ZusammensetzungWirkstoffe
Vericiguat.
Hilfsstoffe
Verquvo 2.5 mg Filmtabletten enthalten: Mikrokristalline Cellulose, Croscarmellose-Natrium, Hypromellose, Lactose-Monohydrat (61.2 mg), Magnesiumstearat, Natriumdodecylsulfat, Überzug: Hypromellose, Talkum, Titandioxid (E171). Natriumgehalt pro Filmtablette: 0.80 mg.
Verquvo 5 mg Filmtabletten enthalten: Mikrokristalline Cellulose, Croscarmellose-Natrium, Hypromellose, Lactose-Monohydrat (58.5 mg), Magnesiumstearat, Natriumdodecylsulfat, Überzug: Hypromellose, Talkum, Titandioxid (E171), Eisenoxid rot (E172). Natriumgehalt pro Filmtablette: 0.81 mg.
Verquvo 10 mg Filmtabletten enthalten: Mikrokristalline Cellulose, Croscarmellose-Natrium, Hypromellose, Lactose-Monohydrat (117.0 mg), Magnesiumstearat, Natriumdodecylsulfat, Überzug: Hypromellose, Talkum, Titandioxid (E171), Eisenoxid gelb (E172). Natriumgehalt pro Filmtablette: 1.65 mg.
Indikationen/AnwendungsmöglichkeitenVerquvo wird angewendet zur Behandlung von symptomatischer, chronischer Herzinsuffizienz bei
erwachsenen Patienten mit reduzierter Auswurffraktion, die nach einer kürzlich aufgetretenen
Dekompensation, welche eine i.v.-Therapie erforderte, stabilisiert wurden.
Verquvo wird in Kombination mit anderen Therapien für Herzinsuffizienz angewendet (siehe Rubrik «Dosierung/Anwendung» und «Eigenschaften/Wirkungen»).
Dosierung/AnwendungVor Initiierung der Behandlung mit Verquvo muss eine ausreichende Stabilisierung nach kürzlich aufgetretener Dekompensation sichergestellt werden, insbesondere bei Patienten mit stark erhöhten NT-proBNP-Spiegeln. Die klinische Stabilisierung schliesst die Behandlung der Volumenüberladung mittels intensivierter (intravenöser) Diuretika-Therapie und die Optimierung der Behandlung mit anderen Standardtherapeutika für Herzinsuffizienz ein (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Die empfohlene Anfangsdosis von Verquvo beträgt 2.5 mg einmal täglich und ist zusammen mit Nahrung einzunehmen. Die Dosis von Verquvo wird je nach Verträglichkeit etwa alle 2 Wochen verdoppelt, um die angestrebte Erhaltungsdosis von 10 mg einmal täglich zu erreichen.
Für Patienten, die keine ganzen Tabletten schlucken können, kann Verquvo zerstossen und unmittelbar vor der Einnahme mit Wasser (ohne Kohlensäure) gemischt werden (siehe Rubrik «Pharmakokinetik»).
Verspätete Dosisgabe
Wird eine Einnahme versäumt, sollte sie am selben Tag nachgeholt werden, sobald sich der Patient daran erinnert. Die Patienten dürfen nicht zwei Dosen von Verquvo am selben Tag einnehmen.
Spezielle Patientengruppen
Eingeschränkte Leberfunktion
Bei Patienten mit leichter oder mässiger Leberfunktionsstörung ist keine Dosisanpassung von Verquvo erforderlich. Die Anwendung von Verquvo bei Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht und wird daher für diese Patientengruppe nicht empfohlen (siehe Rubrik «Pharmakokinetik»).
Eingeschränkte Nierenfunktion
Bei Patienten mit einer geschätzten glomerulären Filtrationsrate (eGFR) von ≥15 ml/min/1.73 m2 (ohne Dialyse) ist keine Dosisanpassung von Verquvo erforderlich. Die Anwendung von Verquvo bei Patienten mit einer eGFR <15 ml/min/1.73 m2 zu Behandlungsbeginn oder unter Dialyse wurde nicht untersucht und wird daher für diese Patientengruppen nicht empfohlen (siehe Rubrik «Pharmakokinetik»).
Ältere Patienten (≥65 Jahre)
Bei älteren Patienten ist keine Dosisanpassung von Verquvo erforderlich (siehe Rubrik «Pharmakokinetik»). Insgesamt wurden zwischen Patienten ab 65 Jahren verglichen mit jüngeren Patienten keine Unterschiede bei der Sicherheit und Wirksamkeit von Vericiguat beobachtet. Eine erhöhte Empfindlichkeit von manchen älteren Personen kann jedoch nicht ausgeschlossen werden
Kinder und Jugendliche
Die Wirksamkeit und Sicherheit von Verquvo wurde für Kinder und Jugendliche unter 18 Jahren nicht untersucht (siehe Rubrik «Pharmakokinetik»).
Kontraindikationen·Verquvo ist bei Patienten, die gleichzeitig andere Stimulatoren der löslichen Guanylatcyclase (sGC) wie Riociguat erhalten, kontraindiziert (siehe Rubrik «Interaktionen»).
·Überempfindlichkeit gegenüber einem der Hilfsstoffe (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
·Verquvo ist kontraindiziert bei Patienten, die gleichzeitig mit einem Phosphodiesterase 5 –Inhibitor (z.B. Sildenafil) behandelt werden (siehe Rubrik «Interaktionen»).
Warnhinweise und VorsichtsmassnahmenPatienten mit stark erhöhten NT-proBNP
Vordefinierte Subgruppenanalysen und post-hoc-Analysen der VICTORIA-Studie lassen darauf schliessen, dass sich die kardiovaskuläre Mortalität und das Risiko einer Hospitalisierung aufgrund von Herzinsuffizienz bei Patienten mit sehr hohen NT-proBNP Spiegeln durch die Behandlung mit Verquvo erhöhen (siehe Rubrik «Klinische Wirksamkeit»). Zusätzliche post-hoc-Analysen weisen auf eine ungenügende Stabilisierung dieser Patienten nach vorangegangener Dekompensation hin. Vor Aufnahme einer Behandlung mit Verquvo muss daher die ausreichende Stabilisierung der betreffenden Patienten sichergestellt werden (siehe Rubrik «Dosierung/Anwendung»).
Symptomatische Hypotonie
Unter der Behandlung mit Verquvo kann eine symptomatische Hypotonie auftreten. In der klinischen Studie VICTORIA traten unerwünschte Ereignisse, die vom Prüfer als Ereignisse symptomatischer Hypotonie festgestellt wurden, bei 9.1% der Patienten unter Verquvo und 7.9% der Patienten unter Placebo auf, die bei 1.2% der Patienten unter Verquvo und 1.5% der Patienten unter Placebo als schwerwiegend angesehen wurden (siehe Rubrik «Unerwünschte Wirkungen»).
Die Anwendung von Verquvo bei Patienten mit einem systolischen Blutdruck unter 100 mmHg oder symptomatischer Hypotonie zu Behandlungsbeginn wurde nicht untersucht.
Bei Patienten mit Hypovolämie, schwerer linksventrikulärer Ausflussobstruktion, Ruhe-Hypotonie, autonomer Dysfunktion, anamnestisch bekannter Hypotonie oder unter gleichzeitiger Behandlung mit Antihypertensiva oder organischen Nitraten muss das Potential für symptomatische Hypotonie berücksichtigt werden (siehe Rubrik «Interaktionen»). Bei Auftreten einer symptomatischen Hypotonie ist eine Dosisanpassung von Diuretika und die Behandlung anderer Ursachen der Hypotonie (z.B. Hypovolämie) zu erwägen. Falls die symptomatische Hypotonie trotz dieser Massnahmen bestehen bleibt, ist eine vorübergehende Reduktion der Dosis oder ein Aussetzen von Verquvo in Betracht zu ziehen.
Hilfsstoffe
Verquvo enthält je nach Dosisstärke 55.59 – 111.15 mg Laktose (als Laktosemonohydrat). Patienten mit der seltenen hereditären Galaktose-Intoleranz, völligem Lactase-Mangel oder Glukose-Galaktose Malabsorption sollten Verquvo nicht anwenden.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro einzunehmende Dosis d.h. es ist nahezu «natriumfrei».
InteraktionenPharmakokinetische Interaktionen
Einfluss anderer Substanzen auf die Pharmakokinetik von Vericiguat
Arzneimittel, die den pH-Wert im Magen erhöhen (z.B. Protonenpumpenhemmer, H2-Rezeptor-Antagonisten, Antazida)
Vericiguat ist bei neutralem pH-Wert weniger löslich als bei saurem pH-Wert. Bei vorausgegangener und gleichzeitiger Behandlung mit Arzneimitteln, welche den pH-Wert im Magen erhöhen, wie z.B. Protonenpumpeninhibitoren oder Antazida, verringert sich die Vericiguat-Exposition (AUC) nach Einnahme unter Nüchternbedingungen um etwa 30%. Eine gleichzeitige Behandlung mit Arzneimitteln, welche den pH-Wert im Magen erhöhen, hatte hingegen keine Auswirkungen auf die Vericiguat-Exposition bei Patienten mit Herzinsuffizienz, wenn Vericiguat wie angewiesen mit Nahrung eingenommen wurde. (siehe Rubrik «Dosierung/Anwendung»).
Vericiguat ist ein Substrat von UGT1A9 und UGT1A1 und der Transporter P-Glycoprotein (Pgp) und Brustkrebs-Resistenz-Protein (BCRP).
Es wurde keine klinisch relevante Wirkung auf die Vericiguat-Exposition beobachtet, wenn Vericiguat zusammen mit Ketoconazol (Multiweg-CYP- und Transporterinhibitor), Mefenaminsäure (UGT1A9-Inhibitor), Rifampicin (Multiweg-UGT, CYP und Transporterinduktor), Digoxin, Warfarin, Aspirin, Sildenafil oder der Kombination Sacubitril/Valsartan verabreicht wurde.
Es wird keine klinisch relevante Wirkung auf die Vericiguat-Exposition erwartet, wenn Vericiguat zusammen mit Atazanavir (UGT1A1-Inhibitor) verabreicht wird.
Basierend auf in vitro Daten ist Vericiguat kein Substrat der organischen Kationentransporter (OCT1) oder organischen Anionen-transportierenden Polypeptide (OATP1B1 und OATP1B3).
Einfluss von Vericiguat auf die Pharmakokinetik anderer Substanzen
In-vitro-Studien deuten darauf hin, dass Vericiguat und sein N-Glucuronid bei klinisch relevanten Konzentrationen weder Inhibitoren der wichtigsten CYP-Isoformen (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 und 3A4) oder UGT-Isoformen (UGT1A1, 1A4, 1A6, 1A9, 2B4 und 2B7) noch Induktoren von CYP1A2, 2B6 und 3A4 sind.
In-vitro-Studien deuten darauf hin, dass Vericiguat und sein N-Glucuronid bei klinisch relevanten Konzentrationen keine Inhibitoren von Arzneimitteltransportern einschliesslich Pgp, BCRP, BSEP, OATP1B1/1B3, OAT1, OAT3, OCT1, OCT2, MATE1 und MATE2K.
Die gleichzeitige Verabreichung von Vericiguat mit Midazolam (CYP3A-Substrat) oder Digoxin (P gp-Substrat) zeigte in In-vivo-Studien keine klinisch relevante Wirkung auf die Exposition von Midazolam, bzw. Digoxin.
Zusammen deuten diese Daten darauf hin, dass bei Verabreichung von Vericiguat keine Beeinflussung der Pharmakokinetik gleichzeitig gegebener Arzneimittel, welche Substrate dieser Enzyme oder Transporter sind, zu erwarten ist.
Pharmakodynamische Interaktionen
Gleichzeitige Anwendung kontraindiziert
Andere Stimulatoren der löslichen Guanylatcyclase
Verquvo ist bei Patienten, die gleichzeitig andere Stimulatoren der löslichen Guanylatcyclase (sGC) wie Riociguat erhalten, kontraindiziert (siehe Rubrik «Kontraindikationen»).
PDE-5-Inhibitoren
Die gleichzeitige Anwendung von Verquvo und Phosphodiesterase-5 (PDE-5) - Inhibitoren wie Sildenafil wurde bei Patienten mit Herzinsuffizienz nicht untersucht und ist aufgrund des potenziell erhöhten Risikos für symptomatische Hypotonie kontraindiziert (siehe «Kontraindikationen»).
Die Zugabe einer Einzeldosis Sildenafil (25, 50 oder 100 mg) zu mehreren Dosen Vericiguat (10 mg) einmal täglich bei gesunden Probanden war mit einer zusätzlichen Senkung des Blutdrucks im Sitzen ≤5,4 mmHg (systolischer/diastolischer Blutdruck, mittlerer arterieller Druck) im Vergleich zur Verabreichung von Vericiguat allein verbunden.
Die gleichzeitige Verabreichung war nicht mit einem klinisch relevanten Effekt auf die Exposition (AUC und Cmax) der beiden Arzneimittel verbunden.
Weitere Interaktionen
Acetylsalicylsäure
Die Verabreichung einer Einzeldosis von Vericiguat 15 mg führte bei gesunden Probanden nicht zu einer Veränderung der Wirkung von 500 mg Acetylsalicylsäure auf die Blutungszeit oder die Thrombozytenaggregation. Blutungszeit und Thrombozytenaggregation blieben unter 15 mg Vericiguat allein unverändert.
Die gleichzeitige Verabreichung von Acetylsalicylsäure war nicht mit einem klinisch relevanten Effekt auf die Exposition (AUC und Cmax) von Vericiguat verbunden.
Warfarin
Die Verabreichung mehrerer Dosen von Vericiguat 10 mg einmal täglich führte bei gesunden Probanden nicht zu einer Veränderung der Wirkung einer Einzeldosis von 25 mg Warfarin auf die Prothrombinzeit und die Aktivität der Faktoren II, VII und X.
Die gleichzeitige Verabreichung war nicht mit einem klinisch relevanten Effekt auf die Exposition (AUC und Cmax) der beiden Arzneimittel verbunden.
Kombination Sacubitril/Valsartan
Die zusätzliche Gabe mehrerer Dosen von Vericiguat 2.5 mg zu mehreren Dosen von Sacubitril/Valsartan 97/103 mg hatte bei gesunden Probanden im Vergleich zur alleinigen Gabe von Sacubitril/Valsartan keine zusätzliche Wirkung auf den im Sitzen gemessenen Blutdruck (BD).
Die gleichzeitige Verabreichung war nicht mit einem klinisch relevanten Effekt auf die Exposition (AUC und Cmax) der beiden Arzneimittel verbunden.
Organische Nitrate
Die gleichzeitige Verabreichung mehrerer auf 10 mg einmal täglich erhöhten Dosen von Vericiguat führte bei Patienten mit koronarer Herzkrankheit nicht zu einer signifikanten Änderung der Wirkung von kurz- und langwirksamen Nitraten (Nitroglycerinspray und Isosorbid-Mononitrat [ISMN] 60 mg mit veränderter Wirkstofffreisetzung) auf den im Sitzen gemessenen BD. Bei Patienten mit Herzinsuffizienz wurde die gleichzeitige Anwendung von kurzwirksamen Nitraten gut vertragen. Zur gleichzeitigen Anwendung von Vericiguat und langwirksamen Nitraten bei Patienten mit Herzinsuffizienz liegen nur begrenzte Erfahrungen vor (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Frauen im gebärfähigen Alter sollten während der Behandlung hochwirksame Verhütungsmethoden anwenden
Schwangerschaft
Es liegen keine Daten zur Anwendung von Verquvo bei Schwangeren vor.
Eine Studie an trächtigen Ratten zeigte, dass Vericiguat über die Plazenta auf den Föten übergeht. Tierexperimentelle Studien haben beim Vorliegen maternaler Toxizität eine Reproduktionstoxizität gezeigt (siehe Rubrik «Präklinische Daten»).
Verquvo sollte während der Schwangerschaft nicht angewendet werden.
Stillzeit
Zum Übergang von Vericiguat in die Muttermilch und zur Wirkung auf das gestillte Kind oder die Milchproduktion liegen keine Daten vor. Vericiguat wurde in der Milch laktierender Ratten nachgewiesen (siehe Rubrik «Präklinische Daten»). Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit Verquvo verzichtet werden soll oder ob die Behandlung mit Verquvo zu unterbrechen ist. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Es liegen keine Daten zur Wirkung von Vericiguat auf die humane Fruchtbarkeit vor. In einer Fertilitätsstudie mit männlichen und weiblichen Ratten zeigte Vericiguat keine Beeinträchtigung der Fertilität und frühen Embryonalentwicklung (siehe Rubrik «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt.
Das Auftreten von symptomatischer Hypotonie wurde berichtet (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»); dies könnte die Fahrtüchtigkeit oder die Fähigkeit Maschinen zu bedienen beeinflussen.
Unerwünschte WirkungenDie Sicherheit von Verquvo bei Patienten mit symptomatischer chronischer Herzinsuffizienz und LVEF ≤45% wurde in der pivotalen Phase-3-Studie VICTORIA evaluiert, bei der Patienten, die mit Verquvo (bis zu 10 mg einmal täglich; n=2519) oder Placebo (n=2515) behandelt wurden. (siehe Rubrik «Eigenschaften/Wirkungen»).
Die mittlere Dauer der Verquvo-Exposition betrug 1 Jahr und die maximale Dauer betrug 2.6 Jahre.
Zu einem Absetzen der Therapie aufgrund einer unerwünschten Wirkung während der Doppelblindphase der Studie VICTORIA kam es bei 167 Patienten unter Verquvo (6.6 %) und bei 158 Patienten unter Placebo (6.3%).
Die am häufigsten berichtete Nebenwirkung unter Behandlung mit Vericiguat war Hypotonie (16,4%). Im Verlauf der VICTORIA-Studie war die mittlere Senkung des systolischen Blutdrucks bei Patienten, die Vericiguat erhielten, im Vergleich zu Placebo um etwa 1 bis 2 mmHg höher. In VICTORIA wurde bei 16,4% der mit Vericiguat behandelten Patienten eine Hypotonie berichtet, verglichen mit 14,9% der mit Placebo behandelten Patienten. Eine symptomatische Hypotonie wurde bei 9,1% der mit Vericiguat behandelten und 7,9% der mit Placebo behandelten Patienten berichtet und bei 1,2% der mit Vericiguat behandelten Patienten und 1,5% der mit Placebo behandelten Patienten als schwerwiegendes unerwünschtes Ereignis angesehen.
Die Gesamtinzidenz unerwünschter Arzneimittelwirkungen (UAW) von Verquvo war mit derjenigen von Placebo vergleichbar. Die einzelnen UAWs von Verquvo und Placebo in den verschiedenen Systemorganklassen unterschieden sich nicht signifikant.
Die unerwünschten Arzneimittelwirkungen sind nach Systemorganklasse und dann entsprechend ihrer Häufigkeit – die am häufigsten auftretenden zuerst – gemäss der folgenden Konvention aufgeführt: Sehr häufig (≥1/10); häufig (≥1/100, <1/10); gelegentlich (≥1/1'000, <1/100); selten (≥1/10'000, <1/1'000), sehr selten (<1/10'000), Häufigkeit nicht bekannt: diese unerwünschten Wirkungen wurden in anderen klinischen Studien als der oben genannten beobachtet, oder sie stammen aus der Postmarketing-Überwachung. Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Wirkungen in absteigender Reihenfolge des Schweregrads angegeben.
In Tabelle 1 sind die unerwünschten Arzneimittelwirkungen aufgelistet, die in VICTORIA bei Patienten unter Verquvo und häufiger als unter Placebo aufgetreten sind.
Tabelle 1: Unerwünschte Arzneimittelwirkungen, die in VICTORIA bei Patienten unter Verquvo und häufiger als unter Placebo aufgetreten sind, nach Systemorganklasse (SOC)
|
Unerwünschte Arzneimittelwirkung
|
Verquvo
N=2519
n (%)
|
Placebo
N=2515
n (%)
| |
Erkrankungen des Blutes und des Lymphsystems
| |
Anämie*
|
243 (9.6)
|
185 (7.4)
| |
Erkrankungen des Gastrointestinaltrakts
| |
Übelkeit
|
96 (3.8)
|
67 (2.7)
| |
Dyspepsie
|
67 (2.7)
|
27 (1.1)
| |
Erbrechen
|
56 (2.2)
|
45 (1.8)
| |
Gastroösophageale Refluxkrankheit
|
44 (1.7)
|
17 (0.7)
| |
Erkrankungen des Nervensystems
| |
Schwindelgefühl
|
169 (6.7)
|
150 (6.0)
| |
Kopfschmerzen
|
86 (3.4)
|
61 (2.4)
| |
Gefässerkrankungen
| |
Hypotonie†
|
412 (16.4)
|
375 (14.9)
|
* Beinhaltet: Anämie, makrozytäre Anämie, Anämie bei chronischer Krankheit, autoimmunhämolytische Anämie, Anämie durch Blutverlust, hämolytische Anämie, hypochrome Anämie, Eisenmangelanämie, mikrozytäre Anämie, nephrogene Anämie, normochrome Anämie, normochrome normozytäre Anämie, normozytäre Anämie, Panzytopenie, perniziöse Anämie, Hämatokrit erniedrigt, Hämoglobin erniedrigt und Erythrozytenzahl erniedrigt
† Beinhaltet: Blutdruck erniedrigt, diastolischer Blutdruck erniedrigt, systolischer Blutdruck erniedrigt, Hypotonie und Orthostasesyndrom
Kardiale Elektrophysiologie
In einer speziellen QT-Studie an Patienten mit stabiler koronarer Herzkrankheit verlängerte die Verabreichung von 10 mg Vericiguat im Steady State das QT-Intervall nicht in klinisch relevantem Ausmass, d.h. die maximale mittlere Verlängerung des QTcF-Intervalls lag nicht über 6 ms (Obergrenze für das 90%-KI <10 ms).
Es wurden keine supratherapeutischen Expositionen getestet. Die integrierte Risikobewertung nicht-klinischer und klinischer Daten zeigte keine klinisch bedeutsame QTc-Verlängerung bei einer Verabreichung von 10 mg Vericiguat.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungSymptome
Für eine Überdosierung bei Patienten, die mit Vericiguat behandelt wurden, liegen nur begrenzte Daten vor. In VICTORIA wurden Dosen bis zu 10 mg untersucht. In einer Studie an HF-Patienten mit erhaltener Auswurfsfraktion (LVEF ≥45%) wurden die untersuchten Mehrfachdosen von Vericiguat 15 mg generell gut vertragen. Eine Überdosierung kann zu Hypotonie führen.
Behandlung
Es ist eine symptomatische Behandlung anzuwenden. Aufgrund seiner hohen Plasmaproteinbindung ist nicht zu erwarten, dass Vericiguat dialysierbar ist.
Eigenschaften/WirkungenATC-Code
C01DX22
Wirkungsmechanismus
Vericiguat ist ein Stimulator der löslichen Guanylatcyclase (sGC). Herzinsuffizienz ist mit einer Störung der Stickstoffmonoxid (NO)-Synthese und einer verminderten Aktivität des NO-Rezeptors sGC assoziiert. Die lösliche Guanylatcyclase katalysiert die Synthese von intrazellulärem zyklischem Guanosinmonophosphat (cGMP), einem wichtigen Signalmolekül, welches wichtige physiologische Prozesse wie Herzkontraktilität, Gefässtonus und kardiales Remodeling reguliert. Ein Mangel an cGMP, das durch sGC katalysiert wird, trägt zur myokardialen und vaskulären Dysfunktion bei. Vericiguat stellt den entsprechenden Ausfall im Signalpfad durch direkte Stimulation der sGC unabhängig von und synergistisch mit NO wieder her, damit die Konzentration an intrazellulärem cGMP ansteigt, um sowohl die myokardiale als auch die vaskuläre Funktion zu fördern. Der zusätzliche kardiovaskuläre Nutzen von Vericiguat bei Patienten mit Herzinsuffizienz ist daher auf die aktive Beseitigung der Störung im NO-sGC-cGMP-Signalweg zurückzuführen, welcher für die Progression der Herzinsuffizienz verantwortlich ist.
Pharmakodynamik
Die pharmakodynamischen Wirkungen von Vericiguat wurden nach einmaliger und wiederholter Gabe an gesunde Probanden und Patienten mit Herzinsuffizienz beurteilt. Die Wirkungen entsprechen dem Wirkmechanismus eines sGC-Stimulators und führen zu einer Relaxation der glatten Muskulatur und Vasodilatation. Im Lauf der VICTORIA-Studie sank der systolische Blutdruck bei Patienten unter Vericiguat um etwa 1 bis 2 mmHg mehr als in der Placebogruppe.
In einer 12wöchigen placebokontrollierten Dosisfindungsstudie (SOCRATES-REDUCED) an Patienten mit Herzinsuffizienz führte Vericiguat als Zusatz zur Standardtherapie im Vergleich zu Placebo zu einer dosisabhängigen Reduktion der NTproBNP-Konzentration, einem Biomarker der Herzinsuffizienz. In VICTORIA war die geschätzte Reduktion der NTproBNP-Konzentration zwischen Baseline und Woche 32 bei Patienten unter Vericiguat grösser als in der Placebogruppe.
Klinische Wirksamkeit
VICTORIA
Bei VICTORIA handelte es sich um eine randomisierte, placebokontrollierte, doppelblinde, ereignisgesteuerte, multizentrische Parallelgruppenstudie zum Vergleich von Verquvo und Placebo an 5050 erwachsenen Patienten mit symptomatischer chronischer Herzinsuffizienz (NYHA -Klasse II-IV) und einer linksventrikulären Auswurfsfraktion ≤45% nach Verschlechterung der Herzinsuffizienz (Hospitalisierung aufgrund von Herzinsuffizienz innerhalb der letzten 6 Monate vor Randomisierung oder ambulante Anwendung von i.v. Diuretika zur Therapie der Herzinsuffizienz innerhalb der letzten 3 Monate vor Randomisierung).
Der zusammengesetzte primäre Endpunkt bestand aus kardiovaskulär (KV) bedingtem Tod oder Hospitalisation aufgrund von Herzinsuffizienz (HF) (Zeit bis zum ersten Auftreten).
Die Behandlung wurde mit 2.5 mg Vericiguat einmal täglich eingeleitet und abhängig von der Verträglichkeit in etwa 2wöchigen Abständen auf 5 mg einmal täglich und anschliessend auf 10 mg einmal täglich erhöht. 90% der Gruppe unter Vericiguat erhielten bis zum Ende der Studie die angestrebte Zieldosis von 10 mg.
Die mediane Nachbeobachtungszeit für den primären Endpunkt betrug 11 Monate.
Das Durchschnittsalter lag bei 67 Jahren. Bei der Randomisierung waren 59% der Patienten in NYHA-Klasse II, 40% in NYHA-Klasse III und 1% in NYHA-Klasse IV. Die mittlere linksventrikuläre Auswurfsfraktion (LVEF) betrug 29%. Zum Zeitpunkt der Randomisierung betrug die mittlere eGFR 62 ml/min/1.73 m2; 10% der Patienten hatten eine eGFR ≤30 ml/min/1.73 m2. In VICTORIA wurden 67% der Patienten innerhalb von 3 Monaten nach dem Index-Ereignis HF-Hospitalisierung aufgenommen; 17% wurden innerhalb von 3 bis 6 Monaten nach einem solchen Ereignis und 16% innerhalb von 3 Monaten nach ambulanter Behandlung einer sich verschlechternden HF mit i.v. Diuretika aufgenommen. Die mediane NTproBNP-Konzentration zum Zeitpunkt der Randomisierung betrug 2816 pg/ml.
Vor der Studienteilnahme waren die Patienten durch eine Therapie entsprechend dem Versorgungsstandard, u.a. mit ACE-Hemmern/ARB (73%), Betablockern (93%), Mineralokortikoidantagonisten (70%) und einer Kombination aus einem Angiotensin-Rezeptor-Blocker und einem Neprilysin-Inhibitor (ARNI; 15%) eingestellt. 28% hatten einen implantierten Defibrillator und 15% einen biventrikulären Schrittmacher. 91% der Patienten wurden mit 2 oder mehreren Arzneimitteln (Betablocker, RAS [Renin-Angiotensin-System] -Inhibitoren oder MRA) gegen Herzinsuffizienz behandelt und 60% erhielten alle 3 Medikationen.
Verquvo senkte gegenüber Placebo das Risiko für KV-Tod oder Hospitalisierung signifikant (Hazard Ratio [HR]: 0.90, 95%-Konfidenzintervall [KI]: 0.82-0.98; p=0.019). Im Verlauf der Studie kam es unter Vericiguat zu einer jährlichen absoluten Risikoreduktion (ARR) von 4.2% im Vergleich zu Placebo (NNT=24 für 1 Jahr); siehe Tabelle 2.
Tabelle 2: Behandlungseffekt für den primären kombinierten Endpunkt, seine Komponenten und die sekundären Endpunkte
|
|
Verquvo
N=2526
|
Placebo
N=2524
|
Therapievergleich
| |
n (%)
|
Jährliche Ereignisrate (%)*
|
n (%)
|
Jährliche Ereignisrate (%)*
|
Hazard Ratio
(95%-KI)†
|
p-Wert‡
|
Jährliche ARR (%)§
| |
Primärer Endpunkt
| |
Kombination aus KV-Tod oder Hospitalisierung aufgrund von Herzinsuffizienz¶
|
897 (35.5)
|
33.6
|
972 (38.5)
|
37.8
|
0.90
(0.82, 0.98)
|
0.019
|
4.2
| |
Kardiovaskulärer Tod
|
206 (8.2)
|
|
225 (8.9)
|
|
|
|
| |
Hospitalisierung aufgrund von Herzinsuffizienz
|
691 (27.4)
|
|
747 (29.6)
|
|
|
|
| |
Sekundäre Endpunkte
| |
Kardiovaskulärer Tod
|
414 (16.4)
|
12.9
|
441 (17.5)
|
13.9
|
0.93
(0.81, 1.06)
|
|
| |
Hospitalisierung aufgrund von Herzinsuffizienz
|
691 (27.4)
|
25.9
|
747 (29.6)
|
29.1
|
0.90
(0.81, 1.00)
|
|
|
*Gesamtzahl der Patienten mit einem Ereignis pro 100 Risikojahren.
†Hazard Ratio (Vericiguat gegenüber Placebo) und Konfidenzintervall aus einem proportionalen Hazard-Modell nach Cox.
‡Aus dem Log-Rank-Test.
§ Jährliche absolute Risikominderung, berechnet als Differenz (Placebo-Vericiguat) in Prozent pro Jahr.
¶Bei Patienten mit mehreren Ereignissen wird nur das erste Ereignis, das zum kombinierten Endpunkt beiträgt, gezählt.
N: Anzahl der Patienten in der Intent-to-Treat (ITT)-Population. n: Anzahl der Patienten mit einem Ereignis.
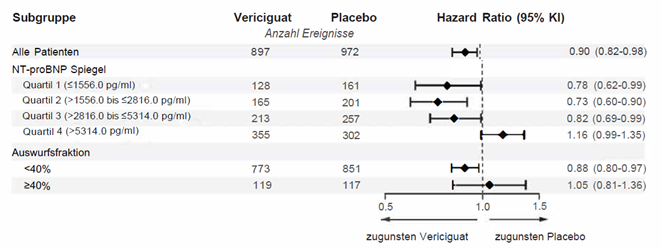
Der Effekt auf den primären kombinierten Endpunkt war weitgehend konsistent für die vordefinierten Subgruppen ausgenommen jene in Abhängigkeit vom NT-proBNP-Spiegel und der linksventrikulären Auswurffraktion zu Studienbeginn (Abbildung 1). Bei Patienten mit einem ausgeprägten Anstieg des NTproBNP (Quartil 4) war das Risiko für den KV-Tod oder Hospitalisierung aufgrund von Herzinsuffizienz in der Vericiguat-Gruppe gegenüber der Placebo-Gruppe erhöht. Auch unterstützen die Subgruppenanalysen die Wirksamkeit von Verquvo uneingeschränkt nur für Patienten mit Herzinsuffizienz und reduzierter linksventrikulärer Auswurffraktion (<40%).
Abbildung 1: Subgruppenanalyse für den primären Endpunkt in Abhängigkeit von Baseline NT-proBNP-Spiegel und Auswurffraktion
PharmakokinetikAbsorption
Vericiguat zeigt eine zeitunabhängige Pharmakokinetik, die etwas weniger als dosisproportional ist und bei Einnahme zu einer Mahlzeit eine niedrige bis mässige intra- und interindividuelle Variabilität aufweist. Die Akkumulation von Vericiguat im Plasma beträgt bis zu 151-171%, und der pharmakokinetische Steady State wird nach etwa 6 Tagen erreicht. Der geometrische Mittelwert der populationspharmakokinetischen (PK) Parameter von Vericiguat im Steady State bei Patienten mit Herzinsuffizienz liegt für Cmax der Dosierungen 2,5mg, 5mg, bzw. 10mg bei 120, 201, bzw. 350 μg/l und für die AUC der Dosierungen 2,5mg, 5mg, bzw. 10mg bei 2300, 3850, bzw. 6680 μg•h/l.
Die absolute Bioverfügbarkeit von Vericiguat ist hoch (93%), wenn die Einnahme mit Nahrung erfolgt. Die Bioverfügbarkeit (AUC) und die maximalen Plasmakonzentrationen (Cmax) von Vericiguat sind bei oraler Verabreichung als zerstossene Tablette in Wasser oder als ganze Tablette vergleichbar (siehe Rubrik «Dosierung/Anwendung»).
Bei Einnahme von Vericiguat mit einer fett- und kalorienreichen Mahlzeit steigt die Tmax von etwa 1 Stunde (nüchtern) auf rund 4 Stunden (nach Nahrungsaufnahme), die PK Variabilität wird reduziert und die Vericiguat-Exposition erhöht sich im Vergleich zum nüchternen Zustand bei der 5-mg-Tablette um 19% (AUC) und 9% (Cmax) und um 44% (AUC) und 41% (Cmax) für die 10-mg-Tablette.
Ähnliche Ergebnisse wurden erzielt, wenn Vericiguat zusammen mit einer fettarmen, kohlenhydratreichen Mahlzeit eingenommen wurde. Vericiguat sollte daher mit Nahrung eingenommen werden (siehe Rubrik «Dosierung/Anwendung»).
Distribution
Das mittlere Verteilungsvolumen von Vericiguat im Steady State beträgt bei gesunden Probanden etwa 44 l. Die Plasmaproteinbindung von Vericiguat beträgt etwa 98%, wobei die Bindung hauptsächlich an Serumalbumin erfolgt.
Metabolismus
Der Abbau von Vericiguat erfolgt überwiegend mittels Glucuronidierung. Dabei entsteht ein N-Glucuronid, das pharmakologisch inaktiv ist und den Hauptmetaboliten im Plasma darstellt. Die N-Glucuronidierung wird vorwiegend durch UGT1A9, lokalisiert in der Niere und in der Leber, sowie UGT1A1, lokalisiert im Darm und in der Leber, katalysiert. Die CYP-vermittelte Metabolisierung ist für die Clearance von geringer Bedeutung (<5%).
Elimination
Vericiguat ist ein Wirkstoff mit niedriger Clearance (1.6 l/h bei gesunden Probanden). Die Halbwertszeit beträgt bei gesunden Probanden ca. 20 Stunden und bei Patienten mit Herzinsuffizienz 30 Stunden. Nach oraler Gabe von [14C] -markiertem Vericiguat wurden bei gesunden Probanden ca. 53% der Dosis über den Urin (vorwiegend als N-Glucuronid) und 45% über die Fäzes (vorwiegend als Vericiguat) ausgeschieden.
Kinetik spezieller Patientengruppen
Nierenfunktionsstörungen
Bei Patienten mit Herzinsuffizienz und leichter, mässiger und schwerer nicht dialysepflichtiger Nierenfunktionsstörung war die mittlere Vericiguat-Exposition (AUC) im Vergleich zu Patienten mit normaler Nierenfunktion um 5%, 13% resp. 20% erhöht. Diese Expositionsunterschiede werden als nicht klinisch relevant eingestuft.
Die Pharmakokinetik von Vericiguat bei Patienten mit einer eGFR <15 ml/min/1.73 m2 zu Behandlungsbeginn oder unter Dialyse wurde nicht untersucht (siehe Rubrik «Dosierung/Anwendung, Spezielle Patientengruppen»).
In einer klinisch-pharmakologischen Studie wiesen ansonsten gesunde Studienteilnehmende mit leichter, mässiger und schwerer Nierenfunktionsstörung nach einer Einmalgabe eine im Vergleich zu gesunden Kontrollen um 8%, 73% resp. 143% höhere mittlere Vericiguat-Exposition (nach Körpergewicht normierte AUC des ungebundenen Wirkstoffs) auf.
Die Plasmaproteinbindung von Vericiguat war bei Nierenfunktionsstörung (eGFR >15 ml/min/1.73 m2) unverändert.
Die offenbare Diskrepanz in Bezug auf den Einfluss einer Nierenfunktionsstörung auf die Vericiguat-Exposition zwischen der klinisch-pharmakologischen Studie und der bei Herzinsuffizienz-Patienten durchgeführten Analyse ist möglicherweise auf Unterschiede in Studiendesign und –grösse zurückzuführen.
Leberfunktionsstörungen
Bei Patienten mit leichter Leberfunktionsstörung (Child-Pugh A) war die mittlere Exposition gegenüber Vericiguat (AUC des ungebundenen Wirkstoffs) um 21% höher als bei gesunden Probanden mit normaler Leberfunktion. Bei Patienten mit mässiger Leberfunktionsstörung (Child-Pugh B) war die mittlere Exposition gegenüber Vericiguat um 47% grösser als bei gesunden Probanden mit normaler Leberfunktion. Die Plasmaproteinbindung von Vericiguat war bei Leberfunktionsstörung (Child-Pugh A und B) unverändert Die Pharmakokinetik von Vericiguat bei Patienten mit schwerer Leberfunktionsstörung (Child-Pugh C) wurde nicht untersucht. (siehe Rubrik «Dosierung/Anwendung»).
Ältere Patienten (≥65 Jahre)
In VICTORIA waren insgesamt 1596 Patienten (63%), die mit Vericiguat behandelt wurden, mindestens 65 Jahre alt, und 783 Patienten (31%), die mit Vericiguat behandelt wurden, waren mindestens 75 Jahre alt. In der populationspharmakokinetischen Analyse wurde kein klinisch bedeutsamer Einfluss des Alters auf die Pharmakokinetik von Vericiguat beobachtet.
Kinder und Jugendliche
An Kindern und Jugendlichen wurden keine Studien mit Vericiguat durchgeführt.
Körpergewicht
In einer populationspharmakokinetischen Analyse von Vericiguat waren die AUC-Werte im Steady State bei HF-Patienten mit einem Körpergewicht <60 kg um etwa 27% höher und bei HF-Patienten mit einem Körpergewicht >90 kg um etwa 20% niedriger als bei HF-Patienten mit einem Körpergewicht zwischen 60 und 90 kg. Die Auswirkung des Körpergewichts auf die Vericiguat-Exposition ist klinisch nicht relevant.
Auswirkung des Geschlechts, der ethnischen Gruppe, der ethnischen Abstammung und der NTproBNP-Konzentration zu Baseline
Basierend auf einer populationspharmakokinetischen Analyse wurde für Geschlecht, ethnische Gruppe und Abstammung und NTproBNP-Konzentration zu Studienbeginn keine klinisch relevante Auswirkung auf die Pharmakokinetik von Vericiguat beobachtet.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktionstoxizität, Genotoxizität, zum kanzerogenen Potential sowie zur männlichen und weiblichen Fertilität lassen die nicht-klinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Mutagenese und Kanzerogenität
Vericiguat war in vitro im mikrobiellen Mutagenitätstest (Ames-Test) und im Maus-Lymphom-Test sowie in vivo im Ratten- und Maus-Mikronukleustest nicht genotoxisch.
Die Beurteilung der Kanzerogenität erfolgte in 2-jährigen Studien an CD1-Mäusen und Wistar-Ratten. Bei den Mäusen zeigte Vericiguat bei Dosierungen im Futter bis zu 150 mg/kg/Tag (männliche Tiere) bzw. bis zu 250 mg/kg/Tag (weibliche Tiere) keine kanzerogene Wirkung. Diese Dosierungen führten zu Expositionen, welche dem 149-Fachen (männliche Tiere) resp. 286-Fachen (weibliche Tiere) der menschlichen Exposition (AUC des ungebundenen Wirkstoffs) bei der maximalen empfohlenen humantherapeutischen Dosis (maximum recommended human dose, MRHD) von 10 mg/Tag entsprach.
In der Kanzerogenität-Studie bei Ratten wurden bei Expositionen, welche dem 12-Fachen der menschlichen Exposition bei der MRHD entsprachen, keine mit Vericiguat in Zusammenhang gebrachten Tumore oder Hyperplasien beobachtet. Ein statistisch nicht signifikanter zahlenmässiger Anstieg gutartiger Phäochromozytome und Leydig-Zell-Tumoren sowie entsprechender Hyperplasien wurde bei männlichen Tieren nach oraler Verabreichung der hohen Dosis von 20 mg/kg/Tag beobachtet, welche zu einer Exposition führte, die dem 41-Fachen der menschlichen Exposition bei der MRHD entsprach. Dies wird als Folge einer kompensatorischen und wiederholten Aktivierung des Renin-Angiotensin-Aldosteron-Systems und des adrenergen Systems durch eine ausgeprägte tägliche Abnahme des Blutdrucks über 2 Jahre eingestuft. Basierend auf der im Gegensatz zum Menschen bekannten Empfindlichkeit von Ratten gegenüber der Entwicklung dieser beiden Tumorarten und basierend auf einem (auch von anderen blutdrucksenkenden Wirkstoffen her bekannten) dokumentierten pharmakologisch bedingten Mechanismus bei supratherapeutischen Dosierungen sowie angesichts ausreichender Sicherheitsmargen werden diese Befunde für Patienten als nicht relevant eingestuft.
Nicht-klinische Daten liessen bei klinischen Dosen von Vericiguat kein kanzerogenes Risiko für den Menschen erkennen.
Reproduktionstoxizität
Eine Studie zur Entwicklungstoxizität bei Ratten, denen Vericiguat während der Organogenese oral verabreicht wurde, zeigte bis zu einer Dosierung von 50 mg/kg/Tag (dem 75-Fachen der AUC des ungebundenen Wirkstoffs beim Menschen bei der MRHD von 10 mg) keine Entwicklungstoxizität. Eine überschiessende pharmakodynamisch vermittelte maternale Toxizität wurde [beim] ≥21-Fachen der AUC des ungebundenen Wirkstoffs beim Menschen bei der MRHD beobachtet; beim 9-Fachen der menschlichen Exposition bei der MRHD fand sich keine maternale Toxizität.
Bei Kaninchen wurde die überschiessende pharmakodynamisch vermittelte maternale Toxizität bei 2.5 mg/kg/Tag und darüber (dem ≥6-Fachen der AUC des ungebundenen Wirkstoffs beim Menschen bei der MRHD) beobachtet, was zu sekundären spontanen Spätaborten und Resorptionen führte. Ausserdem fand sich bei dieser Dosierung eine geringe Inzidenz von Fehlbildungen des Herzens und grosser Gefässe. Während dies nicht eindeutig der Behandlung mit Vericiguat zugeschrieben werden konnte, wurden Anomalien des Herzens und grosser Gefässe auch bei Ratten nach Exposition der Muttertiere gegenüber einer strukturell verwandten Substanz (Riociguat) beobachtet. Nach Verabreichung oraler Dosen von 0.75 mg/kg/Tag (entspricht in etwa der menschlichen Exposition bei der MRHD, basierend auf der AUC des ungebundenen Wirkstoffs) an die Muttertiere fanden sich bei Kaninchen keine maternal-, embryofetal- oder entwicklungstoxischen Wirkungen.
In einer Studie zur prä-/postnatalen Toxizität zeigte Vericiguat bei oraler Verabreichung an Ratten während der Gestation bis zum Ende der Laktationsperiode beim etwa ≥9-Fachen der menschlichen Exposition bei der MRHD eine überschiessende pharmakodynamisch vermittelte maternale Toxizität, welche in der Periode vor dem Absetzen zu einer verminderten Gewichtszunahme der Jungtiere (beim ≥21-Fachen der MRHD) sowie zu Jungtiersterblichkeit (beim 49-Fachen der MRHD) führte.
Sonstige präklinische Befunde
Bei rasch wachsenden adoleszenten Ratten wurden bei Expositionen, die etwa dem ≥20-Fachen der menschlichen Exposition bei der MRHD (AUC des ungebundenen Wirkstoffs) entsprachen
reversible Wirkungen auf die Knochenbildung (Hypertrophie der Wachstumsplatte sowie Hyperostose und metaphysäre und diaphysäre Remodellierung) verzeichnet, die durch einen mit dem Wirkmechanismus zusammenhängenden Anstieg des intrazellulären cGMP-Spiegels vermittelt wurden. Diese Wirkungen wurden nicht nach Langzeitverabreichung von Vericiguat bei erwachsenen Ratten bis zu Expositionen beobachtet, die etwa dem 50-Fachen der humantherapeutischen Exposition unter der MRHD (AUC des ungebundenen Wirkstoffs) entsprachen. Darüber hinaus wurden bei Hunden, die zu Behandlungsbeginn fast ausgewachsen waren, bei Expositionen bis zum 15-Fachen der humantherapeutischen Exposition unter der MRHD keine vergleichbaren Ergebnisse festgestellt (AUC des ungebundenen Wirkstoffs).
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30 °C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer68001 (Swissmedic).
Packungen2.5 mg Filmtabletten: Packungen zu 14 Filmtabletten (B)
5 mg Filmtabletten: Packungen zu 14, 28 oder 98 Filmtabletten (B)
10 mg Filmtabletten: Packungen zu 14, 28 oder 98 Filmtabletten (B)
ZulassungsinhaberinBayer (Schweiz) AG, Zürich.
Stand der InformationSeptember 2021
|