ZusammensetzungWirkstoffe
YESCARTA (Axicabtagen-Ciloleucel) ist eine gegen CD19 gerichtete, genetisch modifizierte autologe T-Zell-Immuntherapie. Zur Herstellung von YESCARTA werden körpereigene T-Zellen des Patienten entnommen und anschliessend ex vivo mittels retroviraler Transduktion modifiziert, um einen chimären Antigenrezeptor (CAR) zu exprimieren, der ein variables Maus-Anti-CD-19-Einzelkettenfragment (scFv) umfasst, das mit der kostimulatorischen CD28-Domäne und der CD3-zeta-Signaldomäne verbunden ist.
Hilfsstoffe
Cryostor CS10 (DMSO; Dextran 40), Natriumchlorid, humanes Serumalbumin (Natriumchlorid, Nacetyl-DL-tryptophan, Caprylsäure, Wasser), 5% DMSO.
YESCARTA enthält ca. 300 mg Natrium pro Infusionsbeutel.
Indikationen/AnwendungsmöglichkeitenYESCARTA ist eine gegen CD19 gerichtete, genetisch modifizierte autologe T-Zell-Immuntherapie und wird angewendet bei erwachsenen Patienten zur Behandlung von
·diffusem grosszelligem B-Zell-Lymphom (DLBCL) oder High-Grade-B-Zell-Lymphom (HGBL), das auf die Erstlinien-Chemoimmuntherapie refraktär ist oder innerhalb von 12 Monaten nach der Erstlinien-Chemoimmuntherapie rezidiviert
·rezidiviertem oder refraktärem (r/r) DLBCL oder primärem mediastinalem grosszelligem B-Zell-Lymphom (PMBCL) nach zwei oder mehr systemischen Therapielinien.
·rezidiviertem oder refraktärem follikulärem Lymphom (FL) nach drei oder mehr systemischen Therapielinien.
Dosierung/AnwendungDie YESCARTA-Therapie ist unter der Anleitung von Ärzten einzuleiten und zu überwachen, die in der Behandlung von hämatologischen Neoplasien erfahren sind und die im Management von Patienten, die mit YESCARTA behandelt wurden, einschliesslich der Behandlung des Zytokin-Freisetzungssyndroms (CRS) und von Neurotoxizität, geschult sind. Die Gabe von YESCARTA muss in einem qualifizierten Behandlungszentrum mit unmittelbarem Zugang zu geeigneten Intensivstationen durch medizinisches Fachpersonal erfolgen, welches für die Gabe von YESCARTA geschult wurde. Vor der Infusion müssen mindestens 4 Dosen Tocilizumab zur Anwendung verfügbar sein.
YESCARTA ist ein Arzneimittel zur einmaligen Infusion und nur für die autologe und intravenöse Anwendung vorgesehen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Verfügbarkeit von YESCARTA muss bestätigt werden, bevor das Behandlungsschema zur Lymphodepletion begonnen wird. Die Herstellung und Freigabe von YESCARTA dauert üblicherweise etwa 3-4 Wochen. Es gibt Gründe, die dazu führen können, dass ein Patient trotz erfolgter Leukapharese nicht mit YESCARTA behandelt werden kann (siehe «Eigenschaften/Wirkungen»).
Vorbehandlung
·Ein Chemotherapieschema zur Lymphodepletion, das aus intravenös verabreichtem Cyclophosphamid 500 mg/m2 und intravenös verabreichtem Fludarabin 30 mg/m2 besteht, sollte am 5., 4. und 3. Tag vor Infusion von YESCARTA verabreicht werden. Es wird eine absolute Neutrophilenzahl (ANC) ≥1000/µl und eine Thrombozytenzahl ≥75'000/µl vor Beginn der Chemotherapie zur Lymphodepletion empfohlen.
Klinische Beurteilung vor der YESCARTA-Infusion
Die Behandlung mit YESCARTA sollte bei bestimmten Risikopatienten aufgeschoben werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Prämedikation
·Es wird empfohlen, die Prämedikation mit oralem Paracetamol 500-1000 mg und intravenösem oder oralem Diphenhydramin 12,5 mg bis 25 mg oder gleichwertige Arzneimittel, ca. 1 Stunde vor der YESCARTA-Infusion zu verabreichen, um die Möglichkeit einer Infusionsreaktion zu verringern.
·Die prophylaktische Anwendung systemischer Steroide wird nicht empfohlen (siehe «Interaktionen»).
Dosierung
Ein patientenspezifischer Einzel-Infusionsbeutel von YESCARTA mit einer Dispersion von Anti-CD19-CAR-T-Zellen in ca. 68 ml für eine Zieldosis von 2 x 106 Anti-CD19-CAR-T-Zellen pro kg Körpergewicht (Spanne: 1,0 x 106 – 2,0 x 106 Zellen/kg), mit maximal 2 x 108 Anti-CD19-CAR-T-Zellen für Patienten von 100 kg und darüber.
Überwachung nach der Infusion
Die Patienten müssen in den ersten 7 Tagen nach der Infusion täglich auf Anzeichen und Symptome eines potenziellen CRS, auf neurologische Ereignisse und andere Toxizitäten in einem qualifizierten Behandlungszentrum überwacht werden. Ärzte können eine Hospitalisierung für die ersten 7 Tage, oder bei ersten Anzeichen oder Symptomen eines CRS und/oder neurologischer Ereignisse, in Erwägung ziehen.
Nach Ablauf der ersten 7 Tage nach der Infusion sollte der Patient nach Ermessen des Arztes bzw. der Ärztin überwacht werden.
Patienten müssen sich nach der Infusion mindestens 4 Wochen lang in der Nähe einer qualifizierten klinischen Einrichtung (maximal 2 Stunden entfernt) aufhalten.
Der Patient muss ebenfalls darauf aufmerksam gemacht werden, dass obwohl die meisten CRS und neurologischen Symptome innerhalb der ersten 4 Wochen nach Infusion auftreten, unerwünschte Wirkungen jederzeit auftreten können, und eine medizinische Unterstützung erfordern können.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Es liegen keine ausreichenden Daten zu Patienten mit Leberfunktionsstörung vor, um Rückschlüsse auf diese Population machen zu können.
Patienten mit Nierenfunktionsstörungen
Es liegen keine ausreichenden Daten zu Patienten mit Nierenfunktionsstörung vor, um Rückschlüsse auf diese Population machen zu können.
Ältere Patienten
28% der Studienpopulation waren Patienten im Alter von 65 Jahren und älter. Wirksamkeit und Sicherheit waren in allen Altersgruppen vergleichbar. Basierend auf diesen Daten ist für ältere Patienten keine Dosierungsanpassung erforderlich.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von YESCARTA bei Kindern und Jugendlichen unter 18 Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Art der Anwendung
Intravenöse Anwendung.
YESCARTA ist ausschliesslich zur autologen Anwendung mittels intravenöser Infusion vorgesehen.
YESCARTA darf nicht bestrahlt werden. Es darf kein Filter zur Leukozytendepletion verwendet werden.
Vorsichtsmassnahmen vor/bei der Handhabung bzw. vor/während der Anwendung des Arzneimittels
Dieses Arzneimittel enthält genetisch modifizierte Zellen. Die Standard-Sicherheitsvorkehrungen zur Handhabung dieser Art von Arzneimitteln sind einzuhalten. Besondere Vorsichtsmassnahmen für die Beseitigung und sonstige Hinweise für die Handhabung, siehe «Sonstige Hinweise».
YESCARTA enthält humane Blutzellen. Medizinische Fachkräfte, die YESCARTA handhaben, müssen daher geeignete Vorsichtsmassnahmen treffen (Handschuhe und Schutzbrille tragen), um eine potenzielle Übertragung von Infektionskrankheiten zu vermeiden.
Vorbereitung von YESCARTA vor der Anwendung
·Es ist zu verifizieren, dass die Identität (ID) des Patienten mit den Patienten-Identifizierungsmerkmalen auf der YESCARTA-Kassette übereinstimmt.
·Der YESCARTA-Infusionsbeutel darf nicht aus der Kassette genommen werden, wenn die Informationen auf dem patientenspezifischen Etikett nicht mit dem vorgesehenen Patienten übereinstimmen.
·Nachdem die Patienten-ID bestätigt wurde, ist der YESCARTA-Infusionsbeutel aus der Kassette zu nehmen.
·Es ist sicherzustellen, dass die Patienteninformationen auf dem Etikett der Kassette mit den Informationen auf dem Etikett des Infusionsbeutels übereinstimmen.
·Vor dem Auftauen ist der Infusionsbeutel auf Unversehrtheit des Behälters zu untersuchen. Wenn der Infusionsbeutel beschädigt ist, sind die vor Ort geltenden Bestimmungen einzuhalten (alternativ kann direkt Kontakt mit Gilead aufgenommen werden).
Auftauen
·Der Infusionsbeutel ist in einen zweiten sterilen Beutel zu geben oder gemäss den vor Ort geltenden Bestimmungen zu handhaben.
·YESCARTA ist bei ca. 37°C unter Verwendung eines Wasserbads oder einer Methode zum trockenen Auftauen aufzutauen, bis im Infusionsbeutel kein Eis mehr sichtbar ist. Der Infusionsbeutel ist vorsichtig durchzumischen, um Klumpen von Zellmaterial aufzulösen. Wenn weiterhin Zellklumpen sichtbar sind, ist der Infusionsbeutel weiter vorsichtig durchzumischen. Kleine Klumpen von Zellmaterial sollten sich durch vorsichtiges manuelles Durchmischen auflösen lassen. YESCARTA darf vor der Infusion nicht gewaschen, zentrifugiert und/oder in einem neuen Medium resuspendiert werden. Das Auftauen sollte ca. 3 bis 5 Minuten dauern.
·Nach dem Auftauen kann YESCARTA bis zu 3 Stunden bei Raumtemperatur (20°C bis 25°C) aufbewahrt werden. Die YESCARTA-Infusion muss jedoch innerhalb von 30 Minuten nach Abschluss des Auftauvorgangs beginnen.
Verabreichung
·Es darf kein Filter zur Leukozytendepletion verwendet werden.
·Vor der Infusion und während der Erholungsphase müssen Tocilizumab und Notfallausrüstung zur Verfügung stehen.
·YESCARTA ist nur zur autologen Anwendung.
·Die Identität des Patienten muss erneut verifiziert werden, um sie mit den Patienten-Identifizierungsmerkmalen auf dem YESCARTA-Infusionsbeutel abzugleichen.
·Für die Anwendung von YESCARTA wird ein zentralvenöser Zugang empfohlen.
·Der Schlauch muss vor der Infusion mit einer sterilen Kochsalzlösung 9 mg/mL (0,9%) (0,154 mmol Sodium per mL) zur Injektion gespült werden.
·Der gesamte Inhalt des YESCARTA-Infusionsbeutels muss innerhalb von 30 Minuten infundiert werden, entweder mittels Schwerkraft oder über eine peristaltische Pumpe.
·Der Infusionsbeutel ist während der YESCARTA-Infusion sanft zu schütteln, um ein Verklumpen der Zellen zu vermeiden.
·Nachdem der gesamte Inhalt des Infusionsbeutels infundiert wurde, müssen der Infusionsbeutel und der Schlauch mit derselben Infusionsrate mit 10 bis 30 mL Kochsalzlösung 9 mg/mL (0,9%) zur Injektion durch Backpriming gespült werden, um sicherzustellen, dass die gesamte YESCARTA-Dosis appliziert wurde.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff, einem der Hilfsstoffe (siehe «Zusammensetzung») oder gegenüber einer der Substanzen, die als Kontraindikationen in der Fachinformation für Fludarabin oder Cyclophosphamid aufgeführt sind.
Warnhinweise und VorsichtsmassnahmenAllgemein
Die Warnhinweise und Vorsichtsmassnahmen der Chemotherapie zur Lymphodepletion müssen berücksichtigt werden.
Gründe für einen Aufschub der Behandlung
Aufgrund der Risiken, die mit der YESCARTA-Behandlung verbunden sind, muss die Infusion verschoben werden, wenn auf den Patienten einer der folgenden Zustände zutrifft:
·Bestehende schwerwiegende unerwünschte Wirkungen (insbesondere Reaktionen bezüglich der Lunge oder des Herzens, oder Hypotonie), einschliesslich jener, die durch vorangegangene Chemotherapien entstanden sind.
·Aktive Entzündung oder unkontrollierte Infektion.
·Aktive Graft-versus-host-disease (GvHD).
·Entwicklung einer klinisch signifikanten Verschlechterung des Lymphoms, welche zu einer medizinisch signifikanten Organfunktionsstörung oder klinischer Verschlechterung führt, nach einer Chemotherapie zur Lymphodepletion.
In manchen Fällen kann die YESCARTA Behandlung nach der Anwendung des Chemotherapieschemas zur Lymphodepletion verschoben werden. Wenn die Infusion um mehr als 2 Wochen, nachdem der Patient die Chemotherapie zur Lymphodepletion erhalten hat, verschoben wird, muss das Chemotherapieschema zur Lymphodepletion erneut angewendet werden (siehe «Dosierung/Anwendung»).
Mit YESCARTA behandelte Patienten sollen kein Blut und keine Organe, Gewebe und Zellen für Transplantationen spenden.
YESCARTA ist ausschliesslich zur autologen Anwendung vorgesehen und darf unter keinen Umständen anderen Patienten verabreicht werden. Vor der Infusion muss die Identität des Patienten mit den Patienten-Identifizierungsmerkmalen auf dem Infusionsbeutel und der Kassette von YESCARTA abgeglichen werden. Infundieren Sie YESCARTA nicht, wenn die Informationen auf dem patientenspezifischen Etikett nicht mit dem vorgesehenen Patienten übereinstimmen.
Überempfindlichkeitsreaktionen
Schwerwiegende und lebensbedrohliche infusionsbedingte Reaktionen wie Anaphylaxie (einschliesslich anaphylaktischer Schock, Herzstillstand und Atemstillstand) wurden mit YESCARTA berichtet. Es können allergische Reaktionen unter einer YESCARTA-Infusion auftreten. Schwerwiegende Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, können auf DMSO oder residuales Gentamicin in YESCARTA zurückzuführen sein.
Hirnbeteiligung des Lymphoms, Lymphom des zentralen Nervensystems und Lymphombeteiligung des Herzens.
Es liegen keine klinischen Erfahrungen zur Anwendung von YESCARTA bei Patienten mit einer Vorgeschichte oder akutem primären ZNS-Lymphom, bei Patienten mit malignen Zellen im Liquor, bei Patienten mit Hirnmetastasen, oder bei Patienten mit Lymphombeteiligung des Herzens vor.
Begleiterkrankungen
Patienten mit aktiver Erkrankung des zentralen Nervensystems (ZNS) oder mitvorbestehender Einschränkung der Organfunktionen, insbesondere der Lunge, des Herzens, der Nieren oder der Leber aufgrund vorbestehender Erkrankungen, sowie Patienten mit Thrombozytopenie oder niedrigem Fibrinogenspiegel, sind wahrscheinlich anfälliger für die Folgen der nachstehend beschriebenen unerwünschten Wirkungen und erfordern besondere Aufmerksamkeit. Zudem liegen keine klinischen Erfahrungen zur Anwendung von YESCARTA bei Patienten mit moderater oder schwerer Einschränkung der Organfunktion vor.
Zytokin-Freisetzungssyndrom
Bei fast allen Patienten ist zu einem gewissen Grad ein CRS aufgetreten. Im Zusammenhang mit YESCARTA wurde ein schweres CRS, einschliesslich lebensbedrohlicher und tödlicher Reaktionen, sehr häufig beobachtet, wobei die Zeit bis zum Einsetzen des Syndroms in ZUMA-1 und ZUMA-7 1 bis 12 Tage und in ZUMA-5 1 bis 15 Tage betrug (siehe «Unerwünschte Wirkungen»).
Für die Diagnose des CRS müssen alternative Ursachen einer systemischen inflammatorischen Reaktion, einschliesslich Infektion, ausgeschlossen werden.
Behandlung des mit YESCARTA-assoziierten Zytokin-Freisetzungssyndroms
Stellen Sie sicher, dass für jeden Patienten vor der YESCARTA-Infusion mindestens 4 Dosen Tocilizumab, (ein Interleukin-6 (IL-6) Rezeptor-Blocker), verfügbar sind.
Es wurden Behandlungsalgorithmen entwickelt, um einige der CRS-Symptome, die bei Patienten unter YESCARTA aufgetreten sind, zu lindern. Dies schliesst die Anwendung von Tocilizumab bzw. Tocilizumab und Kortikosteroiden für mittelgradiges, schweres oder lebensbedrohliches CRS ein (siehe Zusammenfassung in Tabelle 1 unten). Patienten mit CRS Grad 2 oder höher (z.B. nicht auf Flüssigkeitsgabe ansprechende Hypotonie, oder Hypoxie, die eine ergänzende Sauerstoffgabe erfordert) sollten durch kontinuierliches telemetrisches Monitoring der Herzfrequenz sowie Pulsoximetrie überwacht werden. Bei Patienten mit schwerem CRS sollte die Durchführung eines Echokardiogramms zur Beurteilung der Herzfunktion erwogen werden. Bei schwerem oder lebensbedrohlichem CRS ist eine unterstützende intensiv-medizinische Behandlung in Erwägung zu ziehen.
YESCARTA darf nicht bei Patienten mit aktiven Infektionen oder entzündlichen Erkrankungen angewendet werden, bis diese Erkrankungen unter Kontrolle sind.
CRS ist bekanntermassen mit Endorgan-Dysfunktion (z.B. Leber, Nieren, Herz und Lunge) assoziiert. Darüber hinaus kann im Zusammenhang mit CRS eine Verschlechterung zugrunde liegender Organpathologien auftreten. Ein niedriger Fibrinogenspiegel kann, insbesondere im Zusammenhang mit einer Thrombozytopenie, das Risiko für Blutungen erhöhen. Patienten mit medizinisch signifikanter Funktionsstörung des Herzens sollten gemäss den intensiv-medizinischen Standards behandelt werden, und es sind Massnahmen wie eine Echokardiographie in Erwägung zu ziehen.
Eine Untersuchung auf hämophagozytische Lymphohistiozytose/Makrophagen-Aktivierungssyndrom (HLH/MAS) ist bei Patienten mit schwerem oder nicht auf eine Behandlung ansprechenden CRS in Erwägung zu ziehen.
Tabelle 1: Einstufung und Behandlungsleitfaden für CRS
|
CRS-Schweregrada
|
Tocilizumab
|
Steroide
| |
Schweregrad 1
Symptome erfordern nur eine symptomatische Behandlung (z.B. Fieber, Übelkeit, Müdigkeit, Kopfschmerzen, Myalgie, Unwohlsein).
|
Wenn keine Besserung nach 24 Stunden eintritt, Tocilizumab 8 mg/kg intravenös über 1 Stunde verabreichen (maximal 800 mg).
|
n. v./n. z.
| |
Schweregrad 2
Symptome erfordern eine moderate Intervention und sprechen auf diese an.
Sauerstoffbedarf < 40% FiO2 oder Hypotonie, die auf Flüssigkeiten oder einen Vasopressor in geringer Dosis anspricht, oder Organtoxizität 2. Gradesb.
|
Tocilizumabc 8 mg/kg intravenös über 1 Stunde verabreichen (maximal 800 mg).
Tocilizumab bei Bedarf alle 8 Stunden erneut verabreichen, wenn kein Ansprechen auf intravenöse Flüssigkeitsgabe oder auf eine Erhöhung der zusätzlichen Sauerstoffgabe erfolgt. Maximal 3 Dosen über 24-Stunden; maximal 4 Dosen verabreichen, wenn keine klinische Besserung der Anzeichen und Symptome des CRS eintritt.
|
Gemäss Schweregrad 3 behandeln, wenn innerhalb von 24 Stunden nach Beginn der Behandlung mit Tocilizumab keine Besserung eintritt.
| |
Schweregrad 3
Symptome erfordern eine intensive Intervention und sprechen auf diese an.
Sauerstoffbedarf ≥40% FiO2 oder Hypotonie, die hoch dosierte oder mehrere Vasopressoren erfordert, oder Organtoxizität 3. Grades oder Transaminitis 4. Grades.
|
Gemäss Schweregrad 2
|
Methylprednisolon 1 mg/kg intravenös zweimal täglich oder äquivalente Dexamethason-Dosis (z.B. 10 mg intravenös alle 6 Stunden) verabreichen.
Anwendung von Kortikosteroiden fortführen, bis das Ereignis Grad 1 oder geringer erreicht; anschliessend über 3 Tage ausschleichen. Wenn keine Besserung eintritt, gemäss Schweregrad 4 (siehe unten) behandeln.
| |
Schweregrad 4
Lebensbedrohliche Symptome.
Notwendigkeit einer Unterstützung durch mechanische Beatmung oder einer kontinuierlichen venovenösen Hämodialyse (CVVHD) oder Organtoxizität 4. Grades (ausgenommen Transaminitis).
|
Gemäss Schweregrad 2
|
Methylprednisolon 1000 mg über 3 Tage hinweg einmal täglich intravenös verabreichen; bei Besserung wie oben beschrieben behandeln.
Wenn keine Besserung eintritt, Methylprednisolon 1000 mg zwei bis dreimal täglich intravenös verabreichen oder alternative Behandlungd in Betracht ziehen.
|
a Lee et al 2014
b Behandlung neurologischer unerwünschter Wirkungen, siehe Tabelle 2
c Genauere Informationen siehe Fachinformation von Tocilizumab
d Alternative Behandlung umfasst (u.a.): Anakinra, Siltuximab, Ruxolitinib, Cyclophosphamid, intravenöses Immunglobulin (IVIG) und Antithymozytenglobulin (ATG)
Neurologische unerwünschte Wirkungen
Bei mit YESCARTA behandelten Patienten wurden sehr häufig schwere neurologische unerwünschte Wirkungen, auch bekannt als Immuneffektorzell-assoziiertes Neurotoxizitätssyndrom (immune effector cell-associated neurotoxicity syndrome (ICANS)), beobachtet, die lebensbedrohlich oder tödlich verlaufen können (siehe «Unerwünschte Wirkungen»). Die mediane Dauer bis zum Einsetzen betrug 6 Tage (Spanne: 1 bis 133 Tage) in ZUMA-1 und ZUMA-7 und 7 Tage (Spanne: 1 bis 177 Tage) in ZUMA-5. Während der Postmarketingphase sind Fälle mit Status Epilepticus beobachtet worden. Bei Patienten mit Störungen des Zentralnervensystems (ZNS) in der Anamnese, wie z.B. Krampfanfällen oder zerebrovaskulärer Ischämie, besteht möglicherweise ein erhöhtes Risiko. Tödliche und schwerwiegende Fälle von Hirnödemen wurden bei Patienten, die mit YESCARTA behandelt wurden, berichtet. Die Patienten sind auf Anzeichen und Symptome neurologischer unerwünschter Wirkungen/ICANS zu überwachen (Tabelle 2).
Patienten mit neurologischen Toxizitäten/ICANS vom Schweregrad 2 oder höher sollten durch kontinuierliches telemetrisches Monitoring des Herzens und Pulsoximetrie überwacht werden. Bei schweren oder lebensbedrohlichen neurologischen Toxizitäten/ICANS ist eine intensiv-medizinische unterstützende Behandlung anzuwenden.
Levetiracetam kann zur Vorbeugung von Krampfanfällen bei neurologischen Nebenwirkungen in Erwägung gezogen werden. Es wurden Behandlungsalgorithmen entwickelt, um die bei mit YESCARTA behandelten Patienten auftretenden neurologischen unerwünschten Wirkungen zu mildern. Dies schliesst die Anwendung von Tocilizumab (bei gleichzeitig auftretendem CRS) und/oder Kortikosteroiden für milde, mittelgradige, schwere oder lebensbedrohliche neurologische unerwünschte Wirkungen ein (siehe Zusammenfassung in Tabelle 2 unten).
Tabelle 2: Einstufung und Behandlungsleitfaden für neurologische unerwünschte Wirkungen/ICANS
|
Einstufung
Bewertunga
|
Gleichzeitiges CRS
|
Kein gleichzeitiges CRS
| |
Schweregrad 2
|
Tocilizumab gemäss Tabelle 1 zur Behandlung von CRS mit Schweregrad 2 verabreichen.
Wenn innerhalb von 24 Stunden nach Beginn der Behandlung mit Tocilizumab keine Besserung eintritt, Dexamethason 10 mg alle 6 Stunden intravenös verabreichen, wenn nicht bereits andere Kortikosteroide gegeben werden. Anwendung von Dexamethason fortführen, bis das Ereignis Grad 1 oder geringer erreicht; anschliessend über 3 Tage ausschleichen.
|
Dexamethason 10 mg alle 6 Stunden intravenös verabreichen.
Anwendung von Dexamethason fortführen, bis das Ereignis Grad 1 oder geringer erreicht; anschliessend über 3 Tage ausschleichen.
| |
Levetiracetam zur Vorbeugung von Krampfanfällen in Erwägung ziehen.
| |
Schweregrad 3
|
Tocilizumab gemäss Tabelle 1 zur Behandlung von CRS mit Schweregrad 2 verabreichen.
Zusätzlich Dexamethason 10 mg intravenös mit der ersten Dosis Tocilizumab verabreichen und Dosis alle 6 Stunden wiederholen. Anwendung von Dexamethason fortführen, bis das Ereignis Grad 1 oder geringer erreicht; anschliessend über 3 Tage ausschleichen.
|
Dexamethason 10 mg alle 6 Stunden intravenös applizieren.
Anwendung von Dexamethason fortführen, bis das Ereignis Grad 1 oder geringer erreicht; anschliessend über 3 Tage ausschleichen.
| |
Levetiracetam zur Vorbeugung von Krampfanfällen in Erwägung ziehen.
| |
Schweregrad 4
|
Tocilizumab gemäss Tabelle 1 zur Behandlung von CRS mit Schweregrad 2 verabreichen. Methylprednisolon 1000 mg intravenös zusammen mit der ersten Dosis Tocilizumab verabreichen und Methylprednisolon 1000 mg täglich intravenös über 2 weitere Tage fortführen bei Besserung, wie oben beschrieben behandeln.
|
Methylprednisolon 1000 mg über 3 Tage hinweg täglich intravenös verabreichen; bei Besserung wie oben beschrieben behandeln.
Wenn keine Besserung eintritt, Methylprednisolon 1000 mg zwei bis dreimal täglich intravenös verabreichen oder eine alternative Behandlungb in Erwägung ziehen.
| |
Levetiracetam zur Vorbeugung von Krampfanfällen in Erwägung ziehen.
|
aSchweregrad basierend auf Common Terminology Criteria for Adverse Events
bAlternative Behandlung umfasst (u.a.): Anakinra, Siltuximab, Ruxolitinib, Cyclophosphamid, IVIG und ATG
Infektionen und febrile Neutropenie
Schwerwiegende Infektionen wurden sehr häufig im Zusammenhang mit YESCARTA beobachtet (siehe «Unerwünschte Wirkungen»). Bei immunsupprimierten Patienten wurde über lebensbedrohliche und tödlich verlaufende opportunistische Infektionen berichtet, einschliesslich disseminierte Pilzinfektionen.
Patienten müssen während und nach der YESCARTA-Infusion auf Anzeichen und Symptome einer Infektion überwacht und entsprechend behandelt werden. Gemäss den Standardleitlinien der Einrichtung sind antimikrobielle Wirkstoffe prophylaktisch anzuwenden. Nach der YESCARTA-Infusion wurde bei Patienten febrile Neutropenie beobachtet. Diese kann gleichzeitig mit einem CRS auftreten. Im Falle einer febrilen Neutropenie ist eine Infektionsdiagnostik durchzuführen und eine Behandlung mit Breitbandantibiotika, Flüssigkeiten und anderen medizinisch angezeigten unterstützenden Massnahmen, einzuleiten. In klinischen Studien wurden Patienten mit C-reaktivem Protein (CRP)-Anstieg auf >100 mg/L ausgeschlossen.
Bei immunsupprimierten Patienten, einschliesslich mit YESCARTA-behandelten Patienten, wurden Fälle von lebensbedrohlichen und tödlich verlaufenden opportunistischen Infektionen berichtet, einschliesslich disseminierte Pilzinfektionen und Virus-Reaktivierung (z.B. HHV-6 und progressive multifokale Leukoenzephalopathie). Bei Patienten mit neurologischen Ereignissen sollte die Möglichkeit solcher Infektionen berücksichtigt werden und entsprechende diagnostischen Massnahmen durchgeführt werden.
Virusreaktivierung
Eine Reaktivierung des Hepatitis-B-Virus (HBV), die in manchen Fällen zu fulminanter Hepatitis, Leberversagen und zum Tod führt, kann bei Patienten auftreten, die mit gegen B-Zellen gerichteten Arzneimitteln behandelt werden. Patienten mit einer HBV-, HCV- oder HIV-Infektion waren von den klinischen Studien ausgeschlossen, daher liegen keine Daten aus klinischen Studien bei diesen Patienten vor. Vor der Entnahme der Zellen für die Herstellung sollte ein Screening auf HBV, HCV und HIV gemäss den klinischen Leitlinien durchgeführt werden.
Bei Patienten mit HBV- oder HCV-Infektion in der Anamnese oder bei Patienten, die gegen HBV oder HCV behandelt wurden, darf der Virus mittels eines geeigneten sensitiven Nachweistests vor der Behandlung mit YESCARTA nicht detektierbar sein.
Andere lebensbedrohliche und tödlich verlaufende Fälle von Virusreaktivierung mit HHV-6 wurden berichtet.
Länger anhaltende Zytopenien
Nach Chemotherapie zur Lymphodepletion und YESCARTA-Infusion können die Patienten über Wochen und Monate hinweg anhaltende Zytopenien aufweisen und diese müssen gemäss den Standardleitlinien behandelt werden. Nach YESCARTA-Infusion traten sehr häufig länger anhaltende Zytopenien 3. oder höheren Grades auf, einschliesslich Thrombozytopenie, Neutropenie und Anämie. Das Blutbild des Patienten muss nach der YESCARTA-Infusion überwacht werden.
Hypogammaglobulinämie
Da YESCARTA einen Anti-CD19 chimären Antigenrezeptor enthält, kann bei Patienten, die mit YESCARTA behandelt werden, eine B-Zell-Aplasie auftreten, welche zu einer Hypogammaglobulinämie führt (siehe «Unerwünschte Wirkungen»). Eine Hypogammaglobulinämie macht Patienten anfälliger für Infektionen. Hypogammaglobulinämie wurde bei mit YESCARTA behandelten Patienten sehr häufig beobachtet. Überwachen Sie die Immunglobulinkonzentrationen nach der Behandlung mit YESCARTA und ergreifen Sie Massnahmen zur Vorbeugung von Infektionen, antibiotische Prophylaxe und Immunglobulinersatztherapie gemäss den Standardleitlinien im Falle wiederkehrenden Infektionen.
Lebendimpfstoffe
Die Sicherheit einer Immunisierung mit viralen Lebendimpfstoffen während oder nach der YESCARTA-Therapie wurde nicht untersucht. Die Impfung mit viralen Lebendimpfstoffen wird mindestens 6 Wochen lang vor Beginn der Chemotherapie zur Lymphodepletion, während der YESCARTA-Therapie und bis zur Wiederherstellung des Immunsystems nach der Behandlung mit YESCARTA nicht empfohlen.
Sekundäre Malignome
Patienten, die mit YESCARTA behandelt werden, können sekundäre Malignome entwickeln. T-Zell-Malignome wurden nach der Behandlung von hämatologischen Malignomen mit einer auf BCMA- und CD19-gerichteten, genetisch modifizierten autologen T-Zell-Immuntherapie, einschliesslich YESCARTA, berichtet. Reife T-Zell-Malignome, einschliesslich CAR-positiver Tumoren, können innerhalb von einigen Wochen bis Monate nach der Infusion auftreten und tödlich verlaufen (siehe «Unerwünschte Wirkungen»).
Es ist eine lebenslange Überwachung auf sekundäre Malignome, vor allem auf sekundäre Malignome mit hämatologischem Ursprung, durchzuführen. Wenn ein sekundäres Malignom mit hämatologischem Ursprung auftritt, ist das Unternehmen zu kontaktieren, um Anweisungen zur Entnahme von Patientenproben für eine Untersuchung zu erhalten.
Tumorlysesyndrom (TLS)
TLS, das schwerwiegend sein kann, wurde gelegentlich beobachtet. Um das TLS-Risiko zu minimieren, sollten Patienten mit erhöhten Harnsäurewerten oder einer hohen Tumorlast vor der YESCARTA-Infusion Allopurinol oder eine alternative Prophylaxe erhalten. Anzeichen und Symptome eines TLS sollten überwacht und bei einem Auftreten gemäss Standardleitlinien behandelt werden.
Frühere Anti-CD19-Therapie
Es liegen nur begrenzte Erfahrungen zur Anwendung von YESCARTA bei Patienten vor, die zuvor eine gegen CD19 gerichtete Therapie erhalten haben. YESCARTA wird nicht empfohlen, wenn der Patient nach einer früheren Anti-CD19-Therapie ein Rezidiv mit einer CD19-negativen Erkrankung entwickelt hat.
Hilfsstoffe
YESCARTA enthält ca. 300 mg Natrium pro Infusion, entsprechend 15% der von der WHO für einen Erwachsenen empfohlenen maximalen täglichen Natriumaufnahme mit der Nahrung von 2 g.
InteraktionenEs wurden keine pharmakokinetischen oder pharmakodynamischen Studien zur Erfassung von Interaktionen mit YESCARTA durchgeführt.
Die prophylaktische Anwendung systemischer Kortikosteroide kann die Aktivität von YESCARTA beeinflussen und wird daher vor der Infusion nicht empfohlen (siehe «Dosierung/Anwendung»).
Die Gabe von Kortikosteroiden gemäss den Leitlinien zur Behandlung von Toxizitäten hat keinen Einfluss auf die Expansion und Persistenz der CAR-T-Zellen.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Empfängnisverhütung bei Männern und Frauen
Auf der Grundlage der Fachinformation für Fludarabin und Cyclophosphamid sind gebärfähige Frauen anzuweisen, während der Vorbereitung auf die Chemotherapie sowie mindestens 6 Monate lang nach der YESCARTA-Infusion eine wirksame Verhütungsmethode anzuwenden. Informationen zur Notwendigkeit der Anwendung einer wirksamen Verhütungsmethode bei Patientinnen, die eine Chemotherapie zur Lymphodepletion erhalten, können Sie den Fachinformationen zu Fludarabin und Cyclophosphamid entnehmen.
Es liegen unzureichende Expositionsdaten vor, um eine Empfehlung bezüglich der Dauer der Verhütung über 6 Monate hinaus nach einer Behandlung mit YESCARTA auszusprechen.
Männer sind anzuweisen, während der Vorbereitung auf die Chemotherapie sowie mindestens 6 Monate lang nach der YESCARTA-Infusion eine wirksame Verhütungsmethode anzuwenden.
Schwangerschaft
Es wurden keine tierexperimentellen Studien mit YESCARTA durchgeführt, um zu beurteilen, ob es bei Gabe an eine Schwangere schädlich für den Fötus sein kann (siehe «Präklinische Daten»).
Der Schwangerschaftsstatus von gebärfähigen Frauen muss vor Beginn der YESCARTA-Therapie ermittelt werden.
Es ist nicht bekannt, ob YESCARTA potenziell auf den Fötus übergehen kann. Basierend auf dem Wirkungsmechanismus können die transduzierten Zellen, wenn sie plazentagängig sind, zu einer fötalen Toxizität führen, einschliesslich einer B-Zell-Lymphozytopenie. Daher wird YESCARTA bei Schwangeren nicht empfohlen, und eine Schwangerschaft nach der YESCARTA-Infusion ist mit dem behandelnden Arzt bzw. der behandelnden Ärztin zu besprechen.
Stillzeit
Es ist nicht bekannt, ob YESCARTA in die Muttermilch übergeht. Stillende Frauen sollten über das potenzielle Risiko für den gestillten Säugling informiert werden.
Fertilität
Es sind keine klinischen Daten über die Auswirkung von YESCARTA auf die Fertilität verfügbar. Die Auswirkungen auf die männliche und weibliche Fertilität wurden nicht in tierexperimentellen Studien untersucht.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenYESCARTA hat einen mässigen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Nach der YESCARTA-Infusion ist mindestens 8 Wochen lang oder bis zum Abklingen neurologischer unerwünschter Wirkungen vom Führen eines Fahrzeugs oder dem Bedienen schwerer oder potenziell gefährlicher Maschinen abzusehen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die in diesem Abschnitt beschriebenen Sicherheitsdaten stammen von insgesamt 402 erwachsenen Patienten, die im Rahmen von drei multizentrischen, pivotalen klinischen Studien (ZUMA-1, ZUMA-7 und ZUMA-5), mit YESCARTA behandelt wurden, darunter 214 Patienten mit DLBCL, 8 Patienten mit PMBCL, 28 Patienten mit HGBL und 124 Patienten mit FL und zusätzlich weitere 28 Patienten, bei denen die Klassifizierung fehlte, nicht bestätigt wurde oder andere Angaben gemacht wurden, sowie Berichte nach Markteinführung.
Erfahrungen aus den klinischen Studien ZUMA-1, ZUMA-7 und ZUMA-5
Diffuses grosszelliges B-Zell-Lymphom und primäres mediastinales grosszelliges B-Zell-Lymphom
In einer einarmigen Studie wurden Patienten mit rezidiviertem oder refraktärem, aggressivem B-Zell-Non-Hodgkin-Lymphom mit YESCARTA behandelt. Es wurden sieben Patienten in Phase 1 und 101 Patienten in Phase 2 behandelt (N = 108).
Die Sicherheitsdaten aus ZUMA-1 geben die Exposition gegenüber einer Einzeldosis von YESCARTA in einer Phase-1/2-Studie wieder, in der 108 Patienten mit rezidiviertem oder refraktärem DLBCL oder PMBCL nach zwei oder mehr systemischen Therapielinien mit YESCARTA behandelt wurden. Die beschriebenen Daten stammen aus der Analyse der 54-Monats-Nachbeobachtung, bei der die mediane tatsächliche Dauer der Nachbeobachtung 23,5 Monate (Spanne: 0,3 bis 67,8 Monate) betrug.
Die relevantesten und am häufigsten auftretenden unerwünschten Wirkungen sind CRS (93%), Enzephalopathie (60%) und Infektionen (40%). Neutropenie, Thrombozytopenie und Anämie 3. oder höheren Grades, die an Tag 30 oder später noch vorhanden war, traten bei 27%, 23% bzw. 10% der Patienten auf.
Schwerwiegende unerwünschte Wirkungen traten bei 51% der Patienten auf. Die häufigsten (≥5%) schwerwiegenden unerwünschten Wirkungen sind unter anderem Enzephalopathie (22%), Infektionen mit nicht spezifizierten Erregern (15%), bakterielle Infektionen (6%), Virusinfektionen (6%), febrile Neutropenie (5%) und Fieber (5%).
Die häufigsten (≥5%) nicht hämatologischen unerwünschten Wirkungen 3. oder höheren Grades sind unter anderem Enzephalopathie (31%), Infektionen mit nicht spezifizierten Erregern (19%), CRS (11%), bakterielle Infektionen (9%), Virusinfektionen (6%), Delirium (6%), Hypotonie (6%), Transaminasen erhöht (6%) und Hypertonie (6%).
Diffuses grosszelliges B-Zell-Lymphom und High-Grade-B-Zell-Lymphom
Die Sicherheitsdaten aus ZUMA-7 geben die Exposition gegenüber einer Einzeldosis von YESCARTA in einer Phase-3-Studie wieder, in der 170 Patienten mit überwiegend DLBCL oder HGBL, die nach einer Erstlinientherapie rezidiviert oder refraktär sind, mit YESCARTA behandelt wurden. Die im folgenden beschriebenen Daten stammen aus der primären Analyse mit einer Dauer der Nachbeobachtung von 19,6 Monaten (Spanne: 1,5 bis 36,9 Monate).
Die relevantesten und am häufigsten auftretenden unerwünschten Wirkungen waren CRS (92%), Enzephalopathie (49%) und Infektionen (44%). Neutropenie, Thrombozytopenie und Anämie 3. oder höheren Grades, die an Tag 30 oder später noch vorhanden war, traten bei 26%, 6% bzw. 3% der Patienten auf.
Schwerwiegende unerwünschte Wirkungen traten bei 51% der Patienten auf. Die häufigsten (≥5%) schwerwiegenden unerwünschten Wirkungen waren unter anderem CRS (17%), Enzephalopathie (16%), Infektionen mit nicht spezifizierten Erregern (7%) und Fieber (6%).
Die häufigsten (≥5%) nicht hämatologischen unerwünschten Wirkungen 3. oder höheren Grades waren unter anderem Enzephalopathie (19%), Infektionen mit nicht spezifizierten Erregern (8%), CRS (6%) und bakterielle Infektion (5%).
In ZUMA-7 erhielten die Patienten nach Randomisierung entweder eine einzelne YESCARTA-Infusion oder die Standardtherapie (definiert als 2 bis 3 Zyklen Standard-Chemoimmuntherapie [R-ICE, R-DHAP, R-DHAX, R-ESHAP oder R-GDP] gefolgt von hochdosierter Therapie [HDT] und autologer Stammzelltransplantation [ASZT] bei Patienten, deren Erkrankung auf die Behandlung ansprach). Ausgewählte unerwünschte Wirkungen, die im Zusammenhang mit YESCARTA im Vergleich zur Standardtherapie identifiziert wurden, sind in Tabelle 3 angegeben.
Follikuläres Lymphom
Die in diesem Abschnitt beschriebenen Sicherheitsdaten spiegeln die Exposition gegenüber YESCARTA in ZUMA-5 wider, einer Phase-2-Studie, in der 124 Patienten mit rezidiviertem/refraktärem FL CAR-positive T-Zellen in einer empfohlenen, gewichtsbasierten Dosierung erhielten. Die beschriebenen Daten stammen aus der 24-monatigen Nachbeobachtungs-Analyse, in der die mediane tatsächliche Dauer der Nachbeobachtung 26,6 Monate betrug (Spanne: 0,3 bis 44,3 Monate).
Die signifikantesten und am häufigsten auftretenden unerwünschten Wirkungen waren CRS (78%), Infektionen (59%) und Enzephalopathie (47%). Neutropenie, Thrombozytopenie und Anämie 3. oder höheren Grades, die an Tag 30 oder darüber hinaus noch vorlag, trat bei 27%, 10% bzw. 7% der Patienten auf.
Schwerwiegende unerwünschte Wirkungen traten bei 48% der Patienten auf. Die häufigsten schwerwiegenden unerwünschten Wirkungen waren unter anderem Enzephalopathie (19%), Infektionen mit nicht spezifizierten Erregern (14%), CRS (6%), Pyrexie (5%), bakterielle Infektionen (5%), Virusinfektionen (5%), febrile Neutropenie (3%) und Neutropenie (2%).
Die häufigsten unerwünschten Wirkungen 3. oder höheren Grades waren unter anderem Lymphozytopenie (99%), Leukopenie (94%), Neutropenie (92%), Thrombozytopenie (35%), Anämie (35%), Hypophosphatämie (23%) und Hyponatriämie (9%).
Die häufigsten nicht hämatologischen unerwünschten Wirkungen 3. oder höheren Grades waren unter anderem Enzephalopathie (14%), Infektionen mit nicht spezifizierten Erregern (12%), CRS (6%), Hypertonie (4%), Delirium (4%), Virusinfektionen (4%) und Thrombose (4%).
Zusammenfassung der unerwünschten Wirkungen
Die in diesem Abschnitt beschriebenen unerwünschten Wirkungen wurden bei mit YESCARTA behandelten Patienten mit DLBCL oder PMBCL in ZUMA-1 (n = 108), DLBCL oder HGBL in ZUMA-7 (n = 170) und mit FL in ZUMA-5 (n = 124) und anhand von Berichten nach der Markteinführung festgestellt. Diese unerwünschten Wirkungen werden nach Systemorganklasse und Häufigkeit aufgeführt. Die Häufigkeiten sind wie folgt definiert: sehr häufig (≥1/10); häufig (≥1/100, < 1/10); gelegentlich (≥1/1000, < 1/100); selten (≥1/10'000, <1/1000); sehr selten (<1/10'000). Innerhalb der einzelnen Häufigkeitsgruppen werden die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Infektionen und parasitäre Erkrankungen:
Sehr häufig: Infektionen mit nicht spezifizierten Erregern (32%), Virusinfektionen (19%), bakterielle Infektionen (12%).
Häufig: Pilzinfektionen.
Erkrankungen des Blutes und des Lymphsystems:
Sehr häufig: Febrile Neutropenie (10%), Neutropenie (93%)*, Lymphopenie (99%)*, Leukopenie (95%)*, Anämie (45%)*, Thrombozytopenie (37%)*.
Häufig: Koagulopathiea.
Erkrankungen des Immunsystems:
Sehr häufig: CRS (88%), Immunglobuline erniedrigt (15%) b.
Häufig: Infusionsbedingte Reaktionen.
Gelegentlich: Überempfindlichkeit, Hämophagozytische Lymphohistiozytose.
Neubildungen:
Selten: T-Zell-Malignome.
Stoffwechsel- und Ernährungsstörungen:
Sehr häufig: Hyponatriämie (14%)*, Hypophosphatämie (23%)*, Hyperurikämie (14%)*, #, Appetit vermindert (27%) c.
Häufig: Hyperglykämie*, Hypokalämie*, Hypokalzämie*, Hypoalbuminämie*, Dehydratationd, Gewichtsverminderung.
Psychiatrische Erkrankungen:
Sehr häufig: Delirium (13%)e, Schlaflosigkeit (13%).
Häufig: Angst, Affekterkrankungf.
Erkrankungen des Nervensystems:
Sehr häufig: Enzephalopathie (51%)g, Tremor (28%)h, Kopfschmerzen (33%)i, Schwindelgefühl (22%)j.
Häufig: Ataxiek, Krampfanfall, Hemiparese, periphere Neuropathiel, Myoklonus.
Gelegentlich: Status Epilepticus, Quadriplegie, Rückenmarksödem, Myelitis, Gesichtslähmungm, Dyskalkulie.
Herzerkrankungen:
Sehr häufig: Tachykardie (21%)n, Arrhythmie (19%)o.
Häufig: Herzinsuffizienzp.
Gelegentlich: Herzstillstand.
Gefässerkrankungen:
Sehr häufig: Hypotonie (21%)q, Hypertonie (10%).
Häufig: Thromboser.
Gelegentlich: Capillary leak syndrome.
Erkrankungen der Atemwege, des Brustraums und Mediastinums:
Sehr häufig: Husten (26%)s.
Häufig: Hypoxiet, Pleuraerguss, Lungenödem, Dyspnoeu, Rhinorrhoe (einschliesslich allergischer Rhinitis).
Gelegentlich: Lungenversagenv.
Erkrankungen des Gastrointestinaltrakts:
Sehr häufig: Erbrechen (18%), Durchfall (34%)w, Verstopfung (23%), Abdominalschmerz (18%)x, Übelkeit (33%).
Häufig: Dysphagie, Mundtrockenheity.
Leber- und Gallenerkrankungen:
Sehr häufig: Transaminasen erhöht (18%)z.
Häufig: Hyperbilirubinämieaa.
Erkrankungen der Haut und des Unterhautgewebes:
Sehr häufig: Ausschlag (einschliesslich Ausschlag, Ausschlag an der Anwendungsstelle, Dermatitis, allergische Dermatitis, Dermatitis bullös, Erythem, Pruritus, erythematöser Hautausschlag, makulöser Ausschlag, Ausschlag makulo-papulös, Ausschlag Juckreiz, Ausschlag pustulös, Stevens-Johnson-Syndrom, Urtikaria) (15%).
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen:
Sehr häufig: Motorische Funktionsstörung (16%)bb, Schmerzen des Muskel- und Skelettsystems (38%)cc.
Gelegentlich: Rhabdomyolyse.
Erkrankungen der Nieren und Harnwege:
Häufig: Nierenfunktionsbeeinträchtigungdd.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort:
Sehr häufig: Fieber (23%)ee, Ödem (16%)ff, Müdigkeit (44%)gg, Schüttelfrost (12%).
Häufig: Schmerzen.
Gelegentlich: Multiorgandysfunktionssyndrom.
Augenerkrankungen
Häufig: Sehverschlechterunghh.
Fussnoten:
* Häufigkeit basierend auf Laborparametern des Grades 3 oder höher
# Hyperurikämie wurde anhand einer gepoolten Analyse von 232 erwachsenen Patienten in ZUMA-1 und ZUMA-5 festgestellt
a. Koagulopathie umfasst Koagulopathie, erniedrigtes Fibrinogen im Blut, erhöhtes Fibrinogen im Blut, disseminierte intravaskuläre Gerinnung, Hypofibrinogenämie, erhöhter INR-Wert (international normalised ratio), erniedrigte Prothrombinwerte, verlängerte Prothrombinzeit
b. Erniedrigtes Immunglobulin G im Blut, erniedrigte Immunglobuline umfassen Hypogammaglobulinämie
c. Verminderter Appetit umfasst verminderter Appetit, Hypophagie
d. Dehydration umfasst Dehydration, Hypovolämie
e. Delirium umfasst Delirium, Agitation, Wahn, Desorientierung, Halluzination, Unruhe
f. Affekterkrankung umfasst impulsives Verhalten, Manie, Stimmungsänderung, Panikattacke
g. Enzephalopathie umfasst Enzephalopathie, Agraphie, veränderter Bewusstseinszustand, Amnesie, Aphasie, Aphonie, Apraxie, kognitive Störung, Verwirrtheitszustand, getrübter Bewusstseinszustand, Aufmerksamkeitsstörungen, Dysarthrie, Dysgraphie, Dyskinesie, Dyspraxie, Hypersomnie, Immuneffektorzell-assoziiertes Neurotoxizitätssyndrom, Lethargie, Leukenzephalopathie, Verlust des Bewusstseins, eingeschränktes Erinnerungsvermögen, psychische Beeinträchtigung, veränderter Gemütszustand, metabolische Enzephalopathie, Neurotoxizität, langsames Sprechen, Somnolenz, Sprachstörung, Stupor, toxische Enzephalopathie
h. Tremor umfasst Tremor, Titubation des Kopfes
i. Kopfschmerzen umfassen Kopfschmerzen, Kopfbeschwerden, Spannungskopfschmerzen
j. Schwindelgefühl umfasst Schwindelgefühl, Haltungsschwindel, Präsynkope, Synkope, Vertigo
k. Ataxie umfasst Ataxie, Gleichgewichtsstörung, Gangstörung
l. Periphere Neuropathie umfasst periphere Neuropathie, Allodynie, zervikale Radikulopathie, Hyperästhesie, Hypoästhesie, lumbale Radikulopathie, Parästhesie, Parosmie, periphere motorische Neuropathie, periphere sensorische Neuropathie, Lähmung des Peroneusnervs
m. Gesichtslähmung umfasst Gesichtslähmung
n. Tachykardie umfasst Tachykardie, posturales orthostatisches Tachykardie-Syndrom, Sinustachykardie
o. Arrhythmie umfasst Arrhythmie, Vorhofflimmern, Vorhofflattern, atrioventrikulärer Block, atrioventrikulärer Block ersten Grades, Bradykardie, Rechtsschenkelblock, verlängertes QT-Intervall im Elektrokardiogramm, Umkehrung der T-Welle im Elektrokardiogramm, Extrasystolen, erhöhte Herzfrequenz, unregelmässige Herzfrequenz, Sinusbradykardie, supraventrikuläre Extrasystolen, supraventrikuläre Tachykardie, ventrikuläre Arrhythmie, ventrikuläre Extrasystolen, ventrikuläre Tachykardie
p. Herzinsuffizienz umfasst Herzinsuffizienz, akute Linksherzinsuffizienz, reduzierte Ejektionsfraktion, Stress-Kardiomyopathie
q. Hypotonie umfasst Hypotonie, diastolische Hypotonie, Hypoperfusion, orthostatische Hypotonie
r. Thrombose umfasst Thrombose, Thrombose der Axillarvenen, Thrombose der Vena brachiocephalica, tiefe Venenthrombose, Verstopfung eines Medizinprodukts, Embolie, Jugularvenenthrombose, periphere Embolie, periphere Ischämie, Lungenembolie, Thrombose der Milzvene, Thrombose der Vena subclavia, Thrombose im Medizinprodukt, Gefässverschluss
s. Husten umfasst Husten, Husten mit Auswurf, Hustensyndrom der oberen Atemwege
t. Hypoxie umfasst Hypoxie, erniedrigte Sauerstoffsättigung
u. Dyspnoe umfasst Dyspnoe, Belastungsdyspnoe
v. Respiratorische Insuffizienz umfasst respiratorische Insuffizienz, akute respiratorische Insuffizienz
w. Diarrhoe umfasst Diarrhoe, Kolitis, Enteritis
x. Abdominalschmerzen umfassen Abdominalschmerzen, abdominale Beschwerden, Schmerzen im Unterbauch, Schmerzen im Oberbauch, abdominelle (Druck-)Empfindlichkeit, Dyspepsie, epigastrische Beschwerden
y. Mundtrockenheit umfasst Mundtrockenheit, Lippentrockenheit
z. Erhöhte Transaminasen umfassen erhöhte Transaminasen, erhöhte Alanin-Aminotransferase, erhöhte Aspartat-Aminotransferase, erhöhte Leberenzyme
aa. Hyperbilirubinämie umfasst Hyperbilirubinämie, erhöhtes Bilirubin im Blut
bb. Motorische Funktionsstörung umfasst motorische Funktionsstörung, unwillkürliche Muskelkontraktionen, Muskelrigidität, Muskelspasmen, Muskelspastik, Muskelzerrung, Muskelverspannungen, Muskelzuckungen, Muskelschwäche
cc. Schmerzen des Muskel- und Skelettsystems umfassen Schmerzen des Muskel- und Skelettsystems, Arthralgie, Arthritis, Rückenschmerzen, Knochenschmerzen, Flankenschmerzen, Leistenschmerzen, Brustschmerzen die Skelettmuskulatur betreffend, Myalgie, Nackenschmerzen, Osteoarthritis, Schmerzen in den Extremitäten
dd. Nierenfunktionsstörung umfasst akute Nierenschädigung, erhöhtes Kreatinin im Blut, Nierenversagen
ee. Fieber umfasst Hyperthermie, Pyrexie
ff. Ödem umfasst Ödem, Gesichtsödem, generalisiertes Ödem, lokalisiertes Ödem, Genitalödem, peripheres Ödem, periphere Schwellung, Schwellung
gg. Müdigkeit umfasst Müdigkeit, Asthenie, verminderter Aktivitätsgrad, Unwohlsein
hh. Sehstörung umfasst Sehstörung, Hepieanopie, verschwommenes Sehen, verminderte Sehschärfe
Tabelle 3: Unerwünschte Arzneimittelwirkungen in ZUMA-7 mit einem Häufigkeitsunterschied zwischen den Behandlungsarmen von ≥10%
|
Systemorganklasse (SOC)
Unerwünschte Wirkung
|
YESCARTA
(n = 170)
|
Standardtherapie
(n = 168)
| |
Infektionen und parasitäre Erkrankungen
| |
Virusinfektionen
|
15%
|
5%
| |
Erkrankungen des Blutes und des Lymphsystems
| |
Leukopenie ≥3. Grades#
|
95%
|
56%
| |
Lymphopenie ≥3. Grades#
|
99%
|
68%
| |
Neutropenie ≥3. Grades#
|
94%
|
51%
| |
Thrombozytopenie ≥3. Grades#
|
26%
|
63%
| |
Erkrankungen des Immunsystems
| |
Zytokin-Freisetzungssyndrom
|
92%
|
0%
| |
Immunglobuline erniedrigta
|
11%
|
1%
| |
Stoffwechsel- und Ernährungsstörungen
| |
Hyponatriämie ≥3. Grades
|
12%
|
2%
| |
Erkrankungen des Nervensystems
| |
Enzephalopathieb
|
49%
|
8%
| |
Tremorc
|
25%
|
1%
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
| |
Hustend
|
27%
|
11%
| |
Erkrankungen des Gastrointestinaltrakts
| |
Übelkeit
|
34%
|
69%
| |
Erbrechen
|
15%
|
33%
| |
Obstipation
|
20%
|
35%
| |
Leber- und Gallenerkrankungen
| |
Transaminasen erhöhte
|
21%
|
11%
| |
Erkrankungen der Nieren und Harnwege
| |
Nierenfunktionsbeeinträchtigungf
|
9%
|
19%
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Ermüdungg
|
44%
|
57%
| |
Fieberh
|
16%
|
26%
|
In Tabelle 3 sind nur Zytopenien enthalten, die (i) zu neuen oder sich verschlimmernden klinischen Folgeschäden führten oder (ii) eine Therapie erforderten oder (iii) zu einer Anpassung der laufenden Therapie führten.
# Häufigkeit basierend auf Laborparametern
a. Immunglobuline erniedrigt umfasst Hypogammaglobulinämie, Immunglobulin G im Blut erniedrigt.
b. Enzephalopathie umfasst Enzephalopathie, veränderter Bewusstseinszustand, Amnesie, Aphasie, Apraxie, kognitive Störung, Verwirrtheitszustand, getrübter Bewusstseinszustand, Aufmerksamkeitsstörungen, Dysarthrie, Dysgraphie, Dyskinesie, Dyspraxie, Hypersomnie, Lethargie, Leukenzephalopathie, Verlust des Bewusstseins, Erinnerungsvermögen eingeschränkt, geistige Beeinträchtigungen, Gemütszustand verändert, metabolische Enzephalopathie, langsame Sprache, Somnolenz, Stupor, toxische Enzephalopathie.
c. Tremor umfasst Tremor, Wackeltremor des Kopfes.
d. Husten umfasst Husten, Husten mit Auswurf, Hustensyndrom der oberen Atemwege.
e. Transaminasen erhöht umfasst Transaminasen erhöht, Leberenzym erhöht, Alaninaminotransferase erhöht, Aspartataminotransferase erhöht.
f. Nierenfunktionsbeeinträchtigung umfasst Kreatinin im Blut erhöht, akute Nierenschädigung.
g. Ermüdung umfasst Ermüdung, Asthenie, Unwohlsein.
h. Fieber umfasst Pyrexie
Beschreibung ausgewählter Nebenwirkungen aus ZUMA-1, ZUMA-7 und ZUMA-5
Zytokin-Freisetzungssyndrom:
CRS trat bei 92% der Patienten in ZUMA-1 und ZUMA-7 auf. Bei acht Prozent (8%) der Patienten trat ein CRS 3. oder höheren Grades (schwer, lebensbedrohlich oder tödlich) auf. Die mediane Dauer bis zum Einsetzen betrug 3 Tage (Spanne: 1 bis 12 Tage) und die mediane Dauer betrug 7 Tage (mit einer Spanne von 2 bis 29 Tagen mit Ausnahme eines beobachteten Ausreissers von 58 Tagen). 99% der Patienten erholten sich vom CRS.
CRS trat bei 78% der Patienten in ZUMA-5 auf. Bei sechs Prozent (6%) der Patienten trat ein CRS 3. oder höheren Grades (schwer, lebensbedrohlich oder tödlich) auf. Die mediane Dauer bis zum Einsetzen betrug 4 Tage (Spanne: 1 bis 15 Tage) und die mediane Dauer betrug 6 Tage (mit einer Spanne von 1 bis 27 Tagen. Neunundneunzig Prozent (99%) der Patienten erholten sich vom CRS. In ZUMA-5 trat ein CRS-bedingter Todesfall auf.
Die häufigsten (≥20%) unerwünschten Wirkungen, die im Zusammenhang mit CRS auftreten können sind Fieber (89%), Hypotonie (50%), Müdigkeit (50%), Tachykardie (47%), Kopfschmerzen (43%), Diarrhoe (37%), Übelkeit (37%), Schüttelfrost (30%), Hypoxie (25%) und Erbrechen (23%). Schwerwiegende unerwünschte Wirkungen, die im Zusammenhang mit CRS auftreten können, schliessen Fieber (12%), Hypotonie (5%), Hypoxie (2%), Arrhythmie (3%), Herzinsuffizienz (2%), Müdigkeit (2%), Kopfschmerzen (2%), Tachykardie (2%), Herzstillstand (1%) und Dyspnoe (1%). Hinsichtlich eines Leitfadens zur Überwachung und Behandlung siehe «Warnhinweise und Vorsichtsmassnahmen».
Neurologische unerwünschte Wirkungen:
Neurologische unerwünschte Wirkungen traten bei 62% der Patienten in ZUMA-1 und ZUMA-7 auf. Bei fünfundzwanzig (25%) der Patienten traten unerwünschte Wirkungen 3. oder höheren Grades (schwer oder lebensbedrohlich) auf. Neurologische unerwünschte Wirkungen traten bei 75% der Patienten innerhalb der ersten 7 Tage nach der Infusion auf. Die mediane Dauer bis zum Einsetzen betrug 6 Tage (Spanne 1 bis 133 Tage). Die mediane Dauer betrug 10 Tage mit einer Spanne von 1 bis 817 Tagen, wobei die Symptome bei 66% der Patienten innerhalb von 3 Wochen nach der Infusion abklangen. Die Mehrheit der Patienten erholte sich von den neurologischen unerwünschten Wirkungen, mit Ausnahme von 10 Patienten (3,6 % der mit YESCARTA in ZUMA-1 und ZUMA-7 behandelten Patienten). Die neurologischen unerwünschten Wirkungen waren zum Zeitpunkt des Todes noch nicht abgeklungen, aber die Patienten starben aus Gründen, die nicht mit diesen zusammenhingen.
Neurologische unerwünschte Wirkungen traten bei 56% der Patienten in ZUMA-5 auf. Bei fünfzehn (15%) der Patienten traten unerwünschte Wirkungen 3. oder höheren Grades (schwer oder lebensbedrohlich) auf. Neurologische unerwünschte Wirkungen traten bei 66% der Patienten innerhalb der ersten 7 Tage nach der Infusion auf. Die mediane Dauer bis zum Einsetzen betrug 7 Tage (Spanne: 1 bis 177 Tage). Die mediane Dauer betrug 14 Tage, wobei die Symptome bei 60% der Patienten innerhalb von 3 Wochen nach der Infusion abklangen.
Die häufigsten (≥5%) unerwünschten Wirkungen im Zusammenhang mit neurologischen unerwünschten Wirkungen beinhalten Enzephalopathie (51%), Kopfschmerzen (33%), Tremor (28%), Schwindelgefühl (22%), Delirium (13%), Schlaflosigkeit (13%), periphere Neuropathie (9%), Angst (7%) und Ataxie (6%). Bei Patienten, denen YESCARTA verabreicht wurde, wurden schwerwiegende unerwünschte Wirkungen (≥1%), einschliesslich Enzephalopathie (18%), Delirium (2%) und Tremor (1%), berichtet.
Andere neurologische unerwünschte Wirkungen wurden in klinischen Studien seltener berichtet und umfassten Dysphagie (3%), Myelitis (0,2%) und Quadriplegie (0,1%).
Bei einem Patienten in ZUMA-1 trat ein tödliches Ereignis durch eine intrakranielle Blutung auf, welches sich zusammen mit CRS, Neurotoxizität, schwerer Thrombozytopenie, Verabreichung von Heparin zur Deep Vein Thrombosis (DVT)-Prophylaxe und anhaltender bakterieller Sepsis ereignete. Als Todesursache wurde Sepsis angegeben, die der konditionierenden Chemotherapie zugeschrieben wurde.
Zu den nach Markteinführung gemeldeten unerwünschten Wirkungen gehören Status Epilepticus (0,3%), Rückenmarksödem, infusionsbedingte Reaktionen, Blutung (gastrointestinale Blutung, Hirnblutung, Lungenblutung, intrakranielle Blutung, hämorrhagischer Schock, Blutung, hämorrhagische Zystitis, Subarachnoidalblutung), T-Zell-Malignome und ICANS, die im Zusammenhang mit neurologischer Toxizität berichtet wurden.
Hinsichtlich eines Leitfadens zur Überwachung und Behandlung siehe «Warnhinweise und Vorsichtsmassnahmen».
Infektionen und febrile Neutropenie:
Febrile Neutropenie wurde bei 10% der Patienten nach Infusion von YESCARTA beobachtet. Infektionen traten bei 48% der Patienten auf. Infektionen 3. oder höheren Grades (schwer, lebensbedrohlich oder tödlich) traten bei 19% der Patienten auf. Infektionen mit nicht spezifizierten Erregern, bakterielle Infektionen und Virusinfektionen 3. oder höheren Grades traten bei 12%, 6% bzw. 4% der Patienten auf. Der häufigste Ort für Infektionen mit unspezifischen Erregern waren die Atemwege. Hinsichtlich eines Leitfadens zur Überwachung und Behandlung siehe «Warnhinweise und Vorsichtsmassnahmen».
Länger anhaltende Zytopenien:
Neutropenie, Anämie und Thrombozytopenie 3. oder höheren Grades traten bei 68%, 32% bzw. 24% der Patienten auf. Neutropenie (einschliesslich Neutropenie, Neutrophilenzahl erniedrigt und febrile Neutropenie), Thrombozytopenie und Anämie 3. oder höheren Grades, die bis Tag 30 oder länger andauerten, traten bei 27%, 12% bzw. 6% der Patienten auf. Hinsichtlich eines Leitfadens zur Behandlung siehe «Warnhinweise und Vorsichtsmassnahmen».
Hypogammaglobulinämie:
Hypogammaglobulinämie wurde bei 15% der mit YESCARTA behandelten Patienten berichtet. Kumulativ erhielten 36 (33%) von 108 Patienten in ZUMA-1 bis zum Zeitpunkt der 24-Monats-Analyse eine intravenöse Immunglobulintherapie, und 28 (16%) von 170 Patienten in ZUMA-7, die mit YESCARTA behandelt wurden, erhielten bis zum Zeitpunkt der primären Analyse eine intravenöse Immunglobulintherapie. In ZUMA-5 erhielten 35 (28%) von 124 Patienten bis zur 24-monatigen Nachbeobachtungs-Analyse eine intravenöse Immunglobulintherapie. Hinsichtlich eines Leitfadens zur Behandlung siehe «Warnhinweise und Vorsichtsmassnahmen».
Immunogenität
Die Immunogenität von YESCARTA wurde mittels eines enzymgekoppelten Immunadsorptionstests (Enzyme-Linked Immunosorbent Assay, ELISA) zum Nachweis von bindenden Antikörpern gegen FMC63, dem Antikörper, der ursprünglich Anti-CD19-CAR zugrunde liegt, untersucht, gefolgt von einem zellbasierten Bestätigungs-Assay. Patienten wurden bei Baseline, vor der Infusion und bis zu einem Jahr nach der Infusion überwacht. Keiner der mit YESCARTA behandelten Patienten entwickelte nach der Infusion de-novo Antikörper.
Elf von 278 Patienten (4%) wurden vor der Behandlung mit YESCARTA in ZUMA-1 und ZUMA-7 positiv auf Anti-FMC63-Antikörper getestet, und ein Patient (1%) in ZUMA-7, der vor der Behandlung ein negatives Testergebnis hatte, hatte im Screening-ELISA nach der Behandlung ein positives Testergebnis. Die Ergebnisse eines zellbasierten Bestätigungs-Assays, bei dem ein korrekt gefalteter und exprimierter extrazellulärer Teil des CAR (scFv, Gelenkregion und Linker) genutzt wurde, zeigten, dass alle mit YESCARTA behandelten Patienten, die im Screening-ELISA ein positives Ergebnis hatten, zu allen getesteten Zeitpunkten Antikörper-negativ waren. Es gibt keine Hinweise darauf, dass die Kinetik der anfänglichen Expansion und Persistenz von CAR-T Zellen oder die Sicherheit oder Wirksamkeit von YESCARTA bei solchen Patienten beeinflusst wurden.
In ZUMA-5 wurden 14 von 124 FL-Patienten vor der Behandlung mit YESCARTA im Screening-ELISA-Assay positiv auf Antikörper getestet, und 3 Patienten, die vor der Behandlung negative ELISA-Ergebnisse hatten, wiesen nach der Behandlung positive Testergebnisse auf. Die Ergebnisse des zellbasierten Bestätigungstests zeigten, dass alle mit YESCARTA behandelten ZUMA-5-Patienten, die ein positives ELISA-Ergebnis aufwiesen, sowohl vor als auch nach der Behandlung antikörpernegativ waren. Auswirkungen dieser Antikörper auf die klinische Wirksamkeit oder Sicherheit waren nicht zu erkennen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs liegen keine Daten bezüglich der Anzeichen einer Überdosierung mit YESCARTA vor.
Eigenschaften/WirkungenATC-Code
L01XL03
Wirkungsmechanismus
YESCARTA, ein Arzneimittel zur Immuntherapie mit genetisch modifizierten autologen T-Zell-Immuntherapie, bindet an CD19 exprimierende Krebszellen und normale B-Zellen. Nach der Bindung der Anti-CD19-CAR-T-Zellen an die CD19 exprimierenden Zielzellen aktivieren die kostimulierenden Domänen CD28 und CD3-zeta nachgeschaltete Signalkaskaden, die bei den T-Zellen zu Aktivierung, Proliferation, Akquisition von Effektorfunktionen und Sekretion von inflammatorischen Zytokinen und Chemokinen führen. Diese Abfolge von Ereignissen führt zur Apoptose und Nekrose der CD19 exprimierenden Zielzellen.
Pharmakodynamik
Das pharmakodynamische Ansprechen wurde nach der YESCARTA-Infusion untersucht, indem der vorübergehende Anstieg der Zytokine, Chemokine und anderer Moleküle im Blut über einen 4-wöchigen Zeitraum gemessen wurde. Die Konzentrationen der Zytokine und Chemokine wie Interleukin (IL) IL-6, IL-8, IL-10, IL-15, TNF-α, IFN-γ und sIL2Rα wurden analysiert. Der maximale Anstieg wurde in den ersten 14 Tagen nach der Infusion beobachtet, und die Konzentrationen erreichten in der Regel innerhalb von 28 Tagen wieder den Ausgangswert.
Aufgrund der on-target, off-tumour Wirkung von YESCARTA ist nach der Behandlung für einen gewissen Zeitraum eine B-Zell-Aplasie zu erwarten. Unter 73 DLBCL- und PMBCL-Patienten in ZUMA-1 mit auswertbaren Proben zu Studienbeginn hatten 40% nachweisbare B-Zellen; die bei einer Mehrheit der Patienten zu Studienbeginn beobachtete B-Zell-Aplasie wurde auf frühere Therapien zurückgeführt. Nach der YESCARTA-Behandlung sank der Anteil der Patienten mit nachweisbaren B-Zellen: 20% hatten nachweisbare B-Zellen im Monat 3 und 22% nachweisbare B-Zellen im Monat 6.
Die Initiierung der B-Zell-Wiederherstellung wurde erstmals im Monat 9 festgestellt, als 56% der Patienten nachweisbare B-Zellen hatten. Dieser Trend der B-Zell-Wiederherstellung setzte sich im Laufe der Zeit fort, da 64% der Patienten im Monat 18 nachweisbare B-Zellen hatten und 77% der Patienten im Monat 24 nachweisbare B-Zellen hatten.
Unter den 141 Patienten in ZUMA-7 mit auswertbaren Proben zu Studienbeginn hatten 57% nachweisbare B-Zellen. Nach der YESCARTA-Behandlung sank der Anteil der Patienten mit nachweisbaren B-Zellen: 38% hatten nachweisbare B-Zellen im Monat 3 und 41% nachweisbare B-Zellen im Monat 6. Die Initiierung der B-Zell-Wiederherstellung war im Monat 9 zu erkennen, als 58% der Patienten nachweisbare B-Zellen hatten. Dieser Trend der B-Zell-Wiederherstellung setzte sich im Laufe der Zeit fort, da 64% der Patienten im Monat 18 nachweisbare B-Zellen hatten und 85% der Patienten im Monat 24 nachweisbare B-Zellen hatten.
Unter 113 FL-Patienten in ZUMA-5 mit auswertbaren Proben zu Studienbeginn hatten 75% nachweisbare B-Zellen. Nach der YESCARTA-Behandlung sank der Anteil der Patienten mit nachweisbaren B-Zellen: 40% der Patienten hatten nachweisbare B-Zellen im Monat 3. Im Laufe der Zeit wurde eine B-Zell-Wiederherstellung beobachtet, wobei 61% der Patienten im Monat 24 nachweisbare B-Zellen hatten.
Es ist zu beachten, dass Patienten nach dem Fortschreiten der Krankheit nicht weiterverfolgt werden mussten; daher waren die Mehrzahl der Patienten mit auswertbaren Proben Responder.
Klinische Wirksamkeit
Klinische Studie ZUMA-1, Phase 1 und 2 (rezidiviertes oder refraktäres DLBCL, PMBCL und DLBCL, welches aus einem follikulären Lymphom entstanden ist, nach zwei oder mehr systemischen Therapielinien)
Insgesamt wurden 108 Patienten (7 Patienten in Phase 1 und 101 Patienten in Phase 2) mit aggressivem r/r B-Zell-Non-Hodgkin-Lymphom (NHL) in einer offenen, multizentrischen, einarmigen Phase-1/2-Studie mit YESCARTA behandelt.
Basierend auf der WHO-Klassifizierung 2008, welche zum Zeitpunkt der Studie gültig war, wurde die Wirksamkeit an 101 Patienten in Phase 2 mit histologisch bestätigtem DLBCL (N = 77), PMBCL (N = 8), oder DLBCL, welches aus einem follikulären Lymphom (N = 16) entstanden ist, untersucht. Basierend auf der aktuellen WHO-Klassifizierung 2016:
Patienten mit DLBCL in der ZUMA-1 Studie schlossen Patienten mit nicht anderweitig spezifiziertem DLBCL, Patienten mit anderen DLBCL-Subtypen sowie Patienten mit HGBL ein. Dies beruht auf einer retrospektiven, post-hoc Analyse eines Teils der Patienten per «independent pathology review». 47 Patienten konnten im Hinblick auf den MYC-, BCL-2- und BCL-6-Status evaluiert werden. Bei 30 Patienten wurde ein DLBCL mit Doppelexpression (Überexpression von sowohl MYC- als auch BCL-2-Protein) festgestellt; 5 Patienten hatten ein HGBL mit MYC-, BCL-2- oder BCL-6-Gen-Re-Arrangements (Mutation von 2 Genen (double hit) oder 3 Genen (triple hit)); bei 2 Patienten wurde ein nicht anderweitig spezifiziertes HGBL festgestellt. Aufgrund der kleinen Anzahl von HGBL Patienten kann keine Schlussfolgerung zur klinischen Wirksamkeit in dieser Patientenpopulation gezogen werden. 66 Patienten waren im Hinblick auf ihre B-Zell-Subpopulation (germinaler-B-Zell-Typ [GCB] oder aktivierter B-Zell-Typ [ABC]) evaluierbar. 49 dieser Patienten wiesen den GCB-Typ und 17 den ABC-Typ auf.
Geeignete Patienten waren mindestens 18 Jahre alt und wiesen eine refraktäre Erkrankung auf; diese war definiert als progrediente Erkrankung (progressive disease, PD) oder stabile Erkrankung (stable disease, SD) als bestes Ansprechen auf die zuletzt angewendete Therapielinie, oder aber Progression innerhalb von 12 Monaten nach autologer Stammzelltransplantation (ASZT). Patienten, die gegenüber einer Chemotherapie refraktär sind oder nach zwei oder mehr systemischen Therapielinien ein Rezidiv aufweisen, sind im Allgemeinen nicht für eine hämatopoetische Stammzelltransplantation (HSZT) geeignet.
Patienten mussten zuvor mindestens mit einem Anti-CD20-Antikörper sowie einem Anthracyclin enthaltenden Regime behandelt worden sein. Patienten mit einem ZNS-Lymphom, einer vorangegangenen allogenen Stammzelltransplantation oder einer vorherigen Therapie mit Anti-CD19-CAR oder anderen genetisch modifizierten T-Zellen waren ausgeschlossen. Patienten mit ZNS-Erkrankungen (wie z.B. Krampfanfällen oder zerebrovaskulärer Ischämie) in der Anamnese, einer Ejektionsfraktion des Herzens von weniger als 50%, Sauerstoffsättigung von weniger als 92% bei Raumluft oder einer Autoimmunerkrankung, die eine systemische Immunsuppression erfordert, waren ausgeschlossen. Die mediane Dauer der Nachbeobachtung war 27,1 Monate (noch nicht abgeschlossen). ITT war definiert als Anzahl aller Patienten, die einer Leukapharese unterzogen wurden, mITT war definiert als Anzahl aller Patienten, die YESCARTA erhielten.
Demographische Daten der modifizierten intent-to-treat (mITT) Population
Das mediane Alter der mITT-Studienpopulation betrug 58 Jahre (Spanne: 23 bis 76 Jahre); 67% waren Männer, 86% waren weiss, 3% waren asiatischer Abstammung und 4% waren farbig. Der ECOG-Leistungsstatus zum Studienbeginn entsprach bei 42% ECOG 0 und bei 58% ECOG 1. Die mediane Anzahl vorheriger Therapien betrug 3 (Spanne: 1 bis 10); bei 76% der Patienten war die Erkrankung gegenüber einer zweiten oder weiteren Therapielinie refraktär, und 21% waren innerhalb 1 Jahres nach der autologen HSZT rezidiviert. 46% der Patienten wiesen einen Internationalen Prognostischen Index von 3/4 auf, und 85% der Patienten befanden sich im Krankheitsstadium III/IV.
Demographische Daten der intent-to-treat (ITT) Population
Das mediane Alter der ITT-Studienpopulation betrug 58 Jahre (Spanne: 23 bis 76 Jahre), 69% waren Männer, 85% waren weiss, 4% waren asiatischer Abstammung und 4% waren farbig. Der ECOG-Leistungsstatus zum Studienbeginn entsprach bei 41% ECOG 0 und bei 59% ECOG 1. Die mediane Anzahl vorheriger Therapien betrug 3 (Spanne: 1 bis 10); bei 77% der Patienten war die Erkrankung gegenüber einer zweiten oder weiteren Therapielinie refraktär, und 20% waren innerhalb 1 Jahres nach der autologen HSZT rezidiviert. 46% der Patienten wiesen einen Internationalen Prognostischen Index von 3/4 auf, und 85% der Patienten befanden sich im Krankheitsstadium III/IV.
YESCARTA wurde als Einzel-Infusion mit einer Zieldosis von 2 x 106 Anti-CD19-CAR-T-Zellen/kg im Anschluss an ein Chemotherapie-Schema zur Lymphodepletion von 500 mg/m2 intravenösem Cyclophosphamid und 30 mg/m2 intravenösem Fludarabin verabreicht; die Lymphodepletion erfolgte am 5., 4. und 3. Tag vor der Behandlung mit YESCARTA. Alle 108 Patienten, welche mit YESCARTA in ZUMA-1 (Phasen 1 und 2) behandelt wurden, erhielten eine Chemotherapie zur Lymphodepletion. Alle Patienten hatten einen Ausgangswert an weissen Blutkörperchen von ≥1 x 103/μl (d.h. vor der Chemotherapie zur Lymphodepletion). Nur Patienten mit einer absoluten Neutrophilenzahl von ≥1000/μl, einer absoluten Lymphozytenzahl von ≥100/µl und einem Thrombozytenwert von ≥75'000/μl zu Studienbeginn wurden in die Studie einbezogen. Eine Bridging-Chemotherapie zwischen der Leukapherese und der Chemotherapie zur Lymphodepletion war nicht zulässig. Alle Patienten wurden zur Beobachtung und Behandlung von unerwünschten Wirkungen nach der YESCARTA-Infusion für mindestens 7 Tage hospitalisiert.
ZUMA-1 Phase 2
Von 111 Patienten, die einer Leukapherese unterzogen wurden, erhielten 101 YESCARTA. Neun Patienten wurden nicht behandelt, und zwar hauptsächlich aufgrund einer progredienten Erkrankung oder wegen schwerwiegender unerwünschter Wirkungen nach der Aufnahme in die Studie und vor der Lieferung der Zellen. Einer von 111 Patienten erhielt das Arzneimittel wegen eines Herstellungsfehlers nicht. Die mediane Dauer von der Leukapherese bis zur Lieferung des Arzneimittels betrug 17 Tage (Spanne: 14 bis 51 Tage), und die mediane Dauer von der Leukapherese bis zur Infusion betrug 24 Tage (Spanne: 16 bis 73 Tage). Die mediane Dosis betrug 2,0 x 106 Anti-CD19-CAR-T-Zellen/kg.
Der primäre Endpunkt war die objektive Ansprechrate (Objective Response Rate, ORR), bestimmt durch die Prüfärzte. Die sekundären Endpunkte beinhalteten objektive Ansprechrate (ORR), bestimmt durch eine unabhängige Prüfungskommission, Dauer des Ansprechens (Duration of Response, DOR), progressionsfreies Überleben (PFS), Gesamtüberleben (Overall Survival, OS) und die Schwere der unerwünschten Ereignisse. Es wurde vorab festgelegt, dass die ORR bei den ersten 92 Patienten getestet wird; diese war signifikant höher als die vorab festgelegte Rate von 20% (p < 0,0001).
In der primären Analyse (Nachbeobachtung zumindest 6 Monate) betrug die ORR 82%, basierend auf der modifizierten Intention-to-Treat(mITT)-Population, und die Rate des vollständigen Ansprechens (Complete Response, CR) lag bei 54%, ermittelt durch die Prüfärzte (primärer Endpunkt). In der aktualisierten Analyse (Nachbeobachtung zumindest 12 Monate) betrug die ORR 83%, und die CR Rate lag bei 58%, ermittelt durch die Prüfärzte. In der 24-monatigen Nachbeobachtungs-Analyse betrug die ORR 83% und die CR-Rate lag bei 58%, ermittelt durch die Prüfärzte. Die Wirksamkeitsresultate sind in Tabelle 4 unten zusammengefasst. Das mediane OS in der ITT Population betrug 17,4 Monate (95% KI 11,6 – NE). Die 12 und 24 Monats OS Raten waren 59,5% und 47,7%. Das mediane OS wurde in der mITT Population noch nicht erreicht bei beobachteten 50 Ereignissen/101 Patienten. Die 12 und 24 Monats OS Raten waren 60,4% und 50,5%. In einer Analyse über 36 Monate, betrug das Gesamtüberleben in der mITT Population (101 Patienten) 25,8 Monate. In einer Analyse über 60 Monate betrug die Kaplan-Meier Schätzung für die 3-Jahres, 4-Jahres und 5-Jahres OS Raten 47%, 44% bzw. 43%.
Tabelle 4: Zusammenfassung der Wirksamkeitsergebnisse für Phase 2 von ZUMA-1 (24-Monats-Analyse)
|
|
Alle Leukapheresierten (ITT)
Cohort 1 + 2
(N = 111)
|
Alle Behandelten
mITT
Cohort 1 + 2
(N = 101)
| |
|
Bewertung unabhängige Prüfungskommision
|
Bewertung Prüfarzt
|
Bewertung unabhängige Prüfungskommision
|
Bewertung Prüfarzt
| |
ORR (%) [95% KI]
|
68 (58; 76)
|
77 (69; 85)
|
74 (65; 82)
|
83 (74; 90)
| |
CR (%)
|
50
|
55
|
54
|
58
| |
PFS (Monate) [95% KI]
|
9,5 (6,1; 15,4)
|
6,2 (4,0; 12,4)
|
9,1 (5,7; n.a.)
|
5,9 (3,3; 15,0)
| |
DORa, Median (95% KI) in Monaten
|
n.a. (10,9; n.a.)
|
9,0 (3,9; n.a.)
|
n.a. (10,9; n.a.)
|
9,0 (3,9; n.a.)
| |
DOR, CR, Median (95% KI) in Monaten
|
n.a. (n.a.; n.a.)
|
Nicht verfügbar
|
n.a. (n.a.; n.a.)
|
n.a. (12,9; n.a.)
| |
DOR, PR, Median (95% KI) in Monaten
|
2,1 (1,3; 11,1)
|
Nicht verfügbar
|
2,1 (1,3; 11,1)
|
1,9 (1,3; 2,1)
| |
Mediane Nachbeobachtungszeit (Monate)
|
27,1
| |
Minimale Nachbeobachtungszeit (Monate)
|
22,9
| |
OS, Median (Monate) [95% KI]
|
17,4 (11,6; n.a.)
|
n.a. (12,8; n.a.)
| |
6 Monate OS (%) [95% KI]
|
81,1 (72,5; 87,2)
|
79,2 (69,9; 85,9)
| |
12 Monate OS (%) [95% KI]
|
59,5 (49,7; 67,9)
|
60,4 (50,2; 69,2)
| |
24 Monate OS (%) [95% KI]
|
47,7 (38,2; 56,7)
|
50,5 (40,4; 59,7)
|
KI, Konfidenzintervall; CR, vollständiges Ansprechen; DOR, Dauer des Ansprechens; ITT, intention-to-treat; mITT, modifiziertes intention-to-treat); n.a.=Nicht abschätzbar; ORR, objektive Ansprechrate; OS, Gesamtüberleben; PR, partielles Ansprechen.
a Die Dauer des Ansprechens und PFS waren zum Zeitpunkt der SZT für Teilnehmer zensiert, die die SZT während des Ansprechens erhielten
Hinweis: mITT war definiert als alle Patienten, die YESCARTA in einer Mindestdosis von 1x106 CAR-T-Zellen/kg erhielten
Klinische Studie ZUMA-7, Phase 3 (r/r DLCBL und r/r HGBL)
Die Wirksamkeit und Sicherheit von YESCARTA bei erwachsenen Patienten mit r/r DLCBL und r/r HGBL wurden in einer randomisierten, offenen, multizentrischen Phase-3-Studie (ZUMA-7) nachgewiesen. Bei den eingeschlossenen Patienten bestand überwiegend die Diagnose eines nicht anderweitig spezifizierten/nicht weiter klassifizierbaren DLBCLs (69%) und eines High-Grade-B-Zell-Lymphoms (HGBL) (0% mit nicht anderweitig spezifiziertem HGBL und 16% mit MYC / BCL-2 / BCL-6-Gen-Rearrangement (einschliesslich double hit oder triple hit) gemäss Beurteilung des zentralen Labors. 46 Patienten (13%) wurden als «nicht bestätigt» oder «fehlend» kategorisiert, und 10 Patienten (3%) wurden als «andere Lymphome als DLBCL oder HGBL» kategorisiert. Die Patienten wurden nach den molekularen Untergruppen (Keimzentrum-B-Zell-Typ (58%) und aktivierter B-Zell-Typ (7%) sowie «nicht klassifiziert» (9%)) und dem CD19-Expressionsstatus «ja» (77%) und «nein» (7%) gemäss Beurteilung des zentralen Labors weiter kategorisiert. Alle Patienten hatten vorgängig eine Erstlinientherapie mit einer Anthracyclin-haltigen Therapie und monoklonalen Anti-CD20-Antikörpern erhalten, es sei denn, der Tumor war CD20-negativ. Insgesamt erhielten 359 Patienten nach Randomisierung im Verhältnis 1:1 eine einzelne YESCARTA-Infusion oder die Standardtherapie (definiert als 2 bis 3 Zyklen Standard-Chemoimmuntherapie [R-ICE, R-DHAP oder R-DHAX, R-ESHAP oder R-GDP] gefolgt von hochdosierter Therapie [HDT] und autologer Stammzelltransplantation [ASZT] bei Patienten, deren Erkrankung auf die Behandlung ansprach). Die Randomisierung erfolgte stratifiziert 1) gemäss dem Ansprechen auf die Erstlinientherapie (primär refraktär vs. Rezidiv ≤6 Monate vs. Rezidiv > 6 und ≤12 Monate) und gemäss aaIPP («age-adjusted international prognostic index») 0 / 1 vs. 2 / 3. Gründe für den Ausschluss von der Studie waren frühere hämatopoetische Stammzelltransplantation (HSZT), primäres ZNS-Lymphom oder Hirnmetastasen, nachweisbare bösartige Zellen im Liquor und ein Eastern Cooperative Oncology Group (ECOG)-Leistungsstatus von 2 oder höher. Patienten mit aktiven oder schwerwiegenden Infektionen waren ausgeschlossen, jedoch waren Patienten mit einfachen Harnwegsinfektionen und unkomplizierter bakterieller Pharyngitis zugelassen, wenn sie auf eine aktive Behandlung ansprachen. Patienten mit bekannten Infektionen mit dem HIV-, Hepatitis-B- (HBsAG-positiv) oder Hepatitis-C-Virus (Anti-HCV-positiv) waren ausgeschlossen. Der Einschluss von Patienten nach einer erfolgreichen Behandlung von Hepatitis B oder Hepatitis C in der Anamnese war zulässig, wenn keine Viruslast im quantitativen PCR- und/oder Nukleinsäuretest nachweisbar war.
Nach einer Chemotherapie zur Lymphodepletion wurde YESCARTA als einzelne intravenöse Infusion mit einer Zieldosis von 2 x 106 Anti-CD19-CAR-T-Zellen/kg (Höchstdosis: 2 x 108 Zellen) verabreicht. Das Behandlungsschema zur Lymphodepletion bestand aus Cyclophosphamid 500 mg/m² intravenös und Fludarabin 30 mg/m² intravenös, beide verabreicht am 5., 4. und 3. Tag vor YESCARTA. Eine nicht krankheitsmodifizierende Bridging-Therapie, die auf Kortikosteroide beschränkt war, konnte Patienten mit hoher Krankheitslast beim Screening zwischen der Leukapherese und der Chemotherapie zur Lymphodepletion verabreicht werden.
Von den 180 Patienten, die zur Behandlung mit YESCARTA randomisiert wurden, unterzogen sich 178 der Leukapherese, und 170 wurden mit YESCARTA behandelt. Acht Patienten (4%) wurden nach der Leukapherese nicht behandelt, insbesondere wegen progredienter Erkrankung, schwerwiegenden unerwünschten Ereignissen oder Tod. Da eine erneute Gabe für einen Patienten der klinischen Studie einen zweiten Herstellungslauf erforderte, wurden insgesamt 171 Chargen hergestellt. Die mediane Dauer von der Leukapherese bis zur Produktfreigabe betrug 13 Tage (Spanne: 10 bis 24 Tage), und bis zur YESCARTA-Infusion 26 Tage (Spanne: 16 bis 52 Tage). Die mediane Dosis betrug 2,0 x 106 Anti-CD19-CAR-T-Zellen/kg für Patienten mit einem Körpergewicht von < 100 kg (Min: 1,0 x 106 Anti-CD19-CAR-T-Zellen/kg; Max: 2,1, x 106 Anti-CD19-CAR-T-Zellen/kg). Für Patienten mit einem Körpergewicht von > 100 kg betrug die mediane Dosis 200 x 106 Anti-CD19-CAR-T-Zellen. 60 (33%) der behandelten Patienten erhielten eine Bridging-Therapie mit Kortikosteroiden. Alle 170 Patienten, die YESCARTA erhielten, wurden mindestens 7 Tage lang in einer klinischen Einrichtung stationär überwacht. Von den 179 Patienten, die zum Standardtherapie-Studienarm randomisiert wurden, erhielten 64 Patienten (36%) eine HDT-ASZT, einschliesslich 2 Patienten, die eine ASZT ausserhalb der Vorgaben des Prüfplans erhielten.
In der Standardtherapie-Gruppe erhielten 49 Patienten (41%) mit DLBCL und 9 Patienten (35%) mit HGBL eine HD-ASZT.
In der Gesamtpopulation der Studie betrug das mediane Alter 59 Jahre (Spanne: 21 bis 81 Jahre); 66% waren Männer und 83% waren Weisse. Vierundsiebzig Prozent der Patienten hatten ein primäres refraktäres LBCL, und 26% der Patienten hatten innerhalb von 12 Monaten nach der Erstlinientherapie ein Rezidiv erlitten. Die Patienten hatten einen aaIPI-Score von 0–1 (55%) oder 2–3 (45%) und einen ECOG-Leistungsstatus von 0 (54%) oder 1 (46%).
Die mediane tatsächliche Nachbeobachtungszeit betrug 20,07 Monate (Spanne: 0,59 bis 37,75 Monate) in der YESCARTA-Gruppe und 18,23 Monate (Spanne: 0,03 bis 37,26 Monate) in der Gruppe mit Standardtherapie.
Der primäre Endpunkt war das ereignisfreie Überleben (event-free survival, EFS), ermittelt durch verblindete zentrale Auswertung. Das EFS war definiert als die Zeit von der Randomisierung bis zur Krankheitsprogression gemäss Lugano-Klassifikation (Cheson et al., 2014), Beginn einer neuen Lymphom-Therapie oder Tod jeglicher Ursache, je nachdem, was als erstes eintrat. In der Studie wurde eine statistisch signifikante Verbesserung des EFS bei zu YESCARTA randomisierten Patienten im Vergleich zur Standardtherapie gezeigt (HR: 0,398 [95% KI: 0,308; 0,514]). Das ereignisfreie 24-Monats-Überleben betrug 40,5% [95% KI: 33,2; 47,7] im YESCARTA-Arm und 16,3% [95% KI: 11,1; 22,2] im Standardtherapie-Arm.
Die Sekundärendpunkte ORR- und CR-Rate waren bei mit YESCARTA behandelten Patienten signifikant höher, mit einem ORR-Unterschied von 33,1% [95% KI: 23,2; 42,1]) und einem CR-Raten-Unterschied von 33% im YESCARTA-Arm (Tabelle 5). Die mediane DOR betrug im YESCARTA-Arm 26,9 Monate (Spanne: 0 bis 29 Monate), verglichen mit 8,9 Monaten (Spanne: 0 bis 32 Monate) im Standardtherapie-Arm (HR: 0,736 [95% KI: 0,488; 1,108]). Zum Zeitpunkt der primären Analyse des EFS betrug die mediane Studiendauer 24,9 Monate.
Die primäre Analyse des Gesamtüberlebens (OS), ein wichtiger sekundärer Endpunkt, wurde zum im Prüfplan festgelegten Zeitpunkt von fünf Jahren ab Aufnahme des ersten Patienten durchgeführt. Eine statistisch signifikante Verbesserung des OS mit YESCARTA gegenüber der Standardtherapie wurde durch die Reduktion des Sterberisikos von 27,4% (HR: 0,726 [95% KI: 0,540; 0,977]) nachgewiesen. Mit geschätzten 48 Monats-OS-Raten von 54,6% bzw. 46,0% wurde das mediane OS im YESCARTA-Arm im Vergleich zu 31,1 Monaten im Standardtherapie-Arm nicht erreicht. Die mediane Studiendauer liegt bei 47,2 Monaten. Siebenundfünfzig Prozent (57%) der Patienten erhielten eine zelluläre Immuntherapie, nachdem sie auf die Standardtherapie nicht angesprochen oder nach der Randomisierung zur Standardtherapie ein Rezidiv erlitten hatten.
Die Zusammenfassung der Wirksamkeitsergebnisse in der Gesamtpopulation ist in Tabelle 5 aufgeführt. Die Kaplan-Meier Kurve des OS ist in Abbildung 1 dargestellt.
Tabelle 5: Zusammenfassung der Wirksamkeitsergebnisse für ZUMA-7
|
|
YESCARTA
N = 180
|
Standardtherapie
N = 179
| |
EFSa
| |
Anzahl der Ereignisse (%)
|
108 (60)
|
144 (80)
| |
Median, Monate [95% KI]b
|
8,3 [4,5; 15,8]
|
2,0 [1,6; 2,8]
| |
Stratifizierte Hazard Ratio [95% KI]
|
0,398 [0,308; 0,514]
| |
Stratifizierter Log-Rank-p-Wertc
|
< 0,0001
| |
OS
| |
Anzahl der Ereignisse (%)
|
82 (46)
|
95 (53)
| |
Medianes OS, Monate [95% KI]b
|
Nicht erreicht (28,6; n.a.)
|
31,1 (17,1; n.a.)
| |
Stratifizierte Hazard Ratio [95% KI]
|
0,726 (0,540; 0,977)
| |
Stratifizierter Log-Rank-p-Wertc,d
|
0,0168
| |
ORR (%) [95 % KI]a
|
83 [77,1; 88,5]
|
50 [42,7; 57,8]
| |
Odds Ratio [95% KI]
|
5,31 [3,08; 8,90]
| |
p-Wert des stratifizierten CMH-Testsc
|
< 0,0001
| |
Rate des vollständigen Ansprechens (%)
|
65 [57,6; 71,9]
|
32 [25,6; 39,8]
| |
Rate des teilweisen Ansprechens (%)
|
18 [13,0; 24,8]
|
18 [12,6; 24,3]
| |
PFSe
| |
Anzahl der Ereignisse (%)
|
96 (53)
|
103 (58)
| |
Median, Monate [95% KI]b
|
14,7 [5,4; n.a.]
|
3,7 [2,9; 5,3]
| |
Stratifizierte Hazard Ratio [95% KI]
|
0,490 [0,368; 0,652]
|
KI, Konfidenzintervall; CMH, Cochran-Mantel-Haenszel; EFS, Ereignisfreies Überleben; n.a., nicht abschätzbar; OS, Gesamtüberleben; PFS, Progressionsfreies Überleben.
a Ermittelt durch zentrale Auswertung zum Zeitpunkt der primären Analyse des EFS.
b Kaplan-Meier-Methode.
c Einseitige p-Werte. Stratifizierter Log-Rank-Test oder stratifizierter CMH-Test nach Ansprechen auf die Erstlinientherapie (primär refraktär vs. Rezidiv ≤6 Monate nach der Erstlinientherapie vs. Rezidiv > 6 und ≤12 Monate nach der Erstlinientherapie) und dem auf die Zweitlinientherapie bezogenen altersangepassten internationalen prognostischen Index (0 bis 1 vs. 2 bis 3) angepasst.
d Der p-Wert wird mit 0,0249 verglichen, der einseitigen Wirksamkeitsgrenze (Signifikanzniveau) für die primäre OS-Analyse.
e Nach Einschätzung des Prüfarztes.
f Nach Auswertung zum Zeitpunkt der primären Analyse des OS (fünf Jahre ab Aufnahme des ersten Teilnehmers).
Eine anhaltende Wirksamkeit wurde in Subgruppen beobachtet, einschliesslich Ansprechen auf Erstlinientherapie, gemäss Alter bei der Zweitlinientherapie angepasster IPI-Score, ECOG-Leistungsstatus, Alter, Double-Expressor-Lymphom-Status, Erkrankungs-Subtyp HGBL, CD19-Status, B-Symptome, Beteiligung der Milz, extranodal, hohe Tumorlast und Beteiligung des Knochenmarks.
Abbildung 1a. Kaplan-Meier Kurve des Gesamtüberlebens in ZUMA-7 (Vollständiger Analysensatz; primäre Analyse Gesamtüberleben)
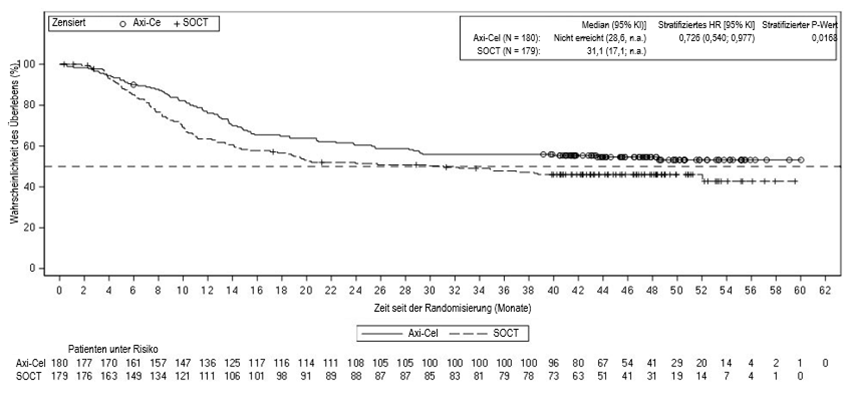
KI, Konfidenzintervall; HR, Hazard Ratio; SOCT, Standardtherapie.
a Patienten, die nicht auf die SOCT ansprachen, konnten ausserhalb der Anforderungen des Protokolls eine anschliessende Lymphom-Behandlung einschliesslich einer Anti-CD19-CAR-T-Zelltherapie erhalten.
Wiederholte Therapie mit YESCARTA
Zum Zeitpunkt der primären OS-Analyse hatten insgesamt 10 Patienten (6%) eine wiederholte Behandlung mit YESCARTA erhalten. Aufgrund der geringen Anzahl an Patienten kann keine Schlussfolgerung bezüglich der Wirksamkeit und Sicherheit gezogen werden.
Nachfolgende Anti-Lymphom-Therapien
Siebenundfünfzig Prozent (57%)der Patienten im Standardtherapiearm erhielten nachfolgend eine zelluläre Immuntherapie ausserhalb des Prüfplans (einschliesslich 55%, die eine autologe Anti-CD19-CAR-Zelltherapie erhielten) nach der Randomisierung.
Acht Prozent (8%) der Patienten im YESCARTA-Arm erhielten nachfolgend eine zelluläre Immuntherapie ausserhalb des Prüfplans (einschliesslich 7%, die eine autologe Anti-CD19-CAR-Zelltherapie erhielten, und einschliesslich der 10 Patienten (6%), die eine wiederholte Behandlung mit YESCARTA erhielten).
Nachfolgende Stammzelltransplantation
Vier Prozent (4%) der Patienten im Standardtherapiearm bzw. 7% der Patienten im YESCARTA-Arm erhielten nachfolgend eine hochdosierte Therapie und autologe Stammzelltransplantation [HDT+ASZT], und 4% der Patienten im Standardtherapiearm bzw. 8% der Patienten im YESCARTA-Arm erhielten nachfolgend eine allogene Stammzelltransplantation (SZT).
Klinische Studie (ZUMA-5), Phase 2, rezidiviertes oder refraktäres FL
Die Wirksamkeit und Sicherheit von YESCARTA bei erwachsenen Patienten mit FL, die mit YESCARTA behandelt wurden, wurden in einer einarmigen, offenen, multizentrischen Phase-2-Studie an Patienten mit rezidiviertem oder refraktärem FL nach WHO-Klassifizierung von 2016 untersucht.
Für die Studie geeignete Patienten waren ≥18 Jahre alt und hatten nach 2 oder mehr vorherigen Therapielinien eine rezidivierte oder refraktäre Erkrankung.
Die vorherige Therapie musste einen gegen CD20 gerichteten monoklonalen Antikörper in Kombination mit einem alkylierenden Mittel umfassen (eine Monotherapie mit einem gegen CD20 gerichteten Antikörper zählte nicht als Therapielinie). Patienten mit SD (ohne Rezidiv) > 1 Jahr nach Abschluss der letzten Therapie waren ausgeschlossen. Patienten mit ZNS-Lymphomen, allogener SZT in der Anamnese oder vorheriger Therapie mit Anti-CD19-CAR- oder anderen genetisch modifizierten T-Zellen waren ausgeschlossen. Patienten mit ZNS-Erkrankungen (wie z.B. Krampfanfälle oder zerebrosvaskuläre Ischämie) in der Anamnese, einer Auswurffraktion des Herzens von weniger als 50% oder einer Sauerstoffsättigung bei Raumluft von weniger als 92% oder mit Autoimmunerkrankungen, die eine systemische Immunsuppression erfordern, waren nicht geeignet. Von der Studie ausgeschlossen waren Patienten mit aktiven oder schwerwiegenden Infektionen, transformierten Lymphomen oder Beteiligung des ZNS.
Zum Zeitpunkt der primären Analyse waren insgesamt 127 FL-Patienten in die Studie aufgenommen worden, einschliesslich 80 Patienten, die 3 oder mehr Therapielinien erhalten hatten, und bei allen war eine Leukapherese durchgeführt worden. Im Zeitraum zwischen dem Datenstichtag zur primären Analyse und dem Datenstichtag zur 24-Monats- Nachbeobachtungs-Analyse wurden keine zusätzlichen FL-Patienten eingeschlossen oder mit YESCARTA behandelt. Die tatsächliche Dauer der Nachbeobachtung betrug 26,55 Monate (Spanne: 0,3 bis 44,3 Monate, Studie noch laufend). Unter den 127 in die Studie aufgenommenen FL-Patienten hatten 80 Patienten 3 oder mehr vorherige Therapielinien erhalten. Die demografischen Daten in der Population, die 3 oder mehr vorherige Therapielinien erhalten hatte, stimmte mit der Gesamtpopulation überein. Eine Zusammenfassung der demografischen Patientendaten ist in Tabelle 6 angegeben.
Tabelle 6: Zusammenfassung der demografischen Daten für FL-Patienten aus ZUMA-5 (24-Monats-Analyse)
|
Kategorie
|
Alle Patienten mit Leukapherese ≥2 vorherige Therapielinien
(N = 127)
|
Alle Patienten mit Leukapherese mit ≥3 vorherige Therapielinien
(N = 80)
| |
Alter (Jahre)
| |
Median (min, max)
|
60 (34; 79)
|
60,5 (34; 79)
| |
≥65
|
31%
|
33%
| |
Männliches Geschlecht
|
59%
|
61%
| |
Ethnische Abstammung
| |
Weisse
|
92%
|
93%
| |
Asiaten
|
2%
|
4%
| |
Farbige
|
3%
|
3%
| |
ECOG-Status
| |
ECOG 0
|
62%
|
58%
| |
ECOG 1
|
38%
|
43%
| |
Hohe Tumorlast gemäss Definition der GELF-Kriterien
|
51%
|
56%
| |
Mediane Anzahl früherer Therapien (min, max)
|
3 (1; 10)
|
4 (3; 10)
| |
Patienten mit refraktärer Erkrankung gegen ≥2 frühere Therapielinien
|
29%
|
24%
| |
Patienten mit Krankheitsstadium III/IV
|
86%
|
88%
| |
Frühere Behandlung mit PI3K-Inhibitor
|
28%
|
43%
| |
Rezidiv < 24 Monate nach erster Anti-CD20-Kombinationschemotherapie
|
56%
|
54%
|
ECOG, Eastern Cooperative Oncology Group; GELF, Groupe d'Étude des Lymphomes Folliculaires.
YESCARTA wurde angewendet als einzelne intravenöse Infusion mit einer Zieldosis von 2 x 106 Anti-CD19-CAR-T-Zellen/kg nach einem Chemotherapieschema zur Lymphodepletion mit Cyclophosphamid 500 mg/m2 intravenös und Fludarabin 30 mg/m2 intravenös, beide verabreicht am 5., 4. und 3. Tag vor YESCARTA. Alle Patienten wurden zur Beobachtung über einen Zeitraum von mindestens 7 Tagen nach der YESCARTA-Infusion hospitalisiert. Die Verabreichung und Überwachung von YESCARTA in ZUMA-5 entspricht dem Vorgehen in ZUMA-1.
Die primäre Analyse wurde durchgeführt, als 84 nacheinander eingeschlossene FL-Patienten ab Beurteilung des ersten Ansprechens mindestens 12 Monate lang nachbeobachtet worden waren. Der primäre Endpunkt war die ORR. Sekundäre Endpunkte waren die CR-Rate, ORR und CR bei Patienten, die 3 oder mehr Therapielinien erhalten hatten, DOR, OS und progressionsfreies Überleben (progression-free survival, PFS) sowie die Inzidenz unerwünschter Ereignisse. Zum Zeitpunkt der primären Analyse wurden drei der 127 eingeschlossenen FL-Patienten nicht behandelt, aufgrund Verletzung der Einschlusskriterien oder Tod vor der Behandlung. Eine 24-Monats-Nachbeobachtungs-Analyse wurde durchgeführt, als 86 FL-Patienten nach der Infusion mindestens 24 Monate lang nachbeobachtet worden waren.
Zum Zeitpunkt der 24-Monats-Nachbeobachtungs-Analyse wurden keine zusätzlichen Patienten einer Leukapherese unterzogen bzw. mit YESCARTA behandelt. Die mediane Zeit von der Leukapherese bis zur Freigabe des Arzneimittels betrug 12 Tage (Spanne: 10 bis 37 Tage), von der Leukapherese bis zur Lieferung des Arzneimittels 17 Tage (Spanne: 13 bis 72 Tage) und von der Leukapherese bis zur YESCARTA-Infusion 27 Tage (Spanne: 19 bis 330 Tage): Die mediane Dosis betrug 2,0 x 106 Anti-CD19-CAR-T-Zellen/kg.
Unter den eingeschlossenen 127 FL-Patienten hatten 80 bereits 3 oder mehr Therapielinien erhalten. Die ORR betrug 91% und die CR-Rate 79%. Die mediane Zeit bis zum Ansprechen betrug 0,99 Monate (Spanne: 0,8 bis 3,1 Monate), die mediane DOR betrug 38,6 Monate und der Anteil der Responder, deren Ansprechen bis Monat 24 anhielt, betrug 55%. Eine Subgruppenanalyse umfasste die ORR bei Patienten, die refraktär waren (89%), sowie Patienten mit einem FLIPI-Score ≥3 (95%), einer hohen Tumorlast (91%), einer Krankheitsprogression innerhalb von 24 Monaten nach der ersten Immuntherapie (91%) und mit einer vorherigen Behandlung mit einem PI3K-Inhibitor (91%). Die wichtigsten Wirksamkeitsergebnisse für FL-Patienten mit 3 oder mehr vorherigen Therapielinien sind in Tabelle 7 zusammengefasst. Neunundzwanzig der 80 FL-Patienten, die 3 oder mehr Therapielinien erhalten hatten, erreichten anfangs eine PR, von denen 19 später eine CR erreichten.
Tabelle 7: Zusammenfassung der Wirksamkeitsergebnisse für FL-Patienten aus ZUMA-5 mit 3 oder mehr vorherigen Therapielinien (24-Monats-Analyse)
|
Kategorie
|
Alle Patienten mit einer Leukapherese
N = 80
| |
ORRa, (%) [95% KI]
|
91% [83; 96]
| |
CR, (%) [95% KI]
|
79% [68; 87]
| |
PR, (%) [95% KI]
|
13% [6; 22]
| |
DORb, Median in Monaten [95% KI]
|
38,6
| |
(Spanne)
|
(24,7; n.a.)
| |
Min, Max, DOR (Monate)
|
(0,0; 38.6)
| |
Anhaltendes Ansprechen (n)
|
44
| |
Rate des anhaltenden Ansprechensc % (95% KI)
|
| |
12 Monate
|
78,1 (66,2; 86,2)
| |
18 Monate
|
72,8 (60,2; 82,0)
| |
24 Monate
|
65,4 (51,2; 76,4)
| |
Progressionsfreies Überleben, Median, Monate [95% KI]
|
40,2 (26,6; n.a.)
| |
Gesamtüberleben, Median (Monate) [95% KI]
|
NE (40,2; n.a.)
| |
Gesamtüberleben, % [95% KI]
|
| |
12 Monate
|
96,2 (88,7; 98,8)
| |
18 Monate
|
91,1 (82,2; 95,7)
| |
24 Monate
|
85,2 (74,7; 91,5)
|
CR, vollständiges Ansprechen; DOR, Dauer des Ansprechens; KI, Konfidenzintervall; n.a., nicht abschätzbar; ORR, objektives Ansprechen; PR, Teilremission.
a. Gemäss der International Working Group Lugano Classification (Cheson 2014), beurteilt durch die unabhängige radiologische Prüfungskommission
b. Unter allen Respondern wird die DOR ab dem Datum des ersten objektiven Ansprechens bis zum Datum der Progression oder des Todes gemessen.
c. Gemessen ab dem Datum des ersten objektiven Ansprechens bis zum Datum der Progression oder des Todes.
PharmakokinetikZelluläre Kinetik
Absorption
YESCARTA besteht aus menschlichen autologen T-Zellen. Die erwarteten Stoffwechselprodukte sind typische zelluläre Abbauprodukte, die aus normalen zellulären Clearance-Mechanismen resultieren. Daher wird erwartet, dass die infundierten CAR-T-Zellen im Laufe der Zeit abgebaut werden.
Nach Infusion von YESCARTA war bei den Anti-CD19-CAR-T-Zellen eine schnelle anfängliche Expansion zu beobachten, gefolgt von einem Rückgang auf Werte, die nach 3 Monaten fast dem Ausgangswert entsprachen.
Die Spitzenkonzentrationen von Anti-CD19-CAR-T-Zellen traten innerhalb der ersten 7–14 Tage nach dem Tag der YESCARTA-Infusion auf. Alter (Spanne: 21 bis 80 Jahre) und Geschlecht hatten keine signifikanten Auswirkungen auf die AUC und die Spitzenkonzentrationen von YESCARTA.
Bei Patienten mit DLBCL in ZUMA 1 lag die mediane Spitzenkonzentration von Anti-CD19-CAR-T-Zellen im Blut (Cmax) bei 38,3 Zellen/µl (Spanne: 0,8-1513,7 Zellen/μl); diese ging nach 1 Monat nach der YESCARTA-Infusion auf einen Median von 2,1 Zellen/µl (Spanne: 0-167,4 Zellen/μl) sowie 3 Monate nach der YESCARTA-Infusion auf einen Median von 0,4 Zellen/µl (Spanne: 0-28,4 Zellen/μl) zurück.
Unter den Patienten in ZUMA-7 lag die mediane Spitzenkonzentration von Anti-CD19-CAR-T-Zellen im Blut bei 25,84 Zellen/µl (Spanne: 0,04 bis 1173,25 Zellen/µl); diese ging bei den auswertbaren Patienten nach 3 Monaten auf das Ausgangsniveau (0,35 Zellen/μl; Spanne: 0,00 bis 28,44 Zellen/µl) zurück, aber die Zellen waren bei 12 von 30 auswertbaren Patienten bis 24 Monate nach der Behandlung noch nachweisbar.
Bei Patienten mit FL lag die mediane Spitzenkonzentration von Anti-CD19-CAR-T-Zellen im Blut (Cmax) bei 39,4 Zellen/µl (Spanne: 0,5-1415,4 Zellen/μl). Die mediane Zeit bis zur Spitzenkonzentration von Anti-CD19-CAR-T-Zellen im Blut betrug nach der Infusion 8 Tage (Spanne: 8 bis 371 Tage). Nach 3 Monaten nahmen die Konzentrationen von Anti-CD19-CAR-T-Zellen fast bis auf das Niveau zu Studienbeginn ab, und zwar auf einen Median von 0,4 Zellen/μl (Spanne: 0 bis 15,8 Zellen/μl).
Bei Patienten mit DLBCL und PMBCL in ZUMA-1 stand die Anzahl der Anti-CD19-CAR-T-Zellen im Blut in einem positiven Zusammenhang mit dem objektiven Ansprechen (CR oder PR). Die medianen Spitzenkonzentrationen der Anti-CD19-CAR-T-Zellen waren bei Respondern (n = 73) um 205% höher als die entsprechenden Konzentrationen bei Non-Respondern (n = 23) (43,6 Zellen/μl vs. 21,2 Zellen/μl). Die mediane AUCTag 0-28 bei ansprechenden Patienten (n = 73) lag bei 251% der entsprechenden Konzentration bei Non-Respondern (n = 23) (557,1 Tage x Zellen/μl vs. 222,0 Tage x Zellen/μl). Die Zeit bis zum Erreichen der Spitzenkonzentration betrug 8 Tage (7 Tage nach dem Tag der YESCARTA-Infusion).
Daten, die während der Nachbeobachtung nach der YESCARTA-Infusion gewonnen wurden, zeigten, dass 13 von 34 Patienten im fortlaufenden Ansprechen nach 24 Monaten keine nachweisbaren genmarkierten Anti-CD19 CAR-T-Zellen hatten, während 21 von 34 Patienten sehr niedrige Werte nachweisbarer genmarkierter CAR-T-Zellen hatten.
Einige Patienten benötigten Tocilizumab/ICANS und Kortikosteroide zur Behandlung von CRS und neurologischen Toxizitäten. Patienten, die mit Tocilizumab behandelt wurden (n = 44), hatten 262% und 232% höhere Anti-CD19 CAR T-Zellen, gemessen mit AUCTag 0-28 bzw. Spitzenkonzentration, im Vergleich zu Patienten, die kein Tocilizumab erhielten (n = 57). Ebenso hatten Patienten, die Kortikosteroide erhielten (n = 26), 217% und 155% mehr AUCTag 0-28 und Spitzenkonzentration als Patienten, die keine Kortikosteroide erhielten (n = 75).
Unter den Patienten in ZUMA-7 stand die Anzahl der Anti-CD19-CAR-T-Zellen im Blut in einem positiven Zusammenhang mit dem objektiven Ansprechen (CR oder PR). Die medianen Spitzenkonzentrationen der Anti-CD19-CAR-T-Zellen waren bei Respondern (n = 142) um etwa 275% höher als die entsprechenden Konzentrationen bei Non-Respondern (n = 20) (28,9 Zellen/μl vs. 10,5 Zellen/ μl). Die mediane AUCTag 0-28 bei ansprechenden Patienten (n = 142) war um etwa 417% höher als die entsprechende Konzentration bei Non-Respondern (n = 20) (292,9 Tage x Zellen/μl vs. 70,1 Tage x Zellen/μl). Die Zeit bis zum Erreichen der Spitzenkonzentration betrug 8 Tage (7 Tage nach dem Tag der YESCARTA-Infusion).
Daten, die während der Nachbeobachtung nach der YESCARTA-Infusion gewonnen wurden, zeigten, dass 18 von 28 Patienten im fortlaufenden Ansprechen und mit auswertbaren CAR-T-Werten nach 24 Monaten keine nachweisbaren genmarkierten Anti-CD19 CAR-T-Zellen hatten, während 10 von 28 Patienten sehr niedrige Werte nachweisbarer genmarkierter CAR-T-Zellen hatten. Einige Patienten benötigten Tocilizumab und Kortikosteroide zur Behandlung von CRS und neurologischen Toxizitäten. Patienten, die mit Tocilizumab behandelt wurden (n = 112), hatten einen 215%- und 274%-fachen Anstieg der Anti-CD19 CAR T-Zellen, gemessen mit AUCTag 0-28 bzw. Spitzenkonzentration, im Vergleich zu Patienten, die kein Tocilizumab erhielten (n = 58). Ebenso hatten Patienten, die Kortikosteroide erhielten (n = 77), 272% und 176% mehr AUCTag 0-28 und Spitzenkonzentration als Patienten, die keine Kortikosteroide erhielten (n = 93).
Bei Patienten mit FL betrug die mediane Spitzenkonzentration von Anti-CD19-CAR-T-Zellen bei Respondern (n = 79) im Vergleich zu Non-Respondern (n = 3) 37,62 Zellen/μ bzw. 35,31 Zellen/μl. Die mediane AUC0-28 betrug bei Respondern im Vergleich zu Non-Respondern 451,17 Zellen/μl•Tage bzw. 247,14 Zellen/μl•Tage.
Distribution
Es liegen keine Daten vor.
Metabolismus
Es liegen keine Daten vor.
Elimination
YESCARTA besteht aus menschlichen autologen T-Zellen, die erwarteten Stoffwechselprodukte sind typische zelluläre Abbauprodukte, die aus normalen zellulären Clearance-Mechanismen resultieren. Daher wird erwartet, dass die infundierten CAR-T-Zellen im Laufe der Zeit abgebaut werden. Anti-CD19-CAR-T-Zellwerte sanken bis 3 Monate nach der Infusion auf Hintergrundniveau (Spanne: 0,000 bis 28,410 Zellen/μl in ZUMA-1 und Spanne: 0,000 bis 28,440 Zellen in ZUMA-7). In ZUMA-1 wurde keine sekundäre Expansion von CAR-T-Zellen beobachtet. In ZUMA-5 wurden 2 Patienten mit sekundärer Expansion beobachtet.
Daten, die während der Nachbeobachtung nach der YESCARTA-Infusion gewonnen wurden, zeigten, dass 13 von 34 Patienten im fortlaufenden Ansprechen nach 24 Monaten keine nachweisbaren genmarkierten Anti-CD19 CAR-T-Zellen hatten, während 21 von 34 Patienten sehr niedrige Werte nachweisbarer genmarkierter CAR-T-Zellen hatten.
Kinetik spezieller Patientengruppen
Alter und Geschlecht
Alter (Spanne: 21–81 Jahre) und Geschlecht hatten keine signifikanten Auswirkungen auf die AUCTag 0-28 sowie auf die Spitzenkonzentration von YESCARTA.
Leberfunktionsstörungen
Untersuchungen zu Leberfunktionsstörungen mit YESCARTA wurden nicht durchgeführt.
Nierenfunktionsstörungen
Untersuchungen zu Nierenfunktionsstörungen mit YESCARTA wurden nicht durchgeführt.
Präklinische DatenYESCARTA enthält genetisch modifizierte humane T-Zellen, welche spezifisch mit dem humanen CD19 reagieren; daher existieren keine repräsentativen In-vitro-Assays, Ex-vivo-Modelle oder In-vivo-Modelle, die den toxikologischen Eigenschaften des humanen Produkts präzise Rechnung tragen können. Folglich wurden keine traditionellen toxikologischen Studien in der Arzneimittel-Entwicklung durchgeführt.
Es wurden keine Studien zur Karzinogenität oder Genotoxizität mit YESCARTA durchgeführt.
Es wurden keine Studien zur Bewertung der Auswirkungen von YESCARTA auf die Fertilität, Fortpflanzung und Entwicklung durchgeführt.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
YESCARTA ist 1 Jahr lang stabil, wenn es gefroren in der Dampfphase von Flüssigstickstoff (≤ -150°C) aufbewahrt wird.
YESCARTA ist nach vollständigem Auftauen bei Raumtemperatur (20°C bis 25°C) bis zu 3 Stunden stabil. Um sicherzustellen, dass die Stabilität nach dem Auftauen aufrechterhalten wird, muss die YESCARTA-Infusion innerhalb von 30 Minuten nach vollständigem Auftauen beginnen, und die Gesamtdauer der Infusion soll 30 Minuten nicht überschreiten (siehe «Dosierung/Anwendung»).
Das aufgetaute Arzneimittel darf nicht wieder eingefroren werden.
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
YESCARTA-Beutel müssen in der Dampfphase von Flüssigstickstoff (≤ -150°C) aufbewahrt werden und gefroren bleiben, bis der Patient für die Behandlung bereit ist, um zu gewährleisten, dass dem Patienten teilungsfähige, lebende autologe Zellen verabreicht werden.
YESCARTA kann einmal bei -80°C (± 10°C) für bis zu 90 Tage gelagert werden. Nach der Lagerung bei -80°C (± 10°C) muss das Arzneimittel innerhalb des 90 Tage-Zeitraums oder bis zum angegebenen Verfalldatum angewendet werden, je nachdem, was zuerst eintritt. Nach diesen Zeitpunkten darf das Arzneimittel nicht mehr verwendet werden und muss entsorgt werden.
Für Kinder unzugänglich aufbewahren.
Für Hinweise zu den Lagerungsbedingungen nach dem Auftauen des Arzneimittels, siehe «Haltbarkeit».
Besondere Vorsichtsmassnahmen für die Beseitigung und sonstige Hinweise für die Handhabung
Eine Bestrahlung könnte zur Inaktivierung des Arzneimittels führen.
Dieses Arzneimittel enthält genetisch modifizierte Zellen. Die für diese Arzneimittel lokal geltenden Bestimmungen zur biologischen Sicherheit sind für nicht verwendete Arzneimittel oder Abfallmaterialien zu beachten. Dieses Arzneimittel kann potenziell infektiös sein (z.B. HIV, HBV oder HCV). Das medizinische Fachpersonal muss Sicherheitsvorkehrungen für die Handhabung und Entsorgung des Arzneimittels treffen. Handschuhe sind erforderliche persönliche Schutzausrüstung für die Verabreichung von YESCARTA. Bei der Dekontamination von Oberflächen und der Beseitigung von ausgelaufenem Produkt im Zusammenhang mit YESCARTA sind die vor Ort geltenden Bestimmungen für die biologische Sicherheit einzuhalten.
Zulassungsnummer66986 (Swissmedic)
PackungenYESCARTA, eine Infusionsdispersion mit maximal 2x108 Zellen/68 ml [A]
YESCARTA wird in einem Ethylenvinylacetat-Beutel für die Kryolagerung geliefert.
Es wird jeweils ein Beutel für die Kryolagerung einzeln in einer Versandkassette verpackt.
ZulassungsinhaberinGilead Sciences Switzerland Sàrl, Zug
Stand der InformationDezember 2025
|