ZusammensetzungWirkstoffe
Avalglucosidase alfa
Avalglucosidase alfa ist eine humane saure α-Glucosidase, die in Eierstockzellen des chinesischen Hamsters (Chinese hamster ovary cells, CHO) durch rekombinante DNA-Technologie hergestellt und anschliessend mit etwa 7 Hexamannose-Strukturen (die jeweils zwei terminale Mannose-6-phosphat (M6P)-Fragmente enthalten) an oxidierte Sialinsäurereste auf dem Molekül konjugiert wird, wodurch die Bis-M6P-Spiegel erhöht werden.
Hilfsstoffe
L-Histidin, L-Histidinhydrochlorid-Monohydrat, Glycin, Mannitol (E 421), Polysorbat 80.
Indikationen/AnwendungsmöglichkeitenNexviadyme (Avalglucosidase alfa) ist zur langfristigen Enzymersatztherapie bei Patienten mit Morbus Pompe mit später Manifestation (Mangel an saurer α-Glucosidase; late-onset Pompe disease, LOPD) indiziert.
Dosierung/AnwendungDie Behandlung mit Nexviadyme sollte von einem Arzt überwacht werden, der Erfahrung in der Behandlung von Patienten mit Morbus Pompe oder anderen hereditären metabolischen oder neuromuskulären Erkrankungen hat.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Dosierung
Die empfohlene Dosis von Nexviadyme beträgt 20 mg/kg Körpergewicht, die alle zwei Wochen als intravenöse Infusion verabreicht wird.
Patienten mit Leberfunktionsstörungen
Die Sicherheit und Wirksamkeit von Nexviadyme wurde bei Patienten mit Leberfunktionsstörungen nicht untersucht.
Folglich kann für diese Patienten keine spezifische Dosierungsempfehlung abgegeben werden.
Patienten mit Nierenfunktionsstörungen
Dosisanpassungen sind bei Patienten mit leichter Nierenfunktionsstörung nicht erforderlich.
Nexviadyme wurde bei Patienten mit mittelschweren oder schweren Nierenfunktionsstörungen nicht untersucht. Folglich kann für diese Patienten mit mittelschwerer und schwerer Nierenfunktionsstörung keine spezifische Dosierungsempfehlung abgegeben werden.
Ältere Patienten
14 Patienten im Alter von 65 bis 74 Jahren sowie 3 Patienten im Alter von 75 Jahren und älter haben an den klinischen Studien mit Nexviadyme teilgenommen. Eine Dosisanpassung für Patienten im Alter von über 65 Jahren wird nicht empfohlen.
Kinder und Jugendliche
Aus klinischen Studien mit LOPD-Patienten liegen nur begrenzte Daten vor. Zwei pädiatrische Patienten im Alter von 9 und 16 Jahren wurden mit Nexviadyme behandelt.
Art der Anwendung
Nexviadyme ist als intravenöse Infusion zu verabreichen. Wie das Arzneimittel vor der Verabreichung zu rekonstituieren und zu verdünnen ist, ist unter «Hinweise für die Handhabung» beschrieben.
Die Infusionsrate ist schrittweise so anzupassen, dass der Patient optimal auf die Infusion anspricht und diese verträgt. Es wird empfohlen, die Infusion mit einer initialen Infusionsrate von 1 mg/kg/Stunde zu beginnen und diese alle 30 Minuten schrittweise um 2 mg/kg/Stunde zu erhöhen, bis eine maximale Infusionsrate von 7 mg/kg/Stunde erreicht ist, sofern keine Anzeichen für infusionsassoziierte Reaktionen (IARs) auftreten (siehe Tabelle 1). Vor jeder schrittweisen Erhöhung der Infusionsrate sind die Vitalzeichen zu überwachen. Die Patienten können vor der Verabreichung von Nexviadyme mit Antihistaminika, Antipyretika und/oder Kortikosteroiden behandelt werden, um das Risiko von allergischen Reaktionen zu verringern.
Tabelle 1 – Infusionsraten
|
Empfohlene
Dosis
|
Infusionsrate (mg/kg/h)
|
Dauer (h)
| |
Stufe 1
|
Stufe 2
|
Stufe 3
|
Stufe 4
| |
20 mg/kg
|
1
|
3
|
5a
|
7a
|
4 bis 5
|
a Bei Patienten mit einer empfohlenen Dosis von 20 mg/kg und einem Körpergewicht von 1,25 - 5 kg kann eine maximale Infusionsrate von 4,8 mg/kg/h angewendet werden.
Während der Verabreichung von Nexviadyme können allergische Reaktionen, einschliesslich Anaphylaxie und Überempfindlichkeitsreaktionen, sowie IARs auftreten (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»).
Wenn eine Anaphylaxie, eine schwere Überempfindlichkeitsreaktion oder IARs auftreten, muss die Verabreichung von Nexviadyme sofort beendet und eine geeignete medizinische Behandlung eingeleitet werden. Wenn leichte oder mittelschwere Überempfindlichkeitsreaktionen oder IARs auftreten, kann die Infusionsrate reduziert oder die Infusion vorübergehend gestoppt und/oder eine geeignete medizinische Behandlung eingeleitet werden. Die Symptome können trotz Unterbrechung der Nexviadyme-Infusion fortbestehen.
Reexposition nach der Unterbrechung der Nexviadyme-Infusion
Falls die Verabreichung von Nexviadyme aufgrund von unerwünschten Wirkungen, wie vorher beschrieben, abgebrochen wurde, kann nach sorgfältiger klinischer Beurteilung eine vorsichtige Reexposition erwogen werden. Die Erfahrungen aus der klinischen Praxis hinsichtlich einer Reexposition sind jedoch begrenzt. Es gilt zu beachten, dass unerwünschte Wirkungen wie die vorher beschriebenen, einschliesslich schwerer Verläufe, auch bei einer Reexposition mit einer geringeren Dosis wieder auftreten können.
Falls eine Reexposition erwogen wird, sollte nach dem Abklingen der Symptome mindestens 30 Minuten gewartet werden. Die Verabreichung von Nexviadyme kann anschliessend unter engmaschiger Überwachung des Patienten wieder aufgenommen werden, wobei die Dosis höchstens der Hälfte derjenigen entsprechen sollte, bei der die Symptome aufgetreten sind. Je nach dem klinischen Ansprechen des Patienten und nach Ermessen des behandelnden Arztes kann die Dosis daraufhin wieder langsam erhöht werden.
KontraindikationenLebensbedrohliche Überempfindlichkeitsreaktion gegen den Wirkstoff oder einen der Hilfsstoffe, nachdem der erneute Versuch einer Infusion fehlgeschlagen ist (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Warnhinweise und VorsichtsmassnahmenÜberempfindlichkeitsreaktionen einschliesslich Anaphylaxie
Bei mit Nexviadyme behandelten Patienten wurden Überempfindlichkeitsreaktionen, einschliesslich Anaphylaxie, berichtet (siehe «Beschreibung ausgewählter Nebenwirkungen»).
Während der Verabreichung von Nexviadyme sollten geeignete medizinische Unterstützungsmassnahmen, einschliesslich Geräte zur kardiopulmonalen Reanimation, zur Verfügung stehen, insbesondere für Patienten mit Herzhypertrophie sowie für Patienten mit stark beeinträchtigter Atemfunktion.
Bei schwerer Überempfindlichkeitsreaktion oder Anaphylaxie muss Nexviadyme sofort abgesetzt und eine geeignete medizinische Behandlung eingeleitet werden. Risiko und Nutzen sind vor einer erneuten Verabreichung von Nexviadyme nach einer Anaphylaxie oder einer schweren Überempfindlichkeitsreaktion abzuwägen. Einige Patienten wurden erneut mit geringeren Infusionsraten bei einer niedrigeren Dosis als der empfohlenen Dosis behandelt. Bei Patienten mit schwerer Überempfindlichkeitsreaktion kann eine Desensibilisierung gegen Nexviadyme in Erwägung gezogen werden. Falls die Entscheidung getroffen wird, Nexviadyme erneut zu verabreichen, sollte mit äusserster Vorsicht vorgegangen werden, wobei geeignete Massnahmen für den Fall einer Reanimation vorzusehen sind. Wenn der Patient die Infusion verträgt, kann die Dosis bis zum Erreichen der empfohlenen Dosis erhöht werden (siehe «Art der Anwendung»).
Wenn eine leichte oder mittelschwere Überempfindlichkeitsreaktion auftritt, kann die Infusionsrate reduziert oder die Infusion vorübergehend gestoppt werden.
Infusionsassoziierte Reaktionen (IARs)
In klinischen Studien wurde berichtet, dass IARs jederzeit während und/oder innerhalb weniger Stunden nach der Infusion von Nexviadyme auftraten, wobei die Wahrscheinlichkeit mit höheren Infusionsraten anstieg (siehe «Beschreibung ausgewählter Nebenwirkungen»).
Patienten mit einer akuten Grunderkrankung zum Zeitpunkt der Nexviadyme-Infusion können ein höheres Risiko für IARs haben. Patienten mit fortgeschrittenem Morbus Pompe können eine beeinträchtigte Herz- und Atemfunktion haben, was sie für ein höheres Risiko für schwere Komplikationen durch IARs prädisponieren kann. Die Gabe von Antihistaminika, Antipyretika und/oder Kortikosteroide vor der Verabreichung von Nexviadyme kann das Risiko von IARs verringern. Dennoch können auch bei vorbehandelten Patienten IARs auftreten.
Im Falle einer schweren IAR sollte die Nexviadyme-Infusion sofort beendet und eine geeignete medizinische Behandlung eingeleitet werden. Risiko und Nutzen sind vor einer erneuten Verabreichung von Nexviadyme nach schweren IARs abzuwägen. Einige Patienten wurden erneut mit geringeren Infusionsraten bei einer niedrigeren Dosis als der empfohlenen Dosis behandelt. Wenn der Patient die Infusion verträgt, kann die Dosis bis zum Erreichen der empfohlenen Dosis erhöht werden (siehe «Art der Anwendung»).
Falls eine leichte oder mittelschwere IAR unabhängig von einer Vorbehandlung auftritt, kann eine Verringerung der Infusionsrate oder ein vorübergehender Abbruch der Infusion zu einer Verbesserung der Symptome führen (siehe «Beschreibung ausgewählter Nebenwirkungen»).
Immunogenität
Sowohl bei therapienaiven (95 %) als auch bei zuvor mit einer anderen Enzymersatztherapie (ERT) behandelten Patienten (62 %) wurden auf die Behandlung zurückzuführende Antikörper gegen das Arzneimittel (anti-drug antibodies, ADA) berichtet.
IARs und Überempfindlichkeitsreaktionen können unabhängig von der Bildung von ADA auftreten. Die meisten IARs und Überempfindlichkeitsreaktionen waren leicht oder mittelschwer und waren mittels klinischer Standardbehandlung therapierbar. Bei therapienaiven Patienten wurde mit steigendem ADA-Titer ein Trend zur Erhöhung der IAR-Inzidenz beobachtet, wobei die höchste IAR-Inzidenz (69,2 %) im Bereich des höchsten ADA-Titers (≥12 800) berichtet wurde, verglichen mit einer Inzidenz von 33,3 % bei Patienten mit mittlerem ADA-Titer (1 600 bis 6 400), einer Inzidenz von 14,3 % bei Patienten mit einem niedrigen ADA-Titer (100 bis 800) und einer Inzidenz von 33,3 % bei Patienten ohne ADA.
Bei den meisten Patienten wurde kein klinisch signifikanter Effekt von ADA auf die Wirksamkeit festgestellt, jedoch werden die pharmakokinetischen und pharmakodynamischen Parameter in Abhängigkeit vom Titer beeinflusst.
Ein Test auf ADA kann bei Patienten, die nicht auf die Behandlung ansprechen, sinnvoll sein. Bei Patienten, die ein Risiko für eine allergische Reaktion aufweisen oder die eine anaphylaktische Reaktion auf Alglucosidase alfa hatten und bei denen ein unerwünschtes Ereignis auftritt, können immunologische Tests, einschliesslich der Bestimmung von ADA IgG- und IgE-Antikörpertiter in Betracht gezogen werden.
Neutralisierende Antikörper (NAb) wurden bei einigen ADA-positiven Patienten nachgewiesen. 14 Patienten (22,6 %) entwickelten NAb, die die Enzymaktivität und die Zellaufnahme hemmten, 5 Patienten (8,6 %) entwickelten NAb, die nur die Enzymaktivität hemmten und 12 Patienten (19,4 %) entwickelten NAb, die nur die Zellaufnahme hemmten. Die Auswirkung von neutralisierenden Antikörpern auf die klinische Wirksamkeit kann auch mithilfe der verfügbaren klinischen Daten nicht schlüssig beurteilt werden. Es kann jedoch nicht ausgeschlossen werden, dass neutralisierende Antikörper einen negativen Einfluss auf die klinische Wirksamkeit ausüben (siehe «Unerwünschte Wirkungen»).
Bei Patienten, die IgE-Antikörper bilden, kann bei wiederholter Gabe des Arzneimittels ein erhöhtes Risiko für schwere IARs vorliegen. Die Entwicklung von IgE-Antikörpern gegen Avalglucosidase alfa wurde bei einem Patienten nachgewiesen.
Risiko eines akuten kardiopulmonalen Versagens
Bei der Verabreichung von Nexviadyme an Patienten, bei denen es zu einer Volumenüberladung kommen kann, oder an Patienten mit bestehender akuter Atemwegserkrankung oder eingeschränkter Herz- und/oder Atemfunktion, für die eine Flüssigkeitsrestriktion indiziert ist, ist Vorsicht geboten. Bei diesen Patienten besteht während der Infusion möglicherweise das Risiko einer schwerwiegenden Verschlechterung ihres kardialen oder respiratorischen Zustands. Geeignete Massnahmen zur medizinischen Unterstützung und Überwachung sollten während der Nexviadyme-Infusion zur Verfügung stehen. Einige Patienten müssen möglicherweise ihren individuellen Bedürfnissen entsprechend über einen längeren Zeitraum hinweg überwacht werden.
Durch Immunkomplexe vermittelte Reaktionen
Wie bei anderen Enzymersatztherapien besteht auch bei der Gabe von Nexviadyme das Risiko von durch Immunkomplexe vermittelten Reaktionen, beispielsweise der Haut oder der Nieren. Obwohl kein solcher Fall bislang beschrieben wurde, sollte dieses Risiko bei Patienten, die mit Nexviadyme behandelt werden, berücksichtigt werden. Bei Patienten mit hohen IgG-Antikörpertiterwerten sollten regelmässig Urintests durchgeführt werden.
InteraktionenEs wurden keine Interaktionsstudien durchgeführt.
Schwangerschaft, StillzeitSchwangerschaft
Es liegen keine Daten zur Anwendung von Nexviadyme bei Schwangeren vor. Tierexperimentelle Studien ergaben hinsichtlich der Reproduktionstoxizität keine Hinweise auf direkte gesundheitsschädliche Wirkungen. Bei Mäusen wurde vermutet, dass die indirekten Auswirkungen auf die Föten mit einer anaphylaktischen Reaktion auf Avalglucosidase alfa in Zusammenhang stehen (siehe «Präklinische Daten»). Das potenzielle Risiko für den Menschen ist nicht bekannt. Nexviadyme darf während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen für die Mutter die potenziellen Risiken, auch für den Fetus, überwiegt.
Stillzeit
Zum Übergang von Nexviadyme in die Muttermilch oder den Auswirkungen auf die Milchbildung oder den gestillten Säugling liegen keine Daten vor. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Nexviadyme verzichtet werden soll. Dabei ist der Nutzen des Stillens für das Kind gegen den Nutzen der Therapie für die Mutter abzuwägen.
Fertilität
Es liegen keine klinischen Daten zu den Auswirkungen von Nexviadyme auf die Fertilität beim Menschen vor. Tierexperimentelle Studien an Mäusen haben keine Beeinträchtigung der männlichen oder weiblichen Fertilität gezeigt (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine Studien zum Einfluss des Arzneimittels auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, durchgeführt. Schwindelgefühl, Hypotonie und Fatigue, die als IARs berichtet wurden, können die die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen am Tag der Infusion beeinträchtigen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Die gepoolte Sicherheitsanalyse aus 4 klinischen Studien (EFC14028/COMET, ACT14132/mini-COMET, TDR12857/NEO sowie LTS13769/NEO-EXT) umfasste insgesamt 142 mit Nexviadyme behandelte Patienten (118 erwachsene und 24 pädiatrische Patienten (ein pädiatrischer Patient wurde direkt in die unverblindete Verlängerungsphase der COMET-Studie aufgenommen)). Unter Nexviadyme wurden die folgenden schwerwiegenden unerwünschten Wirkungen beobachtet: Atemstörung und Schüttelfrost bei 1,4 % der Patienten, und bei jeweils 0,7 % der Patienten traten Kopfschmerzen, Dyspnoe, Hypoxie, Zungenödem, Übelkeit, Pruritus, Urtikaria, Hautverfärbung, Brustkorbbeschwerden, Pyrexie erhöhter oder erniedrigter Blutdruck, erhöhte Körpertemperatur, erhöhte Herzfrequenz und erniedrigte Sauerstoffsättigung auf. Insgesamt haben 4 Patienten, die im Rahmen von klinischen Studien mit Nexviadyme behandelt wurden, die Therapie endgültig abgebrochen, davon 3 Patienten aufgrund eines schwerwiegenden unerwünschten Ereignisses.
Die am häufigsten berichteten unerwünschten Arzneimittelwirkungen (> 5 %) waren Pruritus (13,4 %), Übelkeit (12 %), Kopfschmerzen (10,6 %), Ausschlag (10,6 %), Urtikaria (8,5 %), Schüttelfrost (7,7 %), Fatigue (7,7 %) und Erythem (5,6 %).
In einer gepoolten Sicherheitsanalyse wurden IARs bei 56/142 (39,4 %) Patienten, die in klinischen Studien mit Avalglucosidase alfa behandelt wurden, berichtet. Schwere IARs wurden bei 6/142 (4,2 %) der Patienten berichtet, darunter Symptome wie Atemstörung, Hypoxie, Brustkorbbeschwerden, generalisiertes Ödem, Zungenödem, Dysphagie, Übelkeit, Erythem, Urtikaria und erhöhter oder erniedrigter Blutdruck. Bei mehr als 1 Patienten berichtete IARs waren Atemstörung, Brustkorbbeschwerden, Dyspnoe, Husten, erniedrigte Sauerstoffsättigung, Rachenreizung, Dyspepsie, Übelkeit, Erbrechen, Diarrhö, geschwollene Lippe, geschwollene Zunge, Erythem, Palmarerythem, Ausschlag, erythematöser Ausschlag, Pruritus, Urtikaria, Hyperhidrose, Hautplaque, okuläre Hyperämie, Augenlidödem, Gesichtsödem, erhöhter oder erniedrigter Blutdruck, Tachykardie, Kopfschmerzen, Schwindelgefühl, Tremor, Brennen, Schmerzen (inkl. Schmerzen in den Extremitäten, Oberbauchschmerzen, Schmerzen im Oropharynx und Flankenschmerz), Somnolenz, Trägheit, Fatigue, Pyrexie, grippeähnliche Erkrankung, Schüttelfrost, Flush, Wärme- oder Kältegefühl, Zyanose und Blässe. Die Mehrheit der IARs war von leichter bis mittelschwerer Ausprägung.
Die unerwünschten Wirkungen, die bei mindestens 2 Patienten (≥1 %), die im Rahmen der in die gepoolte Analyse eingeschlossenen klinischen Studien Nexviadyme erhielten, berichtet wurden, sind nachfolgend aufgelistet.
Die unerwünschten Wirkungen sind nach Systemorganklasse aufgeführt. Hierbei gelten die folgenden Häufigkeitsangaben: sehr häufig (≥1/10), häufig (≥1/100, < 1/10), gelegentlich (≥1/1 000, < 1/100), selten (≥1/10 000, < 1/1 000), sehr selten (< 1/10 000) und nicht bekannt (kann aus den verfügbaren Daten nicht abgeschätzt werden).
Da die Patientenzahl (N = 142) gering ist, wird eine bei 2 Patienten gemeldete unerwünschte Wirkung als häufig eingestuft. Innerhalb jeder Häufigkeitskategorie werden die jeweiligen unerwünschten Wirkungen in absteigender Reihenfolge des Schweregrads aufgeführt.
|
Systemorganklasse
|
Häufigkeit
| |
|
Sehr häufig
|
Häufig
|
Gelegentlich
| |
Infektionen und parasitäre Erkrankungen
|
|
|
Konjunktivitis
| |
Erkrankungen des Immunsystems
|
Überempfindlichkeit (60,6 %)
|
Anaphylaxie
|
| |
Erkrankungen des Nervensystems
|
Kopfschmerzen (10,6 %)
|
Schwindelgefühl
Tremor
Somnolenz
Brennen
|
Parästhesie
| |
Augenerkrankungen
|
|
Okuläre Hyperämie
Bindehauthyperämie
Augenjucken
Augenlidödem
|
Verstärkte Tränensekretion
| |
Herzerkrankungen
|
|
Tachykardie
|
Ventrikuläre Extrasystolen
| |
Gefäßerkrankungen
|
|
Hypertonie
Flush
Hypotonie
Zyanose
Hitzewallung
Blässe
|
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
|
Husten
Dyspnoe
Atemstörung
Rachenreizung
Schmerzen im Oropharynx
|
Tachypnoe
Kehlkopfödem
| |
Erkrankungen des Gastrointestinaltrakts
|
Übelkeit (12,0 %)
|
Diarrhö
Erbrechen
Geschwollene Lippe
Geschwollene Zunge
Abdominalschmerz
Oberbauchbeschwerden
Dyspepsie
|
Orale Hypoästhesie
Orale Parästhesie
Dysphagie
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Pruritus (13,4 %)
Ausschlag (10,6 %)
|
Urtikaria
Erythem
Palmarerythem
Hyperhidrose
Erythematöser Hautausschlag
Ausschlag mit Juckreiz
Hautplaque
|
Angioödem
Hautverfärbung
| |
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
|
|
Muskelspasmen
Myalgie
Schmerz in einer Extremität
Flankenschmerz
|
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
|
Fatigue
Schüttelfrost
Pyrexie
Brustkorbbeschwerden
Schmerzen
Grippeähnliche Erkrankung
Schmerzen an der Infusionsstelle
Asthenie
Gesichtsödem
Kältegefühl
Wärmegefühl
Trägheit
|
Gesichtsschmerzen
Hyperthermie
Extravasat an der Infusionsstelle
Gelenkschmerzen an der Infusionsstelle
Ausschlag an der Infusionsstelle
Reaktion an der Infusionsstelle
Urtikaria an der Infusionsstelle
Lokalisiertes Ödem
Periphere Schwellung
| |
Untersuchungen
|
|
Blutdruck erhöht
Sauerstoffsättigung erniedrigt
Körpertemperatur erhöht
|
Herzfrequenz erhöht
Atemgeräusch anomal
Komplementfaktor erhöht
Immunkomplex-Konzentration erhöht
|
Die Tabelle beinhaltet behandlungsbedingte unerwünschte Ereignisse, bei denen aufgrund der Alglucosidase-alfa-Fachinformation ein biologisch plausibler Zusammenhang mit Avalglucosidase alfa möglich erscheint.
Studie EFC14028/COMET
In der Studie EFC14028/COMET wurden 100 Patienten mit LOPD im Alter von 16 bis 78 Jahren, die noch nie eine ERT erhalten hatten, entweder mit 20 mg/kg Nexviadyme (N=51) oder mit 20 mg/kg Alglucosidase alfa (N=49) behandelt. Während der doppelblinden, aktiv kontrollierten Phase von 49 Wochen wurden bei 2 % der mit Nexviadyme und bei 6,1 % der mit Alglucosidase alfa behandelten Patienten schwerwiegende unerwünschte Wirkungen berichtet. 8,2 % der Patienten, die in der Studie mit Alglucosidase alfa behandelt wurden, brachen die Behandlung aufgrund von unerwünschten Wirkungen dauerhaft ab. In der Nexviadyme-Gruppe wurde die Behandlung bei keinem Patienten dauerhaft abgebrochen. Die am häufigsten gemeldeten unerwünschten Wirkungen (> 5 %) bei Patienten, die mit Nexviadyme behandelt wurden, waren Kopfschmerzen, Übelkeit, Pruritus, Urtikaria und Fatigue.
Von den 95 Patienten, die an der unverblindeten Verlängerungsphase von EFC14028/COMET teilnahmen, setzten 51 Patienten die Behandlung mit Nexviadyme fort und 44 Patienten wechselten von Alglucosidase alfa zu Nexviadyme. Während der unverblindeten Verlängerungsphase wurden bei 3 (5,8 %) Patienten, die die Behandlung mit Nexviadyme während der gesamten Studie fortsetzten, und bei 3 (6,8 %) Patienten, die zu Nexviadyme wechselten, schwerwiegende unerwünschte Wirkungen gemeldet. Die am häufigsten gemeldeten unerwünschten Wirkungen (> 5 %) bei Patienten, die die Behandlung mit Nexviadyme während der gesamten Studie fortsetzten, waren Übelkeit, Schüttelfrost, Erythem, Pruritus und Urtikaria. Die von Patienten, die auf Nexviadyme umgestellt wurden, am häufigsten (> 5 %) berichteten unerwünschten Wirkungen waren Pruritus, Ausschlag, Kopfschmerzen, Übelkeit, Schüttelfrost, Fatigue und Urtikaria.
Beschreibung ausgewählter Nebenwirkungen
Überempfindlichkeitsreaktionen (einschliesslich Anaphylaxie)
In einer gepoolten Sicherheitsanalyse zeigten 86/142 Patienten (60,6 %) Überempfindlichkeitsreaktionen. Darunter waren 7/142 Patienten (4,9 %), die von schwerwiegenden Überempfindlichkeitsreaktionen berichteten, und 4/142 Patienten (2,8 %), die eine Anaphylaxie erlitten. Einige der Überempfindlichkeitsreaktionen waren IgE-vermittelt. Die Symptome einer Anaphylaxie umfassten Zungenödem, Hypotonie, Hypoxie, Atemstörung, Brustkorbbeschwerden, generalisiertes Ödem, generalisiertes Erröten, Wärmegefühl, Husten, Schwindelgefühl, Dysarthrie, Engegefühl im Hals, Dysphagie, Übelkeit, Palmarerythem, Schwellung der Unterlippe, anomale Atemgeräusche, Rötung der Füsse, geschwollene Zunge, Juckreiz an Handflächen und Füssen sowie erniedrigte Sauerstoffsättigung. Die Symptome schwerwiegender Überempfindlichkeitsreaktionen umfassten Zungenödem, respiratorische Insuffizienz, Atemstörung, generalisiertes Ödem, Erythem, Urtikaria und Ausschlag. .
Infusionsassoziierte Reaktionen (IARs)
In einer gepoolten Sicherheitsanalyse wurden bei 56/142 (39,4 %) der in den klinischen Studien mit Nexviadyme behandelten Patienten IARs berichtet. In den klinischen Studien berichteten 6 /142 Patienten (4,2 %) über schwere IARs, einschliesslich Symptome wie Atemstörung, Hypoxie, Brustkorbbeschwerden, generalisiertes Ödem, Zungenödem, Dysphagie, Übelkeit, Erythem, Urtikaria und erhöhter oder erniedrigter Blutdruck.
Die bei mehr als einem Patienten berichteten IARs umfassten Symptome wie Atemstörung, Brustkorbbeschwerden, Dyspnoe, Husten, erniedrigte Sauerstoffsättigung, Rachenreizung, Dyspepsie, Übelkeit, Erbrechen, Diarrhö, geschwollene Lippe, geschwollene Zunge, Erythem, Palmarerythem, Ausschlag, erythematöser Ausschlag, Pruritus, Urtikaria, Hyperhidrose, Hautplaque, okuläre Hyperämie, Augenlidödem, Gesichtsödem, erhöhter oder erniedrigter Blutdruck, Tachykardie, Kopfschmerzen, Schwindelgefühl, Tremor, Brennen, Schmerzen (inkl. Schmerzen in den Extremitäten, Oberbauchschmerzen, Schmerzen im Oropharynx und Flankenschmerz), Somnolenz, Trägheit, Fatigue, Pyrexie, grippeähnliche Erkrankung, Schüttelfrost, Flush, Wärme- oder Kältegefühl, Zyanose und Blässe. Die meisten IARs traten während oder innerhalb der ersten 24 Stunden nach der Gabe von Nexviadyme auf und waren von leichter bis mittelschwerer Ausprägung.
Studie EFC14028/COMET
Während der doppelblinden, aktiv kontrollierten Phase von 49 Wochen berichteten in der Avalglucosidase-alfa-Gruppe weniger LOPD-Patienten über mindestens eine IAR (13/51 [25,5 %]) als in der Alglucosidase-alfa-Gruppe (16/49 [32,7 %]). Bei Patienten in der Avalglucosidase-alfa-Gruppe wurden keine schwerwiegenden IARs berichtet, während 2 Patienten in der Alglucosidase-alfa-Gruppe darüber berichteten. Die häufigsten berichteten, während der Infusionstherapie auftretenden IARs (> 2 Patienten) in der Avalglucosidase-alfa-Gruppe waren Pruritus (7,8 %) und Urtikaria (5,9 %), während es in der Alglucosidase-alfa-Gruppe Übelkeit (8,2 %), Pruritus (8,2 %) und Flush (6,1 %) waren.
Während der unverblindeten Verlängerungsphase wurden bei 12 (23,5 %) Patienten, die die Behandlung mit Nexviadyme während der gesamten Studie fortsetzten, IARs berichtet. IARs, die bei mehr als einem Patienten auftraten, waren Übelkeit, Schüttelfrost, Erythem, Pruritus, Pyrexie, Urtikaria, Ausschlag und okuläre Hyperämie. Bei 22 (50 %) Patienten, die auf Nexviadyme umgestellt wurden, wurden IARs berichtet. IARs, die bei mehr als einem Patienten auftraten, waren Pruritus, Kopfschmerzen, Ausschlag, Übelkeit, Schüttelfrost, Fatigue, Urtikaria, Atemstörung, Kältegefühl, Brustkorbbeschwerden, Erythem, erythematöser Ausschlag, Ausschlag mit Juckreiz, Hautplaque, Brennen, geschwollene Lippe und geschwollene Zunge. Die Zahl der IARs nahm in beiden Gruppen im Laufe der Zeit ab.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht ein Immunogenitätsrisiko. Der Nachweis der Antikörperbildung hängt massgeblich von der Sensitivität und Spezifität des Tests ab. Darüber hinaus kann die Anzahl der positiven Ergebnisse, die bei einem Test auf Antikörper (insbesondere neutralisierender Antikörper) beobachtet wird, von mehreren Faktoren beeinflusst werden. Dazu zählen insbesondere die Testmethodik, die Handhabung der Probe, der Zeitpunkt der Probenahme, die Begleitmedikation und die zugrundeliegende Erkrankung. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Nexviadyme in den nachfolgend beschriebenen Studien sowie die Inzidenz von Antikörpern aus anderen Studien oder gegen andere Arzneimittel irreführend sein.
Die Inzidenz der ADA-Antwort auf Avalglucosidase alfa bei mit Nexviadyme behandelten Patienten mit Morbus Pompe ist in Tabelle 3 dargestellt. Die mediane Zeit bis zur Serokonversion betrug 8,3 Wochen.
Bei therapienaiven Erwachsenen wurde das Auftreten von IARs sowohl bei ADA-positiven als auch bei ADA-negativen Patienten beobachtet. Bei höheren ADA-IgG-Titern wurde eine erhöhte Inzidenz der IARs und der Überempfindlichkeit beobachtet. Bei Erwachsenen, die zuvor eine ERT erhalten hatten, waren IARs und Überempfindlichkeitsreaktionen häufiger, sofern sie im Verlauf der Behandlung ADA entwickelten, als bei denjenigen, bei denen keine ADA auftraten. Bei einem therapienaiven Patienten und zwei mit ERT vorbehandelten Patienten trat eine Anaphylaxie auf. IARs traten bei pädiatrischen Patienten mit positivem Antikörperstatus gegen Avalglucosidase alfa ähnlich häufig auf wie bei solchen mit negativem Antikörperstatus. Bei einem mit einer ERT vorbehandelten pädiatrischen Patienten trat ebenfalls eine Anaphylaxie auf.
In der klinischen Studie EFC14028/COMET entwickelten 81 von 96 Patienten (84,4 %) ADA. Die Mehrheit der Patienten hatte ADA-Titer im niedrigen bis mittleren Bereich, wobei 7 Patienten hohe und anhaltende Antikörpertiter (High Sustained Antibody Titers, HSAT) gegen Nexviadyme aufwiesen. Die Auswertung der ADA-Kreuzreaktivität in Woche 49 zeigte, dass 3 Patienten (5,9 %) Antikörper bildeten, die mit Alglucosidase alfa und Nexviadyme kreuzreaktiv sind. Bei den Patienten mit hohen Titern wurden unterschiedliche Auswirkungen auf PK, PD und Wirksamkeitsmessungen beobachtet, jedoch gab es bei den meisten Patienten keine klinisch signifikanten Auswirkungen von ADA auf die Wirksamkeit.
Tabelle 3 - Inzidenz von ADA, die bei Patienten mit Morbus Pompe unter der Behandlung aufgetreten sind
|
|
Nexviadyme
| |
|
ADA gegen Avalglucosidase alfa bei therapienaiven Patientena
|
ADA gegen Avalglucosidase alfa bei vorbehandelten Patientenb
| |
|
Erwachsene
20 mg/kg einmal in zwei Wochen
(N=62)
N (%)
|
Erwachsene
20 mg/kg einmal in zwei Wochen
(N=58)
N (%)
| |
ADA bei Studienbeginn
|
2 (3,3)
|
43 (74,1)
| |
Während der Behandlung aufgetretene ADA
|
59 (95,2)
|
36 (62,1)
| |
Neutralisierende Antikörper
| |
Beide Arten der neutralisierenden NAb
|
14 (22,6)
|
5 (8,6)
| |
Hemmung der Enzymaktivität, ausschliesslich
|
5 (8,1)
|
6 (10,3)
| |
Hemmung der Enzymaufnahme, ausschliesslich
|
12 (19,4)
|
15 (25,9)
|
a Umfasst zwei pädiatrische Patienten
b Patienten, die mit Alglucosidase alfa vorbehandelt wurden, erhielten diese Therapie vor oder während der klinischen Studie über einen Zeitraum von 0,9 - 9,9 Jahren, bevor sie mit Nexviadyme behandelt wurden.
Kinder und Jugendliche
In klinischen Studien berichtete unerwünschte Arzneimittelwirkungen bei Kindern und Jugendlichen (19 Kinder und Jugendliche mit IOPD zwischen 1-12 Jahren [mittleres Alter: 6,8] und zwei pädiatrische Patienten [9 und 16 Jahre alt] mit LOPD) waren vergleichbar mit denen bei Erwachsenen.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungAnzeichen und Symptome
IARs treten mit einer grösseren Wahrscheinlichkeit bei höheren Infusionsraten auf (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Eigenschaften/WirkungenATC-Code
A16AB22
Wirkungsmechanismus
Morbus Pompe (auch bekannt als Glykogenspeicherkrankheit Typ II, saurer Maltasemangel oder Glykogenose Typ II) ist eine seltene, autosomal-rezessiv vererbte metabolische Muskelerkrankung, die durch einen Mangel an saurer α-Glucosidase (GAA) definiert ist, welche für den Abbau von lysosomalem Glykogen erforderlich ist. Die GAA spaltet die Alpha-1,4- und Alpha-1,6-Bindungen von Glykogen in der sauren Umgebung des Lysosoms. Morbus Pompe führt zu einer intralysosomalen Akkumulation von Glykogen in verschiedenen Geweben, insbesondere im Herzmuskel sowie in der Skelettmuskulatur. Dies führt zur Entwicklung einer Kardiomyopathie, zu fortschreitender Muskelschwäche und zu einer Beeinträchtigung der Atemfunktion.
Avalglucosidase alfa ist eine humane rekombinante saure α-Glucosidase (rhGAA), die eine exogene Quelle für GAA liefert. Bei Avalglucosidase alfa handelt es sich um eine Modifikation der Alglucosidase alfa, bei der etwa 7 Hexamannose-Strukturen, die jeweils 2 terminale Fragmente von Mannose-6-Phosphat (Bis-M6P) enthalten, mit oxidierten Sialinsäureresten auf der Alglucosidase alfa konjugiert sind. Avalglucosidase alfa erhöht die Anzahl der Mannose-6-Phosphat (M6P)-Fragmente im Vergleich zu Alglucosidase alfa um das 15-Fache. Die Erhöhung des Bis-m6P-Spiegels bei der rekombinanten humanen sauren Alpha-Glucosidase führt über den kationenunabhängigen M6P-Rezeptor zu einer verstärkten Aufnahme in das Zwerchfell und andere Skelettmuskeln, wo es Glykogen abbauen und die Gewebeschäden reparieren kann. Die Bindung an die M6P-Rezeptoren auf der Zelloberfläche erfolgt über Kohlenhydratgruppen auf dem Molekül der sauren Alpha-Glucosidase, woraufhin sie internalisiert und zu den Lysosomen transportiert wird. Dort wird sie einer proteolytischen Spaltung unterzogen, die zu einer erhöhten Enzymaktivität zum Glykogen-Abbau führt.
Pharmakodynamik
Bei therapienaiven LOPD-Patienten im Alter von 16 bis 78 Jahren betrug die mittlere prozentuale Veränderung (Standardabweichung, SD) von Hexose-Tetrasacchariden im Urin gegenüber den Ausgangswerten -53,90 % (24,03) in der Woche 49 und -53,35 % (72,73) in der Woche 145 bei Patienten, die alle zwei Wochen 20 mg/kg Nexviadyme erhielten.
Bei Patienten, die alle zwei Wochen mit 20 mg/kg Alglucosidase alfa behandelt wurden betrug die mittlere prozentuale Veränderung (SD) von Hexose-Tetrasacchariden im Urin gegenüber den Ausgangswerten -10,80 % (32,33) in der Woche 49, die nach der Umstellung von Alglucosidase alfa auf Nexviadyme auf -48,04 % (41,97) zurückging in Woche 145.
Klinische Wirksamkeit
Die Sicherheit und Wirksamkeit von Nexviadyme wurden in klinischen Studien mit bei Behandlungsbeginn therapienaiven oder mit ERT vorbehandelten Patienten untersucht.
Klinische Studien, die bei LOPD-Patienten durchgeführt wurden
Die Studie 1, EFC14028/COMET, war eine multinationale, multizentrische, randomisierte, doppelblinde Studie, in der die Wirksamkeit und Sicherheit von Nexviadyme und Alglucosidase alfa bei 100 therapienaiven LOPD-Patienten im Alter von 16 bis 78 Jahren, verglichen wurde. Die Patienten wurden im Verhältnis 1:1 basierend auf dem Ausgangswert der forcierten Vitalkapazität (FVC), Geschlecht, Alter und Land randomisiert und erhielten entweder 20 mg/kg Nexviadyme oder Alglucosidase alfa einmal alle zwei Wochen über 12 Monate (49 Wochen).
Die Studie enthielt eine unverblindete Verlängerungsphase, in der alle Patienten, die Alglucosidase alfa erhalten hatten, auf Nexviadyme umgestellt und mindestens bis Behandlungswoche 145 weiterbehandelt wurden. Insgesamt traten 95 Patienten in die unverblindete Verlängerungsphase ein (51 aus dem Nexviadyme-Behandlungsarm und 44 aus dem Alglucosidase-alfa-Behandlungsarm). Ein zusätzlicher pädiatrischer Patient wurde direkt in die unverblindete Verlängerungsphase aufgenommen.
Der primäre Endpunkt in Studie 1 war die Veränderung der FVC (% des Sollwerts) im Stehen nach 12 Monaten gegenüber den Ausgangswerten (Woche 49). In der Woche 49 betrug die Veränderung des Mittelwerts der kleinsten Quadrate (Standardfehler) für die FVC bei Patienten, die mit Nexviadyme und Alglucosidase alfa behandelt wurden, jeweils 2,89 % (0,88) und 0,46 % (0,93). Die klinisch signifikante Differenz der mittleren kleinsten Quadrate von 2,43 % (95-%-KI: -0,13, 4,99) zwischen Nexviadyme und Alglucosidase alfa lag über der Nichtunterlegenheitsschwelle von -1,1 und erreichte eine statistische Nichtunterlegenheit (p=0,0074). Die Studie hat keine statistische Signifikanz für die Überlegenheit aufgezeigt (p=0,0626) und der Test der sekundären Endpunkte wurde ohne Anpassung für Multiplizität durchgeführt.
Die Ergebnisse für den primären Endpunkt sind in Tabelle 4 und Abbildung 1 dargestellt.
Tabelle 4 – Veränderung der mittleren kleinsten Quadrate der FVC (% des Sollwerts) in aufrechter Position zwischen dem Studienbeginn und der Woche 49
|
|
|
Nexviadyme (N=51)
|
Alglucosidase alfa (N=49)
| |
Ausgangswert vor Aufnahme der Behandlung
|
Mittelwert (Standardabweichung)
|
62,5 (14,4)
|
61,6 (12,4)
| |
Woche 49
|
Mittelwert (Standardabweichung)
|
65,49 (17,42)
|
61,16 (13,49)
| |
Geschätzte Veränderung zwischen dem Ausgangswert und der Woche 49 (MMRM)
|
Mittelwert der kleinsten Quadrate (Standardfehler)
|
2,89* (0,88)
|
0,46* (0,93)
| |
Geschätzte Differenz zwischen den Gruppen hinsichtlich der Veränderung zwischen dem Ausgangswert und der Woche 49 (MMRM)
|
Mittelwert der kleinsten Quadrate (95-%-KI)
p-Wert**
p-Wert***
|
2,43* (-0,13; 4,99)
0,0074
0,0626
|
MMRM: gemischtes Modell mit wiederholten Messungen.
*Basierend auf dem MMRM-Modell umfasst das Modell den initialen FVC % des Sollwerts (als kontinuierlichen Effekt), das Geschlecht, das Alter (in Jahren bei der ersten Studienvisite), die Behandlungsgruppe, die Studienvisite, die Interaktion zwischen der Behandlungsgruppe und der Studienvisite als fixe Effekte.
** Nichtunterlegenheitsgrenze von -1,1 %
*** Überlegenheit nicht erreicht
Abbildung 1: Graphische Darstellung der mittleren Veränderung (SF) der kleinsten Quadrate gegenüber dem Studienbeginn hinsichtlich der FVC (% des Sollwerts) in aufrechter Position im Zeitverlauf bei therapienaiven Patienten mit LOPD (Studie 1)
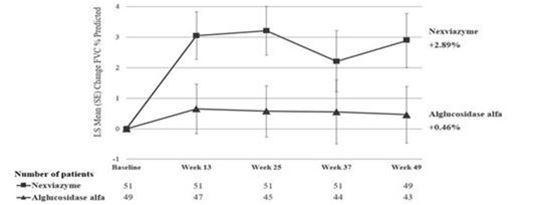
Bei Patienten, die nach Woche 49 von Alglucosidase alfa auf Nexviadyme umgestellt wurden, betrug die mittlere Veränderung des Kleinstquadratmittelwerts der FVC in % des vorhergesagten Normwerts von Woche 49 bis Woche 145 0,81 (1,08) (95 %-KI: -1,32; 2,95). Nach der Umstellung auf Nexviadyme wurde in der Alglucosidase-alfa-Gruppe eine Stabilisierung der FVC in % des vorhergesagten Normwerts mit ähnlichen Werten wie in der Nexviadyme-Gruppe in Woche 145 beobachtet. Bei den Patienten, die weiterhin mit Nexviadyme behandelt wurden, blieb die Verbesserung der FVC in % des vorhergesagten Normwerts im Vergleich zum Ausgangswert erhalten.
Der zentrale sekundäre Endpunkt in Studie 1 war die Veränderung der zurückgelegten Strecke beim 6-Minuten-Walk-Test (6MWT) nach 12 Monaten im Vergleich zum Ausgangswert (Woche 49). In der Woche 49 betrug die Veränderung des Mittelwerts der kleinsten Quadrate (Standardfehler) im Vergleich zum Ausgangswert für die 6MWT bei Patienten, die mit Nexviadyme und Alglucosidase alfa behandelt wurden, jeweils 32,2 Meter (9,93) und 2,19 Meter (10,40). Der klinisch signifikante Unterschied von 30,01 Metern im Mittelwert der kleinsten Quadrate zeigte eine numerische Verbesserung mit Nexviadyme im Vergleich zu Alglucosidase alfa. Die Ergebnisse des 6MWT sowie die anderen sekundären Endpunkte sind in Tabelle 5 ausführlich dargestellt.
Tabelle 5 – Veränderung des Mittelwerts der kleinsten Quadrate zwischen dem Studienbeginn und Woche 49 für die anderen sekundären Endpunkte
|
Endpunkt
|
Nexviadyme
Veränderung des Mittelwerts der kleinsten Quadrate (Standardfehler)
|
Alglucosidase alfa
Veränderung des Mittelwerts der kleinsten Quadrate (Standardfehler)
|
Mittlere Differenz der kleinsten Quadrate
(95-%-KI):
| |
Zurückgelegte Strecke in Metern (6-minute walk test (6MWT))a,b
|
32,21 (9,93)
|
2,19 (10,40)
|
30,01 (1,33;58,69)
| |
Maximaler inspiratorischer Druck (PIM) (% des Sollwerts)c
|
8,70 (2,09)
|
4,29 (2,19)
|
4,40 (-1,63; 10,44)
| |
Maximaler exspiratorischer Druck (PEM) (% des Sollwerts)c
|
10,89 (2,84)
|
8,38 (2,96)
|
2,51 (-5,70; 10,73)
| |
Synthetische Scores für tragbare Kraftmessgeräte (HHD)
|
260,69 (46,07)
|
153,72 (48,54)
|
106,97 (-26,56; 240,5)
| |
Gesamtscore der Messung der motorischen Funktion (QMFT)
|
3,98 (0,63)
|
1,89 (0,69)
|
2,08 (0,22; 3,95)
| |
Untersuchung der gesundheitsbezogenen Lebensqualität (SF-12)
|
PCS-Scored: 2,37 (0,99)
MCS-Scoree: 2,88 (1,22)
|
1,60 (1,07)
0,76 (1,32)
|
0,77 (-2,13;3,67)
2,12 (-1,46; 5,69)
|
a Das MMRM-Modell für die 6MWT-Distanz passt den %-Wert der vorhergesagten FVC zu Beginn und die 6MWT (zurückgelegte Distanz in Metern) zu Beginn, das Alter (in Jahren, zu Beginn), das Geschlecht, die Behandlungsgruppe, die Studienvisite und die Interaktion zwischen Behandlung und der Studienvisite als feste Effekte an.
b Die Veränderung des Mittelwerts zwischen dem Beginn in den Wochen 13, 25 und 37 betrug jeweils 18,02 (8,79), 27,26 (9,98) bzw. 28,43 (9,06) in der Avalglucosidase-alfa-Gruppe und jeweils 15,11 (9,16), 9,58 (10,41) bzw. 15,49 (9,48) in der Alglucosidase-alfa-Gruppe.
c Post-hoc-Sensitivitätsanalyse unter Ausschluss von 4 Patienten (2 in jedem Behandlungsarm) mit supraphysiologischen Ausgangswerten für PIM und PEM.
d Zusammenfassung der physischen Komponente.
e Zusammenfassung der mentalen Komponente.
Bei den Patienten, die nach Woche 49 von Alglucosidase alfa auf Nexviadyme umgestellt wurden, betrug die mittlere Veränderung des Kleinstquadratmittelwerts der 6MWT (zurückgelegte Strecke in Metern) von Woche 49 bis Woche 145 -2,3 m (10,6), 95%-KI: -23,2; 18,7. In Woche 145 wurde eine Stabilisierung der 6MWT nach dem Wechsel von der Alglucosidase-alfa-Gruppe zu Nexviadyme beobachtet. Bei den Patienten im Nexviadyme-Arm wurden die Verbesserung im Vergleich zum Ausgangswert aufrechterhalten.
In einer unverblindeten, nicht kontrollierten Studie mit LOPD-Patienten zeigte sich während der Langzeitbehandlung mit 20 mg/kg Avalglucosidase alfa alle zwei Wochen bis zu 6 Jahre eine anhaltende Wirkung auf die FVC in % des vorhergesagten Normwerts und den 6MWT.
Register für Morbus Pompe Patienten
Angehörige von Gesundheitsberufen bzw. Ärzte werden gebeten, ihre Patienten mit Morbus Pompe im Register einzutragen. Die Patienteninformationen werden in diesem Register anonym erfasst. Mit dem Register sollen das Wissen über Morbus Pompe verbessert und die Patienten sowie ihr Ansprechen auf die Enzymersatztherapie über einen längeren Zeitraum überwacht werden.
PharmakokinetikDie Pharmakokinetik von Avalglucosidase alfa wurde im Rahmen einer Populationsanalyse von 75 Patienten mit LOPD im Alter von 16 bis 78 Jahren beurteilt, die über einen Zeitraum von bis zu 5 Jahren in zweiwöchigen Intervallen zwischen 5 und 20 mg/kg Avalglucosidase alfa erhielten.
Absorption
Bei den Patienten mit LOPD betrugen bei einer vierstündigen intravenösen Infusion von 20 mg/kg, die in zweiwöchigen Intervallen verabreicht wurde, die mittlere Cmax 273 µg/ml (24 %) und die mittlere AUC2W 1 220 µg.h/ml (29 %).
Distribution
Bei Patienten mit LOPD betrug das vom pharmakokinetischen Modell prognostizierte Distributionsvolumen von Avalglucosidase alfa im Zentralkompartiment in der Standardpopulation 3,4 l.
Metabolismus
Nicht zutreffend.
Elimination
Bei Patienten mit LOPD betrug die mit dem pharmakokinetischen Modell prognostizierte lineare Clearance in der Standardpopulation 0,87 l/h. Im Anschluss an die Verabreichung einer Dosis von 20 mg/kg alle zwei Wochen betrug die mittlere Plasma-Eliminationshalbwertszeit 1,55 Stunden.
Linearität/Nicht-Linearität
Die Exposition gegenüber Avalglucosidase alfa stieg bei LOPD-Patienten proportional zur verabreichten Dosis zwischen 5 und 20 mg/kg an. Nach Verabreichung der Dosis alle zwei Wochen wurde keine Akkumulation beobachtet.
Immunogenität
In Studie 1, EFC14028/COMET, entwickelten 96,1 % (49 von 51 Patienten), die Nexviadyme erhielten, Antikörper gegen den Wirkstoff (ADA) während der primären Behandlungsphase bis Woche 49. Angesichts der Tatsache, dass nur 2 Patienten keine ADA entwickelten, wurde die Auswirkung auf die Pharmakokinetik beurteilt, indem die Patienten, die von ADA betroffen waren, in 3 Peak-Titer-Gruppen eingeteilt wurden: ≤800, 1 600 - 6 400 und ≥12 800. Fünf Patienten wiesen im Vergleich zum Studienbeginn in der Woche 49 eine Veränderung der Fläche unter der Kurve (AUC) von 50 % oder mehr auf, wobei sich jedoch kein eindeutiger Trend bezüglich der Titer abzeichnete. Der Vergleich der Fläche unter der Kurve (AUC) am ersten und zweiten Tag und in Woche 49 zwischen den Probanden ging in die Richtung der Gesamtanalyse der prozentualen Veränderung der Fläche unter der Kurve und der ADA-Positivität, die anhand der ADA-Titer kategorisiert wurde. Die in-vitro-Beurteilung von neutralisierenden Antikörpern, die die Enzymaktivität oder die Zellaufnahme hemmten, hat keinen klaren Zusammenhang zwischen der Testpositivität und der AUC aufgezeigt.
Kinetik spezieller Patientengruppen
Populationspharmakokinetische Analysen bei LOPD-Patienten haben aufgezeigt, dass Alter und Geschlecht keinen signifikanten Einfluss auf die pharmakokinetischen Eigenschaften von Avalglucosidase alfa hatten.
Leberfunktionsstörungen
Die pharmakokinetischen Eigenschaften von Avalglucosidase alfa wurden bei Patienten mit Leberfunktionsstörungen nicht untersucht.
Nierenfunktionsstörungen
Zu den Auswirkungen einer Nierenfunktionsstörung auf die Pharmakokinetik von Avalglucosidase alfa wurde keine formelle Studie durchgeführt. Gemäss einer populationspharmakokinetischen Analyse der Daten von 75 LOPD-Patienten, die 20 mg/kg erhielten, darunter 6 Patienten mit leichter Nierenfunktionsstörung (glomeruläre Filtrationsrate: 60 bis 89 ml/min bei der ersten Studienvisite), wurden keine Auswirkungen der Nierenfunktionsstörung auf die Exposition gegenüber Avalglucosidase alfa beobachtet.
Präklinische DatenBasierend auf den konventionellen Studien zur Toxizität bei wiederholter Gabe, die Endpunkte zur Sicherheitspharmakologie beinhalteten, lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. In den meisten Studien an Mäusen wurde vorab Diphenhydramin (DPH) verabreicht, um einer Überempfindlichkeitsreaktion vorzubeugen oder sie abzumildern. Bei den anderen Spezies war DPH nicht erforderlich. Es wurden keine Studien zur Gentoxizität und Kanzerogenität durchgeführt.
Toxizität bei wiederholter Gabe
Wiederholte Dosen von Avalglucosidase alfa bei Mäusen führten zu quantifizierbaren Antikörpern gegen den Wirkstoff und Anzeichen, die mit einer Überempfindlichkeit vereinbar waren. Die chronische Toxizität von Avalglucosidase alfa wurde daher lediglich bei Affen über 26 Wochen untersucht. Bei Affen stellte die verabreichte Dosis von 200 mg/kg Avalglucosidase alfa alle zwei Wochen intravenös die höchste Dosis ohne beobachtete unerwünschte Wirkungen (NOAEL) dar.
Das Expositionsverhältnis (Fläche unter der Kurve [AUC] Exposition gegenüber NOAEL 200 mg/kg einmal in zwei Wochen bei Affen/Exposition gegenüber 20 mg/kg einmal in zwei Wochen bei erwachsenen LOPD-Patienten) beträgt das 23-Fache.
Reproduktionstoxizität
Avalglucosidase alfa führte in einer kombinierten Studie zur Fertilität an männlichen und weiblichen Mäusen in Dosen von bis zu 50 mg/kg i. v. alle zwei Tage (9,4-mal die AUC beim Menschen im Steady State bei empfohlener Dosis von 20 mg/kg alle 2 Wochen bei Patienten mit LOPD) zu keinen schädlichen Wirkungen.
In einer embryofetalen Toxizitätsstudie an Mäusen verursachte die Verabreichung von Avalglucosidase alfa an den Gestationstagen 6 bis 15 in einer Höchstdosis von 50 mg/kg/Tag (das 17-Fache der Steady-State-AUC beim Menschen bei der empfohlenen Dosis von 20 mg/kg alle zwei Wochen für LOPD-Patienten) zu maternalen Toxizität im Zusammenhang mit einer immunologischen Reaktion (einschliesslich einer anaphylaktischen Reaktion). Diese Dosis hatte auch einen höheren Postimplantationsverlust und eine durchschnittliche Anzahl von Spätresorptionen zur Folge.
Avalglucosidase alfa ist bei Mäusen nicht plazentagängig, was darauf hindeutet, dass die unter 50 mg/kg/Tag aufgetretenen embryofetalen Wirkungen auf maternale Toxizität auf die immunologischen Reaktionen zurückzuführen waren. Es wurden weder Missbildungen noch Entwicklungsvariationen beobachtet. Die entwicklungsbezogene NOAEL bei Mäusen betrug 20 mg/kg/Tag (das 4,8-Fache der Steady-state-AUC beim Menschen bei der empfohlenen zweiwöchentlichen Dosis von 20 mg/kg für LOPD-Patienten).
In der Studie zur embryofetalen Toxizität bei Kaninchen führte die intravenöse Verabreichung von Avalglucosidase alfa an den Gestationstagen 6 bis 19 bei Dosen bis zu 100 mg/kg/Tag (das 91-Fache der menschlichen Steady-state-AUC bei einer Dosis von 20 mg/kg alle zwei Wochen, die für LOPD-Patienten empfohlen wird) zu keinen unerwünschten Wirkungen.
In einer Studie zur prä- und postnatalen Entwicklungstoxizität bei Mäusen nach Verabreichung von Avalglucosidase alfa alle zwei Tage vom Gestationstag 6 bis 20 Tage nach der Geburt wurden keine unerwünschten Wirkungen beobachtet. Der NOAEL für die Reproduktion bei den Muttertieren sowie für die Lebensfähigkeit und das Wachstum der Nachkommenschaft betrug 50 mg/kg/Dosis, die intravenös verabreicht wurde.
Toxizitätsprüfungen bei juvenilen Tieren
Bei juvenilen Mäusen hatte die Gabe von Avalglucosidase alfa (bis zu 100 mg/kg alle zwei Wochen intravenös) vom postnatalen Tag (PND) 21 bis PND 77 oder 91 keine Auswirkungen auf Wachstum und Entwicklung. Die Mortalität und die mit der immunologischen Reaktion verbundenen klinischen Anzeichen traten bei allen Dosisstufen auf. Das Expositionsverhältnis (AUC) der Tiere bei einer Dosis von 100 mg/kg alle zwei Wochen war 2,1- bis 3,7-mal höher als die Exposition bei einer Dosis von 40 mg/kg alle zwei Wochen, die bei Patienten mit Morbus Pompe mit Manifestation im Kindesalter (IOPD) angewendet wurde. Da die Behandlung am PND 21 aufgenommen wurde, konnte die Studie das Risiko für Patienten im Alter unter 2 Jahren nicht beurteilen.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2–8 °C) lagern. Avalglucosidase alfa darf nur bis zu dem auf der Durchstechflasche angegebenen Verfalldatum verwendet werden.
Die rekonstituierte und verdünnte Lösung muss umgehend verabreicht werden. Das rekonstituierte Produkt kann bis zu 24 Stunden im Kühlschrank bei 2 °C bis 8 °C und das verdünnte Produkt kann bis zu 24 Stunden im Kühlschrank bei 2 °C bis 8 °C und bis zu 9 Stunden (einschliesslich der Infusionszeit) bei Raumtemperatur (bis zu 25 °C) gelagert werden.
Hinweise für die Handhabung
Die Durchstechflaschen sind zum einmaligen Gebrauch bestimmt. Nicht aufgebrauchte Arzneimittel oder Abfälle müssen gemäss den vor Ort geltenden Bestimmungen entsorgt werden.
Wenden Sie bei der Zubereitung eine aseptische Technik an.
1.Bestimmen Sie die Anzahl der zu rekonstituierenden Durchstechflaschen basierend auf dem Gewicht des Patienten und der empfohlenen Dosis von 20 mg/kg.Gewicht des Patienten (kg) x Dosis (mg/kg) = Patientendosis (in mg). Patientendosis (in mg) geteilt durch 100 mg/Durchstechflasche = Anzahl der zu rekonstituierenden Durchstechflaschen. Falls die Anzahl der Durchstechflaschen keine ganze Zahl ist, runden Sie diese auf die nächste ganze Zahl auf.Beispiel: Gewicht des Patienten (16 kg) x Dosis (20 mg/kg) = Patientendosis (320 in mg). 320 mg geteilt durch 100 mg/Durchstechflasche = 3,2 Durchstechflaschen. Folglich müssen 4 Durchstechflaschen rekonstituiert werden.
2.Entnehmen Sie die für die Infusion benötigte Anzahl an Durchstechflaschen aus dem Kühlschrank und stellen Sie sie für etwa 30 Minuten beiseite, damit sie Raumtemperatur annehmen.
3.Rekonstituieren Sie jede Durchstechflasche, indem Sie nach und nach 10,0 ml steriles Wasser für Injektionszwecke injizieren. Jede Durchstechflasche wird 100 mg/10 ml (10 mg/ml) ergeben. Vermeiden Sie, dass das Wasser für Injektionszwecke auf das Pulver aufprallt und dass Schaum entsteht. Geben Sie hierfür das Wasser für Injektionszwecke langsam und tropfenweise in die Durchstechflasche und nicht direkt auf das lyophilisierte Pulver. Kippen und rollen Sie die Durchstechflasche vorsichtig. Drehen Sie die Durchstechflasche nicht um und schwenken und schütteln Sie sie nicht. Warten Sie, bis sich das Pulver aufgelöst hat. Achten Sie darauf, dass während der Verdünnung des Produkts keine Luft in das Innere des Infusionsbeutels gelangt.
4.Überprüfen Sie die rekonstituierten Durchstechflaschen umgehend auf Partikel und Verfärbungen. Falls die Lösung verfärbt ist oder sichtbare Partikel enthält, darf sie nicht verwendet werden.
5.Die rekonstituierte Lösung sollte mit einer 5%igen Glucoselösung verdünnt werden, um eine Endkonzentration zwischen 0,5 mg/ml und 4 mg/ml zu erhalten. Das empfohlene Gesamtinfusionsvolumen je nach Gewicht des Patienten finden Sie in der Tabelle 6.
6.Entnehmen Sie langsam die rekonstituierte Lösung aus jeder Durchstechflasche (das Volumen wird in Abhängigkeit vom Gewicht des Patienten berechnet).
7.Geben Sie die rekonstituierte Lösung langsam direkt in die 5%ige Glucoselösung. Vermeiden Sie Schaumbildung oder Schütteln des Infusionsbeutels. Achten Sie darauf, dass keine Luft in den Infusionsbeutel gelangt.
8.Drehen Sie den Beutel um oder massieren Sie ihn vorsichtig, um den Inhalt zu vermischen. Nicht schütteln.
9.Es wird empfohlen, zur Verabreichung von Nexviadyme einen Inlinefilter mit einer geringen Proteinbindung von 0,2 μm zu verwenden. Nach Beendigung der Infusion spülen Sie mit einer 5%igen Glucoselösung im Infusionsbeutel nach.
10.Infundieren Sie Nexviadyme nicht zusammen mit anderen Produkten über denselben intravenösen Zugang.
Tabelle 6: Vorgesehenes Volumen für die intravenöse Infusion von Nexviadyme je nach Gewicht des Patienten in der Dosierung von 20 mg.
|
Patientengewichtsbereich
(kg)
|
Gesamtinfusionsvolumen für 20 mg/kg
(ml)
| |
1,25 bis 10
|
50
| |
10,1 bis 20
|
100
| |
20,1 bis 30
|
150
| |
30,1 bis 35
|
200
| |
35,1 bis 50
|
250
| |
50,1 bis 60
|
300
| |
60,1 bis 100
|
500
| |
100,1 bis 120
|
600
| |
120,1 bis 140
|
700
| |
140,1 bis 160
|
800
| |
160,1 bis 180
|
900
| |
180,1 bis 200
|
1000
|
Zulassungsnummer67871 (Swissmedic)
PackungenNexviadyme 100 mg/Durchstechflasche für Konzentrat zur Herstellung einer Infusionslösung.
·Packung mit 1 Durchstechflasche aus Glas zu 100 mg/10 ml zum einmaligen Gebrauch [A]
·Packung mit 5 Durchstechflaschen aus Glas zu 100 mg/10 ml zum einmaligen Gebrauch [A]
·Packung mit 10 Durchstechflaschen aus Glas zu 100 mg/10 ml zum einmaligen Gebrauch [A]
Zulassungsinhaberinsanofi-aventis (schweiz) sa, 1214 Vernier/GE
Stand der InformationJuni 2024
|