ZusammensetzungWirkstoffe
Idecabtagen Vicleucel: Eine genetisch modifizierte autologe T-Zell-Immuntherapie, bestehend aus T-Zellen, die mit einem lentiviralen Vektor (LVV) transduziert wurden, der einen chimären Antigenrezeptor (CAR) kodiert, welcher das B-Zell-Maturations-Antigen erkennt.
Hilfsstoffe
Cryostor CS10 (5% DMSO, Dextran-40), Natriumchlorid, Natriumgluconat, Natriumacetat-Trihydrat, Kaliumchlorid, Magnesiumchlorid und Wasser für Injektionszwecke.
Abecma enthält bis zu 752 mg Natrium und bis zu 274 mg Kalium pro Dosis.
Indikationen/AnwendungsmöglichkeitenAbecma ist für die Behandlung von erwachsenen Patienten mit rezidiviertem und refraktärem multiplem Myelom indiziert, die
·zuvor zwei Therapielinien erhalten haben, inklusive einem immunmodulatorischen Wirkstoff, einem Proteasom-Inhibitor und einem Anti-CD38-Antikörper, und welche eine Progredienz unter oder innerhalb von 60 Tagen nach der letzten Therapie gezeigt haben.
·zuvor mindestens drei Therapielinien erhalten haben, inklusive einem immunmodulatorischen Wirkstoff, einem Proteasom-Inhibitor und einem Anti-CD38-Antikörper, und welche eine Progredienz zur letzten Therapie gezeigt haben.
Dosierung/AnwendungAbecma muss in einem qualifizierten Behandlungszentrum mit unmittelbarem Zugang zu geeigneten intensiv-medizinischen Überwachungsmöglichkeiten verabreicht werden. Die Abecma-Therapie muss unter Leitung und Aufsicht eines Arztes bzw. einer Ärztin eingeleitet werden, der bzw. die Erfahrung in der Behandlung von hämatologischen Malignomen hat und für die Verabreichung und das Management von Patienten, die mit Abecma behandelt werden, einschliesslich der Behandlung des Zytokin-Freisetzungssyndroms (CRS) und von Neurotoxizität, geschult ist.
Vor der Infusion von Abecma müssen mindestens zwei Dosen Tocilizumab zur Anwendung im Falle eines CRS sowie eine Notfallausrüstung zur Verfügung stehen. Das Behandlungszentrum muss innerhalb von 8 Stunden nach jeder vorangegangenen Dosis Zugang zu einer weiteren Dosis Tocilizumab haben.
Abecma ist ausschliesslich zur autologen Anwendung bestimmt.
Es wird als Einzelinfusion bereitgestellt und enthält eine Dispersion von chimären Antigenrezeptor-positiven lebensfähigen T-Zellen in einem oder mehreren Infusionsbeuteln. Die Zieldosis beträgt 420 x 106 CAR-positive lebensfähige T-Zellen in einem Bereich von 260 bis 500 × 106 CAR-positiven lebensfähigen T-Zellen.
Weitere Informationen zur Dosis können dem beiliegenden RFI-Zertifikat entnommen werden.
Vorbehandlung
Die lymphodepletierende Chemotherapie, bestehend aus Cyclophosphamid 300 mg/m2 intravenös (i.v.) und Fludarabin 30 mg/m2 i.v., sollte über 3 Tage verabreicht werden.
Informationen zur Dosisanpassung bei Nierenfunktionsstörungen entnehmen Sie bitte den Fachinformationen von Cyclophosphamid und Fludarabin.
Abecma wird 2 Tage bis maximal 9 Tage nach Abschluss der lymphodepletierenden Chemotherapie verabreicht. Die Verfügbarkeit von Abecma muss vor Beginn des Chemotherapie-Regimes für die Lymphodepletion bestätigt werden. Wenn zwischen dem Abschluss der lymphodepletierenden Chemotherapie und der Infusion mehr als 4 Wochen liegen, sollte der Patient vor der Behandlung mit Abecma erneut mit einer lymphodepletierenden Chemotherapie behandelt werden.
Klinische Beurteilung vor der Infusion
Die Infusion von Abecma ist bis zu 7 Tage aufzuschieben, wenn bei einem Patienten eine der folgenden Situationen zutrifft:
·Noch andauernde schwerwiegende Nebenwirkungen (insbesondere pulmonale oder kardiale Nebenwirkungen oder Hypotonie), einschliesslich solcher nach vorherigen Chemotherapien.
·Aktive Infektionen oder entzündliche Erkrankungen.
·Aktive Graft-versus-host-disease (GVHD).
·Entwicklung einer klinisch signifikanten Verschlechterung des multiplen Myeloms, welche zu einer medizinisch signifikanten Organfunktionsstörung führt.
Prämedikation
Um das Risiko von Infusionsreaktionen zu minimieren, sollte der Patient etwa 30 bis 60 Minuten vor der Abecma-Infusion Paracetamol (Acetaminophen) (500–1000 mg oral) und Diphenhydramin (12,5 mg intravenös oder 25–50 mg oral) oder ein anderes H1-Antihistaminikum erhalten.
Die prophylaktische Anwendung von Dexamethason oder anderen systemischen Kortikosteroiden sollte vermieden werden, da diese die Aktivität von Abecma beeinträchtigen können. Therapeutische Dosen von Kortikosteroiden sollten 72 Stunden vor Beginn der lymphdepletierenden Chemotherapie und nach der Abecma-Infusion vermieden werden, ausser zur Behandlung von CRS, neurologischen Toxizitäten und anderen lebensbedrohlichen Notfällen (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Überwachung
·Die Patienten sollten nach der Abecma-Infusion in der qualifizierten klinischen Einrichtung 10 Tage lang mindestens täglich auf Anzeichen und Symptome eines Zytokinfreisetzungssyndroms und neurologische Toxizitäten überwacht werden.
·Nach Ablauf der ersten 10 Tage nach der Infusion sollte der Patient im Ermessen des Arztes bzw. der Ärztin überwacht werden.
·Die Patienten sollten angewiesen werden, für mindestens 4 Wochen nach der Infusion in der Nähe (maximal 2 Stunden entfernt) einer qualifizierten klinischen Einrichtung zu bleiben.
Spezielle Patientengruppen
Patienten mit einer Infektion des humanen Immundefizienzvirus (HIV), Hepatitis-B-Virus (HBV) und Hepatitis-C-Virus (HCV)
Es liegen keine klinischen Erfahrungen mit Patienten vor, die eine aktive HIV-, HBV- oder HCV-Infektion haben. Vor der Zellentnahme für die Herstellung muss ein Screening auf HBV-Infektion, aktive HIV- und aktive HCV-Infektion gemäss klinischer Leitlinien erfolgen. Leukapheresematerial von Patienten mit aktiver HIV- oder aktiver HCV-Infektion wird nicht für die Herstellung von Abecma akzeptiert (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Patienten mit Leberfunktionsstörungen
Es wurden keine Studien zu Leberfunktionsstörungen mit Abecma durchgeführt.
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien zu Nierenfunktionsstörungen mit Abecma durchgeführt.
Ältere Patienten
Bei Patienten, die über 65 Jahre alt sind, ist keine Dosisanpassung erforderlich (siehe Abschnitt «Sicherheit und Wirksamkeit bei älteren Patienten».
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Abecma bei pädiatrischen oder jugendlichen Patienten (unter 18 Jahren) wurde nicht untersucht.
Art der Anwendung
Abecma ist nur zur intravenösen Anwendung bestimmt.
Vorsichtsmassnahmen, die vor der Handhabung oder Verabreichung des Arzneimittels zu treffen sind
Dieses Arzneimittel enthält genetisch veränderte menschliche Blutzellen. Das medizinische Fachpersonal, das mit Abecma arbeitet, muss geeignete Vorsichtsmassnahmen treffen (Tragen von Handschuhen und Schutzbrille), um eine potenzielle Übertragung von Infektionskrankheiten zu vermeiden.
Vorbereitung von Abecma zur Infusion
·Vor der Vorbereitung von Abecma muss bestätigt werden, dass die Identität des Patienten mit den Angaben zum Patienten auf den Abecma-Kassetten und den Infusionsbeuteln übereinstimmt.
·Der Abecma-Infusionsbeutel darf nicht aus der Kassette herausgenommen werden, wenn die Informationen auf dem patientenspezifischen Etikett nicht mit dem vorgesehenen Patienten übereinstimmen. Bei Unstimmigkeiten zwischen den Etiketten und den Angaben zum Patienten muss die Zulassungsinhaberin kontaktiert werden.
·Der Zeitpunkt des Auftauens von Abecma muss mit dem Zeitpunkt der Infusion koordiniert werden. Es ist im Voraus zu bestätigen, wann die Infusion erfolgen soll. Die Auftauzeit muss dann so angepasst werden, dass Abecma für die Infusion zur Verfügung steht, sobald der Patient bereit ist.
·Der Infusionsbeutel muss vor dem Auftauen auf Beschädigungen wie Brüche oder Risse untersucht werden. Wenn der Beutel beschädigt ist, muss die Zulassungsinhaberin kontaktiert werden.
·Platzieren Sie den Infusionsbeutel gemäss den lokalen Richtlinien in einen zweiten sterilen Beutel.
·Wenn mehr als ein Infusionsbeutel zur Behandlung vorliegt, tauen Sie jeden Infusionsbeutel einzeln auf.
·Tauen Sie Abecma bei ca. 37 °C mit einem zugelassenen Auftaugerät oder im Wasserbad auf, bis im Infusionsbeutel kein Eis mehr zu sehen ist. Den Inhalt des Beutels vorsichtig durchmischen, um sichtbare Klümpchen mit Zellmaterial aufzulösen. Kleine Klümpchen von Zellmaterial können trotz vorsichtigem manuellem Durchmischen bestehen bleiben. Abecma darf vor der Infusion nicht gewaschen, zentrifugiert oder in neuen Medien resuspendiert werden.
·Der Infusionsbeutel ist mit einer transparenten Plastikhülle umwickelt, die auf die Rückseite des Infusionsbeutels gefaltet ist. Entfernen Sie den Infusionsbeutel aus der Plastikhülle, indem Sie die Plastikhülle auf der Rückseite aufschlagen, um den Infusionsbeutel freizulegen. Ziehen sie den Infusionsbeutel vorsichtig aus der Plastikhülle heraus.
Verabreichung
·Verwenden Sie KEINEN Leukodepletionsfilter.
·Stellen Sie sicher, dass Tocilizumab und eine Notfallausrüstung vor der Infusion und während der Erholungsphase zur Verfügung stehen.
·Für die Infusion von Abecma kann ein Zentralvenenkatheter verwendet werden, der für Patienten mit schlechtem peripherem Zugang zu empfehlen ist.
·Bestätigen Sie, dass die Identität des Patienten mit den Angaben zum Patienten auf dem Abecma-Infusionsbeutel übereinstimmt.
·Den Schlauch des Infusionssets vor der Infusion mit 9 mg/ml (0,9%iger) Natriumchloridlösung für Injektionszwecke vorbereiten. Ein Infusionsset mit Inline-Filter (KEIN Leukodepletionsfilter) sollte für aufgetaute Produkte, verwendet werden.
·Abecma innerhalb von 1 Stunde nach Beginn des Auftauens infundieren.
·Nachdem der gesamte Inhalt des Infusionsbeutels infundiert worden ist, spülen Sie mit der gleichen Infusionsgeschwindigkeit den Schlauch, inklusive des Inline-Filters, mit 9 mg/ml (0,9%iger) Natriumchloridlösung für Injektionszwecke, um zu gewährleisten, dass dem Patienten so viele Zellen wie möglich infundiert werden.
·Befolgen Sie das gleiche Verfahren für alle nachfolgenden Infusionsbeutel für den bestimmten Patienten.
Besondere Vorsichtsmassnahmen für die Entsorgung entnehmen Sie bitte dem Abschnitt «Sonstige Hinweise».
KontraindikationenÜberempfindlichkeit gegen den Wirkstoff oder einen der im Abschnitt «Zusammensetzung» aufgeführten Hilfsstoffe.
Die Kontraindikationen der lymphodepletierenden Chemotherapie sind zu beachten.
Warnhinweise und VorsichtsmassnahmenZytokinfreisetzungssyndrom (Cytokine Release Syndrome, CRS)
CRS, einschliesslich tödlicher oder lebensbedrohlicher Reaktionen, sind nach der Behandlung mit Abecma aufgetreten. In klinischen Studien betrug die mediane Zeit bis zum Auftreten von CRS 1 Tag (Bereich: 1 bis 17 Tage) (siehe Abschnitt «Unerwünschte Wirkungen»).
Überwachung und Behandlung von CRS
CRS sollte basierend auf dem klinischen Erscheinungsbild identifiziert werden. Die Patienten sollten auf andere Ursachen von Fieber, Hypoxie und Hypotonie hin untersucht und behandelt werden. Es wurde berichtet, dass CRS mit Befunden von hämophagozytischer Lymphohistiozytose/Makrophagenaktivierungssyndrom (HLH/MAS) assoziiert ist und dass sich die Physiologie der Syndrome überschneiden kann. MAS ist eine potenziell lebensbedrohliche Erkrankung, und die Patienten sollten engmaschig auf Anzeichen von MAS überwacht werden. Die Behandlung von MAS sollte gemäss den institutionellen Standards erfolgen.
Es ist sicherzustellen, dass vor der Infusion von Abecma mindestens zwei Tocilizumab-Behandlungen zur Verfügung stehen. Das Behandlungszentrum muss innerhalb von 8 Stunden nach jeder vorangegangenen Dosis Zugang zu einer weiteren Dosis Tocilizumab haben.
Die Patienten sollten nach der Abecma-Infusion in der qualifizierten klinischen Einrichtung 10 Tage lang mindestens täglich auf Anzeichen und Symptome von CRS überwacht werden. Sie sind nach der Infusion mindestens 4 Wochen lang auf Anzeichen oder Symptome von CRS zu überwachen.
Bei den ersten Anzeichen von CRS muss, falls angezeigt, eine Behandlung mit unterstützenden Massnahmen, Tocilizumab bzw. Kortikosteroiden eingeleitet werden. Bei Verdacht auf CRS ist gemäss den Empfehlungen in Tabelle 1 vorzugehen. Patienten, bei denen ein CRS auftritt, sollten bis zum Abklingen der Symptome hinsichtlich Herz- und Organfunktion engmaschig überwacht werden. Bei schwerem oder lebensbedrohlichem CRS sind eine Überwachung auf der Intensivstation und eine unterstützende Therapie zu erwägen.
Die Patienten sollten informiert und darauf hingewiesen werden, umgehend einen Arzt bzw. eine Ärztin aufzusuchen, wenn zu irgendeinem Zeitpunkt Anzeichen oder Symptome von CRS auftreten.
Eine frühe Eskalation (d.h. höhere Kortikosteroiddosis, alternative Antizytokin-Mittel, Anti-T-Zell-Therapien) empfiehlt sich bei Patienten mit anhaltendem CRS innerhalb von 72 Stunden nach der Abecma-Infusion, das durch anhaltendes Fieber, Endorgantoxizität (z.B. Hypoxie, Hypotonie) und/oder HLH/MAS gekennzeichnet ist und sich nicht innerhalb von 12 Stunden nach Erstlinien-Interventionen verbessert.
Tabelle 1: CRS-Einstufung und Behandlungsleitfaden
|
CRS-Grada
|
Tocilizumab
|
Kortikosteroide
| |
Grad 1
Die Symptome erfordern nur eine symptomatische Behandlung (z.B. Fieber, Übelkeit, Müdigkeit, Kopfschmerzen, Myalgie, Unwohlsein).
|
Wenn die Symptome 72 Stunden oder mehr nach der Infusion auftreten, symptomatisch behandeln.
Wenn die Symptome weniger als 72 Stunden nach der Infusion auftreten, ist Tocilizumab 8 mg/kg i.v. über 1 Stunde (nicht mehr als 800 mg) zu erwägen.
|
─
| |
Grad 2
Die Symptome erfordern eine moderate Intervention und sprechen darauf an.
Sauerstoffbedarf unter 40 % FiO2 oder Hypotonie, die auf Flüssigkeiten oder niedrige Dosisstärken eines Vasopressors anspricht, oder Organtoxizität vom Grad 2.
|
Gabe von Tocilizumab 8 mg/kg i.v. über 1 Stunde (nicht mehr als 800 mg).
|
Dexamethason 10 mg i.v. alle 12 bis 24 Stunden erwägen.
| |
Wenn innerhalb von 24 Stunden keine Besserung oder eine rasche Progression eintritt, die Gabe von Tocilizumab wiederholen und die Dosis und Häufigkeit von Dexamethason erhöhen (20 mg i.v. alle 6 bis 12 Stunden).
Wenn innerhalb von 24 Stunden keine Besserung eintritt bzw. die rasche Progression anhält, Wechsel zu Methylprednisolon 2 mg/kg, gefolgt von 2 mg/kg aufgeteilt auf vier Mal täglich.
Wenn Steroide eingeleitet werden, die Steroide mindestens drei Mal fortsetzen und über maximal 7 Tage ausschleichen.
Nach zwei Tocilizumab-Behandlungen sind andere Antizytokine zu erwägen.
Nicht mehr als drei Tocilizumab-Behandlungen innerhalb von 24 Stunden bzw. vier Tocilizumab-Behandlungen gesamthaft überschreiten.
| |
Grad 3
Die Symptome erfordern eine aggressive Intervention und sprechen darauf an.
Fieber, Sauerstoffbedarf grösser oder gleich 40 % FiO2 oder Hypotonie, die hochdosierte oder mehrere Vasopressoren erfordert, oder Organtoxizität vom Grad 3 oder Transaminaseanstieg vom Grad 4.
|
Gabe von Tocilizumab 8 mg/kg i.v. über 1 Stunde (nicht mehr als 800 mg).
|
Gabe von Dexamethason (z.B. 10 mg i.v. alle 12 Stunden).
| |
Wenn innerhalb von 24 Stunden keine Besserung bzw. eine rasche Progression eintritt, die Gabe von Tocilizumab wiederholen und die Dosis und Häufigkeit von Dexamethason erhöhen (20 mg i.v. alle 6 bis 12 Stunden).
Wenn innerhalb von 24 Stunden keine Besserung eintritt bzw. die rasche Progression anhält, Wechsel zu Methylprednisolon 2 mg/kg, gefolgt von 2 mg/kg aufgeteilt auf vier Mal täglich.
Wenn Steroide eingeleitet werden, die Steroide mindestens drei Mal fortsetzen und über maximal 7 Tage ausschleichen.
Nach zwei Tocilizumab-Behandlungen sind andere Antizytokine zu erwägen.
Nicht mehr als drei Tocilizumab-Behandlungen innerhalb von 24 Stunden bzw. vier Tocilizumab-Behandlungen gesamthaft überschreiten.
| |
Grad 4
Lebensbedrohliche Symptome.
Erforderliche Beatmungsunterstützung, kontinuierliche venovenöse Hämodialyse (CVVHD) oder Organtoxizität vom Grad 4 (ausser Transaminaseanstieg).
|
Gabe von Tocilizumab 8 mg/kg i.v. über 1 Stunde (nicht mehr als 800 mg).
|
Gabe von Dexamethason 20 mg i.v. alle 6 Stunden.
| |
Nach zwei Tocilizumab-Behandlungen sind andere Antizytokine zu erwägen. Nicht mehr als drei Tocilizumab-Behandlungen innerhalb von 24 Stunden bzw. vier Tocilizumab-Behandlungen gesamthaft überschreiten.
Falls innerhalb von 24 Stunden keine Besserung eintritt, Methylprednisolon (1 bis 2 g, bei Bedarf alle 24 Stunden wiederholen; je nach klinischer Indikation ausschleichen) oder Anti-T-Zelltherapien wie Cyclophosphamid 1,5 g/m2 oder andere erwägen.
|
a Lee-Kriterien zur CRS-Einstufung (Lee et al, 2014).
Neurologische Toxizitäten
Neurologische Toxizitäten, einschliesslich Immuneffektorzell-assoziiertes Neurotoxizitätssyndrom (ICANS), die schwerwiegend oder lebensbedrohlich sein können, sind nach der Behandlung mit Abecma aufgetreten, auch gleichzeitig mit CRS, nach Abklingen des CRS und ohne CRS.
Die mediane Zeit bis zum Auftreten des ersten Ereignisses der vom Prüfarzt festgestellten Neurotoxizität betrug 3 Tage (Bereich: 1 bis 317 Tage, ein Patient entwickelte am 317. Tag eine Enzephalopathie als Folge einer sich verschlimmernden Lungenendzündung und einer Clostridium-difficile-Kolitis) (siehe Abschnitt «Unerwünschte Wirkungen»).
Parkinsonismus
Parkinsonismus vom Grad 3 trat nach der Behandlung mit Abecma in einer Studie zum multiplen Myelom auf. Berichtete Symptome beinhalteten Tremor, Dysphasie, Bradykinesie und parkinson-ähnliche Reflexe.
Überwachen Sie die Patienten hinsichtlich Zeichen und Symptomen von neurologischen Toxizitäten, inklusive Symptomen von Parkinsonismus. Es gibt keine ausreichenden Informationen zur Behandlung von Parkinsonismus bei Patienten nach der Behandlung mit Abecma. Die Behandlung von Neurotoxizitäten, einschliesslich Parkinsonismus, sollte sich an der institutionellen oder klinischen Standartpraxis orientieren und vom behandelten Arzt bzw. von der behandelnden Ärztin festgelegt werden.
Überwachung und Behandlung neurologischer Toxizitäten
Die Patienten sollten nach der Abecma-Infusion in der qualifizierten klinischen Einrichtung 10 Tage lang mindestens täglich auf Anzeichen und Symptome neurologischer Toxizitäten überwacht werden (Tabelle 2). Andere Ursachen für neurologische Symptome sollten ausgeschlossen werden. Die Patienten sollten nach der Infusion mindestens 4 Wochen lang auf Anzeichen oder Symptome neurologischer Toxizitäten überwacht und umgehend behandelt werden. Bei Verdacht auf neurologische Toxizität sollte gemäss den Empfehlungen in Tabelle 2 vorgegangen werden, ggf. mit unterstützenden Massnahmen und Kortikosteroiden. Bei schweren oder lebensbedrohlichen neurologischen Toxizitäten sollte eine intensivmedizinische unterstützende Therapie eingeleitet werden.
Wenn während der neurologischen Toxizität ein begleitendes CRS vermutet wird, sollte dieses gemäss den Empfehlungen in Tabelle 1 behandelt werden und für die beiden in Tabelle 1 und 2 genannten Ereignisse die aggressivere Intervention angewendet werden.
Die Patienten sollten angewiesen werden, umgehend einen Arzt bzw. eine Ärztin aufzusuchen, wenn zu irgendeinem Zeitpunkt Anzeichen oder Symptome einer neurologischen Toxizität auftreten.
Tabelle 2: Einstufung von neurologischen Toxizitäten und Behandlungsleitfaden
|
Neurologischer
Toxizitätsgrada
|
Kortikosteroide und Antikonvulsiva
| |
Grad 1
|
Einleitung von nicht sedierenden Antikonvulsiva (z.B. Levetiracetam) zur Vorbeugung von Krampfanfällen.
Falls 72 Stunden oder mehr nach der Infusion, den Patienten beobachten.
Falls weniger als 72 Stunden nach der Infusion, Dexamethason 10 mg i.v. alle 12 bis 24 Stunden für 2 bis 3 Tage erwägen.
| |
Grad 2
|
Einleitung von nicht sedierenden Antikonvulsiva (z.B. Levetiracetam) zur Vorbeugung von Krampfanfällen.
Einleitung von Dexamethason 10 mg i.v. alle 12 Stunden über 2 bis 3 Tage, bei anhaltenden Symptomen auch länger. Bei einer Steroidexposition von gesamthaft über 3 Tagen ein Ausschleichen erwägen. Steroide werden bei isolierten Kopfschmerzen vom Grad 2 nicht empfohlen.
Wenn nach 24 Stunden keine Besserung oder eine Verschlechterung der neurologischen Toxizität eintritt, die Dosis bzw. Häufigkeit von Dexamethason bis zu einem Maximum von 20 mg i.v. alle 6 Stunden erhöhen.
| |
Grad 3
|
Einleitung von nicht sedierenden Antikonvulsiva (z.B. Levetiracetam) zur Vorbeugung von Krampfanfällen.
Einleitung von Dexamethason 10 bis 20 mg i.v. alle 8 bis 12 Stunden. Steroide werden bei isolierten Kopfschmerzen vom Grad 3 nicht empfohlen.
Wenn nach 24 Stunden keine Besserung oder eine Verschlechterung der neurologischen Toxizität eintritt, Eskalation auf Methylprednisolon (2 mg/kg Aufsättigungsdosis, gefolgt von 2 mg/kg aufgeteilt auf vier Mal täglich; innerhalb von 7 Tagen ausschleichen).
Bei Verdacht auf ein Hirnödem, Hyperventilation und hyperosmolare Therapie erwägen. Gabe von hochdosiertem Methylprednisolon (1 bis 2 g, bei Bedarf alle 24 Stunden wiederholen; je nach klinischer Indikation ausschleichen) und Cyclophosphamid 1,5 g/m2.
| |
Grad 4
|
Einleitung von nicht sedierenden Antikonvulsiva (z.B. Levetiracetam) zur Vorbeugung von Krampfanfällen.
Einleitung von Dexamethason 20 mg i.v. alle 6 Stunden.
Wenn nach 24 Stunden keine Besserung oder eine Verschlechterung der neurologischen Toxizität eintritt, Eskalation auf hochdosiertes Methylprednisolon (1 bis 2 g, bei Bedarf alle 24 Stunden wiederholen; je nach klinischer Indikation ausschleichen). Cyclophosphamid 1,5 g/m2 erwägen.
Bei Verdacht auf ein Hirnödem, Hyperventilation und hyperosmolare Therapie erwägen. Gabe von hochdosiertem Methylprednisolon (1 bis 2 g, bei Bedarf alle 24 Stunden wiederholen; je nach klinischer Indikation ausschleichen) und Cyclophosphamid 1,5 g/m2.
|
a Allgemeine Terminologiekriterien für unerwünschte Ereignisse des National Cancer Institute (USA) zur Einstufung neurologischer Toxizitäten.
Überempfindlichkeitsreaktionen
Bei der Infusion von Abecma können allergische Reaktionen auftreten. Schwere Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, können auf Dimethylsulfoxid (DMSO) in Abecma zurückzuführen sein.
Infektionen und febrile Neutropenie
Abecma sollte nicht an Patienten mit aktiven Infektionen oder entzündlichen Erkrankungen verabreicht werden. Schwere, lebensbedrohliche oder tödlich verlaufende Infektionen sind bei Patienten nach der Infusion von Abecma aufgetreten (siehe Abschnitt «Unerwünschte Wirkungen»). Die Patienten müssen vor und nach der Infusion mit Abecma auf Anzeichen und Symptome einer Infektion überwacht und entsprechend behandelt werden. Prophylaktische, präventive bzw. therapeutische Antimikrobiotika sollten gemäss den lokalen institutionellen Richtlinien verabreicht werden.
Febrile Neutropenie wurde bei Patienten nach der Infusion von Abecma beobachtet (siehe Abschnitt «Unerwünschte Wirkungen») und kann gleichzeitig mit CRS auftreten. Bei Auftreten von febriler Neutropenie muss der Patient auf eine Infektion hin abgeklärt und je nach medizinischer Indikation mit Breitbandantibiotika, Flüssigkeiten und anderen unterstützenden Massnahmen behandelt werden.
Virusreaktivierung
Infektionen mit dem Cytomegalovirus (CMV), die zu Lungenentzündung und Tod führten, sind nach der Verabreichung von Abecma aufgetreten. Eine CMV-Reaktivierung ist gemäss den klinischen Leitlinien zu überwachen und zu behandeln.
Eine Reaktivierung des Hepatitis-B-Virus (HBV), die in manchen Fällen zu fulminanter Hepatitis, Leberversagen und Tod führt, kann bei Patienten auftreten, die mit gegen Plasmazellen gerichteten Arzneimitteln behandelt werden.
Vor der Zellgewinnung für die Herstellung sind Screenings auf das CMV, HBV, aktives HIV und aktives HCV gemäss den klinischen Leitlinien durchzuführen.
Erwägen Sie eine antivirale Therapie gemäss den lokalen klinischen Leitlinien /der klinischen Praxis, um eine Virusreaktivierung zu vermeiden.
Länger anhaltende Zytopenien
Die Patienten können nach einer lymphodepletierenden Chemotherapie und Infusion mit Abecma länger anhaltende Zytopenien entwickeln (siehe Abschnitt «Unerwünschte Wirkungen»).
Die Blutwerte sollten vor und nach der Infusion mit Abecma überwacht werden. Eine Zytopenie sollte überwacht werden. Die Überwachung sollte gemäss den lokalen klinischen Leitlinien mit Unterstützung von myeloischen Wachstumsfaktoren und Blutprodukttransfusionen erfolgen.
Hypogammaglobulinämie
Plasmazellaplasie und Hypogammaglobulinämie können bei Patienten auftreten, die mit Abecma behandelt werden (siehe Abschnitt «Unerwünschte Wirkungen»).
Die Immunglobulinspiegel müssen nach der Behandlung mit Abecma überwacht und gemäss den lokalen klinischen Leitlinien gehandhabt werden, einschliesslich Infektionsvorkehrungen, antibiotischer oder antiviraler Prophylaxe und Immunglobulinersatz.
Verwendung von Lebendimpfstoffen
Die Sicherheit der Immunisierung mit viralen Lebendimpfstoffen während und nach der Behandlung mit Abecma wurde nicht untersucht. Eine Impfung mit viralen Lebendimpfstoffen wird für mindestens 6 Wochen vor Beginn der lymphodepletierenden Chemotherapie, während der Behandlung mit Abecma und bis zur Erholung des Immunsystems nach der Behandlung mit Abecma nicht empfohlen.
Sekundäre Malignome
Patienten, die mit Abecma behandelt werden, können sekundäre Malignome entwickeln. Die Patienten sollten lebenslang auf sekundäre Malignome überwacht werden. Falls ein sekundäres hämatologisches Malignom auftritt, sollte die Zulassungsinhaberin kontaktiert werden, um Anweisungen zur Meldung und möglicher Tests zu erhalten.
Spende von Blut, Organen, Gewebe und Zellen
Mit Abecma behandelte Patienten, dürfen kein Blut, Organe, Gewebe und Zellen für Transplantationen spenden.
Frühere Stammzelltransplantation
Es wird nicht empfohlen, dass Patienten Abecma innerhalb von 4 Monaten nach einer allogenen Stammzelltransplantation (SCT) erhalten, da das potenzielle Risiko einer sich verschlimmernden Graft-versus-Host-Disease (GVHD) besteht. Die Leukapherese zur Herstellung von Abecma sollte frühestens 12 Wochen nach einer allogenen SCT erfolgen.
Hilfsstoffe
Abecma enthält bis zu 33 mmol (752 mg) Natrium pro Dosis, was 37,6% der von der WHO empfohlenen maximalen Tagesdosis von 2 g Natrium für einen Erwachsenen entspricht.
Abecma enthält bis zu 7 mmol (274 mg) Kalium pro Dosis. Dies ist bei Patienten mit eingeschränkter Nierenfunktion und Patienten mit einer kontrollierten kaliumarmen Ernährung zu berücksichtigen.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Interaktionen zwischen Arzneimitteln/Laboruntersuchungen
HIV und das Lentivirus, das zur Herstellung von Abecma verwendet wird, haben einen begrenzten, kurzen Bereich identischen genetischen Materials (RNA). Daher können einige kommerzielle HIV-Nukleinsäuretests bei Patienten, die Abecma erhalten haben, falsch-positive Ergebnisse liefern.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter/Verhütungsmethoden bei Männern und Frauen
Der Schwangerschaftsstatus von gebärfähigen Frauen sollte vor Beginn der Behandlung mit Abecma mittels Schwangerschaftstests überprüft werden.
Es liegen nicht genügend Expositionsdaten vor, um eine Empfehlung hinsichtlich der Dauer der Verhütung nach der Behandlung mit Abecma abzugeben.
Informationen über die Notwendigkeit einer wirksamen Verhütungsmethode bei Patientinnen, die eine lymphodepletierende Chemotherapie erhalten, entnehmen Sie bitte der Fachinformationen von Fludarabin und Cyclophosphamid.
Schwangerschaft
Es liegen keine Daten zur Anwendung von Abecma bei schwangeren Frauen vor. Es wurden keine tierexperimentellen Studien zur Reproduktions- und Entwicklungstoxizität mit Abecma durchgeführt, um zu beurteilen, ob Abecma bei Verabreichung an eine schwangere Frau zu einer Schädigung des Fötus führen kann.
Es ist nicht bekannt, ob Abecma das Potenzial hat, auf den Fötus übertragen zu werden. Basierend auf dem Wirkmechanismus können die transduzierten Zellen, falls sie die Plazentaschranke überschreiten, fetale Toxizität verursachen, darunter Plasmazellaplasie oder Hypogammaglobulinämie. Daher wird Abecma nicht für schwangere Frauen empfohlen und eine Schwangerschaft nach der Infusion mit Abecma sollte mit dem behandelnden Arzt bzw. der behandelnden Ärztin besprochen werden. Die Immunglobulinspiegel bei Neugeborenen von Müttern, die mit Abecma behandelt wurden, sollten gemessen werden.
Stillzeit
Es gibt keine Informationen über das Vorhandensein von Abecma in der Muttermilch, die Wirkung auf den gestillten Säugling und die Auswirkungen auf die Milchproduktion. Die Vorteile des Stillens für die Entwicklung und Gesundheit sollten ebenso erwogen werden wie die klinische Notwendigkeit für Abecma seitens der Mutter und die möglichen nachteiligen Auswirkungen auf den gestillten Säugling durch Abecma bzw. durch die zugrunde liegende Erkrankung der Mutter.
Fertilität
Es liegen keine Daten über Beeinflussungen der Fertilität durch Abecma vor.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenDa neurologische Ereignisse, einschliesslich Bewusstseinsveränderungen und Krampfanfälle, auftreten können, besteht für Patienten, die Abecma erhalten, in den 8 Wochen nach der Abecma-Infusion das Risiko einer Veränderung oder Einschränkung des Bewusstseinszustands oder der Koordination. Weisen Sie die Patienten darauf hin, für mindestens 8 Wochen nach der Abecma-Infusion nicht im Strassenverkehr teilzunehmen und keine gefährlichen Arbeiten oder Tätigkeiten wie das Bedienen schwerer oder potenziell gefährlicher Maschinen durchzuführen.
Unerwünschte WirkungenDie in diesem Abschnitt beschriebenen Sicherheitsdaten spiegeln die Exposition gegenüber Abecma in den Studien KarMMa, CRB-401 und KarMMa-3 wider, in denen 409 Patienten mit rezidiviertem und refraktärem multiplem Myelom Abecma erhielten (siehe Abschnitt «Klinische Wirksamkeit»). In KarMMa (N = 128) und CRB-401 (N = 56) betrug die mediane Dauer der Nachkontrollen (von der Abecma-Infusion bis zum Stichtag der Datenerhebung) 20,8 Monate. In KarMMa-3 (N = 225) betrug die mediane Dauer der Nachkontrollen 29,3 Monate.
Zu den häufigsten Nebenwirkungen (Inzidenz ≥20 %) gehörten CRS (84,6 %), Neutropenie (80,0 %), Anämie (63,6 %), Thrombozytopenie (55,0 %), Infektionen – Erreger nicht spezifiziert (43,8 %), Hypophosphatämie (33,3 %), Diarrhö (33,0 %), Leukopenie (32,8 %), Hypokaliämie (32,0 %), Müdigkeit (29,8 %), Übelkeit (28,1 %), Lymphopenie (26,9 %), Pyrexie (24.7 %), Virusinfektionen (23,2 %), Kopfschmerzen (22,5 %) Hypokalzämie (22.0 %), Hypomagnesiämie (21,3 %), Arthralgie (20,0 %) zu den weiteren häufigen Nebenwirkungen, die in einer geringeren Häufigkeit auftraten und als klinisch relevant eingestuft wurden, gehörten, Hypotonie (18,6 %) Infektion der oberen Atemwege (15,6 %), Hypogammaglobulinämie (13,7 %), febrile Neutropenie (11,2 %), Pneumonie (11,0 %), Tremor (5,6 %), Somnolenz (5,6 %), Enzephalopathie (3,4 %), Synkope (3,2 %) und Aphasie (2,9 %).
Schwerwiegende Nebenwirkungen traten bei 57,2 % der Patienten auf. Zu den häufigsten (≥5 %) schwerwiegenden Nebenwirkungen gehörten CRS (10,3 %) und Pneumonie (7,1 %), zu den weiteren schwerwiegenden Nebenwirkungen, die in einer geringeren Häufigkeit auftraten und als klinisch relevant eingestuft wurden, gehörten febrile Neutropenie (4.2 %), Pyrexie (3,7 %), Neutropenie (2,7 %), Sepsis (2,7 %), Verwirrtheitszustand (2,4 %), hämophagozytische Lymphohistiozytose (1,7 %), Thrombozytopenie (1,5 %), Enzephalopathie (1,5 %), Dyspnoe(1,5 %), Krampfanfall (1,0 %), Veränderungen des geistigen Zustands (1,0 %), Hypoxie (0,7 %), und disseminierte intravaskuläre Gerinnung (0,5 %).
Die häufigsten (≥5 %) Nebenwirkungen vom Grad 3 oder 4 waren Neutropenie (77,3 %), Anämie (50.9 %), Thrombozytopenie (42,5 %), Leukopenie (31,5 %), Lymphopenie (25,9 %), Hypophosphatämie (19,8 %), Infektionen (Erreger nicht spezifiziert; 15,2 %), febrile Neutropenie (10,5 %), Virusinfektionen (7,6 %), Pneumonie (6,8 %), Hypertonie (6,6 %), Hypokalzämie (5,6 %) und bakterielle Infektionen (5,4 %).
Beobachtete unerwünschte Arzneimittelwirkungen, die in klinischen Studien bei 409 Patienten unter Abecma innerhalb dem erlaubten Dosierungsbereich von 150 bis 540 x 106 CAR-positiven T-Zellen (siehe Tabelle 3 im Abschnitt «Klinische Wirksamkeit» für den entsprechenden Dosisbereich von CAR-positiven lebensfähigen T-Zellen in KarMMa) auftraten, werden im Folgenden nach MedDRA-Systemorganklasse und nach Häufigkeit dargestellt. Die Häufigkeiten sind definiert als: sehr häufig (≥1/10); häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1'000 bis < 1/100), selten (≥1/10'000 bis < 1/1'000), sehr selten (< 1/10'000) und nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmender Häufigkeit dargestellt.
Infektionen und parasitäre Erkrankungena
Sehr häufig: Infektionen (Erreger nicht spezifiziert) (43,8%), Virusinfektionen (23,2 %), bakterielle Infektionen (15,4%).
Häufig: Pilzinfektionen.
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig: Neutropenie (80,0 %), Anämie (63,6 %), Thrombozytopenie (55,0 %), Leukopenie (32,8 %), Lymphopenie (26,9 %), febrile Neutropenie (11,2 %).
Häufig: Disseminierte intravaskuläre Gerinnung.
Erkrankungen des Immunsystems
Sehr häufig: Zytokinfreisetzungssyndrom (84,6 %), Hypogammaglobulinämie (13,7 %).
Häufig: Hämophagozytische Lymphohistiozytose.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hypophosphatämie (33,3 %), Hypokaliämie (32,0 %), Hypokalzämie (22,0 %), Hypomagnesiämie (21,3 %), verminderter Appetit (17,8 %), Hyponatriämie (13,9 %), Hypoalbuminämie (11,5 %).
Psychiatrische Erkrankungen
Sehr häufig: Schlaflosigkeit (10,3 %).
Häufig: Deliriumb.
Erkrankungen des Nervensystems
Sehr häufig: Enzephalopathied (23,5 %), Kopfschmerzenc (22,5 %), Schwindele (16,1 %).
Häufig: Motorische Dysfunktionf, Tremor, Aphasieg, Ataxieh, Krampfanfall.
Selten: Hemiparese.
Herzerkrankungen
Sehr häufig: Tachykardiei (18,3 %).
Häufig: Vorhofflimmern.
Gefässerkrankungen
Sehr häufig: Hypotoniej (18.6%), Hypertonie (13,9 %).
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Hustenk (18,8 %), Dyspnoel (14,4 %).
Häufig: Hypoxie, Lungenödem.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (33,0 %), Übelkeit (28,1 %), Obstipation (18,3 %), Erbrechen (15,2 %).
Häufig: Gastrointestinale Blutungenm.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Arthralgie (20,0 %).
Häufig: Myalgie.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Müdigkeitn (29,8%), Pyrexie (24,7 %), Ödemeo (19,6 %), Schüttelfrost (10,5 %).
Häufig: Asthenie.
Untersuchungen
Sehr häufig: Aspartat-Aminotransferase erhöht (12,2 %), Alanin-Aminotransferase erhöht (11,2 %).
Häufig: Alkalische Phosphatase im Blut erhöht C-reaktives Protein erhöht.
a Die zur Systemorganklasse der Infektionen und parasitären Erkrankungen gehörenden unerwünschten Arzneimittelwirkungen sind nach Erregertyp und ausgewählten klinischen Syndromen gruppiert.
b Delirium umfasst Delirium, Orientierungslosigkeit, Agitation, Halluzination und Unruhe.
c Kopfschmerzen umfasst Kopfschmerzen und Kopfbeschwerden.
d Enzephalopathie umfasst Amnesie, Bradyphrenie, kognitive Störung, Verwirrtheitszustand, depressive Verstimmung, Störung der Aufmerksamkeit, Dyskalkulie, Dysgraphie, Enzephalopathie, Inkohärenz, Lethargie, Gedächtnisstörung, psychische Beeinträchtigungen, Veränderungen des geistigen Zustands, metabolische Enzephalopathie, Neurotoxizität, Somnolenz, Stupor.
e Schwindel umfasst Schwindel, Präsynkope, Synkope und Vertigo.
f Motorische Dysfunktion umfasst motorische Dysfunktion, Muskelkrämpfe und Muskelschwäche.
g Aphasie umfasst Aphasie und Dysarthrie, langsames Sprechen und Sprechstörungen.
h Ataxie umfasst Ataxie, Dysmetrie und Gangstörung.
i Tachykardie umfasst Sinustachykardie und Tachykardie.
j Hypotonie umfasst Hypotonie und orthostatische Hypotonie.
k Husten umfasst Husten und Hustensyndrom der oberen Atemwege.
l Dyspnoe umfasst Dyspnoe und Belastungsdyspnoe.
m Gastrointestinale Blutungen umfassen gastrointestinale Blutungen, Zahnfleischbluten, Hämatochezie, Hämorrhoidalblutungen, Meläna und Mundblutungen.
n Müdigkeit umfasst Müdigkeit und Unwohlsein.
o Ödeme umfassen Ödeme, periphere Ödeme, Gesichtsödeme, generalisierte Ödeme, und periphere Schwellungen.
Unerwünschte Wirkungen nach Markteinführung
Nicht zutreffend.
Beschreibung ausgewählter unerwünschter Wirkungen
Immunogenität
Abecma hat das Potenzial, Antikörper gegen das Arzneimittel zu induzieren. In klinischen Studien wurde die humorale Immunogenität von Abecma durch Bestimmung der Anti-CAR-Antikörper im Serum vor und nach der Verabreichung gemessen.
In den gepoolten Studien (KarMMa, CRB-401 und KarMMa-3) wurden 3,2 % der Patienten positiv auf Anti-CAR-Antikörper vor der Infusion getestet, und bei 54,0 % der Patienten wurden Anti-CAR-Antikörper nach der Infusion nachgewiesen. Es gibt keine Nachweise darauf, dass das Vorhandensein von Anti-CAR-Antikörpern vor oder nach der Infusion einen Einfluss auf die Zellexpansion, Sicherheit oder Wirksamkeit von Abecma hat.
Zytokinfreisetzungssyndrom
In den gepoolten Studien traten bei 84,6 % der Patienten, die Abecma erhielten, ein CRS auf. Ein CRS vom Grad 3 oder höher (Einstufungssystem nach Lee, 2014) wurde bei 5,1 % der Patienten beobachtet und bei 0,7 % der Patienten verlief es tödlich (Grad 5). Die mediane Zeit bis zum Auftreten (ungeachtet des Grades), betrug 1 Tag (Bereich: 1 bis 17 Tage) und die mediane Dauer des CRS betrug 4 Tage (Bereich: 1 bis 63 Tage).
Zu den häufigsten Manifestationen des CRS (≥10 %) gehörten Pyrexie (82,6 %), Hypotonie (29,1 %), Tachykardie (24,7 %), Schüttelfrost (18,8%), Hypoxie (15,9 %), Kopfschmerzen (11,2 %) und erhöhtes C-reaktives Protein (10,5 %). Zu den Ereignissen vom Grad 3 oder höher, die im Zusammenhang mit CRS beobachtet wurden, gehörten Vorhofflimmern, Kapillarlecksyndrom, Hypotonie, Hypoxie und hämophagozytische Lymphohistiozytose/Makrophagenaktivierungssyndrom (HLH/MAS).
Von den 409 Patienten erhielten 59,7 % der Patienten Tocilizumab; 37,2 % erhielten eine einzelne Verabreichung und 22,5 % mehr als eine Dosis Tocilizumab zur Behandlung des CRS. Insgesamt erhielten 22,7 % der Patienten mindestens eine Dosis Kortikosteroide zur Behandlung des CRS. Von den 92 Patienten in KarMMa und CRB-401 mit der Zieldosis von 450 x 106 CAR-positiven T-Zellen erhielten 54,3 % der Patienten Tocilizumab und 22,8 % mindestens eine Dosis Kortikosteroide zur Behandlung des CRS. Von den 225 Patienten in der KarMMa-3-Studie, die eine Abecma-Infusion erhielten, bekamen 71,6 % der Patienten Tocilizumab und 28,4 % mindestens eine Dosis Kortikosteroide zur Behandlung des CRS.
Neurologische Toxizitäten
In den gepoolten Studien umfassten die häufigsten neurologischen oder psychiatrischen unerwünschten Wirkungen der 409 Patienten (unabhängig davon, ob der Prüfarzt eine Neurotoxizität feststellte) Kopfschmerzen (22,5 %), Schwindel (12,5 %), Verwirrtheitszustand (11,0 %), Schlaflosigkeit (10,3 %), Angstzustände (5,9 %), Tremor (5,6 %) und Somnolenz (5,6 %). Zu den weiteren neurologischen unerwünschten Wirkungen, die weniger häufig auftraten und als klinisch relevant eingestuft wurden, gehörten Enzephalopathie (3,4 %) und Aphasie (2,9 %).
Von den Prüfärzten festgestellte Neurotoxizität, was die primäre Methode zur Beurteilung einer CAR-T-Zellassoziierten Neurotoxizität war, trat in den KarMMa und KarMMa-3 Studien bei 57 (16,1 %) der 353 Patienten auf, die Abecma erhielten, einschliesslich Grad 3 oder 4 bei 3,1 % der Patienten (ohne Ereignisse vom Grad 5). Die mediane Zeit bis zum Auftreten des ersten Ereignisses der Neurotoxizität betrug 3 Tage (Bereich: 1 bis 317, ein Patient entwickelte am 317. Tag eine Enzephalopathie als Folge einer sich verschlimmernden Lungenendzündung und einer Clostridium-difficile-Kolitis). Die mediane Dauer der vom Prüfarzt festgestellten Neurotoxizität betrug 3 Tage (Bereich: 1 bis 252; ein Patient entwickelte 43 Tage nach der ide-cel Infusion Neurotoxizität mit höchstem Grad 3, die nach 252 Tagen abklang). Insgesamt erhielten 7,1 % der Patienten mindestens eine Kortikosteroid-Behandlung als Behandlung von CAR-T-Zell-assoziierter Neurotoxizität, In KarMMa erhielten 7,8 % der Patienten über die Zieldosisstufen hinweg mindestens eine Kortikosteroiddosis zur Behandlung der CAR-T-Zell-assoziierten Neurotoxizität, und bei der Zieldosis von 450 x 106 CAR-positiven T-Zellen 14,8 % der Patienten mindestens eine Kortikosteroid-Behandlung. In der KarMMa-3 Studie erhielten 6,7 % aller Patienten, die eine Abecma-Infusion erhielten, mindestens eine Dosis eines Kortikosteroids zur Behandlung der CAR-T-Zell-assoziierten Neurotoxizität.
Zu den häufigsten Manifestationen der von Prüfärzten festgestellten Neurotoxizität (≥2 %) in den KarMMa und KarMMa-3 Studien (total 353 Patienten), gehörten Verwirrtheitszustand (8,5 %), Enzephalopathie (3,4 %), Aphasie (2,5 %), Tremor (2,3 %), Störung der Aufmerksamkeit (2,0 %) und Dysgraphie (2,0 %).
Das Immuneffektorzell-assoziierte Neurotoxizitätssyndrom (ICANS) wurde als unerwünschte Wirkung während der Anwendung von Abecma nach Markteinführung identifiziert. Da es sich um Meldungen aus einer Population unbekannter Grösse handelt, kann keine Schätzung der Häufigkeit vorgenommen werden.
Infektionen und febrile Neutropenie
In den gepoolten Studien traten Infektionen (bei 62,8% der Patienten auf. Infektionen vom Grad 3 oder 4 traten bei 23,2 % der Patienten auf. Infektionen vom Grad 3 oder 4 mit einem nicht spezifizierten Erreger traten bei 15,2 %, Virusinfektionen bei 7,6 %, bakterielle Infektionen bei 4,6 % und Pilzinfektionen bei 1,2 % der Patienten auf. Tödlich verlaufende Infektionen mit einem nicht spezifizierten Erreger wurden bei 2,0 % der Patienten beobachtet, 0,7 % der Patienten hatten eine tödlich verlaufende Pilzbzw. Virusinfektion und 0,2 % der Patienten hatten eine tödliche verlaufende bakterielle Infektion.
Febrile Neutropenie (Grad 3 oder 4) wurde bei 10,8 % der Patienten nach der Abecma-Infusion beobachtet und kann gleichzeitig mit einem CRS auftreten.
Länger anhaltende Zytopenien
Nach einer Chemotherapie zur Lymphozytendepletion und einer Abecma-Infusion können bei Patienten länger andauernde Zytopenien auftreten. In den gepoolten Studien hatten 151 der 395 Patienten (38,2 %) eine Neutropenie vom Grad 3 oder 4 sowie 164 der 230 Patienten (71,3 %) eine Thrombozytopenie vom Grad 3 oder 4 während des ersten Monats nach der Abecma-Infusion, die bis zur letzten Untersuchung während des ersten Monats nicht zurückgegangen war. Von den 151 Patienten mit einer Neutropenie, die nicht innerhalb des ersten Monats zurückging, erholten sich 88,7 % von der Neutropenie vom Grad 3 oder 4 in einer medianen Zeit bis zur Erholung von 1,9 Monaten nach der Abecma-Infusion. Von den 164 Patienten mit einer Thrombozytopenie, die nicht innerhalb des ersten Monats zurückging, erholten sich 79,9 % von der Thrombozytopenie vom Grad 3 oder 4 in einer medianen Zeit bis zur Erholung von 2,0 Monaten. Angaben zur Überwachung und Behandlung sind im Abschnitt «Warnhinweise und Vorsichtsmassnahmen» aufgeführt.
Hypogammaglobulinämie
Bei 13,7 % der mit Abecma in gepoolten Studien behandelten Patienten wurde über Hypogammaglobulinämie, mit einer durchschnittlichen Zeit bis zum Auftreten von 90 Tagen (Bereich: 1 bis 326), berichtet.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAbecma wird nur durch geschultes medizinisches Personal verabreicht. Die Risiken einer Überdosierung sind nicht bekannt.
Eigenschaften/WirkungenATC-Code
L01XL07
Wirkungsmechanismus
Abecma ist eine chimäre Antigenrezeptor-positive T-Zelltherapie (CAR-positive T-Zelltherapie), die auf das B-Zell-Reifungsantigen abzielt, das auf der Oberfläche von normalen und malignen Plasmazellen exprimiert wird. Das CAR-Konstrukt enthält eine Anti-BCMA-scFv-Zieldomäne für die Antigenspezifität, eine Transmembrandomäne, eine CD3-Zeta-T-Zell-Aktivierungsdomäne und eine 4-1BB kostimulatorische Domäne. Die antigenspezifische Aktivierung von Abecma führt zu CAR-positiver T-Zellproliferation, Zytokinsekretion und anschliessender zytolytischer Abtötung von BCMA-exprimierenden Zellen.
Pharmakodynamik
Nicht zutreffend.
Klinische Wirksamkeit
KarMMa-3
KarMMa-3 ist eine offene, multizentrische, randomisierte, kontrollierte klinische Studie zur Untersuchung der Wirksamkeit und Sicherheit von Abecma im Vergleich zu Standardtherapie-Regimen bei erwachsenen Patienten mit rezidiviertem und refraktärem multiplen Myelom, die zwei bis vier vorangegangene Therapielinien zur Behandlung des multiplen Myeloms erhalten haben, darunter ein immunmodulatorischer Wirkstoff, ein Proteasom-Inhibitor und Daratumumab. Die Patienten waren auf ihre letzte Myelombehandlung refraktär. Jedem Patienten wurde vor der Randomisierung ein Standard-Regime zugewiesen, das sich nach seiner letzten Myelomtherapie richtete. Die Standard-Regimen bestanden aus Daratumumab, Pomalidomid, Dexamethason (DPd); Daratumumab, Bortezomib, Dexamethason (DVd); Ixazomib, Lenalidomid, Dexamethason (IRd); Carfilzomib, Dexamethason (Kd) oder Elotuzumab, Pomalidomid, Dexamethason (EPd). Falls klinisch angezeigt, wurde bei den zum Abecma-Arm randomisierten Patienten das zugewiesene Standard-Regime als Überbrückungstherapie verwendet.
Die Studie schloss Patienten ein, die auf mindestens 1 vorangegangenes Therapie-Regime angesprochen hatten (geringes Ansprechen oder besser) und einen Eastern Cooperative Oncology Group (ECOG) Performance Status von 0 oder 1 aufwiesen. Ausgeschlossen von der Studie waren Patienten mit einer Serumkreatinin-Clearance < 45 ml/min, Serum-Aspartat-Aminotransferase (AST)- oder Serum-Alanin-Aminotransferase (ALT)-Werten > 2,5-fach des oberen Normalwerts und einer linksventrikulären Auswurffraktion (LVEF) < 45 %. Ebenfalls ausgeschlossen waren Patienten mit einer absoluten Neutrophilenzahl < 1000/µl und einer Thrombozytenzahl < 75.000/μl, bei denen < 50 % der kernhaltigen Zellen im Knochenmark Plasmazellen waren, und Patienten mit einer Thrombozytenzahl < 50.000/μl, bei denen ≥50 % der kernhaltigen Zellen im Knochenmark Plasmazellen waren.
Das Durchschnittsalter der Studienpopulation betrug 63 Jahre (Bereich: 30 bis 83 Jahre); 40,9 % waren 65 Jahre oder älter und 60,9 % waren Männer. Der ECOG Performance Status war bei Studienbeginn 0 bei 48,2 %, 1 bei 50,5 % und 2 bei 0,8 % der Patienten.
Neunzig Prozent der Patienten waren refraktär gegenüber eine immunmodulatorischen Medikament (IMiD), 74 % waren refraktär gegenüber einem Proteasom-Inhibitor (PI), und 95 % waren refraktär gegenüber einem monoklonalen Anti-CD38-Antikörper. Sechsundsechzig Prozent waren dreifach refraktär gegenüber den Klassen (refraktär gegenüber einem PI; einem IMiD und einem monoklonalen Anti-CD38 Antikörper).
Die Patienten wurden im Verhältnis 2:1 zu einer Behandlung mit entweder Abecma (n = 254) oder zu Standard-Regime (n = 132) für das rezidivierte und refraktäre multiple Myelom randomisiert. Die Randomisierung erfolgte stratifiziert nach Alter, Anzahl der vorangegangenen Myelomtherapien und zytogenetischen Anomalien mit hohem Risiko. Patienten, die das Standard-Regime erhielten, durften mit Abecma behandelt werden, wenn eine bestätigte Krankheitsprogression vorlag.
Patienten, die zu Abecma randomisiert wurden, sollten eine lymphodepletierende Chemotherapie bestehend aus Cyclophosphamid (300 mg/m2 als i.v.-Infusion täglich über 3 Tage) und Fludarabin (30 mg/m2 als i.v. Infusion täglich über 3 Tage) erhalten, welche 5 Tage vor dem Zieldatum der Infusion von Abecma beginnen sollte. Zwischen der Apherese und bis 14 Tage vor dem Beginn der lymphodepletierenden Chemotherapie war bis zu 1 Zyklus einer Krebstherapie mit DPd, DVd, IRd, Kd oder EPd (Überbrückungstherapie) zur Kontrolle der Erkrankung erlaubt.
Von den 254 zu Abecma randomisierten Patienten unterzogen sich 249 (98 %) einer Leukapherese, und 225 (88,6 %) Patienten erhielten Abecma. Von den 225 Patienten erhielten 192 (85,3 %) Patienten eine Überbrückungstherapie. Bei 29 Patienten wurde aufgrund von Tod (n = 4), unerwünschten Ereignissen (n = 5), Rücktritt (n = 2), Entscheidung des Arztes (n = 7), Nichterfüllung der Kriterien für eine lymphodepletierende Chemotherapie (n = 8) oder Herstellungsausfall (n = 3) kein Abecma angewendet.
Der erlaubte Dosisbereich lag bei 150 bis 540 x 106 CAR-positiven T-Zellen. Die tatsächlich erhaltene mediane Dosis betrug 445,3 x 106 CAR-positive T-Zellen (Bereich: 174,9 bis 529,0 x 106 CAR-positive T-Zellen). Die mediane Zeit von der Leukapherese bis zur Verfügbarkeit des Präparats betrug 35 Tage (Bereich: 24 bis 102 Tage) und die mediane Zeit von der Leukapherese bis zur Infusion betrug 49 Tage (Bereich: 34 bis 117 Tage).
Von den zu den Standard-Regimen randomisierten 132 Patienten erhielten 126 (95,5 %) eine Behandlung. Sechs Patienten brachen die Studie wegen Krankheitsprogression (n = 1), Rücktritt (n = 3) oder Entscheidung des Arztes (n = 2) ab, ohne eine Behandlung erhalten zu haben. Patienten, die ein Standard-Regime erhielten, durften auf Verlangen des Prüfarztes Abecma erhalten, wenn die unabhängige Prüfungskommission (IRC) auf der Grundlage der Kriterien der International Myeloma Working Group (IMWG) eine Krankheitsprogression bestätigte und die Eignung für die Studie bestätigt war. Von den in Frage kommenden Patienten unterzogen sich 69 (54,8 %) einer Leukapherese und 60 (47,6 %) erhielten Abecma.
Der primäre Endpunkt zur Beurteilung der Wirksamkeit war das progressionsfreie Überleben (PFS), definiert nach IMWG Uniform Response Criteria for Mulitple Myeloma und evaluiert durch die IRC.. Weitere Wirksamkeitsendpunkte waren die Gesamtansprechrate (ORR), das Gesamtüberleben (OS) und die von den patient reported outcome (PRO). In der -Zielpopulation (Intent-to-Treat, ITT) betrug die mediane Nachbeobachtungsdauer von der Randomisierung bis zum Stichtag der Datenerhebung 18,6 Monate. Eine Zusammenfassung der Interimsanalyse der Wirksamkeitsergebnisse ist in der Tabelle 3 dargestellt.
Im Abecma-Arm betrug die mediane Ansprechdauer (DOR) bei Patienten mit partiellem Ansprechen (partial response, PR) oder besser 13,9 Monate (KI-95%: 11,2; 17,8). Bei Patienten, die ein vollständiges Ansprechen (complete response, CR) oder besser erreichten, betrug die mediane Dauer des Ansprechens (duration of response, DOR) 20 Monate (KI-95%: 15,8; 24,3).
Tabelle 3: Zusammenfassung der Wirksamkeitsergebnisse basierend auf der KarMMa-3 (Intent-to-Treat-Population)
|
|
Abecma-Arm
(n = 254)
|
Standard-Regime- Arm (n = 132)
| |
Progressionsfreies Überleben (PFS)
| |
Anzahl Ereignisse, n (%)
|
149 (58,7)
|
93 (70,5)
| |
Median, Monate [KI-95%]a
|
13,3 [11,8; 16,1]
|
4,4 [3,4; 5,9]
| |
Hazard ratio [KI-95%]]b
|
0,49 [0,38; 0,65]
| |
Einseitiger p-Wertc
|
< 0,0001
| |
Gesamtansprechrate (ORR)
| |
n (%)
|
181 (71,3)
|
55 (41,7)
| |
KI-95 (%)d
|
(65,7; 76,8)
|
(33,3; 50,1)
| |
Einseitiger p-Werte
|
< 0.0001
| |
CR oder besser (sCR+CR)
|
98 (38,5)
|
7 (5,3)
| |
sCR
|
90 (35,4)
|
6 (4,5)
| |
CR
|
8 (3,1)
|
1 (0,8)
| |
VGPR
|
55 (21,7)
|
13 (9,8)
| |
PR
|
28 (11,0)
|
35 (26,5)
| |
MRD-negativ Status bei NGS and ≥ CR
| |
MRD Negativität-Rate, n (%)f
|
51 (20,1)
|
1 (0,8)
| |
KI-95 (%)d
|
(15,2; 25,0)
|
(0,0; 2,2)
|
KI=Konfidenzintervall; CR=komplettes Ansprechen; MRD=minimal residual disease; PR=Partielles Ansprechen; sCR=stringentes komplettes Ansprechen; VGPR=sehr gutes partielles Ansprechen.
a Kaplan-Meier Schätzung.
b Basierend auf stratifiziertem univariatem Cox proportional hazards model.
c Einseitiger p-Wert basierend auf stratifiziertem log-rank Test.
d Zweiseitiger Forest-Konfidenzintervall.
e Einseitiger p-Wert basierend auf stratifiziertem Cochran-Mantel-Haenszel (CMH) Test.
f MRD-Negativität wurde definiert als der Anteil aller Patienten in der ITT-Population, die eine CR oder stringentes CR und zu einem beliebigen Zeitpunkt innerhalb von drei Monaten vor Erreichen der CR oder stringenten CR bis zum Zeitpunkt der Progression oder des Todes MRD-negativ waren. Basierend auf einem Schwellenwert von 10-5 unter Verwendung von ClonoSEQ, ein next-generation sequencing (NGS) Assay.
Zum Zeitpunkt der finalen PFS-Analyse (Stichtag der Datenerhebung 28.04.2023) mit einer medianen Nachbeobachtungszeit von 30,9 Monaten betrug das mediane PFS für Abecma 13,8 Monate (95% KI: 11,8; 16,1) im Vergleich zu 4,4 Monate für die Standart-Regimen (95% KI: 3,4; 5,8); HR = 0,49 (95% KI: 0,38; 0,63), konsistent mit den Ergebnissen der Interimsanalyse.
Bei der finalen PFS-Analyse wurden 74% der geplanten OS-Ereignisse erreicht. Patienten, die ein Standart-Regime erhielten, konnten bei bestätigtem Fortschreiten der Erkrankung Abecma erhalten. Die OS-Daten sind daher durch 74 (56,1%) Patienten aus dem Standart-Regime-Arm, die Abecma als Folgetherapie erhielten, beeinflusst. Das mediane OS für Abecma betrug 41,4 Monate (95% KI: 30,9; NA) gegenüber 37,9 Monate für die Standart-Regimen (95% KI: 23,4; NA); HR = 1,01 (95% KI: 0,73; 1,40).
Patient reported outcome (PRO)
Deskriptive Analysen
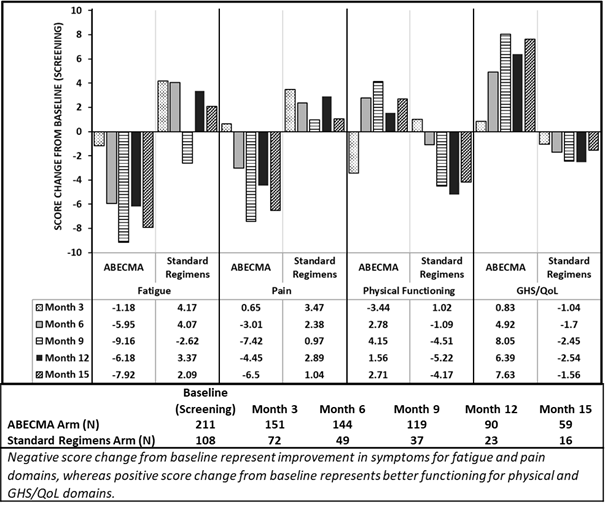
Drei PRO-Messungen (EORTC QLQ-C30, EORTC QLQ-MY20, EQ-5D-5L) wurden zu Studienbeginn (Screening), monatlich bis Monat 24 und danach alle drei Monate durchgeführt. Von den Patienten, die den Fragebogen beantworteten (Abecma n = 211; Standard-Regime n = 108), zeigten sich bei den mit Abecma behandelten Patienten Trends in Richtung Verbesserungen und Unterschiede in den Durchschnittswert-Veränderungen in den meisten PRO-Bereichen, einschliesslich Müdigkeit, Schmerzen, körperliche Funktionsfähigkeit und GHS/QoL, gegenüber dem Ausgangswert im Vergleich zu den mit Standart-Regime behandelten Patienten (Siehe Abbildung 1).
Abbildung 1: EORTC QLQ-C30 - Durchschnittswert-Veränderungen in den Monaten 3, 6, 9, 12 und 15 gegenüber dem Ausgangswert für die Bereiche Müdigkeit, Schmerzen, körperliche Funktionsfähigkeit und GHS/QoL in der KarMMa-3 Studie
Constrained longitudinal Datenanalyse (cLDA)
Beim Vergleich der mittleren Veränderungen der Least Squares (LS) vom Ausgangswert bis zum 25. Monat unter Verwendung von cLDA waren die mittleren LS-Veränderungswerte für die meisten Bereiche der drei PRO-Messungen mit bedeutenden Effektgrössen (Hedge's g > 0.2) zugunsten der mit Abecma behandelten Patienten (Siehe Abbildung 2).
Abbildung 2: Forest plot der Unterschiede zwischen den Gruppen in der gesamten cLDA bei der mittleren Veränderung der LS-Werte gegenüber dem Ausgangswert nach Behandlungsgruppen a,b,c in der KarMMa-3 Studie (Abecma n = 211; Standard-Regimen n = 108)
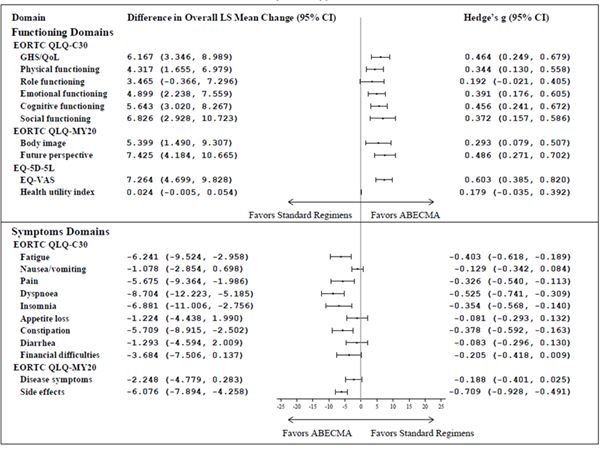
CI = Konfidenzintervall; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life-of-Core 30 Questionnaire; EORTC QLQ-MY20 = European Organization for Research and Treatment of Cancer Quality of Life-of-Questionnaire Multiple Myeloma Module; EQ-VAS = Visual Analogue Scale; GHS = Global health status; LS = Least square; QoL = Quality of Life.
a Guideline von Cohen (1998, 1992) für die Interpretation der Hedge's g values ist 0.20 Indikation für kleine Effekte; 0.50 für mittlere Effekte, und 0.80 für grosse Effekte.
b Primäre Interessensbereiche: EORTC QLQ-C30 Bereiche des globalen Gesundheitszustands/quality of life (QoL), physische Funktion, kognitive Funktion, Müdigkeit und Schmerzen; EORTC QLQ-MY20 Bereiche der Krankheitssymptome und Nebenwirkungen der Behandlung; EQ-5D-5L Gesundheitsindex und EQ-VAS. Die übrigen Bereiche gelten als sekundäre Bereiche.
c Die Analyse beinhaltete keine Multiplikatoren-Bereinigung.
PRO-Time-to-Event Analysen
In der KarMMa-3 Studie wurde die Time-to-Event anhand der Kaplan-Meier-Schätzung ausgewertet. Die Analyse zeigte bei Patienten, die mit Abecma versus Standard-Regime behandelt wurden, die Zeit bis zur Bestätigung einer klinisch bedeutsamen Verbesserung oder Verschlechterung in den PRO Interessensbereichen. Das Eintreten einer Verschlechterung oder Verbesserung wurde als Veränderung gegenüber dem Ausgangswert auf der Grundlage validierter Schwellenwerte definiert und durch eine nachfolgende Bewertung ≥84 Tage nach dem Eintreten bestätigt. Diese wurden in Hazard Ratio (HR) angegeben; HR<1,0 für Verschlechterung und HR>1,0 für Verbesserung, sprach für Abecma. Bei Patienten, die mit Abecma behandelt wurden, verlängerte sich die Zeit bis zur bestätigten Verschlechterung in den meisten Bereichen aller drei PRO's signifikant (HR>1,0). Mit Abecma behandelte Patienten hatten auch eine kürzere Zeitspanne bis zu einer bestätigten Verbesserung in den meisten Bereichen der PRO-Messung (HR>1,0).
KarMMa
KarMMa war eine offene, einarmige, multizentrische Studie zur Beurteilung der Wirksamkeit und Sicherheit von Abecma bei erwachsenen Patienten mit rezidiviertem und refraktärem multiplem Myelom, die mindestens drei vorhergehende Antimyelom-Therapien erhalten hatten, darunter ein immunmodulatorischer Wirkstoff, einen Proteasom-Inhibitor und einen Anti-CD38-Antikörper.
Die Studie bestand aus einer Vorbehandlung (Screening, Leukapherese und Überbrückungstherapie [falls erforderlich]), einer Behandlung (lymphodepletierende Chemotherapie [LDC] und Abecma-Infusion) und einer Nachsorge (laufend) für mindestens 24 Monate nach der Abecma-Infusion oder bis zur dokumentierten Krankheitsprogression, je nachdem, welcher Zeitraum länger war. Der LDC-Abschnitt war ein 3-tägiger Zyklus von Cyclophosphamid (300 mg/m2 i.v. Infusion täglich über 3 Tage) und Fludarabin (30 mg/m2 i.v. Infusion täglich über 3 Tage), der 5 Tage vor dem angestrebten Infusionsdatum von Abecma begann. Die Patienten mussten nach der Abecma-Infusion 14 Tage lang im Spital bleiben und wurden hinsichtlich potenzieller CRS und Neurotoxizität überwacht und behandelt.
Die mit Abecma behandelte Population wies einen hohen Grad an Refraktärität gegenüber früheren Antimyelom-Behandlungen auf: 84,4 % der Studienteilnehmenden waren dreifach refraktär (d.h. refraktär gegenüber einem immunmodulatorischen Wirkstoff, einem Protease-Inhibitor und einem Anti-CD38-Antikörper).
Die Zieldosen in der klinischen Studie waren 150, 300 oder 450 x 106 CAR-positive T-Zellen pro Infusion. Der zulässige Dosisbereich betrug 150 bis 540 x 106 CAR-positive T-Zellen. Die folgende Tabelle 3 zeigt die in der klinischen Studie verwendeten Zieldosen, basierend auf den gesamten CAR-positiven T-Zellen, und den entsprechenden Bereich der tatsächlich verabreichten Dosis, definiert als CAR-positive lebensfähige T-Zellen.
Tabelle 4: Gesamtdosis der CAR-positiven T-Zellen mit dem entsprechenden Dosisbereich der CAR-positiven lebensfähigen T-Zellen (x106)- KarMMa Studie
|
Zieldosis basierend auf den gesamten CAR-positiven T-Zellen, einschliesslich lebensfähiger und nicht lebensfähiger Zellen (x106)
|
CAR-positive lebensfähige T-Zellen (x106)
(min, max)
| |
150
|
133 bis 181
| |
300
|
254 bis 299
| |
450
|
307 bis 485
|
Von 140 Patienten, die einer Leukapherese unterzogen wurden, erhielten 128 Patienten Abecma. Einer der 140 Patienten hat das Produkt aufgrund von Herstellungsmängeln nicht erhalten. 11 weitere Patienten wurden nicht mit Abecma behandelt und zwar aufgrund der Entscheidung des Arztes (n = 3), Abbruch der Teilnahme des Patienten (n = 4), unerwünschter Ereignisse (n = 1), Krankheitsprogression (n = 1) oder Tod (n = 2) vor dem Erhalt von Abecma.
Das mediane Alter der Studienpopulation betrug 60,5 Jahre (Bereich: 33 bis 78 Jahre); 35 % waren mindestens 65 Jahre alt und 59 % waren Männer. Der Eastern Cooperative Oncology Group (ECOG) Performance Status bei Baseline betrug 0 bei 45 %, 1 bei 53 % und 2 bei 2 % der Patienten.
Die meisten Patienten (87,5 %), die mit Abecma behandelt wurden, erhielten eine Überbrückungstherapie zur Kontrolle ihres multiplen Myeloms während dem Herstellungsprozess. Die mediane Zeit von der Leukapherese bis zur Verfügbarkeit des Produkts betrug 32 Tage (Bereich: 24 bis 55 Tage) und die mediane Zeit von der Leukapherese bis zur Infusion 40 Tage (Bereich: 33 bis 79 Tage). Die mediane tatsächliche erhaltene Dosis innerhalb aller Zieldosisstufen lag bei 315,3 x 106 CAR-positiven Zellen (Bereich: 150,5 bis 518,4).
Die Wirksamkeit wurde auf der Grundlage der Gesamtansprechrate (overall response rate, ORR), der Rate des vollständigen Ansprechens (complete response, CR) und der Dauer des Ansprechens (duration of response, DOR) von einem unabhängigen Prüfausschuss (Independent Review Committee, IRC) ermittelt.
Ein weiterer Endpunkt war eine minimale Resterkrankung (minimal residual disease, MRD), die mittels Next-Generation-Sequenzierung (NGS) beurteilt wurde.
Die Wirksamkeitsergebnisse der Zieldosisstufen von 150 bis 450 x 106 CAR-positiven T-Zellen werden in Tabelle 4 dargestellt. In der Primäranalyse, basierend auf der behandelten Population, betrug die ORR 73,4 % (KI 95 %: 65,8; 81,1) und die Rate des vollständigen Ansprechens (CR) betrug 32,8 % (KI 95 %: 24,7; 40,9). Bei Patienten mit partiellem Ansprechen (partial response, PR) oder besser betrug die mediane DOR 10,6 Monate (KI 95 %: 8,0; 11,4). Bei Patienten mit CR oder besser lag die mediane DOR bei 23,3 Monaten (KI 95 %: 11,4; 23,3). Die mediane Nachkontrolldauer betrug bei allen behandelten Patienten 15,4 Monate (Bereich 0,2; 24,2).
Von 140 Patienten in der eingeschlossenen Population betrug die ORR 67,1 % und die CR betrug 30%. Andere Wirksamkeitsergebnisse für die eingeschlossenen Population stimmten mit denen der behandelten Population überein.
Tabelle 5: Zusammenfassung der Wirksamkeitsergebnisse basierend auf der KarMMa-Studie
|
|
Eingeschlossene Population
(n = 140)
|
Behandelte Population
Zieldosis von Abecma (CAR-positive T-Zellen)
| |
[150 x 106]
(n = 4)
|
[300 x 106]
(n = 70)
|
[450 x 106]
(n = 54)
|
[150 bis 450 x 106]
(n = 128)
| |
Gesamtansprechrate (SCR + CR + VGPR + PR), n (%)
|
94 (67,1)
|
2 (50,0)
|
48 (68,6)
|
44 (81,5)
|
94 (73,4)
| |
KI 95 %a
|
59,4, 74,9
|
6,8; 93,2
|
56,4; 79,1
|
68,6; 90,7
|
65,8; 81,1
| |
CR oder besser, n (%)
|
42 (30,0)
|
1 (25,0)
|
20 (28,6)
|
21 (38,9)
|
42 (32,8)
| |
KI 95 %a
|
22,4, 37,6
|
0,6; 80,6
|
18,4; 40,6
|
25,9; 53,1
|
24,7; 40,9
| |
VGPR oder besser, n (%)
|
68 (48,6)
|
2 (50,0)
|
31 (44,3)
|
35 (64,8)
|
68 (53,1)
| |
KI 95 %a
|
40,3, 56,9
|
6,8; 93,2
|
32,4; 56,7
|
50,6; 77,3
|
44,5; 61,8
| |
Patienten mit MRD-negativemb Status und ≥ CR, n
|
|
1
|
17
|
15
|
33
| |
Basierend auf der behandelten Population, %
|
─
|
25,0
|
24,3
|
27,8
|
25,8
| |
KI 95 %a
|
|
0,6; 80,6
|
14,8; 36,0
|
16,5: 41,6
|
18,5; 34,3
| |
Basierend auf Studienteilnehmenden mit ≥ CR, %
|
|
100
|
85,0
|
71,4
|
78,6
| |
KI 95 %a
|
|
2,5; 100,0
|
62,1; 96,8
|
47,8; 88,7
|
63,2; 89,7
| |
Zeit bis zum Ansprechenc, n
|
94
|
2
|
48
|
44
|
94
| |
Median (Monate)
|
1
|
1
|
1
|
1
|
1
| |
Min, Max
|
0,5; 8,8
|
1,0; 1,0
|
0,5; 8,8
|
0,9; 2,0
|
0,5; 8,8
| |
Dauer des Ansprechensc (PR oder besser), n
|
94
|
2
|
48
|
44
|
94
| |
Mediand (Monate)
|
10,6
|
13,0
|
8,5
|
11,3
|
10,6
| |
KI 95 %a
|
8,0; 11,4
|
2,8; 23,3
|
5,4; 10,9
|
10,3; NE
|
8,0; 11,4
| |
Dauer des Ansprechens (CR oder besser), n
|
42
|
1
|
20
|
21
|
42
| |
Mediand (Monate)
|
23,3
|
23,3
|
16,2
|
NE
|
23,3
| |
KI 95 %a
|
11,4; 23,3
|
NE; NE
|
8,0; NE
|
11,4; NE
|
11,4; 23,3
| |
Gesamtüberlebene (OS), Monate, n
|
140
|
4
|
70
|
54
|
128
| |
Median (Monate)
|
21,4
|
18,2
|
NE
|
NE
|
NE
| |
KI 95 %a
|
19,3; NE
|
9,4; NE
|
18,0; NE
|
NE; NE
|
18,9; NE
| |
Ereignisfreie 6-Monats-Rate, %
|
87,4
|
100
|
89,6
|
86,9
|
88,8
| |
Ereignisfreie 12-Monats-Rate, %
|
75,8
|
75,0
|
78,5
|
77,3
|
77,9
|
CAR = chimärer Antigenrezeptor; KI = Konfidenzintervall; CR = complete response (vollständiges Ansprechen); Max = Maximum; Min = Minimum; MRD = Minimal Residual Disease (minimale Resterkrankung); NE = not estimable (nicht abschätzbar); PR = partial response (partielles Ansprechen); sCR = stringent complete response (stringentes vollständiges Ansprechen); VGPR = very good partial response (sehr gutes partielles Ansprechen).
a Für das Total («behandelte Population» und «aufgenommene Population»): Forest-KI; für individuelle Zieldosisstufen: Exaktes KI nach Clopper-Pearson.
b Basierend auf einem Schwellenwert von 10-5 unter Verwendung eines Next-Generation-Sequenzierungsassays.
c Ansprechen ist definiert als das Erreichen von sCR, CR, VGPR oder PR gemäss IMWG-Kriterien.
d Der Median basiert auf der Kaplan-Meier-Schätzung.
e Das OS war definiert als Zeit vom Datum der Leukapherese (aufgenommene Population) oder der Abecma-Infusion (behandelte Population) bis zum Tod aufgrund jeglicher Ursache.
Hinweis: Die Zieldosis beträgt 450 x 106 CAR-positive T-Zellen in einem Bereich von 150 bis 540 × 106 CAR-positiven T-Zellen. Die 150 × 106 CAR-positive T-Zellen-Dosis ist nicht Teil des zugelassenen Dosisbereichs.
Gesundheitsbezogene Lebensqualität (health-related quality of life, HRQoL)
Die HRQoL wurde anhand des C30-Fragebogens zur Lebensqualität (EORTC-QLQ-C30) und des Moduls zu multiplem Myelom (EORTC-QLQ-MY20) der European Organisation for Research and Treatment of Cancer (EORTC) beurteilt, und zwar mit primärem Fokus auf Ermüdung, Schmerzen, körperlicher Funktionsfähigkeit, kognitive Funktionsfähigkeit, allgemeine Gesundheit/Lebensqualität, Nebenwirkungen und Krankheitssymptome auf Subskalen. Gemäss den Ergebnissen aus Daten, die 10 Monate nach der Abecma-Infusion gewonnen wurden, kam es bei den mit Abecma behandelten Patienten kurz nach der Infusion zu klinisch bedeutsamen Verbesserungen in Bezug auf Ermüdung, Schmerzen, körperliche Funktionsfähigkeit und allgemeine Gesundheits-Scores, die zu mehreren Zeitpunkten von Monat 3 bis Monat 9 nach der Behandlung statistisch signifikant (p < 0,05) wurden und zwar ohne Verschlechterung der kognitiven Funktionsfähigkeit, Krankheitssymptome oder Nebenwirkungen. Bei den meisten Endpunkten und Beobachtungspunkten berichtete ein grösserer Prozentsatz der Patienten über eine klinisch bedeutsame Verbesserung und nicht über eine Verschlechterung.
Real-World (RW)-Evidenzstudie
Die RW-Evidenzstudie (Studie NDS-MM-003) war eine retrospektive Beobachtungsstudie, in der Daten über Patienten im Real-World Setting mit rezidiviertem und refraktärem multiplem Myelom (RRMM) gesammelt wurden, die mindestens drei vorgängige Therapien erhalten hatten, darunter einen immunmodulatorischen Wirkstoff, einen PI und einen Anti-CD38-Antikörper. Aus dieser Gruppe wurden Patienten ausgewählt, welche die Auswahlkriterien so nah wie möglich an der KarMMa-Studie erfüllten (d.h. keine Komorbiditäten mit Beginn einer neuen Therapie, nachdem sie gegenüber der letzten Anti-Myelom-Therapie refraktär geworden waren). Die ORR und das Gesamtüberleben (OS) wurden für die beiden Gruppen unter Verwendung der Propensity-Score-Methode bewertet, um die vergleichende Wirksamkeit der mit den verfügbaren Therapien behandelten Patienten im Vergleich zu Abecma in der KarMMa-Studie zu beurteilen. Das relative ORR-Risiko betrug 2,4 (95 % CI: 1,7; 3,3) p < 0,0001. Die OS-Hazard-Ratio betrug 0,41 (95 % CI 0,26, 0,65), was die mit Abecma behandelte Kohorte im Vergleich zu der entsprechenden RRMM-Kohorte, die mit den verfügbaren Therapien behandelt wurde, signifikant begünstigte (p = 0,0002).
Sicherheit und Wirksamkeit bei älteren Patienten
In klinischen Studien zu Abecma waren 163 Patienten (39,9 %) mindestens 65 Jahre alt und 17 (4,2 %) mindestens 75 Jahre alt. Es wurden keine klinisch bedeutsamen Unterschiede bei der Sicherheit oder Wirksamkeit von Abecma zwischen diesen Patienten und Patienten, die jünger als 65 Jahre alt waren, beobachtet.
PharmakokinetikAbsorption
Die Informationen sind für Abecma (ein CAR-T-Zell-Produkt) nicht relevant.
Distribution
Die Informationen sind für Abecma (ein CAR-T-Zell-Produkt) nicht relevant.
Metabolismus
Die Informationen sind für Abecma (ein CAR-T-Zell-Produkt) nicht relevant.
Elimination
Die Informationen sind für Abecma (ein CAR-T-Zell-Produkt) nicht relevant.
Pharmakokinetik
Nach der Abecma-Infusion vermehren sich die CAR-positiven Zellen und durchlaufen eine rasche Multi-Log-Expansion, gefolgt von einem biexponentiellen Rückgang. Die mediane Zeit der maximalen Expansion im peripheren Blut (tmax) trat 11 Tage nach der Infusion ein. Abecma kann im peripheren Blut bis zu 1 Jahr nach der Infusion persistieren. Eine Zusammenfassung von tmax, AUC0-28Tage und Cmax in den KarMMa und KarMMa-3 Studien werden in Tabelle 6 dargestellt.
Tabelle 6: Pharmakokinetische Parameter von Abecma bei Patienten mit rezidiviertem/refraktärem multiplem Myelom
|
Pharmacokinetische Parameter
|
Zusammenfassende Statistik
|
KarMMa Studie
Total [150 to 450 x 106]
CAR-positive T Zellen
(Quantifiziert mit qPCR)a
|
KarMMa-3 Studie
Total [150 to 450 x 106]
CAR-positive T Zellen
(Quantifiziert mit ddPCR)b
| |
Tmax (Tage)
|
Median (Bereich)
|
11 (7-30)
n=127
|
11 (2-31)
n=220
| |
Cmax (Kopien/µg)
|
Geometrisches Mittel (geometrischer CV%)
|
231'278 (178)
n=127
|
115'701 (223)
n=220
| |
AUC0-28days
(Tage*Kopien/ µg)
|
Geometric mean (geometrischer CV%)
|
2'860'340 (197)
n=125
|
1'084'349 (231)
n=218
|
AUC0-28days = Fläche unter der Kurve der Transgenkonzentration vom Zeitpunkt der Dosisgabe bis 28 Tage nach der Infusion; Cmax = maximale Transgenkonzentration; ddPCR = droplet digital polymerase chain reaction; qPCR = quantitative polymerase chain reaction; PK = Pharmakokinetik;
Tmax = Zeit der maximal beobachteten Transgenkonzentration.
a Die PK-Parameter der KarMMa Studie wurden anhand des Zeitverlaufs der Transgenkopien pro Mikrogramm DNA, extrahiert aus CD3+ sortierten Zellen und mittels quantitative polymerase chain reaction (qPCR) quantifiziert.
b Die PK-Parameter der KarMMa-3 Studie wurden anhand des Zeitverlaufs der Transgenkopien pro Mikrogramm DNA, extrahiert aus Vollblut und mit droplet digital PCR (ddPCR) quantifiziert.
Notiz: Die PK-Parameter sollten nicht direkt zwischen KarMMa und KarMMa-3 verglichen werden, da in diesen beiden Studien unterschiedliche PK-Assays verwendet wurden.
Die Transgenkonzentrationen von Abecma wurden positiv mit dem objektiven Tumoransprechen (partielles Ansprechen oder besser) assoziiert. In KarMMa, die medianen Cmax-Werte bei den Respondern (n = 93) waren ca. 4,5 Mal höher als die entsprechenden Werte bei den Non-Respondern (n = 34). Die mediane AUC0-28Tage bei den Respondern (n = 93) war ca. 5,5 Mal höher als bei den Non-Respondern (n = 32). In KarMMa-3 waren die medianen Cmax-Werte bei Respondern (n=180) etwa 5,4-fach höher als die entsprechenden Werte bei Non-Respondern (n=40). Die mediane AUC0-28Tage war bei Respondern (n=180) etwa 5,5-fach höher als bei Non-Respondern (n=38).
Anwendung von Tocilizumab oder Siltuximab und Kortikosteroiden
Einige Patienten benötigten Tocilizumab oder Siltuximab und/oder Kortikosteroide für die Behandlung eines CRS. Abecma kann nach der Gabe von Tocilizumab oder Siltuximab oder Kortikosteroiden weiter expandieren und persistieren (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Patienten in der KarMMa-Studie mit CRS, die mit Tocilizumab behandelt wurden, wiesen höhere Zellexpansionsgrade von Abecma auf, gemessen an der 1,4- bzw. 1,6-fach höheren medianen Cmax (n = 66) bzw. AUC0-28Tage (n = 65), im Vergleich zu Patienten, die kein Tocilizumab erhielten (n = 61 für Cmax und n = 60 für AUC0-28Tage).
Patienten mit CRS, die mit Kortikosteroiden behandelt wurden, wiesen höhere Zellexpansionsgrade von Abecma auf, gemessen an der 1,7- bzw. 2,2-fach höheren medianen Cmax (n = 18) bzw. AUC0-28Tage (n = 18), im Vergleich zu Patienten, die keine Kortikosteroide erhielten (n = 109 für Cmax und n = 107 für AUC0-28Tage).
In der KarMMa-3-Studie wiesen Patienten mit CRS, die mit Tocilizumab oder Siltuximab behandelt wurden, höhere zelluläre Expansionwerte von Abecma auf, gemessen an den 3,1-fachen bzw. 2,9-fach höheren medianen Cmax (n = 156) und AUC0-28Tage (n = 155), im Vergleich zu Patienten, die weder Tocilizumab noch Siltuximab erhielten (n = 64 für Cmax und n = 63 für AUC0-28Tage).
Bei Patienten mit CRS, die mit Kortikosteroiden behandelt wurden, waren die zellulären Expansionswerte von Abecma höher, gemessen am 2,3-fachen und 2,4-fachen Median der Cmax (n = 60) bzw. AUC0-28Tage (n = 60) als bei Patienten, die keine Kortikosteroide erhielten (n = 160 für Cmax und n = 158 für AUC0-28Tage).
Kinetik spezieller Patientengruppen
Leberfunktionsstörung
Es wurden keine Studien zu Leberfunktionsstörungen mit Abecma durchgeführt.
Nierenfunktionsstörung
Es wurden keine Studien zu Nierenfunktionsstörungen mit Abecma durchgeführt.
Ältere Patienten
Das Alter (Bereich: 30 bis 81 Jahre) hatte keinen signifikanten Einfluss auf die Expansionsparameter.
Kinder und Jugendliche
Die Pharmakokinetik von Abecma bei Patienten unter 18 Jahren wurde nicht untersucht.
Sonstige intrinsische Faktoren
Geschlecht, Ethnie und ethnische Herkunft hatten keinen signifikanten Einfluss auf die Expansionsparameter von Abecma. Patienten mit geringem Körpergewicht wiesen eine höhere Expansion auf. Aufgrund der hohen Variabilität der pharmakokinetischen Zellexpansion wird die Gesamtauswirkung des Gewichts auf die Pharmakokinetik von Abecma als nicht klinisch relevant angesehen.
Präklinische DatenAufgrund der Beschaffenheit dieses Produkts wurden herkömmliche Studien zur Toxizität, Fertilität und Pharmakokinetik mit Abecma nicht durchgeführt.
Gentoxizitätsassays und Karzinogenitätsstudien mit Nagetieren sind nicht geeignet, um das Risiko einer Insertionsmutagenese für gentechnisch veränderte Zelltherapieprodukte zu bewerten. Es stehen keine alternativen adäquaten Tiermodelle zur Verfügung.
In-vitro-Expansionsstudien mit CAR-positiven T-Zellen (Abecma) von gesunden Spendern und Patienten zeigten keine Hinweise auf eine Transformation und Immortalisierung von T-Zellen. Eine genomische Insertionsstellenanalyse des lentiviralen Vektors erfolgte mit Abecma-Proben, einschliesslich solchen von Patienten. Es gab keine Belege für eine bevorzugte Integration in der Nähe von bedenklichen Genen oder präferenziellem Wachstum von Zellen, in welchen eine Integration an einer bedenklichen Stelle stattfand.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Dieses Arzneimittel darf nur bis zu dem auf dem Produktetikett angegebenen Verfalldatum («EXP») verwendet werden.
Das für die Infusion vorgesehene Volumen in jedem Beutel muss innerhalb von einer Stunde nach Beginn des Auftauens vollständig infundiert werden.
Besondere Lagerungshinweise
Gefroren in Ethylenvinylacetat-Kryokonservierbeuteln in einem Behälter zur kryogenen Lagerung in der Dampfphase von Flüssigstickstoff (≤ -130 °C) lagern.
Für Kinder unzugänglich aufbewahren.
Hinweise für die Handhabung
Siehe Abschnitt («Dosierung/Anwendung»).
Besondere Vorsichtsmassnahmen für die Handhabung und Entsorgung
Abecma enthält genetisch veränderte menschliche Blutzellen. Es wird aus autologem Blut des Patienten hergestellt, das mittels Leukapherese gewonnen wird. Das Leukapheresematerial der Patienten und Abecma können ein Risiko für die Übertragung infektiöser Viren auf das medizinische Fachpersonal bergen, das mit dem Produkt umgeht. Dementsprechend muss das medizinische Fachpersonal beim Umgang mit Leukapheresematerial oder Abecma angemessene Vorsichtsmassnahmen treffen (Tragen von Handschuhen und Schutzbrille), um eine mögliche Übertragung von Infektionen zu vermeiden.
Arbeitsflächen, die mit Abecma in Berührung gekommen sind oder gekommen sein könnten, müssen mit einem geeigneten Desinfektionsmittel dekontaminiert werden. Nicht verwendetes Arzneimittel oder Materialien, die mit Abecma in Berührung gekommen sind (Fest- und Flüssigabfall), sind als potenziell infektiöser Abfall gemäss den lokalen Biosicherheitsrichtlinien zu handhaben und zu entsorgen.
Zulassungsnummer67575 (Swissmedic)
PackungenDas Fertigprodukt besteht aus einem oder mehreren Infusionsbeuteln, welche eine Gesamtzelldispersion von 260 bis 500 × 106 CAR-positiven lebensfähigen T-Zellen enthalten. Jeder Infusionsbeutel enthält 10–30 ml (50-ml-Beutel), 30–70 ml (250-ml-Beutel) oder 55–100 ml (500-ml-Beutel) Zelldispersion. [A]
Jeder Infusionsbeutel mit Abecma ist mit einer transparenten Plastikhülle umwickelt, die auf die Rückseite des Infusionsbeutels gefaltet und einzeln in einer Metallkassette verpackt ist. Abecma wird in der Dampfphase von Flüssigstickstoff gelagert und in einem Versandbehälter mit Flüssigstickstoff-Trockendampf geliefert.
Dem Versandbehälter liegt ein RFI-Zertifikat bei.
ZulassungsinhaberinBristol-Myers Squibb SA, Steinhausen
Stand der InformationMärz 2025.
|