ZusammensetzungWirkstoffe
Casirivimabum, Imdevimabum (genetisch hergestellt in CHO-Zellen [aus dem Ovar des Chinesischen Hamsters]).
Hilfsstoffe
Durchstechflasche mit Casirivimab: L-histidinum, L-histidini hydrochloridum monohydricum, polysorbatum 80*, saccharum**, aqua ad iniectabile q.s. ad solutionem pro 1 ml.
Durchstechflasche mit Imdevimab: L-histidinum, L-histidini hydrochloridum monohydricum, polysorbatum 80*, saccharum**, aqua ad iniectabile q.s. ad solutionem pro 1 ml.
(*hergestellt aus gentechnisch verändertem Mais, **hergestellt aus gentechnisch veränderter Zuckerrübe).
Indikationen/AnwendungsmöglichkeitenRonapreve wird angewendet zur:
·Behandlung einer bestätigten Coronavirus-Krankheit 2019 (COVID-19) bei Erwachsenen und Jugendlichen (ab dem Alter von 12 Jahren und mit einem Körpergewicht von mindestens 40 kg), die keine Sauerstofftherapie oder Hospitalisierung aufgrund von COVID-19 benötigen und bei denen ein hohes Risiko besteht, einen schweren COVID-19 Verlauf zu entwickeln (siehe «Eigenschaften/Wirkungen»).
·Prävention von COVID-19 bei Erwachsenen und Jugendlichen (ab dem Alter von 12 Jahren und mit einem Körpergewicht von mindestens 40 kg), die nicht fähig sind eine angemessene Immunantwort auf die SARS-CoV-2-Impfung zu erzeugen.
Ronapreve ist nicht als Ersatz für eine Impfung gegen COVID-19 vorgesehen.
Ronapreve sollte gemäss den offiziellen Empfehlungen angewendet werden.
Bei der Entscheidung über die Anwendung von Ronapreve zur Behandlung oder Prophylaxe sollte berücksichtigt werden, was über die Eigenschaften der zirkulierenden SARS-CoV-2-Varianten bekannt ist, einschliesslich regionaler oder geografischer Unterschiede sowie die verfügbaren Informationen über die Suszeptibilität gegenüber Ronapreve (siehe «Eigenschaften/Wirkungen»).
Zum Ausschluss von SARS-CoV-2-Varianten, bei denen eine verringerte Suszeptibilität gegenüber Ronapreve aufgezeigt worden ist, sind bei der Wahl der antiviralen Therapie die Daten aus molekularen Tests oder Sequenzierungsdaten heranzuziehen, sofern diese verfügbar sind.
Dosierung/AnwendungDie Zubereitung und Verabreichung von Ronapreve sollten von einer qualifizierten medizinischen Fachperson eingeleitet und überwacht werden. Die Anwendung sollte in einem Umfeld erfolgen, in dem eine Behandlung schwerer Überempfindlichkeitsreaktionen wie eine Anaphylaxie möglich ist.
Zur Vermeidung von Medikationsfehlern ist es wichtig, die Etiketten auf der Durchstechflasche zu prüfen, um sicherzustellen, dass es sich beim zubereiteten und/oder angewendeten Arzneimittel um Ronapreve handelt.
Um die Rückverfolgbarkeit von biotechnologischen Arzneimitteln zu gewährleisten, wird empfohlen, den Handelsnamen und die Chargennummer bei jeder Behandlung zu dokumentieren.
Übliche Dosierung
Behandlung
Die Dosierung bei Erwachsenen und Jugendlichen ab 12 Jahren und mit einem Körpergewicht von mindestens 40 kg beträgt 600 mg Casirivimab und 600 mg Imdevimab. Die Gabe erfolgt zusammen als einzelne intravenöse Infusion.
Die subkutane Injektion ist eine alternative Art der Verabreichung, falls die intravenöse Infusion nicht durchführbar ist oder zur Verzögerung der Behandlung führen würde.
Die Gabe von Ronapreve sollte so bald wie möglich nach einem positiven Virustest auf SARS-CoV-2 erfolgen (siehe «Eigenschaften/Wirkungen»).
Zur Bestätigung von COVID-19 wird ein Nukleinsäure-Amplifikationstechnik (NAT) Test bevorzugt.
Prävention
Postexpositionelle Prophylaxe
Die Dosierung bei Erwachsenen und Jugendlichen ab 12 Jahren und mit einem Körpergewicht von mindestens 40 kg beträgt 600 mg Casirivimab und 600 mg Imdevimab. Die Gabe erfolgt entweder zusammen als einzelne intravenöse Infusion oder mittels subkutaner Injektion.
Die Gabe von Ronapreve sollte so bald wie möglich nach Exposition gegenüber SARS-CoV-2 erfolgen (siehe «Eigenschaften/Wirkungen»).
Präexpositionelle Prophylaxe
Die Anfangsdosis beträgt 600 mg Casirivimab und 600 mg Imdevimab als intravenöse Infusion oder subkutane Injektion.
Nachfolgende Dosen von 300 mg Casirivimab und 300 mg Imdevimab können als einmalige intravenöse Infusion oder subkutane Injektion alle 4 Wochen verabreicht werden (siehe «Eigenschaften/Wirkungen», «Pharmakokinetik»).
Es gibt keine Daten zu wiederholten Verabreichungen über 24 Wochen hinaus.
Verspätete Dosisgabe
Wenn eine wiederholte Dosis von Ronapreve ausgelassen wurde, ist diese so schnell wie möglich nachzuholen. Um das geeignete Intervall zwischen den Dosen einzuhalten, ist der Dosierungsplan anzupassen.
Dosisanpassung/Titration
Patienten mit Leberfunktionsstörungen
Bei Personen mit leichter Leberfunktionsstörung ist keine Dosisanpassung erforderlich. Für Personen mit mittelschwerer Leberfunktionsstörung sind nur begrenzte Daten verfügbar. Casirivimab und Imdevimab wurden bei Personen mit schwerer Leberfunktionsstörung nicht untersucht (siehe Abschnitt «Kinetik spezieller Patientengruppen»).
Patienten mit Nierenfunktionsstörungen
Bei Personen mit leichter oder mittelschwerer Nierenfunktionsstörung oder mit einer Kreatinin-Clearance (CrCl) < 15 ml/min, einschliesslich Dialysepatienten, ist keine Anpassung der Dosierung erforderlich. Für Personen mit hochgradiger Niereninsuffizienz liegen nur begrenzte Daten vor (siehe Abschnitt «Kinetik spezieller Patientengruppen»).
Ältere Patienten
Bei älteren Personen ist keine Anpassung der Dosis von Casirivimab und Imdevimab erforderlich (siehe Abschnitt «Kinetik spezieller Patientengruppen»).
Kinder und Jugendliche
Bei Kindern und Jugendlichen im Alter von ≥12 Jahren und einem Gewicht ≥40 kg wird keine Anpassung der Dosierung empfohlen (siehe Abschnitt «Unerwünschte Wirkungen», «Eigenschaften/Wirkungen» und «Kinetik spezieller Patientengruppen»).
Die Sicherheit und Wirksamkeit von Casirivimab und Imdevimab bei Kindern < 12 Jahren wurden noch nicht ermittelt. Es liegen keine Daten vor.
Art der Anwendung
Ronapreve ist für die intravenöse Infusion oder die subkutane Injektion bestimmt.
Intravenöse Infusion
Für ausführliche Anweisungen bezüglich der Vorbereitung und Verabreichung von Ronapreve siehe unter «Sonstige Hinweise/Hinweise für die Handhabung» den Abschnitt «Dosisvorbereitung und Dosisgabe».
Tabelle 1: Empfehlungen zur Verdünnung und intravenösen Infusion von 600 mg Casirivimab und 600 mg Imdevimab oder von 300 mg Casirivimab und 300 mg Imdevimab
|
Indikation
|
Grösse des Fertiginfusionsbeutels mit 0,9 % Natriumchlorid oder 5 % Dextrose
|
Ronapreve-Dosis (gesamt)
|
Gesamtvolumen für 1 Dosis
|
Entnahmevolumen aus jeder Durchstechflasche zur Injektion in einen Fertiginfusionsbeutel mit 0,9 % Natriumchlorid oder 5 % Dextrose
|
Mindestdauer der Infusion
| |
·Behandlung
·Postexpositionelle Prophylaxe – Einzeldosis und
·Präexpositionelle Prophylaxe – Anfangsdosis
|
50 ml,
100 ml,
150 ml
|
600 mg Casirivimab und 600 mg Imdevimab (1'200 mg Dosis)
|
10 ml
|
2,5 ml aus zwei 6 ml-Durchstechflaschen mit Casirivimab
2,5 ml aus zwei 6 ml-Durchstechflaschen mit Imdevimab
|
20 Minuten
| |
250 ml
|
5,0 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Casirivimab
5,0 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Imdevimab
|
30 Minuten
| |
·Präexpositionelle Prophylaxe – nachfolgende Dosis
|
50 ml,
100 ml,
150 ml
|
300 mg Casirivimab und 300 mg Imdevimab (600 mg Dosis)
|
5 ml
|
2,5 ml aus einer 6 ml-Durchstechflasche mit Casirivimab
2,5 ml aus einer 6 ml-Durchstechflasche mit Imdevimab
|
20 Minuten
| |
250 ml
|
2,5 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Casirivimab
2,5 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Imdevimab
|
30 Minuten
|
Wenn der Patient/die Patientin Anzeichen infusionsbedingter Ereignisse oder anderer unerwünschter Ereignisse entwickelt, kann die Infusion verlangsamt, unterbrochen oder abgebrochen werden.
Subkutane Injektion
Ausführliche Angaben zum Vorbereiten und Injizieren von Ronapreve siehe unter «Sonstige Hinweise/Hinweise für die Handhabung» im Abschnitt «Dosisvorbereitung und Dosisgabe».
Die subkutanen Injektionen sind jeweils an einer anderen Injektionsstelle zu verabreichen: an den Oberschenkeln, an der Aussenseite der Oberarme oder am Bauch, mit Ausnahme der Fläche von 5 cm um den Nabel (periumbilikal). Die Gürtellinie ist zu vermeiden.
Zur Verminderung von Reaktionen an der Injektionsstelle wird empfohlen, dass die Verabreichung der subkutanen Injektionen durch das medizinische Fachpersonal in verschiedenen Quadranten des Abdomens, der Oberschenkel oder der Aussenseite der Oberarme erfolgen, um die 2,5 ml subkutanen Injektionen von Casirivimab und Imdevimab räumlich voneinander zu trennen (siehe Rubrik «Unerwünschte Wirkungen»).
Tabelle 2: Vorbereitung von Casirivimab und Imdevimab für die subkutane Injektion
|
Indikation
|
Ronapreve-Dosis (gesamt)
|
Gesamtvolumen für 1 Dosis
|
Entnahmevolumen zur Vorbereitung von 4 Spritzen
| |
·Behandlung
·Postexpositionelle Prophylaxe – Einzeldosis und
·Präexpositionelle Prophylaxe – Anfangsdosis
|
600 mg Casirivimab und 600 mg Imdevimab (1'200 mg-Dosis)
|
10 ml
|
2,5 ml aus zwei 6 ml-Durchstechflaschen mit Casirivimab
2,5 ml aus zwei 6 ml-Durchstechflaschen mit Imdevimab
| |
2,5 ml (2x) aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Casirivimab
2,5 ml (2x) aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Imdevimab
| |
·Präexpositionelle Prophylaxe – nachfolgende Dosis
|
300 mg Casirivimab und 300 mg Imdevimab (600 mg Dosis)
|
5 ml
|
2,5 ml aus einer 6 ml-Durchstechflasche mit Casirivimab
2,5 ml aus einer 6 ml-Durchstechflasche mit Imdevimab
| |
2,5 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Casirivimab
2,5 ml aus einer 20 ml-Mehrfachdosen-Durchstechflasche mit Imdevimab
|
KontraindikationenRonapreve ist bei Personen mit Überempfindlichkeit auf die Wirkstoffe oder einen ihrer Hilfsstoffe kontraindiziert.
Warnhinweise und VorsichtsmassnahmenDie Personen sollten nach der Verabreichung gemäss der lokal üblichen medizinischen Praxis überwacht werden. Die Patienten sollten während der intravenösen Verabreichung des Arzneimittels klinisch überwacht und nach Abschluss der Verabreichung mindestens 1 Stunde beobachtet werden.
Schwerwiegende Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie
Es wurde über schwerwiegende Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie bei Anwendung von Ronapreve berichtet. Beim Auftreten von Anzeichen oder Symptomen einer klinisch signifikanten Überempfindlichkeitsreaktion oder Anaphylaxie ist die Verabreichung sofort abzubrechen und es sind adäquate medikamentöse und/oder supportive Massnahmen einzuleiten.
Infusionsbedingte Reaktionen
Bei der intravenösen Gabe von Ronapreve traten infusionsbedingte Reaktionen (IRRs, infusion related reactions) auf. Die in klinischen Studien beobachteten IRRs waren zumeist leicht bis mittelschwer und traten in der Regel während der Infusion oder innerhalb von 24 Stunden danach auf. Die am häufigsten berichteten Anzeichen und Symptome derartiger Reaktionen waren Übelkeit, Schüttelfrost, Schwindel (oder Synkope), Hautausschlag, Urtikaria, Pruritus, Tachypnoe und Hitzewallung (Flushing). Infusionsbedingte Reaktionen können sich aber auch als schwere oder lebensbedrohliche Ereignisse manifestieren und andere Anzeichen und Symptome beinhalten. Bei Auftreten einer IRR ist zu erwägen, die Infusion zu unterbrechen, zu verlangsamen oder abzubrechen, und es sind adäquate medikamentöse und/oder supportive Massnahmen einzuleiten.
Im Rahmen der Zulassung für die Verwendung in Notfallsituationen (Emergency Use Authorization) in den USA wurden Fälle von konvulsiver Synkope beobachtet (siehe «Unerwünschte Wirkungen, Erfahrungen aus der Anwendung in Notfallsituationen»). Konvulsive Synkopen sollten von Krampfanfällen abgegrenzt und der klinischen Indikation entsprechend behandelt werden.
InteraktionenEs wurden keine Studien zu Wechselwirkungen durchgeführt. Casirivimab und Imdevimab sind monoklonale Antikörper, die nicht renal ausgeschieden oder durch Cytochrom P450-Enzyme metabolisiert werden. Daher sind Interaktionen mit Begleitmedikationen, die renal ausgeschieden werden oder Substrate, Induktoren oder Inhibitoren von Cytochrom P450-Enzymen sind, unwahrscheinlich.
COVID-19-Impfstoffe
Casirivimab und Imdevimab binden an Epitope auf dem Spike-Protein, das in allen COVID-19-Impfstoffen als Immunogen verwendet wird. Daher besteht die Möglichkeit, dass Casirivimab und Imdevimab das Ansprechen auf COVID-19-Impfstoffe beeinflussen. Bezüglich des Zeitpunkts der Impfung nach einer Behandlung mit monoklonalen Antikörpern gegen SARS-CoV-2 gelten die aktuellen Impfrichtlinien.
Schwangerschaft, StillzeitSchwangerschaft
Zur Anwendung von Casirivimab und Imdevimab bei Schwangeren liegen keine bzw. Daten in begrenztem Umfang vor. Tierexperimentelle Studien in Bezug auf die Reproduktionstoxizität wurden nicht durchgeführt (siehe «präklinische Daten»). Humane Immunglobulin G1(IgG1)-Antikörper passieren bekanntlich die Plazenta. Es ist nicht bekannt, ob die potenzielle Übertragung von Casirivimab und Imdevimab für den sich entwickelnden Fötus einen Behandlungsnutzen oder ein Risiko bedeutet. Ronapreve sollte während einer Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen das potenzielle Risiko für die Mutter und den Fötus unter Berücksichtigung aller damit einhergehenden Gesundheitsfaktoren rechtfertigt. Falls eine Frau während der Anwendung dieses Arzneimittels schwanger wird, muss sie darüber informiert werden, dass nicht bekannt ist, ob ein potenzielles Risiko für den Fötus besteht.
Stillzeit
Es ist nicht bekannt, ob Casirivimab und Imdevimab in der menschlichen Muttermilch ausgeschieden werden.
Ein Risiko für das Neugeborene/den Säugling kann nicht ausgeschlossen werden.
Da menschliche Muttermilch bekanntlich maternales IgG enthält und das potenzielle Risiko unerwünschter Wirkungen des Arzneimittels beim gestillten Säugling nicht bekannt ist, muss unter Berücksichtigung der positiven Wirkungen des Stillens auf das Kind und des Nutzens der Behandlung für die Frau entschieden werden, ob abgestillt oder die Behandlung mit Ronapreve abgebrochen oder darauf verzichtet wird.
Fertilität
Es wurden keine Fertilitätsstudien durchgeführt.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenRonapreve hat keinen oder einen vernachlässigbaren Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Unerwünschte WirkungenInsgesamt erhielten in klinischen Studien, 8'596 Probanden Ronapreve (6'173 intravenös und 2'423 subkutan). Die am häufigsten berichteten unerwünschte Wirkungen sind Überempfindlichkeitsreaktionen, darunter infusionsbedingte Reaktionen und Reaktionen an der Injektionsstelle.
Zusammenfassung des Sicherheitsprofils
Liste der unerwünschten Wirkungen
Unerwünschte Wirkungen (Tabelle 3) sind nach MedDRA-Systemorganklasse gelistet. Die jeweilige Häufigkeitskategorie für jede unerwünschte Wirkung basiert auf der folgenden Konvention: sehr häufig (≥1/10), häufig (≥1/100 bis < 1/10), gelegentlich (≥1/1000 bis < 1/100), selten (≥1/10'000 bis < 1/1000), sehr selten (<1/10'000).
Tabelle 3: Auflistung unerwünschter Wirkungen, die in klinischen Studien festgestellt wurden
|
Systemorganklasse
|
Unerwünschte Wirkung
|
Häufigkeitskategorie
| |
Intravenöse Gabe1
| |
Erkrankungen der Atemwege, des Brustraums und Mediastinums
|
Tachypnoe*
|
Gelegentlich
| |
Erkrankungen des Immunsystems
|
Anaphylaxie
|
Selten
| |
Überempfindlichkeit
|
Selten
| |
Erkrankungen des Nervensystems
|
Schwindel*
|
Gelegentlich
| |
Gefässerkrankungen
|
Hitzewallung (Flushing)*
|
Gelegentlich
| |
Erkrankungen des Gastrointestinaltrakts
|
Übelkeit2*
|
Gelegentlich
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Pruritus*
|
Gelegentlich
| |
Hautausschlag*
|
Gelegentlich
| |
Urtikaria*
|
Selten
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Schüttelfrost*
|
Gelegentlich
| |
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
|
Infusionsbedingte Reaktionen
|
Gelegentlich
| |
Subkutane Gabe
| |
Erkrankungen des Blutes und des Lymphsystems
|
Lymphadenopathie2 *
|
Gelegentlich
| |
Erkrankungen des Nervensystems
|
Schwindel3
|
Gelegentlich
| |
Erkrankungen der Haut und des Unterhautgewebes
|
Pruritus3*
|
Selten
| |
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
|
Reaktionen an der Injektionsstelle3
|
Häufig
|
1 Bestimmung der Häufigkeit aus den beiden Studien mit i. v.-Gabe (COV-2067 und COV-2066).
2 Bestimmung der Häufigkeit aus der Studie HV-2093 (Studie mit wiederholter subkutaner Gabe).
3 Bestimmung der Häufigkeit aus der Studie COV-2069.
* In manchen Fällen wurden die Symptome von IRRs und ISRs als individuelle UAW gemeldet.
Beschreibung spezifischer unerwünschter Wirkungen und Zusatzinformationen
Überempfindlichkeitsreaktion einschliesslich Anaphylaxie
Im gesamten klinischen Entwicklungsprogramm traten die folgenden Überempfindlichkeitsreaktionen unterschiedlichen Schweregrades auf.
Anaphylaxie/anaphylaktische Reaktion wurde im klinischen Entwicklungsprogramm beobachtet, war aber selten, trat innerhalb von 1 Stunde nach dem Ende der Infusion auf und klang nach einer unterstützenden Behandlung, die Adrenalin einschloss, wieder ab (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Infusionsbedingte Reaktionen (IRRs)
Bei der intravenösen Gabe von Casirivimab und Imdevimab traten in allen Dosisgruppen der klinischen Studien infusionsbedingte Reaktionen auf. Diese Reaktionen waren meist leicht bis mittelschwer, traten in der Regel während der Infusion oder innerhalb von 24 Stunden danach auf und waren entweder ohne Massnahmen oder mit der üblichen Standardbehandlung reversibel. Die am häufigsten berichteten Anzeichen und Symptome von IRRs waren Übelkeit, Schüttelfrost, Schwindel (oder Synkope), Hautausschlag, Urtikaria, Pruritus, Tachypnoe und Hitzewallung (Flushing). Es sind gegebenenfalls auch andere klinische Erscheinungen von IRRs zu erwarten (siehe Rubrik «Warnhinweise und Vorsichtsmassnahmen»).
Reaktionen an der Injektionsstelle (ISRs)
Reaktionen an der Injektionsstelle wurden in allen Studien mit subkutaner Gabe berichtet, d.h. sowohl in Studien mit einmaliger als auch in solchen mit wiederholter Gabe. Alle ISRs waren lokal, leicht bis mittelschwer und entweder ohne Massnahmen oder mit der üblichen Standardbehandlung reversibel. Die am häufigsten berichteten Anzeichen und Symptome derartiger Reaktionen waren Erythem, Pruritus, Ekchymose, Ödem, Schmerzen/Druckempfindlichkeit und Urtikaria. In der Studie mit wiederholter Gabe (HV-2093) zeigte sich auch eine lokalisierte Lymphadenopathie.
Kinder und Jugendliche
Intravenöse Gabe: Zu pädiatrischen Patienten < 18 Jahren liegen keine Daten vor.
Subkutane Gabe: In der Studie COV-2069 erhielten Jugendliche ≥12 und < 18 Jahre eine Behandlung mit Ronapreve, und das beobachtete Sicherheitsprofil war ähnlich wie bei erwachsenen Patienten.
Ältere Patienten
Intravenöse Gabe: In der Studie COV-2067 erhielten Patienten im Alter von ≥ 65 Jahren eine Behandlung mit Ronapreve. Das Sicherheitsprofil war ähnlich wie bei erwachsenen Patienten im Alter < 65 Jahren.
Subkutane Gabe: In den Studien COV-2069 wurden Personen im Alter von ≥65 Jahren mit Ronapreve behandelt, und das Sicherheitsprofil war ähnlich wie bei Erwachsenen im Alter von < 65 Jahren.
Erfahrungen aus der Anwendung in Notfallsituationen
Im Rahmen der Anwendung von Ronapreve in Notfallsituationen in den USA wurden nach intravenöser und subkutaner Gabe Fälle von konvulsiver Synkope beobachtet (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunogenität. Der Nachweis einer Antikörperentwicklung hängt in hohem Mass von der Sensitivität und Spezifität des Tests ab. Zudem kann die beobachtete Inzidenz der Antikörper (einschliesslich neutralisierender Antikörper) in einem Test von mehreren Faktoren beeinflusst werden, z.B. von der Testmethodik, Probenhandhabung, Zeitpunkt der Probengewinnung, Begleitmedikamenten und von einer zugrundeliegenden Krankheit. Aus diesen Gründen kann ein Vergleich der Inzidenz von Antikörpern gegen Casirivimab und Imdevimab in den nachstehend beschriebenen Studien mit der Inzidenz von Antikörpern in anderen Studien oder gegen andere Arzneimittel irreführend sein.
In der Studie COV-2067 wurden nach einer intravenösen Einzeldosis von Ronapreve (1200, 2400 oder 8000 mg) bis Studientag 225 bei 0,8 % (50/6192) bzw. bei 1,8 % (113/6186) der hinsichtlich Antikörpern gegen Arzneimittel (Anti-Drug-Antibodies, ADA) auswertbaren Teilnehmenden behandlungsbedingte oder durch die Behandlung verstärkte Antikörper gegen Casirivimab bzw. gegen Imdevimab nachgewiesen.
Von diesen Patienten wiesen 0,3 % (18/6144) nur neutralisierende Antikörper gegen Imdevimab auf.
In der Studie COV-2069 wurden nach einer subkutanen Einzeldosis von 1200 mg Ronapreve bis Studientag 225 bei 14 % (223/1595) bzw. bei 18,2 % (290/1595) der hinsichtlich ADA auswertbaren Teilnehmenden behandlungsbedingte oder durch die Behandlung verstärkte Antikörper gegen Casirivimab bzw. gegen Imdevimab nachgewiesen. Von diesen Patienten wiesen 2,1 % (33/1593) bzw. 10 % (160/1593) neutralisierende Antikörper gegen Casirivimab bzw. gegen Imdevimab auf.
In der Studie HV-2093 wurden nach einer wiederholten monatlichen subkutanen Dosis von 1200 mg Ronapreve bis Studientag 365 bei 9,1 % (65/717) bzw. bei 16,7 % (120/717) der hinsichtlich ADA auswertbaren Teilnehmenden behandlungsbedingte oder durch die Behandlung verstärkte Antikörper gegen Casirivimab bzw. gegen Imdevimab nachgewiesen. Von diesen Patienten wiesen 1,5 % (11/717) bzw. 6 % (43/716) neutralisierende Antikörper gegen Casirivimab bzw. gegen Imdevimab auf.
Ausgehend von den verfügbaren ADA-Daten waren keine Auswirkungen des ADA-Status auf die Serumkonzentrationen von Casirivimab und Imdevimab oder die Sicherheit von Ronapreve zu erkennen. Aufgrund der geringen Anzahl von Probanden, die ADA-positiv waren, kann jedoch keine eindeutige Schlussfolgerung gezogen werden.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungIn klinischen Studien wurden Dosen bis zu 8'000 mg (je 4'000 mg Casirivimab und Imdevimab, ungefähr die 7-fache empfohlene Dosis) verabreicht, wobei das Sicherheitsprofil jenem der zugelassenen Dosis ähnelte.
Es gibt kein spezielles Antidot gegen eine Überdosierung von Casirivimab und Imdevimab. Bei einer Überdosierung sollte mit allgemeinen unterstützenden Massnahmen behandelt werden, einschliesslich der Überwachung der Vitalzeichen und der Beobachtung des klinischen Zustands des Patienten.
Eigenschaften/WirkungenATC-Code
J06BD07
Wirkungsmechanismus
Casirivimab (IgG1κ) und Imdevimab (IgG1λ) sind zwei rekombinante humane monoklonale Antikörper, die in ihrer jeweiligen Fc-Region nicht modifiziert sind. Casirivimab und Imdevimab binden an nichtüberlappende Epitope der Rezeptor-bindenden Domäne (RBD) des Spike-Proteins von SARS-CoV-2 mit einer Dissoziationskonstante von KD = 45,8 pM bzw. 46,7 pM. Casirivimab, Imdevimab sowie Casirivimab zusammen mit Imdevimab blockierten die RBD-Bindung an den humanen ACE2-Rezeptor mit IC50-Werten von 56,4 pM, 165 pM bzw. 81,8 pM.
In zellbasierten Assays zeigten Imdevimab und Casirivimab Antikörper-abhängige zellvermittelte Zytotoxizität (ADCC) und Antikörper-abhängige zelluläre Phagozytose (ADCP) aber keine komplementabhängige Zytotoxizität.
In-vitro-Untersuchungen an Zellen zeigten keine Aktivität infektionsverstärkender Antikörper (ADE, antibody-dependent enhancement).
Antivirale Aktivität
In einem SARS-CoV-2-Virus-Neutralisationstest in Vero E6-Zellen neutralisierten Casirivimab, Imdevimab sowie Casirivimab zusammen mit Imdevimab SARS-CoV-2 (Isolat USA-WA1/2020) mit IC50-Werten von 37,4 pM (0,005 μg/ml), 42,1 pM (0,006 μg/ml) bzw. 31,0 pM (0,005 μg/ml).
Antivirale Resistenz
Es besteht das potenzielle Risiko eines Therapieversagens aufgrund der Entwicklung von Virusvarianten, die gegen zusammen verabreichtes Casirivimab und Imdevimab resistent sind.
Die Neutralisationsstärke von gemeinsam verabreichtem Casirivimab und Imdevimab wurde mit S-Protein-Varianten, einschliesslich bekannter besorgniserregender Varianten (Variants of Concern; VOC)/Varianten von Interesse (Variants of Interest; VOI), Varianten, die in in vitro Escape-Studien identifiziert wurden, und Varianten aus öffentlich verfügbaren SARS-CoV-2 Genomdaten der Global Initiative on Sharing All Influenza Data (GISAID), untersucht (Tabelle 4).
Tabelle 4: Daten zur In-vitro-Neutralisierung pseudotypisierter virusartiger Partikel mit der vollständigen Sequenz des SARS-CoV-2-S-Proteins oder nach Substitution mit den wichtigsten besorgniserregenden Varianten/Varianten von Interesse des SARS-CoV-2-S-Proteins bzw. authentischer Viren mit Casirivimab und Imdevimab
|
Variante mit Spike-Protein-Substitutionen
|
Wichtige getestete Substitutionen
|
Verringerte Suszeptibilität (Pseudovirus, x-fache Veränderung des IC50)
|
Verringerte Suszeptibilität (authentisches Virus, x-fache Veränderung des IC50)
| |
B.1.1.7 (Alpha)
|
Vollständiges S-Proteina
|
Keine Veränderungb
|
Keine Veränderungb
| |
B.1.351 (Beta)
|
Vollständiges S-Proteinc
|
Keine Veränderungb
|
Keine Veränderungb
| |
P.1 (Gamma)
|
Vollständiges S-Proteind
|
Keine Veränderungb
|
Keine Veränderungb
| |
B.1.617.2 (Delta)
|
Vollständiges S-Proteine
|
keine Veränderungb
|
keine Veränderungb
| |
AY.1
(Delta [+K417N])
|
K417N+L452R+T478K
|
keine Veränderungb
|
keine Veränderungb
| |
B.1.427/B.1.429 (Epsilon)
|
L452R
|
Keine Veränderungb
|
n.b.
| |
B.1.526 (Iota)
|
E484K
|
Keine Veränderungb
|
n.b.
| |
B.1.617.1 (Kappa)
|
Vollständiges S-Proteinf
|
Keine Veränderungb
|
Keine Veränderungb
| |
C.37 (Lambda)
|
L452R+F490S
|
Keine Veränderungb
|
Keine Veränderungb
| |
B.1.621
(Mu)
|
R346K+E484K+N501Y
|
Keine Veränderungb
|
Keine Veränderungb
| |
BA.1 (Omikron)
|
Vollständiges S-Proteing
|
> 1013-fach
|
n.b.
| |
BA.1.1 (Omikron)
|
Vollständiges S-Proteinh
|
>1461-fach
|
n.b.
| |
BA.2 (Omikron)i
|
Vollständiges S-Proteinj
|
325-fach
|
513-fach
| |
BA.2.12.1 (Omikron)i
|
Vollständiges S-Proteink
|
275-fach
|
239-fach
| |
BA.2.75 (Omikron)i
|
Vollständiges S-Proteinl
|
881-fach
|
n.b.
| |
BA.2+R346T (Omikron)i
|
Vollständiges S-Proteinm
|
1147-fach
|
n.b.
| |
BA.3 (Omikron)i
|
Vollständiges S-Proteinn
|
>1457-fach
|
n.b.
| |
BA.4/BA.5 (Omikron)i
|
Vollständiges S-Proteino
|
201-fach
|
n.b.
| |
BA.4.6 (Omikron)i
|
Vollständiges S-Proteinp
|
>1611-fach
|
n.b.
| |
BQ.1 (Omikron)i
|
Vollständiges S-Proteinq
|
>1913-fach
|
n.b.
| |
BQ.1.1 (Omikron)i
|
Vollständiges S-Proteinr
|
>1368-fach
|
n.b.
| |
XBB (Omikron)i
|
Vollständiges S-Proteins
|
>1368-fach
|
n.b.
|
IC50, erforderliche Konzentration für eine 50 % Inhibierung.
a Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante B.1.1.7 (Alpha) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: del69-70, del145, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H.
b Keine Veränderung: ≤5-fache Verringerung der Suszeptibilität.
c Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante B.1.351 (Beta) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: D80Y, D215Y, del241-243, K417N, E484K, N501Y, D614G, A701V.
d Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante P.1 (Gamma) wurden getestet. In der Variante sind die folgenden Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F.
e Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante B.1.617.2 (Delta) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19R, G142D, E156G, F157del, R158del, L452R, T478K, D614G, P681R, D950N.
f Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante B.1.617.1 (Kappa) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T95I, G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H.
g Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.1 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: A67V, del69-70, T95I, G142D/del143-145, del211/L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F.
h Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante von BA.1.1 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: A67V, del69-70, T95I, G142D/del143-145, del211/L212I, ins214EPE, G339D, R346K, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F.
i Bei den angegebenen Werten handelt es sich um den geometrischen Mittelwert von Assay mit mindestens 3 Replikaten.
j Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins von BA.2 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, del24-26, A27S, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
k Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.2.12.1 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452Q, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, S704L, N764K, D796Y, Q954H, N969K.
l Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.2.75 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, G142D, K147E, W152R, F157L, I210V, V213G, G257S, G339H, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, G446S, N460K, S477N, T478K, E484A, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
m Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.2+R346T (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, del24-26, A27S, G142D, V213G, G339D, R346T, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
n Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.3 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: A67V, H69del, V70del, T95I, G142D, V143del, Y144del, Y145del, N211del, L212I, G339D, S371F, S373P, S375F, D405N, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
o Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.4/BA.5 (Omikron) wurden getestet. BA.4 und BA.5 weisen identische S-Protein-Sequenzen auf. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, H69del, V70del, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
p Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BA.4.6 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, H69del, V70del, G142D, V213G, G339D, R346T, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, L452R, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
q Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BQ.1 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, H69del, V70del, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, K444T, L452R, N460K, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
r Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante BQ.1.1 (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, H69del, V70del, G142D, V213G, G339D, R346T, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, K444T, L452R, N460K, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
s Pseudotypisierte VLP mit Expression des gesamten Spike-Proteins der Variante XBB (Omikron) wurden getestet. In der Variante sind folgende Veränderungen des Spike-Proteins gegenüber dem Wildtyp vorhanden: T19I, L24del, P25del, P26del, A27S, V83A, G142D, Y144-, H146Q, Q183E, V213E, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486S, F490S, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K.
* Besorgniserregende Varianten/Varianten von Interesse gemäss Definition der Centers for Disease Control and Prevention (CDC, 2021) {https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html}.
Abkürzungen: del, Deletion; ins, Insertion; n.b., nicht bestimmt.
Für Varianten, bei denen Assays mit Replikaten durchgeführt wurden, sind jeweils Daten aus dem ersten Replikat für jede Variante angegeben (es sei denn, es ist ein geometrischer Mittelwert angegeben, worauf die Fussnote «k» hinweist), mit Ausnahme von Assays, bei denen Replikate aufgrund eines Fehlers im anfänglichen Assay verwendet wurden.
Abschwächung der Immunantwort
Es besteht ein theoretisches Risiko, dass die Gabe von Antikörpern die endogene Immunantwort auf SARS-CoV-2 abschwächt und die Anfälligkeit der Personen für eine Reinfektion erhöht.
Pharmakodynamik
Klinische Wirksamkeit
Behandlung von COVID-19
Studie COV-2067
COV-2067 ist eine randomisierte, doppelblinde, placebokontrollierte klinische Phase-III-Studie zur Untersuchung von Ronapreve (Casirivimab und Imdevimab) zur Behandlung symptomatischer Erwachsener mit RT-qPCR-bestätigter COVID-19-Erkrankung, die nicht hospitalisiert sind oder keine Sauerstofftherapie benötigen. Die Teilnehmenden wurden innert 7 Tagen nach Auftreten der Symptome randomisiert.
In der Phase-III-Kohorte 1 wurden 4'567 Teilnehmende mit mindestens einem Risikofaktor für einen schweren Verlauf von COVID-19, innert 7 Tagen nach Auftreten der Symptome in folgende Gruppen randomisiert: einzelne intravenöse Infusion von 1'200 mg Ronapreve (600 mg Casirivimab und 600 mg Imdevimab) (n = 838), 2'400 mg Ronapreve (1'200 mg Casirivimab und 1'200 mg Imdevimab) (n = 1'529), 8'000 mg Ronapreve (4'000 mg Casirivimab und 4'000 mg Imdevimab) (n = 700) oder Placebo (n = 1'500). Zu Beginn der Phase III wurde Ronapreve in den zwei Dosierungen 8'000 mg und 2'400 mg verabreicht. Da jedoch die Wirksamkeitsanalysen der Daten aus der Phase I/II für die beiden Dosierungen ähnlich ausfielen, wurde der Phase-III-Abschnitt des Prüfplans nachträglich geändert, um sowohl die 2'400 mg-Dosis als auch die 1'200 mg-Dosis mit Placebo zu vergleichen. Die Vergleiche erfolgten zwischen Probanden, die nach der Randomisierung die jeweilige Ronapreve-Dosis erhielten, und Probanden, die gleichzeitig in die Placebogruppe randomisiert wurden.
Das mediane Alter aller randomisierten Teilnehmenden mit mindestens einem Risikofaktor betrug bei Studienbeginn 50 Jahre (13 % der Probanden waren 65 Jahre oder älter), 52 % waren weiblich, 84 % weisser Hautfarbe, 5 % Schwarze oder Afroamerikaner; 36 % hispanisch oder Latinos. Die demografischen Daten und Krankheitsmerkmale bei Aufnahme in die Studie waren in den Behandlungsgruppen mit Casirivimab und Imdevimab und Placebo gut ausgewogen. Die mediane Dauer vom Auftreten der Symptome bis zur Randomisierung betrug in allen Gruppen 3 Tage.
Primärer Endpunkt
Primärer Endpunkt war der Anteil an Probanden mit ≥1 COVID-19-bedingtem Spitalaufenthalt oder Tod jeglicher Ursache bis einschliesslich Tag 29 unter den Probanden mit positivem SARS-CoV-2-Ergebnis in der RT-qPCR von Nasen-Rachen-Abstrichen (NP, nasopharyngeal) bei der Randomisierung und mit mindestens einem Risikofaktor für einen schweren Verlauf von COVID-19 (modifizierte Gesamtanalysegruppe, mFAS). Zu den im Protokoll aufgeführten Risikofaktoren für die Entwicklung einer schweren COVID-19 gehörten Alter > 50 Jahre, Adipositas definiert als BMI ≥30 kg/m2, kardiovaskuläre Erkrankungen einschliesslich Hypertonie, chronische Lungenerkrankungen einschliesslich Asthma, Typ-1- und Typ-2-Diabetes mellitus, chronische Nierenerkrankungen einschliesslich Dialysepatienten, chronische Lebererkrankungen, Schwangerschaft und immunsupprimierte Patienten.
Tabelle 5: Primärer Endpunkt aus der Studie COV-2067
|
|
1'200 mg i. v.
|
Placebo
|
2'400 mg i. v.
|
Placebo
| |
n = 736
|
n = 748
|
n = 1'355
|
n = 1'341
| |
Patienten mit ≥1 COVID-19-bedingter Hospitalisierung oder Tod bis einschliesslich Tag 29
| |
Risikoreduzierung
|
70 %
(p = 0,0024)
|
71 %
(p < 0,0001)
| |
Anzahl Patienten mit Ereignissen
|
7 (1,0 %)
|
24 (3,2 %)
|
18 (1,3 %)
|
62 (4,6 %)
|
Prävention von COVID-19
Bei COV-2069 handelte es sich um eine randomisierte, doppelblinde, placebokontrollierte klinische Studie zum Vergleich von 600 mg Casirivimab und 600 mg Imdevimab zu Placebo, subkutan verabreicht zur Prophylaxe von COVID-19 bei asymptomatischen Haushaltskontakten von symptomatischen Personen, die mit SARS-CoV-2 infiziert sind (Indexfälle). Die Studienteilnehmer waren zuvor nicht gegen das SARS-CoV-2 geimpft worden.
Die Studienteilnehmer erhielten randomisiert im Verhältnis 1:1 entweder Casirivimab und Imdevimab oder Placebo innerhalb von 96 Stunden nach Entnahme der ersten Probe eines Indexfalles mit positivem Ergebnis (RT-qPCR) auf SARS-CoV-2.
Randomisierte Studienteilnehmer mit einem negativen SARS-CoV-2-RT-qPCR-Testergebnis zu Behandlungsbeginn wurden Kohorte A zugeordnet und die Teilnehmer mit einem positiven SARS-CoV-2-RT-qPCR-Testergebnis wurden Kohorte B zugeordnet.
Kohorte A
Die primäre Analysepopulation umfasste Studienteilnehmer, die bei Behandlungsbeginn SARS-CoV-2-RT-qPCR-negativ und seronegativ waren. Studienteilnehmer, die seropositiv waren oder bei denen die Ausgangsserologie unbestimmt war oder fehlte, wurden von der primären Wirksamkeitsanalyse ausgeschlossen. In der primären Analysepopulation bei Behandlungsbeginn betrug das mediane Alter 44 Jahre (davon waren 9 % 65 Jahre oder älter) und 54 % der Studienteilnehmer waren weiblich. Die demografischen Merkmale und die Krankheitscharakteristika waren bei Behandlungsbeginn in den Behandlungsgruppen mit Casirivimab und Imdevimab und mit Placebo ausgewogen.
Der primäre Endpunkt war der Anteil der Studienteilnehmer, die bis Tag 29 eine symptomatische, durch RT-qPCR bestätigte COVID-19-Erkrankung entwickelten. Es kam unter der Behandlung mit Casirivimab und Imdevimab zu einer statistisch signifikanten Risikoreduktion von 81 % für die Entwicklung von COVID-19 im Vergleich zu Placebo. In einer Sensitivitätsanalyse, in die alle bei Studienbeginn RT-qPCR-negativen Studienteilnehmer unabhängig vom serologischen Ausgangsstatus einbezogen wurden, zeigte sich eine statistisch signifikante 82-prozentige Verringerung des Risikos für die Entwicklung von COVID-19 durch die Behandlung mit Casirivimab und Imdevimab im Vergleich zu Placebo.
Tabelle 6: Primäranalyse der Studie COV-2069 – Kohorte A
|
|
Ronapreve
(Einzeldosis zu 1'200 mg)
|
Placebo
| |
Primäre Analysegruppe: Seronegativ bei
Behandlungsbeginn
|
n = 753
|
n = 752
| |
COVID-19-Risiko
| |
Bis einschliesslich Tag 29 (primärer Endpunkt)
| |
Nicht adjustierte Risikoreduzierung
(adjustierte Odds Ratio, p-Wert)
|
81 %
(0,17; p < 0,0001)
| |
Anzahl Personen mit Ereignissen
|
11 (1,5 %)
|
59 (7,8 %)
|
Das Konfidenzintervall (KI) mit p-Wert basiert auf der Odds Ratio (Casirivimab und Imdevimab-Gruppe vs. Placebo-Gruppe) unter Verwendung eines logistischen Regressionsmodells mit festen kategorialen Effekten von Behandlungsgruppe, Altersgruppe (Alter in Jahren: ≥ 12 bis < 50 und ≥ 50) und Region (US vs. nicht-US).
Kohorte B
Die primäre Analysepopulation umfasste asymptomatische Studienteilnehmer, die bei Behandlungsbeginn SARS-CoV-2-RT-qPCR-positiv und seronegativ waren.
In der primären Analysepopulation bei Behandlungsbeginn betrug das mediane Alter 40 Jahre (davon waren 11 % 65 Jahre oder älter) und 55 % der Studienteilnehmer waren weiblich. Die demografischen Merkmale und die Krankheitscharakteristika waren bei Behandlungsbeginn in den Behandlungsgruppen mit Casirivimab und Imdevimab und mit Placebo ausgewogen.
Der primäre Wirksamkeitsendpunkt war der Anteil der Studienteilnehmer, die bis Tag 29 eine durch RT-qPCR bestätigte COVID-19-Erkrankung entwickelten. Das Risiko für die Entwicklung von COVID-19 unter Behandlung mit Casirivimab und Imdevimab vs. Placebo wurde um 31 % gesenkt. Bei einer Sensitivitätsanalyse, in die alle bei Behandlungsbeginn RT-qPCR-positiven Studienteilnehmer, unabhängig vom serologischen Ausgangsstatus einbezogen wurden, zeigte sich unter der Behandlung mit Casirivimab und Imdevimab eine Risikoreduktion um 35 % für RT-qPCR-bestätigte COVID-19-Erkrankung im Vergleich zu Placebo.
Tabelle 7: Primäranalyse der Studie COV-2069, Kohorte B
|
|
Ronapreve
(Einzeldosis zu 1'200 mg)
|
Placebo
| |
Primäre Analysegruppe: Seronegativ bei
Behandlungsbeginn
|
n = 100
|
n = 104
| |
COVID-19-Risiko
| |
Risikoreduzierung insgesamt bis einschliesslich Tag 29 (primärer Endpunkt)
| |
Nicht adjustierte Risikoreduzierung
(adjustierte Odds Ratio, p-Wert)
|
31 %
(0,54; p = 0,0380)
| |
Anzahl Personen mit Ereignissen
|
29 (29 %)
|
44 (42,3 %)
|
Das Konfidenzintervall (KI) mit p-Wert basiert auf der Odds Ratio (Casirivimab und Imdevimab-Gruppe vs. Placebo-Gruppe) unter Verwendung eines logistischen Regressionsmodells mit festen kategorialen Effekten von Behandlungsgruppe, Altersgruppe (Alter in Jahren: ≥ 12 bis < 50 und ≥ 50) und Region (US vs. nicht-US).
PharmakokinetikAbsorption
Sowohl Casirivimab als auch Imdevimab zeigten von 300 mg Ronapreve (150 mg Casirivimab und 150 mg Imdevimab) bis 8'000 mg Ronapreve (4'000 mg Casirivimab und 4'000 mg Imdevimab) nach intravenöser Gabe einer Einzeldosis eine lineare und dosisproportionale Pharmakokinetik (PK) auf. Tabelle 8 enthält eine Zusammenfassung von PK-Parametern nach intravenöser Gabe einer Einzeldosis (600 mg Casirivimab und 600 mg Imdevimab), berechnet anhand eines Populations-PK-Modells für jeden Antikörper auf der Grundlage der Daten von 3'687 (Casirivimab) bzw. 3'716 Probanden (Imdevimab).
Tabelle 8: Zusammenfassung von PK-Parametern für Casirivimab und Imdevimab nach einer Einzeldosis von 1'200 mg Ronapreve i. v.
|
PK-Parameter1
|
Casirivimab
|
Imdevimab
| |
AUC0-28 (mg·Tag/l)2
|
1754,9 (380,50)
|
1600,8 (320,88)
| |
AUCinf (mg·Tag/l)3
|
3563,6 (1239,61)
|
2890,5 (876,31)
| |
Cmax (mg/l)4
|
182,7 (81,45)
|
181,7 (77,78)
| |
C28 (mg/l)5
|
37,9 (10,33)
|
31,0 (8,24)
| |
Halbwertszeit (Tag)
|
31,2 (10,59)
|
27,3 (7,73)
|
1 Mittelwert (SD), wobei SD die Standardabweichung des arithmetischen Mittelwerts ist; 2 AUC0-28 = Fläche unter der Konzentrations-Zeit-Kurve vom Zeitpunkt 0 bis Tag 28 nach der Dosisgabe; 3 AUCinf = Fläche unter der Konzentrations-Zeit-Kurve vom Zeitpunkt 0 bis unendlich; 4 Cmax = Maximale Konzentration im Serum und repräsentiert Konzentration am Ende der Infusion, 5 C28 = Konzentration 28 Tage nach Verabreichung, d.h. an Tag 29.
Tabelle 9 zeigt eine Zusammenfassung von PK-Parametern nach subkutaner Gabe einer Einzeldosis (600 mg Casirivimab und 600 mg Imdevimab) auf der Grundlage des Populations-PK-Modells für jeden Antikörper.
Tabelle 9: Zusammenfassung von PK-Parametern für Casirivimab und Imdevimab nach einer Einzeldosis von 1'200 mg [Ronapreve] s. c.
|
PK-Parameter1
|
Casirivimab
|
Imdevimab
| |
AUC0-28 (mg·Tag/l)2
|
1121,7 (243,12)
|
1016,9 (203,92)
| |
AUCinf (mg·Tag/l)3
|
2559,5 (890,35)
|
2073,3 (628,60)
| |
Cmax (mg/l)4
|
52,2 (12,15)
|
49,2 (11,01)
| |
Tmax (Tag)5, 6
|
6,7 [3,4, 13,6]
|
6,6 [3,4, 13,6]
| |
C28 (mg/l)7
|
30,5 (7,55)
|
25,9 (6,07)
|
1 Mittelwert (SD), wobei SD die Standardabweichung des arithmetischen Mittelwerts ist; 2 AUC0-28 = Fläche unter der Konzentrations-Zeit-Kurve vom Zeitpunkt 0 bis Tag 28 nach der Dosisgabe; 3 AUCinf = Fläche unter der Konzentrations-Zeit-Kurve vom Zeitpunkt 0 bis unendlich; 4 Cmax = Maximale Konzentration im Serum, 5 Tmax = Dauer bis zum Erreichen der Cmax; 6 Median [Minimum, Maximum]; 7 C28 = Konzentration 28 Tage nach Verabreichung, d.h. an Tag 29.
Tabelle 10 zeigt eine Zusammenfassung von PK-Parametern nach einer intravenösen einzelnen Aufsättigungsdosis von 1'200 mg Ronapreve (600 mg Casirivimab und 600 mg Imdevimab), gefolgt von mehreren intravenösen Dosen mit 600 mg Ronapreve alle 4 Wochen (300 mg Casirivimab und 300 mg Imdevimab) auf der Grundlage des Populations-PK-Modells für jeden Antikörper.
Tabelle 10: Zusammenfassung von PK-Parametern für Casirivimab und Imdevimab nach einer einzelnen Aufsättigungsdosis von 1'200 mg Ronapreve i.v. und von Erhaltungsdosen von 600 mg Ronapreve i. v. q4w
|
PK-Parameter1
|
Casirivimab
|
Imdevimab
| |
AUCtau,ss (mg∙Tag/l)2
|
1767,5 (605,79)
|
1436,8 (432,87)
| |
Cmax,ss (mg/l)3
|
133,8 (46,51)
|
122,4 (41,67)
| |
CTal,ss (mg/l)4
|
42,6 (19,72)
|
31,7 (13,56)
| |
C28 (mg/l)5
|
37,9 (10,32)
|
31,0 (8,24)
| |
AR6
|
1,0 (0,241)
|
0,893 (0,174)
|
1 Mittelwert (SD), wobei SD die Standardabweichung des arithmetischen Mittelwerts ist; 2 AUCtau,ss = Fläche unter der Konzentration-Zeit-Kurve in einem Dosierungsintervall im Fliessgleichgewicht; 3 Cmax,ss = Maximale Konzentration im Fliessgleichgewicht; 4 CTal,ss = Talkonzentration im Fliessgleichgewicht; 5 C28 = Konzentration 28 Tage nach der ersten Dosis; 6 Das Akkumulationsverhältnis (AR) berechnet sich aus
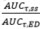
(ED = erste Dosis); q4w = alle 4 Wochen.
Tabelle 11 zeigt eine Zusammenfassung von PK-Parametern nach einer einzelnen subkutanen Aufsättigungsdosis von 1'200 mg Ronapreve (600 mg Casirivimab und 600 mg Imdevimab), gefolgt von mehreren subkutanen Dosen mit 600 mg Ronapreve alle 4 Wochen (300 mg Casirivimab und 300 mg Imdevimab) auf der Grundlage des Populations-PK-Modells für jeden Antikörper.
Tabelle 11: Zusammenfassung von PK-Parametern für Casirivimab und Imdevimab nach einer einzelnen subkutanen Aufsättigungsdosis von 1'200 mg und subkutanen Erhaltungsdosen von 600 mg Ronapreve q4w
|
PK-Parameter1
|
Casirivimab
|
Imdevimab
| |
AUCtau,ss (mg∙Tag/l)2
|
1268,9 (434,68)
|
1030,1 (310,30)
| |
Cmax,ss (mg/l)3
|
56,0 (16,81)
|
47,0 (12,43)
| |
CTal,ss (mg/l)4
|
34,0 (14,56)
|
26,1 (10,17)
| |
C28 (mg/l)5
|
30,5 (7,55)
|
25,9 (6,07)
| |
AR6
|
1,13 (0,288)
|
1,01 (0,213)
|
1 Mittelwert (SD), wobei SD die Standardabweichung des arithmetischen Mittelwerts ist; 2 AUCtau,ss = Fläche unter der Konzentration-Zeit-Kurve in einem Dosierungsintervall im Fliessgleichgewicht; 3 Cmax,ss = Maximale Konzentration im Fliessgleichgewicht; 4 CTal,ss = Talkonzentration im Fliessgleichgewicht; 5 C28 = Konzentration 28 Tage nach der ersten Dosis; 6 Das Akkumulationsverhältnis (AR) berechnet sich aus
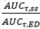
(ED = erste Dosis); q4w = alle 4 Wochen.
In Bezug auf die wiederholte intravenöse und subkutane Gabe zur Vorbeugung kann man ausgehend von populationspharmakokinetischen Simulationen vorhersagen, dass die mediane vorhergesagte CTal,ss von Casirivimab und Imdevimab im Serum ähnlich ist wie die an Tag 29 gemessenen mittleren Serumkonzentrationen nach einer subkutanen Einzeldosis von 1'200 mg Ronapreve (600 mg Casirivimab und 600 mg Imdevimab).
Tabelle 8 und Tabelle 9 enthalten die geschätzte mittlere (Standardabweichung) Cmax und C28 von Casirivimab und Imdevimab nach einer Einzeldosis von 1'200 mg (je 600 mg von jedem monoklonalen Antikörper) i. v. bzw. s. c. aufgrund eines populationspharmakokinetischen Modells. Die mediane (Bereich) Dauer bis zum Erreichen der geschätzten maximalen Serumkonzentration von Casirivimab und Imdevimab (Tmax) nach einer subkutanen Einzeldosis von 1'200 mg (jeweils 600 mg von jedem monoklonalen Antikörper) beträgt 6,7 (3,4-13,6) Tage für Casirivimab und 6,6 (3,4-13,6) Tage für Imdevimab (Tabelle 9).
Nach subkutaner Gabe von Casirivimab und Imdevimab als Einzeldosis von 1'200 mg (je 600 mg von jedem monoklonalen Antikörper) betrug die mittels Population-PK geschätzte Bioverfügbarkeit von Casirivimab 71,8 % und von Imdevimab 71,7 %.
Distribution
Das anhand einer populationspharmakokinetischen Analyse geschätzte Gesamtdistributionsvolumen beträgt 7,161 l für Casirivimab und 7,425 l für Imdevimab.
Metabolismus
Es wurden keine speziellen Studien zum Metabolismus durchgeführt, da es sich bei Casirivimab und Imdevimab um Proteine handelt. Da Casirivimab und Imdevimab humane monoklonale IgG1-Antikörper sind, kann man annehmen, dass sie gleich wie endogenes IgG über katabole Stoffwechselwege zu kleinen Peptiden und Aminosäuren abgebaut werden.
Elimination
Laut einer populationspharmakokinetischen Analyse betrugen die Eliminationshalbwertszeit und die Clearance nach einer einzigen Verabreichung von 600 mg jedes der monoklonalen Antikörper durchschnittlich (5., 95. Perzentil) 29,8 (16,4, 43,1) Tage und 0,188 (0,11, 0,3) l/Tag bei Casirivimab bzw. 26,2 (16,9, 35,6) Tage und 0,227 (0,15, 0,35) l/Tag bei Imdevimab.
Ausscheidung
Casirivimab und Imdevimab sind monoklonale Antikörper, was eine Ausscheidung über die Nieren unwahrscheinlich macht.
Kinetik spezieller Patientengruppen
Der Einfluss aller untersuchten Kovariaten und der beobachteten, statistisch signifikanten Kovariaten (Geschlecht, Körpergewicht und Ausgangswert des Albuminspiegels auf Clearance [CL] und zentrales Verteilungsvolumen [Vc] sowie Ethnie, geringe Leberfunktionsstörung und Ausgangswert der Viruslast auf die CL) auf die Pharmakokinetik von Ronapreve wurde durch eine populationspharmakokinetische Analyse bewertet. Diese Analyse wies auf keinen klinisch relevanten Einfluss auf die Exposition gegenüber Casirivimab und Imdevimab hin: Alter (Median: 42 Jahre, Spanne: 18–96 Jahre), Geschlecht (männlich, weiblich), Körpergewicht (Median: 81,5 kg, Spanne: 35,5–201 kg ), Ethnie (schwarze Hautfarbe, sonstige [weisse Hautfarbe, asiatisch, indigene Einwohner der U.S.A.] ), Albuminspiegel zur Baseline (Median: 43 g/l, Spanne: 26–56 g/l), Nierenfunktionsstörung (normale Funktion, leichte/mässig schwere/hochgradige Niereninsuffizienz) und geringe Leberfunktionsstörung (normale Funktion, geringfügige/mässig schwere Leberinsuffizienz).
Von allen untersuchten Kovariaten hatte das Körpergewicht den grössten Einfluss auf die Exposition gegenüber Casirivimab und Imdevimab. Im Vergleich zu einem Referenzprobanden mit 81 kg ist die vorhergesagte Exposition (AUCTag28, Cmax und CTag28) sowohl von Casirivimab als auch von Imdevimab bei Probanden des 5. Perzentils des Körpergewichts (55,4 kg) um 20 bis 30 % höher und bei Probanden des 95. Perzentils des Körpergewichts (123 kg) um 20 bis 25 % geringer.
Im Vergleich zu einem Referenzprobanden mit 81 kg betragen bei einem Probanden mit der Kovariatenkombination, die zur höchsten vorhergesagten Populationsclearance von Casirivimab und Imdevimab führt (hellhäutig, männlich, Albumin 29 g/l, 151,8 kg), die Quotienten von AUCTag28, Cmax und CTag28 voraussichtlich für Casirivimab 0,48, 0,56 und 0,31 bzw. für Imdevimab 0,47, 0,56 und 0,28.
Leberfunktionsstörungen
Es ist nicht zu erwarten, dass Casirivimab und Imdevimab in signifikantem Umfang über die Leber eliminiert werden. Die Auswirkung einer Leberfunktionsstörung auf die Exposition gegenüber Casirivimab und Imdevimab wurde mithilfe einer Populations-PK-Analyse bei Patienten mit leichter Leberfunktionsstörung (n = 586 für Casirivimab und n = 599 für Imdevimab) beurteilt (Gesamtbilirubin [TB] mehr als das 1,0- bis 1,5-Fache der oberen Normgrenze [ULN] und ein beliebiger Wert für die Aspartataminotransferase [AST]). Es wurden keine klinisch bedeutsamen Unterschiede zwischen Patienten mit leichter Leberfunktionsstörung und Patienten mit normaler Leberfunktion in Bezug auf die Exposition gegenüber Casirivimab und Imdevimab festgestellt. Von Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung sind nur begrenzte Daten (n = 11) verfügbar. Die Pharmakokinetik bei Patienten mit schwerer Leberfunktionsstörung wurde nicht untersucht.
Nierenfunktionsstörungen
Casirivimab und Imdevimab sind monoklonale Antikörper, bei denen aufgrund ihrer Molekülmasse (> 69 kDa) keine wesentliche renale Elimination erwartet wird. Ausgehend von der Populations-PK-Analyse waren die Serum-Talkonzentrationen von Casirivimab und Imdevimab im Fliessgleichgewicht bei Patienten mit leichter oder mittelschwerer Nierenfunktionsstörung und Patienten mit CrCl < 15 ml/min einschliesslich Dialysepatienten vergleichbar mit denen von Personen mit normaler Nierenfunktion. Von Patienten mit schwerer Nierenfunktionsstörung (n = 3) liegen begrenzte Daten vor.
Ältere Patienten
In der Populations-PK-Analyse erwies sich das Alter (18 Jahre bis 96 Jahre) nicht als signifikante Kovariate der PK von Casirivimab und Imdevimab.
Im Vergleich zu Personen im Alter von < 65 Jahren war die Exposition gegenüber Casirivimab und Imdevimab bei Personen im Alter von > 65 Jahren oder ≥75 Jahren nach intravenöser oder subkutaner Gabe ähnlich.
Kinder und Jugendliche
Bei Jugendlichen mit COVID-19-Erkrankung (ab dem Alter von 12 Jahren und mit einem Körpergewicht von mindestens 40 kg in der Studie COV-2067), die intravenös eine Einzeldosis von 1'200 mg erhielten, betrug die mittlere Konzentration ± SD am Ende der Infusion und an Tag 28 nach der Verabreichung 172 ± 96,9 mg/l und 54,3 ± 17,7 mg/l für Casirivimab bzw. 183 ± 101 mg/l und 45,3 ± 13,1 mg/l für Imdevimab.
Für nicht mit SARS-CoV-2 infizierte Jugendliche (ab dem Alter von 12 Jahren und mit einem Körpergewicht von mindestens 40 kg in der Studie COV-2069), die subkutan eine Einzeldosis von 1'200 mg erhielten, betrug die mittlere Konzentration ± SD an Tag 28 nach der Verabreichung 44,9 ± 14,7 mg/l für Casirivimab bzw. 36,5 ± 13,2 mg/l für Imdevimab. Bei Kindern < 12 Jahren wurde die Pharmakokinetik von Casirivimab und Imdevimab noch nicht untersucht.
Präklinische DatenEs wurden keine Studien zur Karzinogenität, Genotoxizität und Reproduktionstoxizität mit Casirivimab und Imdevimab durchgeführt.
In einer toxikologischen Studie an Cynomolgus-Affen wurden leichte, vorübergehende, nicht mit Nebenwirkungen einhergehende Anstiege der Werte von AST and ALT, die wahrscheinlich ihren Ursprung in der Skelettmuskulatur hatten und darum verfahrensbedingt waren, bei einer Exposition beobachtet, die dem 30-Fachen der therapeutischen Exposition beim Menschen entsprach.
In Studien zur Gewebe-Kreuzreaktivität mit Casirivimab und Imdevimab an adultem Gewebe von Menschen und Affen sowie an humanem fötalem Gewebe wurde keine Bindung festgestellt.
Sonstige HinweiseInkompatibilitäten
Da keine Kompatibilitätsstudien vorliegen, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Hinsichtlich der intravenösen Verabreichung wurde die Kompatibilität mit zur Verdünnung verwendeter 0,9%iger Natriumchlorid- und 5%iger Dextrose-Injektionslösung nachgewiesen.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Haltbarkeit nach Anbruch
Zusammen verpackte 20 ml-Mehrfachdosen-Durchstechflaschen
Die Lösung mit Casirivimab bzw. Imdevimab enthält kein Konservierungsmittel. Nach dem ersten Punktieren: Wenn das Produkt nicht sofort verwendet wird, kann es in der Durchstechflasche 4 Stunden bei Raumtemperatur bis 25 °C oder höchstens 24 Stunden gekühlt bei 2 °C bis 8 °C aufbewahrt werden. Die Lagerung des angebrochenen Produkts über längere Zeiträume und unter anderen Bedingungen liegt in der Verantwortung des Anwenders.
Zusammen verpackte 6 ml-Einzeldosis-Durchstechflaschen
Die Lösung mit Casirivimab bzw. Imdevimab enthält kein Konservierungsmittel. Nach dem ersten Punktieren: Das Arzneimittel sollte umgehend angewendet werden, etwaige Restmengen sind zu verwerfen.
Verdünnte Lösung für die intravenöse Gabe
Die Lösung in der Durchstechflasche muss vor der Anwendung verdünnt werden. Die verdünnte Lösung mit Casirivimab bzw. Imdevimab enthält kein Konservierungsmittel. Die fertig zubereitete Infusionslösung ist zur sofortigen Anwendung bestimmt. Die chemische und physikalische Stabilität nach Entnahme aus der Durchstechflasche wurde für 20 Stunden bei Raumtemperatur (bis zu 25 °C) und 72 Stunden bei 2 °C bis 8 °C nachgewiesen. Aus mikrobiologischen Gründen sollte die fertig zubereitete Infusionslösung sofort verwendet werden. Bei nicht sofortiger Verwendung liegen die Aufbewahrungsdauer und -bedingungen vor der Anwendung in der Verantwortung des Anwenders und sollen normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, ausser wenn die Verdünnung unter kontrollierten und validierten aseptischen Bedingungen erfolgte. Bei Kühllagerung den i. v. Infusionsbeutel vor der Infusion etwa 30 Minuten lang die Raumtemperatur annehmen lassen.
Lagerung der Spritzen für die subkutane Gabe
Die Lösung mit Casirivimab bzw. Imdevimab enthält kein Konservierungsmittel, daher sollten die vorbereiteten Spritzen sofort verwendet werden. Die chemische und physikalische Stabilität wurden für 24 Stunden bei Raumtemperatur (bis zu 25 °C) und 72 Stunden bei 2 °C bis 8 °C nachgewiesen. Bei nicht sofortiger Verwendung liegen die Aufbewahrungsdauer und -bedingungen vor der Anwendung in der Verantwortung des Anwenders und sollen normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, ausser wenn die Zubereitung unter kontrollierten und validierten aseptischen Bedingungen erfolgte. Bei Kühllagerung die Spritzen vor der Verabreichung während etwa 10 bis 15 Minuten die Raumtemperatur annehmen lassen.
Besondere Lagerungshinweise
Im Kühlschrank (2–8 °C) lagern.
Nicht einfrieren. Nicht schütteln.
Den Behälter im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Ausser Reichweite von Kindern aufbewahren.
Zu den Lagerungsbedingungen nach Verdünnung des Arzneimittels sowie der Spritzen für die subkutane Gabe siehe Abschnitt «Haltbarkeit nach Anbruch».
Hinweise für die Handhabung
Dosisvorbereitung und Dosisgabe
Allgemeine Vorsichtsmassnahmen
Die Durchstechflaschen mit Casirivimab und Imdevimab sollten vor der Anwendung auf Feststoffe und Farbveränderungen inspiziert werden. Finden sich Feststoffe oder Farbveränderungen, so ist die Durchstechflasche gemäss den örtlichen Entsorgungsrichtlinien zu entsorgen.
Intravenöse Infusion
Vorbereitung der intravenösen Infusion
Ronapreve muss von einer medizinischen Fachperson unter aseptischen Bedingungen vorbereitet werden:
1.Die Durchstechflaschen mit Casirivimab und Imdevimab aus der Kühllagerung nehmen und vor der Zubereitung ungefähr 20 Minuten Raumtemperatur annehmen lassen.• Keiner direkten Wärme aussetzen.• Die Durchstechflaschen nicht schütteln.
2.Die Durchstechflaschen mit Casirivimab und Imdevimab vor der Verabreichung auf Feststoffe und Farbveränderungen inspizieren. Finden sich Feststoffe oder Farbveränderungen, so ist die Durchstechflasche zu verwerfen und stattdessen eine neue Durchstechflasche zu verwenden.• Die Lösung in jeder Durchstechflasche sollte klar bis leicht opaleszent, farblos bis hellgelb sein.
3.Es wird ein i. v. Fertiginfusionsbeutel (PVC oder PO) mit 50 ml, 100 ml, 150 ml oder 250 ml 0,9%iger Natriumchlorid-Injektionslösung oder 5%iger Dextrose-Injektionslösung benötigt.
4.Mithilfe einer sterilen Spritze und Nadel das erforderliche Volumen von Casirivimab und Imdevimab aus jeder Durchstechflasche aufziehen und in einen Fertiginfusionsbeutel mit 0,9%iger Natriumchlorid-Injektionslösung oder 5%iger Dextrose-Injektionslösung injizieren (siehe Rubrik «Dosierung/Anwendung», Tabelle 1).
5.Den Inhalt des Infusionsbeutels durch vorsichtiges Umdrehen mischen. Nicht schütteln.
6.Ronapreve enthält keine Konservierungsstoffe, daher sollte aus mikrobiologischen Gründen die verdünnte Infusionslösung sofort verabreicht werden.• Bei nicht sofortiger Verwendung liegen die Aufbewahrungsdauer und -bedingungen vor der Anwendung in der Verantwortung des Anwenders und sollen normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, ausser wenn die Verdünnung unter kontrollierten und validierten aseptischen Bedingungen erfolgte. Die chemische und physikalische Stabilität nach Entnahme aus der Durchstechflasche wurde für 20 Stunden bei Raumtemperatur (bis zu 25 °C) und 72 Stunden bei 2 °C bis 8 °C nachgewiesen. Bei Kühllagerung den i. v. Infusionsbeutel vor der Infusion etwa 30 Minuten lang die Raumtemperatur annehmen lassen.
Verabreichung als intravenöse Infusion
·Für die Infusion werden die folgenden Materialien empfohlen:
·Infusionsbesteck aus Polyvinylchlorid (PVC), mit Polyethylen (PE) beschichtetem PVC oder Polyurethan (PU).
·Inline- oder Add-on-Endfilter für i. v. Gabe mit 0,2 bis 5 μm Porengrösse aus Polyethersulfon, Polysulfon oder Polyamid.
·Infusionsbesteck am i. v. Infusionsbeutel anschliessen.
·Das Infusionsbesteck vorfüllen.
·Die gesamte Infusionslösung in dem Beutel über eine Pumpe oder durch Schwerkraft über einen intravenösen Zugang mit einem sterilen Inline- oder Add-on-Endfilter mit einer Porengrösse von 0,2 bis 5 μm aus Polyethersulfon, Polysulfon oder Polyamid infundieren (siehe Rubrik «Dosierung/Anwendung»).
·Die zubereitete Infusionslösung sollte nicht gleichzeitig mit anderen Arzneimitteln verabreicht werden. Die Kompatibilität der Casirivimab- und Imdevimab-Injektion mit anderen intravenösen Lösungen und Arzneimitteln als 0,9%iger Natriumchlorid- oder 5%iger Dextrose-Injektionslösung ist nicht bekannt.
·Nach Abschluss der Infusion den Schlauch mit 0,9%iger Natriumchlorid- oder 5%iger Dextrose-Injektionslösung spülen, um sicherzustellen, dass die erforderliche Dosis vollständig infundiert wird.
·Die Patienten bzw. Patientinnen sollten nach der intravenösen Infusion gemäss der vor Ort üblichen medizinischen Praxis überwacht werden.
Subkutane Injektion
Vorbereitung der subkutanen Injektion
Die Durchstechflasche(n) mit Casirivimab und Imdevimab aus der Kühllagerung nehmen und vor der Zubereitung während ungefähr 20 Minuten Raumtemperatur annehmen lassen. Keiner direkten Wärme aussetzen. Die Durchstechflaschen nicht schütteln.
Die Durchstechflasche(n) mit Casirivimab und Imdevimab vor der Verabreichung auf Feststoffe und Farbveränderungen inspizieren. Falls Feststoffe oder Farbveränderungen festgestellt werden, ist die Durchstechflasche zu verwerfen und stattdessen eine neue Durchstechflasche zu verwenden. Die Lösung in jeder Durchstechflasche sollte klar bis leicht opaleszent, farblos bis hellgelb sein.
1.Zur Vorbereitung von Ronapreve sollte die erforderliche Anzahl von Spritzen verwendet werden, (siehe Rubrik «Dosierung/Anwendung», Tabelle 2). Benötigt werden 3 ml- oder 5 ml-Luer-Lock-Spritzen aus Polypropylen mit Luer-Anschluss und 21 Gauge-Transferkanülen.
2.Mithilfe einer sterilen Spritze und Nadel das benötigte Volumen von Casirivimab und Imdevimab aus den jeweiligen Durchstechflaschen in jede Spritze aufziehen (siehe Rubrik «Dosierung/Anwendung», Tabelle 2). Dies ergibt insgesamt 4 Spritzen für die kombinierte Gesamtdosis von 1'200 mg und insgesamt 2 Spritzen für die kombinierte Gesamtdosis von 600 mg. Restmengen des Produkts in den Mehrfachdosen-Durchstechflaschen gemäss Anweisung aufbewahren (siehe Abschnitt «Haltbarkeit nach Anbruch»).
3.Für die subkutane Injektion die 21 Gauge-Transferkanüle durch eine 25 oder 27 Gauge-Kanüle ersetzen.
4.Dieses Arzneimittel enthält keine Konservierungsstoffe, daher sollten die vorbereiteten Spritzen sofort verwendet werden. Bei nicht sofortiger Verwendung liegen die Aufbewahrungsdauer und -bedingungen vor der Anwendung in der Verantwortung des Anwenders und sollen normalerweise 24 Stunden bei 2 °C bis 8 °C nicht überschreiten, ausser wenn die Zubereitung unter kontrollierten und validierten aseptischen Bedingungen erfolgte. Die chemische und physikalische Stabilität wurden für 24 Stunden bei Raumtemperatur (bis zu 25 °C) und 72 Stunden bei 2 °C bis 8 °C nachgewiesen. Bei Kühllagerung die Spritzen vor der Verabreichung während etwa 10 bis 15 Minuten die Raumtemperatur annehmen lassen.
Durchführung der subkutanen Injektion
·Für die subkutane Injektion der 1'200 mg-Dosis von Ronapreve (600 mg Casirivimab und 600 mg Imdevimab) 4 Spritzen bereitlegen (siehe Rubrik «Dosierung/Anwendung», Tabelle 2) und für die subkutanen Injektionen vorbereiten.
·Für die subkutane Injektion der 600 mg-Dosis von Ronapreve (300 mg Casirivimab und 300 mg Imdevimab) 2 Spritzen bereitlegen (siehe Rubrik «Dosierung/Anwendung», Tabelle 2) und für die subkutanen Injektionen vorbereiten.
·Die subkutanen Injektionen sind nacheinander jeweils an einer anderen Injektionsstelle zu verabreichen: Oberschenkel, Aussenseite der Oberarme oder Bauch ausser die Fläche 5 cm um den Nabel (periumbilikal) herum. Die Gürtellinie ist zu vermeiden.
·Die subkutanen Injektionen von je 2,5 ml Casirivimab und Imdevimab sollten jeweils in verschiedene Quadranten des Abdomens oder der Oberschenkel oder der Aussenseite der Oberarme erfolgen, um Abstände voneinander zu wahren. NICHT in empfindliche, geschädigte, blutunterlaufene oder vernarbte Haut injizieren.
Entsorgung von nicht verwendeten/abgelaufenen Medikamenten
Nicht verwendete Arzneimittel oder Abfallmaterial sind gemäss den Vorschriften vor Ort zu entsorgen.
Zulassungsnummer68329 (Swissmedic).
PackungenZusammen verpackte 20 ml-Mehrfachdosen-Durchstechflaschen:
2 Mehrfachdosen-Durchstechflaschen mit je 11,1 ml einer Injektions-/Infusionslösung [A]
Zusammen verpackte 6 ml-Durchstechflaschen zur Einmalverwendung
2 Einzeldosis-Durchstechflaschen mit je 2,5 ml einer Injektions-/Infusionslösung [A]
ZulassungsinhaberinRoche Pharma (Schweiz) AG, Basel.
Stand der InformationOktober 2023.
|