ZusammensetzungWirkstoffe
Siponimod (als Siponimod-Fumarsäure).
Hilfsstoffe
Tablettenkern: Lactose-Monohydrat, mikrokristalline Cellulose, Crospovidon, Glyceryldibehenat und kolloidales wasserfreies Silica.
Eine 0.25-mg-Tablette enthält 62.2 mg Lactose-Monohydrat.
Eine 1-mg-Tablette enthält 61.4 mg Lactose-Monohydrat.
Eine 2-mg-Tablette enthält 60.3 mg Lactose-Monohydrat.
Filmüberzug: Polyvinylalkohol, Titandioxid (E171), rotes Eisenoxid (E172), Talkum, Soja Lecithin (E322), Xanthangummi, schwarzes Eisenoxid (E172, nur 0.25 mg- und 1 mg-Tabletten), gelbes Eisenoxid (E172, nur 2 mg-Tabletten).
Indikationen/AnwendungsmöglichkeitenMayzent wird angewendet zur Behandlung von erwachsenen Patienten mit sekundär progredienter Multipler Sklerose (SPMS) mit entzündlicher Krankheitsaktivität, nachgewiesen durch klinische Schübe oder Bildgebung.
Dosierung/AnwendungDie Behandlung mit Mayzent muss von einem in der Behandlung von MS-Patienten erfahrenen Neurologen begonnen und überwacht werden.
Vor der Einleitung der Behandlung mit Mayzent muss der CYP2C9-Genotyp des Patienten bestimmt werden. Bei Patienten mit einem CYP2C9*3/*3-Genotyp darf Mayzent nicht angewendet werden. (s. «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Bei Patienten mit einem CYP2C9*2/*3 oder *1/*3-Genotyp beträgt die empfohlene Erhaltungsdosis 1 mg einmal täglich (eine Tablette à 1 mg oder vier Tabletten à 0.25 mg) (s. «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Zu anderen seltenen CYP2C9-Allelen, bei denen von einer reduzierten oder fehlenden CYP2C9-Aktivität auszugehen ist, wie insbesondere den CYP2C9-Allelen *5, *6, *8 und *11, können aufgrund fehlender Daten keine Dosierungsempfehlungen gegeben werden (siehe auch Rubriken «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen», «Interaktionen» und «Pharmakokinetik»).
Die empfohlene Erhaltungsdosis von Mayzent bei Patienten mit allen anderen CYP2C9-Genotypen beträgt 2 mg.
Mayzent wird einmal täglich mit oder ohne Mahlzeit eingenommen. Mayzent Filmtabletten sollten ganz mit Wasser eingenommen werden.
Vor Beginn der Behandlung
Ophthalmologische Beurteilung
Es soll eine augenärztliche Untersuchung des Augenhintergrunds, einschliesslich der Makula, durchgeführt werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Dermatologische Beurteilung
Es soll eine dermatologische Untersuchung durchgeführt werden. Verdächtige Hautläsionen müssen umgehend abgeklärt werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Therapieeinleitung
Die Behandlung muss mit einer Starterpackung eingeleitet werden, die für 5 Tage ausreicht.
Patienten mit bestimmten vorbestehenden Herzerkrankungen müssen in den ersten 6 Stunden nach der ersten Dosis von Mayzent auf Zeichen und Symptome einer Bradykardie überwacht werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Die Dosistitration beginnt mit 0.25 mg einmal täglich an Tag 1 und 2, gefolgt von einmal täglich 0.5 mg an Tag 3, 0.75 mg an Tag 4 und 1.25 mg an Tag 5, sodass der Patient ab Tag 6 seine verordnete Erhaltungsdosis von Mayzent erreicht (s. Tabelle 1).
Während der ersten 6 Tage der Behandlung sollte die empfohlene Tagesdosis einmal täglich morgens mit oder unabhängig von einer Mahlzeit eingenommen werden.
Tabelle 1 Dosiertitrationsschema zur Erreichung der Mayzent-Erhaltungsdosis
|
Titration
|
Titrationsdosis
|
Titrationsschema
|
Verpackung
| |
Tag 1
|
0.25 mg
|
1 x 0.25 mg
|
| |
Tag 2
|
0.25 mg
|
1 x 0.25 mg
|
| |
Tag 3
|
0.5 mg
|
2 x 0.25 mg
|
STARTER
| |
Tag 4
|
0.75 mg
|
3 x 0.25 mg
|
| |
Tag 5
|
1.25 mg
|
5 x 0.25 mg
|
| |
Tag 6
|
2 mg°
|
1 x 2 mg°
|
ERHALTUNG für CYP2C9-Genotypen *1/*1, *1/*2 oder *2/*2
|
° Die empfohlene Erhaltungsdosis beträgt 1 mg (1 x 1 mg oder 4 x 0.25 mg) täglich für Patienten mit CYP2C9*2/*3 oder *1/*3-Genotyp. Die Patientensicherheit wird durch eine zusätzliche Exposition von 0.25 mg an Tag 5 nicht beeinträchtigt.
Ausgelassene Dosen während des Behandlungsbeginns
Wird eine Titrationsdosis an einem Tag während der ersten 6 Tage der Behandlung ausgelassen, muss die Behandlung mit einer neuen Starterpackung wiederaufgenommen werden.
Ausgelassene Dosis nach Tag 6
Wird eine Dosis ausgelassen, sollte die verordnete Dosis zur nächsten vorgesehenen Zeit eingenommen werden. Die nächste Dosis darf zum Ausgleich für eine vergessene Tablette nicht doppelt eingenommen werden.
Wiederaufnahme der Erhaltungstherapie nach Behandlungsunterbrechung
Wenn die Erhaltungstherapie mit Mayzent für 4 oder mehr aufeinanderfolgende Tagesdosen unterbrochen wird, muss die Behandlung mit einer neuen Starterpackung wieder aufgenommen werden (s. «Einleitung der Behandlung»). Behandlungsunterbrechungen für bis zu 4 fehlende aufeinanderfolgende Tagesdosen erfordern keine Neutitration und die Behandlung sollte mit der Erhaltungsdosis fortgesetzt werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Siponimod darf bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh-Klasse C) nicht angewendet werden (s. «Kontraindikationen»). Obwohl keine Dosisanpassung bei Patienten mit leichter bis mässiger Leberfunktionsstörung erforderlich ist, ist bei diesen Patienten zu Behandlungsbeginn Vorsicht geboten (s. «Warnhinweise und Vorsichtsmassnahmen», «Pharmakokinetik»).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit Niereninsuffizienz sind keine Dosisanpassungen von Mayzent erforderlich.
Kinder und Jugendliche (unter 18 Jahren)
Es wurden keine Studien bei Kindern und Jugendlichen durchgeführt.
Ältere Patienten
Mayzent wurde bei Patienten ab 65 Jahren nicht untersucht. In klinischen Studien nahmen Patienten bis zu einem Alter von 61 Jahren teil. Bei älteren Patienten sollte Mayzent mit Vorsicht angewendet werden, da keine ausreichenden Daten zur Sicherheit und Wirksamkeit vorliegen.
Kontraindikationen·Überempfindlichkeit gegenüber dem Wirkstoff Siponimod, Erdnüssen, Soja oder einem der Hilfsstoffe gemäss Zusammensetzung
·Immundefizienzsyndrom
·Vorgeschichte einer progressiven multifokalen Leukenzephalopathie (PML) oder Kryptokokkenmeningitis
·Aktive maligne Erkrankungen
·Schwere Leberfunktionsstörung (Child-Pugh-Klasse C)
·Patienten, die in den letzten 6 Monaten einen Myokardinfarkt (MI), instabile Angina pectoris, einen Schlaganfall/eine transitorische ischämische Attacke (TIA), eine dekompensierte Herzinsuffizienz, die eine stationäre Behandlung erforderte, oder eine Herzinsuffizienz der New York Heart Association (NYHA) Klasse III - IV hatten
·Patienten mit einem anamnestisch bekannten AV-Block 2. Grades Mobitz Typ II, einem AV-Block 3. Grades, einer sinusatrialen Blockierung oder Sick-Sinus-Syndrom, wenn sie keinen Herzschrittmacher tragen (s. «Warnhinweise und Vorsichtsmassnahmen»)
·Patienten mit homozygotem Genotyp CYP2C9*3 (CYP2C9*3/*3)
·Während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine zuverlässige Verhütungsmethode anwenden
Warnhinweise und VorsichtsmassnahmenInfektionen
Ein zentraler pharmakodynamischer Effekt von Mayzent ist die dosisabhängige Reduktion der peripheren Lymphozytenzahl bis auf 20 bis 30 % der Ausgangswerte. Dies ist auf das reversible Zurückhalten (Sequestrierung) von Lymphozyten im Lymphgewebe zurückzuführen (s. «Wirkungsmechanismus/Pharmakodynamik»).
Die Wirkung von Mayzent auf das Immunsystem kann das Risiko von Infektionen erhöhen.
Vor Behandlungseinleitung von Mayzent sollte ein aktuelles (d.h. nicht älter als 6 Monate oder nach Absetzen der vorherigen Therapie erstelltes) grosses Blutbild vorliegen. Zusätzlich wird empfohlen, das grosse Blutbild 3 bis 4 Monate nach Beginn der Behandlung und danach mindestens jährlich sowie bei Anzeichen einer Infektion zu kontrollieren. Bei einer bestätigten Gesamtlymphozytenzahl < 0.2 x 109/l sollte die Dosis auf 1 mg reduziert werden, weil in klinischen Studien die Siponimod-Dosis bei Patienten mit einer Gesamtlymphozytenzahl < 0.2 x 109/l reduziert wurde. Bei einer bestätigten Gesamtlymphozytenzahl von < 0.2 x 109/l bei Patienten, die bereits eine Dosis von 1 mg Siponimod erhalten, sollte die Behandlung pausiert werden, bis der Wert 0.6 x 109/l erreicht ist. Dann kann ein erneuter Behandlungsbeginn mit Siponimod in Erwägung gezogen werden.
Bei Patienten mit schweren aktiven Infektionen sollte die Behandlungseinleitung mit Mayzent verschoben werden, bis die Infektion abgeklungen ist. Da verbleibende pharmakodynamische Effekte, wie z.B. die Senkung der peripheren Lymphozytenzahl, bis zu 3 bis 4 Wochen nach Absetzen von Mayzent anhalten können, sollte die Infektionsüberwachung während dieses Zeitraums fortgesetzt werden (s. «Absetzen der Therapie»).
Patienten, die Mayzent erhalten, sollten angewiesen werden, ihrem Arzt Symptome von Infektionen sofort zu melden. Bei Patienten mit Symptomen einer Infektion während der Therapie sind effektive diagnostische und therapeutische Massnahmen anzuwenden. Eine Aussetzung der Behandlung mit Mayzent sollte in Betracht gezogen werden, wenn ein Patient eine schwere Infektion entwickelt.
Im Zusammenhang mit Mayzent wurden Fälle einer Kryptokokkenmeningitis (KM) berichtet. Auch für einen anderen Sphingosin-1-Phospat (S1P)-Rezeptor-Modulator wurden Fälle von KM gemeldet. Ärzte sollten sorgsam auf klinische Symptome oder Anzeichen einer KM achten. Bei Patienten mit derartigen Symptomen und Anzeichen sollte umgehend eine diagnostische Abklärung durchgeführt werden. Die Behandlung mit Mayzent sollte dabei bis zum Ausschluss einer KM ausgesetzt werden. Wenn eine KM diagnostiziert wird, muss umgehend eine geeignete Behandlung eingeleitet werden. Eine spätere Wiederaufnahme der Therapie mit Mayzent ist in diesen Fällen kontraindiziert (s. Rubrik «Kontraindikationen»).
Mit Mayzent wurden Fälle einer Herpesvirusinfektion, einschliesslich durch das Varizella-Zoster-Virus (VZV) verursachte Fälle von Meningitis oder Meningoenzephalitis, berichtet. Patienten ohne eine vom Arzt bestätigte Varizellenanamnese (Windpocken) oder ohne Nachweis einer vollständigen Impfung gegen das Varizella-Zoster-Virus (VZV) sollten auf Antikörper gegen das VZV getestet werden, bevor mit der Einleitung von Mayzent begonnen wird (s. «Impfung»).
Progressive multifokale Leukenzephalopathie
Bei Patienten mit MS, die mit S1P-Rezeptor-Modulatoren behandelt wurden, sind Fälle von progressiver multifokaler Leukenzephalopathie (PML) aufgetreten. PML ist eine opportunistische Virusinfektion des Gehirns, die durch das JC-Virus (JCV) verursacht wird und typischerweise nur bei Patienten mit geschwächtem Immunsystem auftritt und in der Regel zum Tod oder zu schwerer Behinderung führt. PML ist bei mit S1P-Rezeptor-Modulatoren behandelten Patienten aufgetreten, die zuvor nicht mit Natalizumab behandelt worden waren (das bekanntermassen mit PML in Verbindung gebracht wird), die keine anderen immunsuppressiven oder immunmodulierenden Medikamente einnahmen und die keine systemischen Erkrankungen hatten, die zu einer Beeinträchtigung der Funktion des Immunsystems führten. Die meisten Fälle von PML im Zusammenhang mit S1P-Rezeptor-Modulatoren traten bei Patienten auf, die seit mindestens 2 Jahren behandelt wurden. Der Zusammenhang zwischen dem PML-Risiko und der Dauer der Behandlung ist nicht bekannt.
Bei den ersten Anzeichen oder Symptomen, die auf eine PML hindeuten, ist Mayzent abzusetzen und eine angemessene diagnostische Bewertung vorzunehmen. Typische Symptome im Zusammenhang mit PML sind vielfältig, schreiten über Tage bis Wochen voran und umfassen fortschreitende Schwäche auf einer Körperseite oder Schwerfälligkeit der Gliedmassen, Sehstörungen und Veränderungen des Denkens, des Gedächtnisses und der Orientierung, die zu Verwirrtheit und Persönlichkeitsveränderungen führen. MRT-Befunde können vor dem Auftreten klinischer Anzeichen oder Symptome vorliegen. Fälle von PML, die auf der Grundlage von MRT-Befunden und dem Nachweis von JCV-DNA im Liquor in Abwesenheit von klinischen Anzeichen oder spezifischen Symptomen für PML diagnostiziert wurden, wurden bei Patienten berichtet, die mit MS-Arzneimitteln behandelt wurden, die mit einem Risiko für PML assoziiert werden, einschliesslich S1P-Rezeptor-Modulatoren. Bei vielen dieser Patienten traten anschliessend Symptome einer PML auf.
Daher kann die Überwachung mit MRT auf Anzeichen, die auf PML hindeuten, nützlich sein. Im Fall von verdächtigen Befunden sollten weitere Untersuchungen durchgeführt werden, um eine frühzeitige Diagnose der PML zu ermöglichen. Nach dem Absetzen eines anderen MS-Arzneimittels, das mit PML in Verbindung gebracht wird, wurde über eine geringere PML-bedingte Sterblichkeit und Morbidität bei Patienten mit PML berichtet, die zunächst asymptomatisch waren, im Vergleich zu Patienten mit PML, die bei der Diagnose charakteristische klinische Anzeichen und Symptome aufwiesen. Es ist nicht bekannt, ob diese Unterschiede auf die frühzeitige Erkennung und das Absetzen der MS-Behandlung oder auf Unterschiede im Krankheitsverlauf bei diesen Patienten zurückzuführen sind. Bei bestätigter PML sollte die Behandlung mit Mayzent dauerhaft beendet werden.
Entzündliches Immunrekonstitutionssyndrom (engl.: Immune reconstitution inflammatory syndrome, IRIS) wurde bei Patienten berichtet, die mit S1P-Rezeptor-Modulatoren behandelt wurden, bei denen eine PML auftrat und die anschliessend die Behandlung absetzten. IRIS äussert sich in einer möglicherweise schnell eintretenden klinischen Verschlechterung des Zustands des Patienten, kann zu schweren neurologischen Komplikationen oder zum Tod führen und geht häufig mit charakteristischen Veränderungen im MRT einher. Das Auftreten von IRIS bei Patienten mit PML erfolgte meist wenige Monate nach Absetzen des S1P-Rezeptor-Modulators. Es sollte überwacht werden, ob IRIS auftritt, und die damit verbundene Entzündung sollte angemessen behandelt werden.
Impfung
Bei Antikörper-negativen Patienten wird eine vollständige Impfung mit Varizellen-Impfstoff vor Beginn der Behandlung mit Mayzent empfohlen, wonach die Einleitung der Behandlung mit Mayzent um einen Monat verschoben werden sollte, um die volle Wirkung der Impfung zu ermöglichen (s. «Unerwünschte Wirkungen»).
Abgeschwächte Lebendimpfstoffe
Die Anwendung von abgeschwächten Lebendimpfstoffen (z.B. Varizellen-Impfstoff und Gelbfieber-Impfstoff) kann das Risiko einer Infektion mit sich bringen und sollte daher während der Behandlung mit Mayzent und für 4 Wochen nach Beendigung der Behandlung mit Mayzent vermieden werden (s. «Interaktionen»).
Andere Impfstoffarten
Andere Impfstoffarten können weniger wirksam sein, wenn sie während der Behandlung mit Mayzent verabreicht werden. Eine Behandlungsunterbrechung 1 Woche vor bis 4 Wochen nach der geplanten Impfung wird empfohlen. Die Entscheidung, ob die Behandlung mit Mayzent fortgesetzt oder unterbrochen werden soll, sollte auf Grundlage einer Nutzen-Risiko-Bewertung des einzelnen Patienten getroffen werden (s. «Beendigung der Siponimod-Therapie» und «Interaktionen»).
Wenn die Siponimod-Therapie wegen einer Impfung unterbrochen wird, sollte eine mögliche Rückkehr der Krankheitsaktivität in Betracht gezogen werden (s. «Beendigung der Siponimod-Therapie»).
Gleichzeitige Behandlung mit antineoplastischen, immunmodulatorischen oder immunsuppressiven Therapien
Die gleichzeitige Anwendung antineoplastischer, immunmodulatorischer oder immunsuppressiver Therapien (einschliesslich Kortikosteroide) sollte mit Vorsicht erfolgen, da während einer solchen Therapie ein Risiko von additiven Effekten auf das Immunsystem besteht (s. «Interaktionen»).
Makulaödem
Makulaödem (s. «Unerwünschte Wirkungen») mit oder ohne Sehstörungen wurde in der klinischen Phase-3-Studie (A2304) häufiger unter Siponimod (1.8 %) als unter Placebo (0.2 %) gemeldet. Die Mehrzahl der Fälle trat in den ersten 3 bis 4 Monaten der Therapie auf. Aus diesem Grund wird bei allen Patienten vor Behandlungsbeginn sowie 3 bis 4 Monate nach Behandlungsbeginn eine augenärztliche Untersuchung empfohlen. Da auch bei längerfristiger Behandlung Makulaödeme aufgetreten sind, sollten die Patienten während der Therapie mit Mayzent sofort melden, wenn Sehstörungen auftreten. Eine Beurteilung des Fundus, einschliesslich der Makula, wird empfohlen.
Patienten mit einer Vorgeschichte von Diabetes mellitus, Uveitis oder zugrunde liegenden/gleichzeitig bestehenden Netzhauterkrankungen haben ein erhöhtes Risiko, ein Makulaödem zu entwickeln. Es wird empfohlen, dass diese Patienten während der Therapie mit Mayzent eine regelmässige augenärztliche Untersuchung durchführen lassen.
Die Weiterbehandlung mit Mayzent bei Patienten mit Makulaödem wurde nicht untersucht. Es wird empfohlen, Siponimod abzusetzen, wenn der Patient ein Makulaödem entwickelt. Die Entscheidung für oder gegen das Absetzen der Behandlung mit Mayzent muss unter Abwägung des potenziellen Nutzens und der Risiken für den einzelnen Patienten erfolgen.
Bradykardie und Bradyarrhythmie
Herzfrequenz
Wegen des Risikos schwerer Herzrhythmusstörungen oder einer erheblichen Bradykardie sollte Mayzent bei Patienten mit folgenden Erkrankungen nicht angewendet werden:
·Herzstillstand in der Vorgeschichte mehr als 6 Monate vor der Behandlung mit Mayzent,
·zerebrovaskuläre Erkrankung,
·anamnetisch bekannte symptomatische Bradykardie oder wiederkehrende Synkopen,
·unkontrollierte Hypertonie oder
·schwere, unbehandelte Schlafapnoe.
Wenn eine Behandlung in Betracht gezogen wird, sollte vor Beginn der Behandlung der Rat eines Kardiologen eingeholt werden, um die am besten geeignete Überwachungsstrategie zu bestimmen.
Bei diesen Patienten sollte eine Behandlung mit Siponimod nur dann in Betracht gezogen werden, wenn der zu erwartende Nutzen die möglichen Risiken überwiegt (siehe nachfolgende Informationen).
Eine eingehende QT-Studie zeigte keine signifikante direkte Auswirkung auf die Verlängerung des QT-Intervalls und Mayzent geht nicht mit einem arrhythmogenen Potenzial im Zusammenhang mit Verlängerung des QT-Intervalls einher. Die Einleitung einer Behandlung mit Mayzent kann zu einer Verringerung der Herzfrequenz und einer indirekten Verlängerung des QT-Intervalls während der Titrationsphase führen. Mayzent wurde nicht bei Patienten mit einer signifikanten QT-Verlängerung (QTc > 500 msec) oder unter QT-verlängernden Arzneimitteln untersucht. Wenn eine Behandlung mit Mayzent bei Patienten mit bereits bestehender signifikanter QT-Verlängerung oder unter Behandlung mit QT-verlängernden Arzneimitteln mit bekannten arrhythmogenen Eigenschaften in Betracht gezogen wird, sollte vor Einleitung der Behandlung ein Kardiologe hinzugezogen werden, um die am besten geeignete Überwachungsstrategie während der Behandlungseinleitung festzulegen.
Mayzent wurde nicht bei Patienten mit Arrhythmien untersucht, die eine Behandlung mit Antiarrhythmika der Klasse Ia (z.B. Chinidin, Procainamid) oder der Klasse III (z.B. Amiodaron, Sotalol) benötigen. Antiarrhythmika der Klasse Ia und der Klasse III wurden bei Patienten mit Bradykardie mit Fällen von Torsade de pointes in Zusammenhang gebracht. Da die Behandlungseinleitung mit Mayzent zu einer Abnahme der Herzfrequenz führt, sollte Mayzent während der Behandlungseinleitung nicht zusammen mit diesen Arzneimitteln verabreicht werden.
Es gibt nur begrenzte Erfahrung bei Patienten, die gleichzeitig mit herzfrequenzsenkenden Kalziumkanalblockern (wie Verapamil oder Diltiazem) oder anderen Wirkstoffen, die die Herzfrequenz senken können (z.B. Ivabradin oder Digoxin), therapiert wurden. In klinischen Studien mit Mayzent haben Patienten diese Arzneimittel nicht erhalten. Die gleichzeitige Anwendung dieser Wirkstoffe während der Einleitung von Mayzent kann mit schwerer Bradykardie und einem Herzblock verbunden sein. Wegen der möglichen additiven Auswirkung auf die Herzfrequenz sollte die Behandlung mit Mayzent bei Patienten, die gleichzeitig mit diesen Wirkstoffen behandelt werden, in der Regel nicht eingeleitet werden (s. «Interaktionen»).
Wenn bei oben genannten Wirkstoffen eine Begleitbehandlung mit Mayzent dennoch in Betracht gezogen wird, sollte vor der Behandlungseinleitung mit Mayzent ein Kardiologe für die Umstellung auf ein nicht herzfrequenzsenkendes Arzneimittel oder für die Auswahl einer geeigneten Überwachung bei Behandlungseinleitung hinzugezogen werden.
Da die Einleitung der Behandlung mit Mayzent zu einer vorübergehenden Abnahme der Herzfrequenz führt (s. «Unerwünschte Wirkungen»), wird bei Behandlungsbeginn ein Dosistitrationsschema angewendet, um an Tag 6 die Erhaltungsdosis von Mayzent zu erreichen (s. «Dosierung/Anwendung»).
Nach der ersten Titrationsdosis beginnt die Herzfrequenzabnahme innerhalb einer Stunde und der Rückgang an Tag 1 ist nach etwa 3 bis 4 Stunden am höchsten. Bei fortgesetzter Dosistitration werden an den folgenden Tagen weitere Herzfrequenzsenkungen beobachtet, wobei die maximale Abnahme vom Ausgangswert von Tag 1 an Tag 5 bis 6 erreicht wird. Die höchste tägliche Abnahme der absoluten stündlichen mittleren Herzfrequenz nach der Dosierung wird an Tag 1 beobachtet, wobei der Puls im Durchschnitt um 5 bis 6 Schläge pro Minute (bpm) abnimmt. An den Folgetagen sind die Abnahmen nach der Dosierung weniger ausgeprägt. Bei fortgesetzter Dosierung steigt die Herzfrequenz nach Tag 6 an und erreicht innerhalb von 10 Tagen nach Behandlungsbeginn den Placebowert.
Herzfrequenzen unter 40 bpm wurden selten beobachtet. Patienten, bei denen eine Bradykardie auftrat, waren in der Regel asymptomatisch. Einige Patienten hatten leichte bis mittelschwere Symptome wie Schwindel oder Müdigkeit, die innerhalb von 24 Stunden ohne Intervention abklangen (s. «Unerwünschte Wirkung»). Falls nötig kann der durch Siponimod induzierte Abfall der Herzfrequenz durch parenterale Gabe von Atropin oder Isoprenalin rückgängig gemacht werden.
Atrioventrikuläre Überleitungszeit
Die Einleitung der Behandlung mit Mayzent wurde mit vorübergehenden atrioventrikulären Überleitungsverzögerungen in Verbindung gebracht, die einem ähnlichen zeitlichen Muster folgen wie die beobachtete Abnahme der Herzfrequenz während der Dosistitration. Die atrioventrikulären Überleitungsverzögerungen manifestierten sich in den meisten Fällen als atrioventrikulärer (AV) Block 1. Grades (verlängertes PR-Intervall im Elektrokardiogramm). Zum Zeitpunkt der Behandlungseinleitung mit Mayzent wurden bei weniger als 1.7 % der Patienten in klinischen Studien AV-Blöcke 2. Grades, meist Mobitz Typ I (Wenckebach), beobachtet. Die Überleitungsstörungen waren in der Regel vorübergehend, asymptomatisch, innerhalb von 24 Stunden abgeklungen und erforderten kein Absetzen der Behandlung mit Mayzent.
Empfehlung zur Behandlungseinleitung
Als Vorsichtsmassnahme sollten Patienten mit den folgenden Herzerkrankungen für einen Zeitraum von 6 Stunden nach der ersten Dosis von Mayzent auf Anzeichen und Symptome einer Bradykardie überwacht werden:
·Sinusbradykardie (Herzfrequenz (HR) < 55 bpm),
·anamnestisch bekannter atrioventrikulärer Block (AV-Block) 1. oder 2. Grades (Mobitz Typ I),
·anamnestisch bekannter Myokardinfarkt oder anamnestisch bekannte Herzinsuffizienz, wenn nicht kontraindiziert.
Es wird empfohlen, vor der Dosierung und am Ende des Beobachtungszeitraums ein Elektrokardiogramm (EKG) aufzunehmen. Treten nach der Dosierung Bradyarrhythmie- oder überleitungsbedingte Symptome auf oder zeigt das EKG 6 Stunden nach der Dosierung einen erneuten AV-Block 2. Grades oder höher oder ein QTc-Intervall ≥500 msec, sollte eine entsprechende Behandlung eingeleitet und die Überwachung fortgesetzt werden, bis die Symptome/Befunde abgeklungen sind. Wenn eine pharmakologische Behandlung erforderlich ist, sollte die Überwachung über Nacht fortgesetzt werden und die 6-stündige Überwachung sollte nach der zweiten Dosis wiederholt werden.
Bradyarrhythmische Effekte sind ausgeprägter, wenn Mayzent zusätzlich zur Therapie mit Betablockern gegeben wird.
Bei Patienten, die einen Betablocker in stabiler Dosis erhalten, sollte vor der Einleitung der Behandlung mit Mayzent die Ruheherzfrequenz ermittelt werden. Wenn die Ruheherzfrequenz unter laufender Betablocker-Behandlung > 50 bpm beträgt, kann Mayzent eingeleitet werden. Wenn die Ruheherzfrequenz bei ≤50 bpm liegt, sollte die Betablocker-Behandlung unterbrochen werden, bis die Ausgangsherzfrequenz > 50 bpm beträgt. Die Behandlung mit Mayzent kann dann begonnen werden und die Behandlung mit Betablockern kann wieder aufgenommen werden, nachdem Mayzent auf die angestrebte Erhaltungsdosis auftitriert wurde (s. «Interaktionen»).
Ausgelassene Dosis bei Behandlungsbeginn und Wiedereinleitung der Therapie nach Behandlungsunterbrechung
Wird eine Titrationsdosis an einem Tag während der ersten 6 Tage der Behandlung ausgelassen oder werden während der Erhaltungstherapie 4 oder mehr aufeinanderfolgende Tagesdosen ausgelassen, sollten die gleichen anfänglichen Titrations- und Überwachungsempfehlungen gelten (s. «Dosierung/Anwendung»).
Leberfunktion
Vor Beginn der Behandlung mit Mayzent sollten aktuelle (d.h. innerhalb der letzten 6 Monate erhaltene) Transaminase- und Bilirubinwerte vorliegen. In der Studie A2304 wurden bei 5.6 % der mit Mayzent 2 mg behandelten Patienten Alanin-Aminotransferase (ALT)- oder Aspartat-Aminotransferase (AST)-Werte beobachtet, die dreimal über der oberen Normgrenze (ULN) lagen im Vergleich zu 1.5 % der Patienten, die Placebo erhielten (s. «Unerwünschte Wirkungen»). In klinischen Studien wurde Mayzent abgesetzt, wenn die Steigerung das Dreifache überschritt und der Patient Symptome in Verbindung mit der Leberfunktion aufwies, oder wenn der Anstieg eine 5-fache Erhöhung überschritten hatte.
Bei Patienten, die Symptome entwickeln, die auf eine Leberfunktionsstörung hindeuten, wie ungeklärte Übelkeit, Erbrechen, Abdominalschmerz, Ermüdung, Appetitlosigkeit, Ausschlag mit Eosinophilie oder Gelbsucht und/oder dunklem Urin während der Behandlung, sollten die Leberenzyme überprüft und Mayzent abgesetzt werden, wenn sich eine signifikante Leberschädigung bestätigt. Die Wiederaufnahme der Behandlung wird davon abhängen, ob eine andere Ursache der Leberschädigung festgestellt wird und welcher Nutzen für den Patienten von der Wiederaufnahme der Therapie im Verhältnis zu den Risiken eines erneuten Auftretens von Leberfunktionsstörungen erwartet wird.
Obwohl keine Daten vorliegen, die belegen, dass Patienten mit einer bereits bestehenden Lebererkrankung ein erhöhtes Risiko haben, bei der Anwendung von Mayzent erhöhte Leberfunktionswerte (LFT-Werte) zu entwickeln, sollte bei der Anwendung von Mayzent bei Patienten mit einer schweren Lebererkrankung in der Anamnese Vorsicht geübt werden.
Kutane Neoplasien
Basalzellkarzinome (BCC) und andere kutane Neoplasien wie malignes Melanom, Plattenepithelkarzinom, Kaposi-Sarkom und Merkelzellkarzinom wurden bei Patienten berichtet, die mit S1P-Rezeptor-Modulatoren behandelt wurden. In der Studie A2304 war das Basalzellkarzinom (BCC) das häufigste Neoplasma und wurde mit einer ähnlichen Häufigkeit in der Behandlungsgruppe, die Siponimod 2 mg erhielt (1.1 %, 12 Patienten), und in der Placebo-Gruppe (1.3 %, 7 Patienten) berichtet. Die Häufigkeit von Plattenepithelzellkarzinomen (SCC) war in der Studie A2304 bei Mayzent-behandelten Patienten und Placebo-Patienten gleich (0.2 %). In einer Langzeitstudie wurde eine leichte Zunahme der Inzidenz für BCC und SCC bei längerer Einnahme beobachtet.
Bei allen Patienten, insbesondere mit (z.B. Vorgeschichte eines malignen Melanoms), aber auch ohne erhöhtes Risiko für maligne kutane Neoplasien, sollen vor Beginn einer Therapie mit Mayzent und im weiteren Verlauf regelmässige dermatologische Untersuchungen erfolgen. Verdächtige Hautläsionen müssen umgehend abgeklärt werden. Patienten, die mit Siponimod behandelt werden, sollten vor ungeschützter Exposition gegenüber Sonneneinstrahlung gewarnt werden. Patienten unter Mayzent sollten keine gleichzeitige Phototherapie mit UV-B-Strahlung oder PUVA-Photochemotherapie erhalten.
Unerwartete neurologische oder psychiatrische Symptome/Anzeichen
Seltene Fälle des posterioren reversiblen Enzephalopathiesyndroms (PRES) wurden für einen anderen Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulator gemeldet. Solche Ereignisse wurden im Rahmen des Entwicklungsprogramms nicht für Mayzent gemeldet. Sollte ein Patient unter der Behandlung mit Mayzent jedoch unerwartete neurologische oder psychiatrische Symptome/Anzeichen entwickeln (z.B. kognitive Defizite, Verhaltensänderungen, kortikale Sehstörungen oder andere neurologische kortikale Symptome/Anzeichen oder Symptome/Anzeichen, die auf eine Erhöhung des intrakraniellen Drucks hindeuten) oder eine beschleunigte neurologische Verschlechterung auftreten, sollte der Arzt unverzüglich eine vollständige körperliche und neurologische Untersuchung vornehmen und eine Magnetresonanztomographie (MRT) in Betracht ziehen.
Vorherige Behandlung mit immunsuppressiven oder immunmodulierenden Therapien
Bei der Umstellung von anderen krankheitsmodifizierenden Therapien müssen die Halbwertszeit und der Wirkmechanismus der anderen Therapie berücksichtigt werden, um einen additiven Immuneffekt zu vermeiden und gleichzeitig das Risiko einer Krankheitsreaktivierung zu minimieren. Eine Bestimmung der peripheren Lymphozytenzahl (grosses Blutbild) wird vor der Initiierung mit Mayzent empfohlen, um sicherzugehen, dass Immuneffekte der vorherigen Therapie (z.B. Zytopenie) abgeklungen sind.
Auswirkungen auf den Blutdruck
Patienten mit nicht medikamentös eingestellter Hypertonie waren von der Teilnahme an klinischen Studien vor der Zulassung ausgeschlossen. Siponimod ist bei Patienten mit unkontrolliertem Bluthochdruck mit besonderer Vorsicht anzuwenden.
Bluthochdruck wurde in der Studie A2304 bei Patienten mit SPMS unter Siponimod (12.6 %) häufiger berichtet als unter Placebo (9.0 %). Die Behandlung mit Siponimod führte zu einem Anstieg des systolischen und diastolischen Blutdrucks; die Blutdruckwerte stiegen frühzeitig nach Behandlungseinleitung an und erreichten nach etwa 6 Monaten der Behandlung ein Maximum (systolisch 3 mmHg, diastolisch 1.2 mmHg), um anschliessend stabil zu bleiben. Der Effekt blieb bei fortgesetzter Behandlung bestehen.
Der Blutdruck sollte während der Behandlung mit Siponimod regelmässig überwacht und eine arterielle Hypertonie behandelt werden.
CYP2C9 Genotyp
Vor Beginn der Behandlung mit Mayzent muss der CYP2C9-Genotyp der Patienten bestimmt werden, um deren CYP2C9-Metabolisierungsstatus zu bestimmen (s. auch «Kontraindikationen», «Dosierung/Anwendung» und «Pharmakokinetik»). Patienten, die homozygote Träger des CYP2C9*3-Genotyps (CYP2C9*3/*3) (ca. 0.3 bis 0.4 % der kaukasischen Bevölkerung, seltener in anderen Ethnien) sind, dürfen nicht mit Mayzent behandelt werden, da die Anwendung von Mayzent bei diesen Patienten zu deutlich erhöhten Siponimod-Plasmaspiegeln führt (s. auch «Pharmakokinetik» und «Kontraindikationen»).
Die empfohlene Erhaltungsdosis für Träger der Genotypen CYP2C9*2/*3 (1.4 – 1.7 % der Bevölkerung) und CYP2C9*1/*3 (9 – 12 % der Bevölkerung) beträgt 1 mg Mayzent einmal täglich zur Vermeidung einer erhöhten Exposition gegenüber Siponimod (s. auch «Dosierung/Anwendung» und «Pharmakokinetik»).
Die Auswirkungen anderer Varianten als *2 und *3 auf die Pharmakokinetik von Siponimod wurden bislang nicht untersucht. Obwohl keine Untersuchungen zum Einfluss der selteneren Allele CYP2C9 *5, *6, *8 und *11 auf den Siponimod-Metabolismus durchgeführt wurden, können aufgrund der Verringerung oder eines Verlustes der Enzymaktivität bei Trägern dieser CYP2C9-Polymorphismen erhöhte Siponimod-Spiegel nicht ausgeschlossen werden (siehe auch «Interaktionen» und «Pharmakokinetik»). Die Gesamthäufigkeit der vier Allele *5, *6, *8 und *11 beträgt 10 % unter Afrikanern/Afrika-Stämmigen, 2 % unter Hispanics und < 0.4 % unter Kaukasiern und Asiaten. Aufgrund der ungenügenden Datenlage können für diese Genotypen aber bislang keine Empfehlungen hinsichtlich einer Dosisanpassung gemacht werden.
Bei heterozygoten Anlageträgern der CYP2C9-Allele *2 und *3, die sich durch einen reduzierten CYP2C9-Metabolismus auszeichnen, wurden in klinischen Studien unter einer nicht angepassten Tagesdosis von 2 mg Siponimod bislang keine spezifischen und signifikanten klinischen Symptome einer akuten Toxizität beobachtet. Nach Langzeitexposition kam es zu einem geringen Anstieg der Häufigkeit von Makulaödemen (siehe auch «Interaktionen» und «Pharmakokinetik»). Es ist jedoch unklar, ob diese Unterschiede mit einer erhöhten Siponimod-Exposition zusammenhängen.
Frauen im gebärfähigen Alter
Aufgrund des Risikos für den Fötus ist Siponimod während der Schwangerschaft und bei Frauen im gebärfähigen Alter, die keine zuverlässige Verhütungsmethode anwenden, kontraindiziert. Vor Beginn der Behandlung müssen Frauen im gebärfähigen Alter über das Risiko für den Fötus informiert werden, einen negativen Schwangerschaftstest vorweisen und während der Behandlung und für mindestens 10 Tage nach Absetzen der Behandlung eine zuverlässige Verhütungsmethode anwenden (s. «Kontraindikationen», «Schwangerschaft, Stillzeit»).
Beendigung der Siponimod-Therapie
Bei einem anderen S1P-Rezeptor-Modulator wurde nach dem Absetzen in seltenen Fällen über eine schwere Krankheitsverschlimmerung, einschliesslich Rückkehr von Krankheitsaktivität, berichtet. Die Möglichkeit einer schweren Verschlimmerung der Erkrankung nach Beendigung der Siponimod-Behandlung sollte berücksichtigt werden. Nach Absetzen von Siponimod sollten Patienten auf massgebliche Anzeichen einer möglichen schweren Verschlimmerung oder Rückkehr einer hohen Krankheitsaktivität überwacht werden. Bei Bedarf ist eine geeignete Behandlung einzuleiten. Mayzent bleibt nach dem Absetzen der Behandlung noch bis zu 10 Tage lang im Blut nachweisbar. Die Einleitung anderer Therapien in diesem Zeitraum führt zu einer gleichzeitigen Exposition gegenüber Siponimod.
Überwachen Sie PML-Patienten nach dem Absetzen von Mayzent auf das Auftreten eines entzündlichen Immunrekonstitutionssyndroms (PML-IRIS) (siehe Warnhinweis «Progressive multifokale Leukenzephalopathie»).
Bei der überwiegenden Mehrheit (90 %) der SPMS-Patienten kehren die Lymphozytenzahlen innerhalb von 10 Tagen nach Absetzen der Therapie in den Normalbereich zurück. Verbleibende pharmakodynamische Effekte, wie z.B. die Senkung der peripheren Lymphozytenzahl, können bis zu 3 bis 4 Wochen nach der letzten Dosis anhalten. Die Anwendung von Immunsuppressiva innerhalb dieses Zeitraums kann zu einer additiven Wirkung auf das Immunsystem führen und Vorsicht ist daher 3 bis 4 Wochen nach der letzten Dosis angebracht.
Beeinträchtigung hämatologischer Untersuchungen
Da Siponimod die Lymphozytenzahl im Blut über die Umverteilung in sekundäre Lymphorgane reduziert, kann bei Patienten unter Behandlung mit Siponimod die Lymphozytenzahl im peripheren Blut nicht zur Statusbeurteilung der Lymphozyten-Untergruppen herangezogen werden. Da die Anzahl der zirkulierenden Lymphozyten reduziert ist, erfordern Laboruntersuchungen der zirkulierenden mononukleären Zellen grössere Blutmengen.
Sonstige Bestandteile
Die Tabletten enthalten Phospholipide aus Sojabohnen. Patienten, die überempfindlich gegen Erdnüsse oder Soja sind, dürfen dieses Arzneimittel nicht einnehmen (s. «Kontraindikationen»).
Die Tabletten enthalten Lactose. Patienten mit einer seltenen hereditären Galactose-Intoleranz, völligem Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
InteraktionenPharmakokinetische Interaktionen
Potential anderer Arzneimittel die Pharmakokinetik von Siponimod zu beeinflussen
Siponimod wird vorwiegend durch Cytochrom P450 2C9 (CYP2C9) (79.3 %) und in geringerem Masse durch Cytochrom P450 3A4 (CYP3A4) (18.5 %) metabolisiert. CYP2C9 ist ein polymorphes Enzym und der Genotyp hat Einfluss darauf, in welchem Ausmass die beiden oxidativen Stoffwechselwege jeweils zur Gesamtelimination beitragen. PBPK-Modelle deuten auf eine differenzierte, vom CYP2C9-Genotyp abhängige Inhibition und Induktion der CYP3A4-Signalwege hin. Damit hängen die Arzneimittelwirkungen («drug-drug interactions», DDI) in Gegenwart von Substanzen, die CYP3A oder CYP2C9 beeinflussen können, voraussichtlich vom CYP2C9-Genotyp ab (s. «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Die Genotypen CYP2C9 *5, *6, *8 und *11 gehen ebenfalls mit einem teilweisen bis vollständigen Verlust der CYP2C9-Enzymaktivität einher. Pharmakokinetische Untersuchungen zu diesen Polymorphismen wurden bislang nicht durchgeführt. Allerdings wurden bei Trägern dieser Genotypen erhöhte Spiegel anderer CYP2C9-Substrate, wie Phenytoin oder Warfarin, mit der Notwendigkeit einer Dosisanpassung dieser Substrate beobachtet (siehe auch «Warnhinweise und Vorsichtsmassnahmen» sowie «Pharmakokinetik»).
CYP2C9- und CYP3A4-Inhibitoren
Aufgrund einer signifikanten Zunahme der Siponimod-Exposition wird die gleichzeitige Anwendung von Siponimod und Arzneimitteln, die eine mässige CYP2C9- und eine mässige oder starke CYP3A4-Inhibition verursachen, nicht empfohlen. Diese Begleitmedikation kann dabei aus einem mässigen CYP2C9/CYP3A4 dualen Inhibitor (z.B. Fluconazol) oder einem mässigen CYP2C9-Inhibitor in Kombination mit einem separaten mässigen oder starken CYP3A4-Inhibitor bestehen.
Die gleichzeitige Anwendung von Fluconazol (dualer moderater CYP2C9/CYP3A4-Inhibitor) 200 mg täglich bei Steady-State und eine Einzeldosis von Siponimod 4 mg bei gesunden Probanden mit einem CYP2C9*1/*1-Genotyp führte zu einer Verdoppelung der Fläche unter der Kurve (AUC) von Siponimod. Gemäss der Bewertung des Wirkstoffwechselwirkungspotenzials von Medikamenten durch physiologie-basierte pharmakokinetische (PBPK)-Modelle wird für CYP2C9*1/*1, *1/*2, *1/*3 und *2/*3-Genotypen mit jeder Art von CYP3A4- und CYP2C9-Inhibitoren, ein maximal 2-facher Anstieg der AUC von Siponimod erwartet. Bei CYP2C9*2/*2-Patienten wird in Gegenwart von moderaten CYP2C9/CYP3A4-Inhibitoren ein 2.7-facher Anstieg der AUC von Siponimod erwartet. Daten zu Interaktionen mit CYP2C9- und CYP3A4-Inhibitoren stehen bislang für andere CYP2C9-Genotypen mit reduzierter oder fehlender CYP2C9-Aktivität nicht zur Verfügung.
CYP2C9- und CYP3A4-Induktoren
Aufgrund einer klinisch relevanten Verringerung der Siponimod-Exposition ist bei einer gleichzeitigen Anwendung von Mayzent mit Arzneimitteln, die eine mässige CYP2C9- und starke CYP3A4-Induktion verursachen, Vorsicht geboten. Diese Begleitmedikation kann dabei aus einem mässigen CYP2C9/starken CYP3A4 dualen Induktor (wie z.B. Rifampicin oder Carbamazepin) oder einem mässigen CYP2C9-Induktor in Kombination mit einem separaten starken CYP3A4-Induktor bestehen.
Vorsicht ist auch geboten bei gleichzeitiger Anwendung von Mayzent mit mässigen CYP3A4-Induktoren (z.B. Modafinil) oder starken CYP3A4-Induktoren bei Patienten mit CYP2C9*1/*3 oder *2/*3-Genotyp, bei denen eine Dosisanpassung empfohlen wird (s. «Dosierungsempfehlungen»). Daten zu Interaktionen mit CYP2C9- und CYP3A4-Induktoren stehen bislang für andere CYP2C9-Genotypen mit reduzierter oder fehlender CYP2C9-Aktivität nicht zur Verfügung.
Gemäss klinischen Studien zu Arzneimittelwirkungen und In-silico-Tests (Physiologie-basierte Pharmakokinetik) wird erwartet, dass starke CYP3A4-/mässige CYP2C9-Induktoren (z.B. Carbamazepin) und mässige CYP3A4-Induktoren (z.B. Modafinil) die Siponimod-Exposition um bis zu 76 % bzw. bis zu 51 % reduzieren.
Die gleichzeitige Anwendung von Siponimod 2 mg täglich und Rifampicin 600 mg täglich (starker CYP3A4- und mässiger CYP2C9-Induktor) verringerte die AUCtau,ss und Cmax,ss von Siponimod um 57 % bzw. 45 % bei Patienten mit CYP2C9*1/*1-Genotyp.
Siponimod ist kein Substrat der Effluxtransporter P-gp, BCRP oder MRP. Es wird deshalb davon ausgegangen, dass Arzneimittel, die die Aktivität dieser Transporter beeinflussen, keine Wirkung auf die Pharmakokinetik von Siponimod haben.
Die Aufnahme von Siponimod in Hepatozyten erfolgt ausschliesslich durch passive Diffusion. Aus diesem Grund sind Interaktionen von Siponimod mit hepatischen Aufnahmetransportern (OATPs, OCTs, OATs) nicht zu erwarten.
Potential von Siponimod, die Pharmakokinetik anderer Arzneimittel zu beeinflussen
In vitro Studien ergaben, dass Siponimod und seine Metaboliten (M17 und M3) in therapeutisch relevanten Konzentrationen die Aktivität von CYP-Enzymen kaum bzw. gar nicht hemmen (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5) oder aktivieren können (CYP1A2, CYP2B6, CYP2C9 und CYP3A4/5).
Basierend auf in vitro Daten wird keine Hemmwirkung von Siponimod und seinen Metaboliten (M17 und M3) auf die Aufnahme gleichzeitig angewendeter Arzneimittel und/oder biologischer Wirkstoffe erwartet, die von OATP1B1, OATP1B3, OAT1, OAT3, OCT1 oder OCT2 transportiert werden. Es wird davon ausgegangen, dass bei therapeutischen Konzentrationen auch keine Hemmung des Efflux gleichzeitig angewendeter Arzneimittel und/oder biologischer Wirkstoffe erfolgt, die von BCRP, BSEP, MATE1, MATE2K oder über P-gp transportiert werden.
Pharmakodynamische Interaktionen
Antineoplastische, immunmodulatorische oder immunsuppressive Therapien
Mayzent wurde nicht in Kombination mit antineoplastischen, immunmodulatorischen oder immunsuppressiven Therapien untersucht. Bei der gleichzeitigen Verabreichung ist Vorsicht geboten, da das Risiko additiver Immuneffekte während einer solchen Therapie und in den Wochen nach Beendigung der Verabreichung eines dieser Arzneimittel besteht (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei der Umstellung von krankheitsmodifizierenden Therapien müssen die Halbwertszeit und der Wirkmechanismus der anderen Therapie berücksichtigt werden, um einen additiven Immuneffekt zu vermeiden und gleichzeitig das Risiko einer Krankheitsreaktivierung zu minimieren.
Aufgrund der in der Produktinformation beschriebenen Eigenschaften und Dauer der immunsuppressiven Effekte von Alemtuzumab wird die Einleitung einer Behandlung mit Mayzent nach Alemtuzumab nicht empfohlen, es sei denn, der Nutzen der Behandlung mit Mayzent überwiegt deutlich die Risiken für den einzelnen Patienten.
Mit der Einnahme von Mayzent kann sofort nach Absetzen von Beta-Interferon oder Glatirameracetat begonnen werden.
Antiarrhythmika, QT-verlängernde Medikamente und Medikamente, die die Herzfrequenz senken können
Während der Behandlungseinleitung sollte Mayzent wegen möglicher additiver Auswirkungen auf die Herzfrequenz nicht gleichzeitig bei Patienten angewendet werden, die Antiarrhythmika der Klasse Ia (z.B. Chinidin, Procainamid), der Klasse III (z.B. Amiodaron, Sotalol), QT-verlängernde Medikamente mit bekannten arrhythmogenen Eigenschaften, herzfrequenzsenkende Kalziumkanalblocker (wie Verapamil oder Diltiazem) oder andere Wirkstoffe, die die Herzfrequenz senken können (z.B. Ivabradin oder Digoxin) erhalten. Wenn eine Behandlung mit Mayzent in Betracht gezogen wird, sollte vor Beginn der Therapie der Rat eines Kardiologen eingeholt werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Betablocker
Wenn Siponimod bei Patienten eingeleitet wird, die Betablocker erhalten, ist aufgrund der additiven Auswirkungen auf die Senkung der Herzfrequenz Vorsicht geboten (s. «Warnhinweise und Vorsichtsmassnahmen»). Die Behandlung mit Betablockern kann bei Patienten eingeleitet werden, die Mayzent in stabiler Dosis erhalten.
Der negative chronotrope Effekt bei gleichzeitiger Verabreichung von Siponimod und Propranolol wurde in einer speziellen Studie zur Pharmakodynamik und Sicherheit untersucht. Wurde Propranolol zusätzlich zu Siponimod im pharmakokinetischen Steady State gegeben, waren die negativ chronotropen Wirkungen weniger ausgeprägt (geringer als additiv) als bei Gabe von Siponimod zusätzlich zu Propranolol im pharmakokinetischen Steady State (additive Wirkung auf die Herzfrequenz).
Impfung
Abgeschwächte Lebendimpfstoffe
Die Anwendung von abgeschwächten Lebendimpfstoffen (z.B. Varizellen-Impfstoff und Gelbfieber-Impfstoff) kann mit einem Infektionsrisiko verbunden sein und sollte daher während der Behandlung mit Mayzent und für 4 Wochen nach Beendigung der Behandlung mit Mayzent vermieden werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Andere Impfstoffarten
Während und bis zu 4 Wochen nach der Behandlung mit Mayzent kann die Wirksamkeit von Impfungen beeinträchtigt sein. Es wird nicht davon ausgegangen, dass die Wirksamkeit einer Impfung beeinträchtigt wird, wenn die Siponimod-Behandlung 1 Woche vor und bis 4 Wochen nach der Impfung unterbrochen wird (siehe «Wiederaufnahme der Erhaltungstherapie nach Behandlungsunterbrechung»). In einer speziellen Phase-I-Studie mit gesunden Freiwilligen zeigten sich nach maximal 10-tägiger Vorbehandlung mit Siponimod bei Fortführung der Gabe oder während einer kürzeren Behandlungspause von 10 Tagen vor bis 14 Tage nach der Impfung etwa 15 bis 30 % niedrige Ansprechraten auf einen quadrivalenten Influenza-Impfstoff im Vergleich zu Placebo, während sich die Ansprechraten einer PPV-23-Impfung durch die gleichzeitige Behandlung mit Siponimod nicht signifikant gegenüber Placebo veränderten (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Orale Kontrazeptiva
Die Wirksamkeit der untersuchten oralen Kontrazeptiva (Kombination aus Ethinylestradiol und Levonorgestrel) wurde unter Behandlung mit Siponimod aufrechterhalten. Siponimod zeigte keinerlei Einfluss auf die Pharmakodynamik der Kontrazeptiva (Estradiol, Progesteron; FSH, LH, Follikelgrösse, Hoogland score, SHBG). Im Vergleich zur alleinigen Gabe von oralen Kontrazeptiva erhöhte die gleichzeitige Gabe von Siponimod für Levonorgestrel die Fläche unter der Kurve während der Dosierung (AUCtau) um das 1.29-fache (geometric mean ratio (GMR): 1.29, 90 % CI: 1.24-1.34) und die maximale Plasmakonzentration im Gleichgewicht (Cmax.ss) um das 1.18-fache (GMR: 1.18, 90 % CI: 1.11-1.26). Siponimod beeinflusst die Pharmakokinetik von Ethinylestradiol nicht (AUCtau GMR: 1.00, 90 % CI: 0.96-1.05; Cmax,ss GMR: 1.02, 90 %: 0.96-1.08).
Es wurden keine Wechselwirkungsstudien mit oralen Kontrazeptiva, die andere Gestagene enthalten, durchgeführt.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Kontrazeption bei Frauen
Bei Frauen im gebärfähigen Alter, die keine zuverlässige Verhütungsmethode anwenden, ist Mayzent kontraindiziert (s. «Kontraindikationen»).
Frauen im gebärfähigen Alter müssen darauf hingewiesen werden, dass in tierexperimentellen Studien gezeigt wurde, dass Siponimod für den sich entwickelnden Fetus schädlich ist (s. «Präklinische Daten»). Bei Patientinnen im gebärfähigen Alter muss ein negativer Schwangerschaftstest vor Beginn der Siponimod-Behandlung vorliegen. Patientinnen sollten bei der Anwendung von Mayzent und für mindestens zehn Tage nach Beendigung der Behandlung mit Mayzent eine wirksame Verhütung anwenden (Methoden, die zu einer Schwangerschaftsrate von weniger als 1 % führen) (s. «Warnhinweise und Vorsichtsmassnahmen»).
Wird die Siponimod-Therapie zum Zweck der Schwangerschaftsplanung abgesetzt, sollte eine mögliche Rückkehr der Krankheitsaktivität in Betracht gezogen werden.
Schwangerschaft
Es liegen keine Daten über die Anwendung von Mayzent bei schwangeren Frauen vor, um ein arzneimittelbedingtes Risiko unerwünschter Entwicklungsergebnisse aufzuzeigen.
Tierexperimentelle Studien haben eine durch Siponimod hervorgerufene Embryo- und Fetotoxizität bei Ratten und Kaninchen sowie teratogene Wirkungen bei Ratten einschliesslich embryofetaler Todesfälle und skelettaler oder viszeraler Fehlbildungen gezeigt, bei einer Exposition, die mit der Exposition des Menschen bei einer Tagesdosis von 2 mg vergleichbar ist (s. «Präklinische Daten»). Darüber hinaus zeigten die klinischen Erfahrungen mit einem anderen S1P-Rezeptor-Modulator bei Anwendung während der Schwangerschaft ein 2-fach höheres Risiko für schwere angeborene Fehlbildungen im Vergleich zu der in der Allgemeinbevölkerung beobachteten Rate.
Demzufolge ist Siponimod während der Schwangerschaft kontraindiziert (s. «Kontraindikationen»). Siponimod sollte mindestens 10 Tage vor der Planung einer Schwangerschaft abgesetzt werden (s. «Warnhinweise und Vorsichtsmassnahmen»). Wenn eine Frau während der Behandlung schwanger wird, muss Siponimod abgesetzt werden. Es sollte eine medizinische Beratung über das Risiko von schädlichen Auswirkungen auf den Fötus als Folge der Behandlung stattfinden und es sollten Ultraschalluntersuchungen durchgeführt werden.
Stillzeit
Es ist nicht bekannt, ob Siponimod oder seine Hauptmetaboliten beim Menschen in die Muttermilch übergehen. Bei Ratten wurden Siponimod und seine Metaboliten in die Milch ausgeschieden. Siponimod sollte während der Stillzeit nicht angewendet werden.
Fertilität
Die Wirkung von Siponimod auf die Fertilität beim Menschen wurde nicht untersucht. Siponimod wirkte sich nicht auf die männlichen Fortpflanzungsorgane von Ratten und Affen oder auf die Fertilitätsparameter bei Ratten aus.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenMayzent hat keinen, oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen.
Zu Beginn der Therapie mit Siponimod können jedoch Schwindel und Bradyarrhythmien auftreten. Daher sollten Patienten während des ersten Behandlungstags mit Siponimod weder ein Fahrzeug führen noch Maschinen bedienen (s. «Warnhinweise und Vorsichtsmassnahmen»).
Unerwünschte WirkungenIn der klinischen Phase 3 Studie A2304 wurden 1'651 SPMS-Patienten im Verhältnis 2:1 für den Erhalt von Mayzent 2 mg einmal täglich oder Placebo randomisiert. Die mediane Behandlungsdauer betrug 18 Monate (Bereich 0 bis 37 Monate). Zum Zeitpunkt der Zulassung sind Langzeitsicherheitsdaten sehr limitiert. Die häufigsten unerwünschten Wirkungen unter Siponimod 2 mg sind Kopfschmerzen (15.2 %) und Hypertonie (12.6 %).
Unerwünschte Wirkungen aus klinischen Studien wurden hauptsächlich auf der Grundlage der Erfahrungen aus der pivotalen Studie A2304 (Tabelle 2) definiert und sind nach Systemorganklassen gemäss MedDRA geordnet.
Für jede Systemorganklasse werden die unerwünschten Arzneimittelwirkungen nach Häufigkeit aufgelistet, wobei die häufigsten unerwünschten Arzneimittelwirkungen zuerst genannt werden. Zusätzlich basiert die entsprechende Häufigkeitskategorie für jede unerwünschte Arzneimittelwirkung auf den folgenden Häufigkeitsdefinitionen gemäss Konvention (CIOMS III): sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10); gelegentlich (≥1/1'000 bis < 1/100); selten (≥1/10'000 bis < 1/1'000); sehr selten (< 1/10'000), nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Tabelle 2: Tabellarische Auflistung der Nebenwirkungen
|
Infektionen und parasitäre Erkrankungen
| |
Häufig
|
Herpes Zoster
| |
Gelegentlich
|
Kryptokokkenmeningitis*#
| |
Selten
|
Progressive multifokale Leukenzephalopathie*#
| |
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen)
| |
Häufig
|
Melanozytärer Nävus#
Basalzellkarzinom*#
| |
Gelegentlich
|
Plattenepithelzellkarzinom*#
Malignes Melanom
| |
Erkrankungen des Blutes und des Lymphsystems
| |
Häufig
|
Lymphopenie
| |
Erkrankungen des Nervensystems
| |
Sehr häufig
|
Kopfschmerzen
| |
Häufig
|
Schwindel
Krampfanfälle
Tremor
| |
Augenerkrankungen
| |
Häufig
|
Makulaödem
| |
Herzerkrankungen
| |
Häufig
|
Bradykardie
Atrioventrikulärer Block (1. und 2. Grades)
| |
Gefässerkrankungen
| |
Sehr häufig
|
Hypertonie#
| |
Erkrankungen des Gastrointestinaltrakts
| |
Häufig
|
Übelkeit, Diarrhö
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
| |
Häufig
|
Schmerzen in den Extremitäten
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
| |
Häufig
|
Peripheres Ödem
Asthenie
| |
Untersuchungen
| |
Sehr häufig
|
Erhöhte Werte bei Leberfunktionstests
| |
Häufig
|
Verminderte Werte bei Lungenfunktionstests
|
# siehe auch «Warnhinweise und Vorsichtsmassnahmen»
* Unerwünschte Arzneimittelwirkungen aus der offenen Erweiterung der Phase-3-Studie A2304
Beschreibung ausgewählter unerwünschter Arzneimittelwirkungen
Infektionen
In der Studie A2304 war die Gesamtinfektionsrate bei Patienten mit sekundär progredienter Multipler Sklerose (SPMS) zwischen den Patienten, die Siponimod erhielten und denen, die Placebo erhielten, vergleichbar (49.0 % vs. 49.1 %). Allerdings wurde ein Anstieg der Herpes-Zoster-Infektionen unter Siponimod (2.5 %) im Vergleich zu Placebo (0.7 %) berichtet. Bei chronischer Exposition wurde kein weiterer Anstieg der Inzidenzrate (IR) für Varizella-Zoster-Infektionen beobachtet. Unter einer Therapie mit Mayzent wurden auch Fälle einer durch das Varizella-Zoster-Virus verursachten Meningitis oder Meningoenzephalitis, berichtet (s. «Warnhinweise und Vorsichtsmassnahmen»).
Unter Mayzent wurden Fälle von progressiver multifokaler Leukenzephalopathie (PML) und Kryptokokkenmeningitis berichtet (s. «Warnhinweise und Vorsichtsmassnahmen» und «Kontraindikationen»).
Makulaödem
Über ein Makulaödem wurde häufiger bei Patienten unter Siponimod (1.8 %) als unter Placebo (0.2 %) berichtet. Obwohl die meisten Fälle innerhalb von 3 bis 4 Monaten nach der Einleitung von Siponimod auftraten, wurden Fälle auch bei Patienten gemeldet, die mehr als 6 bis 12 Monate lang mit Siponimod behandelt wurden (s. «Warnhinweise und Vorsichtsmassnahmen»). Einige Patienten stellten sich mit verschwommenem Sehen oder einer Abnahme der Sehschärfe vor, andere hingegen waren asymptomatisch und wurden bei einer routinemässigen augenärztlichen Untersuchung diagnostiziert. Nach Absetzen des Medikaments trat im Allgemeinen eine Besserung oder spontane Rückbildung des Makulaödems ein. Das Rezidivrisiko bei erneuter Exposition wurde nicht untersucht.
Bradykardie
Die Einleitung der Behandlung mit Siponimod führt zu einer vorübergehenden Abnahme der Herzfrequenz und kann zudem mit einer Verzögerung der atrioventrikulären Überleitung assoziiert sein (s. «Warnhinweise und Vorsichtsmassnahmen»). Bei 6.2 % der mit Siponimod behandelten Patienten wurde Bradykardie beobachtet, verglichen mit 3.1 % bei Placebo, und bei 1.7 % der mit Siponimod behandelten Patienten wurde AV-Block beobachtet, verglichen mit 0.7 % bei Placebo.
Der maximale Rückgang der Herzfrequenz zeigt sich in den ersten 6 Stunden nach Dosiseinnahme.
Eine vorübergehende, dosisabhängige Abnahme der Herzfrequenz wurde während der Anfangsdosierungsphase beobachtet und pendelte sich bei Dosierungen ≥5 mg ein. Bradyarrhythmische Ereignisse (AV-Blöcke und Sinuspausen) wurden mit einer höheren Inzidenz unter Behandlung mit Siponimod im Vergleich zu Placebo nachgewiesen.
Die meisten AV-Blöcke und Sinuspausen traten oberhalb der therapeutischen Dosis von 2 mg auf. Die Inzidenz war deutlich höher, wenn keine Dosistitration durchgeführt wurde.
Der durch Siponimod hervorgerufene Herzfrequenzabfall kann durch Atropin oder Isoprenalin umgekehrt werden.
Leberfunktionstests
Erhöhte Leberenzyme (meist ALT-Erhöhung) wurden bei MS-Patienten berichtet, die mit Siponimod behandelt wurden. In der Studie A2304 bei Patienten mit SPMS wurde bei Patienten unter Siponimod (11.3 %) ein Anstieg der Werte bei Leberfunktionstests häufiger beobachtet als bei Patienten unter Placebo (3.1 %), hauptsächlich aufgrund von Erhöhungen der Lebertransaminasen (ALT/AST/GGT). Die Mehrzahl der Erhöhungen erfolgte innerhalb von 6 Monaten nach Behandlungsbeginn. Die ALT-Werte normalisierten sich innerhalb von ca. 1 Monat nach Absetzen von Siponimod (s. «Warnhinweise und Vorsichtsmassnahmen»).
Blutdruck
In der klinischen Phase-3-Studie bei Patienten mit SPMS wurde Hypertonie häufiger bei Patienten unter Siponimod (12.6 %) als unter Placebo (9.0 %) gemeldet (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Krampfanfälle
In der Studie A2304 bei Patienten mit SPMS wurden Krampfanfälle häufiger bei Patienten unter Siponimod (1.7 %) als unter Placebo (0.4 %) gemeldet. Es ist nicht bekannt, ob diese Ereignisse mit den Auswirkungen der MS, mit Siponimod oder mit einer Kombination aus beiden zusammenhingen.
Auswirkungen auf die Atemwege
Bei der Behandlung mit Siponimod wurden geringfügige Verringerungen der Werte des forcierten Exspirationsvolumens in 1 Sekunde (FEV1) und der Diffusionskapazität der Lunge für Kohlenmonoxid (DLCO) beobachtet. Nach Monat 3 und Monat 6 der Behandlung in der Studie A2304 bei Patienten mit SPMS betrug die durchschnittliche Veränderung gegenüber dem Ausgangswert in der Siponimod-Gruppe -0.1 Liter (l) am jeweiligen Zeitpunkt, ohne Veränderung in der Placebogruppe. Diese Beobachtungen waren bei Patienten mit Atemwegserkrankungen wie chronisch obstruktiver Lungenerkrankung (COPD) oder Asthma, die mit Siponimod behandelt wurden, geringfügig höher (durchschnittliche Veränderung gegenüber dem Ausgangswert von FEV1 ca. 0.15 l). Bei der Langzeitbehandlung führte diese Reduktion nicht zu klinisch signifikanten unerwünschten Ereignissen und war nicht mit einer Zunahme von Meldungen über Husten oder Dyspnoe verbunden.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungGesunde Teilnehmer erhielten Siponimod als Einzeldosis (0.1 bis 75 mg) oder als Mehrfachdosis (0.25 bis 20 mg). Die maximal verträgliche Einzeldosis wurde basierend auf dem Auftreten einer symptomatischen Bradykardie nach einer Einzeldosis von 75 mg auf 25 mg festgelegt. Die höchste untersuchte Mehrfachdosis von 20 mg über 28 Tage war gut verträglich (9 Teilnehmer erhielten am letzten Tag der Dosierung 100 mg und 5 Teilnehmer erhielten versehentlich bis zu 200 mg täglich für einen Zeitraum von 3 bis 4 Tagen). Einige der 9 Teilnehmer wiesen asymptomatische leichte bis mittelschwere Erhöhungen der Leberfunktionstests auf.
Ein Patient (mit einer Vorgeschichte von Depressionen) nahm 84 mg Siponimod. Abgesehen von einer leichten Erhöhung der Lebertransaminasen traten bei dem Patienten durch die Überdosierung keine weiteren unerwünschten Ereignisse auf.
Wenn die Überdosierung eine erste Exposition gegenüber Mayzent darstellt oder während der Dosis-Titrationsphase von Mayzent auftritt, ist es wichtig, auf Anzeichen und Symptome einer Bradykardie zu achten, die eine Überwachung über Nacht beinhalten können. Regelmässige Messungen der Pulsfrequenz und des Blutdrucks sind erforderlich und es sollten Elektrokardiogramme durchgeführt werden (s. «Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Es gibt kein spezifisches Antidot zu Siponimod. Weder Dialyse noch Plasmaaustausch würden zu einer sinnvollen Ausscheidung von Siponimod aus dem Körper führen.
Eigenschaften/WirkungenATC-Code
L04AE03
Pharmakotherapeutische Gruppe: Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulatoren
Wirkungsmechanismus
Siponimod ist ein Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulator. Siponimod bindet selektiv an zwei von fünf G-Protein-gekoppelten Rezeptoren (GPCRs) für S1P: S1P1 und S1P5. Siponimod wirkt als funktioneller Antagonist auf den S1P1-Rezeptor der Lymphozyten und verhindert so den Austritt der Lymphozyten aus den Lymphknoten. Dies vermindert die Rezirkulation von T-Zellen in das Zentralnervensystem (ZNS), und begrenzt so die Entzündung im ZNS. Siponimod passiert die Blut-Hirn-Schranke. Siponimod hat keinen nachhaltigen Einfluss auf die effektorischen T-Gedächtniszellen im peripheren Gewebe sowie im Blut und beeinträchtigt die Lymphozytenaktivierung nicht.
Pharmakodynamik
In tierexperimentellen Studien wurden direkte Effekte für Siponimod auf Nervenzellen, über S1P1 auf Astrozyten und S1P5 auf Oligodendrozyten nachgewiesen. In einem Mausmodell der experimentellen autoimmunen Enzephalomyelitis wurde eine direkte neuroprotektive Wirkung, unabhängig von Wirkungen auf Lymphozyten, auch für zentral angewendetes Siponimod (über intrazerebroventrikuläre Infusionen) nachgewiesen.
Immunsystem
Mayzent induziert eine dosisabhängige Reduktion der Lymphozytenzahl im peripheren Blut innerhalb von 6 Stunden nach der ersten Dosis, dies ist auf das reversible Zurückhalten (Sequestrierung) von Lymphozyten im Lymphgewebe zurückzuführen.
Bei andauernder täglicher Gabe nimmt die Lymphozytenzahl kontinuierlich ab und erreicht einen medianen Minimalwert (90 % KI) von ungefähr 0.560 (0.271 bis 1.08) Zellen/nl bei einem typischen, nicht aus Japan stammenden CYP2C9*1/*1 oder CYP2C9*1/*2-Patienten mit SPMS, was einer Verringerung von 20 bis 30 % gegenüber dem Ausgangswert entspricht. Bei täglicher Einnahme werden niedrige Lymphozytenzahlen aufrechterhalten.
Die Lymphozytenzahl kehrt bei der überwiegenden Mehrheit (90 %) der SPMS-Patienten innerhalb von 10 Tagen nach Beendigung der Behandlung in den Normalbereich zurück. Nach Beendigung der Mayzent-Behandlung kann die Reduzierung der peripheren Lymphozytenzahl bis zu 3 bis 4 Wochen nach der letzten Dosis anhalten.
Kardialelektrophysiologie
Herzfrequenz und Rhythmus
Mayzent führt bei Behandlungsbeginn zu einer vorübergehenden Abnahme der Herzfrequenz und der atrioventrikulären Überleitung (s. «Unerwünschte Wirkungen»). Dies ist ursächlich verbunden mit einer Aktivierung von G-Protein-gekoppelten, nach innen rektifizierenden Kalium-(GIRK-)Kanälen über die S1P1-Rezeptorstimulation, was zu einer zellulären Hyperpolarisation und verringerter Erregbarkeit führt. Aufgrund ihres funktionellen Antagonismus an S1P1-Rezeptoren desensibilisiert die Anfangstitration von Siponimod nacheinander die GIRK-Kanäle, bis die Erhaltungsdosis erreicht ist.
Potenzial zur Verlängerung des QT-Intervalls
Die Auswirkungen von therapeutischen (2 mg) und supratherapeutischen (10 mg) Siponimod-Dosierungen auf die kardiale Repolarisation wurden in einer ausführlichen QT-Studie untersucht. Die Ergebnisse deuten nicht auf ein arrhythmogenes Potenzial in Zusammenhang mit der QT-Verlängerung mit Siponimod hin, da Siponimod das Placebo-korrigierte Baseline-bereinigte mittlere QTcF-Intervall (ΔΔQTcF) um mehr als 5 ms erhöhte, mit einer maximalen mittleren Wirkung von 7.8 ms (2 mg) bzw. 7.2 ms (10 mg) 3 Stunden nach der Dosisgabe. Die Obergrenze des einseitigen 95 % KI für das ΔΔQTcF-Intervall blieb zu jedem Zeitpunkt unter 10 ms. Die kategorische Analyse ergab keine behandlungsbedingten QTc-Werte über 480 ms, Zunahmen des QTc-Intervalls von mehr als 60 ms gegenüber dem Ausgangswert und kein korrigierter oder unkorrigierter QT/QTc-Wert überstieg 500 ms.
Lungenfunktion
Eine Einzel- oder Mehrfachgabe von Mayzent über 28 Tage ist nicht mit einer klinisch relevanten Erhöhung des respiratorischen Widerstands, gemessen mittels forciertem exspiratorischem Fluss in 1 Sekunde (FEV1) bei 25 und 75 % des Lungenvolumens (FEF25-75 %) verbunden. Bei nicht-therapeutischen Einzeldosen (> 10 mg) wurde ein leichter Trend zu einem reduzierten FEV1 festgestellt. Die gleichzeitige Behandlung mit Mayzent und Propranolol führte zu einem minimalen Rückgang des FEV1 im Vergleich zu Propranolol allein, wobei die Veränderungen mit den einzelnen Medikamenten oder mit der Kombination innerhalb der physiologischen FEV1-Variabilität lagen und klinisch nicht signifikant waren.
Klinische Wirksamkeit
Die Wirksamkeit von Mayzent wurde in einer Phase-3-Studie untersucht, in der eine einmal tägliche Dosis von 2 mg Mayzent bei Patienten mit SPMS bewertet wurde. Eine Phase-2-Dosisfindungsstudie bei Patienten mit RRMS wies eine dosisabhängige Reduktion der entzündlichen Läsionen auf dem MRT nach und zeigte, dass Mayzent 2 mg eine nahezu maximale Wirkung erzielt.
Studie A2304 (EXPAND) bei SPMS
Die Studie A2304 war eine randomisierte, doppelblinde, placebokontrollierte, Ereignis- und Nachbeobachtungsdauer-getriebene Studie der Phase 3 bei Patienten mit SPMS, die in den letzten 2 Jahren in Abwesenheit oder unabhängig von Schüben eine nachgewiesene Progression, keine Hinweise auf einen Schub in den 3 Monaten vor Beginn der Studie und einen medianen EDSS-Score (Expanded Disability Status Scale) von 3.0 bis 6.5 bei Studienbeginn hatten.
Der mediane EDSS-Ausgangswert war 6.0. Patienten älter als 61 Jahre wurden nicht eingeschlossen. Im Hinblick auf die Krankheitsaktivität können die für die Entzündungsaktivität bei SPMS charakteristischen Merkmale Schübe- oder bildgebungsbezogen sein (d.h. Kontrastmittel-anreichernde T1-Läsionen oder aktive [neue oder sich neu vergrössernde] T2-Läsionen).
Die Patienten wurden im Verhältnis 2:1 für den Erhalt von Mayzent 2 mg einmal täglich oder Placebo randomisiert. Die klinischen Bewertungen wurden beim Screening, sowie alle 3 Monate und zum Zeitpunkt eines Schubes durchgeführt. MRT-Bewertungen wurden beim Screening und alle 12 Monate durchgeführt.
Der primäre Endpunkt der Studie war die Zeit bis zur 3-monatigen bestätigten Behinderungsprogression (confirmed disability progression, CDP), die als Zunahme um mindestens 1 Punkt gegenüber dem Ausgangswert des EDSS (0.5-Punkte-Zunahme für Patienten mit einem EDSS von 5.5 oder mehr) über 3 Monate hinweg ermittelt wurde. Wichtige sekundäre Endpunkte waren die Zeit bis zu einer bestätigten Verschlechterung nach 3 Monaten von mindestens 20 % gegenüber Baseline im zeitkontrollierten 25-Fuss-Gehtest (timed 25 foot walk test, T25FW) und die Veränderung gegenüber Baseline im T2-Läsionsvolumen. Weitere sekundäre Endpunkte waren die Zeit bis zur 6-monatigen CDP, prozentuale Veränderung des Gehirnvolumens und Messungen der entzündlichen Krankheitsaktivität (jährliche Schubrate, MRT-Läsionen). Die Veränderung der kognitiven Verarbeitungsgeschwindigkeit beim Symbol Digit Modalities Test war ein explorativer Endpunkt.
Die Studiendauer war für einzelne Patienten variabel (mediane Studiendauer 21 Monate, Bereich 1 Tag bis 37 Monate).
In dieser Studie wurden 1'651 Patienten auf entweder den Erhalt von Mayzent 2 mg (N = 1'105) oder Placebo (N = 546) randomisiert; 82 % der mit Mayzent behandelten Patienten und 78 % der mit Placebo behandelten Patienten schlossen die Studie ab. Das mediane Alter betrug 49.0 Jahre, die mediane Krankheitsdauer 16.0 Jahre und der mediane EDSS-Score 6.0 zu Studienbeginn; 64 % der Patienten hatten in den 2 Jahren vor Studienbeginn keine Schübe und 76 % hatten keine Gadolinium (Gd)-anreichernden Läsionen auf ihrem Ausgangs-MRT-Scan; 78 % der Patienten waren zuvor mit einer Therapie für ihre MS behandelt worden.
Die Zeit bis zum Auftreten der nach 3 und 6 Monaten bestätigten Behinderungsprogression war unter Siponimod signifikant verzögert, mit einer Risikoreduktion für eine nach 3 Monaten bestätigte Behinderungsprogression um 21 % im Vergleich zu Placebo (Hazard Ratio [HR] 0.79, p = 0.0134) und einer Risikoreduktion für eine nach 6 Monaten bestätigte Behinderungsprogression um 26 % gegenüber Placebo (HR 0.74, p = 0.0058).
Die Ergebnisse dieser Studie sind in Tabelle 3 und Abbildung 1 und 2 zusammengefasst.
Tabelle 3 Klinische und MRT-Ergebnisse der Studie A2304
|
Endpunkte
|
A2304 (EXPAND)
| |
Siponimod 2 mg
(n = 1099)
|
Placebo
(n = 546)
| |
Klinische Endpunkte
| |
Primärer Wirksamkeitsendpunkt: Anteil der Patienten mit nach 3 Monaten bestätigter Behinderungsprogression (primärer Endpunkt)
|
26.3 %
|
31.7 %
| |
Risikoreduktion1
|
21 % (p = 0.0134)
| |
Anteil der Patienten mit nach 3 Monaten bestätigter 20%iger Zunahme beim
25-Fuss-Gehtest
|
39.7 %
|
41.4 %
| |
Risikoreduktion1
|
6 % (p = 0.4398)
| |
Anteil der Patienten mit nach 6 Monaten bestätigter Behinderungsprogression
|
19.9 %
|
25.5 %
| |
Risikoreduktion1
|
26 % [(p = 0.0058)] 6
| |
Jährliche Schubrate (ARR)
|
0.071
|
0.152
| |
Verringerung der Rate2
|
55 % [(p < 0.0001)] 6
| |
MRT-Endpunkte
| |
Veränderung des Volumens der
T2-Läsionen (mm3) gegenüber dem Ausgangswert3
|
+184 mm3
|
+879 mm3
| |
Unterschied bei der Änderung des Volumens der T2-Läsionen
|
–695 mm3 (p < 0.0001)7
| |
Prozentuale Veränderung des Gehirnvolumens gegenüber dem Ausgangswert (95%-KI)3
|
-0.497 %
|
-0.649 %
| |
Unterschied bei der prozentualen Veränderung des Gehirnvolumens
|
0.152 % [(p = 0.0002)] 6
| |
Durchschnittliche kumulative Anzahl der Gd-anreichernden, T1-gewichteten Läsionen (95%-KI)4
|
0.081
|
0.596
| |
Verringerung der Rate
|
86 % [(p < 0.0001)] 6
| |
Anteil der Patienten mit einer Verschlechterung um 4 Punkte beim Symbol Digit Modalities Test5
|
16.0 %
|
20.9 %
| |
Risikoreduktion1
|
25 % [(p = 0.0163)] 6
| |
1
Aus Cox-Modellen für die Zeit bis zur Progression
2 Aus einem Modell für wiederkehrende Ereignisse
3 Durchschnitt aus Monat 12 und Monat 24
4 Bis Monat 24
5 Bestätigt nach 6 Monaten
6 [Der nominale p-Wert für Endpunkte, die nicht in den hierarchischen Tests enthalten und nicht an die Multiplizität eingestellt sind]
7 Nicht bestätigender p-Wert; das hierarchische Testverfahren wurde vor Erreichen des Endpunkts beendet
|
Abbildung 1 Patienten mit nach 3 und 6-Monaten bestätigte Behinderungsprogession anhand von EDSS-Kaplan-Meier-Kurven (vollständiger Analysensatz, Studie A2304)
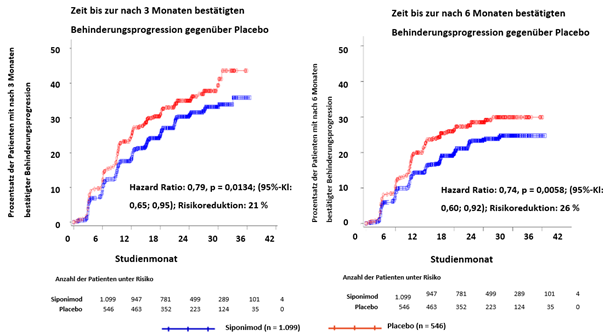
Die Ergebnisse der Studie zeigten eine durchgängige Risikoreduktion bezüglich der Zeit bis zur 3-Monats und 6-Monats-Behinderungsprogression mit Mayzent im Vergleich zu Placebo in Untergruppen, die nach Geschlecht, Alter, vorheriger Therapie der multiplen Sklerose, Schubaktivität vor der Studie, MRT-Krankheitsaktivität bei Baseline, Krankheitsdauer und Behinderungsgrad bei Baseline definiert wurden.
Mayzent hat eine positive Wirkung im Symbol Digit Modalities Test (SDMT) gezeigt. Die Veränderung gegenüber den Basiswerten war für Mayzent stabil oder besser und verschlechterte sich für Placebo mit einem signifikanten Unterschied zwischen den Gruppen von 1.1 Punkten im Monat 12 (p = 0.0132) bzw. 2.3 Punkten im Monat 24 (p = 0.0002). In einer Orientierungsuntersuchung senkte Mayzent das Risiko nach 6 Monaten einer bestätigten Verschlechterung um 4 Punkte im SDMT um 25 % (p = 0.0163) im Placebo-Vergleich. Eine Verschlechterung um 4 Punkte hat sich hierbei schon als klinisch relevant erwiesen.
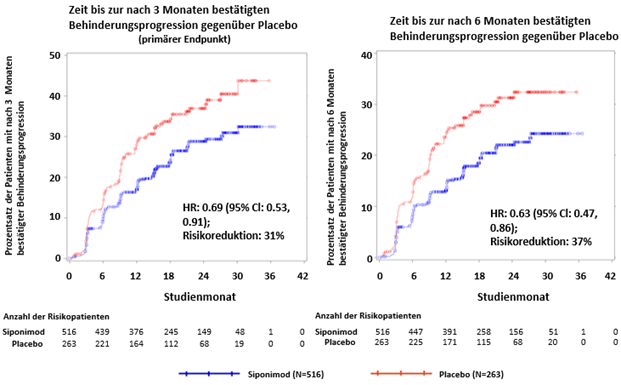
In der Subgruppe der Patienten (47.1 %, n = 779) mit Krankheitsaktivität (definiert als Patienten mit Schub in den 2 Jahren vor der Studie und/oder Vorhandensein von Gd-verstärkenden T1-Läsionen zu Studienbeginn) waren die Baseline-Eigenschaften ähnlich der Gesamtpopulation. Das mediane Alter betrug 47 Jahre, die mediane Krankheitsdauer 15 Jahre und der mediane EDSS-Wert zu Studienbeginn 6.0 (s. «Pharmakokinetik»).
Die Zeit bis zum Auftreten der nach 3 und 6 Monaten bestätigten Behinderungsprogression war bei mit Siponimod behandelten Patienten mit Krankheitsaktivität signifikant verzögert, um 31 % im Vergleich zu Placebo (Hazard Ratio [HR] 0.69; 95%-KI: 0.53; 0.91) und um 37 % im Vergleich zu Placebo (HR 0.63; 95%-KI: 0.47; 0.86). Die ARR (bestätigte Schübe) war im Vergleich zu Placebo um 46 % reduziert (ARR-Verhältnis 0.54; 95%-KI: 0.39; 0.77). Die relative Ratenreduktion der kumulativen Anzahl von Gd-verstärkenden T1-gewichteten Läsionen über 24 Monate betrug 85 % (Ratenverhältnis 0.155; 95%-KI: 0.104; 0.231) im Vergleich zu Placebo. Die Unterschiede in der Veränderung des T2-Läsionsvolumens und im Prozentsatz der Veränderung des Gehirnvolumens (Durchschnitt über die Monate 12 und 24) im Vergleich zu Placebo betrugen 1163 mm3 (95%-KI: 1484, 843 mm3) beziehungsweise 0.141 % (95%-KI: 0.020; 0.261 %).
In der Subgruppe der Patienten (n=827) ohne Anzeichen oder Symptome von Krankheitsaktivität (definiert als Patienten ohne Schub in den 2 Jahren vor der Studie und ohne Vorhandensein von Kontrastmittel anreichernden T1-Läsionen zu Studienbeginn), waren die Auswirkungen auf die nach 3 und 6 Monaten bestätigte Behinderungsprogression gering (die Risikoreduktionen betrugen 7 bzw. 13 %).
Abbildung 2 Patienten mit nach 3 und 6 Monaten bestätigter Behinderungsprogression anhand von EDSS-Kaplan-Meier-Kurven- Subgruppe mit entzündlicher Krankheitsaktivität (vollständiger Analysensatz, Studie A2304)
PharmakokinetikAbsorption
Die Zeit (Tmax) bis zum Erreichen der maximalen Plasmakonzentrationen (Cmax) nach mehrmaliger oraler Gabe von Siponimod betrug etwa 4 Stunden (Bereich 2 bis 12 Stunden). Die absolute orale Bioverfügbarkeit von Siponimod beträgt ca. 84 %. Für 2 mg Siponimod, das einmal täglich über 10 Tage verabreicht wurde, wurden an Tag 10 eine mittlere Cmax von 30.4 ng/ml und eine mittlere AUCtau von 558 h*ng/ml ermittelt. Der Steady-State wurde nach ca. 6 Tagen einer mehrfachen einmal täglichen Verabreichung von Siponimod erreicht.
Eine Nahrungsaufnahme hatte keinen Einfluss auf die systemische Exposition von Siponimod (Cmax und AUC). Daher kann Mayzent ohne Berücksichtigung der Mahlzeiten eingenommen werden.
Distribution
Siponimod wird in Körpergewebe mit einem mittleren Verteilungsvolumen von 124 l verteilt. Der Siponimod-Anteil im Plasma im Verhältnis zum Vollblut liegt beim Menschen bei 68 %. Tierexperimentelle Studien zeigen, dass Siponimod die Blut-Hirn-Schranke ohne weiteres passiert. Die Proteinbindung von Siponimod liegt bei gesunden Personen und bei Leber- und Nierenkranken bei > 99.9 %.
Metabolismus
Siponimod wird extensiv metabolisiert, hauptsächlich über CYP2C9 (79.3 %), gefolgt von CYP3A4 (18.5 %).
Die pharmakologische Aktivität der Hauptmetaboliten M3 und M17 trägt voraussichtlich nicht zur klinischen Wirkung und Sicherheit von Siponimod beim Menschen bei.
Elimination
Eine apparente systemische Clearance (CL/F) von 3.11 l/h wurde bei MS-Patienten geschätzt. Die pharmakokinetisch relevante Eliminationshalbwertszeit liegt bei etwa 30 Stunden.
Siponimod wird hauptsächlich durch den Stoffwechsel und die anschliessende Ausscheidung von Gallenflüssigkeit/Fäzes aus dem systemischen Kreislauf ausgeschieden. Etwa 86.7 % der Siponimod-Dosis wird über die Fäzes ausgeschieden, wovon 9.2 % unverändert bleiben. Nur geringe Mengen der Dosis werden über den Urin ausgeschieden (3.6 %). Im Urin wurde kein unverändertes Siponimod nachgewiesen.
Die mittleren Halbwertszeiten der Siponimod-Metaboliten M17 und M3 nach oraler Verabreichung betragen ca. 155 h bzw. 30 h.
Linearität/Nicht Linearität
Die Siponimod-Konzentration steigt annähernd dosisproportional nach mehreren einmal täglichen Dosen von Siponimod 0.3 mg bis 20 mg.
Steady-State-Plasmakonzentrationen werden bei einmal täglicher Verabreichung nach etwa 6 Tagen erreicht und Steady-State-Spiegel sind etwa 2- bis 3-fach so hoch wie die Werte nach der Initialdosis. Es wird ein Dosistitrationsschema angewendet, um schrittweise die klinische therapeutische Dosis von 2 mg Siponimod nach 6 Tagen zu erreichen und 4 zusätzliche Dosierungstage sind erforderlich, um die Steady-State-Plasmakonzentrationen zu erreichen.
Kinetik spezieller Patientengruppen
CYP2C9-Genotyp
Der CYP2C9-Genotyp hat einen signifikanten Einfluss auf den Siponimod-Stoffwechsel.
Bei Patienten, die homozygot für das CYP2C9*3-Allel sind (CYP2C9*3/*3-Genotyp), ist die Behandlung mit Mayzent kontraindiziert (s. «Kontraindikationen», «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»). Die Anwendung von Mayzent bei diesen Patienten führt zu deutlich erhöhten Siponimod-Plasmaspiegeln. Die empfohlene Erhaltungsdosis von Mayzent beträgt 1 mg täglich bei Patienten mit dem CYP2C9*2/*3 oder *1/*3-Genotyp, um eine erhöhte Exposition gegenüber Siponimod zu vermeiden (s. «Dosierung/Anwendung»).
Es gibt weitere, weniger häufiger vorkommende Polymorphismen für CYP2C9. Die Pharmakokinetik von Siponimod wurde bei Trägern dieser Genotypen nicht untersucht. Einige dieser Polymorphismen, insbesondere die Allele *5, *6, *8 und *11, sind ebenfalls mit einer verminderten oder fehlenden Enzymfunktion verbunden (siehe auch «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»). Nach einer Einzeldosis von 0.25 mg Siponimod waren AUCinf und AUClast bei Probanden mit den Genotypen CYP2C9*2/*3 und CYP2C9*3/*3 etwa 2- bzw. 4-fach höher, während es im Vergleich zu Extensivmetabolisierern (CYP2C9*1/*1) nur einen geringen Anstieg von 21 % bzw. 16 % gab. Die mittlere Halbwertszeit wurde bei CYP2C9*2/*3 und CYP2C9*3/*3-Trägern verlängert (51 h bzw. 126 h).
Nach mehrfacher oraler Gabe von Siponimod an CYP2C9-extensivmetabolisierenden (CYP2C9*1/*1 und CYP2C9*1/*2) SPMS-Patienten wurde eine scheinbare systemische Clearance (Cl/F) von 3.11 l/h geschätzt. Die Cl/F beträgt bei Probanden mit den Genotypen CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3 und CYP2C9*3/*3 jeweils 2.5, 1.9, 1.6 und 0.9 l/h. Die resultierende Zunahme des Siponimod-AUC betrug bei Probanden mit den Genotypen CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3 und CYP2C9*3/*3 im Vergleich zu denen mit dem Genotyp CYP2C9 *1/*1 jeweils 25, 61, 91 bzw. 285 %. Da die geschätzte scheinbare Clearance für Probanden mit dem Genotyp CYP2C9*1/*2 mit derjenigen für Probanden mit dem Genotyp CYP2C9*1/*1 vergleichbar war, wird eine ähnliche Siponimod-Exposition für beide Genotypen erwartet.
Patienten mit Niereninsuffizienz
Bei Patienten mit leichter, mittlerer oder schwerer Niereninsuffizienz sind keine Anpassungen der Siponimod-Dosis erforderlich. Die mittlere Halbwertszeit und Cmax von Siponimod (gesamt und ungebunden) waren zwischen Patienten mit schwerer Niereninsuffizienz und gesunden Probanden vergleichbar. Die gesamten und ungebundenen AUCs waren im Vergleich zu gesunden Probanden nur geringfügig erhöht (um 23 bis 33 %). Die Auswirkungen von terminaler Nierenerkrankung oder Hämodialyse auf die Pharmakokinetik von Siponimod wurden nicht untersucht. Aufgrund der hohen Plasmaproteinbindung (> 99.9 %) von Siponimod wird nicht erwartet, dass die Hämodialyse die gesamte oder ungebundene Siponimodkonzentration verändert, und es werden keine Dosisanpassungen aufgrund dieser Überlegungen erwartet.
Patienten mit Leberinsuffizienz
Bei Patienten mit Leberinsuffizienz sind keine Anpassungen der Siponimod-Dosis erforderlich. Die ungebundene Siponimod AUC ist bei Patienten mit mittlerer bis schwerer Leberinsuffizienz um 15 % bis 50 % höher im Vergleich zu gesunden Probanden für die untersuchte Dosis von 0.25 mg. Bei Patienten mit Leberinsuffizienz blieb die mittlere Halbwertszeit von Siponimod unverändert.
Ältere Patienten
In den klinischen Studien wurden Patienten bis 61 Jahre eingeschlossen (s. «Dosierung/Anwendung»).
Geschlecht
Das Geschlecht hat keinen Einfluss auf die Pharmakokinetik von Siponimod.
Hautfarbe/ethnische Zugehörigkeit
Bei Einzelgabe ergaben sich keine Anzeichen für ethnische Unterschiede im Hinblick auf die PK-Parameter zwischen gesunden japanischen und kaukasischen Patienten, was darauf hindeutet, dass die ethnische Zugehörigkeit keinen Einfluss auf die Pharmakokinetik von Siponimod hat.
Präklinische DatenDas präklinische Sicherheitsprofil von Siponimod wurde in Toxizitätsstudien mit Einzel- und Mehrfachdosen an Mäusen (bis zu 13 Wochen), Ratten (bis zu 26 Wochen) und Affen (bis zu 52 Wochen) untersucht. Dosisbegrenzende Toxizitäten bei Tierarten waren Nephrotoxizität bei Mäusen, Auswirkungen auf die Gewichtszunahme bei Ratten sowie unerwünschte Auswirkungen auf das ZNS und gastrointestinale Auswirkungen bei Affen. Die durch Histopathologie bei Nagetieren identifizierten und durch toxische Auswirkungen am meisten beeinflussten Organe waren u.a. Lunge, Leber, Schilddrüse, Nieren und die Gebärmutter/Vagina. Bei einzelnen Affen wurden Auswirkungen auf Muskeln und Haut beobachtet.
Die NOAEL-Werte (no observed adverse effect levels) wurden für männliche und weibliche Ratten auf 50 bzw. 15 mg/kg/Tag und für Affen beiden Geschlechts auf 10 mg/kg/Tag festgelegt. Die Sicherheitsmargen nach AUC für systemische Effekte (Faktor 171) wurden auf Basis der Erhaltungsdosis von 2 mg/Tag berechnet.
Genotoxizität und Kanzerogenität
In-vitro-Genotoxizitätstests (Bakterienmutation, Mikronukleustest und Chromosomenaberrationstest mit humanen Lymphozyten) und eine In-vivo-Mikronukleus-Studie an Ratten haben kein genotoxisches Potenzial von Siponimod gezeigt.
In Untersuchungen zur Kanzerogenität wurden bei Mäusen Siponimod-induzierte Lymphome, Hämangiome und Hämangiosarkome, bei männlichen Ratten follikuläre Adenome und Karzinome der Schilddrüse identifiziert. Diese Tumorbefunde wurden entweder als mausspezifisch erachtet oder auf metabolische Leberanpassungen bei Ratten zurückgeführt. Die klinische Relevanz für den Menschen ist nicht bekannt.
Die Plasma-Siponimod-Exposition (AUC) bei der niedrigsten getesteten Dosis in der Maus ist etwa 29-mal so hoch wie bei Menschen bei der für Menschen empfohlenen Dosis von 2 mg.
Fruchtbarkeit und Reproduktionstoxizität
Siponimod hatte bis zur höchsten getesteten Dosis keine Auswirkungen auf die Fruchtbarkeitsparameter bei weiblichen und männlichen Ratten. Basierend auf der systemischen Exposition des Menschen (AUC) bei einer täglichen Dosis von 2 mg entspricht dies ungefähr einer 16-fachen Sicherheitsmarge. Es gab keine Auswirkungen auf die Fortpflanzungsorgane bei Ratten und Affen nach chronischer Dosierung.
Reproduktions- und Entwicklungsstudien an trächtigen Ratten und Kaninchen haben gezeigt, dass Siponimod bei Ratten und Kaninchen Embryotoxizität und Fetotoxizität sowie bei Ratten Teratogenität hervorruft. Nach pränataler Exposition gegenüber Siponimod bei einer Dosierung ähnlich der beim Menschen empfohlenen Dosis von 2 mg/Tag wurde bei Ratten eine erhöhte Inzidenz von Post-Implantationsverlusten und fetalen Anomalien (extern, urogenital und am Skelett) sowie bei Kaninchen von embryofetalen Todesfällen, Fehlgeburten und fetalen Veränderungen (Skelett und innere Organe) beobachtet.
Die Exposition bei Ratten und Kaninchen in Fällen, in denen keine Missbildungen bzw. embryofetalen Todesfälle gemeldet wurden, lag unter der klinischen Exposition. Dies deutet darauf hin, dass es bei der Erhaltungsdosis von 2 mg/Tag keine Sicherheitsmarge bezüglich der Auswirkungen auf die embryofetale und prä-/postnatale Entwicklung gibt. Bei laktierenden Ratten, die mit einer einfachen oralen Dosis von 10 mg/kg dosiert wurden, gelangten Siponimod und seine Metaboliten in die Muttermilch.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Nach erstmaligem Öffnen 3 Monate haltbar bei Lagerung nicht über 25°C.
Besondere Lagerungshinweise
Im Kühlschrank lagern (2°C bis 8°C).
In der Originalverpackung aufbewahren.
Arzneimittel für Kinder unzugänglich aufbewahren.
Hinweise für die Handhabung
Keine besonderen Anforderungen.
Besondere Vorsichtsmassnahmen für die Beseitigung
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Zulassungsnummer67230 (Swissmedic)
PackungenStarterpackung mit 12 Filmtabletten à 0.25 mg (B)
Packung mit 120 Filmtabletten à 0.25 mg (B)
Packung mit 28 Filmtabletten à 1 mg (B)
Packung mit 28 Filmtabletten à 2 mg (B)
ZulassungsinhaberinNovartis Pharma Schweiz AG, Risch, Domizil: 6343 Rotkreuz.
Stand der InformationJuni 2025
|