Eigenschaften/WirkungenATC-Code
L04AE01
Pharmakotherapeutische Gruppe: Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulatoren
Wirkungsmechanismus
Fingolimod ist ein Modulator des Sphingosin1-Phosphat-Rezeptors. Der Wirkstoff wird von der Sphingosinkinase zu seinem aktiven Metaboliten Fingolimodphosphat umgesetzt. Fingolimodphosphat bindet im unteren nanomolaren Konzentrationsbereich an die Sphingosin1-Phosphat (S1P)-Rezeptoren 1, 3 und 4 auf Lymphozyten und nach problemloser Überwindung der Blut-Hirn-Schranke an S1P-Rezeptoren 1, 3 und 5 auf Nervenzellen im zentralen Nervensystem (ZNS). Indem es als funktioneller Antagonist des S1PR auf Lymphozyten wirkt, blockiert Fingolimodphosphat die Fähigkeit der Lymphozyten, die Lymphknoten zu verlassen, sodass es zu einer Umverteilung und nicht zu einer Depletion von Lymphozyten kommt. Diese Umverteilung reduziert die Infiltration pathogener Lymphozyten, einschliesslich proinflammatorische Th17-Zellen, in das ZNS, wo sie an Nervenentzündungen und der Schädigung von Nervengewebe beteiligt sind. Tierexperimentelle Untersuchungen und in vitro-Experimente deuten darauf hin, dass die günstigen Auswirkungen von Fingolimod bei Multipler Sklerose auch auf eine Interaktion mit S1P-Rezeptoren auf Nervenzellen zurückzuführen sein könnten. Fingolimod dringt bei Menschen und Tieren in das ZNS ein und es wurde gezeigt, dass Astrogliose, Demyelinisierung und neuronaler Verlust reduziert werden. Ferner erhöht Fingolimod die Levels von BDNF (brain derived neurotrophic factor) im Cortex, Hippocampus und Striatum des Gehirns und unterstützt damit das neuronale Überleben und verbessert die motorischen Funktionen.
Pharmakodynamik
Immunsystem
Auswirkungen auf die Anzahl der Immunzellen im Blut. Innerhalb von 4-6 Stunden nach der ersten Fingolimod-Dosis von 0.5 mg verringert sich die Anzahl der Lymphozyten auf etwa 75% des Baseline-Werts. Wird die tägliche Dosisgabe fortgesetzt, fällt der Lymphozytenwert über einen Zeitraum von zwei Wochen weiter ab und erreicht schliesslich einen Tiefstwert von etwa 500 Zellen/µl bzw. etwa 30% des Baseline-Werts. Achtzehn Prozent der Patienten erreichten mindestens einmal einen Tiefstwert von unter 200 Zellen/µl. Bei anhaltender täglicher Dosisapplikation bleiben die niedrigen Lymphozytenwerte bestehen. Die meisten T- und B-Lymphozyten wandern regelmässig durch lymphoide Organe, sodass Fingolimod auf diese Zellen die stärksten Auswirkungen hat. Etwa 15-20% der T-Lymphozyten haben einen Effektor-Gedächtniszellen-Phänotyp, d.h. diese Zellen sind für die periphere Immunüberwachung wichtig. Da diese Lymphozyten-Untergruppe nicht durch lymphoide Organe wandert, wird sie von Fingolimod auch nicht beeinflusst. Nach dem Absetzen von Fingolimod kommt es innerhalb einiger Tage zu einem Anstieg der Anzahl der peripheren Lymphozyten, und üblicherweise ist innerhalb von ein bis zwei Monaten wieder der Normwert erreicht. Anhaltende Dosisapplikation von Fingolimod führt zu einer leichten Abnahme des Neutrophilenwerts auf etwa 80% des Baseline-Werts. Fingolimod hat keinen Einfluss auf Monozyten.
Herzfrequenz und Herzrhythmus
Fingolimod verursacht zu Beginn der Behandlung eine vorübergehende Verlangsamung der Herzfrequenz und der atrioventrikulären Überleitung (s. «Unerwünschte Wirkungen»). Die Verlangsamung der Herzfrequenz ist 4-5 Stunden nach Dosisgabe am stärksten ausgeprägt, wobei der negative chronotrope Effekt am ersten Tag zu 70% ausgeprägt ist. Die Herzfrequenz kehrt bei kontinuierlicher Behandlung innerhalb eines Monats meist wieder zum Baseline-Wert zurück.
Die Behandlung mit Fingolimod hat keinen Einfluss auf die autonomen Reaktionen des Herzmuskels, auch nicht auf die diurnale Schwankung der Herzfrequenz und die Reaktion auf körperliche Betätigung.
Bei Beginn der Behandlung mit Fingolimod kommt es zu einem Anstieg der Extrasystolen atrialen Ursprungs, aber nicht zu einer erhöhten Rate von Vorhofflimmern/-flattern oder ventrikulären Arrhythmien oder Ektopie. Die Behandlung mit Fingolimod bewirkt keine Verringerung des Herzausstosses.
Der von Fingolimod verursachten Verlangsamung der Herzfrequenz kann mit Atropin, Isoprenalin oder Salmeterol entgegengewirkt werden.
Potenzial zur Verlängerung des QT-Intervalls
In einer eingehenden Studie zum QT-Intervall mit Fingolimoddosen von 1.25 oder 2.5 mg im Steady-State, als der negative chronotrope Effekt von Fingolimod noch vorhanden war, führte die Behandlung mit Fingolimod zu einer Verlängerung des QTcI mit einer Obergrenze des 90% CI von ≤13.0 ms. Es gibt keine Dosis- oder Expositions-Wirkungsbeziehung zwischen Fingolimod und einer QTcI-Verlängerung, und kein einheitliches Signal für eine erhöhte Inzidenz von QTcI-Ausreissern, weder absolut noch als Veränderung gegenüber dem Baseline-Wert, in Zusammenhang mit der Fingolimod-Behandlung. Jedoch sind in Studie 1 im Rahmen der Erstabgabe von 0.5 mg Fingolimod bei 6.6% (Placebo 2.4%) und im weiteren Verlauf bei 13.9% (Placebo 6.7%) der Patienten QTcF-Verlängerungen zwischen 30-60 ms aufgetreten. Die klinische Relevanz ist nicht bekannt.
Lungensystem
Die Behandlung mit einer Einzeldosis oder mit Mehrfachdosen von Fingolimod 0.5 mg oder 1.25 mg für 2 Wochen ist nicht assoziiert mit einer nachweisbaren Erhöhung des Atemwiderstands, gemessen anhand des FEV1 oder FEF25-75. Einzeldosen von ≥5 mg (das 10-Fache der empfohlenen Dosierung) hingegen hatten einen dosisabhängigen Anstieg des Atemwiderstands zur Folge. Die Behandlung mit Mehrfachdosen von Fingolimod 0.5, 1.25 oder 5 mg hat keine beeinträchtigte Sauerstoffzufuhr, Sauerstoff-Entsättigung bei körperlicher Anstrengung oder einen Anstieg der Atemwegsempfindlichkeit auf Methacholin zur Folge. Patienten unter Behandlung mit Fingolimod reagieren mit normaler Empfindlichkeit auf inhalierbare Beta-Agonisten.
Klinische Wirksamkeit
Die Wirksamkeit von Fingolimod Sandoz wurde in zwei Studien mit einmal täglicher Gabe von 0.5 mg und 1.25 mg Fingolimod Sandoz an erwachsenen Patienten mit schubförmig-remittierender Multipler Sklerose aufgezeigt. Bei den Teilnehmern beider Studien handelte es sich um Patienten mit mindestens 2 klinischen Schüben in den 2 Jahren vor der Randomisierung oder mindestens 1 klinischen Schub in dem einen Jahr vor der Randomisierung und mit einem EDSS (Expanded Disability Status Scale)-Score zwischen 0 und 5.5. Eine dritte Studie mit der gleichen Art von Patienten, wurde nach der Registrierung von Fingolimod Sandoz abgeschlossen.
Die Wirksamkeit und Sicherheit der einmal täglichen Gabe von Fingolimod Sandoz in einer Dosis von 0.25 mg bzw. 0.5 mg (Auswahl der Dosis anhand des Körpergewichts und von Expositionsmessungen) wurde bei Kindern und Jugendlichen im Alter von 10 bis unter 18 Jahren mit schubförmig remittierender Multipler Sklerose untersucht.
Studie D2301 (FREEDOMS)
Studie D2301 (FREEDOMS) war eine 2-jährige, randomisierte, doppelblinde und placebokontrollierte Phase-III-Studie bei Patienten mit schubförmig verlaufender Multipler Sklerose, die während mindestens 3 Monaten vor Studienbeginn kein Interferon beta oder Glatirameracetat und während mindestens 6 Monaten vor Studienbeginn kein Natalizumab erhalten hatten.
Der Medianwert des Alters war 37 Jahre, der Medianwert der Krankheitsdauer war 6.7 Jahre und der EDSS-Medianscore zum Baselinezeitpunkt war 2.0. Die Patienten erhielten nach der Randomisierung bis zu 24 Monate lang 0.5 mg Fingolimod Sandoz (n=425) oder 1.25 mg Fingolimod Sandoz (n=429) oder ein Placebo (n=418). Die mittlere Dauer der Behandlung mit der 0.5-mg-Dosis betrug 717 Tage, mit der 1.25-mg-Dosis betrug sie 715 Tage und die mittlere Dauer der Placebogabe war 718.5 Tage.
Der primäre Endpunkt war die jährliche Schubrate.
Die auf das Jahr umgerechnete Schubrate (ARR=annualized relapse rate) war bei mit Fingolimod Sandoz behandelten Patienten statistisch signifikant niedriger als in der Placebogruppe. Der wichtigste sekundäre Endpunkt war der Zeitraum bis zu einer 3-monatigen bestätigten Progression der Behinderung, gemessen anhand einer Erhöhung des EDSS-Scores gegenüber dem Baseline-Wert um mindestens 1 Punkt (bei Patienten mit einem EDSS-Baseline-Wert von 5.5 anhand einer Erhöhung um mindestens 0.5 Punkte), die 3 Monate fortbestand. Der Zeitraum bis zum Einsetzen einer 3-monatigen bestätigten Progression der Behinderung war bei einer Behandlung mit Fingolimod Sandoz im Placebovergleich statistisch signifikant verzögert. Bei keinem Endpunkt ergaben sich statistisch signifikante Unterschiede zwischen der Dosis von 0.5 mg und der Dosis von 1.25 mg.
Die Ergebnisse aus dieser Studie sind in Tabelle 2 und Abbildung 1 gezeigt.
Tabelle 2 Klinische Ergebnisse und MRT-Ergebnis in FREEDOMS Studie
|
|
Fingolimod Sandoz 0.5 mg
|
Placebo
| |
Klinische Endpunkte
|
N=425
|
N=418
| |
Auf das Jahr umgerechnete Schubrate (primärer Endpunkt)
|
0.18
(p<0.001*)
|
0.40
| |
Relative Reduktion (%)
|
54
|
| |
Anteil der schubfrei gebliebenen Patienten zum 24-Monats-Zeitpunkt (%)
|
70.4
(p<0.001*)
|
45.6
| |
Risiko der Progression der Behinderung
| |
Hazard Ratio (95% CI)
(bestätigt, 3 Monate)
|
0.70 (0.52, 0.96)
(p=0.024*)
|
| |
Hazard Ratio (95% CI)
(bestätigt, 6 Monate)
|
0.63 (0.44, 0.90)
(p=0.012*)
|
| |
MRT-Endpunkte
| |
Anzahl der neuen oder neu vergrösserten T2-Läsionen
|
n=370
|
n=339
| |
Median-Wert (Durchschnitt) der Anzahl über 24 Monate
|
0.0 (2.5)
(p<0.001*)
|
5.0 (9.8)
| |
Anzahl der Gd-verstärkten Läsionen
|
n=369
(Monat 24)
|
n=332
(Monat 24)
| |
Median-Wert (Durchschnitt) der Anzahl in
| |
Monat 6
|
0.0 (0.2)
|
0.0 (1.3)
| |
Monat 12
|
0.0 (0.2)
|
0.0 (1.1)
| |
Monat 24
|
0.0 (0.2)
(p<0.001* an
jedem Zeitpunkt)
|
0.0 (1.1)
| |
Prozentuale Veränderung des Gesamtvolumens der T2-Läsionen
|
n=368
|
n=339
| |
Median-Wert (Durchschnitt) der Veränderung über 24 Monate in %
|
-1.7 (10.6)
(p<0.001*)
|
8.6 (33.8)
| |
Veränderung des Volumens der T1 hypointensen Läsionen
|
n=346
|
n=305
| |
Median-Wert (Durchschnitt) der Veränderung über 24 Monate in %
|
0.0 (8.8)
(p=0.012*)
|
1.6 (50.7)
| |
Prozentuale Veränderung des Gehirnvolumens
|
n=357
|
n=331
| |
Median-Wert (Durchschnitt) der Veränderung über 24 Monate in %
|
-0.7 (-0.8)
(p<0.001*)
|
-1.0 (-1.3)
|
Alle Analysen klinischer Endpunkte wurden in der Intent-to-Treat (ITT)-Population durchgeführt. Für die MRT-Analysen wurden auswertbare Datensätze herangezogen.
* Zeigt statistische Signifikanz im Placebovergleich mit zweiseitigem Signifikanzgrad von 0.05.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert
Abbildung 1 Kaplan-Meier-Grafik des Zeitraums bis zum ersten bestätigten Schub bis Monat 24 - FREEDOMS Studie (ITT-Population)
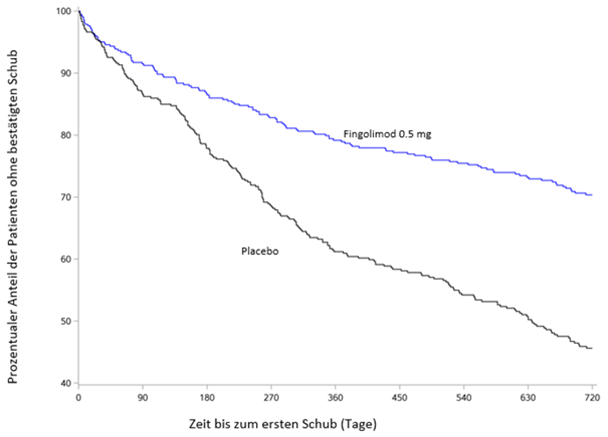
Patienten, welche die Studie FREEDOMS (D2301) abgeschlossen hatten, bot sich die Möglichkeit zur Teilnahme an der dosisverblindeten Verlängerungsstudie D2301E1. In diese Verlängerungsstudie wurden 920 Patienten aus der Hauptstudie aufgenommen, die alle mit Fingolimod behandelt wurden (n=331 setzten die Behandlung in der Dosis von 0.5 mg fort, 289 setzten die Behandlung in der Dosis von 1.25 mg fort, 155 wechselten von Placebo zur Dosis von 0.5 mg und 145 von Placebo zur Dosis von 1.25 mg). Nach 12 Monaten (Monat 36) waren noch 856 Patienten (93%) eingeschlossen. Von insgesamt 811 Patienten (88.2%) lagen Nachuntersuchungsdaten aus mindestens 18 Monaten in der Verlängerungsphase vor.
In Monat 24 der Verlängerungsstudie hatte sich bei den Patienten, die in der Hauptstudie der Placebo-Gruppe angehörten und dann auf 0.5 mg Fingolimod umgestellt wurden, die jährliche Schubrate (Annualized Relapse Rate, ARR) um 55% reduziert (ARR-Verhältnis 0.45, 95% KI 0.32 bis 0.62, p<0.001). Bei Patienten, die bereits in der Hauptstudie mit 0.5 mg Fingolimod behandelt worden waren, blieb die ARR während der Verlängerungsphase niedrig (ARR=0.10).
Zwischen den Monaten 24 und 36 betrug die jährliche Schubrate (ARR) 0.17 für Patienten unter 0.5 mg Fingolimod in der Hauptstudie, die weiterhin 0.5 mg einnahmen (0.21 in der Hauptstudie). Bei Patienten, die von Placebo auf 0.5 mg Fingolimod wechselten, betrug die ARR 0.22 (0.42 in der Hauptstudie).
Studie D2309 (FREEDOMS II)
In der replizierten zweijährigen, randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie mit 1083 Patienten mit schubförmig-remittierender Multipler Sklerose konnten vergleichbare Ergebnisse wie in Studie D2301 erzielt werden. Diese Studie wurde nach der Zulassung von Fingolimod fertiggestellt.
Das mediane Alter betrug 40.5 Jahre, die mediane Dauer der Erkrankung 8.9 Jahre und der mediane EDSS-Score zum Baseline-Zeitpunkt 2.5. Die Ergebnisse dieser Studie sind in Tabelle 3 und Abbildung 2 dargestellt.
Tabelle 3 Klinische Ergebnisse und MRT-Ergebnisse aus der Studie FREEDOMS II
|
|
0.5 mg Fingolimod Sandoz
|
Placebo
| |
Klinische Endpunkte
|
N=358
|
N=355
| |
Jährliche Schubrate (primärer Endpunkt)
|
0.21
(p<0.001*)
|
0.40
| |
Relative Verringerung (in %)
|
48
|
| |
Anteil der nach 24 Monaten schubfreien Patienten (in %)
|
71.5
(p<0.001*)
|
52.7
| |
Risiko des Fortschreitens der Behinderung†
| |
Hazard Ratio (95% KI)
(bestätigt über 3 Monate)
|
0.83 (0.61, 1.12)
(p=0.227)
|
| |
Hazard Ratio (95% KI)
(bestätigt über 6 Monate)
|
0.72 (0.48, 1.07)
(p=0.113)
|
| |
MRT-Endpunkte
| |
Veränderung des Hirnvolumens (in %)
|
n=266
|
n=249
| |
Mediane (durchschnittliche) % Veränderung über 24 Monate
|
-0.7 (-0.9)
(p<0.001*)
|
-1.0 (-1.3)
| |
Anzahl neuer oder sich neu vergrössernder T2-Läsionen
|
n=264
|
n=251
| |
Mediane (durchschnittliche) Anzahl über 24 Monate
|
0.0 (2.3)
(p<0.001*)
|
4.0 (8.9)
| |
Anzahl der Gd-anreichernden Läsionen
|
n=269
(Monat 24)
|
n=256
(Monat 24)
| |
Mediane (durchschnittliche) Anzahl in
| |
Monat 6
|
0.0 (0.2)
|
0.0 (1.1)
| |
Monat 12
|
0.0 (0.2)
|
0.0 (1.3)
| |
Monat 24
|
0.0 (0.4)
(p<0.001* zu
jedem Zeitpunkt)
|
0.0 (1.2)
| |
Veränderung des Gesamtvolumens an T2-Läsionen (in %)
|
n=262
|
n=247
| |
Mediane (durchschnittliche) % Veränderung über 24 Monate
|
-7.1 (13.7)
(p<0.001*)
|
0.8 (25.1)
| |
Veränderung des Volumens an T1-hypointensen Läsionen
|
n=225
|
n=209
| |
Mediane (durchschnittliche) % Veränderung über 24 Monate
|
-9.9 (12.6)
(p=0.372)
|
-8.5 (26.4)
|
Alle Analysen klinischer Endpunkte erfolgten im Intent-to-Treat-Kollektiv. Für die MRT-Analysen wurde der auswertbare Datensatz herangezogen.
* Statistische Signifikanz vs. Placebo auf dem zweiseitigen 0.05-Niveau.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert.
† Weitere Analysen ergaben, dass die Ergebnisse im Gesamtkollektiv aufgrund falschpositiver Progressionen in der Teilgruppe der Patienten mit einem Baseline-EDSS=0 (n=62, 8.7% des Studienkollektivs) nicht statistisch signifikant waren. Bei Patienten mit EDSS>0 (n=651; 91.3% des Studienkollektivs) ergab sich für 0.5 mg Fingolimod eine klinisch bedeutsame und statistisch signifikante Verringerung gegenüber dem Placebo (HR= 0.70; KI (0.50, 0.98); p=0.040), was mit der Studie FREEDOMS übereinstimmt.
Abbildung 2 Kaplan-Meier-Kurven der Zeit bis zum ersten bestätigten Schub bis Monat 24 – Studie FREEDOMS II (ITT-Kollektiv)
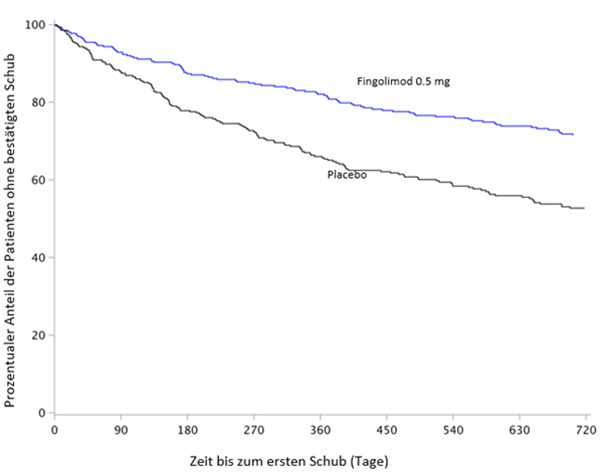
Bei keinem der Endpunkte wurden statistisch signifikante Unterschiede zwischen der 0.5 mg- und der 1.25 mg-Dosis festgestellt.
Studie D2302 (TRANSFORMS)
Bei Studie D2302 (TRANSFORMS) handelte es sich um eine einjährige, randomisierte, doppelblinde, aktiv (Interferon beta-1a, 30 Mikrogramm, intramuskulär, einmal wöchentlich) kontrollierte Phase-III-Studie im Double-Dummy-Design bei Patienten mit schubförmig-remittierend verlaufender MS, die während 6 Monaten vor Studienbeginn kein Natalizumab erhalten hatten. Eine vorhergehende Therapie mit Interferon beta oder Glatirameracetat bis zum Randomisierungszeitpunkt war erlaubt.
Der Medianwert des Alters betrug 36 Jahre, der Medianwert der Krankheitsdauer betrug 5.9 Jahre und der EDSS-Medianscore zum Baselinezeitpunkt war 2.0. Die Patienten erhielten nach der Randomisierung bis zu 12 Monate lang 0.5 mg Fingolimod Sandoz (n=431) oder 1.25 mg Fingolimod Sandoz (n=426) oder 30 Mikrogramm Interferon beta-1a intramuskulär einmal wöchentlich (n=435). Der Medianwert der Dauer der Behandlung mit dem Prüfmedikament war 365 Tage (0.5 mg Fingolimod Sandoz), 354 Tage (1.25 mg Fingolimod Sandoz) und 361 Tage (Interferon beta-1a).
Die Ergebnisse aus dieser Studie sind in Tabelle 4 und in Abbildung 3 gezeigt.
Tabelle 4 Klinische Ergebnisse und MRT-Ergebnisse aus TRANSFORMS Studie
|
|
Fingolimod Sandoz 0.5 mg
|
Interferon
beta-1a, 30 μg
| |
Klinische Endpunkte
|
N=429
|
N=431
| |
Auf das Jahr umgerechnete Schubrate (primärer Endpunkt)
|
0.16
(p<0.001*)
|
0.33
| |
Relative Reduktion (%)
|
52
|
| |
Anteil der schubfrei gebliebenen Patienten zum 12-Monats-Zeitpunkt (%)
|
82.5
(p<0.001*)
|
70.1
| |
Risiko der Progression der Behinderung
| |
Hazard Ratio (95% CI)
(bestätigt, 3 Monate)
|
0.71 (0.42, 1.21)
(p=0.209)
|
| |
MRT-Endpunkte
| |
Anzahl der neuen oder neu vergrösserten T2-Läsionen
|
n=380
|
n=365
| |
Median-Wert (Durchschnitt) der Anzahl über 12 Monate
|
0.0 (1.7)
(p=0.004*)
|
1.0 (2.6)
| |
Anzahl der Gd-verstärkten Läsionen
|
n=374
|
n=354
| |
Median-Wert (Durchschnitt) der Anzahl nach 12 Monaten
|
0.0 (0.2)
(p<0.001*)
|
0.0 (0.5)
| |
Prozentuale Veränderung des Gehirnvolumens
|
n=368
|
n=359
| |
Median-Wert (Durchschnitt) der Veränderung über 12 Monate in %
|
-0.2 (-0.3)
(p<0.001*)
|
-0.4 (-0.5)
|
Alle Analysen klinischer Endpunkte wurden in der Intent-to-Treat (ITT)-Population durchgeführt. Für die MRT-Analysen wurden auswertbare Datensätze herangezogen.
* Zeigt statistische Signifikanz im Vergleich zu Interferon beta-1a, im, mit zweiseitigem Signifikanzgrad von 0.05.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert.
Abbildung 3 Kaplan-Meier-Grafik des Zeitraums bis zum ersten bestätigten Schub bis Monat 12 - TRANSFORMS Studie (ITT-Population)
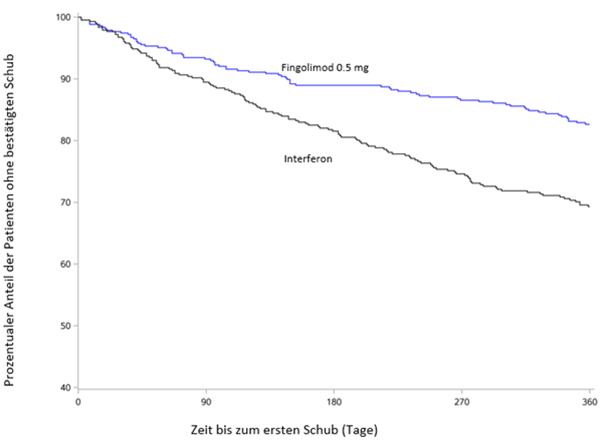
Bei keinem Endpunkt ergaben sich statistisch signifikante Unterschiede zwischen der Dosis von 0.5 mg und der Dosis 1.25 mg.
Patienten, die die Studie TRANSFORMS (D2302) abgeschlossen hatten, bot sich die Möglichkeit zur Teilnahme an der dosisverblindeten Verlängerungsstudie (D2302E1). Insgesamt wurden 1'030 Patienten aus der Hauptstudie aufgenommen und mit Fingolimod behandelt (n=357 setzten die Behandlung in der Dosis von 0.5 mg fort, 330 setzten die Behandlung in der Dosis von 1.25 mg fort, 167 wechselten von Interferon beta-1a zur Dosis von 0.5 mg und 176 von Interferon beta-1a zur Dosis von 1.25 mg). Von 882 dieser Patienten (85.9%) lagen Nachuntersuchungsdaten aus mindestens 12 Monaten in der Verlängerungsphase vor.
In Monat 12 der Verlängerungsstudie hatte sich bei den Patienten, die in der Hauptstudie Interferon beta-1a i.m. erhalten hatten und dann auf 0.5 mg Fingolimod umgestellt wurden, die ARR relativ um 30% reduziert (ARR-Verhältnis=0.70, p=0.06). Bei Patienten, die bereits in der Hauptstudie mit 0.5 mg Fingolimod behandelt worden waren, blieb die ARR über die Haupt- und Verlängerungsphase hinweg niedrig (ARR=0.18 bis Monat 24).
Zwischen den Monaten 12 und 24 betrug die ARR für Patienten unter 0.5 mg Fingolimod in der Hauptstudie, die weiterhin 0.5 mg einnahmen 0.20 (0.19 in der Hauptstudie). Bei Patienten, die von Interferon beta-1a auf 0.5 mg Fingolimod wechselten, betrug die ARR 0.33 (0.48 in der Hauptstudie).
Insgesamt zeigten die Ergebnisse der Studien D2301 (FREEDOMS) und D2302 (TRANSFORMS) in den nach Geschlecht, Alter, vorheriger MS-Therapie, Krankheitsaktivität oder Behinderungsgrad zum Baselinezeitpunkt definierten Untergruppen eine einheitliche Reduktion der auf das Jahr umgerechneten Schubrate im Vergleich zum Vergleichspräparat.
Studie D2311 (PARADIGMS) an Kindern und Jugendlichen ab 10 Jahren
Bei der Studie D2311 (PARADIGMS) handelte es sich um eine doppelblinde, randomisierte, aktiv-kontrollierte, multizentrische Parallelgruppenstudie mit einer flexiblen Dauer von bis zu 24 Monaten zur Untersuchung der Wirksamkeit und Sicherheit von Fingolimod (n=107) im Vergleich mit Interferon beta-1a (n=107) bei Kindern und Jugendlichen mit schubförmig remittierender MS im Alter von 10 bis unter 18 Jahren. Eine Vorbehandlung mit Interferon beta, Dimethylfumarat oder Glatirameracetat bis zum Zeitpunkt der Randomisierung war erlaubt. Eingeschlossen wurden Patienten mit mindestens einem klinischen Schub im Jahr zuvor oder mindestens 2 klinischen Schüben in den 2 Jahren vor der Randomisierung oder Nachweis ≥1 Gd-verstärkenden Läsion im MRT innerhalb 6 Monate vor Randomisierung und mit einem EDSS zwischen 0 bis 5.5. Neurologische Untersuchungen wurden beim Screening sowie danach alle 3 Monate und bei einem vermuteten Schub durchgeführt. Die MRT-Beurteilungen erfolgten beim Screening und dann alle 6 Monate während der gesamten Studie. Der primäre Endpunkt war die auf das Jahr umgerechnete Schubrate.
Das mediane Alter betrug 16 Jahre, die mediane Krankheitsdauer seit dem Auftreten des ersten Symptoms lag bei 1.5 Jahren und der mediane EDSS-Score zur Baseline betrug 1.5. Die Patienten wurden randomisiert und erhielten entweder Fingolimod oder Interferon beta-1a, das einmal wöchentlich intramuskulär verabreicht wurde, für bis zu 24 Monate. In der Altersgruppe ≥10 und ≤12 Jahre (n=13 unter Fingolimod Sandoz) sowie in der Gewichtskategorie ≤40kg (n=9 unter Fingolimod Sandoz) wurden wenige Patienten eingeschlossen, so dass in diesen Patientengruppen nur limitierte Daten zur Beurteilung von Wirksamkeit und Sicherheit vorliegen. Die mediane Behandlungsdauer mit dem Studienmedikament betrug 634 Tage unter Fingolimod und 547 Tage unter Interferon beta-1a.
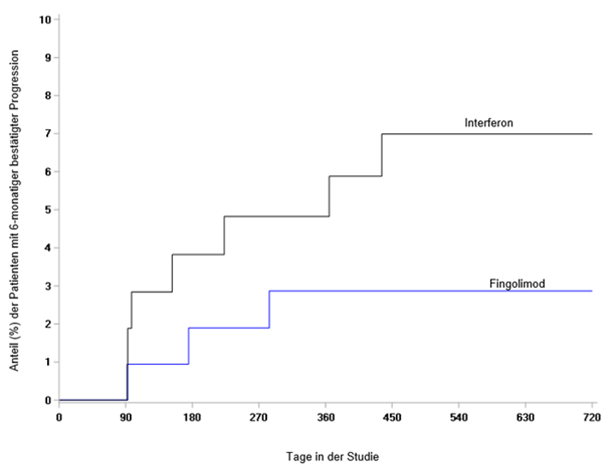
Der primäre Endpunkt, die auf das Jahr umgerechnete Schubrate (Annualized Relapse Rate, ARR) war bei den Patienten, die mit Fingolimod behandelt wurden, statistisch signifikant niedriger als bei denen, die Interferon beta-1a erhielten (relative Reduktion der ARR um 81.9%). Der wichtigste sekundäre Endpunkt, die auf das Jahr umgerechnete Anzahl der neu aufgetretenen bzw. neu vergrösserten T2-Läsionen bis Monat 24, war ausserdem bei den Patienten, die mit Fingolimod behandelt wurden, statistisch signifikant niedriger als bei denen, die Interferon beta-1a erhielten, ebenso wie die Anzahl der vorhandenen Gd-anreichernden T1-Läsionen pro Scan bis Monat 24. Durch Fingolimod wurde zudem die auf das Jahr umgerechnete Hirnatrophie- Rate von der Baseline bis Monat 24 statistisch signifikant reduziert. Eine zusätzliche Post-hoc-Analyse zeigte, dass durch Fingolimod im Vergleich mit Interferon beta-1a der Zeitraum bis zum Einsetzen einer 3-monatigen bestätigten Progression der Behinderung statistisch signifikant verlängert wurde. Die Ergebnisse dieser Studie sind in Tabelle 5, Abbildung 4 und Abbildung 5 dargestellt.
Tabelle 5 Klinische und MRT-Ergebnisse der PARADIGMS-Studie
|
|
Fingolimod
0.25 mg oder 0.5 mg
|
Interferon
beta-1a 30 µg
| |
Klinische Endpunkte
|
N=107
|
N=107#
| |
Auf das Jahr umgerechnete Schubrate (primärer Endpunkt)
|
0.122
(p<0.001*)
|
0.675
| |
Relative Reduzierung (in Prozent)
|
81.9
|
| |
Anteil der bis Monat 24 schubfrei gebliebenen Patienten (in %)
|
85.7
(p<0.001*)
|
38.8
| |
Risiko des Fortschreitens der Behinderung
| |
Hazard-Ratio (95% KI)
(bestätigt über 3 Monate)
|
0.23 (0.08,0.66)
(p=0.007*)
|
| |
Hazard Ratio (95%KI)
(bestätigt über 6 Monate)
|
0.20 (0.04; 0.93)
(p=0.040**)
|
| |
MRT-Endpunkte
| |
Auf das Jahr umgerechnete Anzahl der neu aufgetretenen bzw. neu vergrösserten T2-Läsionen
|
n=106
|
n=102
| |
Bereinigter Mittelwert
|
4.393
(p<0.001*)
|
9.269
| |
Relative Reduzierung (Prozent)
|
52.6
|
| |
Anzahl der Gd-anreichernden T1-Läsionen pro Scan bis Monat 24
|
n=106
|
n=101
| |
Bereinigter Mittelwert
|
0.436
(p<0.001*)
|
1.282
| |
Relative Reduzierung (Prozent)
|
66.0
|
| |
Auf das Jahr umgerechnete Hirnatrophierate von der Baseline bis Monat 24
|
n=96
|
n=89
| |
Kleinste-Quadrate-Mittelwert
|
-0.48
(p=0.014*)
|
-0.80
|
Alle Analysen der klinischen Endpunkte erfolgten am vollständigen Analyseset. Für die MRT-Analysen wurde der auswertbare Datensatz verwendet.# Ein Patient wurde für die Behandlung mit Interferon beta-1a IM, 30 µg wöchentlich randomisiert, war aber nicht in der Lage, die im Rahmen des Double-Dummy-Verfahrens erforderliche zusätzliche Dosis Placebo zu schlucken und wurde aus der Studie herausgenommen. Dieser Patient wurde aus dem vollständigen Analyse- und Sicherheitsset ausgeschlossen.
* Zeigt die statistische Signifikanz im Vergleich mit Interferon beta-1a IM mit einem zweiseitigen Signifikanzniveau von 0.05.
** Post-hoc Analyse, Cox Proportional Hazard Model. p=0.180 im Log-rank Test.
Bestimmung der p-Werte: Aggregierte ARR: durch negative binomiale Regression, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifizierungsfaktor im interaktiven Sprachdialogsystem, IVRS) und Anzahl der Schübe in den vorangegangenen 2 Jahren (Offset: Zeit in der Studie); Prozentsatz der schubfrei gebliebenen Patienten: anhand einer Kaplan-Meier-Schätzung; Risiko der Progression der Behinderung: anhand des proportionalen Hazardmodells nach Cox, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und Anzahl der Schübe in den vorangegangenen 2 Jahren; auf das Jahr umgerechnete Anzahl der neu aufgetretenen/neu vergrösserten T2-Läsionen: durch negative binomiale Regression, angepasst nach Behandlung, Region, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und Anzahl der T2-Läsionen zur Baseline (Offset: Zeit in der Studie); Anzahl der Gd-anreichernden Läsionen pro Scan: durch negative binomiale Regression mit der kumulativen Anzahl der Gd-anreichernden T1-Läsionen auf allen während der Studie planmässig nach der Baseline durchgeführten MRT-Scans als Reaktionsvariable, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und der Anzahl der Gd-anreichernden T1-Läsionen zur Baseline (Offset: Anzahl der MRT-Scans); auf das Jahr umgerechnete Hirnatrophierate: anhand eines ANCOVA-Modells, angepasst nach Behandlung, Region, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und des Gesamt-Hirnvolumens zur Baseline
Abb. 4 Kaplan-Meier-Grafik für den Zeitraum bis zum ersten bestätigten Schub bis Monat 24 - PARADIGMS-Studie (vollständiges Analyseset)
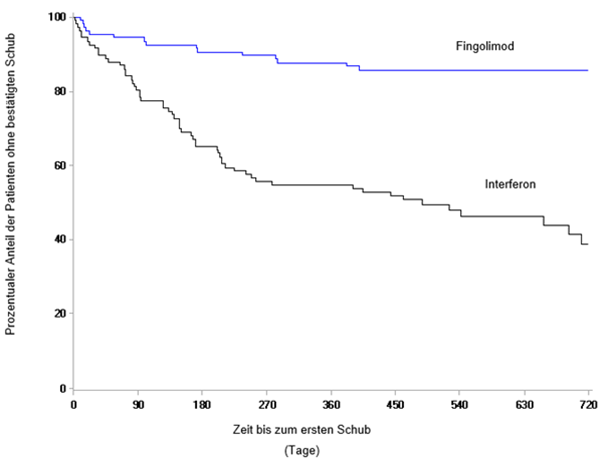
Abb. 5 Kaplan-Meier-Grafik des Zeitraums bis zur 6-monatigen bestätigten Progression der Behinderung - PARADIGMS-Studie (vollständiges Analyseset
|