten mit IGA PN-S von 0 oder 1 in Woche 24b
|
18,4 %
|
48,0 %
|
28,3 %
(13,41; 43,16)
|
15,9 %
|
44,9 %
|
30,8 %
(16,37; 45,22)
| |
Anteil Patienten mit Verbesserung (Reduktion) sowohl auf WI-NRS um ≥4 Punkte gegenüber der Baseline in Woche 24 als auch im IGA PN-S von 0 oder 1 in Woche 24b
|
9,2 %
|
38,7 %
|
29,6 %
(16,42; 42,81)
|
8,5 %
|
32,1 %
|
25,5 %
(13,09; 37,86)
| |
Veränderung in % gegenüber der Baseline auf WI-NRS in Woche 24 (SE)
|
-22,22 (5,74)
|
-48,89 (5,61)
|
-26,67
(-38,44; -14,90)
|
-36,18 (6,21)
|
-59,34 (6,39)
|
-23,16
(-33,81; -12,51)
| |
Veränderung gegenüber der Baseline im DLQI in Woche 24 (SE)
|
-5,77 (1,05)
|
-11,97 (1,02)
|
-6,19
(-8,34; -4,05)
|
-6,77 (1,18)
|
-13,16 (1,21)
|
-6,39
(-8,42; -4,36)
| |
Veränderung gegenüber der Baseline auf Hautschmerz-NRS in Woche 24 (SE)c
|
-2,16 (0,44)
|
-4,33 (0,43)
|
-2,17
(-3,07; -1,28)
|
-2,74 (0,51)
|
-4,35 (0,53)
|
-1,61
(-2,49; -0,73)
| |
Veränderung gegenüber der Baseline auf HADS in Woche 24 (SE)c
|
-2,02 (0,94)
|
-4,62 (0,93)
|
-2,60
(-4,52; -0,67)
|
-2,59 (1,03)
|
-5,55 (1,06)
|
-2,96
(-4,73; -1,19)
|
a Nicht für multiples Testen in PRIME korrigiert.
b Patienten, die zuvor eine Rescue-Therapie erhalten hatten oder bei denen Daten fehlten, wurden als Non-Responder eingestuft.
c Daten von Patienten, die zuvor eine Rescue-Therapie erhalten hatten oder die Studie aufgrund fehlender Wirksamkeit abbrachen, wurden vervollständigt, indem der schlechteste Wert als Ersatzwert verwendet wurde (worst observation carried forward); andere fehlende Daten wurden mittels multipler Imputation vervollständigt.
In den Studien PRIME und PRIME 2 stimmten die Behandlungseffekte in den Untergruppen (Gewicht, Alter, Geschlecht, ethnische Herkunft sowie Hintergrundbehandlung einschliesslich Immunsuppressiva) mit den Ergebnissen in der jeweiligen Studienpopulation überein.
5) Eosinophile Ösophagitis (EoE)
Klinische Wirksamkeit bei Erwachsenen und Jugendlichen mit EoE
Das Entwicklungsprogramm zu eosinophiler Ösophagitis beinhaltete einen dreiteiligen Prüfplan über 52 Wochen (TREET), der aus zwei separat randomisierten, doppelblinden, multizentrischen, placebokontrollierten Parallelgruppenstudien von 24-wöchiger Dauer (TREET Teil A und TREET Teil B), gefolgt von einer 28-wöchigen Verlängerungsstudie mit aktiver Behandlung (TREET Teil C) mit erwachsenen und jugendlichen Patienten im Alter von 12 bis 17 Jahren bestand, wobei Patienten mit einem Gewicht unter 40 kg ausgeschlossen waren. Alle Patienten PPI-refraktär, da bei ihnen nach einer mindestens 8-wöchigen Behandlung mit einem hochdosierten Protonenpumpenhemmer (PPI) entweder vor oder während des Screeningzeitraums die Anzahl der intraepithelialen Eosinophilen (eos) ≥15 pro hochauflösendes Gesichtsfeld (high-power field, hpf) betragen musste. Die Patienten mussten zudem einen Score im Dysphagie-Symptom-Fragebogen (Dysphagia Symptom Questionnaire, DSQ) von ≥10 auf einer Skala von 0 bis 84 aufweisen. Die Patienten wurden stratifiziert anhand ihres Alters zum Zeitpunkt des Screenings (12 bis 17 Jahre versus 18 Jahre und älter) und der Anwendung von PPI zum Zeitpunkt der Randomisierung. TREET Teil A wurde zuerst durchgeführt. Mit TREET Teil B wurde begonnen, nachdem die Aufnahme in TREET Teil A abgeschlossen war. Patienten, welche den 24-wöchigen, doppelblinden Behandlungszeitraum in Teil A oder B durchlaufen hatten, erhielten die Möglichkeit, an einer 28-wöchigen Verlängerungsstudie (TREET Teil C) mit aktiver Behandlung teilzunehmen.
In Teil A wurden insgesamt 81 Patienten, 61 Erwachsene und 20 Jugendliche im Alter von 12 bis 17 Jahren, randomisiert und erhielten entweder 300 mg Dupilumab einmal wöchentlich (n = 42) oder Placebo (n = 39). In Teil B wurden insgesamt 240 Patienten, 161 Erwachsene und 79 Jugendliche im Alter von 12 bis 17 Jahren, randomisiert und erhielten entweder 300 mg Dupilumab einmal wöchentlich (n = 80) oder 300 mg Dupilumab alle zwei Wochen (n = 81; das Dosierungsschema mit 300 mg alle zwei Wochen ist nicht für eosinophile Ösophagitis zugelassen) oder Placebo (n = 79). In Teil C erhielten alle Patienten, die zuvor an Teil A teilgenommen hatten, 300 mg Dupilumab (n = 77) wöchentlich. Von den Patienten, die zuvor an Teil B teilgenommen hatten, erhielten diejenigen, die in Teil B Dupilumab erhalten hatten, in Teil C weiter ihr Dosierungsschema und diejenigen, die Placebo erhalten hatten, wurden randomisiert und erhielten entweder das eine oder das andere Dosierungsschema. Es lag im Ermessen des Prüfarztes, die Anwendung von systemischen und/oder oralen topischen Kortikosteroiden oder eine Notfall-Ösophagusdilatation während der Studie zu veranlassen.
In Teil A hatten insgesamt 74,1 % der Patienten vor Studieneinschluss zur Behandlung der eosinophilen Ösophagitis bereits orale topische Kortikosteroide angewendet und bei 43,2 % war vor Studieneinschluss eine Ösophagusdilatation durchgeführt worden. In Teil B hatten insgesamt 73,3 % der Patienten vor Studieneinschluss zur Behandlung der eosinophilen Ösophagitis bereits orale topische Kortikosteroide angewendet und bei 35,4 % war vor Studieneinschluss eine Ösophagusdilatation durchgeführt worden.
Die koprimären Wirksamkeitsendpunkte in beiden Studien waren der Anteil der Patienten mit erreichter histologischer Remission, definiert als maximaler ösophagealer, intraepithelialer Eosinophilen-Wert von ≤6 eos/hpf in Woche 24, sowie die absolute Veränderung des patientenberichteten DSQ-Scores zwischen Baseline und Woche 24. Die sekundären Endpunkte waren unter anderem folgende Veränderungen gegenüber dem Ausgangswert bei Studieneinschluss: prozentuale Veränderung des maximalen ösophagealen, intraepithelialen Eosinophilen-Werts (eos/hpf), absolute Veränderung des mittleren Schweregrades (Grade) EoE-typischer histologischer Merkmale gemäss EoEHSS (Eosinophilic Esophagitis Histology Scoring System), absolute Veränderung des mittleren Ausmasses (Stage) gemäss EoEHSS, absolute Veränderung des endoskopischen Referenzwerts bei EoE (EoE-Endoscopic Reference Score, EoE-EREFS) sowie Anteil der Patienten, die einen maximalen ösophagealen, intraepithelialen Eosinophilen-Wert von ≤15 eos/hpf erreichen. Die demografischen Angaben und Baseline-Charakteristika für TREET Teil A und Teil B sind in Tabelle 23 dargestellt.
Tabelle 23: Demografische Daten und Baseline-Charakteristika (TREET, Teil A und Teil B)
|
Parameter
|
TREET Teil A
(n = 81)
|
TREET Teil B
(n = 240)
| |
Mittleres Alter in Jahren (SD)
|
31,5 (14,3)
|
28,1 (13,1)
| |
% männlich
|
60,5
|
63,8
| |
% weiss
|
96,3
|
90,4
| |
Gewicht (kg), Mittelwert (SD)
|
77,8 (21,0)
|
76,2 (20,6)
| |
BMI (kg/m2), Mittelwert (SD)
|
26,1 (6,3)
|
25,7 (6,2)
| |
Dauer der EoE (Jahre), Mittelwert (SD)
|
5,01 (4,3)
|
5,57 (4,8)
| |
Vorherige Einnahme oraler Kortikosteroide (%)
|
74,1
|
73,3
| |
Vorherige Ösophagusdilatation (%)
|
43,2
|
35,4
| |
PPI-Anwendung bei Randomisierung (%)
|
67,9
|
72,5
| |
Eliminationsdiät bei Screening (%)
|
40,7
|
37,1
| |
DSQ (0-84a), Mittelwert (SD)
|
33,6 (12,4)
|
36,7 (11,2)
| |
Maximaler ösophagealer, intraepithelialer EOS-Wert aus 3 Bereichen, Mittelwert (SD)
|
89,3 (48,3)
|
87,1 (45,8)
| |
Mittlerer ösophagealer, intraepithelialer EOS-Wert aus 3 Bereichen, Mittelwert (SD)
|
64,3 (37,6)
|
60,5 (32,9)
| |
EoEHSS-Schweregrad (Grade Score) [0-3a], Mittelwert (SD)
|
1,3 (0,4)
|
1,3 (0,4)
| |
EoEHSS-Ausmass (Stage Score) [0-3a], Mittelwert (SD)
|
1,3 (0,4)
|
1,3 (0,3)
| |
EREFS-Gesamtscore [0-18a], Mittelwert (SD)
|
6,3 (2,8)
|
7,2 (3,2)
|
a Höhere Scores weisen auf einen höheren Schweregrad der Erkrankung hin.
SD = Standard Deviation [Standardabweichung]
Die Ergebnisse der TREET.Studien Teil A und Teil B sind in Tabelle 24 dargestellt.
Tabelle 24: Wirksamkeitsergebnisse von Dupilumab in Woche 24 bei Patienten ab 12 Jahren mit eosinophiler Ösophagitis (TREET Teil A und Teil B)
|
|
TREET Teil A
|
TREET Teil B
| |
Dupilumab
300 mg QW
n = 42
|
Placebo
n = 39
|
Unterschied vs. Placebo
(95 %-KI)d
|
Dupilumab
300 mg QW
n = 80
|
Placebo
n = 79
|
Unterschied vs. Placebo
(95 %-KI)d
| |
Koprimäre Endpunkte
| |
Anteil Patienten mit erreichter histologischer Remission (maximaler ösophagealer, intraepithelialer EOS-Wert ≤6 eos/hpf), n (%)
|
25
(59,5)
|
2
(5,1)
|
55,3
(39,58; 71,04)
|
47
(58,8)
|
5
(6,3)
|
53,5
(41,20; 65,79)
| |
Absolute Veränderung gegenüber Ausgangswert im DSQ-Score (Dysphagie-Symptom-Fragebogen) (0-84a), mittlerer LS-Unterschied (SE)
|
-21,92
(2,53)
|
-9,60 (2,79)
|
-12,32
(-19,11; -5,54)
|
-23,78
(1,86)
|
-13,86
(1,91)
|
-9,92
(-14,81; -5,02)
| |
Sekundäre Endpunkte
| |
Prozentuale Veränderung des maximalen ösophagealen, intraepithelialen EOS-Werts gegenüber Baseline, LS-Mittelwert (SE)
|
-71,24
(6,95)
|
-2,98
(7,60)
|
-68,26
(-86,90; -49,62)
|
-80,24
(8,34)
|
8,38
(10,09)
|
-88,62
(-112,19; 65,05)
| |
Absolute Veränderung des mittlerer Schweregrades (Grade Score) des EoEHSS (0-3b) gegenüber Baseline, LS-Mittelwert (SE)
|
-0,76
(0,06)
|
-0,00 (0,06)
|
-0,76
(-0,91; -0,61)
|
-0,83
(0,04)
|
-0,15
(0,05)
|
-0,682
(-0,79; -0,57)
| |
Absolute Veränderung des mittleren Ausmaßes (Stage-Score) des EoEHSS (0-3b) gegenüber Baseline, LS-Mittelwert (SE)
|
-0,75
(0,06)
|
-0,01 (0,06)
|
-0,74
(-0,88, -0,60)
|
-0,80
(0,04)
|
-0,13
(0,04)
|
-0,672
(-0,78; -0,57)
| |
Absolute Veränderung des EoE-EREFS (0-18c) gegenüber Baseline,LS-Mittelwert (SE)
|
-3,2
(0,41)
|
-0,3
(0,41)
|
-2,9
(-3,91; -1,84)
|
-4,5
(0,36)
|
-0,6
(0,38)
|
-3,8
(-4,77; -2,93)
| |
Anteil der Patienten, die einen maximalen ösophagealen, intraepithelialen EOS-Wert von < 15 eos/hpf, erreichen, n (%)
|
27
(64,3)
|
3
(7,7)
|
57
(41,69; 73,33)
|
66
(82,5)
|
6
(7,6)
|
74,9
(64,25; 85,5)
|
a Über zwei Wochen erhobene DSQ-Gesamtscores reichen von 0 bis 84; höhere Scores weisen auf erhöhte Häufigkeit und höheren Schweregrad von Dysphagie hin.
b EoEHSS-Scores reichen von 0 bis 3; höhere Scores weisen auf einen höheren Schweregrad und ein grösseres Ausmass der histologischen Anomalien hin.
c EoE-EREFS-Gesamtscores reichen von 0 bis 18; höhere Scores weisen auf mehr schwerwiegendere endoskopische Befunde von entzündlichen Vorgängen und Remodeling hin.
d Mittlerer LS-Unterschied für kontinuierliche Endpunkte und absoluter Unterschied im Ausmass der kategoriespezifischen Endpunkte.
Die Wirksamkeitsergebnisse der koprimären und wichtigsten sekundären Endpunkte in der Subgruppe der Patienten, die zuvor orale topische Kortikosteroide eingenommen hatten, und bei Patienten, bei denen orale topische Kortikosteroide zu einer unzureichenden Kontrolle oder Unverträglichkeit führten oder kontraindiziert waren, waren konsistent mit der Gesamtpopulation.
Obwohl alle Patienten PPI-refraktär waren, wie dies vor Randomisierung der Studie gezeigt wurde, nahmen bestimmte Patienten während der Studie weiterhin PPI ein. Es gab keine bedeutsamen Unterschiede bei den erreichten koprimären und wichtigsten Wirksamkeitsendpunkten von TREET Teil A und Teil B zwischen den Untergruppen von Patienten, welche die Anwendung von PPI gleichzeitig fortsetzten, und der Untergruppe, die keine PPI anwendeten.
In den Teilen A und B erreichte im Vergleich zu Placebo ein grösserer Anteil an Patienten in der Gruppe, denen randomisiert Dupilumab zugewiesen worden war, eine histologische Remission (maximaler ösophagealer, intraepithelialer Eosinophilen-Wert von ≤6 eos/hpf). Der Anteil an Patienten mit histologischer Remission nach 24-wöchiger Behandlung in den Teilen A und B blieb in Teil C über 52 Wochen erhalten.
Die Behandlung mit Dupilumab führte im Vergleich zu Placebo bereits ab Woche 4 auch zu einer signifikanten Verbesserung der mittleren LS-Veränderung des DSQ-Scores, die bis Woche 24 erhalten blieb. Die Wirksamkeit in Teil C war vergleichbar mit den in den Teilen A und B beobachteten Ergebnissen mit kontinuierlicher Verbesserung im DSQ bis Woche 52 (siehe Abbildung 15 zu Teil A und Teil C der TREET-Studie sowie Abbildung 16 zu Teil B und Teil C der TREET-Studie).
Abbildung 15: Mittlere Veränderung gegenüber Ausgangswert des DSQ-Scores bei Patienten ab 12 Jahren mit eosinophiler Ösophagitis (TREET, Teil A und Teil C)
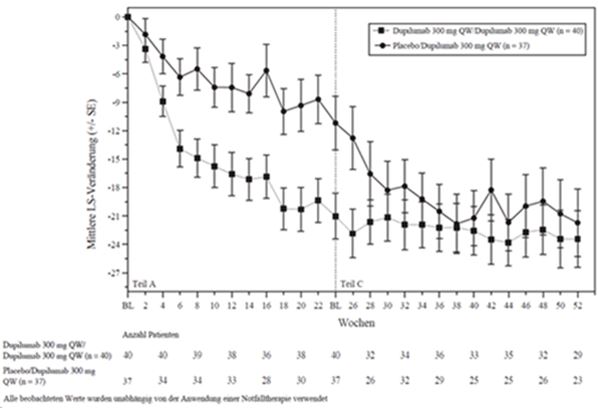
Abbildung 16: Mittlere Veränderung gegenüber Ausgangswert des DSQ-Scores bei Patienten ab 12 Jahren mit eosinophiler Ösophagitis (TREET, Teil B und Teil C)
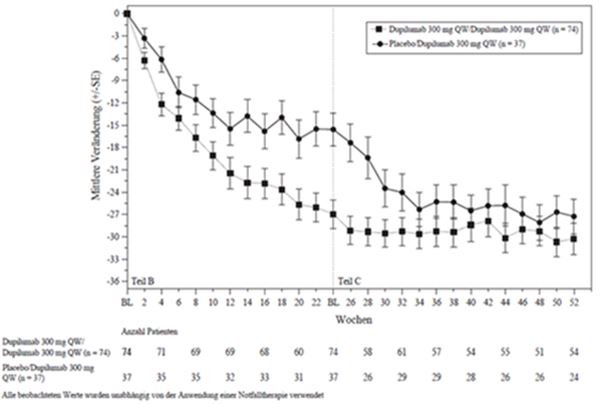
Klinische Wirksamkeit bei Kindern im Alter von 1 bis 11 Jahren mit EoE und einem Körpergewicht von mindestens 15 kg
Die Wirksamkeit und Sicherheit von Dupilumab wurden in einer zweiteiligen Studie (EoE KIDS Teil A und Teil B) bei pädiatrischen Patienten im Alter von 1 bis 11 Jahren mit EoE untersucht. Alle in die Studie aufgenommenen Patienten hatten zuvor auf eine konventionelle medikamentöse Therapie (Protonenpumpenhemmer, PPI) nicht angesprochen, bei 77 % der Patienten wurde ein anderes herkömmliches Arzneimittel (orale topische Kortikosteroide) vor Studieneinschluss angewendet und 54 % der Patienten waren unzureichend mit oralen topischen Kortikosteroiden kontrolliert oder vertrugen diese nicht oder für sie kam eine solche Therapie nicht in Betracht. Bei den Patienten, die für die Studie geeignet waren, betrug die Anzahl der intraepithelialen Eosinophilen (eos) ≥15 pro hochauflösendes Gesichtsfeld (high-power field, hpf) trotz Behandlung mit hochdosiertem Protonenpumpenhemmer vor oder während des Screeningzeitraums und zeigten vorher Anzeichen und Symptome einer EoE. Teil A war eine randomisierte, doppelblinde, multizentrische, placebokontrollierte Parallelgruppenstudievon 16-wöchiger Dauer. Teil B war eine Verlängerungsstudie mit aktiver Behandlung, bei der die Dosierungsschemata von Dupilumab zusätzlich über 36 Wochen untersucht wurden.
In Teil A wurde Dupilumab im Vergleich zu einem Placebo entsprechend den Dosierungsschemata nach dem Körpergewicht ≥15 bis < 30 kg (200 mg alle zwei Wochen) und ≥30 bis < 60 kg (300 mg alle zwei Wochen) untersucht. Das Dosierungsschema von Dupilumab wurde für pädiatrische Patienten im Alter von 1 bis 11 Jahren und mit einem Körpergewicht von ≥40 kg (300 mg wöchentlich) auf der Grundlage von Simulationen mit einem populationspharmakokinetischen Modell festgelegt, um die Expositionen für Erwachsene und pädiatrischen Patienten im Alter von 12 bis 17 Jahren mit EoE einzuhalten, die 300 mg wöchentlich erhielten und bei denen eine histologische symptomatische Wirksamkeit beobachtet worden war (siehe «Klinische Wirksamkeit → Pädiatrische Population» und «Pharmakokinetik»).
Insgesamt wurden 61 Patienten in Teil A aufgenommen. Das mittlere Alter lag bei 8 Jahren (1 bis 11 Jahren), das mittlere Gewicht bei 27,9 kg, 75,4 % der Patienten waren männlich, 85,2 % Weisse, 11,5 % Schwarze 1,6 % Asiaten. Insgesamt 59 Patienten von Teil A nahmen an Teil B teil. Die meisten Patienten erhielten zuvor orale topische Kortikosteroide (OTK) zur Behandlung von EoE (51 Patienten von 61; 83,6 %) und 34,4 % berichteten, dass OTK zuvor bei EoE wirksam gewesen waren. Zu Studienbeginn lag der mittlere maximale ösophageale, intraepitheliale Eosinophilen-Wert bei 97,7 (48,24) bzw. 82,2 (36,14) in den Dupilumab- und Placebo-Gruppen.
Primärer Wirksamkeitsendpunkt in Teil A war der Anteil der Patienten mit erreichter histologischer Remission, die anhand eines maximalen ösophagealen, intraepithelialen Eosinophilen-Werts von ≤6 eos/hpf in der Woche 16 angeben wurde. Sekundäre Endpunkte waren der Anteil der Patienten mit erreichtem ösophagealen, intraepithelialen Eosinophilen-Wert von < 15 eos/hpf und die folgenden Veränderungen gegenüber dem Ausgangswert: ösophagealer, intraepithelialer Eosinophilen-Wert (eos/hpf), absolute Veränderung des mittleren Schweregrades (Grade) EoE-typischer histologischer Merkmale gemäss EoEHSS (Eosinophilic Esophagitis Histology Scoring System), absolute Veränderung des mittleren Ausmasses (Stage) gemäss EoEHSS und absolute Veränderung des endoskopischen Referenzwerts bei EoE (EoE-Endoscopic Reference Score, EoE-EREFS). Die Wirkung auf Anzeichen von EoE wurde mit Hilfe der beobachterberichteten Ergebnisse gemessen; anhand des von den Pflegepersonen ausgefüllten pädiatrischen Fragebogens zu Anzeichen/Symptomen von EoE (Pediatric EoE Sign/Symptom Questionnaire-Caregiver, PESQ-C) wurde der Anteil an Tagen mit einer oder mehreren Anzeichen von EoE und anhand des pädiatrischen Scores der Symptome von eosinophiler Ösophagitis (Pediatric Eosinophilic Esophagitis Symptom Score, PEESS) wurde die Häufigkeit und Schwere der Anzeichen von EoE bewertet.
Die Wirksamkeitsergebnisse in Teil A sind unten und in Tabelle 25 aufgeführt.
Tabelle 25: Ergebnisse der Wirksamkeit von Dupilumab in Woche 16 bei Patienten im Alter von 1 bis 11 Jahren mit eosinophiler Ösophagitis und einem Gewicht von mindestens 15 kg (EoE KIDS, Teil A)
|
|
Dupilumaba
N=32
|
Placebo
N=29
|
Unterschied zu Placebo
(95 % KI)
| |
Primärer Endpunkt
| |
Anteil der Patienten mit erreichter histologischer Remission (maximalem ösophagealen, intraepithelialen Eosinophilen-Wert ≤6 eos/hpf), n (%)b
|
21
(65,6)
|
1
(3,4)
|
62,0
(44,00; 79,95)
| |
Sekundäre Endpunkte
| |
Anteil der Patienten mit erreichtem maximalen ösophagealen, intraepithelialen Eosinophilen-Wert < 15 eos/hpf), n (%)b
|
26 (81,3)
|
1
(3,4)
|
78,0
(63,10; 92,86)
| |
Prozentuale Veränderung des maximalen ösophagealen, intraepithelialen Eosinophilen-Werts gegenüber Baseline (eos/hpf), LS-Mittelwert (SE)c
|
-83,99
(13,25)
|
20,42
(13,67)
|
-104,41
(-142,06; -66,75)
| |
Absolute Veränderung des mittleren Schweregrades(Grade Score) (0-3d) des EoEHSS gegenüber Baseline, LS-Mittelwert (SE)
|
-0,907
(0,05)
|
-0,010
(0,05)
|
-0,897
(-1,04; -0,75)
| |
Absolute Veränderung des mittleren Ausmasses (Stage Score) (0-3d) des EoEHSS gegenüber Baseline, LS-Mittelwert (SE)
|
-0,874
(0,05)
|
-0,002
(0,05)
|
-0,871
(-1,01; -0,73)
| |
Absolute Veränderung des EoE-EREFS (0-18e) gegenüber Baseline, LS-Mittelwert (SE)
|
-3,6
(0,42)
|
0,0
(0,46)
|
-3,6
(-4,89; -2,40)
|
a Dupixent wurde gemäss den progressiven Dosierungsschemata nach Körpergewicht bewertet: ≥15 bis < 30 kg (200 mg alle zwei Wochen) und ≥30 bis < 60 kg (300 mg alle zwei Wochen).
b Für die histologische Remission wurde der prozentuale Unterschied mit Hilfe der Mantel-Haenszel-Methode unter Berücksichtigung der Gewichtsgruppe bei Studieneinschluss (≥15 bis < 30 kg und ≥30 bis < 60 kg) geschätzt.
c Der Unterschied bei der absoluten oder prozentualen Veränderung wurde mit Hilfe des ANCOVA-Modells unter Verwendung der Messung zu Studienbeginn als Kovariate und der Behandlung und der Gewichtsgruppe zu Studienbeginn (≥15 bis < 30 kg und ≥30 bis < 60 kg) als fixe Faktoren geschätzt.
d Die EoEHSS-Scores reichen von 0 bis 3; höhere Scores weisen auf höheren Schweregrad und grösseres Ausmass der histologischen Veränderungen hin. Die mittleren EoEHSS-Schweregrad-Scores bei Studienbeginn betrugen 1,35 (0,42) und 1,30 (0,37) in den Dupilumab- bzw. Placebo-Gruppen und die mittleren EoEHSS-Ausmass-Scores zu Studienbeginn betrugen 1,31 (0,32) und 1,33 (0,37) in den Dupilumab- bzw. Placebo-Gruppen.
e Die EoE-EREFS-Gesamtscores reichen von 0 bis 18; höhere Scores weisen auf schwerwiegendere endoskopische Befunde von entzündlichen Vorgängen und Remodeling hin.
Im Teil B erreichten 17 der 32 Patienten, die mit Dupilumab in Teil A und B behandelt wurden, und 8 der 15 Patienten, die mit Placebo in Teil A und mit Dupilumab in Teil B behandelt wurden, die histologische Remission in Woche 52.
Eine Verbesserung des Anteils der Tage mit einem oder mehreren Anzeichen von EoE (nach PESQ-C) wurde bei den Studienteilnehmern beobachtet, die mit Dupilumab behandelt wurden, im Vergleich zu Placebo nach 16-wöchiger Behandlung in Teil A.
6) Klinische Wirksamkeit bei chronisch obstruktiver Lungenerkrankung (COPD)
Das Entwicklungsprogramm zur chronisch obstruktiven Lungenerkrankung (COPD) umfasste zwei randomisierte, doppelblinde, multizentrische, placebokontrollierte Parallelgruppenstudien (BOREAS und NOTUS) mit einer Behandlungsdauer von 52 Wochen, die insgesamt 1'874 erwachsene COPD-Patienten umfassten, um Dupilumab als Add-on-Erhaltungstherapie zu untersuchen. Die beiden Studien schlossen Patienten ein, die eine COPD-Diagnose mit mittelschwerer bis schwerer Einschränkung des Luftstroms aufwiesen (FEV1/FVC-Verhältnis nach Gabe eines Bronchodilatators < 0,7 und FEV1 nach Gabe eines Bronchodilatators von 30 % bis 70 % des vorhergesagten Wertes), chronisch produktiven Husten über mindestens 3 Monate im Vorjahr sowie eine minimale Blut-Eosinophilenzahl von 300 Zellen/µl bei Aufnahme in die Studie aufwiesen. Die Patienten waren nicht kontrolliert bei einem Dyspnoe-Score des Medical Research Council (MRC) ≥2 (Skala 0–4) und hatten mindestens zwei mittelschwere oder eine schwere Exazerbation im Vorjahr trotz einer Dreifach-Erhaltungstherapie, bestehend aus einem langwirksamen Muskarin-Antagonisten (long-acting muscarinic antagonist, LAMA), einem langwirksamen Beta-Agonisten (long-acting beta agonist, LABA) und einem inhalativen Kortikosteroid (ICS). Die Patienten durften eine Erhaltungstherapie bestehend aus LAMA und LABA erhalten, wenn ICS kontraindiziert war. Exazerbationen wurden als mittelschwer definiert, wenn eine Behandlung mit systemischen Kortikosteroiden und/oder Antibiotika erforderlich war; oder als schwer, wenn sie eine Spitalbehandlung oder eine Beobachtung von mehr als 24 Stunden in einer Notfallstation oder Akutpflegeeinrichtung erforderten.
In beiden Studien wurden die Patienten randomisiert und erhielten über einen Zeitraum von 52 Wochen zusätzlich zur bestehenden Erhaltungstherapie entweder Dupilumab 300 mg alle zwei Wochen (Q2W) oder Placebo. In beiden Studien war der primäre Endpunkt die annualisierte Rate mittelschwerer oder schwerer COPD-Exazerbationen während des 52-wöchigen Behandlungszeitraums. Die sekundären Endpunkte umfassten Veränderungen des FEV1 vor Bronchodilatation zwischen Baseline und Woche 12 und 52, sowohl in der Gesamtpopulation als auch in der Subgruppe der Patienten mit einem initialen FeNO ≥20 ppb, Veränderungen des St.-Georges-Gesamtscores (St. George's Respiratory Questionnaire, SGRQ) in Woche 52 gegenüber Baseline und die annualisierte Rate mittelschwerer oder schwerer COPD-Exazerbationen in der Subgruppe der Patienten mit einem initialen FeNO ≥20 ppb während des 52-wöchigen Behandlungszeitraums.
Die demografischen Daten und Baseline-Charakteristika der Studien BOREAS und NOTUS sind in Tabelle 26 dargestellt.
Tabelle 26: Demografische Daten und Baseline-Charakteristika (BOREAS und NOTUS)
|
Parameter
|
BOREAS
(n = 939)
|
NOTUS
(n = 935)
| |
Mittleres Alter (Jahre) (± SD)
|
65,1 (8,1)
|
65,0 (8,3)
| |
Männlich (%)
|
66,0
|
67,6
| |
Kaukasische Patienten (%)
|
84,1
|
89,6
| |
Mittlere Raucheranamnese (Packungsjahre) (± SD)
|
40,5 (23,4)
|
40,3 (27,2)
| |
Aktive Raucher (%)
|
30
|
29,5
| |
Emphysem (%)
|
32,6
|
30,4
| |
Mittlere COPD-Dauer (Jahre) (± SD)
|
8,8 (6,0)
|
9,3 (6,4)
| |
Mittlere Anzahl mittelschwerera oder schwererb Exazerbationen im Vorjahr (± SD)
|
2,3 (1,0)
|
2,1 (0,9)
| |
Mittlere Anzahl schwerer Exazerbationenb im Vorjahr (± SD)
|
0,3 (0,7)
|
0,3 (0,6)
| |
COPD-Erhaltungstherapie bei Baseline: ICS/LAMA/LABA (%)
|
97,6
|
98,8
| |
Mittleres FEV1/FVC-Verhältnis nach Bronchodilatation (± SD)
|
0,49 (0,12)
|
0,50 (0,12)
| |
Mittlerer FEV1-Wert (l) vor Bronchodilatation (± SD)
|
1,30 (0,46)
|
1,36 (0,50)
| |
Mittlerer FEV1-Wert (l) nach Bronchodilatation (± SD)
|
1,40 (0,47)
|
1,45 (0,49)
| |
Mittlerer Prozentsatz des vorhergesagten FEV1 nach Bronchodilatation (%) (± SD)
|
50,6 (13,1)
|
50,1 (12,6)
| |
Mittlerer SGRQ-Gesamtscore (± SD)
|
48,4 (17,4)
|
51,5 (17,0)
| |
Mittlerer E-RS:COPD-Gesamtscore (± SD)
|
12,9 (7,1)
|
13,3 (7,0)
| |
Mittlerer BODE-Index-Score (± SD)
|
4,1 (1,7)
|
4,0 (1,6)
| |
Mittlerer FeNO-Wert ppb (± SD)
|
24,3 (22,4)
|
24,6 (26,0)
| |
Mittlere Eosinophilenzahl im Blut bei Baseline (Zellen/µl) (± SD)
|
401 (298)
|
407 (336)
|
ICS = inhalatives Kortikosteroid; LAMA = langwirksamer Muskarin-Antagonist; LABA = langwirksamer Beta-Agonist, FEV1 = Einsekundenkapazität (forciertes Exspirationsvolumen (1 s)); FVC = forcierte Vitalkapazität; FeNO = fraktioniertes exhaliertes Stickstoffmonoxid; BODE = Körpermassenindex, Atemwegsobstruktion, Dyspnoe, Kapazität bei Belastung (body-mass index, airflow obstruction, dyspnea, exercise capacity, BODE)
a Exazerbationen, die mit systemischen Kortikosteroiden und/oder Antibiotika behandelt wurden.
b Exazerbationen, die einen Spitalaufenthalt oder eine Überwachung von mehr als 24 Stunden in einer Notfallstation oder Akutpflegeeinrichtung erforderten.
Exazerbationen
In beiden Studien zeigte Dupilumab gegenüber Placebo eine statistisch signifikante Senkung der annualisierten Rate mittelschwerer oder schwerer COPD-Exazerbationen, wenn es mit der Erhaltungstherapie kombiniert wurde (siehe Tabelle 27).
Tabelle 27: Annualisierte Rate mittelschwerera oder schwererb COPD-Exazerbationen in den Studien BOREAS und NOTUS
|
Studie
|
Behandlung
(n)
|
Rate
(Exazerbationen/Jahr)
|
Relatives Risiko vs. Placebo
(95-%-KI)
|
Senkung der Exazerbationsrate vs. Placebo (%)
| |
Primärer Endpunkt: Mittelschwerea oder schwereb COPD-Exazerbationen
| |
BOREAS
|
Dupixent 300 mg alle 2 Wochen
(n = 468)
|
0,78
|
0,705
(0.581, 0.857)c
|
30 %
| |
Placebo
(n = 471)
|
1,10
|
|
| |
NOTUS
|
Dupixent 300 mg alle 2 Wochen
(n= 470)
|
0,86
|
0,664
(0.535, 0.823)d
|
34 %
| |
Placebo
(n = 465)
|
1,30
|
|
| |
Gepoolte Komponente des primären Endpunktese: Schwere COPD-Exazerbationen
| |
BOREAS und NOTUS
|
Dupixent 300 mg alle 2 Wochen
(n = 938)
|
0,08
|
0,674
(0,438 - 1,037)
|
33 %
| |
Placebo
(n = 936)
|
0,12
|
|
|
a Exazerbationen, die mit systemischen Kortikosteroiden und/oder Antibiotika behandelt wurden
b Exazerbationen, die einen Spitalaufenthalt oder eine Beobachtung für > 24 Stunden in einer Notfallstation/Akutpflegeeinrichtung erforderten oder zum Tod führten
c p-Wert = 0,0005
d p-Wert = 0,0002
e Die Analyse der Komponente des primären Endpunktes wurde nicht an die Multiplizität angepasst
In beiden Studien war die über 52 Wochen beobachtete kumulierte mittlere Anzahl mittelschwerer oder schwerer Exazerbationen bei mit Dupilumab behandelten Patienten niedriger im Vergleich zu Placebo (siehe Abbildungen 17 und 18).
Abbildung 17: Kumulierte mittlere Anzahl mittelschwerer oder schwerer COPD-Exazerbationen über 52 Wochen in der BOREAS-Studie
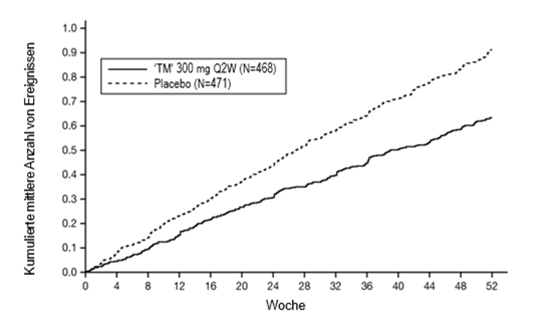
Abbildung 18: Kumulierte mittlere Anzahl mittelschwerer oder schwerer COPD-Exazerbationen über 52 Wochen in der NOTUS-Studie
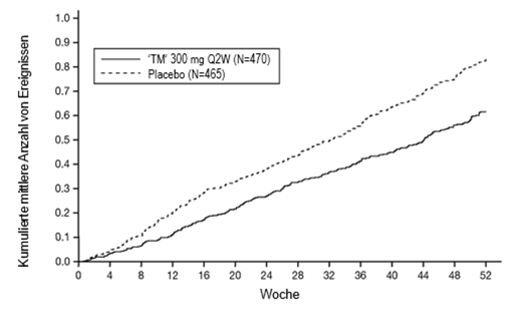
Die Zeitspanne bis zum Auftreten der ersten mittelschweren oder schweren COPD-Exazerbation war bei Patienten unter Dupilumab gegenüber Patienten, die in der BOREAS-Studie Placebo erhielten, verlängert (RR: 0,80; 95-%- KI: 0,66; 0,98) und NOTUS (RR: 0,71; 95 %-KI: 0,57; 0,889).
In der Subgruppenanalyse von Patienten mit einem erhöhten initialen FeNO bei Baseline (≥20 ppb) in BOREAS (n = 383) reduzierte die Behandlung mit Dupilumab die annualisierte Rate mittelschwerer oder schwerer COPD-Exazerbationen im Vergleich zu Placebo statistisch signifikant (RR: 0,625; 95 %-KI: 0,45, 0,869; p = 0,005). In der NOTUS-Studie zeigte die Behandlung mit Dupilumab eine nominal signifikante Reduktion der annualisierten Rate mittelschwerer oder schwerer COPD-Exazerbationen in der Subgruppe der Patienten mit einem erhöhten FeNO (≥20 ppb) bei Baseline (n = 355) im Vergleich zu Placebo (RR: 0,47; 95 %-KI: 0,33, 0,68).
Eine Senkung der annualisierten Rate mittelschwerer oder schwerer COPD-Exazerbationen wurde in allen vordefinierten Subgruppen beobachtet, einschliesslich Alter, Geschlecht, ethnischer Herkunft, Raucherstatus, Eosinophilenzahl im Blut, Anzahl der Exazerbationen im Vorjahr (≤2, 3 und ≥4), hochdosiertem ICS bei Baseline und prozentualem Anteil des vorhergesagten FEV1 nach Bronchodilatation bei Baseline (< 50 %, ≥50 %). Bei Patienten mit Emphysem stimmte die Senkung der Rate mittelschwerer oder schwerer Exazerbationen mit der Gesamtpopulation überein.
Lungenfunktion
In beiden Studien zeigte Dupilumab in Kombination mit einer Erhaltungstherapie eine statistisch signifikante Verbesserung des FEV1 vor Bronchodilatation in Woche 12 und 52 im Vergleich zu Placebo (siehe Tabelle 28). Bei mit Dupilumab behandelten Patienten wurden gegenüber Placebo bereits ab Woche 2 (BOREAS) (erste Beurteilung) und ab Woche 4 (NOTUS) grössere Verbesserungen der Lungenfunktion beobachtet (mittlere LS-Veränderung des FEV1 vor Bronchodilatation gegenüber Baseline) und blieben bis Woche 52 erhalten.
In der BOREAS-Studie wurden bei Behandlung mit Dupilumab gegenüber Placebo schnelle Verbesserungen des FEV1 nach Bronchodilatation, des FEV1/FVC-Verhältnisses nach Bronchodilatation und der FVC vor Bronchodilatation bereits in Woche 2 (erste Beurteilung) beobachtet und bis Woche 52 beibehalten. In der NOTUS-Studie wurden bei Behandlung mit Dupilumab gegenüber Placebo schnelle Verbesserungen des FEV1 nach Bronchodilatation und des FEV1/FVC-Verhältnisses nach Bronchodilatation in Woche 8 bzw. Woche 2 beobachtet und blieben bis Woche 52 erhalten.
Tabelle 28: Mittlere Veränderung der Lungenfunktionsendpunkte gegenüber Baseline in BOREAS und NOTUS
|
|
|
BOREAS
|
|
|
NOTUS
|
| |
|
Dupilumab
(n = 468)
|
Placebo
(n = 471)
|
Differenz (95 %-KI) Dupilumab vs. Placebo
|
Dupilumab
(n = 470)
|
Placebo
(n = 465)
|
Differenz (95 %-KI) Dupilumab vs. Placebo
| |
Mittlere LS-Veränderung des FEV1 vor Bronchodilatation in Woche 12 gegenüber Baseline (SE)
|
0,160 (0,018)
|
0,077
(0,018)
|
0,083
(0,042 - 0,125)a
|
0,139 (0,017)
|
0,057 (0,017)
|
0,082 (0,040 - 0,124)f
| |
Mittlere LS-Veränderung des FEV1 vor Bronchodilatation in Woche 52 gegenüber Baseline (SE)k
|
0,153 (0,019)
|
0,070
(0,019)
|
0,083
(0,038 - 0,128)b
|
0,115 (0,021)
|
0,054 (0,020)
|
0,062
(0,011 - 0,113)g
| |
Mittlere LS-Veränderung des FEV1 nach Bronchodilatation in Woche 12 gegenüber Baseline (SE)
|
0,156 (0,018)
|
0,084
(0,018)
|
0,072
(0,030 - 0,115)c
|
0,136 (0,020)
|
0,064 (0,020)
|
0,072 (0,023 - 0,121)h
| |
Mittlere LS-Veränderung des FEV1/FVC-Verhältnisses nach Bronchodilatation in Woche 12 gegenüber Baseline (SE)
|
0,037 (0,004)
|
0,023
(0,004)
|
0,014
(0,005 - 0,023)d
|
0,030 (0,004)
|
0,013 (0,004)
|
0,017 (0,006 - 0,028)i
| |
Mittlere LS-Veränderung des FVC vor Bronchodilatation in Woche 12 gegenüber Baseline (SE)
|
0,098 (0,022)
|
0,029
(0,022)
|
0,069
(0,016 - 0,121)e
|
0,083 (0,024)
|
0,018 (0,024)
|
0,066 (0,005 - 0,126)j
|
LS = kleinste Quadrate, SE = Standardfehler, FEV1 = Einsekundenkapazität (forciertes Exspirationsvolumen (1 s)), CVF = forcierte Vitalkapazität
ap-Wert < 0,0001; bp-Wert = 0,0003 (alle statistisch signifikant gegenüber Placebo mit Anpassung an die Multiplizität);
cnominaler p-Wert = 0,0010; dnominaler p-Wert = 0,0016; enominaler p-Wert = 0,0103;
fnominaler p-Wert = 0,0001; gnominaler p-Wert = 0,0182 (alle statistisch signifikant gegenüber Placebo mit Anpassung an die Multiplizität);
hnominaler p-Wert = 0,0042; inominaler p-Wert = 0,0020; jnominaler p-Wert = 0,0327;
kWirksamkeitsergebnisse für die mittlere Veränderung des FEV1 vor Bronchodilatation in Woche 52 gegenüber Baseline für 721 von 935 Patienten, die den 52-wöchigen Behandlungszeitraum abgeschlossen haben oder die Studie zum Zeitpunkt der Datenanalyse abgebrochen hatten.
In der Subgruppenanalyse der Patienten mit erhöhtem FeNO (≥20 ppb) bei Baseline verbesserte die Behandlung mit Dupilumab in der BOREAS-Studie (n = 383) den FEV1-Wert vor Bronchodilatation gegenüber Baseline in Woche 12 statistisch signifikant (mittlere LS-Veränderung: 0,232 Dupilumab vs. 0,108 Placebo; mittlere LS-Differenz: 0,124 [95 %-KI: 0,045, 0,203]; p = 0,002) und Woche 52 (mittlere LS-Veränderung: 0,247 Dupilumab vs. 0,120 Placebo; mittlere LS-Differenz: 0,127 [95 %-KI: 0,042, 0,212]; p = 0,003) gegenüber Placebo. In der NOTUS-Studie wurde eine statistisch signifikante Verbesserung gegenüber Baseline in der Subgruppe der Patienten unter Dupilumab mit erhöhtem FeNO (≥20 ppb) bei Baseline in Woche 12 beobachtet (n = 355; mittlere LS-Veränderung: 0,221 Dupilumab vs. 0,081 Placebo; mittlere LS-Differenz: 0,141 [95 %-KI: 0,058–0,223]; p = 0,001).
Die Behandlung mit Dupilumab verbesserte in der NOTUS-Studie den FEV1-Wert vor Bronchodilatation in Woche 52 in der Subgruppe der Patienten mit erhöhtem FeNO (≥20 ppb) bei Baseline gegenüber Placebo (n = 264; mittlere LS-Veränderung: 0,176 Dupilumab vs. 0,095 Placebo; mittlere LS-Differenz: 0,081 [95 %-KI: -0,019; 0,181]), erreichte jedoch keine statistische Signifikanz.
Verbesserungen der Lungenfunktion, gemessen anhand des FEV1 vor Bronchodilatation, wurden in allen vordefinierten Subgruppen beobachtet, einschliesslich Alter, Geschlecht, ethnischer Herkunft, Raucherstatus, Eosinophilenzahl im Blut, Anzahl der Exazerbationen im Vorjahr (≤2, 3 und ≥4), hochdosiertem ICS bei Baseline und prozentualem Anteil des vorhergesagten FEV1 nach Bronchodilatation bei Baseline (< 50 %, ≥50 %). Bei Patienten mit Emphysem stimmte die Verbesserung der Lungenfunktion, gemessen anhand des FEV1 vor Bronchodilatation, mit der Gesamtpopulation überein.
Patientenberichtete Ergebnisse
In beiden Studien wurde die gesundheitsbezogene Lebensqualität anhand der mittleren LS-Veränderung des St. George's Respiratory Questionnaire-(SGRQ)-Gesamtscores in Woche 52 gemessen. In der BOREAS-Studie wurde eine statistisch signifikante Verbesserung des SGRQ-Gesamtscores bei mit Dupilumab behandelten Patienten im Vergleich zu Placebo beobachtet (mittlere LS-Veränderung: -9,73 Dupilumab vs. -6,37 Placebo; mittlere LS-Differenz: -3,36 [95 %-KI: -5,46, -1,27]; p = 0,002). In der NOTUS-Studie verbesserte Dupilumab den SGRQ-Gesamtscore in Woche 52 im Vergleich zu Placebo nominal (mittlere LS-Veränderung: -9,82 bei Dupilumab vs. -6,44 Placebo; mittlere LS-Differenz: -3,37; [95 %-KI: -5,81, -0,93]; p = 0,007).
Pädiatrische Population:
Atopische Dermatitis:
Die Sicherheit und Wirksamkeit von Dupilumab bei pädiatrischen Patienten ab 6 Monaten mit mittelschwerer bis schwerer atopischer Dermatitis wurden nachgewiesen.
Unterstützende Daten zur Anwendung von Dupilumab bei dieser Altersgruppe liefern die Studie AD-1526, in die 251 Jugendliche von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis eingeschlossen wurden, die Studie AD-1652, an der 367 Kinder von 6 bis 11 Jahren mit schwerer atopischer Dermatitis teilnahmen, und die Studie AD-1539, in die 162 Kinder im Alter von 6 Monaten bis 5 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis eingeschlossen waren. Unterstützende Daten zur Langzeitanwendung von Dupilumab liefert die Studie AD-1434, in die 823 pädiatrische Patienten zwischen 6 Monaten und 17 Jahren eingeschlossen waren, darunter 275 Jugendliche, 368 Kinder im Alter von 6 bis 11 Jahren und 180 Kinder im Alter von 6 Monaten bis 5 Jahren. Unterstützende Daten zur Anwendung von Dupilumab liefert die Studie AD-1924, in die 27 Jugendliche im Alter von 12 bis 17 Jahren mit mittelschwerer bis schwerer atopischer Dermatitis der Hände und Füsse eingeschlossen wurden. Die Sicherheit und Wirksamkeit bei pädiatrischen und erwachsenen Patienten waren insgesamt konsistent.
In der Studie AD-1434 wurden keine neuen Sicherheitserkenntnisse gefunden (siehe «Unerwünschte Wirkungen»).
Die Sicherheit und Wirksamkeit bei pädiatrischen Patienten unter 6 Monaten mit atopischer Dermatitis sind nicht erwiesen.
Asthma:
Patienten von 12 bis 17 Jahren
Insgesamt 107 Jugendliche im Alter von 12 bis 17 Jahren mit mittelschwerem bis schwerem Asthma wurden in die QUEST-Studie eingeschlossen und erhielten alle zwei Wochen entweder 200 mg (n = 21) oder 300 mg (n = 18) Dupilumab (oder entweder 200 mg [n = 34] oder 300 mg [n = 34] entsprechendes Placebo). Eine Wirksamkeit hinsichtlich schwerer Asthmaexazerbationen und der Lungenfunktion wurde sowohl bei Jugendlichen als auch bei Erwachsenen beobachtet. Für beide Dosierungen (200 mg und 300 mg alle 2 Wochen) wurden signifikante Verbesserungen des FEV1 (mittlere LS-Veränderung gegenüber der Baseline in Woche 12) beobachtet (0,36 l bzw. 0,27 l). Für die Dosis von 200 mg alle 2 Wochen wurde ebenso wie bei Erwachsenen eine Reduktion der Rate von schweren Exazerbationen beobachtet. Im Allgemeinen war das Nebenwirkungsprofil bei Jugendlichen mit dem von Erwachsenen vergleichbar.
Kinder (6 bis 11 Jahre)
Insgesamt 408 Kinder von 6 bis 11 Jahren mit mittelschwerem bis schwerem Asthma wurden in die VOYAGE-Studie eingeschlossen, in der Dosierungen von 100 mg und 200 mg alle zwei Wochen untersucht wurden. Die Wirksamkeit von Dupilumab 300 mg alle vier Wochen bei Kindern von 6 bis 11 Jahren wurde anhand der VOYAGE-Daten zur Wirksamkeit von 100 mg und 200 mg alle zwei Wochen und der QUEST-Daten zu 200 mg und 300 mg alle zwei Wochen bei Erwachsenen und Jugendlichen extrapoliert. Patienten, die die Behandlungsphase der VOYAGE-Studie vollendet hatten, konnten an der unverblindeten Verlängerungsstudie (EXCURSION) teilnehmen. In dieser Studie erhielten 14 Patienten (≥15 kg bis < 30 kg) von 365 Patienten alle vier Wochen 300 mg, das Sicherheitsprofil war mit dem der VOYAGE-Studie vergleichbar.
Eosinophile Ösophagitis (EoE)
Insgesamt 71 pädiatrische Patienten im Alter von 1 bis 11 Jahren erhielten Dupilumab 100 mg alle zwei Wochen, 200 mg alle zwei Wochen, 300 mg alle zwei Wochen bzw. ein Placebo über 16 Wochen (EoE KIDS Teil A). Davon erhielten 37 Patienten, die mit Dupilumab inTeil A behandelt wurden, eine zusätzliche 36-wöchige Behandlung nach den Dosierungsschemata für Dupilumab (EoE KIDS Teil B). Die Verwendung von Dupilumab bei pädiatrischen Patienten im Alter von 1 bis 11 Jahren mit EoE, die 300 mg wöchentlich (≥40 kg) erhielten, sowie bei pädiatrischen Patienten im Alter von 12 bis 17 Jahren mit einem Körpergewicht von < 40 kg, die die gleiche Dosis nach Gewicht wie die pädiatrischen Patienten im Alter von 1 bis 11 Jahren erhielten, stützt sich ebenfalls auf eine Analyse populationspharmakokinetischer Daten (siehe «Pharmakokinetik»). Die Sicherheit der Anwendung und Wirksamkeit von Dupilumab bei Erwachsenen und pädiatrischen Patienten waren vergleichbar.
|