ZusammensetzungWirkstoffe
Fremanezumab (humanisierter monoklonaler Antikörper, der mittels rekombinanter DNA-Technologie in Ovarialzellen des chinesischen Hamsters [CHO] hergestellt wird).
Hilfsstoffe
Histidin, Histidinhydrochlorid-Monohydrat, Saccharose, Natriumedetat, Polysorbat 80, Wasser für Injektionszwecke.
Der Fertigpen enthält 0.025 mg Natrium pro 1.5 ml
Indikationen/AnwendungsmöglichkeitenProphylaktische Behandlung der Migräne bei Erwachsenen, sofern diese indiziert ist.
Dosierung/AnwendungDie Indikation für die Therapie muss durch einen Arzt oder eine Ärztin mit Erfahrung auf dem Gebiet der Migränebehandlung gestellt und durch diese in der weiteren Behandlung begleitet werden. Bei mangelndem Therapieansprechen, beziehungsweise nach spätestens 12 Monaten, sollte eine Reevaluation zur Fortführung der Therapie vorgenommen werden.
Rückverfolgbarkeit
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Es stehen zwei Behandlungsschemata zur Verfügung:
·225 mg einmal monatlich (monatliche Dosierung) oder
·675 mg alle drei Monate (vierteljährliche Dosierung)
Bei einem Wechsel des Behandlungsschemas sollte die erste Dosis des neuen Schemas am nächsten planmässigen Verabreichungstermin des ursprünglichen Schemas verabreicht werden.
Bei Einleitung der Behandlung mit Fremanezumab kann die Therapie mit einem Arzneimittel zur Migräneprävention begleitend fortgeführt werden, sofern es vom Verordnenden für notwendig erachtet wird («siehe Eigenschaften/Wirkungen»).
Verspätete Dosisgabe
Wird eine Injektion von AJOVY zum vorgesehenen Zeitpunkt versäumt, ist die Verabreichung so bald wie möglich nachzuholen. Die folgenden Injektionen sollen entsprechend der angezeigten Dosierung und dem angezeigten Behandlungsschema ab dem Datum der erneuten Aufnahme der Verabreichung fortgesetzt werden. Es darf keine doppelte Dosis verabreicht werden, um eine ausgelassene Dosis nachzuholen.
Art der Anwendung
AJOVY ist ausschliesslich für die subkutane Injektion bestimmt. AJOVY kann in Bereichen vom Bauch, der Oberschenkel oder der Oberarme injiziert werden, die schmerzunempfindlich und nicht gerötet sind und keine Blutergüsse oder Verhärtungen aufweisen. Werden mehrere Injektionen verabreicht, sollte die Injektionsstelle jeweils gewechselt werden.
AJOVY ist zur Selbstinjektion bestimmt und kann nach einer sorgfältigen Einweisung durch das medizinische Fachpersonal in die korrekte subkutane Selbstinjektion vom Patienten selbst injiziert werden. Ausführliche Hinweise zur Verabreichung sind der Patienteninformation (Packungsbeilage) zu entnehmen.
Spezielle Dosierungsanweisungen
Ältere Patienten
Es liegen nur begrenzte Daten zur Anwendung von AJOVY bei Patienten im Alter von ≥65 Jahren vor. Auf Grundlage der Ergebnisse einer populationspharmakokinetischen Analyse ist eine Dosierungsanpassung nicht erforderlich («siehe Pharmakokinetik»).
Niereninsuffizienz
Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Die pharmakokinetische Populationsanalyse integrierter Daten aus den klinischen Studien mit AJOVY ergab keinen Unterschied zwischen Patienten mit leichter oder mittelschwerer Funktionsstörung und solchen mit normaler Nierenfunktion in Bezug auf die Pharmakokinetik von Fremanezumab. Patienten mit schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1,73 m2) wurden nicht untersucht.
Leberinsuffizienz
Bei Patienten mit leichter bis mittelschwerer Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Die pharmakokinetische Populationsanalyse integrierter Daten aus den klinischen Studien mit AJOVY ergab keinen Unterschied zwischen Patienten mit leichter oder mittelschwerer Leberfunktionsstörung und solchen mit normaler Leberfunktion in Bezug auf die Pharmakokinetik von Fremanezumab. Die Pharmakokinetik in Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht.
Pädiatrie (unter 18 Jahre)
Die Sicherheit und Wirksamkeit von AJOVY bei Kindern und Jugendlichen unter 18 Jahren ist bisher nicht gezeigt. Es liegen keine Daten vor. AJOVY darf daher in dieser Altersgruppe nicht angewendet werden.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff Fremanezumab oder einem der Hilfsstoffe gemäss Zusammensetzung.
Warnhinweise und VorsichtsmassnahmenSchwerwiegende Überempfindlichkeitsreaktionen
Anaphylaktische Reaktionen sind mit Fremanezumab selten berichtet worden (siehe «Unerwünschte Wirkungen»). Die meisten Reaktionen sind innerhalb von 24 Stunden nach der Verabreichung aufgetreten, allerdings waren einige Reaktionen verzögert. Patienten sollten über die Symptome in Verbindung mit Überempfindlichkeitsreaktionen gewarnt werden. Falls eine schwerwiegende Überempfindlichkeitsreaktion auftritt, soll eine geeignete Therapie eingeleitet und die Behandlung mit Fremanezumab nicht fortgesetzt werden (siehe «Kontraindikationen»).
In der Post-Marketing-Phase wurde bei einem Patienten, der mehrere Begleitmedikationen einschliesslich Lamotrigin einnahm und mit Fremanezumab behandelt wurde, über ein Stevens-Johnson-Syndrom berichtet. Über das Auftreten dieser Reaktion wurde in seltenen Fällen auch bei Patienten berichtet, die andere monoklonale Antikörper gegen CGRP zusammen mit Begleitmedikationen einschliesslich Lamotrigin erhielten.
Schwere kardiovaskuläre Erkrankungen
Patienten mit bestimmten schweren kardiovaskulären Erkrankungen waren von einer Teilnahme an klinischen Studien ausgeschlossen (siehe «Eigenschaften/Wirkungen, klinische Wirksamkeit»). Zu diesen Patienten liegen keine Sicherheitsdaten vor.
Pädiatrie
Die Sicherheit und Wirksamkeit von AJOVY bei Kindern und Jugendlichen ist nicht gezeigt worden. AJOVY darf daher bei dieser Altersgruppe nicht angewendet werden.
Natrium
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Fertigpen, d.h. es ist nahezu «natriumfrei».
InteraktionenFremanezumab wird nicht von Cytochrom P450-Enzymen metabolisiert. Daher sind Interaktionen mit Begleitmedikamenten, bei denen es sich um Substrate, Induktoren oder Inhibitoren von Cytochrom P450-Enzymen handelt, unwahrscheinlich.
Es wurden keine klinischen Studien mit AJOVY zur Erfassung von Wechselwirkungen durchgeführt. Aufgrund der Eigenschaften von AJOVY werden keine pharmakokinetischen Wechselwirkungen mit anderen Arzneimitteln erwartet. Darüber hinaus ergab sich durch die begleitende Anwendung von Migräne-Akutbehandlungen (insbesondere Analgetika, Ergotaminderivate und Triptane) und präventiven Migränemedikamenten während der klinischen Studien keine Beeinträchtigung der Pharmakokinetik von Fremanezumab.
Schwangerschaft, StillzeitSchwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung von AJOVY bei Schwangeren. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte schädliche Wirkungen in Bezug auf Reproduktionstoxizität (siehe «Präklinische Daten»). Es ist bekannt, dass humanes Immunglobulin (IgG) plazentagängig ist, daher soll AJOVY während der Schwangerschaft nicht angewendet werden, es sei denn, dies ist eindeutig erforderlich.
Stillzeit
Es ist nicht bekannt, ob Fremanezumab in die menschliche Muttermilch übergeht. Da viele Arzneimittel, darunter Antikörper, in die Muttermilch ausgeschieden werden, kann ein Risiko für das Neugeborene/Kleinkind nicht ausgeschlossen werden. Daher sollte eine Entscheidung darüber getroffen werden, ob abgestillt oder AJOVY abgesetzt wird, wobei der mögliche Nutzen von AJOVY für die Mutter und der mögliche Nutzen des Stillens für den Säugling gegeneinander abzuwägen sind.
Fertilität
Es liegen keine Daten zur Fertilität beim Menschen vor. Die vorliegenden präklinischen Daten lassen keine schädlichen Auswirkungen auf die Fertilität vermuten (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt. Aufgrund der verfügbaren Daten wird jedoch erwartet, dass AJOVY keinen oder einen vernachlässigbaren Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, hat.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Insgesamt wurden in Placebo-kontrollierten Studien über 2500 Patienten (über 1900 Patientenjahre) mit AJOVY behandelt. Über 1400 Patienten wurden mindestens 12 Monate lang behandelt.
Die am häufigsten beobachteten lokalen Reaktionen an der Injektionsstelle waren Schmerzen (24% vs. 22% unter Placebo), Verhärtung (17% vs. 13% unter Placebo) und Erythem (16 % vs. 12 % unter Placebo). Alle lokalen Reaktionen an der Injektionsstelle waren vorübergehend und überwiegend schwach bis mässig ausgeprägt. Schmerzen, Verhärtung und Erythem wurden meist unmittelbar nach der Injektion beobachtet, während Juckreiz (2%) und Ausschlag innerhalb eines medianen Zeitraums von 24 bzw. 48 Stunden auftraten. Alle Reaktionen an der Injektionsstelle klangen zumeist innerhalb weniger Stunden oder Tage ab. Die Reaktionen an der Injektionsstelle erforderten kein Absetzen des Arzneimittels.
Auflistung der in klinischen Studien und nach der Markteinführung berichteten
Nebenwirkungen
Die Häufigkeiten werden wie folgt definiert: Sehr häufig (≥1/10), häufig (< 1/10, ≥1/100), gelegentlich (< 1/100, ≥1/1000), selten (< 1/1000, ≥1/10'000), sehr selten (< 1/10'000), Häufigkeit nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeitsreaktionen wie Hautausschlag, Juckreiz, Urtikaria und Schwellungen.
Selten: Anaphylaktische Reaktion (nach Markteinführung berichtet).
Erkrankungen des Gastrointestinaltrakts
Häufigkeit nicht bekannt: Obstipation*.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Schmerzen (24%), Verhärtungen (17%), Erythem (16%).
Häufig: Juckreiz.
Gelegentlich: Ausschlag.
* Kausalzusammenhang nicht erwiesen.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Schwerwiegende Überempfindlichkeitsreaktionen
Anaphylaktische Reaktionen sind selten berichtet worden. Diese Reaktionen traten meistens innerhalb von 24 Stunden nach der Verabreichung auf; einige Reaktionen waren jedoch verzögert (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunogenität. Der Nachweis der Antikörperbildung ist stark von der Sensitivität und Spezifität des verwendeten Tests abhängig. Darüber hinaus kann die bei einem Test beobachtete Inzidenz positiver Antikörpernachweise (einschliesslich neutralisierender Antikörper) durch mehrere Faktoren beeinflusst werden, darunter Testverfahren, Probenhandhabung, Zeitpunkt der Probenentnahme, Begleitmedikationen und Grunderkrankung. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Fremanezumab in den unten beschriebenen Studien mit der Inzidenz von Antikörpern gegen andere Arzneimittel in anderen Studien irreführend sein. Die klinische Immunogenität von AJOVY wurde durch die Analyse von Anti-Drug-Antikörpern (ADA) und neutralisierenden Antikörpern bei Patienten untersucht, die mit dem Arzneimittel behandelt wurden. Die Daten zeigen den prozentualen Anteil der Patienten, deren Ergebnisse in spezifischen Tests auf Antikörper gegen AJOVY positiv ausfielen.
In placebokontrollierten Studien entwickelten 0,4% der mit Fremanezumab behandelten Patienten (6 von 1701) Anti-Drug-Antikörper (ADA). Die Antikörperantworten wiesen einen niedrigen Titer auf. Einer dieser 6 Patienten entwickelte neutralisierende Antikörper. Nach 12-monatiger Behandlung mit Fremanezumab wurden ADA bei 2.3% der Patienten (43 von 1888) nachgewiesen, wobei 0.95% der Patienten neutralisierende Antikörper entwickelten. Die Sicherheit und Wirksamkeit von Fremanezumab wurden von der ADA-Entwicklung nicht beeinträchtigt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien wurden Dosen von bis zu 2000 mg intravenös ohne dosislimitierende Toxizität verabreicht. Im Fall einer Überdosierung wird empfohlen, den Patienten auf alle Anzeichen oder Symptome von unerwünschten Wirkungen zu überwachen und gegebenenfalls eine angemessene symptomatische Behandlung zu verabreichen.
Eigenschaften/WirkungenATC-Code
N02CD03
Wirkungsmechanismus
Fremanezumab ist ein aus murinen Vorläuferzellen gewonnener humanisierter monoklonaler IgG2Δa/Kappa-Antikörper. Fremanezumab bindet selektiv den Calcitonin Gene-Related Peptide (CGRP)-Liganden und hindert beide CGRP-Isoformen (α- und β-CGRP) an der Bindung an den CGRP-Rezeptor. Der genaue Wirkungsmechanismus von Fremanezumab bei der Prävention von Migräneattacken ist nicht bekannt, es wird angenommen, dass die Migräneprävention durch die bewirkte Modulierung des Trigeminussystems vermittelt wird. Der CGRP-Spiegel steigt während eines Migräneanfalls nachweislich signifikant an und kehrt mit nachlassendem Kopfschmerz auf Normalwerte zurück.
Pharmakodynamik
Fremanezumab ist hochspezifisch für CGRP und bindet nicht an die eng verwandten Mitglieder der Familie (z.B. Amylin, Calcitonin, Intermedin und Adrenomedullin).
Klinische Wirksamkeit
Die Wirksamkeit von Fremanezumab wurde in zwei randomisierten, 12-wöchigen, doppelblinden placebokontrollierten Studien der Phase III an erwachsenen Patienten mit episodischer (Studie 1) und chronischer (Studie 2) Migräne untersucht. Die rekrutierten Patienten wiesen eine mindestens 12monatige Vorgeschichte von Migräne (mit und ohne Aura) gemäss den Diagnosekriterien der International Classification of Headache Disorders (ICHD-III) auf. Ältere Patienten (> 70 Jahre), Patienten, die an mehr als 4 Tagen pro Monat Opioide oder Barbiturate anwendeten, sowie Patienten mit Myokardinfarkt, zerebrovaskulärem Insult oder thromboembolischen Ereignissen in der Vorgeschichte waren ausgeschlossen.
Studie zu episodischer Migräne (Studie 1)
Die Wirksamkeit von Fremanezumab bei episodischer Migräne wurde in einer randomisierten, multizentrischen, 12wöchigen placebokontrollierten doppelblinden Studie (Studie 1) untersucht. Erwachsene mit einer Vorgeschichte episodischer Migräne (weniger als 15 Kopfschmerztage pro Monat) wurden in die Studie eingeschlossen. Insgesamt wurden 875 Patienten (742 Frauen, 133 Männer) einem von drei Behandlungsarmen zugeteilt: 675 mg Fremanezumab alle drei Monate (vierteljährlich, n = 291), 225 mg Fremanezumab einmal pro Monat (monatlich, n = 290) oder monatliche Verabreichung von Placebo (n = 294) als subkutane Injektion. Die demografischen Daten und die Ausgangsmerkmale zu Studienbeginn waren ausgewogen und zwischen den Studienarmen vergleichbar. Die Patienten hatten ein medianes Alter von 42 Jahren (Altersbereich: 18 bis 70 Jahre), 85 % waren weiblich, 80 % waren heller Hautfarbe. Die mittlere Migränehäufigkeit zu Studienbeginn betrug ca. 9 Migränetage pro Monat. Die Patienten durften während der Studie Arzneimittel gegen akute Kopfschmerzen anwenden. Eine Untergruppe von Patienten (21 %) durfte zudem eine übliche präventive Begleitmedikation anwenden (Betablocker, Kalziumantagonisten/Benzocyclohepten, Antidepressiva, Antikonvulsiva). Insgesamt hatten 19 % der Patienten in der Vergangenheit Topiramat eingenommen.
Insgesamt 791 Patienten schlossen den 12-wöchigen doppelblinden Behandlungszeitraum ab.
Der primäre Wirksamkeitsendpunkt war die mittlere Veränderung der durchschnittlichen monatlichen Anzahl von Migränetagen während des 12-wöchigen Behandlungszeitraums. Wichtigste sekundäre Endpunkte waren das Erreichen einer mindestens 50%igen Reduktion der monatlichen Migränetage gegenüber dem Ausgangswert (Responderrate von 50 %) und die Veränderung der durchschnittlichen monatlichen Anzahl von Tagen mit Anwendung von Arzneimitteln gegen akute Kopfschmerzen gegenüber dem Ausgangswert.
Sowohl das monatliche als auch das vierteljährliche Behandlungsschema von Fremanezumab zeigten für die wichtigsten Endpunkte eine statistisch signifikante und klinisch bedeutsame Verbesserung gegenüber dem Ausgangswert im Vergleich zu Placebo (siehe Tabelle 2). Dieser Effekt trat bereits im ersten Behandlungsmonat auf und wurde über den gesamten Behandlungszeitraum aufrechterhalten (siehe Abbildung 1).
Abbildung 1: Mittlere Veränderung (KQ-Mittel) der monatlichen durchschnittlichen Anzahl an Migränetagen gegenüber dem Ausgangswert in Studie 1
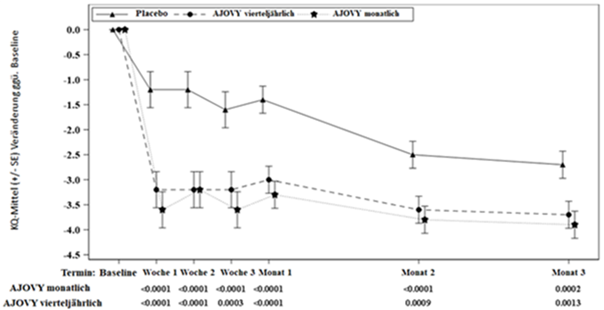
Ausgangsmittelwert (durchschnittliche monatliche Anzahl von Migränetagen): Placebo: 9,1, AJOVY vierteljährlich: 9,2, AJOVY monatlich: 8,9.
Tabelle 2: Wichtigste Wirksamkeitsendpunkte in Studie 1 bei episodischer Migräne
|
Wirksamkeitsendpunkt
|
Placebo
(n = 290)
|
AJOVY
675 mg vierteljährlich
(n = 288)
|
AJOVY
225 mg monatlich
(n = 287)
| |
MMD
| |
Mittlere Veränderunga (95%-KI)
|
-2,2 (-2,68, -1,71)
|
-3,4 (-3,94, -2,96)
|
-3,7 (-4,15, -3,18)
| |
TD (95%-KI)b
|
-
|
-1,2 (-1,74, -0,69)
|
-1,4 (-1,96, -0,90)
| |
Ausgangswert (SD)
|
9,1 (2,65)
|
9,2 (2,62)
|
8,9 (2,63)
| |
p-Wert (vs. Placebo)a
|
-
|
p <0,0001
|
p <0,0001
| |
MHD
| |
Mittlere Veränderunga (95%-KI)
|
-1,5 (-1,88, -1,06)
|
-3,0 (-3,39, -2,55)
|
-2,9 (-3,34, -2,51)
| |
TD (95%-KI)b
|
-
|
-1,5 (-1,95, -1,02)
|
-1,5 (-1,92, -0,99)
| |
Ausgangswert (SD)
|
6,9 (3,13)
|
7,2 (3,14)
|
6,8 (2,90)
| |
p-Wert (vs. Placebo)a
|
-
|
p <0,0001
|
p <0,0001
| |
50%-Responder Rate MMD
| |
Prozentualer Anteil [%]
|
27,9 %
|
44,4 %
|
47,7 %
| |
p-Wert (vs. Placebo)
|
-
|
p <0,0001
|
p <0,0001
| |
MAHMD
| |
Mittlere Veränderunga (95%-KI)
|
-1,6 (-2,04, -1,20)
|
-2,9 (-3,34, -2,48)
|
-3,0 (-3,41, -2,56)
| |
TD (95%-KI)b
|
-
|
-1,3 (-1,73, -0,78)
|
-1,3 (-1,81, 0,86)
| |
Ausgangswert (SD)
|
7,7 (3,60)
|
7,7 (3,70)
|
7,7 (3,37)
| |
p-Wert (vs. Placebo)a
|
-
|
p <0,0001
|
p <0,0001
|
KI = Konfidenzintervall; MAHMD = Monatliche Anzahl an Tagen mit Anwendung von Arzneimitteln gegen akute Kopfschmerzen; MHD = Monatliche Anzahl an Tagen mit mindestens mittelschweren Kopfschmerzen; MMD = monatliche Migränetage; SD = Standardabweichung; TD = Behandlungsunterschied.
a Für alle Endpunkte basieren die mittlere Veränderung und die KI auf dem ANCOVA-Modell, welches Behandlung, Geschlecht, Region und Anwendung von Prophylaktika zu Studienbeginn (ja/nein) als feste Effekte und der entsprechende Ausgangswert und die Jahre seit Auftreten der Migräne als Kovariaten einbezieht.
b Der Behandlungsunterschied basiert auf der MMRM-Analyse mit Behandlung, Geschlecht, Region und Anwendung von Prophylaktika zu Studienbeginn (ja/nein), Monat und Behandlungsmonat als feste Effekte und dem entsprechenden Ausgangswert und den Jahren seit Auftreten der Migräne als Kovariaten.
Bei Patienten mit einer weiteren Begleitmedikation zur Migräneprophylaxe betrug der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Migränetage (MMD) zwischen Fremanezumab 675 mg vierteljährlich und Placebo -1,8 Tage (95%-KI: -2,95, -0,55) und zwischen Fremanezumab 225 mg monatlich und Placebo -2,0 Tage (95%-KI: -3,21, -0,86).
Bei Patienten, die zuvor Topiramat eingenommen hatten, betrug der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Migränetage zwischen Fremanezumab 675 mg vierteljährlich und Placebo -2,3 Tage (95%-KI: -3,64, -1,00) und zwischen Fremanezumab 225 mg monatlich und Placebo -2,4 Tage (95%-KI: -3,61, -1,13).
Studie zu chronischer Migräne (Studie 2)
Fremanezumab wurde bei chronischer Migräne in einer randomisierten, multizentrischen, 12-wöchigen, placebokontrollierten, doppelblinden Studie (Studie 2) untersucht. Die Studienpopulation umfasste Erwachsene mit einer Vorgeschichte chronischer Migräne (mindestens 15 Kopfschmerztage pro Monat). Insgesamt wurden 1130 Patienten (991 Frauen, 139 Männer) randomisiert einem von drei Behandlungsarmen zugeteilt: 675 mg Fremanezumab als Anfangsdosis gefolgt von 225 mg Fremanezumab einmal pro Monat (monatlich, n = 379), 675 mg Fremanezumab alle drei Monate (vierteljährlich, n = 376) oder monatliche Gabe von Placebo (n = 375) als subkutane Injektion. Die demographischen Daten und die Ausgangsmerkmale zu Studienbeginn waren ausgewogen und zwischen den Studienarmen vergleichbar. Die Patienten wiesen ein medianes Alter von 41 Jahren (Altersbereich: 18 bis 70 Jahre) auf, 88 % waren weiblich, 79 % waren heller Hautfarbe. Die mittlere Kopfschmerzhäufigkeit zu Studienbeginn betrug ca. 21 Kopfschmerztage pro Monat (von denen 13 Kopfschmerztage mindestens einen mässigen Schweregrad aufwiesen). Die Patienten durften während der Studie Arzneimittel gegen akute Kopfschmerzen anwenden. Eine Untergruppe von Patienten (21 %) durfte zudem eine verbreitete präventive Begleitmedikation anwenden (Betablocker, Kalziumantagonisten/Benzocyclohepten, Antidepressiva, Antikonvulsiva). Insgesamt hatten 30 % der Patienten zuvor Topiramat und 15 % Onabotulinumtoxin A angewendet. Insgesamt 1034 Patienten schlossen den 12-wöchigen doppelblinden Behandlungszeitraum ab.
Primärer Wirksamkeitsendpunkt war die mittlere Veränderung der durchschnittlichen monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad im 12-wöchigen Behandlungszeitraum gegenüber dem Ausgangswert. Die wichtigsten sekundären Endpunkte waren das Erreichen einer mindestens 50%igen Reduktion der monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad (Responderrate von 50 %) und die Veränderung der durchschnittlichen monatlichen Anzahl von Tagen mit Anwendung von Arzneimitteln gegen akute Kopfschmerzen gegenüber dem Ausgangswert. Sowohl das monatliche als auch das vierteljährliche Behandlungsschema mit Fremanezumab zeigten bei den wichtigsten Endpunkten eine statistisch signifikante und klinisch bedeutsame Verbesserung gegenüber dem Ausgangswert im Vergleich zu Placebo (siehe Tabelle 3). Dieser Effekt trat bereits im ersten Behandlungsmonat auf und wurde über den gesamten Behandlungszeitraum aufrechterhalten (siehe Abbildung 2).
Abbildung 2: Mittlere Veränderung (KQ-Mittel) der durchschnittlichen monatlichen Anzahl von Kopfschmerztagen mit mindestens mässigem Schweregrad gegenüber dem Ausgangswert in Studie 2
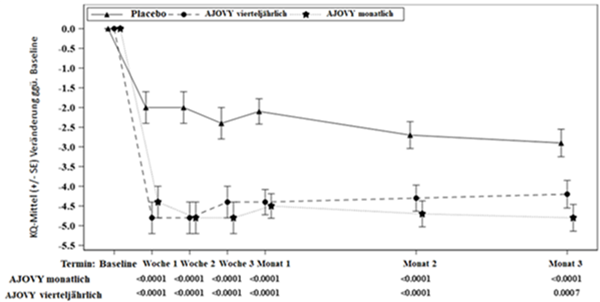
Ausgangsmittelwert (durchschnittliche monatliche Anzahl von Kopfschmerztagen mit mindestens mässigem Schweregrad): Placebo: 13,3, AJOVY vierteljährlich: 13,2, AJOVY monatlich: 12,8.
Tabelle 3: Wichtigste Wirksamkeitsergebnisse in Studie 2 bei chronischer Migräne
|
Wirksamkeitsendpunkt
|
Placebo
(n = 371)
|
Fremanezumab
675 mg vierteljährlich
(n = 375)
|
Fremanezumab
225 mg monatlich mit 675 mg als Anfangsdosis
(n = 375)
| |
MHD
| |
Mittlere Veränderunga (95%-KI)
|
-2,5 (-3,06, -1,85)
|
-4,3 (-4,87, -3,66)
|
-4,6 (-5,16, -3,97)
| |
TD (95%-KI)b
|
-
|
-1,8 (-2,45, -1,13)
|
-2,1 (-2,77, -1,46)
| |
Ausgangswert (SD)
|
13,3 (5,80)
|
13,2 (5,45)
|
12,8 (5,79)
| |
p-Wert (vs. Placebo)a
|
-
|
p<0,0001
|
p<0,0001
| |
MMD
| |
Mittlere Veränderunga (95%-KI)
|
-3,2 (-3,86, -2,47)
|
-4,9 (-5,59, -4,20)
|
-5,0 (-5,70, -4,33)
| |
TD (95%-KI)b
|
-
|
-1,7 (-2,44, -0,92)
|
-1,9 (-2,61, -1,09)
| |
Ausgangswert (SD)
|
16,3 (5,13)
|
16,2 (4,87)
|
16,0 (5,20)
| |
p-Wert (vs. Placebo)a
|
-
|
p <0,0001
|
p <0,0001
| |
50%-Responder Rate MHD
| |
Prozentualer Anteil [%]
|
18,1 %
|
37,6 %
|
40,8 %
| |
p-Wert (vs. Placebo)
|
-
|
p <0,0001
|
p <0,0001
| |
MAHMD
| |
Mittlere Veränderunga (95%-KI)
|
-1,9 (-2,48, -1,28)
|
-3,7 (-4,25, -3,06)
|
-4,2 (-4,79, -3,61)
| |
TD (95%-KI)b
|
-
|
-1,7 (-2,40, -1,09)
|
-2,3 (-2,95, -1,64)
| |
Ausgangswert (SD)
|
13,0 (6,89)
|
13,1 (6,79)
|
13,1 (7,22)
| |
p-Wert (vs. Placebo)a
|
-
|
p <0,0001
|
p <0,0001
|
KI = Konfidenzintervall; MAHMD = Monatliche Anzahl an Tagen mit Anwendung von Arzneimitteln gegen akute Kopfschmerzen; MHD = Monatliche Anzahl an Tagen mit mindestens mittelschweren Kopfschmerzen; MMD = monatliche Migränetage; SD = Standardabweichung; TD = Behandlungsunterschied.
a Für alle Endpunkte basieren die mittlere Veränderung und die KI auf dem ANCOVA-Modell, welches Behandlung, Geschlecht, Region und Anwendung von Prophylaktika zu Studienbeginn (ja/nein) als feste Effekte und der entsprechende Ausgangswert und die Jahre seit Auftreten der Migräne als Kovariaten einbezieht.
b Der Behandlungsunterschied basiert auf der MMRM-Analyse mit Behandlung, Geschlecht, Region und Anwendung von Prophylaktika zu Studienbeginn (ja/nein), Monat und Behandlungsmonat als feste Effekte und dem entsprechenden Ausgangswert und den Jahren seit Auftreten der Migräne als Kovariaten.
Bei Patienten mit genau einer Begleitmedikation zur Migräneprophylaxe betrug der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad zwischen Fremanezumab 675 mg vierteljährlich und Placebo -1,3 Tage (95%-KI: -2,66, 0,03) und zwischen Fremanezumab 225 mg monatlich mit 675 mg als Anfangsdosis und Placebo -2,0 Tage (95%-KI: -3,27, -0,67).
Bei Patienten, die zuvor Topiramat angewendet hatten, betrug der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad zwischen Fremanezumab 675 mg vierteljährlich und Placebo -2,7 Tage (95%-KI: -3,88, -1,51) und zwischen Fremanezumab 225 mg monatlich mit 675 mg als Anfangsdosis und Placebo -2,9 Tage (95%-KI: -4,10, -1,78). Bei Patienten, die zuvor mit Onabotulinumtoxin A behandelt wurden, betrug der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad zwischen Fremanezumab 675 mg vierteljährlich und Placebo -1,3 Tage (95%-KI: -3,01, -0,37) und zwischen Fremanezumab 225 mg monatlich mit 675 mg als Anfangsdosis und Placebo -2,0 Tage (95%-KI: -3,84, -0,22).
Etwa 52 % der Patienten in der Studie zeigten eine übermässige Anwendung von Arzneimitteln gegen akute Kopfschmerzen. Der beobachtete Behandlungsunterschied hinsichtlich der Reduktion der monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad zwischen Fremanezumab 675 mg vierteljährlich und Placebo betrug bei diesen Patienten -2,2 Tage (95%-KI: -3,14, -1,22) und zwischen Fremanezumab 225 mg monatlich mit 675 mg als Anfangsdosis und Placebo -2,7 Tage (95%-KI: -3,71, -1,78).
Langzeitstudie (Studie 3)
Für alle Patienten mit episodischer oder chronischer Migräne in der Langzeitstudie (Studie 3), in der die Patienten Fremanezumab 225 mg monatlich bzw. 675 mg vierteljährlich erhielten, konnte die Wirksamkeit über bis zu 12 zusätzliche Monate aufrechterhalten werden. Insgesamt 79 % der Patienten schlossen den 12-monatigen Behandlungszeitraum von Studie 3 ab. Die gepoolten Daten der beiden Behandlungsschemata zeigten nach 15 Monaten eine Reduktion der monatlichen Migränetage um 6,6 gegenüber den Ausgangswerten von Studie 1 und Studie 2. Insgesamt 61 % der Patienten, die Studie 3 abgeschlossen hatten, erreichten im letzten Studienmonat ein Ansprechen von 50 %. Während der 15-monatigen kombinierten Behandlungsphase wurden keine Sicherheitssignale festgestellt.
Schwer behandelbare Migräne (Studie 4)
Die Wirksamkeit und Sicherheit von Fremanezumab wurde in einer randomisierten Studie (Studie 4) bei insgesamt 838 Patienten mit episodischer und chronischer Migräne mit dokumentiertem unzureichendem Ansprechen auf zwei bis vier Klassen der vorhergehenden Arzneimittel zur Migräneprophylaxe beurteilt. Die Studie bestand aus einer 12 wöchigen doppelblinden, placebokontrollierten Behandlungsphase und einer daran anschliessenden 12 wöchigen unverblindeten Phase.
Der primäre Wirksamkeitsendpunkt war die mittlere Veränderung der durchschnittlichen monatlichen Anzahl von Migränetagen während des 12 wöchigen doppelblinden Behandlungszeitraums. Wichtigste sekundäre Endpunkte waren das Erreichen einer Reduktion der monatlichen Migränetage gegenüber dem Ausgangswert um mindestens 50 %, die mittlere Veränderung der durchschnittlichen monatlichen Kopfschmerztage mit mindestens mässigem Schweregrad gegenüber dem Ausgangswert sowie die Veränderung der durchschnittlichen monatlichen Anzahl von Tagen mit Anwendung eines Akut-Kopfschmerzmedikaments gegenüber dem Ausgangswert. Sowohl das monatliche als auch das vierteljährliche Dosierungsschema von Fremanezumab zeigten bei den wichtigsten Endpunkten eine statistisch signifikante und klinisch bedeutsame Verbesserung gegenüber dem Ausgangswert im Vergleich zu Placebo. Die Ergebnisse der Studie 4 decken sich demnach mit den Hauptergebnissen der vorhergehenden Wirksamkeitsstudien und zeigen zudem eine Wirksamkeit bei schwer behandelbarer Migräne, einschliesslich einer mittleren Reduktion der monatlichen Migränetage (MMD, monthly migraine days) von 3,7 (95 % KI: 4,38; 3,05) bei vierteljährlicher Anwendung von Fremanezumab und 4,1 (95 % KI: 4,73; 3,41) bei monatlicher Anwendung von Fremanezumab gegenüber 0,6 (95 % KI: 1,25; 0,07) bei mit Placebo behandelten Patienten. Während des 12 wöchigen Behandlungszeitraums erreichten 34 % der vierteljährlich mit Fremanezumab behandelten Patienten und 34 % der monatlich mit Fremanezumab behandelten Patienten mindestens eine 50 %ige Reduktion der MMD, verglichen mit 9 % der mit Placebo behandelten Patienten (p < 0,0001). Dieser Effekt trat bereits im ersten Monat auf und wurde über den gesamten Behandlungszeitraum aufrechterhalten. Während des 6 monatigen Behandlungszeitraums wurden keine Sicherheitssignale festgestellt.
PharmakokinetikAbsorption
Nach subkutaner Einmalgabe von 225 mg und 675 mg Fremanezumab betrug die mediane Dauer bis zum Erreichen der Maximalkonzentrationen (tmax) bei gesunden Probanden 5 bis 7 Tage. Die absolute Bioverfügbarkeit von Fremanezumab nach subkutaner Verabreichung von 225 mg und 900 mg an gesunde Probanden betrug 55% (±SD von 23%) bis 66 % (±SD von 26%). Auf der Grundlage der Populationspharmakokinetik wurde eine Dosisproportionalität zwischen 225 mg und 675 mg festgestellt. Der Steady-State wurde bei den Behandlungsschemata mit monatlicher Verabreichung von 225 mg und vierteljährlicher Gabe von 675 mg nach ungefähr 168 Tagen (ca. 6 Monaten) erreicht. Das mediane Akkumulationsverhältnis bei einmal monatlicher und einmal vierteljährlicher Verabreichung beträgt ungefähr 2,4 bzw. 1,2.
Distribution
Ausgehend von der Annahme einer modellierten geschätzten Bioverfügbarkeit von 66 % (± SD von 26 %) für die Patientenpopulation betrug das Verteilungsvolumen für einen typischen Patienten nach subkutaner Verabreichung von 225 mg, 675 mg bzw. 900 mg Fremanezumab 3,6 l (VK 35,1 %).
Metabolismus
Ähnlich wie bei anderen monoklonalen Antikörpern wird erwartet, dass Fremanezumab durch enzymatische Proteolyse in kleine Peptide und Aminosäuren zerfällt.
Elimination
Ausgehend von der Annahme einer modellierten geschätzten Bioverfügbarkeit von 66 % (± SD von 26 %) für die Patientenpopulation lag die zentrale Clearance für einen typischen Patienten nach subkutaner Verabreichung von 225 mg, 675 mg bzw. 900 mg Fremanezumab bei 0,09 l/Tag (VK 23,4 %). Die gebildeten kleinen Peptide und Aminosäuren können im Körper zur de-novo-Synthese von Proteinen wiederverwendet oder über die Nieren ausgeschieden werden. Die Halbwertszeit von Fremanezumab, geschätzt anhand der pharmakokinetischen Populationsanalyse, beträgt 30 Tage.
Kinetik spezieller Patientengruppen
In einer populationspharmakokinetischen Analyse von 2546 Patienten wurden Alter, ethnische Herkunft, Geschlecht und Körpergewicht untersucht. Im Vergleich zum höchsten Körpergewicht-Quartil (84,4 bis 131,8 kg) wird für das niedrigste Körpergewicht-Quartil (43,5 bis 60,5 kg) etwa die doppelte Exposition erwartet. Gemäss den Expositions-Wirkungs-Analysen bei Patienten mit episodischer und chronischer Migräne wurden jedoch keine Auswirkungen des Körpergewichts auf die klinische Wirksamkeit beobachtet. Es sind keine Dosisanpassungen für Fremanezumab erforderlich. Für Patienten mit einem Körpergewicht von > 132 kg liegen keine Daten zum Expositions-Wirkungs-Verhältnis vor.
Eingeschränkte Nieren- oder Leberfunktion
Da bei monoklonalen Antikörpern keine Elimination über die Nieren oder kein Abbau in der Leber bekannt ist, ist nicht zu erwarten, dass eine Nieren- oder Leberfunktionstörung die Pharmakokinetik von Fremanezumab beeinträchtigt.
Patienten mit schwerer Nierenfunktionsstörung (eGFR < 30 ml/min/1,73 m2) wurden nicht untersucht. Eine populationspharmakokinetische Analyse von integrierten Daten aus den klinischen Studien zu Fremanezumab zeigte keinen Unterschied in der Pharmakokinetik von Fremanezumab bei Patienten mit leichter bis mässiger Nieren- oder Leberfunktionsstörung im Vergleich zu Patienten mit normaler Nieren- oder Leberfunktion (siehe «Dosierung/Anwendung»).
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe und zur Reproduktions- und Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Da es sich bei Fremanezumab um einen monoklonalen Antikörper handelt, wurden keine Studien zur Genotoxizität oder zur Karzinogenität durchgeführt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden. Die Fertigspritze und der Fertigpen sind nur zum einmaligen Gebrauch bestimmt.
Besondere Lagerungshinweise
Für Kinder unerreichbar aufbewahren. In der Originalverpackung, vor Licht geschützt und im Kühlschrank (2-8°C) lagern. Nicht einfrieren.
AJOVY kann ungekühlt bis zu 7 Tage lang vor Licht geschützt und bei einer Temperatur von bis zu 30°C aufbewahrt werden. AJOVY ist zu entsorgen, wenn es nicht innerhalb von 7 Tagen nach der Entnahme aus dem Kühlschrank angewendet wird. Nach Aufbewahrung bei Raumtemperatur nicht wieder im Kühlschrank lagern.
Hinweise für die Handhabung
Die in der jeweiligen Packungsbeilage enthaltene Gebrauchsanweisung zur korrekten Handhabung der Fertigspritze oder des Fertigpens ist genau zu befolgen.
AJOVY darf nicht verwendet werden, wenn die Lösung trübe oder verfärbt ist oder Schwebstoffe enthält. AJOVY darf nicht verwendet werden, wenn die Lösung eingefroren wurde.
Die Fertigspritze und der Fertigpen dürfen nicht geschüttelt werden.
Zulassungsnummer67284, 67843 (Swissmedic).
PackungenAJOVY 225 mg/1.5 ml, Injektionslösung in einer Fertigspritze, 1 Fertigspritze [B]
AJOVY 225 mg/1.5 ml, Injektionslösung in einer Fertigspritze, 3 Fertigspritzen [B]
AJOVY 225 mg/1.5 ml, Injektionslösung im Fertigpen, 1 Fertigpen [B]
AJOVY 225 mg/1.5 ml, Injektionslösung im Fertigpen, 3 Fertigpen [B]
ZulassungsinhaberinTeva Pharma AG, Basel.
Stand der InformationApril 2024.
Interne Versionsnummer: 8.1
|