ZusammensetzungWirkstoffe
Fingolimod als Fingolimodhydrochlorid.
Hilfsstoffe
Kapseln zu 0.25 mg:
Carmellose-Calcium, Natriumstearylfumarat (entsprechend 0.030 mg Natrium)
Kapselhülle: Gelatine, Titandioxid (E171), gelbes Eisenoxid (E172)
Drucktinte: Schellack, schwarzes Eisenoxid (E172), Butylalkohol, gereinigtes Wasser, Propylenglycol, wasserfreies Ethanol, Isopropylalkohol, konzentrierte Ammoniak-Lösung.
Kapseln zu 0.5 mg:
Carmellose-Calcium, Natriumstearylfumarat (entsprechend 0.030 mg Natrium)
Kapselhülle: Gelatine, Titandioxid (E171), Tartrazin (E102) (0.028 mg), Gelborange S (E110) (0.003 mg)
Drucktinte: Schellack, schwarzes Eisenoxid (E172), Butylalkohol, gereinigtes Wasser, Propylenglycol, wasserfreies Ethanol, Isopropylalkohol, konzentrierte Ammoniak-Lösung.
Indikationen/AnwendungsmöglichkeitenFingolimod Devatis ist zur Behandlung von Erwachsenen, Jugendlichen und Kindern ab 10 Jahren mit schubförmig remittierend verlaufender Multipler Sklerose (MS) zur Reduzierung der Schubhäufigkeit und zur Verzögerung des Fortschreitens der Behinderung indiziert.
Dosierung/AnwendungAllgemeine Patientengruppen
Dosierung
Bei Erwachsenen beträgt die empfohlene Dosis von Fingolimod Devatis eine Kapsel mit 0.5 mg einmal täglich oral, die mit oder ohne Nahrung eingenommen werden kann.
Bei Kindern und Jugendlichen (ab 10 Jahren) hängt die empfohlene Dosis vom Körpergewicht ab:
·Kinder und Jugendliche mit einem Körpergewicht von 40 kg oder weniger: eine Kapsel zu 0.25 mg einmal täglich oral.
·Kinder und Jugendliche mit einem Körpergewicht von mehr als 40 kg: eine Kapsel zu 0.5 mg einmal täglich oral.
Kinder und Jugendliche, die zu Beginn der Behandlung Kapseln zu 0.25 mg erhalten haben, sollten bei Erreichen eines stabilen Körpergewichts von mehr als 40 kg auf Kapseln zu 0.5 mg umgestellt werden.
Wird eine Dosis ausgelassen, sollte die Behandlung mit der nächsten Dosis wie geplant fortgesetzt werden.
Für Empfehlungen zur Umstellung der Patienten von anderen krankheitsmodifizierenden Behandlungen und von anderen Immunsuppressiva auf Fingolimod Devatis siehe «Warnhinweise und Vorsichtsmassnahmen: Vorherige Behandlung mit Immunsuppressiva oder immun-modulierenden Therapien». Die Dauer der Wirkung dieser Arzneimittel muss bedacht werden, um additive immunsuppressive Effekte zu vermeiden (s. «Warnhinweise und Vorsichtsmassnahmen: Vorherige Behandlung mit immunsuppressiven oder immun-modulierenden Therapien»).
Vor Beginn der Behandlung
Ophthalmologische Beurteilung
Es soll eine augenärztliche Untersuchung des Augenhintergrunds, einschliesslich der Makula, durchgeführt werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Dermatologische Beurteilung
Es soll eine dermatologische Untersuchung durchgeführt werden. Verdächtige Hautläsionen müssen umgehend abgeklärt werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Therapieeinleitung
Bei allen Patienten muss vor Behandlungsbeginn und am Ende der sechsstündigen Überwachungsphase ein EKG durchgeführt werden. Mindestens während 6 Stunden nach Einnahme der Erstdosis sollten alle Patienten mit stündlichen Messungen von Puls und Blutdruck auf Hinweise für Bradykardie sowie atrioventrikuläre Überleitungsstörungen überwacht werden. Die Möglichkeit einer kardiologischen Notfallbehandlung muss gegeben sein. Eine kontinuierliche Echtzeit-EKG Überwachung während dieser ersten sechs Stunden nach Erstgabe von Fingolimod Devatis wird empfohlen.
Bei einer Umstellung der Tagesdosis von 0.25 mg auf 0.5 mg wird empfohlen, die Verabreichung der ersten erhöhten Dosis wie bei der Ersteinnahme bei Therapiebeginn zu überwachen.
Nach Therapieunterbruch wird eine Dosisüberwachung wie bei Einleitung der Behandlung empfohlen (s. «Warnhinweise und Vorsichtsmassnahmen»).
Eine verlängerte kardiale Überwachung über die ersten sechs Stunden nach Therapiebeginn hinaus ist für bestimmte Patienten erforderlich (s. auch nachstehende «Tabellarische Zusammenfassung – Überwachung nach Erstgabe» in dieser Rubrik und unter «Warnhinweise und Vorsichtsmassnahmen»). Darüber hinaus liegt es in der Verantwortung des behandelnden Arztes zu entscheiden, inwieweit ein Monitoring der Vitalparameter/des EKGs auch bei nachfolgenden Gaben erfolgen muss (s. «Warnhinweise und Vorsichtsmassnahmen»).
Die folgende Tabelle fasst die Massnahmen zur kardialen Überwachung nach Ersteinnahme von Fingolimod Devatis zusammen (s. auch «Warnhinweise und Vorsichtsmassnahmen»).
Tabelle 1 Überwachung nach Erstgabe von Fingolimod Devatis – Tabellarische Zusammenfassung
|
Bei allen Patienten
| |
sollte eine 6-stündige Überwachung auf Symptome einer Bradykardie sowie auf atrioventrikuläre Überleitungsstörungen wie folgt erfolgen:
·Stündliche Messungen von Puls und Blutdruck
·EKG vor Behandlungsbeginn und nach der 6-stündigen Überwachung
·Möglichkeit einer kardiologischen Notfallbehandlung
·Eine kontinuierliche (Echtzeit-) EKG-Überwachung wird empfohlen.
| |
Bei Patienten mit Auffälligkeiten in den ersten 6 Stunden nach Ersteinnahme
| |
Bei Auftreten von symptomatischen Bradyarrhythmien
|
ist der Patient nach der 6-stündigen Überwachungsphase bis zum vollständigen Abklingen der Symptome weiter zu überwachen.
| |
Wenn die Herzfrequenz 6 Stunden nach Ersteinnahme den niedrigsten Wert erreicht,
|
sollte das kardiale Monitoring bis zur Erholung der Herzfrequenz, mindestens jedoch um 2 Stunden verlängert werden.
| |
Wenn im EKG 6 Stunden nach der ersten Dosis einer der folgenden Befunde vorliegt:
·Herzfrequenz <45 Schläge pro Minute
·Persistierender neuer AV-Block 2.Grades oder höhergradiger AV-Block
·QTc-Intervall ≥500 ms
Wenn folgender EKG-Befund zu jedwedem Zeitpunkt während der Überwachungsphase nach Ersteinnahme vorliegt:
·neu aufgetretener AV-Block 3. Grades
|
ist die Verlängerung des kardialen Monitorings mindestens über Nacht angezeigt.
| |
Falls während der ersten Dosisgabe eine medikamentöse Behandlung Bradyarrhythmie-bedingter Symptome erforderlich ist, sollte der Patient bzw. die Patientin in einer medizinischen Einrichtung über Nacht beobachtet werden. Bei der Gabe der zweiten Dosis sollte die initiale Überwachungsstrategie angewendet werden.
| |
Bei Patienten mit kardialen Vorerkrankungen
| |
Fingolimod Devatis sollte für bestimmte Patientengruppen nur dann in Betracht gezogen werden, wenn der erwartete Nutzen die potentiellen Risiken überwiegt.
| |
Für prädisponierte Patienten mit:
·bekannter ischämischer Herzkrankheit (einschliesslich Angina pectoris)
·Stauungsinsuffizienz
·zerebrovaskulärer Krankheit
·unkontrolliertem Bluthochdruck
·schwerer unbehandelter Schlafapnoe
Sowie bei Patienten mit folgenden Erkrankungen in der Vorgeschichte:
·Myokardinfarkt
·Herzstillstand
·Rezidivierende Synkopen
·Symptomatische Bradykardie
|
sollte vor Behandlungsbeginn:
·ein Kardiologe konsultiert werden
·das geeignete kardiale Monitoring (mindestens über Nacht) festgelegt werden
| |
Bei Patienten unter Herzfrequenz-verlangsamenden Therapie
| |
Bei Patienten unter
·Betablockern
·Calciumkanalblockern (mit verlangsamender Wirkung auf die Herzfrequenz wie Verapamil, oder Dilitiazem)
·anderen Substanzen, die die Herzfrequenz reduzieren (z.B. Ivabradin, Digoxin, Azetylcholinesterasehemmer, Pilocarpin)
|
sollte, vor Behandlungsbeginn:
·ein Kardiologe konsultiert werden
zwecks Prüfung, ob eine Umstellung des Arzneimittels auf eine Substanz ohne Herzfrequenz verlangsamende Wirkung bzw. verzögernde Wirkung auf die AV-Überleitung möglich ist.
oder
·Falls eine Umstellung der Medikation nicht möglich ist, sollte das geeignete kardiale Monitoring, (inkl. kontinuierlichem EKG) mindestens über Nacht durchgeführt werden.
| |
Bei Patienten mit QT-Intervall Verlängerungen
| |
Bei Patienten mit:
·signifikanter QTc-Verlängerung (QTc >470 ms bei Frauen, QTc >450 ms bei Männern) vor Behandlungsbeginn
·zusätzlichen Risikofaktoren für das Auftreten einer QT-Verlängerung (wie z.B. Hypokaliämie, Hypomagnesiämie oder angeborenes Long-QT-Syndrom)
|
sollte vor Behandlungsbeginn:
·ein Kardiologe konsultiert werden
und
·das geeignete kardiale Monitoring (inkl. kontinuierliches EKG-Monitoring mindestens über Nacht in einer medizinischen Einrichtung) festgelegt werden.
|
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Bei Patienten mit leichter Leberinsuffizienz (Child-Pugh-Klasse A) sind keine Anpassungen der Fingolimod Devatis-Dosis notwendig, jedoch sollte die Behandlung mit Vorsicht erfolgen (s. «Warnhinweise und Vorsichtsmassnahmen, Leberfunktion» und «Pharmakokinetik»). Bei mittlerer (Child-Pugh-Klasse B) und schwerer Leberinsuffizienz (Child-Pugh-Klasse C) soll Fingolimod Devatis nicht verabreicht werden (s. «Kontraindikationen»).
Patienten mit Nierenfunktionsstörungen
Es stehen keine klinischen Daten zu Wirkung und Sicherheit bei Patienten mit Niereninsuffizienz zur Verfügung.
Ältere Patienten
Es liegen nur sehr limitierte klinische Daten für Patienten mit Multipler Sklerose im Alter von über 55 Jahren vor.
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Fingolimod bei Kindern unter 10 Jahren wurde bisher nicht untersucht. Fingolimod Devatis soll bei Kindern unter 10 Jahren nicht angewendet werden. In der Altersgruppe ≥10 und ≤12 Jahre stehen nur limitierte Daten zur Verfügung (s. «Klinische Wirksamkeit»).
Ethnische Zugehörigkeit
Es sind keine Anpassungen der Fingolimod Devatis-Dosis aufgrund der ethnischen Zugehörigkeit notwendig (s. «Pharmakokinetik»).
Geschlecht
Es sind keine Anpassungen der Fingolimod Devatis-Dosis aufgrund des Geschlechts notwendig (s. «Pharmakokinetik»).
Kontraindikationen·Patienten mit Myokardinfarkt, instabiler Angina pectoris, Schlaganfall/TIA, dekompensierter Herzinsuffizienz (stationär behandlungspflichtig), oder Herzinsuffizienz NYHA-Klasse III/IV in den vorangehenden sechs Monaten.
·Patienten mit schweren Herzrhythmusstörungen, die einer antiarrhythmischen Behandlung mit Klasse Ia und III Antiarrhythmika bedürfen (s. «Warnhinweise und Vorsichtsmassnahmen», «Interaktionen»).
·Patienten mit AV-Block 2. Grades vom Typ Mobitz II, oder AV-Block 3. Grades oder Sick-Sinus-Syndrom, sofern nicht Schrittmacher-versorgt.
·Patienten mit QTc-Intervall ab 500 ms bei Baseline (s. «Warnhinweise und Vorsichtsmassnahmen»).
·Patienten mit einem bestehenden Immundefizienzsyndrom.
·Patienten mit einem erhöhten Risiko für opportunistische Infektionen, einschliesslich derer, die derzeit eine immunsuppressive Therapie erhalten oder immungeschwächt sind.
·Patienten mit schweren aktiven Infektionen oder aktiven chronischen bakteriellen, mykotischen oder viralen Infektionen (z.B. Hepatitis, Tuberkulose).
·Patienten mit bestehenden aktiven malignen Erkrankungen, ausgenommen Patienten mit Basalzellkarzinom der Haut.
·Patienten mit mittlerer und schwerer Leberinsuffizienz/Leberzirrhose (entsprechend Child-Pugh-Klasse B und C).
·Patienten mit bestehendem Makulaödem.
·Fingolimod Devatis ist bei Patientinnen im gebärfähigen Alter ohne ausreichende Kontrazeption, sowie bei Schwangerschaft und in der Stillzeit kontraindiziert.
·Bekannte Überempfindlichkeit gegenüber Fingolimod oder einem der Hilfsstoffe.
Warnhinweise und VorsichtsmassnahmenBradyarrhythmie
Die Einleitung der Behandlung mit Fingolimod Devatis bewirkt eine vorübergehende Verlangsamung der Herzfrequenz und kann zudem mit einer Verzögerung der atrioventrikulären Überleitung assoziiert sein (s. «Unerwünschte Wirkungen» und «Pharmakodynamik»). Nach der ersten Dosis beginnt sich die Herzfrequenz innerhalb einer Stunde zu verringern, wobei der niedrigste Wert innert den ersten 6 Stunden bzw. bei einigen Patienten innerhalb von 24 Stunden erreicht wird. Aus diesem Grund sollten nach der Ersteinnahme von Fingolimod Devatis alle Patienten mindestens während den ersten 6 Stunden auf Symptome einer Bradykardie überwacht werden. Im weiteren Verlauf kehrt die Herzfrequenz bei kontinuierlicher Behandlung innerhalb eines Monats auf den Ausgangswert zurück (s. «Pharmakodynamik», Unterabschnitt «Herzfrequenz und Herzrhythmus»). Bei Patienten, die 0.5 mg Fingolimod erhalten, verlangsamt sich die Herzfrequenz um etwa 8 Schläge pro Minute (/Min.). Selten wurden Herzfrequenzen unter 40/Min. (bei Erwachsenen) und unter 50/Min. (bei Kindern und Jugendlichen) festgestellt (s. «Unerwünschte Wirkungen»). Patienten, bei denen eine Bradykardie auftrat, waren im Allgemeinen asymptomatisch, jedoch entwickelten einige Patienten leichte bis moderate Symptome wie Hypotension, Schwindel, Müdigkeit, Palpitationen und Schmerzen im Brustbereich, die üblicherweise innerhalb der ersten 24 Stunden der Behandlung wieder abklangen. Falls erforderlich, kann die Bradykardie mit der parenteralen Gabe von Atropin oder Isoprenalin behandelt werden.
Die Einleitung einer Behandlung mit Fingolimod wurde mit verzögerter atrioventrikulärer (AV-) Überleitung in Verbindung gebracht, in der Regel als AV-Block ersten Grades (verlängertes PR-Intervall im Elektrokardiogramm). Weniger als 0.2% der erwachsenen Patienten, die 0.5 mg Fingolimod erhielten, entwickelten einen atrioventrikulären Block zweiten Grades, üblicherweise vom Mobitz-Typ I (Wenckebach). Die Überleitungsanomalien waren meistens vorübergehend, asymptomatisch, erforderten in der Regel keine Behandlung und klangen innerhalb der ersten 24 Stunden der Behandlung wieder ab. Seit Markteinführung von Fingolimod sind einzelne Fälle eines vorübergehenden, spontan abklingenden, kompletten AV-Blocks berichtet worden (s. «Unerwünschte Wirkungen» und «Pharmakodynamik»).
Kardiale Überwachungsmassnahmen bei Erstgabe (s. auch «Tabellarische Zusammenfassung unter Dosierung/Anwendung»)
Bei allen Patienten sollte ein EKG vor der ersten Dosisgabe und am Ende des 6-stündigen Beobachtungszeitraums erstellt werden. Zu Beginn der Behandlung mit Fingolimod Devatis sollten alle Patienten über einen Zeitraum von 6 Stunden mit stündlichen Puls- und Blutdruckmessungen auf Symptome einer Bradykardie überwacht werden. Zudem wird eine kontinuierliche (Echtzeit-) EKG-Überwachung während der ersten sechs Stunden empfohlen.
Falls nach der Ersteinnahme symptomatische Bradyarrhythmien auftreten, sind angemessene Massnahmen zu ergreifen, und der Patient bzw. die Patientin ist über die sechsstündige Überwachungsphase hinaus weiter zu beobachten, bis die Symptome vollständig abgeklungen sind.
Falls ein Patient bzw. eine Patientin während des Beobachtungszeitraums nach der ersten Dosisgabe medikamentös behandelt werden muss, sollte er/sie über Nacht zur Beobachtung in einer medizinischen Einrichtung bleiben, und nach Gabe der zweiten Dosis Fingolimod Devatis sollte die gleiche Überwachungsstrategie wie nach der ersten Dosis angewendet werden.
Falls am Ende der sechsstündigen Überwachungsphase nach Erstgabe die Herzfrequenz den niedrigsten Wert nach Dosisgabe erreicht (was darauf schliessen lässt, dass sich die maximale pharmakodynamische Wirkung auf das Herz noch nicht eingestellt hat), sollte die Überwachung bis zur Erholung der Herzfrequenz, jedoch um mindestens zwei Stunden, verlängert werden.
Wenn bei Kindern und Jugendlichen die Tagesdosis von 0.25 mg auf 0.5 mg umgestellt wird, sollten dieselben Vorsichtsmassnahmen wie bei der Erstgabe ergriffen werden.
Darüber hinaus ist ein verlängertes kardiales Monitoring, jedoch mindestens über Nacht, bei Vorliegen eines der folgenden Kriterien angezeigt:
·neu aufgetretener AV-Block 3. Grades zu jedwedem Zeitpunkt während der Überwachungsphase nach Therapiebeginn
·Zum Zeitpunkt von 6 Stunden nach Therapiebeginn das Vorhandensein von:
·Herzfrequenz <45 Schläge pro Minute bei Erwachsenen, < 55 Schläge pro Minute bei Jugendlichen und Kindern ab 12 Jahren oder < 60 Schläge pro Minute bei Kindern ab 10 oder 11 Jahren der niedrigsten Herzfrequenz seit Beginn der Monitorisierung, so dass der maximale pharmakodynamische Effekt noch nicht eingetreten ist
·persistierender neuer AV-Block 2.Grades oder höhergradiger AV-Block
·QTc-Intervall ≥500 ms.
Fingolimod Devatis sollte in bestimmten Patientenpopulationen nur dann in Betracht gezogen werden, wenn der erwartete Nutzen die potentiellen Risiken überwiegt. Patienten mit bekannter ischämischer Herzkrankheit (einschliesslich Angina pectoris), Myokardinfarkt in der Vorgeschichte, Stauungsinsuffizienz und zerebrovaskulärer Krankheit können eine Bradykardie gegebenenfalls schlecht tolerieren. Falls eine Behandlung mit Fingolimod Devatis in Betracht gezogen wird, sollte vor Behandlungsbeginn ein Kardiologe konsultiert werden, um das geeignete kardiale Monitoring (mindestens über Nacht) festzulegen. (s. «Interaktionen»).
Aufgrund des Risikos für schwere Herzrhythmusstörungen sollte Fingolimod Devatis bei Patienten mit einem sinuatrialen Block bzw. mit symptomatischer Bradykardie oder rezidivierenden Synkopen in der Anamnese nicht angewendet werden.
Da eine signifikante Bradykardie von Patienten mit einem Herzstillstand, einer unkontrollierten Hypertonie oder einer schweren unbehandelten Schlafapnoe in der Anamnese möglicherweise schlecht toleriert wird, sollte Fingolimod Devatis bei diesen Patienten nicht angewendet werden.
Für Patienten, die mit Betablockern, Calciumkanalblockern mit verlangsamender Wirkung auf die Herzfrequenz (beispielsweise Verapamil oder Diltiazem) oder anderen Substanzen behandelt werden, die die Herzfrequenz reduzieren können (z.B. Ivabradin, Digoxin, Azetylcholinesterasehemmern, Pilocarpin), ist die Erfahrung mit Fingolimod begrenzt. Da die Einleitung der Behandlung mit Fingolimod Devatis ebenfalls mit einer Verlangsamung der Herzfrequenz einhergeht (s. «Bradyarrhythmie»), kann es bei gleichzeitiger Anwendung dieser Substanzen während der Therapieeinleitung mit Fingolimod Devatis zu einer schweren Bradykardie und Herzblock kommen. Wegen der potenziell additiven Wirkung auf die Herzfrequenz sollte bei Patienten, die gleichzeitig mit solchen Substanzen behandelt werden, generell von der Einleitung einer Behandlung mit Fingolimod Devatis abgesehen werden. Falls eine Behandlung mit Fingolimod Devatis in Erwägung gezogen wird, sollte ein Kardiologe konsultiert werden, um die Möglichkeit einer Umstellung auf Arzneimittel ohne verlangsamende Wirkung auf die Herzfrequenz bzw. verzögernde Wirkung auf die AV-Überleitung zu überprüfen bzw. die optimalen Überwachungsmassnahmen bei Therapieeinleitung festzulegen. Patienten, für die eine solche medikamentöse Umstellung nicht in Frage kommt, sollten mit kontinuierlichem EKG-Monitoring mindestens über Nacht überwacht werden (s. «Interaktionen»).
Die Auswirkung auf die Herzfrequenz und die atrioventrikuläre Überleitung können sich bei Wiederaufnahme der Fingolimod Devatis-Behandlung je nach Länge des Therapieunterbruchs und der Länge der zuvor durchgeführten Fingolimod Devatis-Behandlung wiederholen.
Dieselben Vorsichtsmassnahmen wie bei der Erstgabe werden empfohlen, bei Unterbruch der Therapie für:
·einen oder mehrere Tage während der ersten zwei Wochen der Behandlung
·mehr als sieben Tage während der dritten und vierten Behandlungswoche
·mehr als zwei Wochen nach dem ersten Monat der Behandlung
·Ist der Zeitraum der Behandlungsunterbrechung kürzer als oben genannt, sollte die Behandlung wie geplant mit der nächsten Dosis fortgesetzt werden.
QT-Verlängerung
Bei einigen Patienten wurden unter Fingolimod-Exposition Verlängerungen des QT-Intervalls beobachtet (Einzelne Patienten mit QTcF-Verlängerung zwischen 30 und 60 ms, keine QTcF-Verlängerung >60 ms und keine individuellen Werte von >500 ms). Patienten mit einem Risiko für eine QTc-Verlängerung wurden in den klinischen Studien nicht untersucht. Die klinische Relevanz dieser Befunde ist unklar.
Da die Einleitung einer Behandlung mit Fingolimod Devatis die Herzfrequenz verringert und das QT-Intervall verlängert, ist die Behandlung mit Fingolimod Devatis bei Patienten mit Baseline-QTc-Intervall ab 500 ms kontraindiziert (s. «Kontraindikationen»).
Bei folgenden Patientengruppen sollte eine Anwendung mit Fingolimod Devatis möglichst vermieden werden. Falls dennoch eine Behandlung mit Fingolimod Devatis in Betracht gezogen wird, sollte vorrangig ein Kardiologe konsultiert werden, um das geeignete kardiale Monitoring (inkl. kontinuierlicher EKG- Überwachung mindestens über Nacht in einer medizinischen Einrichtung) festzulegen:
·Patienten mit signifikanter QTc-Verlängerung (QTc >470 ms bei erwachsenen Frauen, QTc >460 ms bei Mädchen, QTc >450 ms bei erwachsenen Männern und bei Jungen) vor Behandlungsbeginn.
·Patienten mit zusätzlichen Risikofaktoren für das Auftreten einer QT-Verlängerung (wie z.B. Hypokaliämie, Hypomagnesiämie oder angeborenes Long-QT-Syndrom) (s. «Pharmakodynamik» und «Interaktionen»).
Eine Verlängerung des kardialen Monitorings ist mindestens über Nacht angezeigt bei Patienten mit einem QTc-Intervall ≥500 ms am Ende der sechsstündigen Überwachungsphase nach Therapiebeginn (s. «Dosierung/Anwendung»).
Fingolimod wurde nicht untersucht bei Patienten mit Arrhythmien, die eine Behandlung mit Antiarrhythmika der Klasse Ia (z.B. Chinidin, Procainamid) oder Klasse III (z.B. Amiodaron, Sotalol) benötigen. Antiarrhythmika der Klasse Ia und Klasse III sind unter anderem bei Bradykardie-Patienten mit Fällen von Torsades de Pointes in Zusammenhang gebracht worden. Da der Beginn einer Behandlung mit Fingolimod Devatis die Herzfrequenz verringert, darf Fingolimod Devatis nicht gemeinsam mit solchen Arzneimitteln verabreicht werden (s. «Kontraindikationen»).
Infektionen
Ein zentraler pharmakodynamischer Effekt von Fingolimod Devatis ist eine dosisabhängige Reduktion der peripheren Lymphozytenzahl auf 20-30% des Baseline-Wertes infolge der reversiblen Speicherung von Lymphozyten in den lymphatischen Geweben (s. «Pharmakokinetik»).
Aufgrund der Auswirkungen von Fingolimod Devatis auf das Immunsystem (s. «Pharmakokinetik») kann sich das Infektionsrisiko (einschliesslich opportunistischer Infekte) erhöhen (s. «Unerwünschte Wirkungen»).
Vor Einleitung einer Behandlung mit Fingolimod Devatis sollte ein kürzlich durchgeführtes grosses Blutbild (d.h. innerhalb von 6 Monaten oder nach Abbruch einer früheren Therapie) vorhanden sein.
Zusätzlich wird empfohlen, das grosse Blutbild inklusive Differentialblutbild in Monat 3 und danach regelmässig - mindestens jährlich - während der Behandlung sowie bei Anzeichen einer Infektion zu kontrollieren. Bei einer bestätigten Gesamtlymphozytenzahl < 0.1x109/l sollte die Behandlung bis zur Besserung pausiert werden. Bei einer Gesamtlymphozytenzahl < 0.2x109/l sollten engmaschige Kontrollen des Differentialblutbildes mindestens alle 3 Monate erfolgen.
Bei Patienten mit schweren aktiven Infektionen oder aktiven chronischen Infektionen sollte keine Therapie mit Fingolimod Devatis begonnen (s. «Kontraindikationen») bzw. mit einer Einleitung der Therapie bis zum Abklingen der Infektion abgewartet werden.
Bei Patienten, die während der Therapie Anzeichen einer Infektion entwickeln, sind daher unverzüglich geeignete diagnostische und therapeutische Massnahmen anzuwenden, insbesondere auch bei Verdacht auf das Vorliegen einer Infektion mit Viren der Herpesgruppe (u.a. Herpes simplex (HSV) und Varizella Zoster (VZV)) (s. «Unerwünschte Wirkungen»). Weil die Elimination von Fingolimod nach Therapieende bis zu zwei Monate andauern kann, muss die Überwachung hinsichtlich einer Infektion während dieses Zeitraums fortgeführt werden (s. nachstehenden Unterabschnitt: «Absetzen der Therapie»). Aufgrund des Risikos additiver Auswirkungen auf das Immunsystem sollte keine gleichzeitige Anwendung von antineoplastischen, immunsupprimierenden oder immunmodulierenden Therapien erfolgen. Spezifische Entscheidungen hinsichtlich der Dosierung und Dauer der Behandlung mit Kortikosteroiden sind nach klinischem Ermessen zu treffen. In den klinischen Phase-III-Studien hatte ein begleitend zu Fingolimod applizierter kurzer Kortikosteroidzyklus (bis zu 5 Tage je nach Studienprotokoll) im Placebovergleich keine Erhöhung der Infektionsgesamthäufigkeit zur Folge. Ausgehend von diesen Daten können in Kombination mit Fingolimod Devatis kurze Kortikosteroidzyklen (bis zu 5 Tage) durchgeführt werden (s. «Unerwünschte Wirkungen» und «Interaktionen»).
Patienten, die Fingolimod Devatis erhalten, sind anzuhalten, Infektionssymptome ihrem Arzt zu melden. Entwickelt ein Patient eine schwerwiegende Infektion, sollte eine Unterbrechung der Behandlung mit Fingolimod Devatis in Betracht gezogen und vor der Fortsetzung der Therapie eine Nutzen-Risiko-Abwägung vorgenommen werden.
Infektionen durch Viren der Herpes-Gruppe
In placebokontrollierten klinischen Studien entwickelten 9% der erwachsenen Patienten unter Fingolimod gegenüber 7% unter Placebo eine Herpesinfektion. Seit der Marktzulassung wurden teilweise schwerwiegende, lebensbedrohliche Fälle von Meningitis/Enzephalitis durch Varizella-Zoster-(VZV)- und Herpes-simplex-(HSV)-Viren, berichtet, die zu jedem Zeitpunkt der Therapie mit Fingolimod auftraten. Bei Auftreten lebensbedrohlicher Infektionen durch Viren der Herpes-Gruppe, wie einer Enzephalitis/Meningitis oder eines Multiorganversagens infolge einer disseminierten Infektion soll die Therapie mit Fingolimod Devatis abgesetzt und umgehend die entsprechende Diagnostik und Therapie in die Wege geleitet werden.
Vor der Behandlung mit Fingolimod Devatis sind die Patienten hinsichtlich ihrer Immunität gegenüber Varizella (Windpocken) zu beurteilen. Es wird empfohlen, bei Patienten ohne ärztlich bestätigte Windpocken in der Krankengeschichte oder Nachweis eines vollen Impfzyklus mit einem Varizella-Impfstoff vor Einleitung der Fingolimod Devatis-Therapie einen Test auf Antikörper gegen den Varizella-Zoster-Virus (VZV) durchzuführen. Bei antikörpernegativen Patienten wird vor Beginn der Behandlung mit Fingolimod Devatis ein vollständiger Immunisierungszyklus mit Varizella-Impfstoff empfohlen (s. «Unerwünschte Wirkungen»). Die Behandlung mit Fingolimod Devatis soll erst einen Monat nach der Impfung begonnen werden, um eine volle Wirkung der Impfung zu gewährleisten.
Darüber hinaus wurden seit der Marktzulassung Fälle von Kaposi-Sarkom, ausgelöst durch eine Infektion mit dem humanen Herpes Virus Typ 8 (HHV-8), beobachtet. Patienten mit Symptomen oder Zeichen eines Kaposi-Sarkoms sollen zeitnah einer weiteren Abklärung und Therapie zugeführt werden.
Progressive multifokale Leukoenzephalopathie
Seit Markteinführung wurden Fälle progressiver multifokaler Leukoenzephalopathie (PML) dokumentiert (s. «Unerwünschte Wirkungen»). PML ist eine opportunistische Infektion, die vom JC-Virus verursacht wird und tödlich verlaufen oder zu schwerer Behinderung führen kann.
PML kann nur bei einer vorliegenden JCV-Infektion auftreten. Wird ein JCV-Test durchgeführt, sollte berücksichtigt werden, dass der Einfluss einer Lymphopenie auf die Genauigkeit der anti-JCV-Antikörper-Tests bei Fingolimod-behandelten Patienten nicht untersucht wurde.
Es sollte auch darauf hingewiesen werden, dass ein negativer anti-JCV-Antikörper-Test die Möglichkeit einer anschliessenden JCV-Infektion nicht ausschliesst.
Zu Beginn einer Fingolimod-Behandlung wird eine MRI-Aufnahme (im Allgemeinen nicht älter als 3 Monate) als Referenz empfohlen. Bei Routine-MRI-Untersuchungen (gemäss den nationalen und lokalen Empfehlungen) sollten Ärzte auf Läsionen achten, die auf eine PML hindeuten könnten. Die MRI-Bildgebung sollte als Teil einer engen Überwachung von Patienten mit einem erhöhten PML-Risiko erwogen werden.
Ärzte sollten auf Symptome, wie Sprach- und Gangstörungen sowie Wesensveränderungen bzw. MRI-Befunde achten, die auf PML hindeuten.
Wird eine PML-Infektion vermutet, sollte zu diagnostischen Zwecken sofort eine MRI-Untersuchung durchgeführt und die Behandlung mit Fingolimod ausgesetzt werden, bis eine PML-Infektion ausgeschlossen werden konnte. Bei bestätigter PML sollte die Behandlung mit Fingolimod Devatis dauerhaft beendet werden.
MRI-Befunde, die auf eine PML hindeuten, können bereits vor dem Auftreten klinischer Anzeichen oder Symptome sichtbar sein. Fälle von PML, die auf der Grundlage von MRI-Befunden und dem Nachweis von JCV-DNA im Liquor bei Abwesenheit von klinischen Anzeichen oder spezifischen Symptomen für PML diagnostiziert wurden, wurden bei Patienten berichtet, die mit MS-Arzneimitteln behandelt wurden, die mit einem Risiko für eine PML assoziiert werden, wozu auch Fingolimod Devatis gehört.
PML-Fälle traten ohne Vorbehandlung mit Natalizumab nach einer ungefähren Behandlungsdauer von 2-3 Jahren auf. Das geschätzte Risiko scheint nach kumulierter Exposition im Laufe der Zeit zu steigen, ein genauer Zusammenhang mit der Behandlungsdauer ist aber nicht bekannt. Zusätzlich gab es PML Fälle in Patienten, welche zuvor mit Natalizumab behandelt wurden (Natalizumab ist mit einem erhöhten Risiko für eine PML assoziiert).
Die Inzidenzrate für PML scheint für Patienten in Japan höher zu sein; die Gründe sind zurzeit nicht bekannt.
Entzündliches Immunrekonstitutionssyndrom (engl.: Immune reconstitution inflammatory syndrome, IRIS) wurde bei Patienten berichtet, die mit S1P-Rezeptor-Modulatoren, darunter Fingolimod, behandelt wurden, bei denen eine PML auftrat und die anschliessend die Behandlung absetzten. IRIS äussert sich in einer möglicherweise schnell eintretenden klinischen Verschlechterung des Zustands des Patienten, kann zu schweren neurologischen Komplikationen oder zum Tod führen und geht häufig mit charakteristischen Veränderungen im MRT einher. Das Auftreten von IRIS bei Patienten mit PML erfolgte meist wenige Monate nach Absetzen des S1P-Rezeptor-Modulators. Es sollte überwacht werden, ob IRIS auftritt, und die damit verbundene Entzündung sollte angemessen behandelt werden.
Kryptokokkeninfektionen
Seit Markteinführung sind Fälle von Kryptokokkeninfektionen einschliesslich Kryptokokkenmeningitis berichtet worden (s. «Unerwünschte Wirkungen»). Die meisten Fälle traten nach einer Behandlung von ungefähr 2-3 Jahren auf. Ein genauer Zusammenhang mit der Behandlungsdauer ist jedoch nicht bekannt. Kryptokokkenmeningitis kann tödlich verlaufen. Aus diesem Grund sollten Patienten mit Symptomen und Anzeichen, die einer Kryptokokkenmeningitis entsprechen (Kopfschmerzen begleitet von Nackenstarre, Lichtempfindlichkeit, Übelkeit und/oder Verwirrtheit) umgehend diagnostisch beurteilt werden. Wird eine Kryptokokkenmeningitis diagnostiziert, ist eine geeignete Behandlung einzuleiten.
Infektionen mit humanem Papillomavirus
Infektionen mit humanem Papillomavirus (HPV), einschliesslich Papillom, Dysplasie, Warzen und HPV-bedingter Krebserkrankungen, wurden bei mit Fingolimod behandelten Patienten nach der Markteinführung berichtet (s. «Unerwünschte Wirkungen»). Aufgrund der immunsuppressiven Eigenschaften von Fingolimod sollte vor Beginn der Behandlung mit Fingolimod Devatis eine Impfung gegen HPV unter Berücksichtigung der Impfempfehlungen in Betracht gezogen werden. Eine Krebsvorsorge, einschliesslich Pap-Test wird gemäss Versorgungsstandard empfohlen.
Impfung
Während der Einnahme von Fingolimod Devatis und bis zu zwei Monate lang nach Absetzen der Behandlung kann die Wirksamkeit von Impfungen eingeschränkt sein (s. nachstehenden Unterabschnitt: «Absetzen der Therapie»). Die Anwendung von attenuierten Lebendimpfstoffen ist in den ersten zwei Monaten nach Absetzen der Behandlung mit Fingolimod Devatis zu vermeiden.
Zu Kinder und Jugendlichen beachten Sie bitte auch den Unterabschnitt «Kinder und Jugendliche».
Makulaödem
Bei 0.5% der mit 0.5 mg Fingolimod behandelten Patienten wurde über Makulaödem (s. «Unerwünschte Wirkungen») mit oder ohne visuelle Symptome berichtet, das hauptsächlich in den ersten 3-4 Therapiemonaten auftrat. Vor Beginn und 3-4 Monate nach Beginn einer Therapie mit Fingolimod Devatis muss eine augenärztliche Untersuchung mit Beurteilung des Augenhintergrundes, einschliesslich der Makula, durchgeführt werden. Alle 6 Monate sollte eine Visusuntersuchung durch den behandelnden Neurologen erfolgen. Wenn die Patienten zu einem beliebigen Zeitpunkt während der Fingolimod Devatis-Therapie über Sehstörungen klagen, sollte eine Untersuchung des Augenhintergrundes, einschliesslich der Makula, erfolgen. Patienten mit Diabetes mellitus oder Uveitis in der Krankengeschichte sowie Patienten mit einem Makulaödem in der Krankengeschichte sollten während ihrer Therapie mit Fingolimod Devatis regelmässig ophthalmologisch untersucht werden (s. «Kontraindikationen»).
Leberfunktion
Eine Erhöhung der Leberwerte, insbesondere der Alanin-Aminotransaminasen (ALT), aber auch der Gamma-Glutamyltransferase (γGT) und der Aspartat-Transaminase (AST), trat unter der Behandlung mit Fingolimod auf. So kam es in klinischen Studien bei erwachsenen MS Patienten in 8% (Placebo 1.9%) zu einer Erhöhung der ALT um mehr als das 3-Fache der oberen Normgrenze (ONG). Eine Erhöhung um mehr als das 5-Fache der ONG trat in den klinischen Studien unter Fingolimod bei 1.8% auf (Placebo 0.9%). Die Behandlung wurde in diesen Fällen abgesetzt. Bei erneuter Exposition gegenüber Fingolimod traten bei einigen Patienten erneut erhöhte Lebertransaminasen-Werte auf, was auf einen ursächlichen Zusammenhang mit Fingolimod hindeutet. Auch seit der Markteinführung wurden bei Patienten, die mit Fingolimod behandelt wurden, klinisch bedeutsame Leberschäden beobachtet. So wurde über Fälle von akutem Leberversagen berichtet, bei denen eine Lebertransplantation erforderlich wurde (s. «Unerwünschte Wirkungen»). Anzeichen einer Leberschädigung, einschliesslich deutlich erhöhter Serumtransaminasen und eines erhöhten Gesamtbilirubins, traten in einigen Fällen bereits 10 Tage nach der ersten Dosis auf, wurden aber auch nach längerer Anwendung gemeldet.
Vor Einleitung einer Behandlung mit Fingolimod Devatis soll daher eine Bestimmung der Transaminasen- und Bilirubinspiegel vorliegen, die nicht länger als 6 Monate zurückliegt. Während der Behandlung und bis zwei Monate nach dem Absetzen von Fingolimod Devatis sind auch bei fehlenden klinischen Hinweisen auf eine Leberschädigung regelmässige Kontrollbestimmungen angezeigt: Nach Beginn einer Therapie mit Fingolimod Devatis sollen nach 1, 3, 6, 9 und 12 Monaten Leberwertkontrollen durchgeführt werden; im weiteren Verlauf sollen Bestimmungen der Leberwerte auch bei Fehlen von klinischen Symptomen periodisch bis einschliesslich 2 Monate nach Beendigung der Therapie erfolgen.
Bei einem signifikanten Anstieg der Lebertransaminasen ohne begleitende klinische Symptome um mehr als das 3-Fache, aber weniger als das 5-Fach der oberen Normgrenze (ONG) und OHNE begleitende Bilirubinerhöhung sind häufigere laborchemische Leberwertkontrollen einschliesslich des Serumbilirubins und der alkalischen Phosphatase (AP) angezeigt, um einen weiteren Anstieg nachzuweisen. Alternative Ursachen für die Leberschädigung sollen begleitend abgeklärt werden. Bei einer Erhöhung der Transaminasen auf mindestens das 5-Fache der ONG oder einer Erhöhung auf das 3-Fache der ONG MIT begleitender Erhöhung des Bilirubins soll die Behandlung mit Fingolimod Devatis zunächst ausgesetzt und engmaschige Laborkontrollen der Leberwerte durchgeführt werden.
Die Behandlung soll nach Normalisierung der Leberwerte nur nach sorgfältiger Risiko-Nutzen-Analyse wiederaufgenommen werden, z.B., wenn eine plausible alternative Ätiologie für die Anzeichen und Symptome der Leberschädigung nachgewiesen werden konnte.
Die Patienten sollen zudem klinisch auf Anzeichen und Symptome einer Leberschädigung hin überwacht werden. Bei Patienten mit Symptomen, die auf eine Leberschädigung hinweisen können, wie z.B. eine ungeklärte Übelkeit, Erbrechen, Bauchschmerzen, Beschwerden im rechten Oberbauch, neu aufgetretene oder sich verschlimmernde Müdigkeit, Konzentrationsstörungen, Anorexie oder Gelbsucht und/oder dunkel verfärbter Urin soll umgehend eine Bestimmung der Leberenzyme und des Bilirubins erfolgen. Die Behandlung mit Fingolimod Devatis soll bei Bestätigung einer signifikanten Leberschädigung unterbrochen und nur bei Bestätigung einer plausiblen alternativen Ätiologie der Leberschädigung wiederaufgenommen werden. Obwohl keine Daten vorliegen, die belegen, dass Patienten mit vorbestehender Leberschädigung einem erhöhten Risiko für den Anstieg der Leberfunktionswerte unter Einnahme von Fingolimod unterliegen, ist bei Patienten mit anamnestisch bekannter schwerer Lebererkrankung Vorsicht geboten. Leberwertbestimmungen sollen auch bei Fehlen von klinischen Symptomen periodisch bis einschliesslich 2 Monate nach Beendigung der Therapie erfolgen. Die zusätzliche Einnahme potentiell lebertoxischer Medikamente/Substanzen (einschliesslich alkoholischer Getränke) soll vermieden werden. Patienten mit einer Leberzirrhose und Leberinsuffizienz (Child-Pugh-Klasse B und C) sollen nicht mit Fingolimod Devatis behandelt werden. Ebenso soll keine Behandlung von Patienten mit einer akuten oder chronisch aktiven Hepatitis B Infektion erfolgen, da die Gefahr einer Exazerbation der viralen Lebererkrankung besteht (s.a. «Kontraindikationen»).
Blutdruck
In klinischen Studien zu MS zeigten Patienten, die mit Fingolimod 0.5 mg behandelt wurden, einen durchschnittlichen Anstieg des systolischen Blutdrucks um etwa 3 mmHg und um etwa 1 mmHg des diastolischen Drucks, der erstmals ca. 1 Monat nach Behandlungsbeginn festzustellen war und während der Behandlung anhielt. Der Blutdruck sollte während der Behandlung mit Fingolimod Devatis regelmässig kontrolliert werden.
Posteriores reversibles Enzephalopathie-Syndrom
In klinischen Studien und im Rahmen der Anwendungsbeobachtung nach der Markteinführung ist in Zusammenhang mit der 0.5-mg-Dosis über seltene Fälle des posterioren reversiblen Enzephalopathie-Syndroms (PRES) bei Erwachsenen berichtet worden (s. «Unerwünschte Wirkungen»). Zu den berichteten Symptomen zählten plötzliches Auftreten starker Kopfschmerzen, Übelkeit, Erbrechen, veränderter Geisteszustand, Sehstörungen und Krämpfe. Die Symptome eines PRES sind in der Regel reversibel, können sich aber zu einem ischämischen Schlaganfall oder einer zerebralen Hämorrhagie weiterentwickeln. Eine verzögerte Diagnose und Behandlung kann zu chronischen neurologischen Folgeschäden führen. Bei Verdacht auf PRES darf Fingolimod Devatis nicht weiter angewendet werden.
Lungenfunktion
Eine dosisabhängige Reduktion von FEV1 und DLCO-Werten (Diffusionskapazität) wurde bereits im ersten Monat nach Therapiebeginn mit Fingolimod beobachtet, wobei die Werte danach stabil reduziert blieben. Nach 24-monatiger Therapie betrug die Reduktion des erwarteten FEV1 in Prozent der Basiswerte 2.7% für Fingolimod 0.5 mg und 1.2% für Placebo. Für DLCO betrugen die Reduktionen verglichen mit Baseline nach 24-monatiger Therapie 3.3% für Fingolimod 0.5 mg und 2.7% für Placebo. Die Veränderungen von FEV1 erscheinen nach Abbrechen der Therapie reversibel. Es gibt nur beschränkte Daten über die Reversibilität der DLCO Veränderung nach Abbrechen der Therapie. In kontrollierten klinischen Studien mit MS-Patienten trat Dyspnoe bei 5% unter Fingolimod 0.5 mg und bei 4% unter Placebo auf. Einige Patienten brachen die Behandlung mit Fingolimod aufgrund ungeklärter Dyspnoe in den (unkontrollierten) Extensionsstudien ab. Fingolimod wurde nicht in MS-Patienten mit eingeschränkter Lungenfunktion untersucht. Bei Auftreten von Symptomen, die auf eine pulmonologische Störung hinweisen, muss eine fachärztliche Untersuchung (inklusive Spirometrie und Bestimmung der DLCO) erfolgen.
Kutane Malignome
Basalzellkarzinome (BCC) und andere kutane Neoplasien wie malignes Melanom, Plattenepithelkarzinom, Kaposi-Sarkom und Merkelzellkarzinom wurden bei Patienten berichtet, die mit Fingolimod behandelt wurden (s. «Unerwünschte Wirkungen»).
Bei allen Patienten, insbesondere mit aber auch ohne erhöhtem Risiko für maligne kutane Neoplasien, sollen vor Beginn einer Therapie mit Fingolimod Devatis und im weiteren Verlauf regelmässige dermatologische Untersuchungen erfolgen. Verdächtige Hautläsionen müssen umgehend abgeklärt werden.
Da ein potenzielles Risiko für maligne kutane Neoplasien besteht, sollten Patienten, die mit Fingolimod Devatis behandelt werden, vor ungeschützter Exposition gegenüber Sonnenlicht gewarnt werden. Diese Patienten sollten keine gleichzeitige Phototherapie mit UV-B-Strahlung oder PUVA.Photochemotherapie erhalten.
Lymphome
Patienten, die Immunsuppressiva erhalten, sind in der Regel einem erhöhten Risiko ausgesetzt, an einem Lymphom oder anderen malignen Neoplasien zu erkranken. In klinischen Studien und nach der Markteinführung wurden Fälle von Lymphom berichtet. Die berichteten Fälle waren heterogener Natur, es handelte sich vor allem um Non-Hodgkin-Lymphome, einschliesslich B-Zell- und T-Zell-Lymphomen. Auch Fälle von kutanen T-Zell-Lymphomen (Mycosis fungoides) wurden beobachtet (siehe «Unerwünschte Wirkungen»).
Veränderung der Lymphozytenzahl
Basierend auf dem Wirkungsmechanismus führen 0.5 mg Fingolimod Devatis zur reversiblen Reduktion der Lymphozytenzahl um 70% des Steady-State-Wertes. Es sollten regelmässig Blutbildkontrollen durchgeführt werden.
Vorherige Behandlung mit immunsuppressiven oder immunmodulierenden Therapien
Es wurden keine klinischen Studien zur Beurteilung der Sicherheit und Wirksamkeit von Fingolimod nach Umstellung von Teriflunomid, Dimethylfumarat oder Alemtuzumab auf Fingolimod durchgeführt.
Bei der Umstellung von anderen krankheitsmodifizierenden Behandlungen müssen die Eliminationshalbwertszeit und die Wirkungsweise der anderen Therapie berücksichtigt werden, um einen additiven Immuneffekt zu vermeiden und gleichzeitig das Risiko einer Reaktivierung der Krankheit zu vermindern. Vor Einleitung einer Behandlung mit Fingolimod Devatis sollte ein kürzlich erstelltes grosses Blutbild (d.h. nach Absetzen der vorherigen Therapie) verfügbar sein, um sicher zu stellen, dass allfällige Immunwirkungen (d.h. Zytopenie) abgeklungen sind.
Interferon beta, Glatirameracetat oder Dimethylfumarat
Mit Fingolimod Devatis kann üblicherweise unmittelbar nach Absetzen von Interferon beta, Glatirameracetat oder Dimethylfumarat begonnen werden
Natalizumab oder Teriflunomid
Infolge der langen Eliminationshalbwertszeit von Natalizumab oder Teriflunomid ist bei der Umstellung der Patienten von diesen Therapien auf Fingolimod Devatis in Bezug auf mögliche additive Immunwirkungen Vorsicht geboten. Es wird empfohlen, den Zeitpunkt der Therapieeinleitung mit Fingolimod Devatis nach sorgfältiger Beurteilung von Fall zu Fall festzulegen.
Die Elimination von Natalizumab dauert nach Absetzen üblicherweise bis zu 2-3 Monate.
Teriflunomid wird ebenfalls langsam vom Plasma ausgeschieden. Ohne ein beschleunigtes Eliminationsverfahren kann die Clearance von Teriflunomid aus dem Plasma mehrere Monate bis zu 2 Jahre dauern. Ein beschleunigtes Eliminationsverfahren ist in der Arzneimittelfachinformation von Teriflunomid beschrieben.
Alemtuzumab
Aufgrund der Eigenschaften und der Dauer der immunsuppressiven Wirkung von Alemtuzumab, die in der Fachinformation beschrieben sind, wird der Beginn einer Behandlung mit Fingolimod Devatis nach Alemtuzumab nicht empfohlen, es sei denn, der Nutzen einer Behandlung mit Fingolimod Devatis überwiegt eindeutig die Risiken für den individuellen Patienten.
Rückkehr der Krankheitsaktivität (Rebound) nach Absetzen von Fingolimod Devatis
Nach der Markteinführung wurden Fälle von schwerer Exazerbation der Krankheit mit zum Teil fulminant verlaufenden Schubereignissen nach dem Absetzen von Fingolimod berichtet. Dies wurde in der Regel innerhalb von 12 Wochen nach dem Absetzen von Fingolimod beobachtet, aber auch bis zu 24 Wochen nach dem Absetzen von Fingolimod und darüber hinaus berichtet. Daher ist Vorsicht geboten, wenn die Behandlung mit Fingolimod Devatis abgesetzt wird (s. unten «Absetzen der Therapie»). Nach Absetzen von Fingolimod Devatis müssen die Patienten auf relevante Anzeichen und Symptome einer verstärkten Krankheitsaktivität überwacht werden. Bei Bedarf muss eine entsprechende Behandlung eingeleitet werden.
Überwachen Sie PML-Patienten nach dem Absetzen von Fingolimod Devatis auf das Auftreten eines entzündlichen Immunrekonstitutionssyndroms (PML-IRIS) (siehe Warnhinweis «Progressive multifokale Leukoenzephalopathie»).
Tumefaktive Läsionen
Nach der Markteinführung wurden seltene Fälle von tumefaktiven Läsionen im Zusammenhang mit MS-Schüben berichtet. Bei schweren Schüben sollte eine MRI-Untersuchung erfolgen, um tumefaktive Läsionen auszuschliessen. Das Absetzen der Behandlung mit Fingolimod Devatis sollte vom Arzt bzw. von der Ärztin im Einzelfall unter Berücksichtigung des Nutzens und der Risiken für den betroffenen Patienten geprüft werden.
Absetzen der Therapie
Wenn entschieden wird, die Behandlung mit Fingolimod Devatis abzusetzen, muss bedacht werden, dass Fingolimod nach der letzten Dosis bis zu zwei Monate lang im Blut bleibt und pharmakodynamische Auswirkungen hat, wie beispielsweise verringerte Lymphozytenwerte. Üblicherweise kehren die Lymphozytenwerte innerhalb von 1-2 Monaten nach Ende der Therapie wieder in den Normbereich zurück (s. «Pharmakokinetik»). Auch eine Überwachung auf Infektionen sollte über bis zu 2 Monate fortgesetzt werden und Patienten sollte in diesem Zeitraum weiterhin Anzeichen einer Infektion melden. Werden in diesem Zeitraum andere Therapien begonnen, kommt es zu einer gleichzeitigen Exposition gegenüber Fingolimod. Die Anwendung von Immunsuppressiva kurz nach Absetzen von Fingolimod Devatis kann zu einem additiven Effekt auf das Immunsystem führen, daher ist Vorsicht geboten (s. auch «Rückkehr der Krankheitsaktivität (Rebound) nach Absetzen von Fingolimod Devatis»). Auch die Kontrolle der Transaminasen und des Bilirubins soll bis einschliesslich 2 Monate nach Absetzen von Fingolimod Devatis fortgeführt werden.
Kinder und Jugendliche (ab 10 Jahren)
Das Sicherheitsprofil bei Kindern und Jugendlichen ist mit dem bei Erwachsenen vergleichbar, weshalb die Warnhinweise und Vorsichtsmassnahmen für Erwachsene auch für Kinder und Jugendliche gelten. Bei Verordnung von Fingolimod Devatis an Kinder und Jugendliche sollte insbesondere Folgendes beachtet werden:
·Bei der Gabe der ersten Dosis sind Vorsichtsmassnahmen zu befolgen (siehe «Bradyarrhythmie»). Dieselben Vorsichtsmassnahmen wie bei der ersten Einnahme werden auch empfohlen, wenn Patienten von der 0,25 mg Tagesdosis auf die 0,5 mg Tagesdosis umgestellt werden.
·In der kontrollierten pädiatrischen Studie D2311 wurden Krampfanfälle, Angstzustände, depressive Verstimmung und Depressionen bei den mit Fingolimod behandelten Patienten häufiger berichtet als bei den mit Interferon beta-1a behandelten Patienten. Daher ist in dieser Untergruppe von Patienten besondere Vorsicht geboten (siehe «Kinder und Jugendliche» in «Unerwünschte Wirkungen»).
·Bei Kindern und Jugendlichen unter Fingolimod wurden leichte isolierte Anstiege der Bilirubinwerte festgestellt.
·Es wird empfohlen, bei Kindern und Jugendlichen erst dann mit der Behandlung mit Fingolimod Devatis zu beginnen, wenn alle vorgesehenen Impfungen in Übereinstimmung mit den geltenden Impfrichtlinien durchgeführt wurden.
·Es liegen nur sehr begrenzte Daten zur Anwendung bei Kindern im Alter von 10 – 12 Jahren, Kindern mit weniger als 40 kg oder Kindern im Tanner-Stadium < 2 vor (siehe «Unerwünschte Wirkungen» und «Klinische Wirksamkeit»). Aufgrund der sehr begrenzten Erkenntnisse aus der klinischen Studie ist bei diesen Untergruppen von Kindern und Jugendlichen besondere Vorsicht geboten.
·Langzeitsicherheitsdaten bei Kindern und Jugendlichen liegen nicht vor.
Kinder und Jugendliche (unter 10 Jahren)
Die Sicherheit und Wirksamkeit bei Patienten unter 10 Jahren wurde nicht untersucht. Fingolimod Devatis soll bei Kindern unter 10 Jahren daher nicht angewendet werden.
Schwangerschaft, Frauen im gebärfähigen Alter, Risiko für den Fetus und Empfängnisverhütung
Fingolimod Devatis ist während der Schwangerschaft, bei Frauen im gebärfähigen Alter ohne ausreichende Kontrazeption und in der Stillzeit kontraindiziert (s. «Kontraindikationen»). Aufgrund des potenziellen hohen Risikos für den Fetus sollte bei Frauen im gebärfähigen Alter vor Beginn der Behandlung mit Fingolimod Devatis ein Schwangerschaftstest durchgeführt werden und negativ sein. Es sollte eine ärztliche Beratung bezüglich des Risikos schädlicher Auswirkungen auf den Fetus im Zusammenhang mit der Behandlung stattfinden. Während der Behandlung mit Fingolimod Devatis sollten Frauen nicht schwanger werden und die Anwendung einer wirksamen Methode zur Empfängnisverhütung ist während der Behandlung und für 2 Monate nach Beendigung der Behandlung durchzuführen. (s. «Kontraindikationen», «Schwangerschaft/Stillzeit» sowie den Abschnitt oben: «Rückkehr der Krankheitsaktivität (Rebound) nach Absetzen von Fingolimod Devatis»).
Fingolimod Devatis 0.5 mg enthält die Azofarbstoffe Tartrazin (E102) und Gelborange S (E110), welche allergische Reaktionen auslösen können.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Kapsel, d.h. es ist nahezu «natriumfrei».
InteraktionenPharmakokinetische Interaktionen
Fingolimod wird in erster Linie durch das Cytochrom-P450-4F2 (CYP4F2) und möglicherweise anderen CYP4F-Isoenzymen abgebaut. In vitro-Studien an Hepatozyten zeigten, dass CYP3A4 zum Metabolismus von Fingolimod beitragen kann, falls CYP3A4 stark stimuliert wird.
Potenzial von Fingolimod und Fingolimodphosphat zur Hemmung des Metabolismus gleichzeitig applizierter Arzneimittel:
In vitro-Inhibitionsstudien mit gepoolten humanen Lebermikrosomen und bestimmten metabolischen Sondensubstraten ergaben, dass Fingolimod und Fingolimodphosphat die Aktivität von CYP-Enzymen (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, oder CYP4A9/11 (nur Fingolimod)) kaum bzw. gar nicht hemmen können. Es ist daher unwahrscheinlich, dass Fingolimod und Fingolimodphosphat die Clearance von Wirkstoffen, die primär von den wichtigsten CYP Isoenzymen verstoffwechselt werden, verringern.
Potenzial von Fingolimod und Fingolimodphosphat zur Stimulation ihres eigenen Metabolismus und/oder des Metabolismus gleichzeitig applizierter Arzneimittel:
Fingolimod wurde hinsichtlich seines Potenzials zur Stimulation humaner CYP3A4-, CYP1A2-, CYP4F2- und ABCB1 (P-gp)-mRNA und der Aktivität von CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 und CYP4F2 in primären humanen Hepatozyten untersucht. Fingolimod bewirkte gegenüber der Vehikelkontrolle keine Stimulation der mRNA oder der Aktivität der verschiedenen CYP450-Enzyme und ABCB1. Deshalb wird bei therapeutischen Konzentrationen keine klinisch relevante Induktion der getesteten CYP450-Enzyme oder von ABCB1 (P-gp) durch Fingolimod erwartet. In vitro- Experimente zeigten keinen Hinweis auf CYP Induktion durch Fingolimodphosphat.
Potenzial von Fingolimod und Fingolimodphosphat zur Hemmung des aktiven Transports gleichzeitig applizierter Arzneimittel:
Basierend auf in vitro Daten, dürften Fingolimod wie auch Fingolimodphosphat keine Hemmwirkung auf die Aufnahme gleichzeitig applizierter Arzneimittel und/oder biologischer Wirkstoffe ausüben, die von Organo-Anion-Transporter (OATP1B1, OATP1B3) oder Natriumtaurocholate co-transportierende Polypeptide (NTCP) transportiert werden. Entsprechend wird davon ausgegangen, dass bei therapeutischen Konzentrationen auch keine Hemmung des Ausstroms gleichzeitig applizierter Arzneimittel und/oder biologischer Wirkstoffe erfolgt, die von BCRP (Breast Cancer Resistant-Protein), BSEP (Gallensalzexportpumpe) oder MRP2 (Multidrug Resistance-Associated-Protein 2) transportiert oder über P-glycoprotein (P-gp) herausgeschleust werden.
Orale Kontrazeptiva
Die gleichzeitige Anwendung von 0.5 mg/Tag Fingolimod und oralen Kontrazeptiva (Ethinylestradiol und Levonorgestrel) bewirkte keine Veränderung der Exposition gegenüber den oralen Kontrazeptiva. Die Exposition gegenüber Fingolimod und Fingolimodphosphat stimmte mit den in früheren Studien gemessenen Werten überein. Mit oralen Kontrazeptiva, die andere Progestagene enthalten, wurden keine Interaktionsstudien durchgeführt; eine Wirkung von Fingolimod auf die Exposition gegenüber diesen Substanzen wird jedoch nicht erwartet.
Cyclosporin
Die Pharmakokinetik einer Fingolimod-Einzeldosis war bei gleichzeitiger Applikation mit Cyclosporin im Steady-State unverändert. Auch umgekehrt ergaben sich bei Applikation einer Dosis oder mehrerer Dosen (28 Tage) von Fingolimod keine Veränderungen der Pharmakokinetik von Cyclosporin im Steady-State. Diese Daten deuten darauf hin, dass Fingolimod die Clearance von hauptsächlich über CYP3A4 eliminierten Arzneimitteln weder reduziert noch erhöht, und dass es unwahrscheinlich ist, dass die Inhibition von CYP3A4 die Clearance von Fingolimod reduziert. Die starke Inhibition der Transportermoleküle P-gp, MRP2 und OATP1B1 OATP-C beeinflusst die Disposition von Fingolimod nicht.
Ketoconazol
Die gleichzeitige Anwendung von Ketoconazol 200 mg zweimal täglich im Steady-State und einer Fingolimod-Einzeldosis von 5 mg führte zu einem Anstieg des AUC-Werts von Fingolimod und von Fingolimodphosphat (Anstieg um das 1.7-Fache) durch die Inhibition von CYP4F2.
Isoproterenol, Atropin, Atenolol und Diltiazem
Bei gleichzeitiger Applikation einer Einzeldosis von Fingolimod und Fingolimodphosphat und Isoproterenol oder Atropin wurden keine Veränderungen festgestellt. Entsprechend blieben die Pharmakokinetik von Fingolimod- und Fingolimodphosphat-Einzeldosen und die Steady-State-Pharmakokinetik von Atenolol und Diltiazem bei gleichzeitiger Anwendung der letztgenannten beiden Arzneimittel mit Fingolimod unverändert.
Carbamazepin
Die gleichzeitige Anwendung von Carbamazepin 600 mg zweimal täglich im Steady-State und einer Fingolimod- Einzeldosis von 2 mg, führte zu einem schwachen Effekt auf die AUC-Werte von Fingolimod und Fingolimod-Phosphat (bei beiden eine Abnahme von ca. 40%), was darauf hindeutet, dass die gleichzeitige Einnahme von Carbamazepin die Wirksamkeit von Fingolimod reduzieren kann.
Andere starke CYP3A4 Enzyminduktoren, z.B. Rifampicin, Phenobarbital, Phenytoin, Oxcarbazepin, Efavirenz und echtes Johanniskraut, können die AUC von Fingolimod und seinem Metaboliten mindestens in vergleichbarem Ausmass reduzieren und bei gleichzeitiger Einnahme entsprechend die Wirksamkeit von Fingolimod beeinträchtigen.
Labortests
Da Fingolimod die Anzahl der Lymphozyten im Blut durch Umverteilung in sekundäre lymphoide Organe verringert, können die peripheren Blutlymphozytenwerte nicht zur Beurteilung des Lymphozyten-Untergruppenstatus eines mit Fingolimod Devatis behandelten Patienten herangezogen werden.
Labortests, für die zirkulierende mononukleäre Zellen benötigt werden, erfordern wegen der Verringerung der Anzahl der Lymphozyten im Blutkreislauf grössere Blutmengen.
Pharmakodynamische Interaktionen
Eine gleichzeitige Anwendung antineoplastischer, immunmodulierender oder immunsupprimierender Therapien (inklusive Kortikosteroide) sollte wegen des Risikos additiver Auswirkungen auf das Immunsystem unterbleiben (s. «Warnhinweise und Vorsichtsmassnahmen»). Spezifische Entscheidungen über die Dosierung und die Dauer der gleichzeitigen Behandlung mit Kortikosteroiden, sollten auf einer klinischen Einschätzung beruhen. In den klinischen Phase-III-Studien hatte ein begleitend zu Fingolimod applizierter kurzer Kortikosteroidzyklus (bis zu 5 Tage je nach Studienprotokoll) im Placebovergleich keine Erhöhung der Infektionsgesamthäufigkeit zur Folge (s. «Warnhinweise und Vorsichtsmassnahmen und «Unerwünschte Wirkungen»).
Bei der Umstellung der Patienten von lang wirkenden Therapien mit Immun-Effekten wie Natalizumab, Teriflunomid oder Mitoxantron auf Fingolimod Devatis ist Vorsicht geboten (s. «Warnhinweise und Vorsichtsmassnahmen, Vorherige Behandlung mit immunsuppressiven oder immunmodulierenden Therapien»).
Wird Fingolimod zusammen mit Atenolol angewendet, kommt es nach Beginn der Behandlung mit Fingolimod zu einer zusätzlichen Verlangsamung der Herzfrequenz um 15%. Dieser Effekt wird mit Diltiazem nicht beobachtet.
Bei Patienten, die Betablocker, Calciumkanalblocker mit verlangsamender Wirkung auf die Herzfrequenz (beispielsweise Verapamil oder Diltiazem) oder andere Substanzen erhalten, die die Herzfrequenz verlangsamen können (z.B. Ivabradin, Digoxin, Azetylcholinesterasehemmer, Pilocarpin), sollte aufgrund der potenziell additiven Wirkung auf die Herzfrequenz keine Behandlung mit Fingolimod Devatis eingeleitet werden. Falls eine Behandlung mit Fingolimod Devatis in Erwägung gezogen wird, sollte hinsichtlich einer Umstellung auf Arzneimittel ohne verlangsamende Wirkung auf die Herzfrequenz bei Therapieeinleitung ein Kardiologe konsultiert werden. Patienten, welche nicht umgestellt werden können, sollten mindestens über Nacht mit einem kontinuierlichen EKG überwacht werden (s. «Warnhinweise und Vorsichtsmassnahmen» und «Dosierung/Anwendung»). Bei Einnahme von Klasse Ia - oder Klasse III-Antiarrhythmika ist Fingolimod Devatis kontraindiziert (s. «Kontraindikationen»).
Während und bis zu zwei Monate lang nach der Behandlung mit Fingolimod Devatis kann die Wirksamkeit von Impfungen eingeschränkt sein. Die Anwendung von attenuierten Lebendimpfstoffen kann ein Infektionsrisiko bergen und ist daher auch während der Behandlung mit Fingolimod Devatis und bis zu 2 Monate nach Abschluss der Behandlung mit Fingolimod Devatis zu vermeiden. (s. «Unerwünschte Wirkungen» und «Warnhinweise und Vorsichtsmassnahmen»).
Wirkung von Fingolimod Devatis auf andere Arzneimittel
Populationspharmakokinetische Analyse möglicher Arzneimittelwechselwirkungen
Eine bei Patienten mit Multipler Sklerose durchgeführte Untersuchung der Populationspharmakokinetik ergab keine Hinweise auf einen wesentlichen Einfluss von Fluoxetin und Paroxetin (starke CYP2D6-Inhibitoren) auf die Konzentrationen von Fingolimod oder Fingolimodphosphat. Durch die Gabe von Carbamazepin wird die Konzentration von Fingolimodphosphat um weniger als 30% reduziert. Darüber hinaus hatten die folgenden, häufig verschriebenen Substanzen keinen klinisch relevanten Effekt (≤20%) auf die Konzentrationen von Fingolimod oder Fingolimodphosphat: Baclofen, Gabapentin, Oxybutynin, Amantadin, Modafinil, Amitriptylin, Pregabalin, Kortikosteroide und orale Kontrazeptiva.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter / Empfängnisverhütung für Frauen
Fingolimod ist bei Frauen im gebärfähigen Alter ohne ausreichende Kontrazeption kontraindiziert (s. «Kontraindikationen»). Daher muss bei Frauen im gebärfähigen Alter vor Beginn einer Behandlung mit Fingolimod Devatis ein negatives Ergebnis eines Schwangerschaftstests vorliegen. Eine Aufklärung über die möglicherweise schwerwiegenden Folgen für das ungeborene Kind und die Notwendigkeit wirksamer Empfängnisverhütung während der Behandlung und für 2 Monate nach Beendigung der Behandlung mit Fingolimod Devatis muss erfolgen. Da die Elimination von Fingolimod Devatis aus dem Körper nach Abbruch der Behandlung etwa 2 Monate dauert (s. «Warnhinweise und Vorsichtsmassnahmen»), kann das Risikopotenzial für den Fetus andauern. Daher muss während dieses Zeitraums die Empfängnisverhütung fortgesetzt werden (s. «Kontraindikationen»).
Wird die Behandlung mit Fingolimod Devatis wegen einer Schwangerschaft oder geplanten Schwangerschaft abgebrochen siehe Abschnitte («Warnhinweise und Vorsichtsmassnahmen», Unterabschnitte «Rückkehr der Krankheitsaktivität (Rebound) nach Absetzen von Fingolimod Devatis» und «Absetzen der Therapie»).
Schwangerschaft
Während der Behandlung sollten Frauen nicht schwanger werden und eine effektive Methode zur Empfängnisverhütung muss angewandt werden (s. «Kontraindikationen»). Wenn eine Frau während der Einnahme von Fingolimod Devatis schwanger wird, soll Fingolimod Devatis nicht mehr eingenommen werden. In tierexperimentellen Studien zeigte sich Reproduktionstoxizität, einschliesslich Verlust und Organdefekte des Fetus, d.h. persistierender Truncus arteriosus und Ventrikelseptumdefekt (s. «Präklinische Daten»). Darüber hinaus ist der von Fingolimod beeinflusste Rezeptor (Sphingosin1-Phosphat-Rezeptor) bekanntermassen an der Gefässbildung während der Embryogenese beteiligt. Es ist derzeit nicht bekannt, ob beim Menschen kardiovaskuläre Fehlbildungen auftreten.
Es gibt keine adäquaten und gut-kontrollierten Studien mit Fingolimod in schwangeren Frauen.
Die verfügbaren Daten für die Anwendung beim Menschen (Daten nach der Markteinführung und Informationen aus dem Schwangerschaftsregister) deuten darauf hin, dass die Anwendung von Fingolimod mit einer erhöhten Prävalenz schwerer angeborener Fehlbildungen im Vergleich zur Allgemeinbevölkerung verbunden ist. Während der Behandlung sollten Frauen nicht schwanger werden und die Anwendung einer wirksamen Methode zur Empfängnisverhütung wird empfohlen. Sollte eine Frau während der Einnahme von Fingolimod Devatis schwanger werden, muss die Behandlung mit Fingolimod Devatis beendet werden. Die Patientinnen müssen über die schädlichen Wirkungen auf den Fetus aufgeklärt werden und eine ärztliche Nachsorgeuntersuchung sollte durchgeführt werden (z.B. Ultraschall). Dabei sollte auch die Möglichkeit einer schwerwiegenden Exazerbation der Erkrankung berücksichtigt werden, wenn Patientinnen die Behandlung mit Fingolimod Devatis aufgrund einer Schwangerschaft oder einer geplanten Schwangerschaft abbrechen. Die betroffenen Patientinnen sollten den behandelnden Arzt bzw. die behandelnde Ärztin bezüglich möglicher alternativer Behandlungen um Rat fragen (s. «Vorsichtsmassnahmen und Warnhinweise»).
Daten von Schwangerschaftsregistern in Kanada, Ländern der EU und Südamerika zeigen bislang, dass das Risiko von Geburtsfehlern in der MS Population ähnlich zum Risiko in der Gesamtbevölkerung ist. Aus den Daten eines Schwangerschaftsregisters in den USA erscheint das Risiko für Fehlgeburten und Totgeburten vergleichbar in MS Population und Allgemeinbevölkerung zu sein.
Bei über 600 prospektiven Schwangerschaften mit Lebendgeburten, Totgeburten oder Schwangerschaftsabbruch aufgrund einer fetalen Anomalie infolge einer Exposition der Mutter gegenüber Fingolimod während der Schwangerschaft, die nach der Markteinführung gemeldet wurden, lag der Anteil der schwerwiegenden angeborenen Fehlbildungen bei ca. 5 %. Die Prävalenz schwerwiegender angeborener Fehlbildungen in der Allgemeinbevölkerung liegt bei 2 bis 4 %. Das Muster der im Zusammenhang mit Fingolimod gemeldeten Fehlbildungen ist mit dem der Allgemeinbevölkerung vergleichbar. Die häufigsten schwerwiegenden Fehlbildungen sind:
·Angeborene Herzerkrankungen wie atriale und ventrikuläre Septumdefekte, Fallot-Tetralogie
·Nierenanomalien
·Anomalien des Bewegungsapparates
Es gibt keine Hinweise auf eine Häufung spezifischer Geburtsfehler mit Fingolimod Devatis.
Wehentätigkeit und Geburtsvorgang
Es liegen keine Daten zu den Auswirkungen von Fingolimod auf die Wehentätigkeit und den Geburtsvorgang vor.
Stillzeit
Fingolimod ist während der Stillzeit kontraindiziert (s. «Kontraindikationen»). Fingolimod geht während der Laktation in die Milch behandelter Tiere über. Es liegen keine Daten zu den Auswirkungen von Fingolimod auf gestillte Kinder oder auf die Milchproduktion vor. Da viele Wirkstoffe in die Muttermilch übergehen und wegen der Möglichkeit schwerwiegender unerwünschter Arzneimittelreaktionen beim gestillten Kind infolge der Fingolimodexposition dürfen Frauen, die mit Fingolimod Devatis behandelt werden, nicht stillen.
Schwangerschaftstest
Bei Frauen im gebärfähigen Alter sollte vor Beginn der Behandlung mit Fingolimod Devatis überprüft werden, ob eine Schwangerschaft vorliegt.
Fertilität
Tierexperimentelle Studien geben keinen Hinweis, dass Fingolimod mit einem erhöhten Risiko einer reduzierten Fruchtbarkeit verbunden wäre (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenFingolimod Devatis hat keinen oder vernachlässigbaren Einfluss auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen.
Unerwünschte WirkungenZusammenfassung des Sicherheitsprofils
Das zur Charakterisierung des Sicherheitsprofils von Fingolimod herangezogene Kollektiv stammt aus zwei placebokontrollierten klinischen Phase-III-Studien und einer aktiv kontrollierten klinischen Phase-III-Studie an erwachsenen Patienten mit schubförmig remittierender multipler Sklerose. Es umfasst insgesamt 2'431 erwachsene Patienten unter Behandlung mit Fingolimod (0.5-oder 1.25-mg-Dosis). Bei Studie D2301 (FREEDOMS) handelte es sich um eine placebokontrollierte klinische Studie über 2 Jahre bei 854 erwachsenen Patienten mit Multipler Sklerose, die mit Fingolimod behandelt wurden (Placebogruppe: 418 erwachsene Patienten). Studie D2309 (FREEDOMS II) war eine 2 Jahre laufende placebokontrollierte klinische Studie an 728 erwachsene Patienten mit multipler Sklerose unter Behandlung mit Fingolimod (Placebo: 355). In den gepoolten Daten aus diesen beiden Studien waren die schwerwiegendsten unerwünschten Reaktionen bei Anwendung der empfohlenen therapeutischen Dosis von 0.5 mg Infektionen, Makulaödem und transiente atrioventrikuläre Blocks nach Behandlungsbeginn. Die häufigsten unerwünschten Reaktionen (Inzidenz ≥10%) bei Gabe der 0.5-mg-Dosis waren Kopfschmerzen, Anstieg der Leberenzymwerte, Diarrhö, Husten, Influenza, Sinusitis und Rückenschmerzen. Das häufigste unerwünschte Ereignis, das bei Gabe von 0.5 mg Fingolimod mit einer Inzidenz über 1% auftrat und zu einem Behandlungsabbruch führte, waren ALT-Erhöhungen (2.2%).
Die unerwünschten Reaktionen in Studie D2302 (TRANSFORMS), einer aktiv kontrollierten Studie über 1 Jahr, in der 849 erwachsene Patienten mit Multipler Sklerose mit Fingolimod oder dem Vergleichspräparat Interferon beta-1a behandelt wurden, waren generell ähnlich wie in den placebokontrollierten Studien, wenn man die unterschiedliche Studiendauer berücksichtigt.
Häufigkeiten der unerwünschten Wirkungen aus den gepoolten Daten der beiden placebokontrollierten Studien FREEDOMS und FREEDOMS II
Die unerwünschten Reaktionen sind nach MedDRA-Systemorganklasse aufgeführt. Die Häufigkeiten waren wie folgt definiert: Sehr häufig (≥1/10); häufig (≥1/100 bis <1/10); gelegentlich (≥1/1'000 bis <1/100); selten (≥1/10'000 bis <1/1'000); «sehr selten» (<1/10'000). Innerhalb jeder Häufigkeitsgruppe sind die unerwünschten Reaktionen nach abnehmendem Schweregrad angegeben.
Infektionen und parasitäre Erkrankungen
Sehr häufig: Grippale Virusinfektionen (11%, Placebo: 8%), Sinusitis (11%, Placebo: 8%).
Häufig: Bronchitis, Herpes Zoster, Tinea versicolor.
Gelegentlich: Pneumonie.
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen)
Häufig: Basaliom (oder Basalzellkarzinom).
Gelegentlich: Malignes Melanom.
Selten: Lymphom, Plattenepithelkarzinom.
Sehr selten: Kaposi-Sarkom.
Erkrankungen des Blutes und des Lymphsystems
Häufig: Leukopenie, Lymphopenie.
Gelegentlich: Thrombozytopenie.
Psychiatrische Erkrankungen
Häufig: Depressionen.
Gelegentlich: Depressive Verstimmungen.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (25%, Placebo: 23%).
Häufig: Schwindel, Migräne.
Gelegentlich: Krampfanfälle.
Selten: Posteriores reversibles Enzephalopathie-Syndrom (PRES)*.
Augenerkrankungen
Häufig: Verschwommensehen.
Gelegentlich: Makulaödem.
Herzerkrankungen
Häufig: Bradykardie, atrioventrikuläre Blocks.
Gefässerkrankungen
Häufig: Hypertonie.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Husten (12%, Placebo: 11%).
Häufig: Atemnot.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Durchfall (13%, Placebo: 10%).
Leber– und Gallenerkrankungen
Sehr häufig: erhöhte Leberenzyme (erhöhte ALT, GGT, AST) (15%, Placebo: 4%).
Erkrankungen der Haut und des Unterhautgewebes
Häufig: Ekzem, Pruritus.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Rückenschmerzen (10%, Placebo: 9%).
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Asthenie.
Untersuchungen
Häufig: erhöhte Bluttriglyzeride.
* Nicht berichtet in den FREEDOMS, FREEDOMS II und TRANSFORMS Studien. Die Häufigkeitskategorie basiert auf der Inzidenz bei den ungefähr 10'000 Fingolimod-Patienten in allen klinischen Studien.
Unerwünschte Wirkungen nach Markteinführung
Infektionen und parasitäre Erkrankungen
Progressive multifokale Leukoenzephalopathie (PML).
Kryptokokkeninfektionen (einschliesslich Kryptokokkenmeningitis).
Infektionen einschl. Meningitis/Enzephalitis durch Viren der Herpesgruppe (v.a. Varizella-Zoster-Virus (VZV) und Herpes-simplex-Virus (HSV)).
Gutartige, bösartige und nicht spezifizierte Neubildungen (einschl. Zysten und Polypen)
Merkelzellkarzinom.
Erkrankungen des Immunsystems
Überempfindlichkeitsreaktionen wie u.a. Ausschlag, Nesselsucht und Angioödeme bei Einleitung der Behandlung, autoimmunhämolytische Anämie.
Erkrankungen des Nervensystems
Schwere Exazerbation der Krankheit nach Absetzen von Fingolimod Devatis (s. «Warnhinweise und Vorsichtsmassnahmen»).
Erkrankungen des Gastrointestinaltrakts
Übelkeit.
Leber- und Gallenerkrankungen
Leberschaden bis zum Leberversagen mit Transplantationspflichtigkeit.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Myalgie, Arthralgie.
Untersuchungen
Gewichtsabnahme.
Beschreibung ausgewählter Nebenwirkungen
Infektionen
In klinischen Studien bei Multipler Sklerose (MS) war die Gesamthäufigkeit von Infektionen (65.1%) bei Gabe der Dosis zu 0.5 mg ähnlich wie bei Placebogabe. Bronchitis, Herpes Zoster und Pneumonie traten jedoch bei mit Fingolimod behandelten Patienten häufiger auf. Schwerwiegende Infektionen traten in der mit 0.5 mg Fingolimod behandelten Gruppe mit einer Häufigkeit von 1.6% und in der Placebogruppe mit einer Häufigkeit von 1.4% auf.
In den klinischen Phase-III-Studien hatte ein begleitend zu Fingolimod angewendeter kurzer Kortikosteroidzyklus (bis zu 5 Tage gemäss Studienprotokollen) im Placebovergleich keine Erhöhung der Infektionsgesamthäufigkeit zur Folge (s. «Warnhinweise und Vorsichtsmassnahmen» und «Interaktionen»).
Infektionen mit dem humanen Papillomavirus (HPV), einschliesslich Papillomen, Dysplasien, Warzen und HPV-bedingten Krebserkrankungen, wurden während der Behandlung mit Fingolimod nach der Markteinführung berichtet (s. «Warnhinweise und Vorsichtsmassnahmen»).
Seit Markteinführung wurden Fälle von Infekten mit opportunistischen Erregern mit teilweise tödlichem Ausgang berichtet. Neben viralen Infektionen (z.B. PML durch das JC-Virus, Meningitis/Enzephalitis durch Viren der Herpes-Gruppe (v.a. Herpes simplex-Virus (HSV) und Varizella-Zoster-Virus (VZV), Kaposi-Sarkom durch humanes Herpes Virus Typ 8 (HHV8)) wurden mykotische (z.B. Meningitis/Enzephalitis durch Kryptokokken) und bakterielle Infektionen (z.B. atypische Mycobakterien) berichtet (s. «Warnhinweise und Vorsichtsmassnahmen»).
Makulaödem
In klinischen Studien traten Makulaödeme bei 0.5% der Patienten mit der empfohlenen Fingolimod-Dosis von 0.5 mg auf und bei 1.1% der Patienten, welche die höhere Dosis zu 1.25 mg erhielten.
Die meisten Fälle in den klinischen Studien bei Multipler Sklerose traten innerhalb der ersten 3-4 Therapiemonaten auf. Einige Patienten wurden mit Verschwommensehen oder verringerter Sehschärfe vorstellig, andere waren jedoch asymptomatisch und die Diagnosestellung erfolgte im Rahmen der augenärztlichen Routineuntersuchung. Das Makulaödem verbesserte sich im Allgemeinen oder klang nach Absetzen des Medikaments spontan ab. Das Rezidivrisiko nach Wiederaufnehmen der Behandlung wurde nicht untersucht.
Die Inzidenz von Makulaödem ist bei MS-Patienten mit Uveitis in der Anamnese erhöht (etwa 20% mit Uveitis in der Anamnese gegenüber 0.6% ohne Uveitis in der Anamnese).
Bradyarrhythmie
Die Einleitung einer Behandlung mit Fingolimod Devatis führt zu einer vorübergehenden Verlangsamung der Herzfrequenz und kann darüber hinaus mit einer Verzögerung der atrioventrikulären Überleitung einhergehen (s. «Dosierung/Anwendung», «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakodynamik»).
In klinischen Studien bei Multipler Sklerose erreichte die mittlere maximale Verlangsamung der Herzfrequenz innert 6 Stunden nach Gabe der ersten Dosis einen Höchstwert, wobei sich bei Gabe von 0.5 mg Fingolimod die mittlere Herzfrequenz um 8 Schläge pro Minute verlangsamte. Nach der zweiten Dosis kann eine weitere schwächere Verlangsamung eintreten. Herzfrequenzen unter 40 Schlägen/Minute waren bei Patienten, die Fingolimod 0.5 mg erhielten, selten. Innerhalb des ersten Monats unter konstanter Dosisgabe kehrte die Herzfrequenz wieder auf den Ausgangswert zurück.
In klinischen Studien trat nach Einleitung der Behandlung ein AV-Block ersten Grades (verlängertes PR-Intervall im Elektrokardiogramm) bei 4.7% der Patienten, die 0.5 mg Fingolimod erhielten, bei 2.8% der Patienten, die intramuskuläres Interferon beta-1a erhielten, und bei 1.6% der Patienten in der Placebogruppe auf. Ein AV-Block zweiten Grades wurde bei weniger als 0.2% der mit 0.5 mg Fingolimod behandelten Patienten festgestellt. Nach Markteinführung wurden isolierte Fälle eines transienten, spontan abklingenden, kompletten AV-Blocks während des sechsstündigen Beobachtungszeitraums nach Gabe von Fingolimod berichtet. Die in klinischen Studien und im Rahmen der Anwendung seit Markteinführung beobachteten Überleitungsanomalien waren in der Regel vorübergehend, asymptomatisch und klangen innerhalb von 24 Stunden nach Beginn der Behandlung ab. Bei den meisten Patienten war keine medizinische Intervention erforderlich, aber ein Patient, der die Dosis von 0.5 mg in der klinischen Studie erhalten hatte, wurde wegen eines asymptomatischen atrioventrikulären Blocks zweiten Grades vom Typ Mobitz I mit Isoprenalin behandelt.
Nach Markteinführung wurden isolierte Fälle von verzögert (innerhalb 24 Stunden) nach der ersten Dosis einsetzenden Ereignissen einschliesslich transienter Asystolie und Tod aus ungeklärter Ursache beobachtet. Die abschliessende Beurteilung dieser Fälle wurde aufgrund Begleitmedikationen und/oder Vorerkrankungen erschwert. Der Kausalzusammenhang solcher Ereignisse mit Fingolimod ist ungeklärt.
Blutdruck
In klinischen Studien bei Multipler Sklerose war 0.5 mg Fingolimod mit einem leichten Anstieg des mittleren arteriellen Drucks um durchschnittlich 1 mmHg assoziiert, der etwa 2 Monate nach Behandlungsbeginn festgestellt wurde und bei Fortsetzung der Behandlung persistierte. Hypertonie wurde bei 6.5% der mit 0.5 mg Fingolimod behandelten Patienten und bei 3.3% der Patienten in der Placebogruppe festgestellt.
Leberfunktion
Bei Multiple Sklerose Patienten, die mit Fingolimod behandelt wurden, wurden erhöhte Leberenzymwerte, grösstenteils der Alanin-Aminotransaminase (ALT), festgestellt. In klinischen Studien bei Multipler Sklerose entwickelten 8.0% bzw. 1.8% der mit 0.5 mg Fingolimod behandelten Patienten eine asymptomatische Erhöhung des Serumspiegels der ALT um mindestens das 3-Fache bzw. um mindestens das 5-Fache der oberen Normgrenze (ONG), in den meisten Fällen innerhalb von 6-9 Monaten. Nach Absetzen von Fingolimod kehrte der Serumspiegel der ALT innerhalb von etwa 2 Monaten in den Normbereich zurück. Bei einer kleinen Zahl von Patienten, d.h. bei 10 Patienten, die Fingolimod 1.25 mg erhielten, und bei 2 Patienten, die 0.5 mg Fingolimod erhielten, bei denen die Lebertransaminasen um ≥5x ONG anstiegen und welche die Fingolimod-Therapie fortsetzten, dauerte es etwa 5 Monate, bis sich die Werte wieder normalisierten.
Auch nach der Markteinführung wurden bei Patienten, die mit Fingolimod behandelt wurden, klinisch bedeutsame Leberschäden beobachtet (s. «Warnhinweise und Vorsichtsmassnahmen»). Anzeichen einer Leberschädigung, einschliesslich deutlich erhöhter Serumtransaminasen und eines erhöhten Gesamtbilirubins, traten in einigen Fällen bereits 10 Tage nach der ersten Dosis auf, wurden aber auch nach längerer Anwendung gemeldet. Es wurde auch über Fälle von akutem Leberversagen berichtet, bei denen eine Lebertransplantation erforderlich wurde.
Atmungsorgane
Unter der Therapie mit Fingolimod wurde eine dosisabhängige Reduktion von FEV1 und DLCO-Werten (Diffusionskapazität) beobachtet (s. «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Krampfanfälle
Sowohl in klinischen Studien als auch im Rahmen von Anwendungsbeobachtungen nach der Marktzulassung wurde über das Auftreten von Krampfanfällen inklusive Status epilepticus unter der Behandlung mit Fingolimod berichtet. Es ist nicht bekannt, ob diese Ereignisse allein mit den Symptomen der Multiplen Sklerose zusammenhingen oder mit Fingolimod, oder mit einer Kombination aus beidem.
Vaskuläre Ereignisse
In klinischen Phase-III-Studien traten bei Patienten, die mit höheren Fingolimod-Dosen (1.25 oder 5.0 mg) behandelt wurden, seltene Fälle von peripherer arterieller Verschlusskrankheit auf. Ausserdem wurden in klinischen Studien und im Rahmen der Anwendungsbeobachtung nach der Zulassung unter 0.5 mg Fingolimod seltene Fälle ischämischer oder hämorrhagischer Schlaganfälle beobachtet; ein Kausalzusammenhang wurde jedoch nicht nachgewiesen.
Kutane Neoplasien
In den gepoolten Daten der beiden placebokontrollierten klinischen Studien sind Basalzellkarzinome bei 14/783 (1.8%) unter Fingolimod 0.5 mg und bei 5/773 (0.6%) unter Placebo aufgetreten. Daneben sind andere Fälle von malignen, kutanen Neoplasien (z.B. Melanome, Kaposi-Sarkome) bei Patienten aufgetreten, die Fingolimod in klinischen Studien und nach der Markteinführung eingenommen haben. Bei Risikopatienten für maligne kutane Neoplasien sollten vor Beginn einer Therapie mit Fingolimod und im weiteren Verlauf regelmässige dermatologische Untersuchungen erfolgen.
Lymphom
In den klinischen Studien und nach der Markteinführung wurden Fälle von Lymphomen (einschliesslich eines tödlich verlaufenden Falles des Epstein-Barr-Virus (EBV)-positiven B-Zell-Lymphoms) beobachtet. Die berichteten Fälle waren heterogen, sie beinhalteten hauptsächlich Non-Hodgkin-Lymphome sowie B-Zell- und T-Zell-Lymphome. Die Inzidenz von B- und T-Zell-Lymphomen war in den klinischen Studien höher als in der Allgemeinbevölkerung erwartet. Es wurden Fälle von kutanem T-Zell-Lymphom (Mycosis fungoides) beobachtet.
Kinder und Jugendliche (ab 10 Jahren)
In einer kontrollierten pädiatrischen Studie war das Sicherheitsprofil bei Kindern und Jugendlichen (10 bis unter 18 Jahre), die Fingolimod 0.25 mg oder 0.5 mg einmal täglich erhielten, weitestgehend ähnlich wie bei erwachsenen Patienten.
Allerdings traten in der pädiatrischen Studie bei 5.6% der mit Fingolimod behandelten Patienten und bei 0.9% der mit Interferon beta-1a behandelten Patienten Krampfanfälle auf.
Es ist bekannt, dass Depressionen und Angstzustände bei Patienten mit Multipler Sklerose vermehrt auftreten. Bei pädiatrischen Patienten, die Fingolimod erhielten, wurden ebenfalls Depressionen und Angstzustände berichtet.
Leichte isolierte Anstiege der Bilirubinwerte wurden bei pädiatrischen Patienten unter Fingolimod festgestellt.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungEinzeldosen bis zum 80-Fachen der empfohlenen Dosen (0.5 mg) wurden bei gesunden Freiwilligen untersucht. Unter 40 mg berichteten 5 von 6 Probanden über ein leichtes Engegefühl oder Beschwerden im Brustkorb, die klinisch mit einer Reaktivität der kleinen Luftwege übereinstimmten.
Fingolimod kann eine Bradykardie hervorrufen und die AV-Überleitung verlangsamen. In der Regel beginnt die Abnahme der Herzfrequenz innerhalb einer Stunde nach der ersten Dosis und erreicht meist innerhalb von 6 Stunden ein Maximum. Im weiteren Verlauf kehrt bei kontinuierlicher Behandlung die Herzfrequenz in der Regel innerhalb eines Monats auf den Ausgangswert zurück (s. «Warnhinweise und Vorsichtsmassnahmen»). Es liegen Berichte über verlangsamte atrioventrikuläre Überleitung mit isolierten Berichten eines transienten, spontan abklingenden kompletten AV-Blocks vor (s. «Warnhinweise und Vorsichtsmassnahmen» und «Unerwünschte Wirkungen»).
Anzeichen und Symptome
Wenn eine Überdosierung mit Fingolimod Devatis erfolgt, ist es wichtig, auch auf Symptome einer Bradykardie/-arrhythmie zu achten. Erfolgte die Überdosierung zu Therapiebeginn ist es wichtig, die Patienten mindestens während der ersten sechs Stunden mit einem kontinuierlichen (Echtzeit-) EKG und stündlichen Messungen von Puls und Blutdruck zu überwachen, wobei die gleichen Massnahmen wie bei einer Erstdosisüberwachung gelten (s. «Tabelle 1 Überwachung nach Erstgabe von Fingolimod Devatis, Rubrik Dosierung/Anwendung» und «Warnhinweise und Vorsichtsmassnahmen»).
Behandlung
Weder eine Dialyse noch ein Plasma-Austausch führen zu einer relevanten Entfernung von Fingolimod aus dem Körper.
Eigenschaften/WirkungenATC-Code
L04AA27
Pharmakotherapeutische Gruppe: Sphingosin-1-Phosphat (S1P)-Rezeptor-Modulatoren
Wirkungsmechanismus
Fingolimod ist ein Modulator des Sphingosin1-Phosphat-Rezeptors. Der Wirkstoff wird von der Sphingosinkinase zu seinem aktiven Metaboliten Fingolimodphosphat umgesetzt. Fingolimodphosphat bindet im unteren nanomolaren Konzentrationsbereich an die Sphingosin1-Phosphat (S1P)-Rezeptoren 1, 3 und 4 auf Lymphozyten und nach problemloser Überwindung der Blut-Hirn-Schranke an S1P-Rezeptoren 1, 3 und 5 auf Nervenzellen im zentralen Nervensystem (ZNS). Indem es als funktioneller Antagonist des S1PR auf Lymphozyten wirkt, blockiert Fingolimodphosphat die Fähigkeit der Lymphozyten, die Lymphknoten zu verlassen, sodass es zu einer Umverteilung und nicht zu einer Depletion von Lymphozyten kommt. Diese Umverteilung reduziert die Infiltration pathogener Lymphozyten, einschliesslich proinflammatorischer Th17-Zellen, in das ZNS, wo sie an Nervenentzündungen und der Schädigung von Nervengewebe beteiligt sind. Tierexperimentelle Untersuchungen und in vitro-Experimente deuten darauf hin, dass die günstigen Auswirkungen von Fingolimod bei Multipler Sklerose auch auf eine Interaktion mit S1P-Rezeptoren auf Nervenzellen zurückzuführen sein könnten. Fingolimod dringt bei Menschen und Tieren in das ZNS ein und es wurde gezeigt, dass Astrogliose, Demyelinisierung und neuronaler Verlust reduziert werden. Ferner erhöht Fingolimod die Levels von BDNF (brain derived neurotrophic factor) im Cortex, Hippocampus und Striatum des Gehirns und unterstützt damit das neuronale Überleben und verbessert die motorischen Funktionen.
Pharmakodynamik
Immunsystem
Auswirkungen auf die Anzahl der Immunzellen im Blut. Innerhalb von 4-6 Stunden nach der ersten Fingolimod-Dosis von 0.5 mg verringert sich die Anzahl der Lymphozyten auf etwa 75% des Baseline-Werts. Wird die tägliche Dosisgabe fortgesetzt, fällt der Lymphozytenwert über einen Zeitraum von zwei Wochen weiter ab und erreicht schliesslich einen Tiefstwert von etwa 500 Zellen/µl bzw. etwa 30% des Baseline-Werts. Achtzehn Prozent der Patienten erreichten mindestens einmal einen Tiefstwert von unter 200 Zellen/µl. Bei anhaltender täglicher Dosisapplikation bleiben die niedrigen Lymphozytenwerte bestehen. Die meisten T- und B-Lymphozyten wandern regelmässig durch lymphoide Organe, sodass Fingolimod auf diese Zellen die stärksten Auswirkungen hat. Etwa 15-20% der T-Lymphozyten haben einen Effektor-Gedächtniszellen-Phänotyp, d.h. diese Zellen sind für die periphere Immunüberwachung wichtig. Da diese Lymphozyten-Untergruppe nicht durch lymphoide Organe wandert, wird sie von Fingolimod auch nicht beeinflusst. Nach dem Absetzen von Fingolimod kommt es innerhalb einiger Tage zu einem Anstieg der Anzahl der peripheren Lymphozyten, und üblicherweise ist innerhalb von ein bis zwei Monaten wieder der Normwert erreicht. Anhaltende Dosisapplikation von Fingolimod führt zu einer leichten Abnahme des Neutrophilenwerts auf etwa 80% des Baseline-Werts. Fingolimod hat keinen Einfluss auf Monozyten.
Herzfrequenz und Herzrhythmus
Fingolimod verursacht zu Beginn der Behandlung eine vorübergehende Verlangsamung der Herzfrequenz und der atrioventrikulären Überleitung (s. «Unerwünschte Wirkungen»). Die Verlangsamung der Herzfrequenz ist 4-5 Stunden nach Dosisgabe am stärksten ausgeprägt, wobei der negative chronotrope Effekt am ersten Tag zu 70% ausgeprägt ist. Die Herzfrequenz kehrt bei kontinuierlicher Behandlung innerhalb eines Monats meist wieder zum Baseline-Wert zurück.
Die Behandlung mit Fingolimod hat keinen Einfluss auf die autonomen Reaktionen des Herzmuskels, auch nicht auf die diurnale Schwankung der Herzfrequenz und die Reaktion auf körperliche Betätigung.
Bei Beginn der Behandlung mit Fingolimod kommt es zu einem Anstieg der Extrasystolen atrialen Ursprungs, aber nicht zu einer erhöhten Rate von Vorhofflimmern/-flattern oder ventrikulären Arrhythmien oder Ektopie. Die Behandlung mit Fingolimod bewirkt keine Verringerung des Herzausstosses.
Der von Fingolimod verursachten Verlangsamung der Herzfrequenz kann mit Atropin, Isoprenalin oder Salmeterol entgegengewirkt werden.
Potenzial zur Verlängerung des QT-Intervalls
In einer eingehenden Studie zum QT-Intervall mit Fingolimoddosen von 1.25 oder 2.5 mg im Steady-State, als der negative chronotrope Effekt von Fingolimod noch vorhanden war, führte die Behandlung mit Fingolimod zu einer Verlängerung des QTcI mit einer Obergrenze des 90% CI von ≤13.0 ms. Es gibt keine Dosis- oder Expositions-Wirkungsbeziehung zwischen Fingolimod und einer QTcI-Verlängerung, und kein einheitliches Signal für eine erhöhte Inzidenz von QTcI-Ausreissern, weder absolut noch als Veränderung gegenüber dem Baseline-Wert, in Zusammenhang mit der Fingolimod-Behandlung. Jedoch sind in Studie 1 im Rahmen der Erstabgabe von 0.5 mg Fingolimod bei 6.6% (Placebo 2.4%) und im weiteren Verlauf bei 13.9% (Placebo 6.7%) der Patienten QTcF-Verlängerungen zwischen 30-60 ms aufgetreten. Die klinische Relevanz ist nicht bekannt.
Lungensystem
Die Behandlung mit einer Einzeldosis oder mit Mehrfachdosen von Fingolimod 0.5 mg oder 1.25 mg für 2 Wochen ist nicht assoziiert mit einer nachweisbaren Erhöhung des Atemwiderstands, gemessen anhand des FEV1 oder FEF25-75. Einzeldosen von ≥5 mg (das 10-Fache der empfohlenen Dosierung) hingegen hatten einen dosisabhängigen Anstieg des Atemwiderstands zur Folge. Die Behandlung mit Mehrfachdosen von Fingolimod 0.5, 1.25 oder 5 mg hat keine beeinträchtigte Sauerstoffzufuhr, Sauerstoff-Entsättigung bei körperlicher Anstrengung oder einen Anstieg der Atemwegsempfindlichkeit auf Methacholin zur Folge. Patienten unter Behandlung mit Fingolimod reagieren mit normaler Empfindlichkeit auf inhalierbare Beta-Agonisten.
Klinische Wirksamkeit
Die Wirksamkeit von Fingolimod wurde in zwei Studien mit einmal täglicher Gabe von 0.5 mg und 1.25 mg Fingolimod an erwachsenen Patienten mit schubförmig-remittierender Multipler Sklerose aufgezeigt. Bei den Teilnehmern beider Studien handelte es sich um Patienten mit mindestens 2 klinischen Schüben in den 2 Jahren vor der Randomisierung oder mindestens 1 klinischen Schub in dem einen Jahr vor der Randomisierung und mit einem EDSS (Expanded Disability Status Scale)-Score zwischen 0 und 5.5. Eine dritte Studie mit der gleichen Art von Patienten, wurde nach der Registrierung von Fingolimod abgeschlossen.
Die Wirksamkeit und Sicherheit der einmal täglichen Gabe von Fingolimod in einer Dosis von 0.25 mg bzw. 0.5 mg (Auswahl der Dosis anhand des Körpergewichts und von Expositionsmessungen) wurde bei Kindern und Jugendlichen im Alter von 10 bis unter 18 Jahren mit schubförmig remittierender Multipler Sklerose untersucht.
Studie D2301 (FREEDOMS)
Studie D2301 (FREEDOMS) war eine 2-jährige, randomisierte, doppelblinde und placebokontrollierte Phase-III-Studie bei Patienten mit schubförmig verlaufender Multipler Sklerose, die während mindestens 3 Monaten vor Studienbeginn kein Interferon-beta oder Glatirameracetat und während mindestens 6 Monaten vor Studienbeginn kein Natalizumab erhalten hatten.
Der Medianwert des Alters war 37 Jahre, der Medianwert der Krankheitsdauer war 6.7 Jahre und der EDSS-Medianscore zum Baselinezeitpunkt war 2.0. Die Patienten erhielten nach der Randomisierung bis zu 24 Monate lang 0.5 mg Fingolimod (n=425) oder 1.25 mg Fingolimod (n=429) oder ein Placebo (n=418). Die mittlere Dauer der Behandlung mit der 0.5-mg-Dosis betrug 717 Tage, mit der 1.25-mg-Dosis betrug sie 715 Tage und die mittlere Dauer der Placebogabe war 718.5 Tage.
Der primäre Endpunkt war die jährliche Schubrate.
Die auf das Jahr umgerechnete Schubrate (ARR=annualized relapse rate) war bei mit Fingolimod behandelten Patienten statistisch signifikant niedriger als in der Placebogruppe. Der wichtigste sekundäre Endpunkt war der Zeitraum bis zu einer 3-monatigen bestätigten Progression der Behinderung, gemessen anhand einer Erhöhung des EDSS-Scores gegenüber dem Baseline-Wert um mindestens 1 Punkt (bei Patienten mit einem EDSS-Baseline-Wert von 5.5 anhand einer Erhöhung um mindestens 0.5 Punkte), die 3 Monate fortbestand. Der Zeitraum bis zum Einsetzen einer 3-monatigen bestätigten Progression der Behinderung war bei einer Behandlung mit Fingolimod im Placebovergleich statistisch signifikant verzögert. Bei keinem Endpunkt ergaben sich statistisch signifikante Unterschiede zwischen der Dosis von 0.5 mg und der Dosis von 1.25 mg.
Die Ergebnisse aus dieser Studie sind in Tabelle 2 und Abbildung 1 gezeigt.
Tabelle 2 Klinische Ergebnisse und MRT-Ergebnis in FREEDOMS Studie
|
|
Fingolimod 0.5 mg
|
Placebo
| |
Klinische Endpunkte
|
N=425
|
N=418
| |
Auf das Jahr umgerechnete Schubrate
(primärer Endpunkt)
|
0.18
(p<0.001*)
|
0.40
| |
Relative Reduktion (%)
|
54
|
| |
Anteil der schubfrei gebliebenen Patienten
zum 24-Monats-Zeitpunkt (%)
|
70.4
(p<0.001*)
|
45.6
| |
Risiko der Progression der Behinderung
| |
Hazard Ratio (95% CI)
(bestätigt, 3 Monate)
|
0.70 (0.52, 0.96)
(p=0.024*)
|
| |
Hazard Ratio (95% CI)
(bestätigt, 6 Monate)
|
0.63 (0.44, 0.90)
(p=0.012*)
|
| |
MRT-Endpunkte
| |
Anzahl der neuen oder neu vergrösserten T2-Läsionen
|
n=370
|
n=339
| |
Median-Wert (Durchschnitt) der Anzahl über 24 Monate
|
0.0 (2.5)
(p<0.001*)
|
5.0 (9.8)
| |
Anzahl der Gd-verstärkten Läsionen
|
n=369
(Monat 24)
|
n=332
(Monat 24)
| |
Median-Wert (Durchschnitt) der Anzahl in
| |
Monat 6
|
0.0 (0.2)
|
0.0 (1.3)
| |
Monat 12
|
0.0 (0.2)
|
0.0 (1.1)
| |
Monat 24
|
0.0 (0.2)
(p<0.001* an
jedem Zeitpunkt)
|
0.0 (1.1)
| |
Prozentuale Veränderung des Gesamtvolumens
der T2-Läsionen
|
n=368
|
n=339
| |
Median-Wert (Durchschnitt) der Veränderung
über 24 Monate in %
|
-1.7 (10.6)
(p<0.001*)
|
8.6 (33.8)
| |
Veränderung des Volumens der T1 hypointensen Läsionen
|
n=346
|
n=305
| |
Median-Wert (Durchschnitt) der Veränderung
über 24 Monate in %
|
0.0 (8.8)
(p=0.012*)
|
1.6 (50.7)
| |
Prozentuale Veränderung des Gehirnvolumens
|
n=357
|
n=331
| |
Median-Wert (Durchschnitt) der Veränderung
über 24 Monate in %
|
-0.7 (-0.8)
(p<0.001*)
|
-1.0 (-1.3)
|
Alle Analysen klinischer Endpunkte wurden in der Intent-to-Treat (ITT)-Population durchgeführt. Für die MRT-Analysen wurden auswertbare Datensätze herangezogen.
* Zeigt statistische Signifikanz im Placebovergleich mit zweiseitigem Signifikanzgrad von 0.05.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert
Abbildung 1 Kaplan-Meier-Grafik des Zeitraums bis zum ersten bestätigten Schub bis Monat 24 - FREEDOMS Studie (ITT-Population)
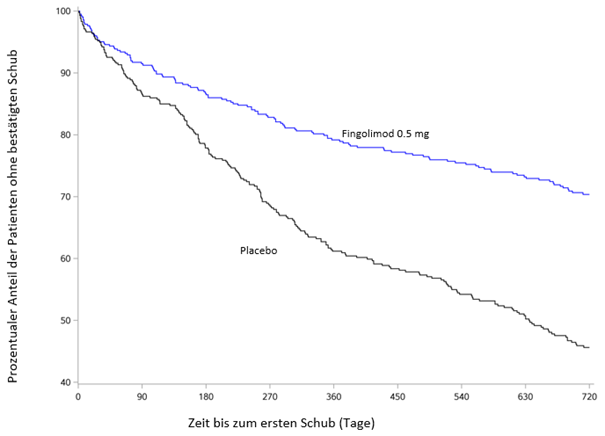
Patienten, welche die Studie FREEDOMS (D2301) abgeschlossen hatten, bot sich die Möglichkeit zur Teilnahme an der dosisverblindeten Verlängerungsstudie D2301E1. In diese Verlängerungsstudie wurden 920 Patienten aus der Hauptstudie aufgenommen, die alle mit Fingolimod behandelt wurden (n=331 setzten die Behandlung in der Dosis von 0.5 mg fort, 289 setzten die Behandlung in der Dosis von 1.25 mg fort, 155 wechselten von Placebo zur Dosis von 0.5 mg und 145 von Placebo zur Dosis von 1.25 mg). Nach 12 Monaten (Monat 36) waren noch 856 Patienten (93%) eingeschlossen. Von insgesamt 811 Patienten (88.2%) lagen Nachuntersuchungsdaten aus mindestens 18 Monaten in der Verlängerungsphase vor.
In Monat 24 der Verlängerungsstudie hatte sich bei den Patienten, die in der Hauptstudie der Placebo-Gruppe angehörten und dann auf 0.5 mg Fingolimod umgestellt wurden, die jährliche Schubrate (Annualized Relapse Rate, ARR) um 55% reduziert (ARR-Verhältnis 0.45, 95% KI 0.32 bis 0.62, p<0.001). Bei Patienten, die bereits in der Hauptstudie mit 0.5 mg Fingolimod behandelt worden waren, blieb die ARR während der Verlängerungsphase niedrig (ARR=0.10).
Zwischen den Monaten 24 und 36 betrug die jährliche Schubrate (ARR) 0.17 für Patienten unter 0.5 mg Fingolimod in der Hauptstudie, die weiterhin 0.5 mg einnahmen (0.21 in der Hauptstudie). Bei Patienten, die von Placebo auf 0.5 mg Fingolimod wechselten, betrug die ARR 0.22 (0.42 in der Hauptstudie).
Studie D2309 (FREEDOMS II)
In der replizierten zweijährigen, randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie mit 1083 Patienten mit schubförmig-remittierender Multipler Sklerose konnten vergleichbare Ergebnisse wie in Studie D2301 erzielt werden. Diese Studie wurde nach der Zulassung von Fingolimod fertiggestellt.
Das mediane Alter betrug 40.5 Jahre, die mediane Dauer der Erkrankung 8.9 Jahre und der mediane EDSS-Score zum Baseline-Zeitpunkt 2.5. Die Ergebnisse dieser Studie sind in Tabelle 3 und Abbildung 2 dargestellt.
Tabelle 3 Klinische Ergebnisse und MRT-Ergebnisse aus der Studie FREEDOMS II
|
|
0.5 mg Fingolimod
|
Placebo
| |
Klinische Endpunkte
|
N=358
|
N=355
| |
Jährliche Schubrate (primärer Endpunkt)
|
0.21
(p<0.001*)
|
0.40
| |
Relative Verringerung (in %)
|
48
|
| |
Anteil der nach 24 Monaten schubfreien Patienten (in %)
|
71.5
(p<0.001*)
|
52.7
| |
Risiko des Fortschreitens der Behinderung†
| |
Hazard Ratio (95% KI)
(bestätigt über 3 Monate)
|
0.83 (0.61, 1.12)
(p=0.227)
|
| |
Hazard Ratio (95% KI)
(bestätigt über 6 Monate)
|
0.72 (0.48, 1.07)
(p=0.113)
|
| |
MRT-Endpunkte
| |
Veränderung des Hirnvolumens (in %)
|
n=266
|
n=249
| |
Mediane (durchschnittliche) % Veränderung
über 24 Monate
|
-0.7 (-0.9)
(p<0.001*)
|
-1.0 (-1.3)
| |
Anzahl neuer oder sich neu vergrössernder T2-Läsionen
|
n=264
|
n=251
| |
Mediane (durchschnittliche) Anzahl über 24 Monate
|
0.0 (2.3)
(p<0.001*)
|
4.0 (8.9)
| |
Anzahl der Gd-anreichernden Läsionen
|
n=269
(Monat 24)
|
n=256
(Monat 24)
| |
Mediane (durchschnittliche) Anzahl in
| |
Monat 6
|
0.0 (0.2)
|
0.0 (1.1)
| |
Monat 12
|
0.0 (0.2)
|
0.0 (1.3)
| |
Monat 24
|
0.0 (0.4)
(p<0.001* zu
jedem Zeitpunkt)
|
0.0 (1.2)
| |
Veränderung des Gesamtvolumens an T2-Läsionen (in %)
|
n=262
|
n=247
| |
Mediane (durchschnittliche) % Veränderung
über 24 Monate
|
-7.1 (13.7)
(p<0.001*)
|
0.8 (25.1)
| |
Veränderung des Volumens an T1-hypointensen Läsionen
|
n=225
|
n=209
| |
Mediane (durchschnittliche) % Veränderung
über 24 Monate
|
-9.9 (12.6)
(p=0.372)
|
-8.5 (26.4)
|
Alle Analysen klinischer Endpunkte erfolgten im Intent-to-Treat-Kollektiv. Für die MRT-Analysen wurde der auswertbare Datensatz herangezogen.
* Statistische Signifikanz vs. Placebo auf dem zweiseitigen 0.05-Niveau.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert.
† Weitere Analysen ergaben, dass die Ergebnisse im Gesamtkollektiv aufgrund falschpositiver Progressionen in der Teilgruppe der Patienten mit einem Baseline-EDSS=0 (n=62, 8.7% des Studienkollektivs) nicht statistisch signifikant waren. Bei Patienten mit EDSS>0 (n=651; 91.3% des Studienkollektivs) ergab sich für 0.5 mg Fingolimod eine klinisch bedeutsame und statistisch signifikante Verringerung gegenüber dem Placebo (HR= 0.70; KI (0.50, 0.98); p=0.040), was mit der Studie FREEDOMS übereinstimmt.
Abbildung 2 Kaplan-Meier-Kurven der Zeit bis zum ersten bestätigten Schub bis Monat 24 – Studie FREEDOMS II (ITT-Kollektiv)
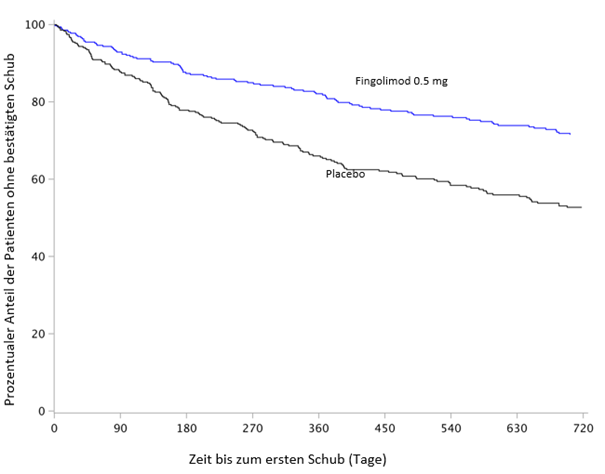
Bei keinem der Endpunkte wurden statistisch signifikante Unterschiede zwischen der 0.5 mg- und der 1.25 mg-Dosis festgestellt.
Studie D2302 (TRANSFORMS)
Bei Studie D2302 (TRANSFORMS) handelte es sich um eine einjährige, randomisierte, doppelblinde, aktiv (Interferon beta-1a, 30 Mikrogramm, intramuskulär, einmal wöchentlich) kontrollierte Phase-III-Studie im Double-Dummy-Design bei Patienten mit schubförmig-remittierend verlaufender MS, die während 6 Monaten vor Studienbeginn kein Natalizumab erhalten hatten. Eine vorhergehende Therapie mit Interferon-beta oder Glatirameracetat bis zum Randomisierungszeitpunkt war erlaubt.
Der Medianwert des Alters betrug 36 Jahre, der Medianwert der Krankheitsdauer betrug 5.9 Jahre und der EDSS-Medianscore zum Baselinezeitpunkt war 2.0. Die Patienten erhielten nach der Randomisierung bis zu 12 Monate lang 0.5 mg Fingolimod (n=431) oder 1.25 mg Fingolimod (n=426) oder 30 Mikrogramm Interferon beta-1a intramuskulär einmal wöchentlich (n=435). Der Medianwert der Dauer der Behandlung mit dem Prüfmedikament war 365 Tage (0.5 mg Fingolimod), 354 Tage (1.25 mg Fingolimod) und 361 Tage (Interferon beta-1a).
Die Ergebnisse aus dieser Studie sind in Tabelle 4 und in Abbildung 3 gezeigt.
Tabelle 4 Klinische Ergebnisse und MRT-Ergebnisse aus TRANSFORMS Studie
|
|
Fingolimod 0.5 mg
|
Interferon beta-1a, 30 μg
| |
Klinische Endpunkte
|
N=429
|
N=431
| |
Auf das Jahr umgerechnete Schubrate (primärer Endpunkt)
|
0.16
(p<0.001*)
|
0.33
| |
Relative Reduktion (%)
|
52
|
| |
Anteil der schubfrei gebliebenen Patienten
zum 12-Monats-Zeitpunkt (%)
|
82.5
(p<0.001*)
|
70.1
| |
Risiko der Progression der Behinderung
| |
Hazard Ratio (95% CI)
(bestätigt, 3 Monate)
|
0.71 (0.42, 1.21)
(p=0.209)
|
| |
MRT-Endpunkte
| |
Anzahl der neuen oder neu vergrösserten T2-Läsionen
|
n=380
|
n=365
| |
Median-Wert (Durchschnitt) der Anzahl über 12 Monate
|
0.0 (1.7)
(p=0.004*)
|
1.0 (2.6)
| |
Anzahl der Gd-verstärkten Läsionen
|
n=374
|
n=354
| |
Median-Wert (Durchschnitt) der Anzahl nach 12 Monaten
|
0.0 (0.2)
(p<0.001*)
|
0.0 (0.5)
| |
Prozentuale Veränderung des Gehirnvolumens
|
n=368
|
n=359
| |
Median-Wert (Durchschnitt) der Veränderung
über 12 Monate in %
|
-0.2 (-0.3)
(p<0.001*)
|
-0.4 (-0.5)
|
Alle Analysen klinischer Endpunkte wurden in der Intent-to-Treat (ITT)-Population durchgeführt. Für die MRT-Analysen wurden auswertbare Datensätze herangezogen.
* Zeigt statistische Signifikanz im Vergleich zu Interferon beta-1a, im, mit zweiseitigem Signifikanzgrad von 0.05.
Bestimmung von p-Werten: Analyse der ARR-Gesamtsumme durch negative binomiale Regression, angepasst nach Behandlung, Land (gepoolt), Anzahl der Schübe in den vorhergehenden 2 Jahren und EDSS-Baseline-Wert.
Abbildung 3 Kaplan-Meier-Grafik des Zeitraums bis zum ersten bestätigten Schub bis Monat 12 - TRANSFORMS Studie (ITT-Population)

Bei keinem Endpunkt ergaben sich statistisch signifikante Unterschiede zwischen der Dosis von 0.5 mg und der Dosis 1.25 mg.
Patienten, die die Studie TRANSFORMS (D2302) abgeschlossen hatten, bot sich die Möglichkeit zur Teilnahme an der dosisverblindeten Verlängerungsstudie (D2302E1). Insgesamt wurden 1'030 Patienten aus der Hauptstudie aufgenommen und mit Fingolimod behandelt (n=357 setzten die Behandlung in der Dosis von 0.5 mg fort, 330 setzten die Behandlung in der Dosis von 1.25 mg fort, 167 wechselten von Interferon beta-1a zur Dosis von 0.5 mg und 176 von Interferon beta-1a zur Dosis von 1.25 mg). Von 882 dieser Patienten (85.9%) lagen Nachuntersuchungsdaten aus mindestens 12 Monaten in der Verlängerungsphase vor.
In Monat 12 der Verlängerungsstudie hatte sich bei den Patienten, die in der Hauptstudie Interferon beta-1a i.m. erhalten hatten und dann auf 0.5 mg Fingolimod umgestellt wurden, die ARR relativ um 30% reduziert (ARR-Verhältnis=0.70, p=0.06). Bei Patienten, die bereits in der Hauptstudie mit 0.5 mg Fingolimod behandelt worden waren, blieb die ARR über die Haupt- und Verlängerungsphase hinweg niedrig (ARR=0.18 bis Monat 24).
Zwischen den Monaten 12 und 24 betrug die ARR für Patienten unter 0.5 mg Fingolimod in der Hauptstudie, die weiterhin 0.5 mg einnahmen 0.20 (0.19 in der Hauptstudie). Bei Patienten, die von Interferon beta-1a auf 0.5 mg Fingolimod wechselten, betrug die ARR 0.33 (0.48 in der Hauptstudie).
Insgesamt zeigten die Ergebnisse der Studien D2301 (FREEDOMS) und D2302 (TRANSFORMS) in den nach Geschlecht, Alter, vorheriger MS-Therapie, Krankheitsaktivität oder Behinderungsgrad zum Baselinezeitpunkt definierten Untergruppen eine einheitliche Reduktion der auf das Jahr umgerechneten Schubrate im Vergleich zum Vergleichspräparat.
Studie D2311 (PARADIGMS) an Kindern und Jugendlichen ab 10 Jahren
Bei der Studie D2311 (PARADIGMS) handelte es sich um eine doppelblinde, randomisierte, aktiv-kontrollierte, multizentrische Parallelgruppenstudie mit einer flexiblen Dauer von bis zu 24 Monaten zur Untersuchung der Wirksamkeit und Sicherheit von Fingolimod (n=107) im Vergleich mit Interferon beta-1a (n=107) bei Kindern und Jugendlichen mit schubförmig remittierender MS im Alter von 10 bis unter 18 Jahren. Eine Vorbehandlung mit Interferon-beta, Dimethylfumarat oder Glatirameracetat bis zum Zeitpunkt der Randomisierung war erlaubt. Eingeschlossen wurden Patienten mit mindestens einem klinischen Schub im Jahr zuvor oder mindestens 2 klinischen Schüben in den 2 Jahren vor der Randomisierung oder Nachweis ≥1 Gd verstärkenden Läsion im MRT innerhalb 6 Monate vor Randomisierung und mit einem EDSS zwischen 0 bis 5.5. Neurologische Untersuchungen wurden beim Screening sowie danach alle 3 Monate und bei einem vermuteten Schub durchgeführt. Die MRT-Beurteilungen erfolgten beim Screening und dann alle 6 Monate während der gesamten Studie. Der primäre Endpunkt war die auf das Jahr umgerechnete Schubrate.
Das mediane Alter betrug 16 Jahre, die mediane Krankheitsdauer seit dem Auftreten des ersten Symptoms lag bei 1.5 Jahren und der mediane EDSS-Score zur Baseline betrug 1.5. Die Patienten wurden randomisiert und erhielten entweder Fingolimod oder Interferon beta-1a, das einmal wöchentlich intramuskulär verabreicht wurde, für bis zu 24 Monate. In der Altersgruppe ≥10 und ≤12 Jahre (n=13 unter Fingolimod) sowie in der Gewichtskategorie ≤40kg (n=9 unter Fingolimod) wurden wenige Patienten eingeschlossen, so dass in diesen Patientengruppen nur limitierte Daten zur Beurteilung von Wirksamkeit und Sicherheit vorliegen. Die mediane Behandlungsdauer mit dem Studienmedikament betrug 634 Tage unter Fingolimod und 547 Tage unter Interferon beta-1a.
Der primäre Endpunkt, die auf das Jahr umgerechnete Schubrate (Annualized Relapse Rate, ARR) war bei den Patienten, die mit Fingolimod behandelt wurden, statistisch signifikant niedriger als bei denen, die Interferon beta-1a erhielten (relative Reduktion der ARR um 81.9 %). Der wichtigste sekundäre Endpunkt, die auf das Jahr umgerechnete Anzahl der neu aufgetretenen bzw. neu vergrösserten T2-Läsionen bis Monat 24, war ausserdem bei den Patienten, die mit Fingolimod behandelt wurden, statistisch signifikant niedriger als bei denen, die Interferon beta-1a erhielten, ebenso wie die Anzahl der vorhandenen Gd-anreichernden T1-Läsionen pro Scan bis Monat 24. Durch Fingolimod wurde zudem die auf das Jahr umgerechnete Hirnatrophie- Rate von der Baseline bis Monat 24 statistisch signifikant reduziert. Eine zusätzliche Post-hoc-Analyse zeigte, dass durch Fingolimod im Vergleich mit Interferon beta-1a der Zeitraum bis zum Einsetzen einer 3-monatigen bestätigten Progression der Behinderung statistisch signifikant verlängert wurde.
Die Ergebnisse dieser Studie sind in Tabelle 5, Abbildung 4 und Abbildung 5 dargestellt.
Tabelle 5 Klinische und MRT-Ergebnisse der PARADIGMS-Studie
|
|
Fingolimod
0.25 mg oder 0.5 mg
|
Interferon
beta-1a, 30 µg
| |
Klinische Endpunkte
|
N=107
|
N=107#
| |
Auf das Jahr umgerechnete Schubrate (primärer Endpunkt)
|
0.122
(p<0.001*)
|
0.675
| |
Relative Reduzierung (in Prozent)
|
81.9
|
| |
Anteil der bis Monat 24 schubfrei gebliebenen Patienten
(in %)
|
85.7
(p<0.001*)
|
38.8
| |
Risiko des Fortschreitens der Behinderung
| |
Hazard-Ratio (95% KI)
(bestätigt über 3 Monate)
|
0.23 (0.08,0.66)
(p=0.007*)
|
| |
Hazard Ratio (95%KI)
(bestätigt über 6 Monate)
|
0.20 (0.04; 0.93)
(p=0.040**)
|
| |
MRT-Endpunkte
| |
Auf das Jahr umgerechnete Anzahl der neu aufgetretenen bzw. neu vergrösserten T2-Läsionen
|
n=106
|
n=102
| |
Bereinigter Mittelwert
|
4.393
(p<0.001*)
|
9.269
| |
Relative Reduzierung (Prozent)
|
52.6
|
| |
Anzahl der Gd-anreichernden T1-Läsionen pro Scan
bis Monat 24
|
n=106
|
n=101
| |
Bereinigter Mittelwert
|
0.436
(p<0.001*)
|
1.282
| |
Relative Reduzierung (Prozent)
|
66.0
|
| |
Auf das Jahr umgerechnete Hirnatrophierate von der Baseline bis Monat 24
|
n=96
|
n=89
| |
Kleinste-Quadrate-Mittelwert
|
-0.48
(p=0.014*)
|
-0.80
|
Alle Analysen der klinischen Endpunkte erfolgten am vollständigen Analyseset. Für die MRT-Analysen wurde der auswertbare Datensatz verwendet.# Ein Patient wurde für die Behandlung mit Interferon beta-1a IM, 30 µg wöchentlich randomisiert, war aber nicht in der Lage, die im Rahmen des Double-Dummy-Verfahrens erforderliche zusätzliche Dosis Placebo zu schlucken und wurde aus der Studie herausgenommen. Dieser Patient wurde aus dem vollständigen Analyse- und Sicherheitsset ausgeschlossen.
* Zeigt die statistische Signifikanz im Vergleich mit Interferon beta-1a IM mit einem zweiseitigen Signifikanzniveau von 0.05.
** Post-hoc Analyse, Cox Proportional Hazard Model. p=0.180 im Log-rank Test.
Bestimmung der p-Werte: Aggregierte ARR: durch negative binomiale Regression, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifizierungsfaktor im interaktiven Sprachdialogsystem, IVRS) und Anzahl der Schübe in den vorangegangenen 2 Jahren (Offset: Zeit in der Studie); Prozentsatz der schubfrei gebliebenen Patienten: anhand einer Kaplan-Meier-Schätzung; Risiko der Progression der Behinderung: anhand des proportionalen Hazardmodells nach Cox, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und Anzahl der Schübe in den vorangegangenen 2 Jahren; auf das Jahr umgerechnete Anzahl der neu aufgetretenen/neu vergrösserten T2-Läsionen: durch negative binomiale Regression, angepasst nach Behandlung, Region, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und Anzahl der T2-Läsionen zur Baseline (Offset: Zeit in der Studie); Anzahl der Gd-anreichernden Läsionen pro Scan: durch negative binomiale Regression mit der kumulativen Anzahl der Gd-anreichernden T1-Läsionen auf allen während der Studie planmässig nach der Baseline durchgeführten MRT-Scans als Reaktionsvariable, angepasst nach Behandlung, Land, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und der Anzahl der Gd-anreichernden T1-Läsionen zur Baseline (Offset: Anzahl der MRT-Scans); auf das Jahr umgerechnete Hirnatrophierate: anhand eines ANCOVA-Modells, angepasst nach Behandlung, Region, Pubertätsstatus (der Stratifikationsfaktor im IVRS) und des Gesamt-Hirnvolumens zur Baseline
Abb. 4 Kaplan-Meier-Grafik für den Zeitraum bis zum ersten bestätigten Schub bis Monat 24 - PARADIGMS-Studie (vollständiges Analyseset)
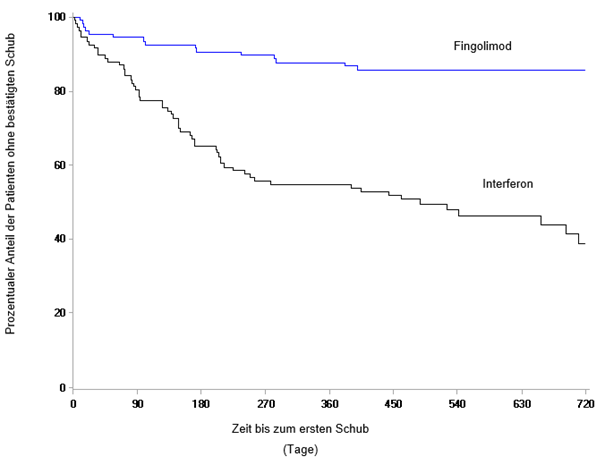
Abb. 5 Kaplan-Meier-Grafik des Zeitraums bis zur 6-monatigen bestätigten Progression der Behinderung - PARADIGMS-Studie (vollständiges Analyseset
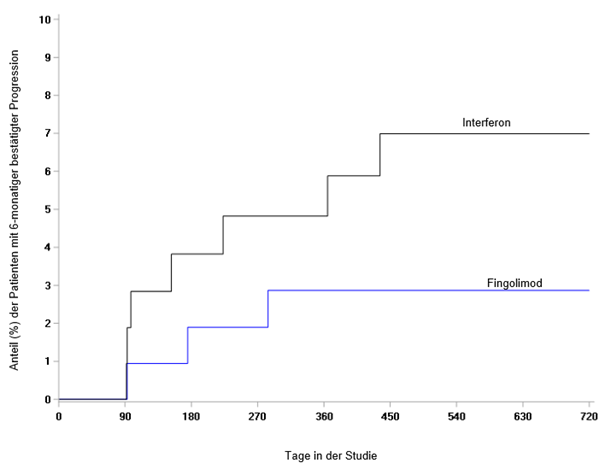
PharmakokinetikAbsorption
Fingolimod wird langsam (tmax von 12-16 Stunden) und in hohem Mass (≥85%, ausgehend von der im Urin ausgeschiedenen Menge an Radioaktivität und der in den Fäzes ausgeschiedenen Menge der Metaboliten, auf Unendlichkeit extrapoliert) absorbiert. Die scheinbare absolute orale Bioverfügbarkeit ist hoch (93%).
Nahrungsmittelaufnahme verändert weder den Cmax noch den Expositionswert (AUC) von Fingolimod, Fingolimod Devatis kann daher unabhängig von den Mahlzeiten eingenommen werden (s. «Dosierung/Anwendung»).
Die Steady-State-Blutkonzentrationen sind bei einmal täglicher Applikation innerhalb von 1 bis 2 Monaten erreicht und etwa 10 Mal höher als nach der Anfangsdosis.
Distribution
Fingolimod wird in hohem Mass in Erythrozyten verteilt, wobei die Fraktion in Blutzellen 86% beträgt. Fingolimodphosphat wird nur zu <17% in Blutzellen aufgenommen. Fingolimod und Fingolimodphosphat sind weitgehend proteingebunden (>99.7%). Die Proteinbindung von Fingolimod und Fingolimodphosphat wird durch Nieren- oder Leberinsuffizienz nicht verändert.
Fingolimod wird in hohem Mass in die Körpergewebe verteilt, wobei das Verteilungsvolumen etwa 1'200±260 l beträgt. Eine Studie mit vier gesunden Freiwilligen, die mit radioaktivem Iod markiertes Fingolimod als intravenöse Einzeldosis erhielten, ergab, dass Fingolimod ins Gehirn übergeht. In einer Studie an 13 männlichen Patienten mit multipler Sklerose, die 0.5 mg Fingolimod pro Tag im Steady-State erhielten, war die Menge an Fingolimod (und Fingolimodphosphat) im Ejakulat über 10'000 Mal niedriger als die verabreichte Dosis (0.5 mg).
Metabolismus
Bei Menschen gibt es drei Hauptwege zur Biotransformation von Fingolimod: durch reversible, stereoselektive Phosphorylierung zum pharmakologisch aktiven (S)-Enantiomer von Fingolimodphosphat, durch oxidative Biotransformation, hauptsächlich katalysiert durch CYP4F2 und möglicherweise anderen CYP4F Isoenzymen, und anschliessenden fettsäureähnlichen Abbau zu inaktiven Metaboliten und schliesslich durch Bildung von pharmakologisch inaktiven unpolaren Ceramidanalogen von Fingolimod.
Nach oraler Einzelgabe von [14C]-Fingolimod sind Fingolimod selber (23.3%), Fingolimodphosphat (10.3%) und inaktive Metaboliten (M3-Carbonsäuremetabolit (8.3%), M29-Ceramidmetabolit (8.9%) und M30-Ceramidmetabolit (7.3%)) die primären Fingolimod-bedingten Komponenten im Blut, wie anhand ihres Beitrags zum AUC bis zu 816 Stunden nach Dosisapplikation relativ zum Gesamtbeitrag aller radioaktiv markierten Komponenten festgestellt wurde.
Elimination
Die Blutclearance von Fingolimod beträgt 6.3±2.3 l/Std., und die durchschnittliche scheinbare terminale Eliminationshalbwertszeit (t1/2) beträgt 6-9 Tage. Die Blutkonzentration von Fingolimodphosphat geht in der terminalen Phase parallel zu der von Fingolimod zurück, was ähnliche Halbwertszeiten für die beiden Substanzen ergibt.
Nach oraler Gabe werden etwa 81% der applizierten Dosis als inaktive Metaboliten langsam im Urin ausgeschieden. Fingolimod und Fingolimodphosphat werden im Urin nicht intakt ausgeschieden, sind aber die primären Komponenten in den Fäzes, wobei die Mengen jeweils weniger als 2.5% der Dosis betragen. Nach 34 Tagen beträgt die Wiederfindung der applizierten Dosis 89%.
Linearität/Nicht Linearität
Nach mehrmaliger einmal täglicher Applikation einer Fingolimod-Dosis von 0.5 mg oder 1.25 mg steigen die Konzentrationen von Fingolimod und Fingolimodphosphat scheinbar proportional zur Dosis an.
Nach mehrmaliger einmal täglicher Gabe einer Fingolimod-Dosis von 0.25 mg oder 0.5 mg steigt bei Kindern und Jugendlichen die Konzentration von Fingolimod Phosphat offenbar proportional zur Dosis an.
Kinetik spezieller Patientengruppen
Ethnische Zugehörigkeit
Die Auswirkungen der ethnischen Zugehörigkeit auf die Pharmakokinetik von Fingolimod und Fingolimodphosphat sind klinisch nicht relevant.
Geschlecht
Das Geschlecht hat keinen Einfluss auf die Pharmakokinetik von Fingolimod und Fingolimodphosphat.
Leberfunktionsstörungen
Die Pharmakokinetik einer Einzeldosis Fingolimod (1 oder 5 mg) war bei Patienten mit leichter, moderater und schwerer Leberinsuffizienz (Child-Pugh Klasse A, B und C) hinsichtlich des Cmax-Werts von Fingolimod unverändert, jedoch erhöhte sich der AUC-Wert um 12%, 44% bzw. 103%. Die scheinbare Eliminationshalbwertszeit bleibt bei leichter Leberinsuffizienz unverändert, verlängert sich aber bei moderater und schwerer Leberinsuffizienz um 49-50%. Bei Patienten mit schwerer Leberinsuffizienz (Child-Pugh Klasse C) wurde von Fingolimodphosphat der Cmax um 22% reduziert und der AUC-Wert um 38% erhöht. Die Pharmakokinetik von Fingolimodphosphat wurde bei Patienten mit leichter oder mittelschwerer Leberinsuffizienz nicht bestimmt.
Nierenfunktionsstörungen
Bei schwerer Niereninsuffizienz erhöhen sich Cmax und AUC von Fingolimod um 32% bzw. 43% und Cmax und AUC von Fingolimodphosphat um 25% bzw. 14%. Die scheinbare Eliminationshalbwertszeit beider Analyten bleibt unverändert. Bei niereninsuffizienten Patienten sind keine Anpassungen der Fingolimod Devatis-Dosis erforderlich.
Ältere Patienten
Der Eliminationsmechanismus und die Ergebnisse aus den populationspharmakokinetischen Untersuchungen legen nahe, dass bei älteren Patienten keine Dosisanpassung erforderlich wäre. Allerdings sind die klinischen Erfahrungen bei Patienten über 55 Jahren begrenzt.
Kinder und Jugendliche
Die Konzentration von Fingolimod-Phosphat im Steady-State ist bei Kindern und Jugendlichen niedriger als bei Erwachsenen.
Die Sicherheit und Wirksamkeit von Fingolimod bei Kindern und Jugendlichen unter 10 Jahren ist nicht untersucht worden.
Präklinische DatenDas vorklinische Sicherheitsprofil von Fingolimod wurde in Mäusen, Ratten, Hunden und Affen untersucht. Die Hauptzielorgane waren das lymphoide System (Lymphopenie und lymphoide Atrophie), Lunge (erhöhtes Gewicht, Glattmuskelhypertrophie am bronchioalveolären Übergang) und Herz (negativer chronotroper Effekt, Blutdruckanstieg, perivaskuläre Veränderungen und Degeneration des Herzmuskels) bei mehreren Arten; Blutgefässe (Vaskulopathie) nur bei der Ratte und Hypophyse, Vormagen, Leber, Nebennieren, Magendarmtrakt und Nervensystem nur bei hohen Dosen (häufig in Zusammenhang mit Anzeichen einer allgemeinen Toxizität) bei mehreren Arten.
Mutagenität und Kanzerogenität
Fingolimod war in einem Ames-Test und in einer L5178Y-Mauslymphomzelllinie in vitro nicht mutagen. In V79-Lungenzellen des Chinesischen Hamsters ergaben sich in vitro keine klastogenen Effekte. In V79-Zellen induzierte Fingolimod bei Konzentrationen von 3.7 µg/ml und höher numerische chromosomale Aberrationen (Polyploidie). Im Mikrokerntest bei Mäusen und Ratten in vivo war Fingolimod nicht klastogen.
In einem 2-jährigen Bioassay bei der Ratte mit oralen Fingolimoddosen bis zur maximal verträglichen Dosis von 2.5 mg/kg, was ausgehend von dem AUC-Wert der systemischen Exposition des Menschen bei Gabe der 0.5-mg-Dosis einer ungefähr 50-fach höheren Grenze entspricht, ergaben sich keine Hinweise auf Karzinogenität. In einer über 2 Jahre laufenden Studie bei Mäusen wurde in einer Dosis ab 0.25 mg/kg, was ausgehend von dem AUC-Wert der systemischen Exposition des Menschen bei täglicher Gabe einer 0.5-mg-Dosis einer ungefähr 6-fach höheren Grenze entspricht, ein Anstieg der Inzidenz maligner Lymphome festgestellt.
Reproduktionstoxizität
Fingolimod hatte bis zur höchsten getesteten Dosis (10 mg/kg) (ausgehend von dem AUC-Wert der systemischen Exposition des Menschen bei Gabe der 0.5-mg-Dosis entspricht dieser Wert einer ungefähr 150-fach höheren Grenze) weder Auswirkungen auf die Spermienzahl/-motilität noch auf die Fruchtbarkeit männlicher und weiblicher Ratten.
Bei Applikation in einer Dosis von 0.1 mg/kg oder höher war Fingolimod bei Ratten teratogen. Zu den häufigsten viszeralen Fehlbildungen beim Fetus zählten persistierender Truncus arteriosus und Ventrikelseptumdefekt. Bei Dosen von 1 mg/kg und höher wurde ein Anstieg der Häufigkeit des Verlusts von Feten nach der Implantation beobachtet. Bei Dosen von 3 mg/kg wurde ein Rückgang der Zahl der lebensfähigen Feten festgestellt. Bei Kaninchen war Fingolimod nicht teratogen, wobei sich bei Dosen von 1.5 mg/kg und höher ein Anstieg der embryofetalen Mortalität einstellte sowie bei 5 mg/kg ein Rückgang der Zahl der lebensfähigen Feten und ein verzögertes Wachstum der Feten.
Bei der Ratte war bei Dosen, die beim Muttertier keine Toxizität verursachten, das Überleben der F1-Nachkommen in der frühen nachgeburtlichen Phase verringert. Die Behandlung mit Fingolimod hatte jedoch keinen Einfluss auf das Körpergewicht, die Entwicklung, das Verhalten und die Fruchtbarkeit der F1-Nachkommen.
Fingolimod wurde während der Laktation in die Milch behandelter Tiere ausgeschieden. Bei trächtigen Kaninchen passierten Fingolimod und seine Metabolite die Plazentaschranke.
Tierexperimentelle Studien an Jungtieren
Ergebnisse aus zwei Toxizitätsstudien an jungen Ratten zeigten geringgradige Auswirkungen auf die Knochenmineraldichte und die neurobehaviorale Reaktion sowie eine leichte Verzögerung der sexuellen Reifung und eine geringgradige Verminderung der Immunantwort auf wiederholte Stimulationen mit Schlitzschnecken-Hämocyanin (KLH). Diese Effekte wurden nicht als unerwünschte Ereignisse angesehen. Insgesamt waren die behandlungsbedingten Wirkungen von Fingolimod bei Jungtieren vergleichbar mit denen, die bei erwachsenen Ratten in ähnlicher Dosierung beobachtet wurden, mit Ausnahme des Fehlens einer Hypertrophie der glatten Muskulatur in den Lungen der jungen Ratten. Die NOAEL (no observed adverse effect level)-Werte bei Jungtieren wurden hauptsächlich durch unspezifische Wirkungen auf das Körpergewicht oder den Futterverzehr und weniger durch offenkundige Toxizität bestimmt.
Sonstige HinweiseInkompatibilitäten
Nicht zutreffend.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30°C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer68406 (Swissmedic)
PackungenKapseln zu 0.25 mg: 7 und 28 [B].
Kapseln zu 0.5 mg: 7, 28, 84 und 98 [B].
ZulassungsinhaberinDevatis AG, 6330 Cham.
Stand der InformationDezember 2024.
|