ZusammensetzungWirkstoffe
Risankizumab, aus gentechnisch veränderten CHO (Chinese Hamster Ovary)-Zellen hergestellt.
Hilfsstoffe
Skyrizi 150 mg, Injektionslösung, im Fertigpen:
Natriumacetat-Trihydrat, Essigsäure 99%, Trehalose-Dihydrat, Polysorbat 20 und Wasser für Injektionszwecke q.s. ad solutionem pro 1 ml corresp. Natrium 0,21 mg.
Skyrizi 150 mg, Injektionslösung in einer Fertigspritze:
Natriumacetat-Trihydrat, Essigsäure 99%, Trehalose-Dihydrat, Polysorbat 20 und Wasser für Injektionszwecke q.s. ad solutionem pro 1 ml corresp. Natrium 0,21 mg.
Skyrizi 75 mg, Injektionslösung in einer Fertigspritze:
Dinatriumsuccinat-Hexahydrat, Succinylsäure, 34,0 mg Sorbitol (E420), Polysorbat 20 und Wasser für Injektionszwecke q.s. ad solutionem pro 0,83 ml corresp. Natrium 0,15 mg.
Indikationen/AnwendungsmöglichkeitenSKYRIZI ist indiziert zur Behandlung mittelschwerer bis schwerer Plaque-Psoriasis bei erwachsenen Patienten, die auf andere systemische Therapien wie beispielsweise Cyclosporin, Methotrexat (MTX) oder PUVA (Psoralen und UV-A) unzureichend angesprochen haben oder bei denen eine Kontraindikation oder Unverträglichkeit gegenüber solchen Therapien besteht.
Dosierung/AnwendungDie Anwendung von SKYRIZI sollte unter Aufsicht eines Arztes mit Erfahrung in der Diagnose und Behandlung der Plaque-Psoriasis erfolgen. Vor Beginn der Therapie muss der Arzt sicherstellen, dass die Patienten verstanden haben, dass es sich bei SKYRIZI um eine neuartige Therapie mit limitierter Erfahrung über ein Jahr hinaus und unbekannten Langzeitrisiken handelt.
Nach geeigneter Schulung in der Technik der subkutanen Injektion und Entsorgung können die Patienten SKYRIZI selbst injizieren, wenn ein Arzt dies für angemessen erachtet. Jedoch sollte der Arzt für eine geeignete Nachbeobachtung der Patienten sorgen. Patienten und deren Betreuer sollen auf die Anwendungshinweise im entsprechenden Beipackzettel hingewiesen werden.
Um die Rückverfolgbarkeit von biotechnologisch hergestellten Arzneimitteln sicherzustellen, wird empfohlen, Handelsname und Chargennummer bei jeder Behandlung zu dokumentieren.
Empfohlene Dosierung
Die empfohlene Dosis beträgt 150 mg als subkutane Injektion in Woche 0, Woche 4 und danach alle 12 Wochen.
Bei Patienten, die nach 16-wöchiger Behandlung kein Ansprechen zeigen, sollte der Abbruch der Behandlung in Erwägung gezogen werden. Manche Patienten mit initial partiellem Ansprechen können bei Weiterführung der Behandlung über 16 Wochen hinaus eine weitere Verbesserung zeigen.
Spezielle Dosierungsanweisungen
Patienten mit Nieren- oder Leberinsuffizienz
Es wurden keine spezifischen Studien zur Beurteilung der Auswirkung einer Leber- oder Niereninsuffizienz auf die Pharmakokinetik von SKYRIZI durchgeführt. Allgemein wird nicht davon ausgegangen, dass diese Erkrankungen einen signifikanten Einfluss auf die Pharmakokinetik von monoklonalen Antikörpern haben, weshalb Dosisanpassungen für nicht erforderlich gehalten werden (siehe «Pharmakokinetik»).
Ältere Patienten
Es ist keine Dosisanpassung erforderlich (siehe «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von SKYRIZI bei Kindern und Jugendlichen unter 18 Jahren sind noch nicht erwiesen.
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Klinisch relevante aktive Infektionen (z.B. aktive Tuberkulose).
Warnhinweise und VorsichtsmassnahmenInfektionen
SKYRIZI kann das Risiko von Infektionen erhöhen. In klinischen Studien traten Infektionen während einer 16- wöchigen Behandlung bei 22.1% der Patienten aus der SKYRIZI-Gruppe versus 14.7% der Patienten aus der Placebo-Gruppe auf. Die Raten schwerwiegender Infektionen in der SKYRIZI-Gruppe war 0.4% (5/1306) und in der Placebo-Gruppe 0.3% (1/300). Bei Patienten mit einer klinisch relevanten aktiven Infektion sollte die Behandlung mit SKYRIZI nicht eingeleitet werden, bevor die Infektion abgeklungen oder angemessen behandelt ist. Patienten, die in Bezug auf HCV- oder HIV seropositiv waren, solche, die positiv auf eine Hepatitis-B-Infektion getestet wurden, und solche mit chronischen oder rezidivierenden Infektionen in der Vorgeschichte, waren von den klinischen Studien ausgeschlossen.
Bei Patienten mit chronischen Infektionen oder anamnestisch bekannten rezidivierenden Infektionen sollte vor der Einleitung einer Therapie die Infektion abgeklungen oder angemessen therapiert sein.
Die Patienten sollten angewiesen werden, ärztlichen Rat einzuholen, wenn es bei ihnen zu Zeichen oder Symptomen einer klinisch bedeutsamen Infektion kommt. Falls sich bei einem Patienten eine solche Infektion entwickelt, oder falls dieser nicht auf eine Standardtherapie anspricht, sollte der Patient engmaschig überwacht werden und die Behandlung mit SKYRIZI sollte unterbrochen werden, bis die Infektion abgeklungen ist.
Tuberkulose
In den klinischen Phase-3-Studien zu Psoriasis entwickelte sich während des mittleren Nachbeobachtungszeitraums von 61 Wochen bei keinem der 72 Patienten mit latenter Tuberkulose (TB), die während der Studien gleichzeitig SKYRIZI und eine geeignete TB-Prophylaxe erhielten, auf SKYRIZI eine aktive TB. Vor der Einleitung einer Behandlung sind die Patienten bezüglich einer TB-Infektion zu beurteilen. Bei Patienten mit latenter TB sollte vor der Verabreichung von SKYRIZI eine TB-Therapie eingeleitet werden. SKYRIZI darf bei Patienten mit aktiver TB nicht angewendet werden.
Patienten unter Behandlung mit SKYRIZI sollten während und nach der Behandlung auf Anzeichen und Symptome einer aktiven TB überwacht werden.
Impfungen
Vor Beginn einer Behandlung mit SKYRIZI sollte erwogen werden, alle geeigneten Impfungen entsprechend den aktuellen Leitlinien für Impfungen vorzunehmen. SKYRIZI sollte nicht zusammen mit Lebendimpfstoffen angewendet werden. Es liegen keine Daten zum Ansprechen auf Lebend- oder inaktivierte Impfstoffe vor. Ein genügender zeitlicher Abstand zwischen Impfungen mit Lebendimpfstoffen und dem Beginn der Therapie gemäss aktuellen Impfrichtlinien zu immunsuppressiven Wirkstoffen ist einzuhalten. Informationen zur Anwendung von immunsuppressiven Wirkstoffen mit spezifischen Impfstoffen können auch den jeweiligen Fachinformationen entnommen werden.
Maligne Erkrankungen
In klinischen Studien mit einer Beobachtungszeit über ein Jahr liegen keine Hinweise vor für ein erhöhtes Risiko für maligne Erkrankungen.
Psoriasis-Patienten, die zuvor eine UV-Therapie erhalten haben, sollten vor und während der Behandlung mit SKYRIZI gründlich auf das Vorliegen von Hauttumoren untersucht werden.
Begleitende Therapien mit anderen systemischen Immunsuppressiva oder Phototherapie
Die Sicherheit und Wirksamkeit von SKYRIZI in Kombination mit Immunsuppressiva, einschliesslich Biologika, oder Phototherapie sind nicht untersucht worden.
Hypersensitivität
Wenn eine schwerwiegende Überempfindlichkeitsreaktion auftritt, sollte die Verabreichung von SKYRIZI unverzüglich abgebrochen werden und eine geeignete Therapie eingeleitet werden.
Sonstige Bestandteile mit bekannter Wirkung
SKYRIZI, Injektionslösung im Fertigpen bzw. in einer Fertigspritze à 150 mg und SKYRIZI, Injektionslösung in einer Fertigspritze à 75 mg enthalten weniger als 1 mmol Natrium (23 mg) pro 150 mg Dosis, d.h. es ist nahezu «natriumfrei».
SKYRIZI, Injektionslösung in einer Fertigspritze à 75 mg enthält 68,0 mg Sorbitol pro 150 mg Dosis.
InteraktionenEs ist nicht zu erwarten, dass SKYRIZI durch Leberenzyme metabolisiert oder renal ausgeschieden wird. Arzneimittelinteraktionen zwischen SKYRIZI und Inhibitoren/Induktoren von Wirkstoff-metabolisierenden Enzymen sind nicht zu erwarten.
Angesichts der Ergebnisse einer Arzneimittelinteraktionsstudie an Patienten mit Plaque-Psoriasis und von populationspharmakokinetischen Analysen dürfte Risankizumab weder Arzneimittelinteraktionen verursachen noch durch solche beeinflusst werden (siehe «Pharmakokinetik»).
Bei gleichzeitiger Anwendung von Risankizumab und Cytochrom-P450-Substraten ist keine Dosisanpassung erforderlich.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Frauen im gebärfähigen Alter sollen während und für mindestens 20 Wochen nach der Behandlung eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Die begrenzten Daten zur Anwendung von SKYRIZI bei Schwangeren reichen für eine Beschreibung arzneimittelassoziierter Risiken nicht aus. Tierexperimentelle Studien deuten nicht auf direkte oder indirekte schädliche Wirkungen in Bezug auf Reproduktionstoxizität hin (siehe «Präklinische Daten»). Als Vorsichtsmassnahme soll eine Anwendung von SKYRIZI während der Schwangerschaft vermieden werden.
Stillzeit
Es liegen keine Daten zum Übergang von Risankizumab in die Muttermilch beim Menschen, zu den Auswirkungen auf das gestillte Kind oder zu den Wirkungen auf die Milchproduktion vor. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen zu unterbrechen ist oder ob auf die Behandlung mit SKYRIZI verzichtet werden soll. Dabei soll sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie für die Frau berücksichtigt werden.
Fertilität
Es liegen keine klinischen Daten zur Wirkung von Risankizumab auf die Fertilität vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte Wirkung auf die Fertilität (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenEs wurden keine entsprechenden Studien durchgeführt.
Unerwünschte WirkungenKlinische Studien
Im Rahmen der klinischen Entwicklung wurden insgesamt 2'234 Patienten in Studien zur Plaque-Psoriasis mit SKYRIZI behandelt; dies entspricht einer Exposition von 2'167 Patientenjahren. Davon wurden 1'208 Patienten mit Psoriasis mindestens ein Jahr lang mit SKYRIZI behandelt. 1590 Patienten erhielten 150 mg SKYRIZI nach Randomisierung oder nach 16 Wochen Behandlung mit Placebo, dies entspricht einer Exposition von 1'688 Patientenjahren. Davon wurden 1091 Patienten mindestens ein Jahr lang mit SKYRIZI behandelt.
Die Daten aus placebo- und aktiv kontrollierten Studien wurden zusammengefasst, um die Sicherheit von SKYRIZI über bis zu 16 Wochen zu beurteilen. In der Gruppe unter SKYRIZI 150 mg wurden die Daten von insgesamt 1'306 Patienten ausgewertet. Zu schwerwiegenden unerwünschten Ereignissen kam es bei 2,4% der Patienten in der Gruppe unter SKYRIZI (9,9 Ereignisse pro 100 Patientenjahre), verglichen mit 4,0% in der Placebogruppe (17,4 Ereignisse pro 100 Patientenjahre), 5,0% in der Ustekinumab-Gruppe (18,4 Ereignisse pro 100 Patientenjahre) und 3,0% in der Adalimumab-Gruppe (14,7 Ereignisse pro 100 Patientenjahre).
Die unerwünschten Wirkungen von SKYRIZI in klinischen Studien (Tabelle 1) sind nach MedDRA-Systemorganklasse und entsprechend folgender Konvention aufgeführt: sehr häufig (≥1/10); häufig (<1/10, ≥1/100); gelegentlich (<1/100, ≥1/1'000); selten (<1/1'000, ≥1/10'000); sehr selten (<1/10'000).
Tabelle 1: Unerwünschte Arzneimittelwirkungen in klinischen Studien
|
Systemorganklasse
|
Häufigkeit
|
Unerwünschte Wirkungen
| |
Infektionen und parasitäre Erkrankungen
|
Sehr häufig
|
Infektion der oberen Atemwegea (13%)
| |
Häufig
|
Tineab
| |
Gelegentlich
|
Follikulitis
| |
Erkrankungen des Nervensystems
|
Häufig
|
Kopfschmerzenc
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Häufig
|
Müdigkeitd
Reaktionen an der Injektionsstellee
| |
a
Beinhaltet: Atemwegsinfektion (viral, bakteriell oder nicht spezifiziert), Sinusitis (auch akute), Rhinitis, Nasopharyngitis, Pharyngitis (auch virale), Tonsillitis
b Beinhaltet: Tinea pedis, Tinea cruris, Tinea corporis, Tinea versicolor, Tinea manuum, Onychomykose
c Beinhaltet: Kopfschmerzen, Spannungskopfschmerzen, Sinus-Kopfschmerzen
d Beinhaltet: Müdigkeit, Asthenie
e Beinhaltet: Blauer Fleck, Erythem, Hämatom, Blutung, Reizung, Schmerzen, Juckreiz, Reaktion und Schwellung an der Injektionsstelle
|
Beschreibung ausgewählter Nebenwirkungen
Infektionen
In den ersten 16 Wochen kam es bei 22,1% der Patienten in der Gruppe unter SKYRIZI zu Infektionen (90,8 Ereignisse pro 100 Patientenjahre), verglichen mit 14,7% in der Placebogruppe (56,5 Ereignisse pro 100 Patientenjahre), 20,9% in der Ustekinumab-Gruppe (87,0 Ereignisse pro 100 Patientenjahre) und 24,3% in der Adalimumab-Gruppe (104,2 Ereignisse pro 100 Patientenjahre). Die Infektionen waren in der Mehrzahl der Fälle nicht schwerwiegend, wiesen einen leichten bis mässigen Schweregrad auf und führten nicht zum Absetzen von SKYRIZI.
Über das gesamte Psoriasis-Programm hinweg, die Langzeitexposition gegenüber SKYRIZI mit eingeschlossen, fiel die Infektionsrate (75,5 Ereignisse pro 100 Patientenjahre) ähnlich aus wie in den ersten 16 Behandlungswochen.
Langfristige Sicherheit
Die Häufigkeit von unerwünschten Wirkungen bis einschliesslich Woche 52 stand mit dem Sicherheitsprofil während der ersten 16 Behandlungswochen in Einklang. Die nach Exposition adjustierte Rate von schwerwiegenden unerwünschten Ereignissen pro 100 Patientenjahre belief sich bis einschliesslich Woche 52 bei Patienten unter SKYRIZI auf 9,4 und bei Patienten unter Ustekinumab auf 10,9. Bei Patienten, die bis zu maximal 77 Wochen gegenüber SKYRIZI exponiert waren, wurden im Vergleich zu den ersten 16 Behandlungswochen keine neuen unerwünschten Wirkungen erfasst.
Immunogenität
Wie alle therapeutischen Proteine besitzt auch SKYRIZI eine potentielle Immunogenität. Der Nachweis einer Antikörperbildung hängt in hohem Mass von der Sensitivität und Spezifität des Assays ab. Darüber hinaus kann die beobachtete Inzidenz der Antikörperpositivität (gilt auch für neutralisierende Antikörper) in einem Assay von mehreren Faktoren beeinflusst werden, darunter der Assay-Methode, der Handhabung der Proben, dem Zeitpunkt der Probenahme, Begleitmedikationen und der Grunderkrankung. Aus diesen Gründen ist ein Vergleich der Inzidenz von Antikörpern gegen Risankizumab mit der Inzidenz von Antikörpern gegen andere Biologika mit Vorsicht zu interpretieren.
Bei Patienten, die in klinischen Psoriasis-Studien bis zu 52 Wochen lang SKYRIZI in der empfohlenen klinischen Dosis erhalten haben, wurden bei 24% (263/1'079) der beurteilten Patienten unter Behandlung aufgetretene Antikörper gegen den Wirkstoff und bei 14% (150/1'079) neutralisierende Antikörper nachgewiesen.
Antikörper gegen Risankizumab, auch neutralisierende Antikörper, waren bei den meisten Patienten nicht mit Veränderungen des klinischen Ansprechens oder der Sicherheit assoziiert. Höhere Antikörpertiter bei etwa 1% der Patienten, welche mit SKYRIZI behandelt wurden, waren assoziiert mit geringeren Konzentrationen an Risankizumab und reduziertem klinischen Ansprechen.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIm Fall einer Überdosierung wird empfohlen, den Patienten auf Zeichen und Symptome von unerwünschten Wirkungen zu überwachen und unverzüglich eine geeignete symptomatische Behandlung einzuleiten.
Eigenschaften/WirkungenATC-Code
L04AC
Risankizumab, ein Interleukin-23-Blocker, ist ein humanisierter Immunoglobulin G1 (IgG1) monoklonaler Antikörper. Risankizumab wird in einer Säugerzelllinie produziert unter Einsatz rekombinanter DNA-Technologie.
Wirkungsmechanismus
Risankizumab ist ein humanisierter Immunoglobulin G1 (IgG1) monoklonaler Antikörper, der selektiv mit hoher Affinität an die p19-Untereinheit des humanen Zytokins Interleukin 23 (IL-23) bindet und dessen Interaktion mit dem IL-23-Rezeptorkomplex hemmt. IL-23 ist ein natürlich vorkommendes Zytokin, das an Entzündungs- und Immunreaktionen beteiligt ist. IL-23 unterstützt die Proliferation, die Erhaltung und die Aktivierung von Th17-Zellen, die IL-17A, IL-17F und IL-22 sowie weitere proinflammatorische Zytokine produzieren und eine zentrale Rolle bei der Pathogenese von entzündlichen Autoimmunkrankheiten wie Psoriasis spielen. Bei Patienten mit Plaque-Psoriasis findet sich in Hautläsionen gegenüber nicht betroffenen Arealen eine Hochregulation von IL-23. Indem Risankizumab die Bindung von IL-23 an seinen Rezeptor blockiert, hemmt Risankizumab die IL-23-abhängige zelluläre Signaltransduktion und die Freisetzung proinflammatorischer Zytokine.
Risankizumab bindet nicht an humanes IL-12, welches wie das IL-23 auch eine p40-Untereinheit besitzt.
Pharmakodynamik
In einer Studie mit Psoriasis-Patienten wurde nach einer Einzeldosis Risankizumab eine reduzierte Expression von Genen, die mit der IL-23/IL-17-Axis assoziiert sind, in der Haut beobachtet. Darüber hinaus wurden in Psoriasis-Läsionen Verminderungen in der Epidermis-Dicke, Infiltration von Entzündungszellen und Expression von Psoriasis-Krankheitsmarkern festgestellt.
Klinische Wirksamkeit
Die Wirksamkeit und Sicherheit von SKYRIZI wurden bei 2'109 Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis in vier multizentrischen, randomisierten Placebo- und/oder- aktiv- kontrollierten Doppelblindstudien beurteilt (ULTIMMA-1, ULTIMMA-2, IMMHANCE und IMMVENT). In den ULTIMMA-1 und ULTIMMA-2 Studien wurde Ustekinumab als aktiver Komparator eingesetzt. In der IMMHANCE Studie wurde zudem bei Patienten mit erfolgreichem Ansprechen das Absetzen von Risankizumab und die Wiederaufnahme der Therapie untersucht. In der IMMVENT Studie wurde Adalimumab als aktiver Komparator eingesetzt. Nach Abschluss der Studien konnten die Patienten in die offene Fortsetzungsstudie, LIMITLESS, eingeschlossen werden. Die eingeschlossenen Patienten waren mindestens 18 Jahre alt und litten an Plaque-Psoriasis mit betroffener Körperoberfläche (body surface area, BSA) von ≥10%, einem Score der statischen Beurteilung durch den Arzt (static Physician Global Assessment, sPGA) von ≥3, der Psoriasis auf einer Schweregrad-Skala von 0 bis 4, sowie einem Psoriasis Area and Severity Index (PASI) von ≥12. Patienten mit erythrodermischer Psoriasis, Psoriasis guttata oder pustulöser Psoriasis waren ausgeschlossen.
Insgesamt wiesen die Patienten einen medianen Baseline PASI-Score von 17,8 und eine mediane BSA-Beteiligung von 20,0% auf. Der Baseline sPGA-Score entsprach bei 19,3% der Patienten einer schweren Erkrankung. Bei insgesamt 9,8% der Studienpatienten lag anamnestisch eine diagnostizierte Psoriasis-Arthritis vor.
Über alle Studien hinweg hatten 30,9% der Patienten zuvor keine nicht-biologische systemische oder biologische Therapie, 38,1% eine Phototherapie, 48,3% eine nicht-biologische systemische Therapie und 42,1% eine biologische Therapie (23.7% aller Patienten in diesen Studien erhielten mindestens einen TNF-alpha-Blocker) zur Behandlung der Psoriasis erhalten. Um eine Beurteilung der Wirksamkeit von SKYRIZI in der Behandlung der Psoriasis ohne Verzerrung zu ermöglichen, war eine Begleitbehandlung mit systemischer oder topischer Therapie (mit Ausnahme milder topischer Kortikosteroide im Gesicht, Achseln und/oder Genitalien) oder Phototherapie in den Studien nicht zulässig.
ULTIMMA-1 und ULTIMMA-2
In die Studien ULTIMMA-1 und ULTIMMA-2 wurden 997 Patienten eingeschlossen (598 Patienten wurden zu SKYRIZI 150 mg, 199 Patienten zu Ustekinumab 45 mg oder 90 mg [nach Ausgangskörpergewicht] und 200 Patienten zu Placebo randomisiert). Die Behandlung wurde in Woche 0, Woche 4 und anschliessend alle 12 Wochen verabreicht. Die Ergebnisse sind in Tabelle 2 und Abbildung 1 dargestellt.
Tabelle 2: Wirksamkeitsergebnisse bei Erwachsenen mit Plaque-Psoriasis in ULTIMMA-1 und ULTIMMA-2
|
|
ULTIMMA-1
|
ULTIMMA-2
| |
|
SKYRIZI
(N=304)
n (%)
|
Ustekinumab
(N=100)
n (%)
|
Placebo
(N=102)
n (%)
|
SKYRIZI
(N=294)
n (%)
|
Ustekinumab
(N=99)
n (%)
|
Placebo
(N=98)
n (%)
| |
sPGA «frei von» oder «nahezu frei von» (0 oder 1)
| |
Woche 16
|
267 (87,8)a
|
63 (63,0)
|
8 (7,8)
|
246 (83,7)a
|
61 (61,6)
|
5 (5,1)
| |
Woche 52
|
262 (86,2)
|
54 (54,0)
|
--
|
245 (83,3)
|
54 (54,5)
|
--
| |
sPGA «frei von» (0)
| |
Woche 16
|
112 (36,8)
|
14 (14,0)
|
2 (2,0)
|
150 (51,0)
|
25 (25,3)
|
3 (3,1)
| |
Woche 52
|
175 (57,6)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
PASI 75
| |
Woche 12
|
264 (86,8)
|
70 (70,0)
|
10 (9,8)
|
261 (88,8)
|
69 (69,7)
|
8 (8,2)
| |
Woche 52
|
279 (91,8)
|
70 (70,0)
|
--
|
269 (91.5)
|
76 (76,8)
|
--
| |
PASI 90
| |
Woche 16
|
229 (75,3)a
|
42 (42,0)
|
5 (4,9)
|
220 (74,8)a
|
47 (47,5)
|
2 (2,0)
| |
Woche 52
|
249 (81,9)
|
44 (44,0)
|
--
|
237 (80,6)
|
50 (50,5)
|
--
| |
PASI 100
| |
Woche 16
|
109 (35,9)
|
12 (12,0)
|
0 (0,0)
|
149 (50,7)
|
24 (24,2)
|
2 (2,0)
| |
Woche 52
|
171 (56,3)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
Bei allen Vergleichen von SKYRIZI mit Ustekinumab und Placebo wurde ein p-Wert von < 0,001 erreicht, ausser für den PASI 75 in Woche 52 in ULTIMMA-2 (p = 0,001).
a co-primäre Endpunkte versus Placebo
|
Abbildung 1: Zeitlicher Verlauf der mittleren prozentualen Änderung von der Baseline ausgehend des PASI in ULTIMMA-1 und ULTIMMA-2
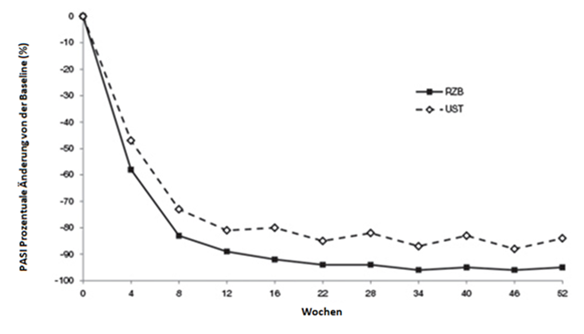
RZB = Risankizumab
UST = Ustekinumab
P < 0,001 zu jedem Zeitpunkt
Eine Analyse nach Alter, Geschlecht, ethnische Abstammung, Körpergewicht, Baseline PASI-Score, gleichzeitiger Psoriasis-Arthritis, vorheriger nicht-biologischer systemischer Behandlung, vorheriger Biologika-Therapie und vorherigem Versagen eines Biologikums erbrachte zwischen diesen Teilgruppen keine Unterschiede hinsichtlich des Ansprechens auf SKYRIZI.
Bei Patienten, die mit SKYRIZI behandelt wurden, zeigte sich in Woche 16 und Woche 52 eine Besserung der Psoriasis im Bereich der Kopfhaut, der Nägel, der Handflächen und der Fusssohlen.
IMMHANCE
In die Studie IMMHANCE wurden 507 Patienten eingeschlossen (407 Patienten wurden zu SKYRIZI 150 mg und 100 Patienten zu Placebo randomisiert). Die Behandlung wurde in Woche 0, Woche 4 und anschliessend alle 12 Wochen verabreicht.
In Woche 16 erwies sich SKYRIZI gegenüber Placebo hinsichtlich der co-primären Endpunkte sPGA «frei von» oder «nahezu frei von» (83,5% SKYRIZI vs. 7,0% Placebo) und PASI 90 (73,2% SKYRIZI vs. 2,0% Placebo) als überlegen. In Woche 16 wiesen mehr Patienten unter SKYRIZI ein erscheinungsfreies Hautbild [sPGA 0 (46,4% SKYRIZI vs. 1,0% Placebo) bzw. einen PASI 100 (47,2% SKYRIZI vs. 1,0% Placebo)] auf. Zudem erreichten Patienten unter SKYRIZI mit höherer Wahrscheinlichkeit ein PASI-75-Ansprechen als Patienten unter Placebo (88,7% SKYRIZI vs. 8,0% Placebo).
Bei keinem der 31 Patienten der IMMHANCE-Studie, die eine latente Tuberkulose (TB) aufwiesen und während der Studie keine Prophylaxe erhielten, entwickelte sich während des mittleren Nachbeobachtungszeitraums auf SKYRIZI von 55 Wochen eine aktive TB. Jedoch sollte bei Patienten mit latenter TB mit einer TB-Therapie vor Verabreichung von SKYRIZI begonnen werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
IMMVENT
In die Studie IMMVENT wurden 605 Patienten eingeschlossen (301 Patienten wurden zu SKYRIZI und 304 Patienten zu Adalimumab randomisiert). Patienten, die zu SKYRIZI randomisiert wurden, erhielten in Woche 0, Woche 4 und anschliessend alle 12 Wochen eine Dosis von 150 mg. Zu Adalimumab randomisierte Patienten erhielten 80 mg in Woche 0, 40 mg in Woche 1 und 40 mg alle zwei Wochen bis einschliesslich Woche 15. Bei den mit Adalimumab behandelten Patienten erfolgte ab Woche 16, je nach Ansprechen, eine Weiterbehandlung oder eine Therapieumstellung:
·< PASI 50: Umstellung auf SKYRIZI
·PASI 50 bis < PASI 90: Re- Randomisierung für Weiterbehandlung mit 40 mg Adalimumab alle 2 Wochen oder Umstellung auf SKYRIZI
·PASI 90: Weiterbehandlung mit 40 mg Adalimumab alle 2 Wochen.
In IMMVENT zeigten sich unter SKYRIZI in Woche 16 ähnliche Ergebnisse wie in den anderen klinischen Studien (Tabelle 3 und Abbildung 2).
Tabelle 3: Wirksamkeitsergebnisse in Woche 16 bei Erwachsenen mit Plaque-Psoriasis in IMMVENT
|
|
SKYRIZI
(N=301)
n (%)
|
Adalimumab
(N=304)
n (%)
| |
sPGA «frei von» oder «nahezu frei von»a
|
252 (83,7)
|
183 (60,2)
| |
PASI 75
|
273 (90,7)
|
218 (71,7)
| |
PASI 90a
|
218 (72,4)
|
144 (47,4)
| |
PASI 100
|
120 (39,9)
|
70 (23,0)
| |
Bei allen Vergleichen wurde ein p-Wert von < 0,001 erreicht.
a co-primäre Endpunkte
|
Bei Patienten, die in Woche 16 unter Adalimumab einen PASI 50 bis < PASI 90 aufwiesen und re-randomisiert wurden, zeigten sich bereits 4 Wochen nach der Re-Randomisierung zwischen Patienten mit Therapieumstellung auf SKYRIZI und Patienten, die mit Adalimumab weiterbehandelt wurden, Unterschiede in Bezug auf die PASI-90-Ansprechraten (49,1% vs. 26,8%). Insgesamt 66,0% (35/53) der Patienten erreichten nach Behandlung mit SKYRIZI über 28 Wochen einen PASI 90, verglichen mit 21,4% (12/56) der mit Adalimumab weiterbehandelten Patienten. Andere Parameter des Ansprechens fielen nach Umstellung auf SKYRIZI ebenfalls besser aus: 39,6% PASI 100, 39,6% sPGA «frei von» und 73,6% sPGA «frei von» oder «nahezu frei von» nach Umstellung auf SKYRIZI, verglichen mit 7,1% PASI 100, 7,1% sPGA «frei von» und 33,9% sPGA «frei von» oder «nahezu frei von» unter Weiterbehandlung mit Adalimumab.
Abbildung 2: Zeitlicher Verlauf des PASI 90 nach Re-Randomisierung in IMMVENT
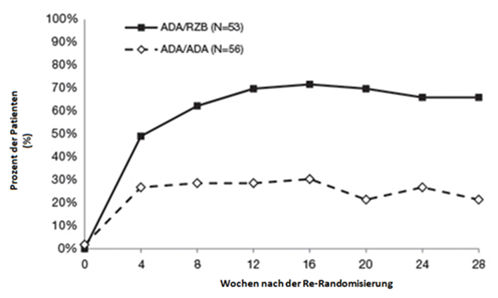
ADA/ADA: Patienten, die ursprünglich zu Adalimumab randomisiert und mit Adalimumab weiterbehandelt wurden
ADA/RZB: Patienten, die ursprünglich zu Adalimumab randomisiert und auf SKYRIZI umgestellt wurden
p < 0,05 in Woche 4 und p < 0,001 zu jedem Zeitpunkt ab Woche 8
Erhalt und Dauerhaftigkeit des Ansprechens
In einer kombinierten Analyse von Patienten, die innerhalb von ULTIMMA-1 und ULTIMMA-2 SKYRIZI erhielten und in Woche 16 ein PASI-100-Ansprechen zeigten, blieb das Ansprechen bei 79,8% (206/258) der mit SKYRIZI weiterbehandelten Patienten bis Woche 52 erhalten. 88,4% (398/450) der Patienten mit PASI-90-Ansprechen in Woche 16 zeigten bis Woche 52 ein anhaltendes Ansprechen.
Patienten, die innerhalb von IMMHANCE ursprünglich SKYRIZI erhalten hatten und in Woche 28 ein sPGA-Ansprechen «frei von» oder «nahezu frei von» aufwiesen, wurden nach Re-Randomisierung entweder mit SKYRIZI alle 12 Wochen bis einschliesslich Woche 88 (N=111) weiterbehandelt oder die Behandlung wurde abgesetzt (N=225).
In Woche 104 (16 Wochen nach der letzten SKYRIZI Verabreichung) erreichten 81,1% (90/111) der mit SKYRIZI weiterbehandelten Patienten ein sPGA-Ansprechen «frei von» oder «nahezu frei von» verglichen mit 7,1% (16/225) der Patienten, bei denen SKYRIZI abgesetzt wurde. Ein Verlust des sPGA-Ansprechens «frei von» oder «nahezu frei von» wurde bei Patienten, bei denen SKIRIZI abgesetzt wurde, bereits 12 Wochen nach einer verpassten Dosis beobachtet.
Die sPGA-Ansprechrate «frei von» in Woche 104 war 63,1% (70/111) in mit SKYRIZI weiterbehandelten Patienten verglichen mit 2,2% (5/225) in Patienten, bei welchen die SKYRIZI Behandlung abgesetzt wurde.
Es gab einen Anstieg zwischen Woche 28 und 88 im Anteil der mit SKYRIZI weiterbehandelten Patienten im PASI-100 und sPGA-Ansprechen «frei von».
Von denjenigen Patienten, welche ein sPGA-Ansprechen von «frei von» oder «nahezu frei von» in Woche 28 erreichten und ein Rezidiv (sPGA ≥3) nach Absetzen von SKYRIZI vorwiesen, erreichten 83,7% (128/153) wieder ein sPGA-Ansprechen «frei von» oder «nahezu frei von» nach 16 Wochen Wiederaufnahme der Behandlung.
Lebensqualität/Von Patienten berichtete Ergebnisse
Signifikant mehr Patienten, welche mit SKYRIZI behandelt wurden, erreichten einen Dermatology Life Quality Index (DLQI) Score von 0 oder 1 [keine Auswirkungen auf die gesundheitsbezogene Lebensqualität] in Woche 16 verglichen zu Placebo, Adalimumab oder Ustekinumab behandelten Patienten. Die Verbesserung hielt bis einschliesslich Woche 52 in den ULTIMMA-1 und ULTIMMA-2 Studien an.
In ULTIMMA-1 und ULTIMMA-2 zeigte sich in Woche 16 unter SKYRIZI eine signifikant stärkere Besserung der Psoriasis-Symptome (Juckreiz, Schmerzen, Rötung und Brennen, gemessen anhand des Psoriasis Symptom Score [PSS]) als unter Placebo. Verglichen mit Ustekinumab und Placebo erreichte ein signifikant höherer Anteil der Patienten unter SKYRIZI in Woche 16 einen PSS von 0 (Symptomfreiheit). Bis Woche 52 berichteten 55,7% (333/598) der Patienten unter SKYRIZI keinerlei Juckreiz, Schmerzen, Rötung oder Brennen.
PharmakokinetikAbsorption
Risankizumab zeigte in einem Dosierungsbereich von 18 bis 300 mg und 0,25 bis 1 mg/kg subkutan sowie von 200 bis 1'200 mg und 0,01 bis 5 mg/kg intravenös eine lineare Pharmakokinetik mit einem dosisproportionalen Anstieg der Exposition.
Nach subkutaner Anwendung von Risankizumab wurden 3 bis 14 Tage nach Verabreichung maximale Plasmakonzentrationen mit einer geschätzten absoluten Bioverfügbarkeit von 89% erreicht. Unter dem Dosierungsschema für Psoriasis-Patienten (150 mg in Woche 0, Woche 4 und anschliessend alle 12 Wochen) belaufen sich die geschätzte maximale Steady-State-Konzentrationen und Talkonzentrationen im Plasma auf 12 bzw. 2 µg/ml.
Mit der Fertigspritze wurde die Bioäquivalenz zwischen einer einzelnen Injektion à 150 mg Risankizumab und zwei Injektionen à 75 mg Risankizumab nachgewiesen. Auch wurde die Bioäquivalenz zwischen der Risankizumab 150 mg Fertigspritze und dem Fertigpen nachgewiesen.
Distribution
Bei einem typischen Psoriasis-Patienten mit einem Körpergewicht von 90 kg lag das Distributionsvolumen im Steady State (Vss) bei 11,2 l, was auf eine primär auf den vaskulären und interstitiellen Raum begrenzte Distribution von Risankizumab hinweist.
Metabolismus
Therapeutische monoklonale IgG-Antikörper werden ähnlich wie endogene IgG typischerweise über katabolische Stoffwechselwege zu kleinen Peptiden und Aminosäuren abgebaut. Es ist nicht zu erwarten, dass Risankizumab über Cytochrom-P450-Enzyme metabolisiert wird.
Elimination
Bei einem typischen Psoriasis-Patienten mit einem Körpergewicht von 90 kg betrugen die systemische Clearance (CL) von Risankizumab 0,31 l/Tag und die terminale Eliminationshalbwertszeit 28 Tage.
Als monoklonaler IgG1-Antikörper dürfte Risankizumab weder in den Nieren glomerulär filtriert noch als intaktes Molekül über den Urin ausgeschieden werden.
Arzneimittelinteraktionen
Es wurde eine Arzneimittelinteraktionsstudie an Patienten mit Plaque-Psoriasis durchgeführt, um die Wirkung einer wiederholten Anwendung von Risankizumab auf die Pharmakokinetik von Cytochrom-P450-(CYP-)empfindlichen Testsubstraten zu bewerten. Die Exposition gegenüber Koffein (CYP1A2-Substrat), Warfarin (CYP2C9-Substrat), Omeprazol (CYP2C19-Substrat), Metoprolol (CYP2D6-Substrat) und Midazolam (CYP3A4-Substrat) war nach der Behandlung mit Risankizumab ähnlich wie vor der Behandlung mit Risankizumab, was auf keine klinisch bedeutsamen Arzneimittelinteraktionen über diese Enzyme hindeutet.
In populationspharmakokinetischen Analysen zeigte sich, dass die Risankizumab-Exposition nicht durch Begleitmedikationen (Metformin, Atorvastatin, Lisinopril, Amlodipin, Ibuprofen, Acetylsalicylsäure und Levothyroxin), die einige Patienten mit Plaque-Psoriasis während der klinischen Studien anwendeten, beeinflusst wurde (siehe «Interaktionen»).
Kinetik spezieller Patientengruppen
Niereninsuffizienz oder Leberinsuffizienz
Es wurden keine spezifischen Studien zur Beurteilung der Auswirkung einer Nieren- oder Leberinsuffizienz auf die Pharmakokinetik von Risankizumab durchgeführt. Basierend auf populationspharmakokinetischen Analysen hatten Serumkreatininspiegel, Kreatinin-Clearance oder Leberfunktionswerte (ALT/AST/Bilirubin) bei Psoriasis-Patienten keinen bedeutenden Einfluss auf die Clearance von Risankizumab.
Als monoklonaler IgG1-Antikörper wird Risankizumab vorwiegend über intrazellulären Katabolismus eliminiert und durchläuft erwartungsgemäss keine Metabolisierung durch hepatische Cytochrom-P450-Enzyme oder renale Elimination (siehe «Dosierung/Anwendung»).
Ältere Patienten
Von 2'234 Patienten mit Plaque-Psoriasis, die mit SKYRIZI behandelt wurden, waren 243 Patienten mindestens 65 Jahre alt und 24 Patienten mindestens 75 Jahre alt. Zwischen älteren und jüngeren Patienten, die mit SKYRIZI behandelt wurden, zeigten sich hinsichtlich der Exposition, Sicherheit und Wirksamkeit von Risankizumab allgemein keine Unterschiede (siehe «Dosierung/Anwendung»).
Kinder und Jugendliche
Die Pharmakokinetik von Risankizumab bei Kindern und Jugendlichen wurde nicht untersucht.
Körpergewicht
Die Clearance und das Distributionsvolumen von Risankizumab nehmen mit steigendem Körpergewicht zu. Es wurde keine Korrelation des Körpergewichts mit klinisch bedeutsamen Veränderungen der Wirksamkeit und Sicherheit von Risankizumab beobachtet, weshalb keine gewichtsentsprechende Dosisanpassung erforderlich ist.
Geschlecht oder ethnische Abstammung
Bei erwachsenen Patienten mit Plaque-Psoriasis hatte das Geschlecht oder die ethnische Abstammung keine bedeutenden Auswirkungen auf die Clearance von Risankizumab. In einer klinischen Studie zur Pharmakokinetik wurden zwischen chinesischen oder japanischen Probanden und kaukasischen Probanden keine klinisch bedeutsamen Unterschiede hinsichtlich der Risankizumab-Exposition beobachtet.
Präklinische DatenBasierend auf den Studien zur Toxizität bei wiederholter Gabe, in denen auch Endpunkte der Sicherheitspharmakologie untersucht wurden, sowie einer Studie zur Reproduktions- und Entwicklungstoxizität an Cynomolgus-Affen mit Dosen von bis zu 50 mg/kg/Woche (entsprechend einer Exposition in Höhe des ca. 70-Fachen der klinischen Exposition unter der für den Menschen empfohlenen Höchstdosis [MRHD, maximum recommended human dose]) liessen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Mutagenität
Es wurden keine Studien zur Beurteilung der Mutagenität von SKYRIZI durchgeführt.
Karzinogenität
Es wurden keine Studien zur Beurteilung der Karzinogenität von SKYRIZI durchgeführt. In einer 26-wöchigen Studie zur chronischen Toxizität an Cynomolgus-Affen mit Dosen von bis zu 50 mg/kg/Woche (ungefähr das 70-Fache der klinischen Exposition unter der MRHD) wurden keine präneoplastischen oder neoplastischen Läsionen beobachtet.
Beeinträchtigung der Fertilität
Studien mit SKYRIZI an Cynomolgus-Affen mit Dosen von bis zu 50 mg/kg/Woche (ungefähr das 70-Fache der klinischen Exposition unter der MRHD) ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen auf die männliche oder weibliche Fertilität. In der 26-wöchigen Studie zur Toxizität bei wiederholter Gabe zeigten sich bei der histopathologischen Untersuchung der Reproduktionsorgane von männlichen und weiblichen Cynomolgus-Affen keinerlei relevante Befunde. In einer 26-wöchigen Studie mit wiederholter Gabe an geschlechtsreifen männlichen Cynomolgus-Affen wurden keine Wirkungen auf Parameter der männlichen Fertilität beobachtet.
Pharmakologie und/oder Toxikologie bei Tieren
In einer 26-wöchigen toxikologischen Studie mit wöchentlichen subkutanen Dosen von bis zu 50 mg/kg wurden bei männlichen und weiblichen Cynomolgus-Affen unter Expositionen, die etwa dem 70-Fachen der klinischen Exposition unter der MRHD entsprachen, keine unerwünschten Wirkungen festgestellt.
Es wurde eine erweiterte Studie zur prä- und postnatalen Entwicklungstoxizität an Cynomolgus-Affen durchgeführt. Trächtige Cynomolgus-Affen wurden ab Gestationstag 20 bis zur Geburt wöchentlich mit subkutanen Risankizumab-Dosen von 5 oder 50 mg/kg behandelt. Die Cynomolgus-Affen (Muttertiere und Jungtiere) wurden nach der Geburt 6 Monate (180 Tage) lang beobachtet. Diese Dosen führten zu Expositionen, die bis zu etwa dem 70-Fachen der klinischen Exposition unter der für den Menschen empfohlenen Höchstdosis (MRHD) entsprachen. Es wurden keine wirkstoffbedingten Todesfälle und/oder Missbildungen bei den Föten oder Jungtieren beobachtet. Bei der Beurteilung von externen, viszeralen, skelettbezogenen und verhaltensneurologischen Parametern und der Entwicklungs-Immunotoxikologischen Endpunkten wurden keine Wirkungen auf das Wachstum und die Entwicklung festgestellt. Die mittleren Serumkonzentrationen bei den Jungtieren stiegen dosisabhängig an und entsprachen etwa 20 - 90% der jeweiligen maternalen Konzentrationen. Nach der Geburt wiesen die meisten adulten weiblichen Cynomolgus-Affen und alle Jungtiere in den mit Risankizumab behandelten Gruppen bis zu 91 Tage postpartal messbare Risankizumab-Konzentrationen im Serum auf. Die Serumkonzentrationen lagen 180 Tage nach der Geburt unterhalb der Nachweisgrenze.
Sonstige HinweiseInkompatibilitäten
Da keine Verträglichkeitsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis und mit dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Im Kühlschrank (2 - 8 °C) lagern. Nicht einfrieren. In der Originalverpackung aufbewahren, um den Inhalt vor Licht zu schützen. Bewahren Sie dieses Arzneimittel nicht in Reichweite von Kindern auf.
Hinweise für die Handhabung
Die Lösung im 150 mg Fertigpen und in der 150 mg Fertigspritze ist farblos bis gelb und klar bis leicht opaleszent. Die Lösung in der 75 mg Fertigspritze ist farblos bis gelblich und klar bis leicht opaleszent.
Die Lösung im 150 mg Fertigpen und in der 150 mg Fertigspritze sowie in der 75 mg Fertigspritze kann wenige transluzente bis weisse produktbezogene Partikel enthalten. SKYRIZI darf nicht verwendet werden, wenn die Lösung trübe oder verfärbt ist oder grosse Partikel enthält.
Patienten können sich SKYRIZI selbst injizieren, nachdem sie in der Technik der subkutanen Injektion geschult wurden. Patienten sollten vor der Anwendung die Anwendungshinweise lesen.
Bei Verwendung der SKYRIZI 75 mg Fertigspritze sind Patienten anzuweisen, 2 Injektionen (2 Fertigspritzen) vorzunehmen, um sich die volle Dosis von 150 mg zu verabreichen. Jeder Fertigpen und jede Fertigspritze ist nur zum einmaligen Gebrauch bestimmt.
Patienten sollten nicht in Bereiche injizieren, in denen die Haut empfindlich, blutunterlaufen, gerötet, verhärtet oder von Psoriasis betroffen ist. Eine Injektion von SKYRIZI an der Aussenseite des Oberarms darf nur durch medizinisches Fachpersonal oder einen Betreuer erfolgen.
Bei Verwendung des Fertigpens sollte der Karton vor der Injektion aus dem Kühlschrank genommen werden und Raumtemperatur annehmen lassen (30 bis 90 Minuten, vor direktem Sonnenlicht geschützt), ohne dabei den Fertigpen aus dem Karton zu nehmen.
Für eine angenehmere Injektion, können Patienten bei Verwendung der 75 mg bzw. 150 mg Fertigspritze den Karton vor der Injektion aus dem Kühlschrank nehmen und Raumtemperatur annehmen lassen (15 bis 30 Minuten, vor direktem Sonnenlicht geschützt), ohne dabei die Fertigspritzen aus dem Karton zu nehmen.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den lokalen Vorschriften zu entsorgen.
Zulassungsnummer66944, 68118 (Swissmedic)
PackungenSKYRIZI 150 mg, Injektionslösung im Fertigpen
Jeder Karton enthält 1 Fertigpen. (B)
SKYRIZI 150 mg Injektionslösung in einer Fertigspritze
Jeder Karton enthält 1 Fertigspritze mit Nadelschutz. (B)
SKYRIZI 75 mg, Injektionslösung in einer Fertigspritze
Jeder Karton enthält 2 Fertigspritzen mit Nadelschutz und 2 Alkoholtupfer. (B)
ZulassungsinhaberinAbbVie AG, 6330 Cham, Schweiz.
Stand der InformationMai 2021
|