ZusammensetzungWirkstoffe
Natalizumab, gentechnologisch in NS/0 Mauszellen hergestellt.
Hilfsstoffe
Natriumdihydrogenphosphat-Monohydrat, Dinatriumhydrogenphosphat-Heptahydrat, Natriumchlorid, Polysorbat 80, Wasser für Injektionszwecke.
Eine Fertigspritze (150 mg/ml) enthält 3,45 mg Natrium.
Indikationen/AnwendungsmöglichkeitenBei der Indikationsstellung bzw. vor Therapiebeginn ist das PML-Risiko (PML= progressive multifokale Leukenzephalopathie) zu berücksichtigen (siehe unter «Warnhinweise und Vorsichtsmassnahmen»).
Tysabri ist für die krankheitsmodifizierende Monotherapie von hochaktiver, schubförmig remittierend verlaufender Multipler Sklerose (RRMS) bei folgenden Patientengruppen indiziert:
·Patienten mit hoher Krankheitsaktivität trotz Behandlung mit einem vollständigen und angemessenen Zyklus mit mindestens einer krankheitsmodifizierenden Therapie (Ausnahmen und Informationen zu Auswaschphasen siehe «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakodynamik»).
oder
·Patienten mit rasch fortschreitender schubförmig remittierend verlaufender Multipler Sklerose, definiert durch 2 oder mehr Schübe mit Behinderungscharakter in einem Jahr, und mit 1 oder mehr Gadolinium anreichernden Läsionen im MRI des Gehirns oder mit einer signifikanten Erhöhung der T2-Läsionen im Vergleich zu einem kürzlich durchgeführten MRI.
Tysabri ist für die krankheitsmodifizierende Monotherapie von Patienten mit aktiver, schubförmig remittierend verlaufender MS mit einem negativen anti-JCV Antikörperstatus indiziert.
Dosierung/AnwendungDie Therapie muss von – in der Diagnosestellung und Behandlung von neurologischen Erkrankungen erfahrenen – Spezialisten (Fachärzten) in Zentren mit raschem Zugang zu MRI eingeleitet und kontinuierlich überwacht werden.
Eine Behandlung Zuhause darf nicht erfolgen.
Die Verabreichung muss von medizinischem Fachpersonal vorgenommen werden und die Patienten müssen hinsichtlich früher Anzeichen und Symptome einer progressiven multifokalen Leukenzephalopathie (PML) beobachtet werden.
Es müssen Möglichkeiten zur Behandlung von Überempfindlichkeitsreaktionen zur Verfügung stehen.
Patienten sind in einem für Tysabri vorgesehenen Monitoringprogramm aufzunehmen.
Patienten, die mit Tysabri behandelt werden, muss der Patientenpass ausgehändigt werden und sie müssen über die mit Tysabri verbundenen Risiken aufgeklärt werden. Nach zweijähriger Behandlung sollten die Patienten erneut über die mit Tysabri verbundenen Risiken, insbesondere über das erhöhte Risiko für eine PML, aufgeklärt und gemeinsam mit ihren Pflegepersonen über frühe Zeichen und Symptome einer PML instruiert werden.
Manche Patienten haben möglicherweise zuvor Immunsuppressiva (z.B. Mitoxantron, Cyclophosphamid, Azathioprin) erhalten. Diese Wirkstoffe können zu einer anhaltenden Immunsuppression führen, auch wenn ihre Gabe bereits beendet wurde. Der Arzt muss sich daher vor Einleitung der Therapie mit Tysabri vergewissern, dass diese Patienten nicht mehr immungeschwächt sind (siehe unter «Warnhinweise und Vorsichtsmassnahmen»).
Bei Patienten, die nach 6-monatiger Behandlung noch keinerlei Hinweise auf einen Behandlungserfolg zeigen, ist die Fortsetzung der Therapie sorgfältig zu überdenken.
Aus kontrollierten Doppelblindstudien liegen Daten zur Sicherheit und Wirksamkeit von Natalizumab über einen Behandlungszeitraum von 2 Jahren vor. Eine Fortsetzung der Therapie über diesen Zeitraum hinaus sollte nur dann in Betracht gezogen werden, wenn zuvor eine erneute Nutzen-Risiko-Abwägung vorgenommen wurde.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Erwachsene
Die subkutane Darreichungsform von Tysabri ist nicht für die intravenöse Gabe bestimmt. Etwaige Umstellungen der Art der Anwendung von Tysabri sollten 4 Wochen nach der vorgängigen Tysabri-Dosis stattfinden.
Die empfohlene Dosis von Tysabri für die subkutane Injektion beträgt 300 mg alle 4 Wochen. Die beiden Injektionen (jeweils 150 mg) sind nacheinander ohne wesentlichen Zeitverzug durchzuführen. Die zweite Injektion sollte spätestens 30 Minuten nach der ersten Injektion erfolgen.
Die Patienten müssen während der Zeit der subkutanen Injektionen und danach 1 Stunde lang auf Anzeichen und Symptome von Injektionsreaktionen, einschliesslich Überempfindlichkeitsreaktionen, überwacht werden. Nach den ersten 6 Dosen sollten die Patienten weiterhin gemäss klinischem Ermessen nach den subkutanen Injektionen überwacht werden. Die einstündige Beobachtungszeit kann nach klinischem Ermessen verkürzt oder aufgehoben werden, sofern bei den Patienten keine Injektionsreaktionen, einschliesslich Überempfindlichkeitsreaktionen, aufgetreten sind.
Die subkutane Injektion sollte in den Oberschenkel, die Bauchdecke oder die Rückseite des Oberarms erfolgen. Sie darf nicht in einen Bereich des Körpers injiziert werden, an dem die Haut in irgendeiner Weise gereizt, gerötet, geprellt, infiziert oder vernarbt ist. Beim Entfernen der Spritze aus der Injektionsstelle muss der Kolben losgelassen werden und die Nadel gleichzeitig in einer geraden Linie herausgezogen werden. Durch Loslassen des Kolbens wird der Nadelschutz aktiviert und kann die Nadel bedecken. Die zweite Injektion sollte mindestens 3 cm von der ersten Injektionsstelle entfernt erfolgen.
Nach zweijähriger Behandlung müssen die Patienten erneut über Risikofaktoren für eine PML, wie die Dauer der Behandlung, die Anwendung von Immunsuppressiva vor der Behandlung mit Tysabri und das Vorhandensein von anti-JCV Antikörpern aufgeklärt werden (siehe unter «Warnhinweise und Vorsichtsmassnahmen»).
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Es wurden keine Studien zum Einfluss von Leberschädigung durchgeführt.
Der Eliminationsmechanismus und die Erkenntnisse aus der Populations-Pharmakokinetik lassen vermuten, dass eine Dosisanpassung bei Patienten mit Leberfunktionsstörung nicht notwendig ist.
Patienten mit Nierenfunktionsstörungen
Es wurden keine Studien zum Einfluss von Nierenschädigung durchgeführt.
Der Eliminationsmechanismus und die Erkenntnisse aus der Populations-Pharmakokinetik lassen vermuten, dass eine Dosisanpassung bei Patienten mit Nierenfunktionsstörung nicht notwendig ist.
Ältere Patienten
Die Anwendung von Tysabri bei Patienten über 65 Jahre wird nicht empfohlen, da keine Daten über diese Patientengruppe vorliegen.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Tysabri sind bei Kindern und Jugendlichen im Alter von bis zu 18 Jahren nicht erwiesen. Es kann keine Dosierungsempfehlung gegeben werden. Die zurzeit vorliegenden Daten sind in den Abschnitten «Unerwünschte Wirkungen» und «Eigenschaften/Wirkungen» beschrieben.
Art der Anwendung
Für Anweisungen zur Verabreichung von Tysabri, Injektionslösung in einer Fertigspritze, siehe unter «Sonstige Hinweise».
Wiederholte Verabreichung
Siehe unter «Warnhinweise und Vorsichtsmassnahmen».
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe gemäss Zusammensetzung.
Progressive multifokale Leukenzephalopathie (PML).
Patienten mit einem erhöhten Risiko für opportunistische Infektionen, wie immungeschwächte Patienten (einschliesslich solcher Patienten, die aktuell eine immunsuppressive Behandlung erhalten oder durch frühere Therapien, z.B. Mitoxantron oder Cyclophosphamid, immungeschwächt sind (siehe auch unter «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»)).
Kombination mit anderen krankheitsmodifizierenden Therapien.
Bekannte aktive Malignome mit Ausnahme von Patienten mit einem Basaliom.
Warnhinweise und VorsichtsmassnahmenProgressive multifokale Leukenzephalopathie (PML)
Die Anwendung von Tysabri geht mit einem erhöhten Risiko für die Entwicklung einer PML einher, einer durch das John Cunningham Virus (JCV) verursachten opportunistischen Infektion, die tödlich verlaufen oder zu schwerer Behinderung führen kann. Aufgrund des erhöhten Risikos für die Entwicklung einer PML sollten Nutzen und Risiken einer Behandlung mit Tysabri in jedem Einzelfall regelmässig im Rahmen von klinischen Kontrolluntersuchungen erneut durch den Facharzt bzw. die Fachärztin mit den Patienten abgewogen werden. Patienten müssen gemeinsam mit ihren Pflegepersonen über frühe Zeichen und Symptome einer PML instruiert und diesbezüglich regelmässig beobachtet werden.
JCV kann auch eine Granularzellneuropathie (GCN) verursachen, die bei Patienten unter Behandlung mit Tysabri festgestellt worden ist. Die Symptome einer JCV-bedingten GCN gleichen denen einer PML (d.h. zerebelläres Syndrom) und die Diagnose und das Management der JCV-bedingten GCN sollten sich nach den für PML geltenden Richtlinien richten.
Die folgenden Risikofaktoren werden mit einem erhöhten Risiko für die Entwicklung einer PML in Zusammenhang gebracht:
·Vorhandensein von anti-JCV Antikörpern. Untersuchungen auf Vorhandensein von anti-JCV Antikörpern sollten nur mit dem STRATIFY JCV-Assay durchgeführt werden.
·Behandlungsdauer, insbesondere länger als zwei Jahre.
·Behandlung mit einem Immunsuppressivum vor der Therapie mit Tysabri.
Nach zwei Jahren müssen alle Patienten erneut über das mit Tysabri verbundene Risiko für eine PML aufgeklärt werden.
Ein Algorithmus zur Abschätzung des PML-Risikos fasst das PML-Risiko nach den oben aufgeführten Risikofaktoren zusammen und stratifiziert dieses Risiko nach dem ermittelten anti-JCV Antikörpertiters (Indexwert). Ärzte sind angehalten, weitere Zusatzinformationen zum Management des PML-Risikos bei mit Tysabri behandelten Patienten den «Arzt-Information und Management-Richtlinien» zu entnehmen.
Patienten mit positivem Nachweis von anti-JCV Antikörpern haben ein erhöhtes Risiko für die Entwicklung einer PML verglichen mit Patienten mit negativem Nachweis von anti-JCV Antikörpern. Patienten, bei denen alle drei Risikofaktoren für eine PML vorliegen (d.h. bei denen anti-JCV Antikörper nachgewiesen wurden und die über einen Zeitraum von mehr als 2 Jahren mit Tysabri behandelt worden sind und eine vorausgegangene immunsuppressive Therapie erhalten haben), tragen ein signifikant höheres Risiko für eine PML.
Bei Patienten ohne vorausgegangene immunsuppressive Therapie kann bei nachgewiesenen anti-JCV-Antikörpern der anti-JCV Antikörpertiter (Indexwert) zur weiteren Stratifizierung des PML-Risikos verwendet werden. Nach aktuellem Kenntnisstand ist bei Indexwerten von 0,9 oder darunter das PML-Risiko eher niedrig. Bei Patienten mit Indexwerten über 1,5 und mehr als 2-jähriger Behandlung mit Tysabri steigt das PML-Risiko an (siehe «Arzt-Information und Management-Richtlinien»).
Im Vergleich zum zugelassenen Dosierungsintervall wird bei anti-JCV-Antikörper-positiven Patienten ein verlängertes Tysabri-Dosierungsintervall (durchschnittlich etwa 6 Wochen) in Zusammenhang mit einem geringeren Risiko für PML gebracht (siehe «Eigenschaften/Wirkungen», Klinische Wirksamkeit, «Verlängertes Dosierungsintervall»). Bei Anwendung eines verlängerten Dosierungsintervalls ist Vorsicht geboten, da die Wirksamkeit eines verlängerten Dosierungsintervalls im Vergleich zum zugelassenen Q4W-Dosierungsschema nicht formal durch eine non-inferiority-Studie nachgewiesen wurde. Die Verringerung des PML-Risikos basiert auf Daten, die sich auf die intravenöse Anwendung beziehen. Zur Sicherheit oder Wirksamkeit dieses verlängerten Dosierungsintervalls bei der subkutanen Anwendung sind keine klinischen Daten verfügbar.
Bei Patienten, deren Risiko als erhöht eingestuft wird (z.B. alle drei Risikofaktoren erfüllt oder Patienten ohne vorausgegangene immunsuppressive Therapie jedoch mit einem Indexwert von über 1,5 und mehr als 2-jähriger Behandlung) sollte die Behandlung mit Tysabri nur dann fortgesetzt werden, wenn der Nutzen die Risiken überwiegt.
Die aus klinischen Daten und Daten aus Beobachtungsstudien abgeleiteten Abschätzungen des PML-Risikos korrelierten mit den Daten nach der Markteinführung.
Patienten mit aktiver RRMS mit einem negativen anti-JCV Antikörperstatus
Patienten mit negativem anti-JCV Antikörperstatus haben ein geringeres Risiko, eine PML zu entwickeln (siehe auch «Warnhinweise und Vorsichtsmassnahmen», Abschnitt «Progressive multifokale Leukenzephalopathie (PML)» weiter oben, sowie «Arzt-Information und Management-Richtlinien»).
Bei Patienten, die anti-JCV Antikörper negativ sind, kann dennoch ein PML-Risiko vorliegen, beispielsweise infolge einer neuen JCV-Infektion, wegen eines fluktuierenden Antikörperstatus oder eines falsch-negativen Testergebnisses. In einer Phase IV Studie mit Untersuchung des langfristigen Antikörperstatus über 18 Monate gab es eine jährliche Veränderung des anti-JCV Antikörperstatus von negativ zu positiv von ungefähr 11 %. Untersuchungen sollten nur mit dem STRATIFY JCV-Assay durchgeführt werden, einem zweistufigen anti-JCV Antikörper-Assay (ELISA) mit einer falsch-negativen Rate von 3 %, der für die Anwendung bei MS-Patienten validiert worden ist.
Eine Wiederholung des anti-JCV Antikörper-Tests wird bei Patienten mit negativem anti-JCV Antikörperstatus empfohlen, da sich der Antikörperstatus ändern kann. Es wird daher empfohlen, Patienten mit aktiver RRMS und einem negativen anti-JCV Antikörperstatus alle 3 Monate erneut zu testen (siehe «Anti-JCV-Antikörper-Tests» unten).
Die Dauer der Behandlung, insbesondere über 2 Jahre hinaus, ist mit einem erhöhten Risiko für PML verbunden.
Das Nutzen-Risiko-Verhältnis eines frühen Behandlungsbeginns muss vor dem Start einer Therapie individuell mit dem Patienten besprochen werden, wobei auch weitere schwerwiegende unerwünschte Wirkungen wie opportunistische Infektionen, Überempfindlichkeitsreaktionen und unerwünschte hepatische Ereignisse zu berücksichtigen sind (siehe spezifische Abschnitte unter «Warnhinweise und Vorsichtsmassnahmen»).
Eine mögliche Krankheitsexazerbation nach Absetzen des Arzneimittels sollte berücksichtigt werden.
Die Patienten sollten über Therapiemöglichkeiten informiert werden, falls sich ihr Anti-JCV-Antikörperstatus während der Behandlung zu positiv ändert.
Anti-JCV-Antikörper-Tests
Es wird empfohlen, bei Patienten vor Beginn einer Therapie mit Tysabri oder bei Patienten, die bereits Tysabri erhalten, bei denen jedoch der Antikörperstatus nicht bekannt ist, eine entsprechende Untersuchung durchzuführen. Aufgrund einer neuen JCV-Infektion, einem schwankenden Antikörperstatus oder einem falsch-negativen Testergebnis können jedoch auch anti-JCV Antikörper-negative Patienten ein Risiko für eine PML tragen. In einer Phase IV-Studie, in welcher der Antikörperstatus longitudinal während 18 Monaten untersucht wurde, wechselte pro Jahr bei etwa 11 % der Patienten der Serostatus von anti-JCV Antikörper-negativ zu anti-JCV Antikörper-positiv. Die Untersuchung sollte ausschliesslich mit einem anti-JCV Antikörper-Assay, der bei Patienten mit MS analytisch validiert wurde, wie dem STRATIFY JCV® oder dem STRATIFY JCV® DxSelectTM Assay, durchgeführt werden. Bei hochaktiver RRMS wird eine Wiederholung des Anti-JCV-Antikörper-Tests mindestens alle 6 Monate empfohlen bei 1) Patienten mit negativem Nachweis von anti-JCV Antikörpern sowie bei 2) Patienten mit anti-JCV-Antikörper-positivem Status und niedrigerem Indexwert ohne vorausgegangene Therapie mit Immunsuppressiva (sobald die zweijährige Behandlungsdauer erreicht ist), da sich der Antiköperstatus oder der Indexwert ändern können. Bei Patienten mit aktiver RRMS und negativem anti-JCV Antikörperstatus wird eine Testwiederholung alle 3 Monate empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen» / Patienten mit aktiver RRMS mit einem negativen anti-JCV Antikörperstatus).
Der anti-JCV Antikörper-Assay (ELISA) ist jedoch nicht für die Diagnose einer PML geeignet.
Zusatzinformationen zu den Anti-JCV-Antikörper-Tests ist den «Arzt-Information und Management-Richtlinien» zu entnehmen.
MRI-Untersuchung auf PML
Vor Beginn der Behandlung mit Tysabri sollte eine aktuelle MRI-Aufnahme als Referenzaufnahme vorliegen (gewöhnlich nicht älter als 3 Monate) und auf Routinebasis mindestens einmal jährlich wiederholt werden, um diese Referenz zu aktualisieren. Die Patienten müssen durchgehend in regelmässigen Abständen kontrolliert werden.
Bei Patienten mit höherem PML-Risiko sollten häufigere MRI-Kontrollen (z.B. alle 3 bis 6 Monate) in Betracht gezogen werden. Zu diesen zählen die folgenden Patientengruppen:
·Patienten, bei denen alle drei Risikofaktoren für PML vorliegen (d.h. solche, die anti-JCV Antikörper-positiv sind und eine mehr als 2jährige Therapie mit Tysabri und eine vorausgegangene immunsuppressive Therapie erhalten haben),
oder
·Patienten mit einem anti-JCV Antikörper-Indexwert von über 1,5 ohne vorausgegangene immunsuppressive Therapie und mehr als 2-jähriger Behandlung mit Tysabri.
Für den Fall, dass eine PML oder JCV-GCN vermutet wird, muss die Gabe von Tysabri solange ausgesetzt werden, bis eine PML oder JCV-GCN ausgeschlossen werden kann.
Der Facharzt bzw. die Fachärztin sollte den Patienten untersuchen, um entscheiden zu können, ob die Symptome auf eine neurologische Dysfunktion hindeuten, und falls ja, ob diese Symptome typisch für die MS sind oder möglicherweise auf eine PML oder JCV-GCN hindeuten. Wenn irgendwelche Zweifel bestehen, sind weitergehende Untersuchungen einschliesslich einer MRI-Untersuchung, vorzugsweise mit Kontrastmittel (zum Abgleich mit dem MRI-Befund, der vor Behandlungsbeginn erhoben wurde), Liquortests auf DNA des JC-Virus und wiederholte neurologische Kontrolluntersuchungen in Erwägung zu ziehen (siehe unter «Fachliche Unterstützung - Arzt-Information und Management-Richtlinien»). Sobald der Facharzt bzw. die Fachärztin eine PML und/oder JCV-GCN ausgeschlossen hat (gegebenenfalls durch Wiederholung der klinischen und bildgebenden Untersuchungen und/oder der Laboruntersuchungen, falls der klinische Verdacht bestehen bleibt), kann die Gabe von Tysabri wieder aufgenommen werden.
Der Arzt sollte insbesondere auf Symptome achten, die auf eine PML oder JCV-GCN hindeuten, die der Patient möglicherweise nicht bemerkt (z.B. kognitive, psychiatrische Symptome oder Zeichen eines zerebellären Syndroms). Ausserdem sollte den Patienten empfohlen werden, ihren Partner oder Pfleger über ihre Behandlung zu informieren, da diese Symptome feststellen könnten, die der Patient nicht bemerkt.
Es wurde auch über Fälle von asymptomatischer PML berichtet. Asymptomatische PML kann mit einem MRI erkannt werden und muss durch Vorhandensein von JCV DNA im Liquor oder mit einer Gehirnbiopsie bestätigt werden.
Darüber hinaus sollten medizinische Fachpersonen auf alle neuen Anzeichen (z.B. im MRI) oder Symptome achten, die auf eine PML schliessen lassen könnten. In solchen Fällen sollte Tysabri unverzüglich abgesetzt werden. Wenn die ersten Untersuchungen ein negatives Ergebnis liefern, der klinische Verdacht auf PML aber weiter besteht, sollte die Anwendung von Tysabri nicht fortgesetzt werden, und die Untersuchungen sollten wiederholt werden.
PML ist auch nach Absetzen von Tysabri bei Patienten aufgetreten, die zum Zeitpunkt der Beendigung der Behandlung keine Anzeichen einer möglichen PML aufwiesen. Patienten und Ärzte müssen daher auch nach Beendigung der Behandlung mit Tysabri etwa 6 Monate lang auf alle neuen Anzeichen oder Symptome einer möglichen PML achten und die MRI-Kontrollen über diesen Zeitraum fortführen.
Entwickelt ein Patient eine PML oder JCV-GCN, muss die Gabe von Tysabri dauerhaft abgesetzt werden.
Nach Wiederherstellung der Immunabwehr bei immungeschwächten Patienten mit PML wurde ein besserer Behandlungserfolg beobachtet.
PML und IRIS (Immune Reconstitution Inflammatory Syndrome)
Ein IRIS tritt bei fast allen Patienten, die unter Tysabri eine PML entwickelt haben, nach Absetzen oder Entfernung von Tysabri, z.B. durch Plasmaaustausch (PLEX) (siehe unter «Pharmakokinetik»), auf. Es wird angenommen, dass das IRIS eine Folge der Wiederherstellung der Immunfunktion bei Patienten mit PML ist. Das IRIS kann zu einer schnell verlaufenden Verschlechterung des neurologischen Status, zu schwerwiegenden neurologischen Komplikationen führen und tödlich verlaufen. Es sollten sowohl eine Überwachung hinsichtlich der Entwicklung eines IRIS, das bei mit Tysabri behandelten Patienten mit PML innerhalb von Tagen bis einigen Wochen nach einem Plasmaaustausch aufgetreten ist, als auch eine angemessene Behandlung der damit verbundenen Entzündung während der Erholung von einer PML erfolgen.
Eine retrospektive Analyse von Natalizumab nach der Zulassung konnte keinen Unterschied innert 2 Jahren hinsichtlich der Überlebensrate nach PML Diagnose zwischen Patienten mit oder ohne PLEX Behandlung feststellen. Die Verwendung von PLEX zur PML Behandlung soll vom Arzt medizinisch beurteilt werden. Die Anwendung von Plasmapherese/Plasmaaustausch (PLEX) oder von intravenösem Immunglobulin (IVIg) kann eine aussagekräftige Interpretation von Untersuchungen auf anti-JCV Antikörper im Serum beeinträchtigen. Die Untersuchung auf anti-JCV Antikörper sollte während und mindestens 2 Wochen nach einem Plasmaaustausch aufgrund der Entfernung von Antikörpern aus dem Serum nicht durchgeführt werden oder innerhalb von 6 Monaten nach der IVIg-Gabe (d.h. 6 Monate = 5 Halbwertszeiten des Immunglobulins) nicht durchgeführt werden.
Infektionen, einschliesslich sonstiger opportunistischer Infektionen
Unter der Anwendung von Tysabri wurde über sonstige opportunistische Infektionen berichtet, vorwiegend bei Patienten mit Morbus Crohn (nicht zugelassene Indikation), bei Patienten, die immungeschwächt waren oder bei denen eine relevante Komorbidität vorlag. Jedoch kann ein erhöhtes Risiko für sonstige opportunistische Infektionen unter der Anwendung von Tysabri bei Patienten ohne diese Komorbiditäten derzeit nicht ausgeschlossen werden. Opportunistische Infektionen wurden auch bei MS-Patienten festgestellt, die mit Tysabri als Monotherapie behandelt wurden (siehe unter «Unerwünschte Wirkungen»).
Tysabri erhöht das Risiko eine durch Herpes-simplex- oder Varizella-Zoster Viren hervorgerufene Enzephalitis und Meningitis zu entwickeln. In der Post-Marketing Erfahrung wurden bei MS-Patienten unter der Anwendung von Tysabri schwerwiegende, lebensbedrohliche und zum Teil tödlich verlaufende Fälle von Enzephalitis und Meningitis, hervorgerufen durch Herpes-simplex- oder Varizella-Zoster-Viren, berichtet. Die Dauer der Behandlung mit Tysabri vor dem Auftreten der Enzephalitis bzw. Meningitis betrug zwischen wenigen Monaten und mehreren Jahren. Bei Auftreten einer Herpes-Enzephalitis bzw. Herpes-Meningitis ist die Gabe von Tysabri zu beenden und eine angemessene Behandlung der Herpes-Enzephalitis bzw. Herpes-Meningitis vorzunehmen (siehe «Unerwünschte Wirkungen nach Markteinführung/Infektionen»).
Akute Retinanekrose (ARN) ist eine fulminante virale Infektion der Retina, welche durch Herpes-Viren (z.B. Varicella zoster) verursacht wird. ARN wurde bei Patienten unter Behandlung mit Tysabri beobachtet und kann potenziell zu Erblindung führen. Patienten mit Symptomen wie reduzierter Sehschärfe, gerötete und schmerzende Augen sollten zur Untersuchung der Retina auf ARN überwiesen werden. Bei einer klinischen Diagnose von ARN sollte eine Unterbrechung der Tysabri-Therapie in Erwägung gezogen werden (siehe «Unerwünschte Wirkungen nach Markteinführung/Infektionen»).
Der verschreibende Arzt sollte sich über die Möglichkeit von sonstigen opportunistischen Infektionen während der Tysabri-Therapie bewusst sein und diese in die Differentialdiagnose von Infektionen, die bei einem mit Tysabri behandelten Patienten auftreten, mit einbeziehen. Wenn eine opportunistische Infektion vermutet wird, muss die Gabe von Tysabri so lange ausgesetzt werden, bis diese durch weitere Untersuchungen ausgeschlossen werden kann.
Ausser über das Risiko der Reaktivierung von JC-Polyoma-Virus liegen keine Daten über die Reaktivierung anderer latenter Virusinfektionen (Hepatitis B oder C) vor.
Wenn ein mit Tysabri behandelter Patient eine opportunistische Infektion entwickelt, muss die Gabe von Tysabri dauerhaft abgesetzt werden.
Fachliche Unterstützung
Alle Ärzte, die Tysabri verordnen, müssen mit den «Arzt-Information und Management-Richtlinien» vertraut sein.
Ärzte müssen Nutzen und Risiken der Tysabri-Therapie mit dem Patienten besprechen und ihm die Patienteninformationsmaterialien, einschliesslich des Patientenpasses, aushändigen. Die Patienten sollten angewiesen werden, ihren Arzt darüber zu informieren, dass sie mit Tysabri behandelt werden, sollte es bei ihnen zu einer Infektion kommen.
Ärzte sollten die Patienten über die Wichtigkeit einer kontinuierlichen Verabreichung von Tysabri insbesondere in den Anfangsmonaten der Behandlung beraten (siehe auch unter «Überempfindlichkeit»).
Überempfindlichkeit
Es sind Überempfindlichkeitsreaktionen mit Tysabri in Zusammenhang gebracht worden, darunter auch schwerwiegende systemische Reaktionen bei intravenöser Infusion (siehe unter «Unerwünschte Wirkungen»).
Bei subkutaner Anwendung von Natalizumab wurden keine schwerwiegenden Überempfindlichkeitsreaktionen festgestellt, wobei die Datenlage limitiert ist (siehe unter «Eigenschaften/Wirkungen» und «Pharmakokinetik»).
Bei der intravenösen Anwendung von Tysabri traten Überempfindlichkeitsreaktionen gewöhnlich während der Infusion oder bis zu 1 Stunde nach Infusionsende auf. Allerdings können diese auch noch in einem Zeitraum von mehr als vier Stunden nach der Infusion auftreten. Das Risiko einer Überempfindlichkeit war bei den ersten Infusionen am grössten. Patienten, die nach einer kurzzeitigen Exposition (eine oder zwei Infusionen) und einer längeren Zeitdauer ohne Behandlung (drei Monate oder länger) wieder mit Tysabri in Kontakt gebracht wurden, zeigten ebenfalls ein erhöhtes Risiko einer Überempfindlichkeitsreaktion. Dennoch sollte bei jeder Anwendung das Risiko einer Überempfindlichkeitsreaktion in Betracht gezogen werden (siehe unter «Dosierung/Anwendung»).
Die Patienten sind während der subkutanen Injektionen sowie danach 1 Stunde lang auf Anzeichen und Symptome von Injektionsreaktionen, einschliesslich Überempfindlichkeitsreaktionen, zu überwachen (siehe unter «Dosierung/Anwendung» und «Unerwünschte Wirkungen»). Es sollten Ressourcen zur Behandlung von Überempfindlichkeitsreaktionen bereitstehen.
Patienten mit Symptomen von Überempfindlichkeitsreaktionen wie z.B. Urtikaria sollten diese umgehend ihrem Arzt melden.
Vor jeder Anwendung muss bestimmt werden, ob die letzte Injektion zu unerwünschten Symptomen geführt hat, die möglicherweise auf eine allergische/Überempfindlichkeitsreaktion hinweisen, wie zum Beispiel Brustschmerzen oder –beschwerden, Atemnot, signifikante Veränderungen des Blutdrucks, Angioödem, Hautreaktionen und/oder Pruritus.
Bei den ersten Anzeichen oder Symptomen einer Überempfindlichkeit muss die Gabe von Tysabri beendet und eine entsprechende Therapie eingeleitet werden.
Patienten mit einer Überempfindlichkeitsreaktion müssen dauerhaft von einer Behandlung mit Tysabri ausgeschlossen werden.
Begleitende Behandlung mit Immunsuppressiva
Die Sicherheit und Wirksamkeit von Tysabri in Kombination mit anderen Immunsuppressiva und antineoplastischen Therapien sind nicht ausreichend belegt. Die begleitende Anwendung dieser Substanzen neben Tysabri kann das Risiko für Infektionen, auch für opportunistische Infektionen, erhöhen und stellt daher eine Kontraindikation dar (siehe unter «Kontraindikationen»).
In klinischen MS-Studien der Phase III war die begleitende kurzdauernde Behandlung von Schüben mit Kortikosteroiden nicht mit einer erhöhten Infektionsrate verbunden. Kurzzeitige Kortikoidsteroidgaben können zusammen mit Tysabri verabreicht werden.
Vorbehandlung mit immunsupprimierenden oder immunmodulatorischen Therapien
Patienten, die bereits mit Immunsuppressiva behandelt wurden, haben ein erhöhtes Risiko für die Entwicklung einer PML.
Es wurden keine Studien durchgeführt, in denen die Wirksamkeit und Sicherheit von Tysabri bei der Umstellung von Patienten von krankheitsmodifizierenden Therapien mit immunsuppressiver Wirkung auf Tysabri untersucht wurden. Es ist nicht bekannt, ob bei Patienten, die von diesen Therapien auf Tysabri umgestellt werden, ein erhöhtes PML-Risiko besteht. Diese Patienten sollten deshalb engmaschiger überwacht werden (d.h. ähnlich häufig wie Patienten, die von Immunsuppressiva auf Tysabri umgestellt werden; siehe «MRI-Untersuchung auf PML»).
Bei diesen Patienten muss dafür gesorgt werden, dass dem Immunsystem ausreichend Zeit gegeben wird, um sich wieder zu erholen. Der behandelnde Arzt muss jeweils im Einzelfall beurteilen, ob Hinweise auf einen immungeschwächten Status vorliegen, bevor er mit der Gabe von Tysabri beginnt (siehe unter «Kontraindikationen»).
Bei der Umstellung von Patienten von einer anderen krankheitsmodifizierenden Therapie auf Tysabri müssen die Halbwertszeit und das Wirkprinzip der bisherigen Therapie berücksichtigt werden, um einerseits eine additive Immunwirkung zu vermeiden und andererseits das Risiko einer Krankheitsreaktivierung zu minimieren. Vor Behandlungsbeginn mit Tysabri wird ein grosses Blutbild (Complete Blood Count, CBC, einschliesslich Lymphozyten) empfohlen, um sicherzustellen, dass sich Immunwirkungen der Vortherapie (d.h. Zytopenie) zurückgebildet haben.
Patienten können direkt von Interferon beta oder Glatirameracetat auf Tysabri umgestellt werden, sofern keine Anzeichen für relevante behandlungsbedingte Auffälligkeiten, wie z.B. eine Neutropenie oder Lymphopenie, vorliegen.
Bei der Umstellung von Dimethylfumarat sollte die Auswaschphase so gestaltet sein, dass sich die Lymphozytenwerte vor Behandlungsbeginn mit Tysabri erholen können.
Nach Absetzen von Fingolimod normalisieren sich die Lymphozytenwerte innerhalb von 1 bis 2 Monaten nach Beendigung der Therapie allmählich wieder. Die Auswaschphase sollte so gestaltet sein, dass sich die Lymphozytenwerte vor Behandlungsbeginn mit Tysabri erholen können.
Teriflunomid wird langsam aus dem Plasma eliminiert. Ohne ein Verfahren zur beschleunigten Elimination kann die Plasmaclearance von Teriflunomid mehrere Monate bis zu 2 Jahre dauern. Ein beschleunigtes Eliminationsverfahren, wie in der Fachinformation von Teriflunomid beschrieben, wird empfohlen, andernfalls sollte die Auswaschphase mindestens 3,5 Monate betragen. Bei der Umstellung von Patienten von Teriflunomid auf Tysabri ist im Hinblick auf mögliche gleichzeitige Immunwirkungen Vorsicht geboten.
Alemtuzumab besitzt ausgeprägte und lang anhaltende immunsupprimierende Wirkungen. Da die tatsächliche Dauer dieser Wirkungen nicht bekannt ist, wird die Einleitung einer Behandlung mit Tysabri nach Vorbehandlung mit Alemtuzumab nicht empfohlen, es sei denn, der Nutzen überwiegt im Einzelfall eindeutig die Risiken.
Impfungen und in vivo-Hauttests
In einer randomisierten open-label Immunisierungsstudie bei Patienten mit schubförmiger MS zeigte sich beim Vergleich von Patienten einer unbehandelten Kontrollgruppe (n= 23) mit Patienten, die während 6 Monaten mit Tysabri behandelt wurden (n= 19), kein signifikanter Unterschied in der Verdoppelung der Antikörperspiegel gegen ein Neoantigen (Keyhole-Limpet-Hämocyanin, KLH) oder ein Recall-Antigen (Tetanustoxoid). Die Mittelwerte der jeweiligen Antikörperspiegel waren unter Tysabri jedoch deutlich niedriger als unter der Kontrollgruppe. Ein Patient unter Tysabri war non-Responder gegen Tetanustoxoid, zwei Patienten unter Tysabri waren non-Responder gegen KLH. Lebendvakzine wurden nicht untersucht.
Über die Aussagekraft diagnostischer Hauttests (z.B. mit Tuberkulin) während und nach der Behandlung mit Tysabri ist nichts bekannt.
Immunogenität
Eine Verschlechterung der Erkrankung oder injektionsbedingte Ereignisse können auf die Bildung von Antikörpern gegen Natalizumab hindeuten. In diesen Fällen und bei Patienten mit einer kurzzeitigen Exposition gegenüber Tysabri und einer folgenden längeren Zeitdauer ohne Behandlung sollte das Vorhandensein von Antikörpern untersucht werden. Patienten mit anfänglich wenigen Behandlungen, insbesondere bei 1 bis 2 Anwendungen mit Tysabri, und einem anschliessenden längeren behandlungsfreien Zeitraum, haben bei neuerlicher Aufnahme der Behandlung ein höheres Risiko für die Entwicklung von Antikörpern und/oder Überempfindlichkeitsreaktionen. Die Behandlung sollte abgesetzt und nicht wieder aufgenommen werden, falls Antikörper in einem Bestätigungstest nach mindestens 6 Wochen positiv bleiben, da persistierende Antikörper mit einer verminderten Wirksamkeit von Tysabri und einer erhöhten Häufigkeit von Überempfindlichkeitsreaktionen einhergehen (siehe unter «Unerwünschte Wirkungen»).
Unerwünschte hepatische Ereignisse
Nach der Markteinführung wurde selten von spontanen schwerwiegenden unerwünschten Ereignissen einer Leberschädigung berichtet.
Die Leberschäden können zu einem beliebigen Zeitpunkt während der Behandlung auftreten, selbst nach der ersten Dosis. In einigen Fällen trat die Reaktion nach Wiederaufnahme der Behandlung mit Tysabri erneut auf. Bei einigen Patienten mit pathologischem Lebertest in der Anamnese kam es unter Behandlung mit Tysabri zur Verschlechterung des pathologischen Lebertests.
Patienten sollten hinsichtlich einer eingeschränkten Leberfunktion überwacht und instruiert werden, ihren Arzt zu kontaktieren, falls Anzeichen oder Symptome auftreten, die auf eine Leberschädigung hindeuten, z.B. Gelbsucht oder Erbrechen.
Beim Vorliegen einer signifikanten Leberschädigung sollte Tysabri abgesetzt werden.
Beendigung der Tysabri-Therapie
Wenn entschieden wird, die Behandlung mit Tysabri zu beenden, muss sich der behandelnde Arzt darüber im Klaren sein, dass Tysabri entsprechend seiner Halbwertszeit von 16±4 Tagen (siehe «Pharmakokinetik») noch im Blut vorhanden ist und bis zu etwa 12 Wochen nach der letztmaligen Gabe noch pharmakodynamische Wirkungen (z.B. eine erhöhte Lymphozytenzahl) zeigt. Die Einleitung anderer Therapien in dieser Zeit wird zwangsläufig mit einer begleitenden Exposition von Tysabri verbunden sein. Bei Wirkstoffen wie Interferon und Glatirameracetat war eine begleitende Exposition über diesen Zeitraum in klinischen Studien nicht mit Sicherheitsrisiken assoziiert. Es liegen keine Informationen für MS-Patienten im Hinblick auf die begleitende Exposition gegenüber Immunsuppressiva vor. Der Einsatz dieser Arzneimittel kurz nach dem Absetzen von Tysabri kann aber einen additiven immunsupprimierenden Effekt zur Folge haben. Dies sollte in jedem Einzelfall individuell abgewogen werden, gegebenenfalls könnte eine Washout-Phase von Tysabri angebracht sein. Kurzzeitige Steroidgaben zur Behandlung von Schüben waren in klinischen Prüfungen nicht mit häufigeren Infektionen assoziiert.
Natriumgehalt
Eine Fertigspritze (150 mg/ml) enthält 3,45 mg Natrium. Eine volle Dosis (300 mg/2 ml) enthält 6,9 mg Natrium.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Tysabri ist bei Kindern und Jugendlichen im Alter von bis zu 18 Jahren nicht erwiesen. Der Nutzen der Behandlung von Kindern und Jugendlichen mit Tysabri sollte gegenüber dem Risiko für die Entwicklung einer PML und sonstigen opportunistischen Infektionen abgewogen werden (siehe «Unerwünschte Wirkungen» und «Eigenschaften Wirkungen»).
InteraktionenSiehe unter «Kontraindikationen».
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Wenn eine Frau unter der Behandlung mit Natalizumab schwanger wird, sollte die Beendigung der Therapie mit Tysabri in Erwägung gezogen werden.
Schwangerschaft
Es liegen keine hinreichenden Daten für die Verwendung von Natalizumab bei Schwangeren vor. Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt (siehe unter «Präklinische Daten»). Das potenzielle Risiko für den Menschen ist nicht bekannt.
Nach Markteinführung gab es Berichte über Fälle von Thrombozytopenie und Anämie bei Säuglingen, deren Mütter während der Schwangerschaft (v.a. nach dem ersten Trimenon) Natalizumab ausgesetzt waren. Die Anämie des Neugeborenen war ausgeprägter als der erwartete physiologische Abfall des Hämoglobins nach der Geburt und erforderte in einigen Fällen eine spezifische Therapie. Es wird empfohlen, hämatologische Parameter, insbesondere die Thrombozytenzahl, Hämoglobin und Hämatokrit, von Neugeborenen, deren Mütter während der Schwangerschaft Natalizumab ausgesetzt waren, postpartal und ggf. bis zur Normalisierung zu überwachen.
Natalizumab darf nicht während der Schwangerschaft verwendet werden, es sei denn, der klinische Befund der Patientin macht eine Behandlung mit Tysabri erforderlich.
Stillzeit
Tysabri geht in die Muttermilch über. Die Wirkung von Natalizumab auf Neugeborene/Kleinkinder ist nicht bekannt. Das Stillen sollte daher während der Behandlung mit Tysabri beendet werden.
Fertilität
Angaben zur Fertilität siehe unter «Präklinische Daten».
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenMit Tysabri wurden keine Studien zu den Wirkungen auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt. Da jedoch Schwindel häufig berichtet wurde, sollte Patienten, bei denen diese unerwünschte Wirkung auftritt, empfohlen werden, bis zum Abklingen des Schwindels keine Fahrzeuge zu führen oder Maschinen zu bedienen.
Unerwünschte WirkungenDas Sicherheitsprofil von subkutan gegebenem Natalizumab entsprach weitestgehend dem bekannten Sicherheitsprofil von intravenös gegebenem Natalizumab, mit Ausnahme der unerwünschten Wirkung «Schmerz an der Injektionsstelle». «Schmerz an der Injektionsstelle» trat bei Studienteilnehmern, die alle 4 Wochen 300 mg Natalizumab subkutan erhielten, mit einer Gesamthäufigkeit von 4 % (3/71) auf (siehe unter «Eigenschaften/Wirkungen», klinische Wirksamkeit und «Pharmakokinetik»).
In Placebo-kontrollierten Studien mit 1'617 MS-Patienten, die bis zu 2 Jahre mit Natalizumab (intravenöse Infusion) behandelt wurden (Placebo: 1'135), traten zu einem Abbruch der Therapie führende unerwünschte Ereignisse bei 5,8 % der mit Natalizumab behandelten Patienten auf (Placebo: 4,8 %). Im Verlauf des 2-jährigen Studienzeitraums berichteten 43,5 % der mit Natalizumab behandelten Patienten über unerwünschte Wirkungen (Placebo: 39,6 %).
Die höchste Inzidenz von unerwünschten Wirkungen, die in Placebo-kontrollierten klinischen Prüfungen bei MS-Patienten unter der intravenösen Anwendung von Natalizumab in der empfohlenen Dosierung beobachtet wurden, wird angegeben für Schwindel, Übelkeit, Urtikaria und Rigor im Zusammenhang mit den Infusionen.
Unerwünschte Wirkungen, die unter der Behandlung mit intravenös verabreichtem Natalizumab mit einer gegenüber der Placebo-Gruppe um 0,5 % höheren Inzidenz berichtet wurden, sind im Folgenden aufgeführt.
Sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1'000 bis < 1/100), selten (≥1/10'000 bis < 1/1'000).
Innerhalb jeder Häufigkeitsgruppe werden die unerwünschten Wirkungen nach abnehmendem Schweregrad angegeben.
Infektionen und parasitäre Erkrankungen
Sehr häufig: Harnwegsinfektionen, Nasopharyngitis.
Gelegentlich: Opportunistische Infektionen, PML, Herpes (Varicella-Zoster-, Herpes-simplex-Encephalitis/-Meningitis).
Erkrankungen des Immunsystems
Häufig: Urtikaria.
Gelegentlich: Anaphylaktische/anaphylaktoide Reaktionen während der Infusion oder innerhalb der ersten Stunde nach Infusionsende.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen, Schwindel.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Übelkeit.
Häufig: Erbrechen.
Affektionen der Leber und Gallenblase
Vereinzelt: Hyperbilirubinämie, Transaminasenerhöhung.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Gelenkschmerzen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Abgeschlagenheit.
Häufig: Rigor, Fieber.
Infusionsbedingte Reaktionen
Sehr häufig: Infusionsreaktionen (23,1 %; Placebo: 18,7 %) wie Schwindel, Übelkeit, Erbrechen, Urtikaria, Schüttelfrost, Flushing und Rigor.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Überempfindlichkeitsreaktionen
Überempfindlichkeitsreaktionen traten üblicherweise innerhalb von 1 Stunde nach Abschluss der subkutanen Injektionen auf.
Bei subkutaner Anwendung von Natalizumab wurden keine schwerwiegenden Überempfindlichkeitsreaktionen festgestellt, wobei die Datenlage limitiert ist (siehe unter «Eigenschaften/Wirkungen», Klinische Wirksamkeit und «Pharmakokinetik»).
Immunogenität
Häufig: Antikörper gegen Natalizumab bei 10 %, davon persistierende anti-Natalizumab-Antikörper (ein positives Testergebnis, das mindestens 6 Wochen später in einem erneuten Test reproduzierbar sein muss) bei circa 6 % der Patienten, die Natalizumab intravenös erhielten. Persistierende Antikörper sind mit einem erheblichen Rückgang der Wirksamkeit von Tysabri und einer erhöhten Inzidenz für Überempfindlichkeitsreaktionen assoziiert. Weitere infusionsbedingte Reaktionen im Zusammenhang mit persistierenden Antikörpern können Rigor, Übelkeit, Erbrechen und Flushing sein.
In der 32wöchigen DELIVER-Studie, die an MS-Patienten ohne vorherige Exposition gegenüber Natalizumab durchgeführt wurde, traten persistierende Antikörper gegen Natalizumab bei 1 Patienten (4 %) von 26 Patienten, die Natalizumab subkutan erhielten, auf. Antikörper wurden nur bei einer Gelegenheit bei weiteren 5 Patienten gefunden (19 %). In der 60wöchigen REFINE-Studie, die an MS-Patienten durchgeführt wurde, wurden bei keinem Patienten (136 Patienten), die von der intravenösen Natalizumab-Gabe zu einer subkutanen Gabe wechselten, nachweisbare Antikörper gegen den Wirkstoff während der Studie gefunden (siehe Abschnitt «Eigenschaften/Wirkungen»).
Wenn nach ungefähr 6monatiger Therapie persistierende Antikörper vermutet werden (z.B. aufgrund verminderter Wirksamkeit oder Hypersensitivität), können diese mit einem 6 Wochen nach dem ersten positiven Test durchgeführten Anschlusstest nachgewiesen und bestätigt werden. Die Behandlung sollte bei Patienten, die persistierende Antikörper entwickeln, beendet werden.
Infektionen
In klinischen Studien sind bei MS-Patienten unter Behandlung mit Tysabri Fälle von PML beschrieben worden. PML ist mit Behinderung oder Tod in Zusammenhang gebracht worden (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
In klinischen Studien bei Patienten mit Multipler Sklerose betrug die Infektionsrate sowohl bei Patienten unter Behandlung mit Tysabri als auch bei den Patienten in den Placebogruppen ungefähr 1,5 je Patientenjahr. Es wurde sowohl über einen Fall einer durch Cryptosporidium ausgelösten Durchfallerkrankung als auch über Fälle weiterer opportunistischer Infektionen berichtet, von denen einige tödlich verliefen. Die Art der Infektionen war in Tysabri und Placebo behandelten Patienten ähnlich. Die meisten Patienten unterbrachen ihre Behandlung mit Tysabri während der Infektionen nicht, und die Infektionen konnten mit angemessener Behandlung zum Abklingen gebracht werden.
Wirkungen auf Labortests
In 2-jährigen kontrollierten klinischen Studien mit MS-Patienten war die Behandlung mit Natalizumab mit einer Erhöhung der Anzahl zirkulierender Lymphozyten, Monozyten, Eosinophilen, Basophilen und kernhaltigen roten Blutzellen assoziiert.
Eine Erhöhung der Neutrophilenzahl wurde nicht beobachtet. Während der Behandlung mit Natalizumab wurde ein geringer Abfall der Werte für Hämoglobin (mittlerer Rückgang 0,6 g/dl), Hämatokrit (mittlerer Rückgang 2 %) und rote Blutzellen (mittlerer Rückgang der Zellzahl 0,1×106/l) beobachtet, was zumeist vorübergehend war. Sämtliche Veränderungen bei den Blutwerten erreichten gewöhnlich innerhalb von 16 Wochen nach der letztmaligen Gabe von Natalizumab wieder das Ausgangsniveau der Werte vor Behandlungsbeginn und die Veränderungen waren nicht mit klinischen Symptomen assoziiert.
Unerwünschte Wirkungen aus der Postmarketingphase
Nach der Markteinführung wurde über Eosinophilie (Eosinophilenzahl > 1500/mm3) ohne klinische Symptome berichtet. Wenn in solchen Fällen die Behandlung mit Tysabri abgebrochen wurde, normalisierten sich die erhöhten Eosinophilenzahlen. Es gab nach Markteinführung Berichte über Thrombozytopenien und Immunthrombozytopenien (ITP). Diese waren zum Teil unter Steroidgabe reversibel.
In Anwendungsbeobachtungen nach Markteinführung wurden seltene Fälle von schwerer Anämie und hämolytischer Anämie bei Patienten unter der Behandlung mit Tysabri berichtet.
Infektionen
Nach der Markteinführung gab es Meldungen über PML bei Patienten unter Monotherapie mit Tysabri, einschliesslich Fälle mit PML ohne klinische Symptome bei Krankheitsbeginn. Einige Fälle wurden in einem Zeitraum von bis zu 6 Monaten nach Absetzen der Monotherapie mit Tysabri berichtet (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»). Die Untersuchung auf anti-JCV Antikörper im Serum liefert unterstützende Informationen für die Stratifizierung des Risikos der Tysabri-Behandlung (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»). Bei Patienten, die anti-JCV Antikörper–negativ sind, kann dennoch ein PML Risiko vorliegen, beispielsweise infolge einer neuen JCV-Infektion, wegen eines fluktuierenden Antikörperstatus oder eines falsch-negativen Testergebnisses. In einer Phase IV Studie mit Untersuchung des langfristigen Antikörperstatus über 18 Monate gab es eine jährliche Veränderung des anti-JCV Antikörper-Status von negativ zu positiv von ungefähr 11 %. Untersuchungen sollten nur mit dem STRATIFY JCV-Assay durchgeführt werden, einem zweistufigen anti-JCV Antikörper-Assay (ELISA) mit einer Testspezifität von 97 %, der für die Anwendung bei MS-Patienten validiert worden ist. Fälle einer JCV-bedingten GCN wurden auch nach der Markteinführung von Tysabri berichtet. Die Symptome einer JCV-bedingten GCN können denen einer PML ähnlich sein (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Bei Patienten mit Multipler Sklerose unter Behandlung mit Natalizumab sind nach der Markteinführung schwerwiegende, lebensbedrohliche und mitunter tödlich verlaufene Fälle von Enzephalitis und Meningitis infolge einer Infektion mit Herpes simplex oder Varicella zoster berichtet worden (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Nach der Markteinführung trat bei Patienten unter Behandlung mit Natalizumab mit grösserer Häufigkeit eine akute Retinanekrose (ARN) auf. In einigen Fällen handelte es sich dabei um Patienten mit Herpesinfektionen des Zentralnervensystems (ZNS) (z.B. Herpes-Meningitis und -Enzephalitis). In gravierenden Fällen führte die ARN, in einem oder in beiden Augen vorliegend, bei einigen Patienten zur Erblindung. Diese Fälle wurden zum Beispiel mit einer antiviralen Therapie und mitunter operativ behandelt (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
Pädiatrische Population
Es wurde eine Beurteilung schwerwiegender unerwünschter Ereignisse bei 621 pädiatrischen MS-Patienten aus einer Metaanalyse-Studie durchgeführt (siehe Abschnitt «Klinische Wirksamkeit»). Innerhalb der Beschränkungen dieser Daten wurden in diesem Patientenkollektiv keine Sicherheitsrisiken und keine neuen Sicherheitssignale identifiziert. Im Rahmen dieser Meta-Analyse wurde ein Fall von Herpes-Meningitis dokumentiert, es wurden keine PML-Fälle festgestellt. Jedoch wurden nach Markteinführung von Tysabri Fälle von PML-Erkrankungen auch bei mit Natalizumab behandelten pädiatrischen Patienten gemeldet.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEs wurden keine Fälle von Überdosierung berichtet.
Eigenschaften/WirkungenATC-Code
L04AG03
Wirkungsmechanismus
Natalizumab ist ein selektiver Adhäsionsmolekül-Inhibitor und bindet an die α4-Untereinheit von humanen Integrinen, die in hohem Masse auf der Oberfläche aller Leukozyten mit Ausnahme der Neutrophilen exprimiert werden. Natalizumab bindet spezifisch an das α4β1-Integrin, wobei es die Wechselwirkung mit dessen Rezeptor, dem vaskulären Zelladhäsionsmolekül-1 (VCAM-1), dem Liganden Osteopontin oder einer alternativ gesplicten Domäne von Fibronektin, dem Connecting Segment-1 (CS-1), blockiert. Natalizumab blockiert die Wechselwirkung des α4β7-Integrins mit dem Adhäsionsmolekül MadCAM-1 (mucosal addressin cell adhesion molecule-1). Durch die Unterbindung dieser molekularen Interaktionen wird die transendotheliale Migration von mononukleären Leukozyten in entzündetes Parenchymgewebe verhindert. Ein weiterer Wirkungsmechanismus von Natalizumab liegt möglicherweise in der Unterdrückung von bestehenden Entzündungsreaktionen in erkrankten Geweben durch Hemmung der Wechselwirkung von α4-Integrin-exprimierenden Leukozyten mit ihren Liganden in der extrazellulären Matrix und auf den Parenchymzellen. So unterdrückt Natalizumab möglicherweise auch eine bestehende Entzündungsaktivität in erkrankten Bereichen und hemmt eine weitere Rekrutierung von Immunzellen in entzündete Gewebe.
Es wird angenommen, dass es bei der MS zu Läsionen kommt, wenn aktivierte T-Lymphozyten die Blut-Hirn-Schranke (BHS) passieren. Die Migration der Leukozyten durch die BHS beinhaltet eine Interaktion zwischen Adhäsionsmolekülen auf Entzündungszellen und Rezeptoren auf den Endothelzellen der Gefässwand. Die Interaktion zwischen α4β1-Integrin und seinen Zielzellen ist eine wichtige Komponente der pathologischen Entzündung im Gehirn und die Unterbindung dieser Wechselwirkungen führt zu einer Reduktion der Entzündung. Unter normalen Bedingungen wird VCAM-1 im Hirnparenchym nicht exprimiert. In Anwesenheit von pro-inflammatorischen Zytokinen wird VCAM-1 jedoch auf Endothelzellen und möglicherweise auch auf Gliazellen in der Nähe des Entzündungsgeschehens hochreguliert. Wenn bei der MS das Zentralnervensystem (ZNS) von dem Entzündungsgeschehen betroffen ist, vermittelt die Interaktion von α4β1-Integrin mit VCAM-1, CS-1 und Osteopontin die feste Adhäsion und transendotheliale Migration von Leukozyten in das Gehirnparenchym und kann möglicherweise die Entzündungskaskade in zentralnervösem Gewebe fortsetzen. Die Blockade der molekularen Interaktionen von α4β1-Integrin mit seinen Zielzellen reduziert die bei MS im Gehirn vorhandene Entzündungsaktivität und hemmt die weitere Rekrutierung von Immunzellen in entzündliches Gewebe, wodurch die Bildung oder Vergrösserung von MS-Läsionen eingeschränkt wird.
Der EC50-Wert der Bindung von Natalizumab an α4β1-Integrin wird auf der Grundlage eines populationspharmakokinetischen/pharmakodynamischen Modells auf 2,04 mg/l geschätzt. Was die α4β1-Integrin-Bindung anbelangte, gab es zwischen der subkutanen und der intravenösen Gabe von 300 mg Natalizumab alle 4 Wochen keinen Unterschied. Die mittlere PD (Alpha-4-Sättigung auf mononukleären Lymphozyten) war bei der intravenösen Verabreichung alle 6 Wochen (Q6W) und der intravenösen Verabreichung alle 4 Wochen (Q4W) ähnlich, wobei der Unterschied in der mittleren prozentualen Alpha-4-Sättigung zwischen 9 % und 16 % lag.
Pharmakodynamik
Siehe «Wirkungsmechanismus».
Klinische Wirksamkeit
Klinische AFFIRM-Studie
Die Wirksamkeit als Monotherapie wurde in einer randomisierten, doppelblinden, Placebo-kontrollierten Studie über 2 Jahre (AFFIRM-Studie) bei Patienten mit schubförmig remittierender Multipler Sklerose untersucht, die im Jahr vor der Aufnahme in die Studie mindestens einen klinischen Schub erlebt hatten und einen «Expanded Disability Status Scale» (EDSS)-Score nach Kurtzke zwischen 0 und 5 aufwiesen. Das mediane Alter lag bei 37 Jahren (von 18 bis 50 Jahren), wobei die mediane Krankheitsdauer 5 Jahre betrug. Die Patienten wurden im Verhältnis 2:1 zu den Behandlungsarmen mit Tysabri 300 mg i.v. (n= 627) bzw. Placebo (n= 315) alle 4 Wochen mit bis zu 30 Infusionen randomisiert. Neurologische Untersuchungen wurden alle 12 Wochen und bei Verdacht auf einen Schub durchgeführt. MRI-Untersuchungen für T1gewichtete Gadolinium (Gd)anreichernde Läsionen und T2hyperintense Läsionen wurden auf jährlicher Basis durchgeführt.
Die Studienmerkmale und -ergebnisse sind in der nachstehenden Tabelle aufgeführt.
|
AFFIRM-Studie: Hauptmerkmale und Ergebnisse
| |
Design
|
Monotherapie; randomisierte doppelblinde Placebo-kontrollierte Parallelgruppenstudie über 120 Wochen
| |
Patienten
|
RRMS (McDonald-Kriterien)
| |
Behandlung
|
Placebo / Natalizumab 300 mg i.v. alle 4 Wochen
| |
1-Jahres-Endpunkt
|
Schubrate
| |
2-Jahres-Endpunkt
|
Progression auf der EDSS
| |
Sekundäre Endpunkte
|
Schubratenabhängige Variablen / MRI-abhängige Variablen
| |
Patienten
|
Placebo
|
Natalizumab
| |
Randomisiert
|
315
|
627
| |
1 Jahr abgeschlossen
|
296
|
609
| |
2 Jahre abgeschlossen
|
285
|
589
| |
Alter in Jahren, Median (Bereich)
|
37 (19-50)
|
36 (18-50)
| |
MS-Dauer in Jahren, Median (Bereich)
|
6,0 (0-33)
|
5,0 (0-34)
| |
Zeit seit Diagnose, Jahre, Median (Bereich)
|
2,0 (0-23)
|
2,0 (0-24)
| |
Schübe in den letzten 12 Monaten, Median (Bereich)
|
1,0 (0-5)
|
1,0 (0-12)
| |
EDSS-Ausgangsscore, Median (Bereich)
|
2 (0-6,0)
|
2 (0-6,0)
| |
ERGEBNISSE
| |
Jährliche Schubrate
| |
Nach einem Jahr (primärer Endpunkt)
|
0,805
|
0,261
| |
Nach zwei Jahren
|
0,733
|
0,235
| |
Ein Jahr
|
Rate ratio 0,33 CI95 % 0,26; 0,41
| |
Zwei Jahre
|
Rate ratio 0,32 CI95 % 0,26; 0,40
| |
Schubfrei
| |
Nach einem Jahr
|
53 %
|
76 %
| |
Nach zwei Jahren
|
41 %
|
67 %
| |
Behinderung
| |
Anteil mit Progression1 (bestätigt nach 12 Wochen; primärer Endpunkt)
|
29 %
|
17 %
| |
|
Hazard ratio 0,58, CI95 % 0,43; 0,73, p< 0,001
| |
Anteil mit Progression (bestätigt nach 24 Wochen)
|
23 %
|
11 %
| |
|
Hazard ratio 0,46, CI95 % 0,33; 0,64, p< 0,001
| |
MRI (0-2 Jahre)
| |
Mediane prozentuale Veränderung des T2-hyperintensen Läsionsvolumens
|
+8,8 %
|
-9,4 %
(p< 0,001)
| |
Mittlere Anzahl neu auftretender oder sich neu vergrössernder T2-hyperintenser Läsionen
|
11,0
|
1,9
(p< 0,001)
| |
Mittlere Anzahl von T1-hypointensen Läsionen
|
4,6
|
1,1
(p< 0,001)
| |
Mittlere Anzahl Gd anreichernder Läsionen
|
1,2
|
0,1
(p< 0,001)
| |
1
Progression der Behinderung war definiert als eine Erhöhung von mindestens 1,0 Punkten auf der EDSS von einem Ausgangs-EDSS ≥1,0, die mindestens 12 oder 24 Wochen bestehen blieb, oder eine Erhöhung von mindestens 1,5 Punkten auf der EDSS von einem Ausgangs-EDSS=0, die mindestens 12 oder 24 Wochen bestehen blieb.
|
In der Subgruppe der Patienten mit Indikation zur Behandlung wegen rasch fortschreitender schubförmig remittierender MS (Patienten mit 2 oder mehr Schüben und 1 oder mehr Gd+-Läsionen) betrug die jährliche Schubrate 0,282 in der mit Tysabri behandelten Gruppe (n= 148) und 1,455 in der Placebogruppe (n= 61) (p< 0,001). Die Hazard-Ratio für die Behinderungsprogression betrug 0,36 (95% KI: 0,17; 0,76) p= 0,008. Diese Ergebnisse stammen aus einer post-hoc-Analyse und sollten mit Vorsicht interpretiert werden. Es sind keine Angaben zur Schwere der Schübe vor Aufnahme in die Studie verfügbar.
Tysabri Observational Program (TOP)
Eine Interimsanalyse von Ergebnissen (Stand: Mai 2015) aus dem noch laufenden Tysabri Observational Program (TOP), einer multizentrischen, einarmigen Phase IV-Studie (n= 5770), zeigte, dass von Interferon beta (n= 3255) oder Glatirameracetat (n= 1384) auf Tysabri i.v. umgestellte Patienten eine nachhaltige, statistisch signifikante Abnahme der jährlichen Schubrate aufwiesen (p< 0,0001). Die mittleren EDSS-Werte blieben über 5 Jahre stabil. In Übereinstimmung mit den Wirksamkeitsergebnissen, die bei von Interferon beta oder Glatirameracetat auf Tysabri umgestellten Patienten beobachtet wurden, wurde auch bei von Fingolimod (n= 147) auf Tysabri umgestellten Patienten eine statistisch signifikante Abnahme der jährlichen Schubrate beobachtet, die über 2 Jahre stabil blieb. Ferner blieben die mittleren EDSS-Werte vom Ausgangswert bis Jahr 2 auf vergleichbarem Niveau. Bei der Interpretation dieser Daten sollten die begrenzte Kohortengrösse und die kürzere Dauer der Tysabri-Exposition in dieser Patientenuntergruppe berücksichtigt werden.
Verlängertes Dosierungsintervall
Eine vorab festgelegte retrospektive Analyse (TOUCH Verschreibungsprogramm in den USA, n= 15'120) hat gezeigt, dass eine verlängerte Intervalldosierung (Extended Interval Dosing bzw. EID, ungefähr alle 6 Wochen) von intravenös verabreichtem Tysabri bei Anti-JCV-Antikörper-positiven Patienten im Vergleich zu der zugelassenen Standardintervalldosierung mit einem geringeren PML-Risiko verbunden ist (Hazard Ratio= 0,06, 95 %-KI = 0,01–0,22). Die meisten dieser Patienten wurden vor der Umstellung auf die EID mit der zugelassenen Dosierung ≥1 Jahr behandelt.
Modelle und Simulationen zeigen, dass das Risiko einer MS-Krankheitsaktivität bei Patienten, die auf längere Dosierungsintervalle umgestellt werden, nachdem sie ≥1 Jahr lang die zugelassene Dosierung erhalten hatten, bei solchen mit Dosierungsintervallen von ≥7 Wochen eventuell höher ist.
Die Wirksamkeit von Tysabri bei Verabreichung alle 6 Wochen (Q6W) wurde nicht formal durch eine non-inferiority Studie im Vergleich zur zugelassenen Q4W-Dosierung nachgewiesen.
Zur Sicherheit oder Wirksamkeit dieses verlängerten Dosierungsintervalls bei der subkutanen Anwendung sind keine klinischen Daten verfügbar.
Klinische Studie REFINE (subkutane Anwendungsart, Population für mindestens 12 Monate mit Natalizumab [intravenöse Infusion] vorbehandelt)
Die Wirksamkeit und Sicherheit von Tysabri für die subkutane Anwendung wurden in einer randomisierten, verblindeten Parallelgruppenstudie der Phase II (REFINE, 101MS206) zur Sicherheit, Verträglichkeit und Wirksamkeit mehrerer Natalizumab-Regimes (300 mg intravenös alle 4 Wochen, 300 mg subkutan alle 4 Wochen, 300 mg intravenös alle 12 Wochen, 300 mg subkutan alle 12 Wochen, 150 mg intravenös alle 12 Wochen und 150 mg subkutan alle 12 Wochen) bei erwachsenen Teilnehmern (n= 290) mit schubförmig remittierend verlaufender multipler Sklerose über einen Zeitraum von 60 Wochen untersucht.
Der primäre Endpunkt dieser Studie war die kumulative Anzahl kombinierter, einzelner, aktiver (Combined Unique Active, CUA) MRI-Läsionen (Summe der neuen Gd+-Läsionen im Hirn-MRI und der neuen oder sich neu vergrössernden T2hyperintensen Läsionen, die in T1gewichteten Scans nicht mit Gd+ assoziiert waren). Die mittlere, kumulative Anzahl der CUA-Läsionen in dem alle 4 Wochen mit 300 mg s.c. behandelten Arm war niedrig (0,02) und vergleichbar mit dem Wert in dem alle 4 Wochen mit 300 mg i.v. behandelten Arm (0,23). Die mittlere, kumulative Anzahl der CUA-Läsionen in den alle 12 Wochen behandelten Armen war signifikant höher als in den alle 4 Wochen behandelten Armen und führte zur vorzeitigen Beendigung der alle 12 Wochen behandelten Arme.
Klinische Studie DELIVER (subkutane Anwendungsart, Natalizumab-naive Population)
Die Wirksamkeit und Sicherheit von Natalizumab zur subkutanen Anwendung bei den nicht mit Natalizumab vorbehandelten Patienten mit MS wurden in einer randomisierten, offenen Dosisbereichsstudie der Phase I (DELIVER) untersucht. In die Arme mit subkutaner Behandlung wurden 12 Patienten mit RRMS und 14 Patienten mit sekundär progredienter MS aufgenommen.
Ein exploratorischer Endpunkt dieser Studie schloss die Anzahl der neuen Gd+ Läsionen im Gehirn-MRT zwischen Baseline und Woche 32 ein. Keiner der mit Natalizumab behandelten Probanden wies nach der Baseline Gd+ Läsionen auf, unabhängig vom Erkrankungsstadium (RRMS oder sekundär progrediente MS), der zugewiesenen Art der Anwendung oder dem Vorhandensein von Gd+ Läsionen zur Baseline.
In der Gesamtpopulation aller Patienten mit RRMS und sekundär progressiver MS erlitten insgesamt 2 Patienten in der Gruppe, die Natalizumab 300 mg subkutan erhielt, Schübe, verglichen mit 3 Patienten in der Gruppe, die Natalizumab 300 mg intravenös erhielt. Die geringe Populationsgrösse und die Inter- und Intra-Patienten-Variabilität verhindern aussagekräftige Vergleiche der Wirksamkeitsdaten zwischen den Gruppen.
Pädiatrie
Im Rahmen einer Meta-Analyse nach Markteinführung wurden die Daten von 621 pädiatrischen MS-Patienten (medianes Alter 17 Jahre, Altersspanne 7-18 Jahre, 91 % ≥14 Jahre alt), die mit Tysabri behandelt wurden, untersucht. In dieser Analyse wurde bei einer kleinen Subgruppe von Patienten, bei denen Daten zur jährlichen Schubrate vor der Behandlung verfügbar waren (158 der 621 Patienten), eine Reduktion der jährlichen Schubrate von 1,466 (95 % KI 1,337; 1,604) vor der Behandlung auf 0,110 (95 % KI 0,094; 0,128) belegt.
PharmakokinetikAbsorption
Die Pharmakokinetik von Natalizumab nach subkutaner Gabe wurde in 2 Studien (DELIVER, REFINE) bewertet. Die Studie DELIVER (101MS102) war eine randomisierte, offene Dosisbereichsstudie zur Bewertung der Pharmakokinetik von subkutanem und intramuskulärem Natalizumab bei Patienten mit multipler Sklerose (n= 76) (siehe «klinische Wirksamkeit» für eine Beschreibung der Studie REFINE (101MS206)). Nach subkutaner Gabe von 300 mg Natalizumab wurde eine Verzögerung der maximalen Natalizumabkonzentration (Cmax) im Plasma von 5,8 Tagen (Bereich: 2 bis 7,9 Tage) festgestellt, danach korrelierte die Disposition von Natalizumab mit der bei intravenöser Gabe. Die mittlere Cmax für Teilnehmer mit RRMS betrug 35,44 μg/ml (Bereich 22,0 bis 47,8 μg/ml) und betrug 33% der nach intravenöser Gabe erreichten Spitzenwerte.
Die wiederholte Gabe subkutaner Dosen von 300 mg alle 4 Wochen ergab einen ähnlichen CTal-Wert wie nach intravenöser Gabe von 300 mg alle 4 Wochen. Sowohl bei intravenöser als auch bei subkutaner Gabe von Natalizumab alle 4 Wochen führten die CTal-Werte zu einer vergleichbaren α4β1-Integrin-Bindung.
Wie die aktualisierte Analyse der Populationspharmakokinetik ergab, betrug die Bioverfügbarkeit von Natalizumab nach subkutaner Gabe schätzungsweise 84 %. Die Absorption aus der Injektionsstelle in den systemischen Kreislauf fand als Absorption erster Ordnung mit einer modellgeschätzten Verzögerung von 3 Stunden statt. Für die Absorption wurden keine Kovariaten festgestellt. Bei der intravenösen und bei der subkutanen Art der Anwendung ergaben sich dieselben pharmakokinetischen Dispositionsparameter (CL, Vss und t½) und dieselben Gruppen von Kovariaten, wie sie in der aktualisierten Analyse der Populationspharmakokinetik beschrieben sind.
Distribution
Der Median des Steady State Verteilungsvolumens betrug 5,96 L (5,59-6,38 L, 95 % Konfidenzintervall).
Metabolismus
Siehe «Absorption».
Elimination
Eine Analyse (intravenöse Gabe) zur Populations-Pharmakokinetik umfasst 12 Studien mit 1781 Individuen, welche Dosen von 1 bis 6 mg/kg Natalizumab oder fixe Dosen von 150/300 mg als Monotherapie erhielten. Der für die lineare Clearance geschätzte Populationsmedian betrug 6,08 ml/h (5,75-6,33 ml/h, 95 % Konfidenzintervall). Der geschätzte Median der Halbwertszeit betrug 28,2 Tage. Die Populations-Analyse mit 1781 Patienten untersuchte die Wirkungen ausgewählter Kovariaten wie Körpergewicht, Alter, Geschlecht, das Vorliegen von anti-Natalizumab-Antikörpern und die Formulierung auf die pharmakokinetischen Eigenschaften. Es wurde festgestellt, dass nur das Körpergewicht, das Vorliegen von anti-Natalizumab-Antikörpern und die in Phase 2 Studien verwendete Formulierung die Disposition von Natalizumab beeinflussen. Es wurde festgestellt, dass das Körpergewicht die Clearance unterproportional beeinflusst, so dass eine ±43 %ige Veränderung des Körpergewichts nur zu einer -33 %igen bis 30 %igen Veränderung der Clearance führte. Das Vorliegen von persistierenden anti-Natalizumab-Antikörpern erhöhte die Natalizumab-Clearance um das circa 2,45fache, was mit den reduzierten Natalizumabkonzentrationen im Serum übereinstimmt, die bei persistierend Antikörper-positiven Patienten beobachtet werden (siehe unter «Unerwünschte Wirkungen»).
Die Auswirkungen eines Plasmaaustauschs auf die Clearance und die Pharmakodynamik von Natalizumab wurden in einer Studie mit 12 MS-Patienten untersucht. Die geschätzte Wirkstoffentfernung nach 3 Behandlungen mit Plasmaaustausch (über einen Zeitraum von 5-8 Tagen) lag bei etwa 70-80 %. In früheren Studien, in denen die Messung nach Absetzen des Wirkstoffes ohne Plasmaaustausch über einen ähnlichen Beobachtungszeitraum hinweg erfolgte, ergab sich dagegen ein Wert von etwa 40 %. Die Bedeutung eines Plasmaaustauschs für die Wiederherstellung der Lymphozytenmigration und letztendlich sein klinischer Nutzen sind nicht bekannt. Eine retrospektive Analyse von Natalizumab nach der Zulassung konnte keinen Unterschied innert 2 Jahren hinsichtlich der Überlebensrate nach PML Diagnose zwischen Patienten mit oder ohne PLEX Behandlung feststellen (siehe auch «Warnhinweise und Vorsichtsmassnahmen», PML und IRIS (Immune Reconstitution Inflammatory Syndrome)).
Kinetik spezieller Patientengruppen
Die Pharmakokinetik von subkutan verabreichtem Natalizumab bei pädiatrischen MS-Patienten wurde nicht untersucht.
Die Pharmakokinetik von Natalizumab bei Patienten mit Nieren- oder Leberinsuffizienz wurde nicht untersucht.
Präklinische DatenBasierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Gentoxizität, Kanzerogenität und Reproduktionstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Übereinstimmend mit der pharmakologischen Wirkung von Natalizumab trat in den meisten invivo-Studien ein verändertes Lymphozyten-Trafficking in Form einer Zunahme der Zahl von weissen Blutzellen sowie eines erhöhten Milzgewichts in Erscheinung. Diese Veränderungen waren reversibel und schienen keine nachteiligen toxikologischen Konsequenzen zu haben.
Kanzerogenität
In bei Mäusen durchgeführten Studien wurden das Wachstum und die Metastasierung von Melanomzellen und lymphoblastischen leukämischen Tumorzellen durch die Gabe von Natalizumab nicht erhöht.
Im Ames-Test bzw. humanen Chromosomenaberrationstests wurden keine klastogenen oder mutagenen Wirkungen von Natalizumab beobachtet. Natalizumab zeigte keine Wirkungen auf invitro-Testverfahren zur α4-Integrin-positiven Tumorzelllinienproliferation oder -zytotoxizität.
Reproduktionstoxizität
In einer Studie mit Dosierungen, die höher als die Dosis beim Menschen waren, wurde eine Reduktion der Fertilität weiblicher Meerschweinchen beobachtet; Natalizumab beeinträchtigte die männliche Fertilität nicht. Es wird als unwahrscheinlich erachtet, dass Natalizumab bei Einhaltung der empfohlenen Höchstdosis die Fertilität von Menschen beeinträchtigt.
Die Wirkung von Natalizumab auf die Reproduktion wurde in 5 Studien beurteilt, 3 bei Meerschweinchen und 2 bei Cynomolgus-Affen. Diese Studien zeigten keinen Hinweis auf teratogene Wirkungen oder Wirkungen auf das Wachstum der Nachkommen. In einer Studie bei Meerschweinchen wurde ein geringer Rückgang bei der Zahl der lebenden Nachkommen festgestellt. In einer Studie bei Affen verdoppelte sich die Zahl der Aborte in den mit 30 mg/kg Natalizumab behandelten Gruppen im Vergleich zu den entsprechenden Kontrollgruppen. Dies war die Folge einer hohen Inzidenz von Aborten in beiden behandelten Gruppen der ersten Kohorte, die in der zweiten Kohorte nicht beobachtet wurde. In keiner anderen Studie wurden Auswirkungen auf die Abortrate festgestellt. Eine Studie bei trächtigen Cynomolgus-Affen zeigte Natalizumab bedingte fetale Veränderungen, die eine leichte Anämie, eine reduzierte Thrombozytenzahl, ein erhöhtes Milzgewicht sowie ein reduziertes Leber- und Thymusgewicht beinhalteten. Diese Veränderungen waren assoziiert mit einer gesteigerten extramedullären Hämatopoese in der Milz, einer Thymusatrophie sowie einer verminderten Hämatopoese in der Leber. Bei Nachkommen von Muttertieren, die bis zur Geburt mit Natalizumab behandelt worden waren, wurde ebenfalls eine reduzierte Thrombozytenzahl festgestellt, jedoch fanden sich bei diesen Jungtieren keine Hinweise auf eine Anämie. Sämtliche Veränderungen wurden bei Dosen beobachtet, die über denjenigen lagen, die beim Menschen zum Einsatz kommen, und waren nach der Clearance von Natalizumab reversibel.
Bei Cynomolgus-Affen, die bis zur Geburt Natalizumab erhielten, wurde Natalizumab in geringer Konzentration in der Milch einiger Muttertiere nachgewiesen.
Sonstige HinweiseInkompatibilitäten
Tysabri zur subkutanen Injektion wird nicht verdünnt.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Die Fertigspritzen können bis zu 24 Stunden bei Raumtemperatur in ihrer Originalverpackung aufbewahrt werden. Die Fertigspritzen sollten nicht zurück in die Kühllagerung verbracht werden. Keine externen Wärmequellen wie heisses Wasser verwenden, um die Fertigspritzen zu erwärmen.
Besondere Lagerungshinweise
Im Kühlschrank (2-8 °C) lagern. Nicht einfrieren.
Die Fertigspritzen in der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
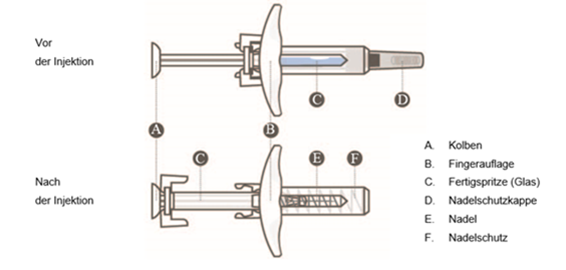
Die Fertigspritze weist ein Nadelschutzsystem auf, das bei vollständigem Drücken des Kolbens automatisch aktiviert wird. Beim Loslassen des Kolbens wird der Nadelschutz über die offenliegende Nadel geschoben.
1. Die Dosispackung aus dem Kühlschrank nehmen und vor der Durchführung der Injektionen Raumtemperatur annehmen lassen. Die empfohlene Dauer für das Erwärmen beträgt 30 Minuten.
·Keine externen Wärmequellen wie heisses Wasser verwenden, um die Fertigspritzen zu erwärmen.
2. BEIDE Fertigspritzen aus der Schale nehmen. Überprüfen, ob das Arzneimittel in jeder Fertigspritze als farblose bis leicht gelbe, leicht opaleszierende Lösung vorliegt, die im Wesentlichen frei von sichtbaren Partikeln ist. Möglicherweise sind in den Sichtfenstern Luftbläschen zu sehen. Dies ist normal und hat keinen Einfluss auf die Dosis.
·Die Fertigspritzen nicht verwenden, wenn:
·ihr Haltbarkeitsdatum abgelaufen ist.
·Farbe und Klarheit der Flüssigkeit nicht den vorstehenden Angaben entsprechen oder die Flüssigkeit Schwebstoffe enthält.
·es Anzeichen einer Beschädigung (Risse, Absplitterungen usw.) gibt.
·Wenn einer der vorstehend beschriebenen Umstände zutrifft, ist unverzüglich die zuständige Apotheke zu kontaktieren.
3. Eine volle Dosis entspricht zwei innerhalb von 30 Minuten hintereinander verwendeten Spritzen.

4.Bei der Injektion aseptisch (sauber und keimfrei) vorgehen und auf einer flachen Arbeitsfläche arbeiten.
5.Für die erste subkutane Injektion eine Stelle am Oberschenkel, der Bauchdecke oder der Rückseite des Oberarms wählen.
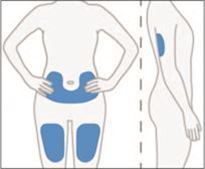
·Nicht in einen Bereich des Körpers injizieren, an dem die Haut in irgendeiner Weise gereizt, gerötet, geprellt, infiziert oder vernarbt ist.
6. Die erste Injektion geben und dabei die empfohlenen Standardverfahren für subkutane Injektionen befolgen.
7. Den Kolben in einer langsamen, gleichmässigen Bewegung drücken, bis die Spritze vollständig leer ist. Den Kolben nicht zurückziehen.

8. Beim Entfernen der Spritze aus der Injektionsstelle den Kolben loslassen und die Nadel GLEICHZEITIG in einer geraden Linie herausziehen. Beim Loslassen des Kolbens wird der Nadelschutz über die offenliegende Nadel geschoben.
·Zur Vermeidung einer versehentlichen Nadelstichverletzung die Nadel nicht berühren.
·Zur Vermeidung einer versehentlichen Nadelstichverletzung die Schutzkappe nicht wieder auf die Nadel aufsetzen.
9. Die Injektionen nacheinander ohne wesentlichen Zeitverzug durchführen. Für den Fall, dass die zweite Injektion nicht unmittelbar nach der ersten Injektion durchgeführt werden kann, sollte die zweite Injektion spätestens 30 Minuten nach der ersten Injektion stattfinden. Die zweite Injektion sollte in einem Abstand von mindestens 3 cm von der ersten Injektionsstelle gegeben werden.
Die Patienten zwischen den Injektionen und nach der letzten Injektion eine Stunde lang beobachten. Die Injektion ist sofort zu beenden, wenn Anzeichen oder Symptome auftreten, die auf eine Reaktion vom Überempfindlichkeitstyp hindeuten (siehe unter «Warnhinweise und Vorsichtsmassnahmen»).

10. Die benutzte Spritze nach den vor Ort geltenden Bestimmungen entsorgen.
Zulassungsnummer68008 (Swissmedic).
PackungenDie Dosis von 300 mg Tysabri zur subkutanen Injektion befindet sich in zwei Fertigspritzen für den Einmalgebrauch, die jeweils 150 mg/1 ml enthalten. B
ZulassungsinhaberinBiogen Switzerland AG, 6340 Baar.
Stand der InformationNovember 2024
|